

SUMMARY
DNA double-strand breaks (DSBs) arise during physiological transcription, DNA replication, and antigen receptor diversification. Mistargeting or misprocessing of DSBs can result in pathological structural variation and mutation. Here we describe a sensitive method (END-seq) to monitor DNA end resection and DSBs genome-wide at base-pair resolution in vivo. We utilized END-seq to determine the frequency and spectrum of restriction-enzyme-, zinc-finger-nuclease-, and RAG-induced DSBs. Beyond sequence preference, chromatin features dictate the repertoire of these genome-modifying enzymes. END-seq can detect at least one DSB per cell among 10,000 cells not harboring DSBs, and we estimate that up to one out of 60 cells contains off-target RAG cleavage. In addition to site-specific cleavage, we detect DSBs distributed over extended regions during immunoglobulin class-switch recombination. Thus, END-seq provides a snapshot of DNA ends genome-wide, which can be utilized for understanding genome-editing specificities and the influence of chromatin on DSB pathway choice.
Graphical Abstract
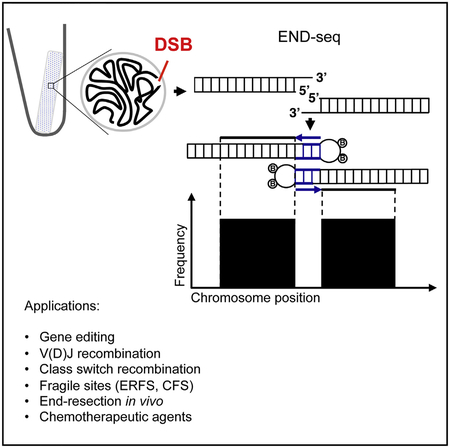
In Brief
Canela et al. develop a sensitive and quantitative method that provides a landscape of DNA double-strand breaks and end resection in vivo prior to DNA repair. This opens up the possibility for better understanding the causes and consequences of genome instability.
INTRODUCTION
RAG1 and RAG2 (recombination-activating gene, RAG) proteins recognize conserved recombination signal sequences (RSSs) positioned adJαcent to V (variable), D (diversity), and J (joining) gene segments, where they introduce double-strand breaks (DSBs) (Fugmann et al., 2000). RAG cleavage generates a pair of blunt, broken RSS signal ends (SEs) and a pair of coding ends (CEs). In addition to antigen receptor loci, RAG binds to several thousand genomic sites corresponding to active promoters and enhancers, some of which bear RSS-related sequences (Ji et al., 2010). Although such cryptic RSSs (c-RSSs) are documented to contribute to lymphomagenesis (Mijušković et al., 2015; Papaemmanuil et al., 2014), only a few areas of RAG binding are at risk of RAG-mediated damage (Teng et al., 2015). Thus, methods that discriminate between RAG binding and RAG activity would enable us to better understand the rules underlying target specificity.
RAG cleavage is restricted to the G1 phase of the cell cycle (Schlissel et al., 1993). The non-homologous end joining (NHEJ) pathway subsequently fuses pairs of SEs and CEs (Helmink and Sleckman, 2012). The joining of SE is normally precise and leads to RSS joins, which are susceptible to re-cleavage (Neiditch et al., 2002). In contrast, hairpin-terminated CEs are opened and further processed, leading to small deletions or insertions surrounding the DSB site (Pannunzio et al., 2014). When cells progress into S and G2 phases of the cell cycle, DSBs can be channeled into the homologous recombination (HR) pathway. Rather than minimal end trimming associated with NHEJ, HR requires extensive 5′−3′ processing to generate 3′ single-strand DNA (ssDNA). The ssDNA generated during 5′DNA end resection is utilized for homologous pairing and strand invasion and is also inhibitory to NHEJ. Thus, DSB resection is a major determinant in the choice between utilization of HR or NHEJ pathways (Bunting and Nussenzweig, 2013).
