

Abstract
Dehalococcoides mccartyi are obligate organohalide-respiring bacteria that play an important detoxifying role in the environment. They have small genomes (~1.4 Mb) with a core region interrupted by two high plasticity regions (HPRs) containing dozens of genes encoding reductive dehalogenases involved in organohalide respiration. The genomes of eight new strains of D. mccartyi were closed from metagenomic data from a related set of enrichment cultures, bringing the total number of genomes to 24. Two of the newly sequenced strains and three previously sequenced strains contain CRISPR-Cas systems. These D. mccartyi CRISPR-Cas systems were found to primarily target prophages and genomic islands. The genomic islands were identified either as integrated into D. mccartyi genomes or as circular extrachromosomal elements. We observed active circularization of the integrated genomic island containing vcrABC operon encoding the dehalogenase (VcrA) responsible for the transformation of vinyl chloride to non-toxic ethene. We interrogated archived DNA from established enrichment cultures and found that the CRISPR array acquired three new spacers in 11 years. These data provide a glimpse into dynamic processes operating on the genomes distinct to D. mccartyi strains found in enrichment cultures and provide the first insights into possible mechanisms of lateral DNA exchange in D. mccartyi.
Subject terms: Environmental microbiology, Metagenomics
Introduction
Dehalococcoides mccartyi are hydrogen-utilizing, obligate organohalide-respiring bacteria. They are of interest because of their unique dehalogenating metabolism catalyzed by highly specific reductive dehalogenase enzymes that have widespread application in bioremediation and detoxification [1]. D. mccartyi are remarkably small (diameter <500 nm) disk-shaped, strictly anaerobic microbes belonging to the Chloroflexi [1] that are difficult to isolate and have not been successfully grown on agar media. They have highly streamlined genomes under 1.4 Mbp, while individual strains have been shown to contain up to 36 reductive dehalogenase homologous genes (rdhAB) some of which are known to be used for organohalide respiration. Many genomes contain contiguous regions or clusters of genes that appear to have been acquired horizontally and are referred to as genomic islands (GIs) [2]. McMurdie et al. (2011) identified the first D. mccartyi genomic island carrying the vcrABC operon coding for a functional vinyl chloride (VC) reductase [3]. The VcrA enzyme catalyzes the conversion of VC to non-toxic ethene and is critical for effective clean-up of sites contaminated with chlorinated ethenes. The vcrA gene is the target of monitoring tools to assess remediation progress during in situ bioremediation [4, 5]. Previous analysis of D. mccartyi genomes revealed the presence of two high plasticity regions (HPRs) flanking the origin, separated by a highly conserved core region common to all strains [6]. These HPRs contain a majority of a strain’s rdhAB genes. Lateral transfer of these reductive dehalogenase genes is thought to be a fundamental ecological strategy used by D. mccartyi [6, 7] to adapt to naturally occurring and anthropogenic halogenated compounds, although no mechanism has been identified. Many D. mccartyi genomes harbor prophages that may contribute to the spread of these reductive dehalogenase genes between different strains via lateral gene transfer. Some strains of D. mccartyi also encode CRISPR-Cas (clustered regularly interspaced short palindromic repeats—CRISPR associated) systems involved in adaptive defense mechanisms that protect the host from invading mobile elements including plasmids, phages, and transposons [8–10]. The role of phages and CRISPR-Cas systems in facilitating or blocking lateral transfer of rdhAB genes among D. mccartyi strains has never been investigated.
CRISPR-Cas adaptive immune systems are encoded by a genetic locus that is comprised of one or more CRISPR arrays and several CRISPR associated (cas) genes. They are divided into two main classes, six types and 21 subtypes based on the cas genes present. The CRISPR arrays contain identical 21–48 base pair (bp) repeats interspaced by unique spacer sequences (26–76 bp), some of which are homologous to sequences in mobile genetic elements. The Cas proteins are involved in all stages of CRISPR immunity, including adaptation, maturation, and interference [8]. During adaptation, fragments of an invading genetic element, known as spacers, are added to the CRISPR array. In maturation, the CRISPR array is first transcribed into pre-CRISPR RNA and then separated into short pieces each containing an individual recognition sequence between repeats (crRNAs) [11]. During interference, Cas proteins and crRNAs form effector complexes that use the crRNAs as a guide to target and destroy invading DNA in a sequence-specific manner [10, 12]. To date the function and targets of D. mccartyi CRISPR-Cas systems have not been described.
A D. mccartyi-containing enrichment culture, referred to as KB-1, has been used commercially for the past 15 years to bioaugment sites contaminated with chlorinated ethenes to accelerate detoxification [13–18]. The KB-1 mixed culture was originally enriched from contaminated soil, and contains multiple strains of D. mccartyi that couple growth to the sequential dechlorination of tetrachloroethene (PCE) and trichloroethene (TCE) via cis-dichloroethene (cDCE) and VC to non-toxic ethene [19]. KB-1 can also couple growth to the dechlorination of 1,2-dichloroethane (1,2-DCA) to ethene [20]. We closed the genomes of eight new and distinct strains of D. mccartyi from established subcultures of the parent KB-1 culture enriched on different chlorinated electron acceptors [21]. Two strains were found to contain CRISPR-Cas systems. Sequence data suggested the existence of circular extrachromosomal elements, some of which appeared to contain reductive dehalogenase genes. The purpose of this study was to verify the existence of these circular extrachromosomal elements and their relationship with CRISPR-Cas systems of these strains. The CRISPR array was found to target prophage and circular extrachromosomal elements. The CRISPR array itself was found to adapt over time acquiring three new spacers over 11 years. Active circularization of extrachromosomal elements, particularly the island containing the vcrABC operon, was inferred from sequence data and confirmed by polymerase chain reaction (PCR), providing the first clues to a mechanism for lateral transfer of DNA in D. mccartyi. The existence of a circular form of the genomic island (GI) containing the vcrABC operon also explains the periodic observation of higher vcrA to 16S rRNA gene copies in DNA samples from field sites [22, 23]. We have gained new insights to lateral transfer of dehalogenase genes, discovered new mobile elements and their connection with CRISPR-Cas systems in D. mccartyi.
Materials and methods
Enrichment cultures
The KB-1 set of enrichment cultures originated from microcosms prepared with aquifer materials from a TCE-contaminated site in southern Ontario in 1996 as described previously [13]. The KB-1 parent enrichment culture, KB-1/TCE-MeOH, has been maintained with ~100 mg/L TCE as electron acceptor and methanol (MeOH) as electron donor, added at 5× the electron equivalents (eeq) required for complete dechlorination, as previously described [13, 14, 19]. The parent culture was used to inoculate several sub-cultures that were established between 2001 and 2003 and maintained on different chlorinated acceptors, including daughter products cDCE (KB-1/cDCE-MeOH) and VC (KB-1/VC-MeOH), as well as 1,2-DCA (KB-1/1,2-DCA-MeOH). For further details on culturing please refer to supplemental information.
