

Viruses are the most abundant biological entity on the planet and interact with microbial communities with which they associate. The virome of animals is often dominated by bacterial viruses, known as bacteriophages or phages, which can (re)structure bacterial communities potentially vital to the animal host. Beta diversity relationships of animal-associated bacterial communities in laboratory and wild populations frequently parallel animal phylogenetic relationships, a pattern termed phylosymbiosis. However, little is known about whether viral communities also exhibit this eco-evolutionary pattern. Metagenomics of purified viruses from recently diverged species of Nasonia parasitoid wasps reared in the lab indicates for the first time that the community relationships of the virome can also exhibit complete phylosymbiosis. Therefore, viruses, particularly bacteriophages here, may also be influenced by animal evolutionary changes either directly or indirectly through the tripartite interactions among hosts, bacteria, and phage communities. Moreover, we report several new bacteriophage genomes from the common gut bacteria in Nasonia.
KEYWORDS: Nasonia, Proteus, Providencia, Morganella, bacteriophage, microbiome, microbiota, phage, phylosymbiosis, virome
ABSTRACT
Phylosymbiosis was recently proposed to describe the eco-evolutionary pattern whereby the ecological relatedness (e.g., beta diversity relationships) of host-associated microbial communities parallels the phylogeny of the host species. Representing the most abundant biological entities on the planet and common members of the animal-associated microbiome, viruses can be influential members of host-associated microbial communities that may recapitulate, reinforce, or ablate phylosymbiosis. Here we sequence the metagenomes of purified viral communities from three different parasitic wasp Nasonia species, one cytonuclear introgression line of Nasonia, and the flour moth outgroup Ephestia kuehniella. Results demonstrate complete phylosymbiosis between the viral metagenome and insect phylogeny. Across all Nasonia contigs, 69% of the genes in the viral metagenomes are either new to the databases or uncharacterized, yet over 99% of the contigs have at least one gene with similarity to a known sequence. The core Nasonia virome spans 21% of the total contigs, and the majority of that core is likely derived from induced prophages residing in the genomes of common Nasonia-associated bacterial genera: Proteus, Providencia, and Morganella. We also assemble the first complete viral particle genomes from Nasonia-associated gut bacteria. Taken together, results reveal the first complete evidence for phylosymbiosis in viral metagenomes, new genome sequences of viral particles from Nasonia-associated gut bacteria, and a large set of novel or uncharacterized genes in the Nasonia virome. This work suggests that phylosymbiosis at the host-microbiome level will likely extend to the host-virome level in other systems as well.
IMPORTANCE Viruses are the most abundant biological entity on the planet and interact with microbial communities with which they associate. The virome of animals is often dominated by bacterial viruses, known as bacteriophages or phages, which can (re)structure bacterial communities potentially vital to the animal host. Beta diversity relationships of animal-associated bacterial communities in laboratory and wild populations frequently parallel animal phylogenetic relationships, a pattern termed phylosymbiosis. However, little is known about whether viral communities also exhibit this eco-evolutionary pattern. Metagenomics of purified viruses from recently diverged species of Nasonia parasitoid wasps reared in the lab indicates for the first time that the community relationships of the virome can also exhibit complete phylosymbiosis. Therefore, viruses, particularly bacteriophages here, may also be influenced by animal evolutionary changes either directly or indirectly through the tripartite interactions among hosts, bacteria, and phage communities. Moreover, we report several new bacteriophage genomes from the common gut bacteria in Nasonia.
INTRODUCTION
Ecological similarity of host-associated microbial communities between species can often mirror phylogenetic similarity of hosts across a wide range of animal taxa (1–6). This eco-evolutionary pattern, termed phylosymbiosis (1, 7), can arise from a variety of biotic or abiotic factors. Resultantly, phylosymbiosis does not a priori presume stable or long-term, transgenerational associations between microbial communities and their hosts. Phylosymbiosis may change with environments, lifestyles, or multipartite interactions that shift assembly of microbial communities. For example, phages (i.e., bacteriophages; viruses that infect bacteria) can outnumber bacteria in both free-living and host-associated communities (8, 9), represent the majority of viruses within animal microbiomes (8, 10–13), and may drive or ablate bacterial phylosymbiosis as they prey on bacteria.
A phage can exhibit two main life cycles: lytic and temperate. A lytic phage infects its bacterial host and immediately replicates and lyses the bacterial cell. A temperate phage, however, can integrate into and replicate as part of the bacterial genome until a biotic or abiotic trigger causes it to excise and enter the lytic cycle. In mammalian host-associated phage communities, the temperate life cycle dominates (10, 14–16), presumably due to environmental parameters such as host density (17) and mucosal tissue structure (18). Phage integration into animal-associated bacterial genomes (i.e., prophage) can alter the phenotype of the host bacterium through lysogenic conversion (19, 20), as well as enhance biofilm formation and thereby horizontal gene transfer among co-occurring bacteria (21, 22). The prevalence of temperate phages in host-associated microbiomes suggests that these phages may more intimately evolve with their bacterial hosts and/or shape the composition of the bacterial communities. Additionally, the discovery of intraspecific and interspecific core viromes dominated by phages across animal systems is often reflective of the core bacterial communities described in these same organisms (11, 23–25). Although it has been suggested previously (24), phylosymbiosis at the viral level has yet to be explicitly demonstrated, and evidence for this tripartite association pattern could underpin new ecological and functional interactions between an animal host, its bacterial community, and the viruses infecting both.
RESULTS
Virome samples and assemblies.
Viral purifications from adults of three species of Nasonia, a Nasonia introgression line, and the Mediterranean flour moth Ephestia kuehniella were sequenced. Each of the pure Nasonia species (N. vitripennis, N. longicornis, and N. giraulti) maintains their natural Wolbachia infections from supergroup A. The introgression line IntG has the genome of N. giraulti and the cytoplasm of N. vitripennis, including the maternally inherited supergroup A Wolbachia strain wVitA from N. vitripennis (26). E. kuehniella harbors a supergroup B Wolbachia strain named wCauB (27). Viral particle sequencing and single sample assembly statistics are outlined in Table S1 in the supplemental material.
Assembly statistics. Download Table S1, PDF file, 0.05 MB (52.5KB, pdf) .
Copyright © 2018 Leigh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylosymbiosis: host relatedness reflects compositional similarity of viral metagenomes.
Phylosymbiosis describes a significant host phylogenetic signal on host-associated microbiome communities (1). Bacterial communities frequently, but not universally, exhibit this relationship under wild and laboratory conditions (1, 7). For viromes, there is no a priori reason to expect that phylosymbiosis will occur because inducible proviruses and/or lytic viruses, i.e., the targets of this study, may constitute a small subset of the total viral DNA in bacterial and eukaryotic genomes, and active viral particles have the potential to lyse and shift bacterial communities that may disrupt phylosymbiosis. Here we evaluate if the Nasonia viromes form phylosymbiotic community relationships.