Unlike programmed DSBs initiated by RAG, DNA damage associated with transcriptional activity and replication errors is more sporadic. For example, some genomic loci such as early-replication fragile sites and common fragile sites are inherently unstable and DSBs span large megabase-sized domains (Barlow et al., 2013). Recurrent DSBs have also been detected at genes whose transcription is induced by a variety of stimuli (Bunch et al., 2015; Haffner et al., 2010; Ju et al., 2006; Madabhushi et al., 2015; Williamson and Lees-Miller, 2011). Although DSBs are usually repaired with high fidelity by NHEJ and HR, occasional errors in these pathways produce collateral damage that can lead to pathological conditions such as cancer, aging, and neurological disorders. Therefore, there is great interest in determining the genomic location, structure, and frequencies of recurrent low-level DSBs.
Several methods have been developed that assess the location, persistence, end structure, and rate of DSB generation (Hu et al., 2016). For example, “BLESS” maps un-joined broken ends in an unbiased manner (Crosetto et al., 2013), but is associated with high background and does not provide information about end structure (Hu et al., 2016). HCoDES reveals high-resolution information about DNA end structure (Dorsett et al., 2014), but does not map the location of DSBs genome-wide. High-throughput, genome-wide translocation sequencing (HTGTS) and similar techniques map translocations induced by DSBs (Frock et al., 2015). Since translocation is dependent on close nuclear proximity and ligation, HTGTS underestimates the frequency of DSBs (Hu et al., 2016). Finally, none of these techniques have been tested outside of tissue culture conditions, and therefore it is important to develop a simple method to interrogate DSB formation and repair in vivo.
Here we describe a method to quantitatively determine the DSB initiation landscape and end resection prior to DSB repair. This enabled us to monitor end resection in vivo and map cleavage sites for various genome-editing enzymes.
RESULTS
Nucleotide Resolution Mapping of DSBs
To avoid artificial generation of DSBs as a result of mechanical shearing or fixation, we embedded live cells in low melting agarose (Figure 1A). To remove proteins bound to DNA ends and perform enzymatic manipulations, agarose plugs were treated with Proteinase K and RNase A. After blunting and A-tailing the DNA ends, we captured them with a biotinylated hairpin adaptor containing a 3′ T overhang and Illumina’s p5 sequence. Subsequently, the agarose plug was melted and DNA was extracted and sheared, after which fragments containing the adaptor (and the DNA end) were captured with streptavidin-coated beads. The new ends created by sonication were also end repaired and A-tailed, allowing ligation of a second hairpin adaptor containing Illumina’s p7 sequence. PCR amplification resulted in a ready-to-use library in which the first base sequenced (read number 1) corresponded to the first base of the blunted DSBs. The number of reads mapping to the DSB is proportional to the frequency of DNA ends in the cell population (see below).
Figure 1. DNA Breaks Measured by END-Seq.

(A) Schematic overview of END-seq.
(B) Top panel shows END-seq read coverage for chromosome 8 in G1-arrested WT pre-B cells after 4 hr AsiSI induction. Predicted AsiSI target sites are indicated above by black bars. The y axis corresponds to the number of reads. Middle panel shows END-seq reads corresponding to an AsiSI DSB generated in vivo in pre-B cells or in vitro with purified recombinant AsiSI. Lower panel shows close-up magnification of the cut site, revealing absence of reads at AT overhangs generated by enzymatic digestion.
(C) Comparison between END-seq and BLESS in detecting AsiSI DSBs in LIG4−/− pre-B G1-arrested cells. Left panel shows percentage of total reads mapped at AsiSI sites, and the right panel shows the total reads at individual sites plotted on a log scale. Dashed diagonal red line is indicative of the same number of reads detected by both methods.
(D) Comparison of END-seq and BLESS detection of AsiSI DSBs on chromosome 8. Filled triangles indicate peaks detected by END-seq.
(E) Two examples showing the differences in the symmetry of AsiSI sites detected by END-seq and BLESS.