Metagenomic sequencing and genome assembly
DNA for metagenome sequencing was extracted from larger samples (40–615 mL) taken from the four stable enrichment cultures described above (and in supplemental methods): KB-1/VC-H2 (40 mL culture sample), KB-1/TCE-MeOH (500 mL sample), KB-1/cDCE-MeOH (300 mL culture), and KB-1/1, 2-DCA-MeOH (615 mL sample). Extractions were conducted between February and May 2013. Cultures were filtered using Sterivex™ filters (Millipore 0.2 µm) and the DNA was extracted using the CTAB method (JGI bacterial genomic DNA isolation using CTAB protocol v.3). DNA was sequenced at the Genome Quebec Innovation Sequencing Center using Illumina HiSeq 2500 technology. Paired-end sequencing with an insert size of ~400 bp and read length of ~150 bp provided roughly 50 million reads per culture. Additional mate-pair sequencing with insert size of ~8000 bp and read length of ~100 bp was conducted for the KB-1/TCE-MeOH and KB-1/1, 2-DCA-MeOH cultures where we had more culture DNA available. A random subset of reads (30 million mate-pair and paired-end total) was quality trimmed using Trimmomatic [24]. D. mccartyi genomes were closed from metagenomic data using the same methodology described in Tang et al. (2012) [25]. In brief, reads were assembled using ABySS v. 1.3.2 [26] (for paired-end) and ALLPATHS-LG [27] (for paired-end with mate-pair). Several assemblies were used in order to maximize length of D. mccartyi contigs. Scaffolding of ABySS contigs was conducted using SSPACE v. 2.0 [28]. All gaps were resolved using an in-house automatic gap resolution program [25]. Each genome was polished by read mapping in Geneious v. 6.1 [29] with a 90% cut off for single nucleotide polymorphisms (SNPs). The origin of replication was identified using Oriloc in R [30]. Each genome was annotated using RAST [31], with hypothetical annotations resubmitted to BASys [32] some of which were assigned an annotation. Microbial Genomes Check from NCBI assisted in finding errors produced from automatic annotation. Results were manually inspected and corrected where required using Geneious ORF finder. NCBI automatically annotated the ref_seq version using PGAP [33]. Additional searches for conserved domains were conducted using NCBI conserved domain search (e-value threshold of 0.01). Multiple D. mccartyi genomes could be readily distinguished and closed from each of the four enrichment cultures because the different strains in each culture had different abundances (as measured by read depth). Two genomes were closed from the KB-1/VC-H2 culture. We named the D. mccartyi strains from this culture KBVC1 and KBVC2 using a naming convention indicating the electron acceptor of subculture of provenance (in this case VC) and rank abundance (number 1 for highest abundance and number 2 for the second most abundant strain). Three genomes were closed from KB-1/1, 2-DCA-MeOH enrichment culture and are referred to as strains KBDCA1, KBDCA2, and KBDCA3. Three genomes were closed from the KB-1/TCE-MeOH culture (KBTCE1, KBTCE2, and KBTCE3). Unassembled contigs indicated that additional low abundance strains whose genomes could not be closed were also present in these cultures. A detailed comparative analysis of all eight genomes is the subject of a separate manuscript [21]; this study focuses primarily on the two CRISPR-containing strains KBDCA3 and KBVC1 and associated extrachromosomal elements. All eight genome sequences are available in NCBI. Genome images were created using BRIG 0.95 [34] with coverage information from read-mapping in Geneious 8.1 (98% similarity only if paired read matched nearby). An overview of relevant KB-1 enrichment cultures and associated closed genomes, including NCBI accession numbers, rank abundance as inferred from read depth, number of rdhA genes identified, presence of prophage, and CRISPR-Cas is provided in Fig. 1.
Fig. 1.
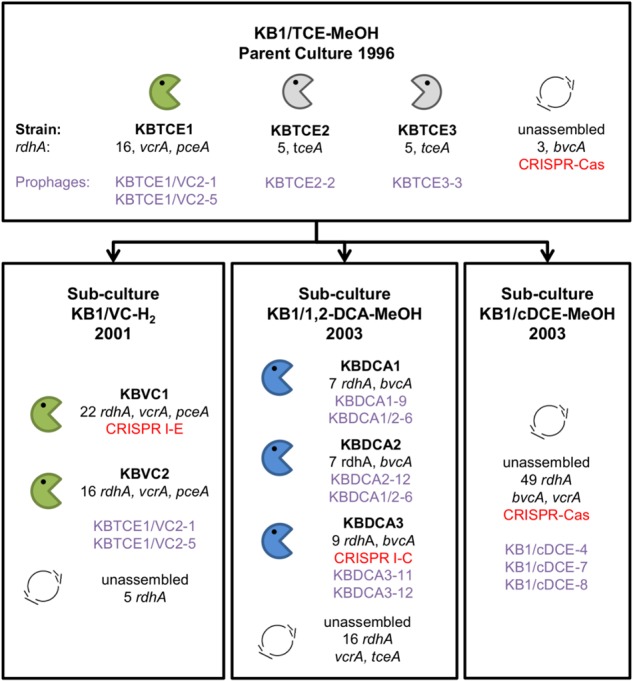
Overview of the eight Dehalococcoides mccartyi genomes closed from the metagenomes of KB-1 enrichment cultures. Enrichment sub-cultures are named by electron acceptor, electron donor, and the date the sub-culture was first created. Electron acceptors include trichloroethene (TCE), vinyl chloride (VC), 1,2-dichloroethane (1,2-DCA), and cis-dichloroethene (cDCE) and donors are either methanol (MeOH) or hydrogen. Closed D. mccartyi genomes were assigned a strain name based on electron acceptor and rank abundance in the mixed culture. The number of reductive dehalogenase homologous genes (rdhA) per strain genome is indicated and any rdhA that has been functionally characterized is listed by name (vcrA, pceA, tceA, and bvcA). Prophages identified in each genome are listed by name in purple. CRISPR-Cas systems are also indicated in red
PCR amplification of CRISPR-Cas array and sequencing
DNA samples dating back to 2002 had been taken periodically from the KB-1/VC-H2 culture and stored at −80 °C. DNA from samples archived from 2002 to 2012 was extracted using the UltraClean Soil DNA kit (Mo Bio Laboratories, Inc.) and DNA extracts were stored at −80 °C. After 2012, DNA was extracted using the PowerSoil DNA kit (Mo Bio Laboratories Inc.). Primers were designed to anneal just outside of the array region in the I-E type D. mccartyi CRISPR region using Geneious Pro 5.5.4 Primer Design Feature. Primers were searched against the NCBI nr bacteria database and against KB-1 metagenomes using Primer-BLAST to confirm that primers had one unique target. PCR reactions were amplified using a high-fidelity polymerase to increase speed and improve product quality (ThermoFisher Phusion high-fidelity DNA polymerase) using an MJ Research PTC-200 Peltier Thermal Cycler (98 °C for 30 s, 30 cycles of: 98 °C for 10 s, 65 or 59 °C for 30 s, 72 °C for 1 min, with a final extension of 72 °C for 5 min). PCR products were run on 1% TAE agarose gel to estimate product size. Additional primers were designed in order to sequence entire PCR products (all primers are in Supplemental Information Table S1). PCR products were sequenced (Sanger) at the SickKids Center for Applied Genomics (Toronto, Ontario).
Prophage sequence identification and CRISPR array sequence alignments
A nucleotide BLAST search of metagenomics contigs was conducted in order to identify all phage sequences from the metagenomes, closed genomes and D. mccartyi genomes in NCBI (BLASTN 2.2.29) by searching previously identified D. mccartyi prophage [35], and any prophage regions identified by RAST [31] in closed genomes. Contigs with BLAST hits were submitted to RAST for annotation. Prophage regions were defined using PHAST[36] and Phage Finder [37]. Additional annotation was carried out using our in-house Phage Annotation Toolkit (PAT), which uses HMMs and genomic position to accurately annotate the morphogenetic genes of phage genomes. The KB-1 metagenomic contigs were trimmed to only include putative prophage regions to create a KB-1 putative prophage database containing 12 new prophages. Other strains of D. mccartyi in the NCBI database were also searched for prophages and seven additional prophages were identified in the genomes of strains 195, 11a5, CG1, CG3, WBC-2, and BTF08. Prophages were named by strain of origin followed by a number in the case of multiple prophages. If the prophage sequence came from a KB-1 metagenome contig where strain name is unknown, the prophage was named by the culture it originated from followed by a number. The naming convention excludes the prophage found in strain 11a5 which is named pg11a5 first described in Zhao et al. (2017) [38].
The spacers from CRISPR arrays from five different D. mccartyi strains (KBVC1, KBDCA3, CBDB1, DCMB5, and GT) were aligned (BLASTN) against sequences from a database of D. mccartyi prophages (KB-1 and from NCBI genomes), against KB-1 metagenome contigs, and all D. mccartyi genomes available in NCBI (Discontiguous Megablast with max e-value of <0). If spacers hit regions of D. mccartyi genomes that were not prophages, those regions were further inspected to see whether the hit involved a putative mobilizable element. GIs were identified with the assistance of IslandViewer [39]. Islands were manually inspected for indicative features, such as sequence composition bias, location near a tRNA gene, flanked by direct repeats, and over-representation of mobility, virulence, phage-related, and unknown genes.
Construction of phylogenetic trees
A database of 227 NCBI cas1 genes was updated with all D. mccartyi cas1 genes pulled from NCBI (KBVC1, KBDCA3, GT, CBDB1, DCMB5 strains). Sequences were aligned using MUSCLE (MUSCLE plugin for Geneious 8.1.8) and a maximum likelihood (ML) tree was constructed with 100 bootstraps (RAxML plugin for Geneious 8.18 using Gamma BLOSUM62 substitution matrix and rapid bootstrapping with searching for best-scoring ML tree). Clades were highlighted using FigTree 1.4.2.