The phylogeny of Nasonia spp. rooted with the outgroup E. kuehniella is based on DNA sequences of the cytochrome oxidase I (COI) gene as previously shown (1, 7, 28–31). It resulted in the same branching pattern as the dendrogram generated from Bray-Curtis beta diversity of the viral metagenomes across the host species (Fig. 1). The matching cluster and Robinson-Foulds tree metrics were utilized to calculate host phylogenetic and virome dendrogram topological congruence, which is highly significant based on both metrics with 100,000 randomly bifurcating trees to simulate stochastic virome assembly (1) (P value = 0.00451). Additionally, using the same methodology, matching cluster and Robinson-Foulds metrics were evaluated by the Binary Jaccard beta diversity index, which produced identical results using viral presence and absence within each sample. Taken together, these findings comprise one of the first lines of evidence for phylosymbiosis in host-associated viral communities. We next evaluated the number and types of viruses that comprise these phylosymbiotic communities.
FIG 1.

Phylosymbiosis occurs between insects and their viral communities. The host phylogeny is constructed with PHYML from 385 bp of the cytochrome oxidase I gene, and the UPGMA hierarchical cluster relationships of the viromes are based on Bray-Curtis beta diversity distances. Significance of topological congruence was determined using a previously described method (1) based on the rooted Robinson-Foulds (P value: 0.00451) and rooted matching cluster (P value: 0.00451) metric with a total of 100,000 randomized topologies simulating a null hypothesis of stochastic virome ecological assembly.
Characterizing the Nasonia virome: host genetic effects, the virome core, and toxins.
Unlike many environmental viral metagenomes, the majority of the viral contigs from the insects studied here had at least one gene with BLASTx similarity to either known lytic viruses or genes from their potential respective hosts. An average of 30.9% of the genes identified in each of the samples have a predicted annotation and function (Fig. 2A). Therefore, to identify groups of proteins independent of the database annotations, unique protein clusters, defined as groups of proteins with significant sequence similarity (>70%), were determined in each of the samples by the protein clustering tool vContact (Fig. 2B). The protein cluster networks identified N. giraulti and IntG as the most diverse viromes, which share a N. giraulti genetic background but vary in the origin of their cytotype. This result suggests that host genotype rather than cytotype more strongly impacts diversity of the host-associated viral metagenome, either through interactions with phage directly or through interactions with the bacteria harboring these phages. N. longicornis and N. vitripennis yielded approximately 50% fewer unique protein groups in their viromes.
FIG 2.

Nasonia species harbor a modest core virome. (A) Percent viral contigs with at least one functionally annotated gene as determined by Pfam analysis. (B) Viral protein cluster analysis illustrating diversity of viral proteins within each virome. Each dot represents a unique viral protein and connecting lines indicate >70% sequence similarity between two proteins. (C) Venn diagram illustrating the viral contigs unique within and shared between the Nasonia species. (D) Taxonomic affiliation of the 219 members of the identified core virome as determined by BLASTx against the nr database. Shading indicates the relative abundance of each member within single viromes and was determined by read mapping to viral contigs.
To assess identity and diversity of proteins with predicted function in the viromes, contigs from all samples were compared to the protein family (Pfam) database. Each Nasonia species virome maintained a small, host-specific set of Pfams ranging from 6.7% to 14.8% of the Pfams (Table S2). Precisely 24.4% of the Pfams (173) were shared among all of the Nasonia samples, which parallels the 21% of the total contigs described as the core virome below. Across all species, the most abundant Pfam (4.7% of total Pfam predictions) was the helix-turn-helix (HTH) DNA-binding motif (PF01381) followed by the phage integrase Pfam (PF00589, 2.9% of total Pfam predictions).
Pfam assignments in viral metagenomes. Download Table S2, PDF file, 0.3 MB (288.1KB, pdf) .
Copyright © 2018 Leigh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To further explore the protein content of the viromes and the interactions that could underpin phylosymbiosis between hosts and their viromes, we assessed if domains similar to known toxins or domains that interact with eukaryotic hosts were present in these viruses using the Pfam annotations. Proteins identified as toxins and eukaryotic-interacting domains span immunoglobulin peptidases, virulence genes, lysins, and others (indicated by boldface in Table S2). Domains identified within these groups were found in viral contigs isolated from N. giraulti and IntG where 36 and 34 unique identifiable toxin and eukaryotic-interacting proteins spanned 0.045% and 0.067% of the total contigs, respectively. N. vitripennis and N. longicornis maintained 17 and 25, which spanned 0.025% and 0.098% of the contigs, respectively. One identified domain is the hemolysin-encoding XhlA (PF10779) detected in Bacilli class-associated contigs in all of the samples, which was also the most abundant in the N. giraulti and the IntG introgression samples. This family of hemolysins, first observed in the entomopathogenic Xenorhabdus nematophila, notably lyses insect immune cells (32).
Next, core viral contigs shared among all samples were determined by read mapping to the assembled contigs using the iVirus pipeline (33). Across the Nasonia samples, the core was comprised of 219 viral contigs or 21% of total Nasonia viral contigs (Fig. 2C). Of these core viral contigs, the majority (84%) are homologous to members infecting species of the most abundant bacterial genera found within the Nasonia gut microbiome: Morganella, Proteus, and Providencia (Fig. 2D). Additionally, 14 of the core viral contigs are homologous to sequences from the Bacilli class, all of which are relatively more abundant in N. giraulti and IntG. Two core viral contigs showed amino acid similarity to sequences in the genome of the entomopathogenic Xenorhabdus innexi (34); they contain phage structural genes typical of active phage particles. Additionally, the complete genome of wVitA phage WO, a prophage of the obligate intracellular bacterium Wolbachia that infects each of these aforementioned Nasonia species (35), was detected only in N. vitripennis. The genome of this prophage was described previously (36) and produces viral particles as seen in transmission electron microscopy in N. vitripennis (35).
Viral diversity among Nasonia species.
The number of reads mapped to each viral contig adjusted for contig size varied among species, highlighting distinct relative abundance differences of Proteus, Providencia, Morganella, and Bacilli phages among the Nasonia species (Fig. 3). Proteus phages dominate the N. giraulti virome at 34.3% of the total contigs, and Morganella phages make up the next largest portion at 30.8%. Morganella phages dominate the N. longicornis virome at 45.9%, and Providencia dominates the N. vitripennis virome at 41.4%. Phages with similarity to the Bacillaceae family outnumber all other groups in introgression line IntG (38.7%), followed by Proteus (26.8%). Thus, a different family of phages dominates each individual host genotype as was similarly shown for bacterial communities associated with these wasps (1). For example, Providencia bacteria dominate the N. vitripennis microbiome (1), which correlates with the highest abundance of Providencia phages in the sequenced virome.