See also Figures S1and S2 and Table S1.
A restriction enzyme (RE), zinc-finger endonuclease (ZFN), or RAG endonuclease was used to generate DSBs in an inducible manner in pre-B cell lines transformed by v-abl kinase (Figure S1A, available online): (1) the AsiSI RE generates site-selective DSBs across the genome upon co-treatment with doxycycline (DOX) and 4-hydroxytamoxifen (4-OHT) (Iacovoni et al., 2010), (2) the ZFN targets a region downstream of the mouse T cell receptor beta (TCRβ) enhancer upon treatment with DOX (Dorsett et al., 2014), and (3) RAG endonuclease targets RSSs that lie adJαcent to antigen receptor gene segment (e.g., Igκ, Igλ, and IgH) (Schatz and Ji, 2011). Treatment of pre-B cell lines with the v-abl kinase inhibitor imatinib leads to G1 cell-cycle arrest, RAG induction, and the initiation of V(D)J recombination, marked by one to two γ-H2AX foci (Chen et al., 2000) in the majority of cells (Figure S1B). Treatment with DOX and imatinib leads to simultaneous RAG and ZFN expression, and the combination of DOX, imatinib, and 4-OHT was used to produce all three types of DNA breaks in G1-arrested cells (Figure S1A).
A retrovirus encoding AsiSI (pTRE3G-HA-ER-AsiSI) was stably transduced into the v-abl kinase transformed pre-B cells that also contained the inducible ZFN (Figure S1A). DOX treatment led to AsiSI accumulation in the cytoplasm, after which cells were treated for 4 hr with 4-OHT, resulting in nuclear translocation of AsiSI and robust induction of γ-H2AX in almost 100% of cells (Figure S1C). As shown in the snapshot view of chromo-some 8, end sequencing (END-seq) detected several peaks in wild-type (WT) cells whose summits were well above noise (Figure 1B). A close-up view of the peaks revealed sequence reads on either side of a gap corresponding to the AT overhang, which was removed by the end repair (Figures 1B and S1D). Two blocks of reads have opposite divergent orientation corresponding to sequencing of the 3′ ends of the two-ended DSB (Figure S1E). A very similar structure consisting of two symmetrical blocks of reads was produced when DNA was digested with recombinant AsiSI in vitro within the plug (Figure 1B). AsiSI induced breakage was detected in both G1-arrested cells and proliferating cultures (Figure S2A). Besides the RAG breaks produced in G1, peak calling indicated that the majority of peaks (90%) were precisely at sites of AsiSI-induced breakage (Figure S2B; Experimental Procedures). Thus, END-seq readily detects AsiSI DSBs in both cycling and non-cycling conditions.
Comparison with BLESS
A recently developed approach to map DSBs (called BLESS) involves in situ break labeling after formaldehyde fixation, followed by enrichment on streptavidin (Crosetto et al., 2013). Although this method has successfully characterized aphidicolin-sensitive regions and off-target CRISPR DSBs (Ran et al., 2015), it is limited by relatively high background (Hu et al., 2016). We performed END-seq and BLESS in parallel, using G1-arrested, LIG4 knockout cells to prevent DSB repair by NHEJ. Both methods detected peaks at AsiSI sites (Figures 1C and 1D). However, END-seq exhibited an average of 319-fold increase in the number of reads at the RE recognition sequence, and a 36-fold increase in the proportion of reads mapped to AsiSI sites (Figure 1C; Table S1). Moreover, the structure of the DNA ends was altered using BLESS, evidenced by the significant number of reads starting at a distance away from the break site and associated with asymmetry of the peaks (Figure 1E). We speculate that the decrease in sensitivity, specificity, and alteration in end structure may be introduced during handling or formaldehyde fixation. Since formaldehyde damages DNA (Ross and Shipley, 1980), this may interfere with end repair and adaptor ligation.