GIs from D. mccartyi whose sequences matched to CRISPR spacers were extracted and aligned, and a phylogenetic tree was made based on this alignment using the same technique as above. Prophage sequences were aligned using kalign and a phylogenetic tree was made from these sequences using the same technique as above. The genoplotR package in R 3.2.5 [40] was used to create figures from these alignments and ML trees.
Quantitative PCR (qPCR) and PCR to track vcrA genomic island (vcrA-GI)
The abundance of D. mccartyi genes was measured by qPCR using 16S rRNA gene primers Dhc1f and Dhc264r [41] and primers vcrA670f and vcrA440r targeting the vcrA gene [42]. All D. mccartyi have a single 16S rRNA gene per genome. Reactions were prepared in a PCR cabinet (ESCO Technologies, Gatboro, PA) and each qPCR reaction was run in triplicate. A concatenated DNA sequence comprised of four D. mccartyi gene fragments corresponding to the 16S rRNA gene and three partial reductive dehalogenase genes (including vcrA) was designed and synthesized (IDT technologies). Geneious 8.1.8 DNA fold feature was used to choose the best orientation of gene fragments to reduce DNA folding, especially where primers were expected to anneal. The synthesized plasmid (Figure S1, Additional methods in Supplemental Information) was cloned into Escherichia coli using Invitrogen TOP10 cells. This single concatenated gene plasmid served as the standard for qPCR targeting both 16S rRNA and vcrA genes to obtain accurate ratios of vcrA to 16S rRNA gene copies.
All qPCR analyses were conducted using a CFX96 real-time PCR detection system, with a C1000 Thermo Cycler (Bio-Rad Laboratories, Hercules, CA). Each 20 µL qPCR reaction was prepared in sterile UltraPure distilled water containing 10 µL of EvaGreen® Supermix (Bio-Rad Laboratories, Hercules, CA), 0.5 µL of each primer (forward and reverse, each from 10 μM stock solutions), and 2 μL of diluted template (DNA extract at 1:10 or standard plasmid dilution series). The thermocycling program was as follows: initial denaturation at 95 °C for 2 min, followed by 40 cycles of denaturation at 98 °C for 5 s, annealing at 60 °C followed by extension for 10 s at 72 °C. A final melting curve analysis was conducted at the end of the program. Calibration R2 values were 0.99 or greater and efficiencies were 80–110%.
PCR reactions were designed to amplify the vcrA-GI in either of two states (genomic or circular). The first set of primers targeted conserved genomic regions outside of the vcrA-GI yielding an amplicon of predicted length 19,361 bp (PCR 1). The second set of primers faced outwards from the vcrA gene targeting a circular version of the island, outward in both directions from the vcrA gene (PCR 2) of predicted length 10,426 bp. Primers were designed using Geneious 8.1.8 Primer Design feature. PCR reactions were performed using a high-fidelity polymerase (ThermoFisher Phusion) using an MJ Research PTC-200 Peltier Thermal Cycler. All reactions started with an initial denaturation at 98 °C for 30 s, followed by 30 cycles of 10 s of denaturation at 98 °C, primer-specific annealing and elongation at 72 °C, and a final extension of 5 min at 72 °C. For the genomic primers (PCR 1), the annealing temperature was 59.5 °C and elongation was 10 min at 72 °C. For the vcrA-to-vcrA primers (PCR 2), the annealing temperature was 65 °C and elongation was 2 min at 72 °C. PCR products were separated on 1% TAE agarose gel to estimate product size and check for non-specific amplifications. PCR products were sequenced (Sanger) at the SickKids Center for Applied Genomics (TCAG) sequencing/synthesis facility (Toronto, Ontario). The sequences of all qPCR, PCR and internal sequencing primers are provided in Table S1.
Nucleotide sequence accession numbers
KB-1 D. mccartyi closed genome nucleotide accession numbers: strain KBDCA1 CP019867, strain KBDCA2 CP019868, strain KBDCA3 CP019946, strain KBVC1 CP019968, strain KBVC2 CP19969, strain KBTCE1 CP01999, strain KBTCE2 CP019865, and strain KBCTCE3 CP019866.
Results and discussion
Two new CRISPR-containing D. mccartyi genomes
Metagenomes were sequenced from four related KB-1 mixed microbial cultures that have been grown and maintained since 2003 or earlier on TCE, cDCE, VC, or 1,2-DCA. The metagenomes obtained from these cultures enabled the closure of eight new D. mccartyi genomes as illustrated and named in Fig. 1. These closed genomes were found to contain as few as five, and as many as 22 reductive dehalogenase homologous (rdhA) genes (Table S2). Herein we will focus on two of the eight closed genomes that were found to contain CRISPR-Cas systems: strains KBVC1 and KBDCA3 (Figure S2). The genome of strain KBVC1 was found to contain 22 rdhA genes including a gene that encodes for the functionally characterized VC reductase, VcrA. The genome of strain KBDCA3 was found to contain only nine reductive dehalogenase genes, including a gene for a second functionally characterized VC reductase, BvcA [43].
D. mccartyi strains encode type I CRISPR-Cas systems
In addition to the genomes of strain KBVC1 and strain KBDCA3, the genomes of three previously described strains, namely GT [44], CBDB1 [45], and DCMB5 [46], also encode CRISPR-Cas loci, bringing the total to five out of 24 sequenced genomes (Table S2). The genomes of strains KBVC1, GT, and CBDB1 contain Class I type I-E systems, while strain KBDCA3 contains a Class I type I-C system. Strain DCMB5 was found to encode both type I-E and I-C CRISPR-Cas systems. The hallmark of the Class I CRISPR-Cas systems is the presence of a multi-subunit crRNA-effector complex. This ribonucleoprotein complex mediates the processing and interference stages of CRISPR-Cas activity. The signature gene for type I systems is cas3, which contains helicase and nuclease domains that unwind and cleave target DNA [47–49]. Both type I-E and I-C CRISPR-Cas subtypes in D. mccartyi, have cas3 genes, and the order and presence of other cas genes does not differ from common examples of I-E and I-C systems across many different bacterial genera [8, 48] (Figure S3).
In general, CRISPR-Cas systems are considered to be polyphyletic with many rearrangements and lateral gene transfer events occurring between organisms [48]. The type I-E and I-C CRISPR-Cas systems identified in D. mccartyi likely arose from independent acquisition events. This is especially evident in strain DCMB5, which contains both type I systems (Figure S4). Each system sub-type is well conserved among different strains. The type I-E cas genes from strains KBVC1, GT, CBDB1, and DCMB5 share 99.5% pairwise nucleotide identity, while the type I-C cas genes of KBDCA3 and DCMB5 share 99.6% pairwise nucleotide identity. The CRISPR arrays themselves show low sequence identity due to the high variability in the spacer sequences. The CRISPR arrays contain between 19 and 54 spacer sequences (Table S2 and Table 1). The unique sets of spacer sequences reflect adaptation of the CRISPR locus to the specific conditions experienced by each strain. We investigated the targets of CRISPR spacers in D. mccartyi to understand the nature and history of invasion by mobile genetic elements. We were able to identify likely targets of 45–98% of the CRISPR spacers in the six D. mccartyi CRISPR arrays. These spacers target primarily phages and GIs (Tables 1 and 2).
Table 1.
Summary of D. mccartyi CRISPR-Cas system targets
| D. mccartyi strain | CRISPR type | Number of spacers that match specific types of mobile DNAa | Total number of spacers | ||||
|---|---|---|---|---|---|---|---|
| Prophage | IME1 | Other IME | rdhA IME | Unknown | |||
| KBDCA3 | I-C | 15 | 9 | 0 | 3 | 10 | 37 |
| 41% | 24% | 0% | 8% | 27% | |||
| DCMB5 | I-C | 17 | 5 | 2 | 1 | 29 | 54 |
| 31% | 9% | 4% | 2% | 54% | |||
| DCMB5 | I-E | 18 | 3 | 2 | 1 | 4 | 28 |
| 64% | 11% | 7% | 4% | 14% | |||
| CBDB1 | I-E | 11 | 3 | 0 | 0 | 5 | 19 |
| 58% | 16% | 0% | 0% | 26% | |||
| GT | I-E | 18 | 3 | 2 | 1 | 13 | 37 |
| 49% | 8% | 5% | 3% | 35% | |||
| KBVC1 | I-E | 12 | 6 | 1 | 1 | 21 | 41 |
| 29% | 15% | 2% | 2% | 51% | |||
IME integrative and mobilizable element
aIME1 refer to type 1 IMEs as defined in the text, rdhA IME are IMEs that contain an rdhA gene
Table 2.