FIG 3.
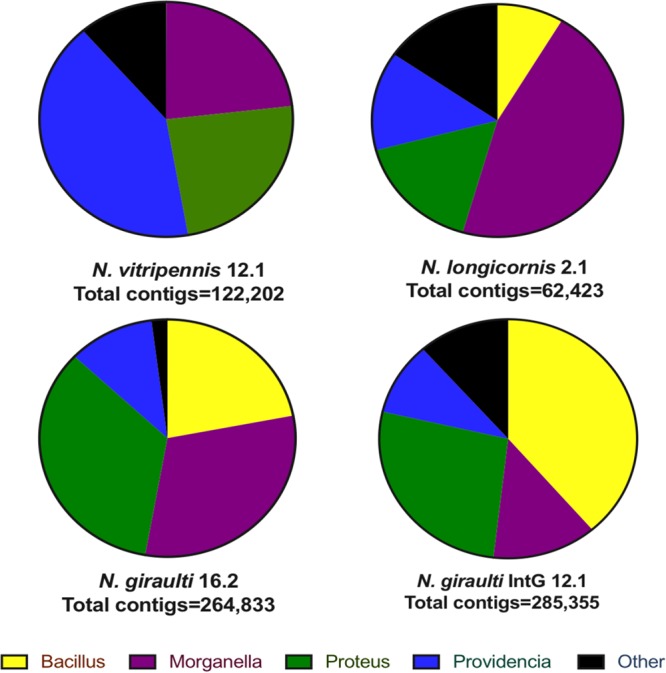
Viral communities are distinguishable between Nasonia species and dominated by a few taxa. The assigned taxa are bacterial genera that harbor sequences, presumably prophages, homologous to the viral protein sequences. The relative abundance of viral contigs within each species is variable. Taxonomy is determined by highest similarity through tBLASTx against the nr database.
Complete and abundant viral genomes.
Six putative circular phage genomes in the core virome with moderate amino acid similarity (>70% homology) to sequences in members of Proteus and Morganella were identified using the viral classification program VirSorter and annotated using BLASTx against the RefSeq database (Fig. 4). None of the circular phage genomes were previously reported as prophages in the bacterial genomes from which they were identified, nor have they been previously described as forming lytic phage particles. Genes in five of these circular phage particle genomes have closest matches in the Proteus bacterial genus. The other, phage NG54, contains genes most homologous to Morganella spp. Thus, these six newly assembled phage genomes, as well as most contigs recovered here, establish the hypothesis that homologous regions in close bacterial relatives of those that colonize Nasonia are prophages with the potential to form phage particles.
FIG 4.

Taxonomy and annotated function of circular genomes and most abundant linear viral contigs in Nasonia. Six complete and circular viral genomes were part of the core Nasonia virome, five of which consisted mostly of open reading frames (ORFs) with similarity to Proteus proteins as determined via BLASTx through NCBI against the nr database (denoted by colored line of inner circles). A total of six viral contigs (circular phage NV18 and five linear contigs above) composed >50% of reads from each of the samples. Each of these dominant viruses was shared among all of the samples with the exception of phage NGI95, which was present only in N. giraulti and N. giraulti IntG. Colored arrows indicate predicted gene function, and colored inner circles represent the genus of the closest BLASTx hit of each gene to the nr database.
To determine the most abundant phage variants within the Nasonia virome, reads were mapped to each of the viral contigs, and six contigs with total read coverage over 2,000× were identified as the most abundant. These six phage genomes, one of which was circular (phage NV18, Fig. 4) and five of which were incomplete genomes (contigs), represented 26% of the total reads in N. longicornis and over 50% of reads in the other three samples. Five of these six most abundant phages were dominated by ORFs with similarity to Proteus, Providencia, and Morganella as well. However, phage NGI95 (Fig. 4) shared the most similarity with Bacilli proteins and was detected only in the introgression line IntG and N. giraulti. Again, a large number of these genes encode unannotated hypothetical proteins, and of these abundant linear viral contigs, three of the five maintain identifiable integrase genes.
Last, two additional novel circular phage genomes recovered from N. vitripennis (phage NV11X) and N. giraulti (phage NG24X) are composed of Xenorhabdus genes, have 94% nucleotide similarity to each other, and maintain predicted phage structural and hypothetical proteins (Fig. S1). These two Xenorhabdus phages show an average of 64% amino acid identity and complete genome synteny to predicted proteins of Xenorhabdus innexi and KK7.4, suggesting that prophages are present within these two bacterial genomes. Xenorhabdus bacteria are insect pathogens that suppress the immune system and produce numerous virulence factors such as hemolysin and cytotoxin that result in insect lethality (37–39). Although hemolysins were found in these viromes, they were associated with Bacilli phages and not these Xenorhabdus phages, consistent with previous reports that the Xenorhabdus bacteria themselves encode these toxins (37–39).
Complete Xenorhabdus phage genomes. Two similar (94% nucleotide similarity) circular Xenorhabdus phage genomes were recovered, one from N. vitripennis (phage NV11X) and the other from N. giraulti (phage NG24X). Download FIG S1, PDF file, 0.05 MB (55.7KB, pdf) .
Copyright © 2018 Leigh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
Phylosymbiosis between host and bacterial communities is emerging as a trend in microbiome studies of the animal world, across both vertebrate and invertebrate species (1, 40–42). While the genetic and biochemical mechanisms underlying phylosymbiosis require more study, animal performance or fitness is often highest when animals contain a homospecific microbiome in comparison to a heterospecific microbiome (1, 43). These findings imply that there are mechanisms by which animals differentially respond to the membership of the microbiome and/or vice versa. Animal-associated viromes, often composed of mostly phages, have generally received much less study than bacterial microbiomes, and there is no a priori reason to expect that phylosymbiosis will occur in phage metagenomes because animals are not expected to directly exert influence on membership, nor is the phage community expected to directly determine which animal it occurs in. However, evidence for direct phage protein interactions within insect hosts is found in endosymbionts where a stable association among the phage, bacterium, and animal has been established (44–46). The bacterial endosymbionts of Nasonia, Wolbachia and its prophage (WO), represent another potential case as the phage-encoded Cif proteins cause (47) and rescue (48) reproductive parasitism phenotypes in arthropod hosts. Additionally, phage particles can bind animal mucus on epithelial tissues via immunoglobulin domains found on the surface of some phage capsids, providing a form of immunity against colonizing bacteria (49, 50). The phages in this environment can also be transcytosed across the epithelial membrane and trafficked through the Golgi apparatus via the endomembrane system (51), further highlighting a direct interaction between phages and animals.