Low-Level DSBs
To determine the sensitivity of END-seq, we introduced a single DSB near the TCRβ enhancer using the DOX-inducible ZFN (Dorsett et al., 2014) (Figure 2A). In contrast to AsiSI, which makes sequence-specific DSBs, ZFN is coupled to FOK1, which, once bound to DNA, produces non-specific DNA cleavage. This resulted in a round symmetrical peak surrounding the break site (Figure 2B). To maximize the total number of DSBs produced by ZFN, we inserted the DOX-inducible ZFN in LIG4−/− cells, which are unable to repair DSBs. We mixed DOX-treated, G1-arrested cells in serial dilutions (up to 1 in 10,000) with cells that had not been treated with DOX and therefore did not harbor DSBs at the TCRβ locus. Peak heights ranged from 153,000 accumulated sequence reads (in undiluted samples) to 100 reads in samples that had been diluted by 1/10,000 (Figures 2B and 2C). The lowest detectable signal, still significantly above background, corresponded to 2,000 cells with ZFN breaks among 20 million cells without DSBs. We also induced cells with DOX + 4OHT to simultaneously activate AsiSI DSBs across the genome. Similar to ZFN breaks, we observed a decrease in extent of breakage directly proportional to cell dilution at all genomic sites (Figure 2D). We conclude that END-seq is sensitive enough to detect a single DSB if it occurs in 10,000 cells, and that the relative number of reads at a specific position is proportional to the fraction of cells carrying the DSB. In addition to site-specific DSBs, we could also detect DSBs during class-switch recombination in WT B cells that spread over the 9 kb switch μ and 13 kb switch γ1 regions (Figure S2C).
Figure 2. Sensitivity of END-Seq.

(A) Schematic of a dilution experiment used to determine the limits of END-seq sensitivity using a pair of zinc-finger nucleases (ZFNs).
(B) END-seq tracks for undiluted and 10-fold serially diluted samples. y axis represents number of reads.
(C) Number of reads within a 240 bp window surrounding the ZFN break in each dilution library normalized by the total number of mapped reads.
(D) Comparison between the normalized reads at AsiSI sites in undiluted versus serially diluted samples. ZFN and AsiSI breaks were produced simultaneously in LIG4−/− cells after treating cells with DOX and 4OHT.
Besides on-target DSB at the TCRβ locus, we detected eight “off-target” ZFN DSBs (Figure S2D; Table S2). On average, the number of reads at off-target ZFN breaks in the undiluted sample was 368-fold less than the level detected at the on-target site. Consistent with this reduction in cleavage activity, only three off-target ZFN break sites were detectable when cells were diluted to 1:1,000. Sequences surrounding the off-target ZFN DSBs were similar, but not identical, to the intended target sequence, exhibiting up to eight base-pair mismatches on either side (Table S2). We also detected cases of cleavage by the homodimeric ZFN, in which only one of the pairs binds to the selected site (Table S2) (Cathomen and Joung, 2008). Despite harboring significant sequence mismatches, seven of the eight off-targets were in accessible chromatin and associated with actively transcribed genes (Table S2). The reduction in cleavage activity and divergence from the consensus sequence at off-target sites are consistent with previous studies of ZFN specificities (Cathomen and Joung, 2008; Gabriel et al., 2011).
Variation in AsiSI Targeting across the Genome
Only 221 out of the predicted 1,088 genomic AsiSI sites were cleaved. In addition, we detected seven DSBs in which a SNP created an AsiSI site not present in the reference genome (Figures 3A and S2B; Table S2). Among the cut sites, there were considerable differences in peak intensity, which was reproducible in multiple experiments (Figure 3B). This variation in breakage across the genome was generally independent of cell cycle, although some cell-cycle-related genes were differentially cut between resting and cycling cells (Figure 3C). Although the overall DSB accumulation was higher in the repair-deficient LIG4−/− background relative to WT, evidenced by the increased number of reads at all broken sites (Figure 3D), the relative breakage at any given AsiSI site was independent of genotype. This suggests that the fluctuation in DSBs (Figure 3A) reflects differences in RE targeting rather than differences in DNA repair.