General features of mobile DNA found in Dehalococcoides mccartyi targeted by CRISPR-Cas system
| Type | Prophage/phage | Integrative and mobilizable elements (IMEs) | ||
|---|---|---|---|---|
| Class | Prophage/phage | IME1 | IME with rdhA gene | IME2 |
| Size (kbp) | 6–40 | 21–23 | 4–11 | 8–9 |
| GC content | 49% | 44% | 41% | 38% |
| Integration sites | At ~800 and ~300 kbp | tRNA-Ala, Ile or lys | Multiple tRNA or tmRNA | At ~780 kbp |
| Naming convention | strain name - number | IME1-number | IME-rdhA present | IME2-number |
| General description | Siphoroviridae type virus with lytic/lysogenic cycle. | Contain phage-like integration and excision machinery. Observed to replicate as a high copy circular element or integrated within genome. | Contain phage-like integration and excision machinery. Observed to replicate as a high copy circular element or integrated within genome. Carry a reductive dehalogenase (rdhA) gene. | Predicted genomic island. Targeted by CRISPR function unknown |
D. mccartyi CRISPR-Cas systems target phages and GIs
Phages were the most common target of D. mccartyi CRISPR spacers (Table 1 and Fig. 2). We identified a total of 20 prophage sequences in D. mccartyi genomes. We identified 12 new prophage sequences in the KB-1 cultures sequenced in this work, and seven additional sequences were found in published D. mccartyi genomes from other strains, available in NCBI. One had previously been identified in KB-1 (prophage KB/TCE-0) through an earlier sequencing project [35] and another in 11a5 [38]. In the current study, we found a prophage sequence (prophage KBTCE1/KBVC2-1) that has 99.9% pairwise nucleotide identity with the KB-1 prophage previously reported and is presumably the same prophage. The few nucleotide substitutions observed were all aggregated in one region of the prophage (Figure S5) where the phage tail proteins are encoded (Table S4). Phage tail fibers interact with the cell surface and thus are typically subject to stronger adaptive selection pressure than the rest of the phage genome [50]. D. mccartyi prophages appear to fall into three different groups based on open reading frames: (i) prophage sequences similar to Escherichia coli phage HK97; (ii) prophage sequences similar to Bacillus subtilus SPP1 phage; and (iii) hybrid prophages with HK97-like heads and SPP1 tails (Table S3). The CRISPR spacers found in strain KBVC1 had matches to all 12 prophages found in KB-1 D. mccartyi genomes, such that interference would be expected without the addition of new spacers. The spacers in strain KBDCA3 matched seven KB-1 prophage sequences (Fig. 2). An example of spacer match to a prophage sequence is depicted in Figure S6.
Fig. 2.

Maximum likelihood phylogenetic tree of prophages identified in D. mccartyi closed genomes including those from KB-1. Most likely tree of 100 bootstraps, scale indicates number of nucleotide substitutions per site. Spacer matches are highlighted with red bars (KBDCA3 I-C system) or blue bars (KBVC1 I-E system) with spacer number indicated below hit. Matches from all D. mccartyi spacers are provided in Table S3. Two incomplete and two highly similar D. mccartyi prophages were omitted but can be found in Table S4. Nucleotide sequences are provided as supplemental File S3. Sequences corresponding to prophage morphological proteins are abbreviated by name in first instance and subsequently color coded: ST small terminase, LT large terminase, PO portal, HP head protease, HD head decoration, MH major head, PC packaging chaperone, TC tail connector, TR tail terminator, TT tail tube, TG tail assembly chaperone, TM tail tape measure, HN HTH endonuclease, SA recombinase, DT tail protein, BH baseplate, HS head scaffold, 6F T6SS IcmF, RP replication protein, VV DSMS3 protein, IN phage integrase, CR HTH repressor, LM tail tip, PG PG hydrolase, C1 connector 1, EP encapsulin packaged protein, S4 PMR-associated domain, HC hcp1 Type VI SS
Eight of 41 spacers in the strain KBVC1 type I-E CRISPR-Cas system and 12 of 37 in the KBDCA3 type I-C system were found to match to regions of D. mccartyi genomes that appear to have been laterally acquired based on sequence information. These GIs are clearly distinct from prophage sequences because standard phage morphogenetic proteins are absent. What is common is the presence of genes coding for integration, excision, and replication. These types of GIs, often referred to as integrative and mobilizable elements or IMEs [51, 52], encode their own integration, excision, and replication proteins but rely on other helper proteins to be transferred outside of the cell. An alignment was created of all IMEs that were targeted by CRISPR-Cas systems (Fig. 3). This alignment revealed one group of IMEs, referred to as IME1, which were similar at the nucleotide and predicted protein level. Integration of IME1 sequences is site-specific, mediated by a tyrosine recombinase and insertion is either at tRNA-Ala (seen in strains BAV1, KBDCA2, KBTCE1), tRNA-Ile (strains KBVC1, KBDCA2, WBC-2, and KBDCA1) or tRNA-Lys (strain 195). IME1 in the chromosome is flanked by repeats. Spacers from all D. mccartyi CRISPR-Cas systems had matches to IME1 family sequences.
Fig. 3.

Maximum likelihood phylogenetic tree of integrative mobilizable elements (IMEs) targeted by KBVC1 and KBDCA3 CRISPR-Cas systems. KBVC1 CRISPR spacer matches as blue bars with spacer number underneath, KBDCA3 spacer matches as red bars. a D. mccartyi (Dmc) IME1 family and b other IMEs. Most likely tree of 100 bootstraps, scale indicates number of nucleotide substitutions per site. Coding regions are shown as arrows and are identified by name, or by number as follows: 1—transcriptional regulator; 2—phage antirepressor; 3—recombinase/integrase; 4—Phage DNA primase/polymerase; 6—DNA segregation protein; 7—transposase. A star (*) indicates flanking repeat region. See Table S3 for tabular version of spacer matches to targets. Gray arrows are hypothetical coding regions
Mobilization of IME1 sequences
Illumina mate-pair and paired-end sequencing data suggested that IME1 sequences are capable of mobilization. The IME1 found in strain KBCV1 (Figure S2) had twice the read depth as the rest of the genome. Furthermore, mate-pair and paired-end information mapped the reads back to the genome or back to itself. Searching through metagenomic contigs, we found IME1 sequences that had 700× and 900× higher read depth than the highest abundance D. mccartyi genome closed from that metagenome (Figure S7), indicating that IME1 sequences integrated into the genome can simultaneously occur as circular extrachromosomal elements. A review of the literature on IMEs revealed that such mobile elements have been observed to replicate autonomously using rolling circle replication or by hijacking integrative and conjugative element machinery [53] and that they can be either mutualistic or opportunistic [54]. D. mccartyi strain 11a5 is reported to carry a plasmid-like circular extrachromosomal element (eDhc6) [55], however this element was found as single copy per cell and is not targeted by any of the D. mccartyi CRISPR-Cas systems. The IMEs identified herein have no sequence or predicted protein similarity to sequences in this plasmid-like element (eDhc6) and thus appear to be the first example of CRISPR-targeted IMEs in D. mccartyi.
Genes found in D. mccartyi IME1 sequences
IME1 sequences (Fig. 3a) are the second most frequent target of CRISPR-Cas systems (Table 1). These IMEs have a 70.5% pairwise nucleotide identity when aligned and contain similar coding regions. They are 21–23-kbp long, with an average GC content of 44% (Fig. 3a). The most conserved protein that could be annotated is D. mccartyi integrase with 97.2% pairwise nucleotide identity, which is similar to integrase (dsiB) that is found on the vcrA-GI [7]. However, none contain rdhA homologous genes. Other protein sequences that could be identified in IME1 include a transcriptional regulator from either XRE, LexA, or Cro/IC family, a bifunctional primase/polymerase and a protein thought to be involved in DNA repair (Fig. 3a). Proteins that could not be annotated were checked for the presence of conserved domains to help predict their function. Transcriptional regulators from the XRE, LexA, or Cro/CI families are typically used by phages to regulate lytic growth [56–58]. Bifunctional primase/polymerase is related to the phage P4 family of primases/polymerases. These are encoded in a single multifunctional gene with primase, helicase, and specific DNA-binding activities, and are used by phage to replicate as plasmids. The P4 plasmid phage uses this type of primase/polymerase to replicate as a high copy plasmid in its host E. coli. P4 relies on helper phage P2 for morphological functions related to an infectious state, such as packaging and cell lysis [59–61]. Double-stranded DNA repair proteins, although used in the maintenance of genomes, can also be used by prokaryotes during homologous recombination [62, 63].