While bacteriophages may simply exhibit phylosymbiosis in a passive manner by association with phylosymbiotic bacterial communities, inducible prophages and/or lytic phages that are the subject of study here may only constitute a small subset of the phage DNA in bacterial genomes. Moreover, active phage particles have the potential to lyse and shift bacterial communities that may disrupt phylosymbiosis. Thus, there is no preferred reason to expect the metagenome of the purified community of virus particles will exhibit phylosymbiosis. Similarly to other animal viromes (8, 10–12, 14, 24), the majority of viruses within Nasonia species are phages, and they appear to be derived mainly from prophages predicted in the most prevalent bacterial genera in Nasonia: Proteus, Providencia, and Morganella. Previous reports in Hydra also showed that viromes were host species specific, composed mostly of phages, and partially phylosymbiotic, although congruence of the host and virome topologies was not investigated (24). Interestingly, wild-caught and lab strains of the same species (Hydra vulgaris) harbor significantly different bacterial communities (52, 53) and therefore maintained unique viral communities as well (24).
Here we describe the first report of phylosymbiosis among host-associated viromes in the parasitoid wasp genus Nasonia. Members of this genus diverged very recently, between 200,000 and 1 million years ago (31), and controlled rearing of each species leads to distinguishable, phylosymbiotic microbiomes that significantly impact development and survival (1, 7). Indeed, interspecific microbiota transplantation causes 25 to 42% decreases in Nasonia survival to adulthood compared to intraspecific microbial transplantations (1). Moreover, hybrid death in the F2 generation is due to a breakdown in phylosymbiosis whereby inoculations of resident gut bacterial species into germfree hybrids recapitulate hybrid lethality (7).
The results here are consistent with the model that if bacterial communities show phylosymbiosis with animal hosts, so too will their viromes. More simply put, viral phylosymbiosis appears to emerge as a by-product of host-bacterium phylosymbiosis. From a methodological perspective, the result is striking given that the sequencing methods to build the bacterial and viral community dendrograms are fundamentally different: 16S amplicon sequencing versus shotgun viral metagenomics. Machine learning on 16S amplicon data previously specified that three of the major distinguishing bacterial genera in Nasonia are closely related symbionts from the Enterobacteriaceae family (genera Proteus, Providencia, and Morganella) (1). Interestingly, abundant phages of Proteus, Providencia, and Morganella dominate the virome identified within all of the pure Nasonia species (Fig. 2D and Fig. 3). Nonetheless, distinguishability of the viromes between Nasonia species is evident through at least two observations: (i) one of the most abundant viruses, phage NGI95, is solely found in the Nasonia giraulti genotype and (ii) the majority of the phage particle genetic diversity within N. giraulti and IntG is represented by a shared group of abundant Bacillaceae phages (Fig. 2B and C). Similarities between the samples with an N. giraulti genetic background support the hypothesis that host genotype, rather than cytotype, plays a role in shaping elements of the phage community structure.
Many of the dominant bacteria present within Nasonia are related to well-studied human pathogens present in enteric diseases (54–60) in addition to other insects (61–63), and genomes are therefore available (64–66). However, most prophage genomes present within these bacteria have not yet been described, and 69% of the genes remain annotated as encoding hypothetical proteins. Thus, the majority of the viruses found in this study were active, unannotated phages of the most prevalent types of bacteria found in Nasonia.
We assembled five complete Proteus phages and one Morganella phage (Fig. 3). Four of these phages (phages NGI4, NV18, and NG55 [Proteus] and phage NG54 [Morganella]) maintained an integrase gene, indicating likely integration into their host’s genome as a prophage. One of the circular Proteus phages maintaining an integrase, phage NV18, was by far the most prevalent phage in all of the samples with over 20,000-fold read coverage from N. vitripennis compared to the 10- to 200-fold coverage of most other viral contigs. This phage genome is composed of mostly hypothetical proteins and proteins with domains of unknown function. Many of these phages show amino acid similarity to sequences within the Proteus, Providencia, and Morganella genera (Fig. 3). These similarities suggest that the described phages may be able to infect members across these sister genera, integrating and acquiring or leaving behind genes in the process.
The discovery of animal-bacterial-viral phylosymbiosis provides a new insight into the tritrophic relationships between animal evolution, bacterial communities, and their phage communities. We note that phylosymbiosis does not equate to coevolution, codiversification, or cospeciation because these are evolutionary processes that assume divergence from a common ancestor. Phylosymbiosis is an eco-evolutionary pattern whereby ecological similarities in the microbiome, or virome in this case, parallel phylogenetic relationships of the host. These patterns are not necessarily ones that occur long term, and they can change rapidly in time or space. However, the detection of phylosymbiosis of the virome is consistent with host identity providing either a direct or indirect influence that partitions clustering relationships of viral particle communities in a manner that reflects animal evolution among closely related species. Whether these patterns hold in wild populations will require future study.
The microbiome has now been widely recognized as a key component of many animal functions, and alterations of this bacterial community can result in performance or fitness reductions (67–69). Prophages are more common than lytic phages in stable host-associated microbial communities (8, 10), outnumber bacteria ∼3:1, and represent a potential structural force for establishment and maintenance of a microbiome (70–72). Intimate associations among phages, bacteria, and their animal hosts are complex, and further studies investigating phylosymbiotic phage communities throughout the animal kingdom are necessary to gain a fuller understanding of the role that microbiomes and viromes play in animal functions and evolution.
MATERIALS AND METHODS
Sample collection and sequencing.
Nasonia species were reared as previously described (7). Four strains were used in this study: Nasonia vitripennis (strain 12.1), N. longicornis (2.1), N. giraulti (16.2), and N. giraulti (IntG 12.1). Each strain maintains Wolbachia infections of the A supergroup. The IntG line was generated by repeatedly backcrossing N. vitripennis 12.1 females to uninfected N. giraulti RV2R males for nine generations to generate a line that contains wVitA-infected cytoplasm of N. vitripennis in the genetic background of N. giraulti (26). Each strain was maintained under constant light at 25°C and raised on flesh fly pupae (Sarcophaga bullata). The transfected line of the Mediterranean flour moth Ephestia kuehniella harboring Wolbachia strain wCauB was obtained from Takema Fukatsu and Tetsuhiko Sasaki (27). Moths were maintained at 24°C and 70% humidity on a diet consisting of wheat bran, glycerol, and dried yeast (20:2:1 [wt/wt]).