Figure 3. Variation in AsiSI Targeting across the Genome.

(A) Number of reads at each AsiSI cut site in the genome in G1-arrested LIG4−/− pre-B cells. A total of 1,088 non-overlapping AsiSI sites are sorted by chromosome position.
(B) Comparison of number of reads at each AsiSI site between biological replicas.
(C) Comparison of number of reads at each AsiSI site between G1-arrested versus cycling cells. Blue circles highlight AsiSI cut sites with relatively more reads in cycling versus non-cycling cells, and red circles highlight those sites that are more cut in resting cells. Cell-cycle-related genes (Jun, Trps1), genes that are activated in proliferating cells (Dusp5, Mapkk14, Glrx2), or genes with a role in mitosis (Syde2) are blue; genes that inhibit growth or proliferation (Ell2, Gps1, Aurkaip1, Samhd1) or modify histones upon cell-cycle exit (Prmt7) are in red; and AsiSI cut sites are found near the promoters of these genes.
(D) Comparison of number of reads at each AsiSI site between WT versus LIG4−/− cycling cells.
(E) Number of END-seq reads for each AsiSI site that is cut in vivo (left panel) or in vitro (middle panel) in accordance with the methylation status of the two CpGs within the AsiSI site. Right panels show examples of END-seq reads at a non-methylated (top) and methylated (bottom) AsiSI site in vitro and in vivo. A black circle indicates a methylated CpG and the white circle indicates a non-methylated CpG.
(F) Overlap of AsiSI sites produced in vivo or in vitro.
(G–K) Correlation between the number of END-seq reads with chromatin marks H3K4me3 (G), H3K27ac (H), ATACseq (I), transcription of the closest gene (J), and H3K9me2 (K).
Since AsiSI is sensitive to DNA methylation (Iacovoni et al., 2010), we compared the extent of DNA methylation, measured in a pre-B cell line, with DNA cleavage across the genome detected by END-seq. Overall, DNA methylation correlated inversely with AsiSI cleavage both in vivo and in vitro with purified enzyme (Figure 3E), and the majority of DSB sites overlapped (93%) under these two conditions (Figure 3F).
Despite the significant coincidence between in vitro and in vivo cleavage (Figure 3F), there was no correlation between the efficiency of cutting under the two conditions (Figure S2E). Since histones and chromatin-associated proteins are removed by Proteinase K prior to in vitro digestion, this suggested that DNA sequence per se does not determine the variability of the RE activity. Instead, we hypothesized that epigenetic features might contribute to AsiSI cutting efficiency in vivo. Consistent with this, we found that sites that were cut efficiently in vivo correlated with marks of open chromatin (H3K4me3, H3K27ac, transposase-accessible chromatin) and transcription, and inversely correlated with closed chromatin marks (H3K9me2) (Figures 3G–3K). Thus, beyond the DNA sequence surrounding the AsiSI site, chromatin accessibility contributes to AsiSI targeting.
Nucleotide Resolution Mapping of End Resection
END-seq signals generated by cutting DNA extracted in agarose plugs in vitro with purified AsiSI enzymes produced a perfectly regular block on either side of the DSB (Figure 1B). In contrast, when the DSB was generated in vivo in WT cells, we observed at a low frequency that the first nucleotide read from the proximal adaptor was at a distance from the restriction enzyme target site (Figures S3A–S3C). This suggested that in a small proportion of cells, DSB processing occurred in vivo prior to linker ligation. To test whether END-seq could capture resected DSB ends, we induced AsiSI for 4 hr in cycling WT and LIG4−/− 53BP1−/− pre-B cells. LIG4−/− 53BP1−/− cells are deficient in NHEJ and exhibit hyper-resection (Dorsett et al., 2014). Simultaneously, we performed chromatin immunoprecipitation sequencing (ChIP-seq) with RPA, which binds and stabilizes resected 3′ssDNA tails. Relative to WT cells, which showed little resection at the AsiSI site, there was a dramatic increase in END-seq reads at a distance from the RE site in LIG4/53BP1-deficient cells (Figure 4A). END-seq signals spread away from the initial RE site to a similar extent as ssDNA bound by RPA (Figure 4A). Moreover, in several regions, resection was detectable by END-seq, but not by RPA ChIP-seq (Figure 4B), suggesting an increased sensitivity of our sequencing-based method.