All IME1 sequences contain a putative bifunctional primase/polymerase which belongs to a large family of proteins, referred to as the “D5 protein superfamily”, containing five subfamilies (NCBI). The D. mccartyi IME1 primase/polymerase belongs to the subfamily PRK07078 along with 355 other proteins in NCBI without any citing publications at this time. Upon closer inspection of PRK07078 proteins found in other bacteria with closed genomes, the primase/polymerase protein was also found in the vicinity of the same predicted coding regions as a D. mccartyi IME1, including an integrase and transcriptional regulator. Functionally speaking, it is possible that other bacteria also have IME1-like sequences; noting that the similarities are found here at the amino acid level not the nucleotide level. IME1-like constructs were found in a wide range of different bacteria genomes, such as Ralstonia, Lysobacter, Desulfovibrio, Geobacter, and Nitrospira (supplemental Table S6 for full list). Twenty-five of 29 IME1-like sequences occurred directly adjacent to a complete phage, possibly functioning as a satellite phage much like P4 does in E. coli. Eight of 29 genomes also contained a CRISPR-Cas system, and two of eight targeted their own IME1-like sequences (Table S6). In Ralstonia solanacearum ([64] for review), the construct is present in different strains and targeted by CRISPR-Cas, which is the same case as D. mccartyi. This type of element possibly exists in a wide variety of bacteria, not just D. mccartyi, but has received little attention. The IME1s in D. mccartyi are important because of their similarity to the vcrA-genomic island and provide the first hint to the mechanism of lateral gene transfer in this species. We defined a naming scheme to identify the different IME1 sequences discovered in this research (Fig. 3a). The name begins with Dmc (for host D. mccartyi), IME1, followed by a number (Table S5).
Integrative and mobilizable elements that contain reductive dehalogenase genes are also targets of CRISPR-Cas systems
The CRISPR-Cas systems in D. mccartyi were found to primarily target prophages (Fig. 2) and IME1s (Fig. 3a; Table 1). However, a few spacer sequences were found to target GIs that contain reductive dehalogenase genes (Fig. 3b). The previously identified vcrA [7] and tceA [65] GIs were targeted, as well as GIs were newly identified in strains BAV1 (rdhA OG 24) and DCMB5 (rdhA OG 49), where OG refers to the ortholog group to which the RdhA sequence belongs, as defined by Hug et al.(2013) [66]. Several spacers were found to target the integrase gene, dsiB, which is similar to integrase found in IME1 sequences. Others were found to match regions unique to rdhA-GIs (Fig. 3b, Table S2), suggesting that these islands could be specifically targeted by CRISPR-Cas systems. It is interesting to note that strain KBDCA3 that contains bvcA has a CRISPR spacer (#9 Fig. 3b) that would be expected to interfere with the acquisition of the vcrA IME, and that no strain to date has both vcrA and bvcA in their genome. CRISPR-Cas systems are complex and may have many roles in the cell.
IME1s and the vcrA-GI both replicate via a circular intermediate
While assembling contigs from the metagenome of the KB-1/VC-H2 culture, we found that the vcrA-containing genomic island had twice the read depth than the rest of the genome (Fig. 4), similar to high read depths we found over IME1 regions in strain KBVC1 (Figure S2). Furthermore, half of the mate-pair and paired-end sequencing information linked the island to itself, suggesting the simultaneous occurrence of a circular intermediate and a chromosomal copy which would explain high read depth. To further investigate this possibility, triplicate bottles with sterile medium were inoculated with KB1/VC-H2 and grown to stationary phase with methanol, ethanol, and VC. The abundance of vcrA gene copies relative to 16S rRNA gene copies over three successive dechlorination cycles was measured using qPCR calibrated with a single concatenated gene plasmid standard to yield accurate relative abundances. In all three replicate culture bottles, we found that the ratio of vcrA to 16S rRNA gene copies was not constant, it varied from 1:1 to 2:1 repetitively (Fig. 5a and Figure S8). Amplification of vcrA-GI using genomic primers on either side of the island was always successful (PCR 1), indicating a stable single copy of the integrated island per genome (Fig. 5b). We were also able to obtain the PCR amplicon for the circular element using outward-facing primers (PCR 2, Fig. 5c), confirming that the element does exist in its circular form as well. The circular vcrA-GI was detected by PCR even when the ratio of vcrA to 16SrRNA was close to one, as measured by qPCR. It seems that a fraction of the D. mccartyi population is always producing circular vcrA-GI, and that endpoint PCR is sensitive enough to detect this—especially after 30 cycles as used in this study.
Fig. 4.

Illustration of the circularization of the vcrA genomic island (GI). PCR reactions used to verify sequencing results are shown targeting the vcrA genomic island integrated within the genome (PCR 1) or in circular form (PCR 2). Read depth observed over the vcrA island compared to the rest of the genome is shown in blue, where darker blue indicates read depth >120
Fig. 5.

Evidence of circularization of the vcrA genomic island. Panel a shows vinyl chloride (VC) and ethene concentrations (left axis), and gene abundances (right axis) as VC is dechlorinated to ethene in a KB-1 sub-culture. The culture was purged and re-fed on days indicated by black arrows. The gene copies per mL of culture of D. mccartyi 16S rRNA (Dhc, gray diamonds) and vinyl chloride reductase (vcrA, black squares) were tracked using qPCR, and the copy number ratio is shown above each point. Similar results were found in two additional replicate cultures (Figure S8). Panels b and c show the agarose gel images with amplification products generated using reactions PCR 1 and PCR 2 (illustrated in Fig. 4) on the same DNA samples (days indicated above gel) used for qPCR analyses shown in panel a. The PCR1 amplicon size (panel b) is always the expected 19,361 bp indicating one copy of the island integrated in the genome; this gel was run with the Lambda/HindII ladder (L). The PCR2 amplicon (panel c) confirms simultaneous detection of the circular vcrA genomic island with expected 10,426 bp fragment size; this gel was run with the 10 kbPlus ladder (L). Expected fragment sizes are indicated with black arrows. PCR products were verified by Sanger sequencing with sequences provided in supplemental File S4
Four observations could be made from the DNA analyses in these bottles: [1] the ratio of vcrA to D. mccartyi 16S copies was not associated with any particular time point during batch-style feeding dechlorination; [2] when the ratio was greater than 1, it is because of a higher number of copies of vcrA not a lower number copies of 16S rRNA genes; [3] vcrA copies doubled within a time span of 2–3 days or less, faster than typical D. mccartyi growth in these cultures; and [4] amplification of the circular state was successful. Sequence annotation cues suggest that the vcrA-GI is mobilizable [7] and several studies have reported the puzzling result of finding many more D. mccartyi vcrA copies than 16S rRNA gene copies [22, 23, 67] without an explanation other than PCR error, since vcrA has never been found in any species other than D. mccartyi. Periodic replication of the vcrA island as an independent extrachromosomal circular element now explains these observations. These data indicate that the vcrA-GI and possibly other rdhA-containing GIs can excise from the genome and circularize as integrative and mobilizable elements (IMEs).
Type I-E CRISPR-Cas is active in KB-1 and adapted to invading DNA over time
Through a combination of PCR and sequencing of archived frozen DNA, we were able to determine that the type I-E CRISPR-Cas system in strain KBVC1 actively recruited three new spacers over 11 years of maintenance of this batch culture in our laboratory (Fig. 6). Two spacers were added at the promoter region of the array, and one integrated in the middle of the array, which is less common but has been observed in other bacteria [68–70]. The new spacer occurring in the middle of the array had similarity (BLASTN) to a phage portal protein from a D. mccartyi prophage (Fig. 6). The second new spacer occurring at the promoter region of the array was a match to a D. mccartyi prophage capsid. We could not identify the target for the third spacer (Fig. 6).
Fig. 6.