Whole insects were suspended in sterile SM buffer and homogenized to release the viruses from the animal tissue. Viral particles were PEG precipitated as previously described (36) and filtered through an 0.22-μm filter. Viral DNA was extracted using the Qiagen MinElute Virus Spin kit, amplified using the Qiagen REPLI-g minikit, and sequenced on the Illumina HiSeq 2000 platform with paired-end reads (2 × 100 bp).
Bioinformatics.
Mate-pair reads from the viromes were analyzed using the iVirus pipeline (33). First, the sequences were trimmed using Trimmomatic 0.35.0 (73) and quality checked using FastQC. De novo assembly of mate-pair reads was completed using SPAdes 3.6.0 (74) with a k-mer value of 63 and default parameters. Assembly quality was determined by QUAST (75) and is reported in Table S1 in the supplemental material. All samples were coassembled with SPAdes 3.6.0 with a k-mer of 63 to generate a single reference file and run through VirSorter (76) in addition to the single assemblies. Viral contigs less than 500 bp and with coverage of less than five were removed from further analysis. Reads were then mapped back to the VirSorter viral contig outputs to estimate the relative abundance of each viral contig for each sample. BowtieBatch (33) was used to run bowtie2 on all samples of the coassembled contigs and produced BAM output files read by Read2RefMapper to generate relative abundance and coverage plots for each viral contig within each metagenome. To consider a contig present within an individual sample, reads from that sample needed to cover 75% of the viral contig from the coassembled virome. Venn diagrams were generated using the VennDiagram package (77) through R v 3.3.2 software to display the overlap of contigs in different gut compartments using the mapping data from Read2RefMapper (33). Relative abundance plots were illustrated using GraphPad’s Prism v 7.0c. Viral protein cluster diversity was determined using vContact through the iVirus pipeline (33, 78) and visualized using Gephi 0.9.2 (79).
The VirSorter output for single assemblies was used for taxonomic classification against the NCBI nr protein database. All taxonomic classifications were determined using the top BLASTx hit, with a threshold score of 50 on BLAST bitscore. Open reading frames (ORFs) were predicted and annotated using Prokka v1.12.0 (80). Additionally, protein families (PFAMs) within the ORFs were identified with InterProScan v5.26.65 (81). jModelTest v2.1.7 (82) was performed to determine the optimal model of host gene evolution and, using this model (PHYML with the JC69 substitution model), a phylogenetic tree was constructed for the host species (as previously described in reference 1) from a nucleotide alignment of the mitochondrial cytochrome oxidase subunit I (COI) genes. Virome similarities as determined by Bray-Curtis beta diversity unweighted pair group method with arithmetic mean (UPGMA) clustering were determined using read coverage counts of viral contigs. These count profiles were rarefied 10 times to a depth of 16,400 counts for each host virome to normalize for differential sequencing coverage. Bray-Curtis beta diversity and resulting UPGMA clustergrams between host viromes were calculated, and UPGMA trees were averaged to generate a consensus clustergram across the rarefied community profiles. Phylosymbiosis as measured through topological similarity between the host phylogeny and the virome clustergram was evaluated using the rooted Robinson-Foulds and rooted matching cluster methods previously described (1). Significance was determined by comparing the observed degree of congruence to the congruence obtained across 100,000 randomized tree topologies using a custom script with methods previously described (1).
Data accessibility.
Assembled contigs from each viral metagenome have been submitted to the WGS database of NCBI under BioProject PRJNA481165. Additionally, each circular genome has been submitted to the NCBI nr database under the accession numbers MK047638 to MK047643.
ACKNOWLEDGMENTS
This work was supported by National Science Foundation award 1456778, National Institutes of Health R01 AI132581, and a Vanderbilt Microbiome Trans-Institutional Initiative Award to Seth R. Bordenstein. A. W. Brooks was supported by T32 National Institutes of Health training grants 6424T32GM08017810, 5T32GM08017809, and 5T32GM0817808. A. Mikaelyan was supported by a Deutsche Forschungsgemeinschaft (DFG) postdoctoral fellowship (MI 2242/1-1). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Brooks AW, Kohl KD, Brucker RM, van Opstal EJ, Bordenstein SR. 2016. Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol 14:e2000225. doi: 10.1371/journal.pbio.2000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Easson CG, Thacker RW. 2014. Phylogenetic signal in the community structure of host-specific microbiomes of tropical marine sponges. Front Microbiol 5:532. doi: 10.3389/fmicb.2014.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanders JG, Powell S, Kronauer DJ, Vasconcelos HL, Frederickson ME, Pierce NE. 2014. Stability and phylogenetic correlation in gut microbiota: lessons from ants and apes. Mol Ecol 23:1268–1283. doi: 10.1111/mec.12611. [DOI] [PubMed] [Google Scholar]
- 4.Phillips CD, Phelan G, Dowd SE, McDonough MM, Ferguson AW, Delton Hanson J, Siles L, Ordonez-Garza N, San Francisco M, Baker RJ. 2012. Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Mol Ecol 21:2617–2627. doi: 10.1111/j.1365-294X.2012.05568.x. [DOI] [PubMed] [Google Scholar]
- 5.Moeller AH, Li Y, Mpoudi Ngole E, Ahuka-Mundeke S, Lonsdorf EV, Pusey AE, Peeters M, Hahn BH, Ochman H. 2014. Rapid changes in the gut microbiome during human evolution. Proc Natl Acad Sci U S A 111:16431–16435. doi: 10.1073/pnas.1419136111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ochman H, Worobey M, Kuo CH, Ndjango JB, Peeters M, Hahn BH, Hugenholtz P. 2010. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol 8:e1000546. doi: 10.1371/journal.pbio.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brucker RM, Bordenstein SR. 2013. The hologenomic basis of speciation: gut bacteria cause hybrid lethality in the genus Nasonia. Science 341:667–669. doi: 10.1126/science.1240659. [DOI] [PubMed] [Google Scholar]
- 8.Reyes A, Haynes M, Hanson N, Angly FE, Heath AC, Rohwer F, Gordon JI. 2010. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 466:334–338. doi: 10.1038/nature09199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suttle CA. 2005. Viruses in the sea. Nature 437:356–361. doi: 10.1038/nature04160. [DOI] [PubMed] [Google Scholar]