Figure 4. Nucleotide Resolution Mapping of End Resection.

(A) Top panel shows END-seq tracks surrounding an AsiSI DSB generated in Lig4−/− 53BP1−/−cycling pre-B cells. The accumulation of reads away from the DSB is indicative of end resection. Bottom panel shows the read coverage for RPA ChIP-seq for the same interval
(B) END-seq and RPA ChIP-seq reads for an AsiSI site distinct from that shown in (A). RPA ChIP-seq is not detectable above background for this site.
(C) End resection in G1-arrested versus cycling pre-B cells. The bottom track shows RPA ChIP-seq reads for the same interval. End resection is greater in cycling cells than arrested cells.
See also Figure S3.
End resection is dependent on cyclin-dependent kinase activity, which peaks in cycling cells (Ira et al., 2004). Consistent with this, we observed that resection tracks extended further in cycling compared to G1-arrested cells in both WT and LIG4−/− 53BP1−/− genotypes (Figure 4C). Cycling LIG4−/− 53BP1−/− cells exhibited a greater resection length compared to WT (7.8 kb versus 3.4 kb) and, additionally, an increased integrated number of reads mapping away from the RE site (Figure 4C). This suggests that a greater fraction of LIG4/53BP1-deficient cells undergo resection at this site.
RAG Endonuclease On- and Off-Target Activity
To examine the spectrum and frequency of RAG-mediated DSBs, we performed END-seq in LIG4−/− pre-B cell lines in which DSB repair is prevented (Figures 5A and S4A–S4D). Libraries prepared from RAG1−/− B cells were used as a control (Figures 5A and S4A–S4D). Peaks were readily detectable at the endogenous Igκ, Igλ, IgH, TCRα/δ, and TCRγ loci in LIG4−/− cells that were not present in RAG1−/− cells (Figures 5A and S4A–S4D). Breaks formed precisely at RSS heptamer cleavage sites (Figure S4E). In addition, we detected RAG-dependent DSBs at a lower frequency outside of antigen receptor loci (Figure 5B). By comparing LIG4−/− and RAG2−/− libraries, and restricting our analyses to peaks containing the CAC/GTG motif of the heptamer RSS (Lewis et al., 1997) (see Experimental Procedures), we identified 202 off-target, RAG-dependent DSB sites (Figure 5C; Table S3).
Figure 5. RAG Endonuclease On- and Off-Target Activity.

(A) END-seq reads at the Igκ locus for LIG4−/− (top data track) and RAG1−/− (bottom data track) cells. Position of all the V and J gene segments are displayed at the top.
(B) A pair of RAG off-target DSBs on chromosome 1 detected by END-seq at a convergent pair of c-RSSs, previously identified by HTGTS in ATM−/− cells (top right cartoon) (Hu et al., 2015). Both off-target DSBs are highlighted in blue; blue triangles represent cryptic RSSs, and dashed red line indicates RAG cleavage. Middle and lower panels show the magnified view of the SE and CE breaks associated with the cryptic RSSs (triangle) whose sequence is indicated.
(C) Venn diagram comparing the number of RAG off-targets identified by END-seq versus HTGTS.
(D) Consensus sequence logo at cryptic RSSs classified as those that do not contain a nonamer (top) and those that do (bottom).