Evidence for the extension of D. mccartyi KBVC1 CRISPR array and new targets acquired. a Agarose gel showing PCR product of CRISPR array from KB-1/VC-H2 amplified from DNA extracted over time. DNA extracted in different years was stored at −80 °C. Amplified products were sequenced with internal primers; primers used are provided in Table S1, and corresponding sequences in File S1. Expected 1725 bp PCR product produced (2013–2016). Smaller 1542 bp PCR product produced from 2002 DNA. b Schematic of partial CRISPR array in 2016 compared to 2002 indicating the addition of three new spacers as determined from sequencing of PCR products. Repeats are shown as black diamonds. Blue rectangles indicate spacer match to prophage. Yellow rectangles indicate spacer match to D. mccartyi IME1. Only 24 spacers of the 41 spacers are shown in the region where new spacers were added; cross-hatches indicate where the other spacer-repeat sequences occur. New spacers are identified by spacer number (1, 2, and 20). c New spacers have best matches to two different D. mccartyi prophages indicated with black arrows. Prophages are annotated using same abbreviations as in Fig. 3 and Table S4. All spacer sequences are in supplemental File S2
Further evidence to the functioning of the CRISPR-Cas system can be inferred from the sequences of multiple strains inhabiting the same enrichment cultures. The highest abundance strain (KBVC1) in the KB-1/VC-H2 enrichment culture contains a CRISPR-Cas system, while the lower abundance strain (KBVC2) does not. Strain KBVC2 without a CRISPR-Cas system contains two prophages (KBTCE1/KBVC2-1 and KBTCE1/KBVC2-5) in its genome, while strain KBVC1 does not, and its I-E CRISPR-Cas system has spacer matches to these prophages (Table S3, Fig. 2). Similarly, in the KB-1/1,2-DCA-MeOH enrichment culture, the prophage found in the two most abundant strains (KBDCA1/KBDCA2-6) is not found in the lower abundance strain KBDCA3 that harbors the I-C CRISPR-Cas system and a spacer match to this prophage (Table S3, Fig. 2).
Implications for lateral gene transfer in D. mccartyi
This analysis of the CRISPR-Cas system in D. mccartyi has led to the discovery of circular extrachromosomal elements, defined by the IME1 family that shares commonalities with vital rdhA-containing IMEs. Both IME1 and the vcrA-genomic island were found to replicate independently via a circular intermediate. Considering that all D. mccartyi genomes contain predicted comEA competence genes, it is possible that lateral gene transfer by transformation occurs in this genus. The mechanism by which D. mccartyi exports IME1 and rdhA-containing IMEs is yet to be determined, however all D. mccartyi genomes also harbor genes for Ftsk/SpoIIIE domain translocases involved in DNA export in other genera. Specifically, Actinobacteria use a single FtsK/SpoIIIE-like translocation protein to move double-stranded plasmids outside the cell [71–74]. Integrative and conjugative elements in Actinobacteria, once excised from the chromosome, also replicate autonomously before translocation [75, 76].
It is interesting to consider why CRISPR-Cas-containing D. mccartyi strains would reject incoming DNA that encodes new reductive dehalogenase genes that may be favorable to growth, especially since D. mccartyi is so reliant on these genes. Other bacteria that maintain CRISPR-Cas systems have also sometimes been found to target beneficial or conjugative DNA, suggesting that these systems are in a constant state of flux between beneficial or negative consequences of CRISPR-Cas maintenance [77–79]. From our strains, we did not find any relationships between CRISPR-Cas and strain abundance, suggesting that in our cultures at this time there is no clear benefit or cost. Bacteria may have other ways to resist phage predation, so the CRISPR system may not need to play a significant role thus may not be selected. One explanation for maintaining a CRISPR-Cas could be related to the energetic cost of IMEs, and the fact that our laboratory cultures are constantly maintained on the same chlorinated substrates and do not require new genes to respire them. Considering that the average genome size of D. mccartyi is 1.4 Mbp and some of these IMEs, such as IME1 are up to 23 kbp in length, if replicated six times (using an average of six from read depths of IMEs identified from metagenomic sequencing), D. mccartyi is expending ~8% of the energy required to replicate their entire genome on replicating IMEs. This energy requirement is even higher for strains that have more than one IME per genome.
The analysis of the CRISPR-Cas systems and mobile elements across a growing set of Dehalococcoides genomes has enabled the discovery of different types of actively replicating extrachromosomal elements and the demonstration of a growing CRISPR array. This study has begun to explain observations of higher ratios of vcrA and other rdhA genes to 16S rRNA gene copies in the experimental data and has expanded our understanding of population dynamics and mobile DNA in D. mccartyi, thus pointing toward avenues of future research to decipher mechanisms behind lateral gene transfer, especially of reductive dehalogenase genes.
Electronic supplementary material
Acknowledgements
Support was provided by the Government of Canada through Genome Canada and the Ontario Genomics Institute (no. 2009-OGI-ABC-1405 to E.A.E.), Natural Sciences and Engineering Research Council (NSERC) of Canada (Student Scholarships to O.M. and S.T. and Discovery grant funding to E.A.E.), and the Canadian Institutes for Health Research (MOP-136845 to K.L.M). Support was also provided by the Government of Ontario through the ORF-GL2 program and the Unites States Department of Defense through the strategic Environmental Research and Development Program (SERDP) under the contract (W912HQ-07-C-0036 project ER-1586).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41396-018-0254-2) contains supplementary material, which is available to authorized users.
References
- 1.Löffler FE, Yan J, Ritalahti KM, Adrian L, Edwards EA, Konstantinidis KT, et al. Dehalococcoides mccartyi gen. nov., sp. nov., obligately organohalide-respiring anaerobic bacteria relevant to halogen cycling and bioremediation, belong to a novel bacterial class, Dehalococcoidia classis nov., order Dehalococcoidales ord. nov. and family Dehalococcoidaceae fam. nov., within the phylum Chloroflexi. Int J Syst Evol Microbiol. 2013;63:625–35. doi: 10.1099/ijs.0.034926-0. [DOI] [PubMed] [Google Scholar]
- 2.Langille MGI, Hsiao WWL, Brinkman FSL. Detecting genomic islands using bioinformatics approaches. Nat Rev Microbiol. 2010;8:373–82. doi: 10.1038/nrmicro2350. [DOI] [PubMed] [Google Scholar]
- 3.Müller JA, Rosner BM, Von Abendroth G, Meshulam-Simon G, McCarty PL, Spormann AM. Molecular identification of the catabolic vinyl chloride reductase from Dehalococcodies sp. strain VS and its environmental distribution. Appl Environ Microbiol. 2004;70:4880–8. doi: 10.1128/AEM.70.8.4880-4888.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ritalahti KM, Hatt JK, Lugmayr V, Henn K, Petrovskis EA, Ogles DM, et al. Comparing on-site to off-site biomass collection for Dehalococcoides biomarker gene quantification to predict in situ chlorinated ethene detoxification potential. Environ Sci Technol. 2010;44:5127–33. doi: 10.1021/es100408r. [DOI] [PubMed] [Google Scholar]
- 5.Lee PK, Johnson DR, Holmes VF, He J, Alvarez-Cohen L. Reductive dehalogenase gene expression as a biomarker for physiological activity of Dehalococcoides spp. Appl Environ Microbiol. 2006;72:6161–8. doi: 10.1128/AEM.01070-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McMurdie PJ, Behrens SF, Müller JA, Goke J, Ritalahti KM, Wagner R, et al. Localized plasticity in the streamlined genomes of vinyl chloride respiring Dehalococcoides. PLoS Genet. 2009;5:e1000714. doi: 10.1371/journal.pgen.1000714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McMurdie PJ, Hug LA, Edwards EA, Holmes S, Spormann AA. Site-specific mobilization of vinyl-chloride respiration islands by a mechanism common in Dehalococcoides. BMC Genom. 2011;12:287–302. doi: 10.1186/1471-2164-12-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E, Horvath P, et al. Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol. 2011;9:467–77. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJJ. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci. 2009;34:401–7. doi: 10.1016/j.tibs.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Fineran PC, Gerritzen MJH, Suárez-Diez M, Künne T, Boekhorst J, van Hijum SAFT, et al. Degenerate target sites mediate rapid primed CRISPR adaptation. Proc Natl Acad Sci USA. 2014;111:E1629–38. doi: 10.1073/pnas.1400071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hatoum-Aslan A, Maniv I, Marraffini LA. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc Natl Acad Sci USA. 2011;108:21218–22. doi: 10.1073/pnas.1112832108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fineran PC, Charpentier E. Memory of viral infections by CRISPR-Cas adaptive immune systems: acquisition of new information. Virology. 2012;434:202–9. doi: 10.1016/j.virol.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 13.Duhamel M, Wehr SD, Yu L, Rizvi H, Seepersad D, Dworatzek S, et al. Comparison of anaerobic dechlorinating enrichment cultures maintained on tetrachloroethene, trichloroethene, cis-dichloroethene and vinyl chloride. Water Res. 2002;36:4193–202. doi: 10.1016/S0043-1354(02)00151-3. [DOI] [PubMed] [Google Scholar]
- 14.Duhamel M, Edwards EA. Growth and yields of dechlorinators, acetogens, and methanogens during reductive dechlorination of chlorinated ethenes and dihaloelimination of 1, 2-dichloroethane. Environ Sci Technol. 2007;41:2303–10. doi: 10.1021/es062010r. [DOI] [PubMed] [Google Scholar]
- 15.Major DJ, McMaster ML, Cox EE, Edwards EA, Dwortzek SM, Hendrickson ER, et al. Field demonstration of sucessful bioaugmentation to achieve dechlorination of tetrachloroethene to ethene. Environ Sci Technol. 2000;36:5106–16. doi: 10.1021/es0255711. [DOI] [PubMed] [Google Scholar]
- 16.Major DW, McMaster ML, Cox EE, Edwards EA, Dworatzek SM, Hendrickson ER, et al. Field demonstration of successful bioaugmentation to achieve dechlorination of tetrachloroethene to ethene. Environ Sci Technol. 2002;36:5106–16. doi: 10.1021/es0255711. [DOI] [PubMed] [Google Scholar]
- 17.Stroo HF, Leeson A, Ward CH. Bioaugmentation for Groundwater Remediation. New York, NY: Springer; 2012. [Google Scholar]
- 18.SiREM. http://www.siremlab.com. Accessed 14-June-2018.