- 10.Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, Wu GD, Lewis JD, Bushman FD. 2011. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res 21:1616–1625. doi: 10.1101/gr.122705.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leigh BA, Djurhuus A, Breitbart M, Dishaw LJ. 2018. The gut virome of the protochordate model organism, Ciona intestinalis subtype A. Virus Res 244:137–146. doi: 10.1016/j.virusres.2017.11.015. [DOI] [PubMed] [Google Scholar]
- 12.Fawaz M, Vijayakumar P, Mishra A, Gandhale PN, Dutta R, Kamble NM, Sudhakar SB, Roychoudhary P, Kumar H, Kulkarni DD, Raut AA. 2016. Duck gut viral metagenome analysis captures snapshot of viral diversity. Gut Pathog 8:30. doi: 10.1186/s13099-016-0113-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sachsenroder J, Twardziok SO, Scheuch M, Johne R. 2014. The general composition of the faecal virome of pigs depends on age, but not on feeding with a probiotic bacterium. PLoS One 9:e88888. doi: 10.1371/journal.pone.0088888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim MS, Bae JW. 2018. Lysogeny is prevalent and widely distributed in the murine gut microbiota. ISME J 12:1127–1141. doi: 10.1038/s41396-018-0061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD, Bushman FD. 2013. Rapid evolution of the human gut virome. Proc Natl Acad Sci U S A 110:12450–12455. doi: 10.1073/pnas.1300833110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reyes A, Wu M, McNulty NP, Rohwer FL, Gordon JI. 2013. Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc Natl Acad Sci U S A 110:20236–20241. doi: 10.1073/pnas.1319470110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knowles B, Silveira CB, Bailey BA, Barott K, Cantu VA, Cobian-Guemes AG, Coutinho FH, Dinsdale EA, Felts B, Furby KA, George EE, Green KT, Gregoracci GB, Haas AF, Haggerty JM, Hester ER, Hisakawa N, Kelly LW, Lim YW, Little M, Luque A, McDole-Somera T, McNair K, de Oliveira LS, Quistad SD, Robinett NL, Sala E, Salamon P, Sanchez SE, Sandin S, Silva GG, Smith J, Sullivan C, Thompson C, Vermeij MJ, Youle M, Young C, Zgliczynski B, Brainard R, Edwards RA, Nulton J, Thompson F, Rohwer F. 2016. Lytic to temperate switching of viral communities. Nature 531:466–470. doi: 10.1038/nature17193. [DOI] [PubMed] [Google Scholar]
- 18.Silveira CB, Rohwer FL. 2016. Piggyback-the-Winner in host-associated microbial communities. NPJ Biofilms Microbiomes 2:16010. doi: 10.1038/npjbiofilms.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gama JA, Reis AM, Domingues I, Mendes-Soares H, Matos AM, Dionisio F. 2013. Temperate bacterial viruses as double-edged swords in bacterial warfare. PLoS One 8:e59043. doi: 10.1371/journal.pone.0059043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brussow H, Canchaya C, Hardt WD. 2004. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev 68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madsen JS, Burmolle M, Hansen LH, Sorensen SJ. 2012. The interconnection between biofilm formation and horizontal gene transfer. FEMS Immunol Med Microbiol 65:183–195. doi: 10.1111/j.1574-695X.2012.00960.x. [DOI] [PubMed] [Google Scholar]
- 22.Molin S, Tolker-Nielsen T. 2003. Gene transfer occurs with enhanced efficiency in biofilms and induces enhanced stabilisation of the biofilm structure. Curr Opin Biotechnol 14:255–261. doi: 10.1016/S0958-1669(03)00036-3. [DOI] [PubMed] [Google Scholar]
- 23.Manrique P, Bolduc B, Walk ST, van der Oost J, de Vos WM, Young MJ. 2016. Healthy human gut phageome. Proc Natl Acad Sci U S A 113:10400–10405. doi: 10.1073/pnas.1601060113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grasis JA, Lachnit T, Anton-Erxleben F, Lim YW, Schmieder R, Fraune S, Franzenburg S, Insua S, Machado G, Haynes M, Little M, Kimble R, Rosenstiel P, Rohwer FL, Bosch TC. 2014. Species-specific viromes in the ancestral holobiont Hydra. PLoS One 9:e109952. doi: 10.1371/journal.pone.0109952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soffer N, Zaneveld J, Vega Thurber R. 2015. Phage-bacteria network analysis and its implication for the understanding of coral disease. Environ Microbiol 17:1203–1218. doi: 10.1111/1462-2920.12553. [DOI] [PubMed] [Google Scholar]
- 26.Chafee ME, Zecher CN, Gourley ML, Schmidt VT, Chen JH, Bordenstein SR, Clark ME, Bordenstein SR. 2011. Decoupling of host-symbiont-phage coadaptations following transfer between insect species. Genetics 187:203–215. doi: 10.1534/genetics.110.120675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujii Y, Kubo T, Ishikawa H, Sasaki T. 2004. Isolation and characterization of the bacteriophage WO from Wolbachia, an arthropod endosymbiont. Biochem Biophys Res Commun 317:1183–1188. doi: 10.1016/j.bbrc.2004.03.164. [DOI] [PubMed] [Google Scholar]
- 28.Campbell BC, Steffen-Campbell JD, Werren JH. 1993. Phylogeny of the Nasonia species complex (Hymenoptera: Pteromalidae) inferred from an internal transcribed spacer (ITS2) and 28S rDNA sequences. Insect Mol Biol 2:225–237. [DOI] [PubMed] [Google Scholar]
- 29.Bordenstein SR, O’Hara FP, Werren JH. 2001. Wolbachia-induced incompatibility precedes other hybrid incompatibilities in Nasonia. Nature 409:707–710. doi: 10.1038/35055543. [DOI] [PubMed] [Google Scholar]
- 30.Bordenstein SR, Werren JH. 2007. Bidirectional incompatibility among divergent Wolbachia and incompatibility level differences among closely related Wolbachia in Nasonia. Heredity (Edinb) 99:278–287. doi: 10.1038/sj.hdy.6800994. [DOI] [PubMed] [Google Scholar]
- 31.Werren JH, Richards S, Desjardins CA, Niehuis O, Gadau J, Colbourne JK, Nasonia Genome Working Group. 2010. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 327:343–348. doi: 10.1126/science.1178028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cowles KN, Goodrich-Blair H. 2005. Expression and activity of a Xenorhabdus nematophila haemolysin required for full virulence towards Manduca sexta insects. Cell Microbiol 7:209–219. doi: 10.1111/j.1462-5822.2004.00448.x. [DOI] [PubMed] [Google Scholar]