(E) Boxplot representing the number of reads at each RAG on- and off-target site for LIG4−/− cells. Red triangle indicates the mean value and black solid line is median in each group.
See also Figures S4 and S5 and Table S3.
Remarkably, 54 out of 107 of the DSBs deduced by HTGTS in an independently generated ATM−/− pre-B cell line (Hu et al., 2015) were also found by END-seq in LIG4-deficient cells (Figure 5C). Based on the 202 off-target sites, sequence logos were used to determine a consensus sequence, which matched 12 of 13 canonical heptamer and nonamer positions (Figure 5D). In summary, END-seq detected approximately twice as many breaks as HTGTS, perhaps because only a subset of these is repaired by chromosomal translocation.
Among the 202 RAG off-targets, 30 consisted of pairs (15 pairs) containing convergent c-RSSs within 100 kb in the same chromatin loop defined by ChIP-seq profiles of CTCF/RAD21, 149 were isolated single peaks in which no other off-target was detectable in the vicinity (<100 kb) or in the same chromatin loop, and 23 off-target DSBs were contained within a series of 3 or more c-RSS clusters (average 5 c-RSSs) in which at least one was oriented in a convergent orientation with respect to the others (Figures S5A and S5B; Table S3). This is consistent with the predominance of the convergent positioning of targeted c-RSSs deduced by HTGTS and copy-number variation in thymic lymphomas (Hu et al., 2015; Mijušković et al., 2015). Interestingly, the paired DSBs located within a chromatin loop tended to exhibit similar peak intensities, indicating that these DSBs may be coupled together (Figure 5B).
Each LIG4−/− pre-B cell had at least one distinct γ-H2AX focus after induction with imatinib, indicating that most cells are active for V(D)J recombination (Figure S1B). Consistent with this, it has been estimated by southern blot analyses that 50%–100% of Igκ alleles are cut in LIG4−/− pre-B cell lines (Dorsett et al., 2014). On average, we found that RAG off-target DSBs were 30-fold less abundant than Igκ on-target breakage (Figure 5E). Therefore, assuming that at least 50% of cells are active for Vκ-Jκ recombination, we estimate that up to one out of 60 cells harbors a DSB at a c-RSS.
DSB Repertoire in Thymocytes
Since END-seq does not rely on transfection or exogenously introduced “bait” DSBs, the technique should permit direct detection of DSBs in vivo. To test this, we examined the distribution of DSBs in thymocytes actively undergoing V(D)J recombination (Figure 6A). We isolated unfractionated whole thymocytes from WT, ATM−/−, and RAG2−/− mice (Figures 6A and S6A–S6C). The RAG2−/− mouse expressed a TCRβ transgene, allowing for cellular expansion without recombination (Figures 6A and S6A–S6C). Numerous RAG-dependent DSBs were detected within the (1.6 Mb Vα and 64 Kb Jα) TCRα locus, precisely at annotated RSSs (Figure 6A; Table S4). Among all RSSs annotated to be functional, 99% carried DSBs (Table S4). We also detected DSBs outside of annotated RSSs and in gene segments that have been classified as non-functional (Table S4). In addition to TCRα, DSBs were detected at a lower frequency at TCRβ, TCRγ, and IgH D-J loci (Figures S6A–S6C) (e.g., 80-fold fewer reads at TCRβ relative to TCRα), consistent with rearrangement at these segments occurring at the CD4−CD8−double-negative (DN) stage of development in less than 1% of thymocytes.
Figure 6. DSB Repertoire of Freshly Isolated Thymocytes.

(A) Top panel: END-seq reads at TCRα locus from WT (top track) and RAG2−/− TCRβ (bottom track) thymocytes. The position of all V and J gene segments is displayed as bars above, and TCRα J segments are highlighted in blue. Bottom panel: magnification of the TCRα J region, with the Jα31 highlighted in blue.
(B) END-seq reads at TCRα J31 gene segment.