- 19.Duhamel M, Mo K, Edwards EA. Characterization of a highly enriched Dehalococcoides-containing culture that grows on vinyl chloride and trichloroethene. Appl Environ Microbiol. 2004;70:5538–45. doi: 10.1128/AEM.70.9.5538-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waller AS, Krajmalnik-Brown R, Loeffler FE, Edwards EA. Multiple reductive-dehalogenase-homologous genes are simultaneously transcribed during dechlorination by Dehalococcoides-containing cultures. Appl Environ Microbiol. 2005;71:8257–64. doi: 10.1128/AEM.71.12.8257-8264.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molenda O, Tang S, Lomheim L, Edwards EA. Eight new genomes of organohalide-respiring Dehalococcoides mccartyi reveal evolutionary trends in reductive dehalogenases. 2018;bioRxiv 345173; 10.1101/345173.
- 22.Kocur CMD, Lomheim L, Molenda O, Weber KP, Austrins LM, Sleep BE, et al. Long-term field study of microbial community and dechlorinating activity following carboxymethyl cellulose-stabilized nanoscale zero-valent iron injection. Environ Sci Technol. 2016;50:7658–70. doi: 10.1021/acs.est.6b01745. [DOI] [PubMed] [Google Scholar]
- 23.van der Zaan B, Hannes F, Hoekstra N, Rijnaarts H, de Vos WM, Smidt H, et al. Correlation of Dehalococcoides 16S rRNA and chloroethene-reductive dehalogenase genes with geochemical conditions in chloroethene-contaminated groundwater. Appl Environ Microbiol. 2010;76:843–50. doi: 10.1128/AEM.01482-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang Shuiquan, Gong Yunchen, Edwards Elizabeth A. Semi-Automatic In Silico Gap Closure Enabled De Novo Assembly of Two Dehalobacter Genomes from Metagenomic Data. PLoS ONE. 2012;7(12):e52038. doi: 10.1371/journal.pone.0052038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–23. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gnerre SMI, Przybylski D, Ribeiro F, Burton J, Walker B, Sharpe T, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA. 2011;108:1513–8. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–9. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 29.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frank A, Lobry J. Oriloc: prediction of replication boundaries in unannotated bacterial chromosomes. Bioinformatics. 2000;16:566–7. doi: 10.1093/bioinformatics/16.6.560. [DOI] [PubMed] [Google Scholar]
- 31.Aziz Ramy K, Bartels Daniela, Best Aaron A, DeJongh Matthew, Disz Terrence, Edwards Robert A, Formsma Kevin, Gerdes Svetlana, Glass Elizabeth M, Kubal Michael, Meyer Folker, Olsen Gary J, Olson Robert, Osterman Andrei L, Overbeek Ross A, McNeil Leslie K, Paarmann Daniel, Paczian Tobias, Parrello Bruce, Pusch Gordon D, Reich Claudia, Stevens Rick, Vassieva Olga, Vonstein Veronika, Wilke Andreas, Zagnitko Olga. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genomics. 2008;9(1):75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Domselaar GH, Stothard P, Shrivastava S, Cruz JA, Guo A, Dong X, et al. BASys: a web server for automated bacterial genome annotation. Nucleic Acids Res. 2005;33:W455–W9. doi: 10.1093/nar/gki593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–24. doi: 10.1093/nar/gkw569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genom. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waller AS, Hug LA, Mo K, Radford DR, Maxwell KL, Edwards EA. Transcriptional analysis of a Dehalococcoides-containing microbial consortium reveals prophage activation. Appl Environ Microbiol. 2012;78:1178–86. doi: 10.1128/AEM.06416-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–W52. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fouts DE. Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006;34:5839–51. doi: 10.1093/nar/gkl732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao S, Ding C, He J. Genomic characterization of Dehalococcoides mccartyi strain 11a5 reveals a circular extrachromosomal genetic element and a new tetrachloroethene reductive dehalogenase gene. FEMS Microbiol Ecol. 2017;93:fiw235-fiw. doi: 10.1093/femsec/fiw235. [DOI] [PubMed] [Google Scholar]
- 39.Dhillon BK, Laird MR, Shay JA, Winsor GL, Lo R, Nizam F, et al. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 2015;43:W104–W8. doi: 10.1093/nar/gkv401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guy L, Kultima JR, Andersson SGE. genoPlotR: comparative gene and genome visualization in R. Bioinformatics. 2010;26:2334–5. doi: 10.1093/bioinformatics/btq413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hendrickson ER, Payne JA, Young RM, Starr MG, Perry MP, Fahnestock S, et al. Molecular analysis of Dehalococcoides 16S Ribosomal DNA from chloroethene-contaminated sites throughout North America and Europe. Appl Environ Microbiol. 2002;68:485–95. doi: 10.1128/AEM.68.2.485-495.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molenda O, Quaile AT, Edwards EA. Dehalogenimonas sp. strain WBC-2 genome and identification of its trans-dichloroethene reductive dehalogenase, TdrA. Appl Environ Microbiol. 2016;82:40–50. doi: 10.1128/AEM.02017-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krajmalnik-Brown R, Holscher T, Thomson IN, Saunders FM, Ritalahti KM, Löffler FE. Genetic identification of a putative vinyl chloride reductase in Dehalococcoides sp. strain BAV1. Appl Environ Microbiol. 2004;70:6347–51. doi: 10.1128/AEM.70.10.6347-6351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sung Y, Ritalahti KM, Apkarian RP, Löffler FE. Quantitative PCR confirms purity of strain GT, a novel trichloroethene-to-ethene-respiring Dehalococcoides isolate. Appl Environ Microbiol. 2006;72:1980–7. doi: 10.1128/AEM.72.3.1980-1987.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kube M, Beck A, Zinder SH, Kuhl H, Reinhardt R, Adrian L. Genome sequence of the chlorinated compound-respiring bacterium Dehalococcoides species strain CBDB1. Nat Biotech. 2005;23:1269–73. doi: 10.1038/nbt1131. [DOI] [PubMed] [Google Scholar]
- 46.Pöritz M, Goris T, Wubet T, Tarkka MT, Buscot F, Nijenhuis I, et al. Genome sequences of two dehalogenation specialists - Dehalococcoides mccartyi strains BTF08 and DCMB5 enriched from the highly polluted Bitterfeld region. FEMS Microbiol Lett. 2013;343:101–4. doi: 10.1111/1574-6968.12160. [DOI] [PubMed] [Google Scholar]
- 47.Gong B, Shin M, Sun J, Jung CH, Bolt EL, van der Oost J, et al. Molecular insights into DNA interference by CRISPR-associated nuclease-helicase Cas3. Proc Natl Acad Sci USA. 2014;111:16359–64. doi: 10.1073/pnas.1410806111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Micro. 2015;13:722–36. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huo Y, Nam KH, Ding F, Lee H, Wu L, Xiao Y, et al. Structures of CRISPR Cas3 offer mechanistic insights into Cascade-activated DNA unwinding and degradation. Nat Struct Mol Biol. 2014;21:771–7. doi: 10.1038/nsmb.2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Canchaya C, Proux C, Fournous G, Bruttin A, Brüssow H. Prophage genomics. Microbiol Mol Biol Rev. 2003;67:238–76. doi: 10.1128/MMBR.67.2.238-276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wozniak RA, Waldor MK. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat Rev Microbiol. 2010;8:552–63. doi: 10.1038/nrmicro2382. [DOI] [PubMed] [Google Scholar]