- 33.Bolduc B, Youens-Clark K, Roux S, Hurwitz BL, Sullivan MB. 2017. iVirus: facilitating new insights in viral ecology with software and community data sets imbedded in a cyberinfrastructure. ISME J 11:7–14. doi: 10.1038/ismej.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lengyel K, Lang E, Fodor A, Szallas E, Schumann P, Stackebrandt E. 2005. Description of four novel species of Xenorhabdus, family Enterobacteriaceae: Xenorhabdus budapestensis sp. nov., Xenorhabdus ehlersii sp. nov., Xenorhabdus innexi sp. nov., and Xenorhabdus szentirmaii sp. nov. Syst Appl Microbiol 28:115–122. doi: 10.1016/j.syapm.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 35.Bordenstein SR, Marshall ML, Fry AJ, Kim U, Wernegreen JJ. 2006. The tripartite associations between bacteriophage, Wolbachia, and arthropods. PLoS Pathog 2:e43. doi: 10.1371/journal.ppat.0020043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bordenstein SR, Bordenstein SR. 2016. Eukaryotic association module in phage WO genomes from Wolbachia. Nat Commun 7:13155. doi: 10.1038/ncomms13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chaston JM, Suen G, Tucker SL, Andersen AW, Bhasin A, Bode E, Bode HB, Brachmann AO, Cowles CE, Cowles KN, Darby C, de Léon L, Drace K, Du Z, Givaudan A, Herbert Tran EE, Jewell KA, Knack JJ, Krasomil-Osterfeld KC, Kukor R, Lanois A, Latreille P, Leimgruber NK, Lipke CM, Liu R, Lu X, Martens EC, Marri PR, Médigue C, Menard ML, Miller NM, Morales-Soto N, Norton S, Ogier J-C, Orchard SS, Park D, Park Y, Qurollo BA, Sugar DR, Richards GR, Rouy Z, Slominski B, Slominski K, Snyder H, Tjaden BC, van der Hoeven R, Welch RD, Wheeler C, Xiang B, Barbazuk B, Gaudriault S, Goodner B, Slater SC, Forst S, Goldman BS, Goodrich-Blair H. 2011. The entomopathogenic bacterial endosymbionts Xenorhabdus and Photorhabdus: convergent lifestyles from divergent genomes. PLoS One 6:e27909. doi: 10.1371/journal.pone.0027909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goodrich-Blair H, Clarke DJ. 2007. Mutualism and pathogenesis in Xenorhabdus and Photorhabdus: two roads to the same destination. Mol Microbiol 64:260–268. doi: 10.1111/j.1365-2958.2007.05671.x. [DOI] [PubMed] [Google Scholar]
- 39.Sicard M, Ferdy JB, Pages S, Le Brun N, Godelle B, Boemare N, Moulia C. 2004. When mutualists are pathogens: an experimental study of the symbioses between Steinernema (entomopathogenic nematodes) and Xenorhabdus (bacteria). J Evol Biol 17:985–993. doi: 10.1111/j.1420-9101.2004.00748.x. [DOI] [PubMed] [Google Scholar]
- 40.Groussin M, Mazel F, Sanders JG, Smillie CS, Lavergne S, Thuiller W, Alm EJ. 2017. Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat Commun 8:14319. doi: 10.1038/ncomms14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kohl KD, Varner J, Wilkening JL, Dearing MD. 2018. Gut microbial communities of American pikas (Ochotona princeps): evidence for phylosymbiosis and adaptations to novel diets. J Anim Ecol 87:323–330. doi: 10.1111/1365-2656.12692. [DOI] [PubMed] [Google Scholar]
- 42.Kohl KD, Dearing MD, Bordenstein SR. 2018. Microbial communities exhibit host species distinguishability and phylosymbiosis along the length of the gastrointestinal tract. Mol Ecol 27:1874–1883. doi: 10.1111/mec.14460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shultz LD, Ishikawa F, Greiner DL. 2007. Humanized mice in translational biomedical research. Nat Rev Immunol 7:118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 44.Degnan PH, Moran NA. 2008. Diverse phage-encoded toxins in a protective insect endosymbiont. Appl Environ Microbiol 74:6782–6791. doi: 10.1128/AEM.01285-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toh H, Weiss BL, Perkin SA, Yamashita A, Oshima K, Hattori M, Aksoy S. 2006. Massive genome erosion and functional adaptations provide insights into the symbiotic lifestyle of Sodalis glossinidius in the tsetse host. Genome Res 16:149–156. doi: 10.1101/gr.4106106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moran NA, Degnan PH, Santos SR, Dunbar HE, Ochman H. 2005. The players in a mutualistic symbiosis: insects, bacteria, viruses, and virulence genes. Proc Natl Acad Sci U S A 102:16919–16926. doi: 10.1073/pnas.0507029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.LePage DP, Metcalf JA, Bordenstein SR, On J, Perlmutter JI, Shropshire JD, Layton EM, Funkhouser-Jones LJ, Beckmann JF, Bordenstein SR. 2017. Prophage WO genes recapitulate and enhance Wolbachia-induced cytoplasmic incompatibility. Nature 543:243–247. doi: 10.1038/nature21391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shropshire JD, On J, Layton EM, Zhou H, Bordenstein SR. 2018. One prophage WO gene rescues cytoplasmic incompatibility in Drosophila melanogaster. Proc Natl Acad Sci U S A 115:4987–4991. doi: 10.1073/pnas.1800650115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barr JJ, Auro R, Furlan M, Whiteson KL, Erb ML, Pogliano J, Stotland A, Wolkowicz R, Cutting AS, Doran KS, Salamon P, Youle M, Rohwer F. 2013. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc Natl Acad Sci U S A 110:10771–10776. doi: 10.1073/pnas.1305923110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barr JJ, Youle M, Rohwer F. 2013. Innate and acquired bacteriophage-mediated immunity. Bacteriophage 3:e25857. doi: 10.4161/bact.25857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen S, Baker K, Padman BS, Patwa R, Dunstan RA, Weston TA, Schlosser K, Bailey B, Lithgow T, Lazarou M, Luque A, Rohwer F, Blumberg RS, Barr JJ. 2017. Bacteriophage transcytosis provides a mechanism to cross epithelial cell layers. mBio 8:e01874-17. doi: 10.1128/mBio.01874-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fraune S, Bosch TC. 2007. Long-term maintenance of species-specific bacterial microbiota in the basal metazoan Hydra. Proc Natl Acad Sci U S A 104:13146–13151. doi: 10.1073/pnas.0703375104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Franzenburg S, Fraune S, Altrock PM, Kunzel S, Baines JF, Traulsen A, Bosch TC. 2013. Bacterial colonization of Hydra hatchlings follows a robust temporal pattern. ISME J 7:781–790. doi: 10.1038/ismej.2012.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muller HE. 1986. Occurrence and pathogenic role of Morganella-Proteus-Providencia group bacteria in human feces. J Clin Microbiol 23:404–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McDermott C, Mylotte JM. 1984. Morganella morganii: epidemiology of bacteremic disease. Infect Control 5:131–137. doi: 10.1017/S0195941700059993. [DOI] [PubMed] [Google Scholar]
- 56.Hola V, Peroutkova T, Ruzicka F. 2012. Virulence factors in Proteus bacteria from biofilm communities of catheter-associated urinary tract infections. FEMS Immunol Med Microbiol 65:343–349. doi: 10.1111/j.1574-695X.2012.00976.x. [DOI] [PubMed] [Google Scholar]