- 52.Roberts AP, Chandler M, Courvalin P, Guedon G, Mullany P, Pembroke T, et al. Revised nomenclature for transposable genetic elements. Plasmid. 2008;60:167–73. doi: 10.1016/j.plasmid.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daccord A, Ceccarelli D, Rodrigue S, Burrus V. Comparative analysis of mobilizable genomic islands. J Bacteriol. 2013;195:606–14. doi: 10.1128/JB.01985-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rankin DJ, Rocha EPC, Brown SP. What traits are carried on mobile genetic elements, and why? Heredity. 2011;106:1–10. doi: 10.1038/hdy.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao S, Ding C, He J. Genomic characterization of Dehalococcoides mccartyi strain 11a5 reveals a circular extrachromosomal genetic element and a new tetrachloroethene reductive dehalogenase gene. FEMS Microbiol Ecol. 2016;93:fiw235. [DOI] [PubMed]
- 56.McDonnell GE, McConnell DJ. Overproduction, isolation, and DNA-binding characteristics of Xre, the repressor protein from the Bacillus subtilis defective prophage PBSX. J Bacteriol. 1994;176:5831–4. doi: 10.1128/jb.176.18.5831-5834.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ibarra JA, Pérez-Rueda E, Carroll RK, Shaw LN. Global analysis of transcriptional regulators in Staphylococcus aureus. BMC Genom. 2013;14:126. doi: 10.1186/1471-2164-14-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barragán María J. L., Blázquez Blas, Zamarro María T., Mancheño José M., García José L., Díaz Eduardo, Carmona Manuel. BzdR, a Repressor That Controls the Anaerobic Catabolism of Benzoate in Azoarcus sp. CIB, Is the First Member of a New Subfamily of Transcriptional Regulators. Journal of Biological Chemistry. 2005;280(11):10683–10694. doi: 10.1074/jbc.M412259200. [DOI] [PubMed] [Google Scholar]
- 59.Tocchetti A, Galimberti G, Dehò G, Ghisotti D. Characterization of the oriI and oriII Origins of Replication in Phage-Plasmid P4. J Virol. 1999;73:7308–16. doi: 10.1128/jvi.73.9.7308-7316.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ziegelin G, Scherzinger E, Lurz R, Lanka E. Phage P4 alpha protein is multifunctional with origin recognition, helicase and primase activities. EMBO J. 1993;12:3703–8. doi: 10.1002/j.1460-2075.1993.tb06045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Briani F, Deho G, Forti F, Ghisotti D. The plasmid status of satellite bacteriophage P4. Plasmid. 2001;45:1–17. doi: 10.1006/plas.2000.1497. [DOI] [PubMed] [Google Scholar]
- 62.Rojowska A, Lammens K, Seifert FU, Direnberger C, Feldmann H, Hopfner KP. Structure of the Rad50 DNA double-strand break repair protein in complex with DNA. EMBO J. 2014;33:2847–59. doi: 10.15252/embj.201488889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ayora S, Carrasco B, Cardenas PP, Cesar CE, Canas C, Yadav T, et al. Double-strand break repair in bacteria: a view from Bacillus subtilis. FEMS Microbiol Rev. 2011;35:1055–81. doi: 10.1111/j.1574-6976.2011.00272.x. [DOI] [PubMed] [Google Scholar]
- 64.Peeters N, Guidot A, Vailleau F, Valls M. Ralstonia solanacearum, a widespread bacterial plant pathogen in the post-genomic era. Mol Plant Pathol. 2013;14:651–62. doi: 10.1111/mpp.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krajmalnik-Brown R, Sung Y, Ritalahti KM, Saunders FM, Löffler FE. Environmental distribution of the trichloroethene reductive dehalogenase gene (tceA) suggests lateral gene transfer among Dehalococcoides. FEMS Microbiol Ecol. 2007;59:206–14. doi: 10.1111/j.1574-6941.2006.00243.x. [DOI] [PubMed] [Google Scholar]
- 66.Hug LA, Maphosa F, Leys D, Löffler FE, Smidt H, Edwards EA, et al. Overview of organohalide-respiring bacteria and a proposal for a classification system for reductive dehalogenases. Philos Trans R Soc Biol Sci. 2013;368:1–10. doi: 10.1098/rstb.2012.0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maphosa F, Smidt H, de Vos WM, Röling WFM. Microbial community- and metabolite dynamics of an anoxic dechlorinating bioreactor. Environ Sci Technol. 2010;44:4884–90. doi: 10.1021/es903721s. [DOI] [PubMed] [Google Scholar]
- 68.Biswas A, Staals RHJ, Morales SE, Fineran PC, Brown CM. CRISPRDetect: a flexible algorithm to define CRISPR arrays. BMC Genom. 2016;17:356. doi: 10.1186/s12864-016-2627-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–12. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 70.Nuñez JK, Harrington LB, Kranzusch PJ, Engelman AN, Doudna JA. Foreign DNA capture during CRISPR–Cas adaptive immunity. Nature. 2015;527:535–8. doi: 10.1038/nature15760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ghinet MG, Bordeleau E, Beaudin J, Brzezinski R, Roy S, Burrus V. Uncovering the prevalence and diversity of integrating conjugative elements in Actinobacteria. PLOS One. 2011;6:e27846. doi: 10.1371/journal.pone.0027846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Poele EMt, Bolhuis H, Dijkhuizen L. Actinomycete integrative and conjugative elements. Antonie Leeuwenhoek. 2008;94:127–43. doi: 10.1007/s10482-008-9255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Possoz C, Ribard C, Gagnat J, Pernodet JL, Guerineau M. The integrative element pSAM2 from Streptomyces: kinetics and mode of conjugal transfer. Mol Microbiol. 2001;42:159–66. doi: 10.1046/j.1365-2958.2001.02618.x. [DOI] [PubMed] [Google Scholar]
- 74.Vogelmann J, Ammelburg M, Finger C, Guezguez J, Linke D, Flötenmeyer M, et al. Conjugal plasmid transfer in Streptomyces resembles bacterial chromosome segregation by FtsK/SpoIIIE. EMBO J. 2011;30:2246–54. doi: 10.1038/emboj.2011.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sezonov G, Duchene AM, Friedmann A, Guerineau M, Pernodet JL. Replicase, excisionase, and integrase genes of the Streptomyces element pSAM2 constitute an operon positively regulated by the pra gene. J Bacteriol. 1998;180:3056–61. doi: 10.1128/jb.180.12.3056-3061.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sezonov G, Hagege J, Pernodet JL, Friedmann A, Guerineau M. Characterization of pra, a gene for replication control in pSAM2, the integrating element of Streptomyces ambofaciens. Mol Microbiol. 1995;17:533–44. doi: 10.1111/j.1365-2958.1995.mmi_17030533.x. [DOI] [PubMed] [Google Scholar]
- 77.Jiang W, Maniv I, Arain F, Wang Y, Levin BR, Marraffini LA. Dealing with the evolutionary downside of CRISPR immunity: bacteria and beneficial plasmids. PLoS Genet. 2013;9:e1003844. doi: 10.1371/journal.pgen.1003844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Westra ER, Staals RHJ, Gort G, Høgh S, Neumann S, de la Cruz F, et al. CRISPR-Cas systems preferentially target the leading regions of MOBF conjugative plasmids. RNA Biol. 2013;10:749–61. doi: 10.4161/rna.24202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weinberger AD, Gilmore MS. CRISPR-Cas: to take up DNA or not, that is the question. Cell Host Microbe. 2012;12:125–6. doi: 10.1016/j.chom.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.