- 57.Albert MJ, Alam K, Ansaruzzaman M, Islam MM, Rahman AS, Haider K, Bhuiyan NA, Nahar S, Ryan N, Montanaro J. 1992. Pathogenesis of Providencia alcalifaciens-induced diarrhea. Infect Immun 60:5017–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Hara CM, Brenner FW, Miller JM. 2000. Classification, identification, and clinical significance of Proteus, Providencia, and Morganella. Clin Microbiol Rev 13:534–546. doi: 10.1128/CMR.13.4.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Armbruster CE, Smith SN, Johnson AO, DeOrnellas V, Eaton KA, Yep A, Mody L, Wu W, Mobley HL. 2017. The pathogenic potential of Proteus mirabilis is enhanced by other uropathogens during polymicrobial urinary tract infection. Infect Immun 85:e00808-16. doi: 10.1128/IAI.00808-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Armbruster CE, Smith SN, Yep A, Mobley HL. 2014. Increased incidence of urolithiasis and bacteremia during Proteus mirabilis and Providencia stuartii coinfection due to synergistic induction of urease activity. J Infect Dis 209:1524–1532. doi: 10.1093/infdis/jit663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Galac MR, Lazzaro BP. 2011. Comparative pathology of bacteria in the genus Providencia to a natural host, Drosophila melanogaster. Microbes Infect 13:673–683. doi: 10.1016/j.micinf.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vicente CS, Nascimento FX, Espada M, Barbosa P, Hasegawa K, Mota M, Oliveira S. 2013. Characterization of bacterial communities associated with the pine sawyer beetle Monochamus galloprovincialis, the insect vector of the pinewood nematode Bursaphelenchus xylophilus. FEMS Microbiol Lett 347:130–139. doi: 10.1111/1574-6968.12232. [DOI] [PubMed] [Google Scholar]
- 63.Wei T, Miyanaga K, Tanji Y. 2014. Persistence of antibiotic-resistant and -sensitive Proteus mirabilis strains in the digestive tract of the housefly (Musca domestica) and green bottle flies (Calliphoridae). Appl Microbiol Biotechnol 98:8357–8366. doi: 10.1007/s00253-014-5846-9. [DOI] [PubMed] [Google Scholar]
- 64.Pearson MM, Sebaihia M, Churcher C, Quail MA, Seshasayee AS, Luscombe NM, Abdellah Z, Arrosmith C, Atkin B, Chillingworth T, Hauser H, Jagels K, Moule S, Mungall K, Norbertczak H, Rabbinowitsch E, Walker D, Whithead S, Thomson NR, Rather PN, Parkhill J, Mobley HL. 2008. Complete genome sequence of uropathogenic Proteus mirabilis, a master of both adherence and motility. J Bacteriol 190:4027–4037. doi: 10.1128/JB.01981-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clifford RJ, Hang J, Riley MC, Onmus-Leone F, Kuschner RA, Lesho EP, Waterman PE. 2012. Complete genome sequence of Providencia stuartii clinical isolate MRSN 2154. J Bacteriol 194:3736–3737. doi: 10.1128/JB.00615-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Olaitan AO, Diene SM, Gupta SK, Adler A, Assous MV, Rolain JM. 2014. Genome analysis of NDM-1 producing Morganella morganii clinical isolate. Expert Rev Anti Infect Ther 12:1297–1305. doi: 10.1586/14787210.2014.944504. [DOI] [PubMed] [Google Scholar]
- 67.Cho I, Blaser MJ. 2012. The human microbiome: at the interface of health and disease. Nat Rev Genet 13:260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Greenblum S, Turnbaugh PJ, Borenstein E. 2012. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci U S A 109:594–599. doi: 10.1073/pnas.1116053109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, Bousvaros A, Korzenik J, Sands BE, Xavier RJ, Huttenhower C. 2012. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Backhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, Li Y, Xia Y, Xie H, Zhong H, Khan MT, Zhang J, Li J, Xiao L, Al-Aama J, Zhang D, Lee YS, Kotowska D, Colding C, Tremaroli V, Yin Y, Bergman S, Xu X, Madsen L, Kristiansen K, Dahlgren J, Wang J. 2015. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17:852. doi: 10.1016/j.chom.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 71.Breitbart M, Haynes M, Kelley S, Angly F, Edwards RA, Felts B, Mahaffy JM, Mueller J, Nulton J, Rayhawk S, Rodriguez-Brito B, Salamon P, Rohwer F. 2008. Viral diversity and dynamics in an infant gut. Res Microbiol 159:367–373. doi: 10.1016/j.resmic.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 72.Lim ES, Zhou Y, Zhao G, Bauer IK, Droit L, Ndao IM, Warner BB, Tarr PI, Wang D, Holtz LR. 2015. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat Med 21:1228–1234. doi: 10.1038/nm.3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Roux S, Enault F, Hurwitz BL, Sullivan MB. 2015. VirSorter: mining viral signal from microbial genomic data. PeerJ 3:e985. doi: 10.7717/peerj.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen H, Boutros PC. 2011. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12:35. doi: 10.1186/1471-2105-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bolduc B, Jang HB, Doulcier G, You ZQ, Roux S, Sullivan MB. 2017. vConTACT: an iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 5:e3243. doi: 10.7717/peerj.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bastian M, Heymann S, Jacomy M. 2009. Gephi: an open source software for exploring and manipulating networks. International AAAI Conference on Weblogs and Social Media 8:361–362. [Google Scholar]
- 80.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 81.Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R. 2005. InterProScan: protein domains identifier. Nucleic Acids Res 33:W116–W120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Assembly statistics. Download Table S1, PDF file, 0.05 MB (52.5KB, pdf) .
Copyright © 2018 Leigh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Pfam assignments in viral metagenomes. Download Table S2, PDF file, 0.3 MB (288.1KB, pdf) .
Copyright © 2018 Leigh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Complete Xenorhabdus phage genomes. Two similar (94% nucleotide similarity) circular Xenorhabdus phage genomes were recovered, one from N. vitripennis (phage NV11X) and the other from N. giraulti (phage NG24X). Download FIG S1, PDF file, 0.05 MB (55.7KB, pdf) .
Copyright © 2018 Leigh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
Assembled contigs from each viral metagenome have been submitted to the WGS database of NCBI under BioProject PRJNA481165. Additionally, each circular genome has been submitted to the NCBI nr database under the accession numbers MK047638 to MK047643.