(C) Boxplot representing the distribution of the number of END-seq reads for each SE and CE in WT thymocytes. Red triangle designates the mean value and black solid line indicates median value in each group.
(D) END-seq reads along the 64 kb TCR Jα locus for three independent WT, three ATM−/−, and two RAG2−/− TCRβ transgenic thymocytes.
See also Figures S6 and S7 and Tables S4 and S5.
In WT thymocytes, SEs dominated the DSB repertoire (Figure 6B), exceeding CE peak intensities by 21-fold (Figure 6C), consistent with previous studies (Roth et al., 1992; Schlissel et al., 1993). SEs are thought to accumulate in WT cells either because they are repaired slower than CEs, or because they are re-cleaved by RAG (Neiditch et al., 2002). After cleavage in vitro, RAG stays associated with the SE in an extra-chromosomal post-cleavage complex (Schatz and Swanson, 2011). We compared the DSB repertoire detected by END-seq to RAG1/2 binding, as determined previously from published ChIP-seq data (Ji et al., 2010; Teng et al., 2015) (Figure 6B). Interestingly, the RAG1 and RAG2 ChIP signals abruptly disappeared precisely at the RSS heptamer sequence, at the border of the SE DSB detected by END-seq (Figure 6B). This indicates that the majority of RAG binding at the TCRα locus in WT thymocytes reflects post-cleavage association with the broken SE.
The DSB repertoire was strikingly similar in thymocytes derived from three independent WT and ATM−/− mice, although the repertoire differed among the two genotypes (Figure 6D). By integrating the total number of reads across the TCRα locus, we estimated that the number of DNA ends in ATM−/− thymocytes was 2.5-fold lower than WT. Since ATM is critical for DSB repair, it was surprising that ATM−/− thymocytes harbored less DNA damage than WT. One possible reason could be because ATM−/− thymocytes harbored DSBs focused mainly in the 5′ Jα segments, whereas DSBs in WT were distributed throughout the Jα cluster (Figure 6D). This is likely because Jα recombination proceeds in a 5′−3′ direction until in-frame, productive VJ joints are positively selected in the thymus (Carico and Krangel, 2015). Since ATM is required for efficient V(D)J recombination (Bredemeyer et al., 2006), we hypothesize that some of the initial 5′ Jα CE breaks are left unrepaired, and therefore fewer cells are able to undergo further rearrangements at the 3′ end of the locus.
The efficiency with which each RSS mediates recombination depends on its sequence. Although the CAC of the heptamer is highly conserved, the remaining positions in the RSS show less conservation. A statistical model to calculate recombination potential of different RSSs and their contribution to the pre-selection repertoire was developed (Cowell et al., 2002). In this algorithm, each RSS is given an “RIC” (recombination information content) score, which is predicted to be proportional to DSB frequency. We observed no correlation between the RIC score and the repertoire of either CEs or SEs in WT thymocytes along the TCRα locus (Figures S7A and S7B). Even in an LIG4−/− background, which should better reflect recombination initiation frequencies, there was no correlation between RIC scores and the DSB repertoire throughout the Jκ locus (Figures S7C and S7D). This highlights the fact that similar to restriction enzyme cleavage (above), chromatin structure, rather than sequence alone, is likely to be a major determinant of RAG targeting in the genome (Teng and Schatz, 2015).
Distinct DSB Ends in WT and ATM−/− Thymocytes
Whereas ATM−/− thymocytes accumulated resected CEs, SEs dominated the WT repertoire (Figures 7A and 7B). Similarly, whereas SE-associated peaks along the Igκ locus tended to be higher than CEs in WT pre-B cells, the opposite pattern emerged (CE > SE accumulation) when the same cells were treated with ATM inhibitor (ATMi) (Figures 7C and 7D). Like ATM-deficient cells, LIG4-deficient cells also showed lower levels of SE versus CE abundance (Figure 5B). We speculate that when SEs join in ATM−/− cells (or occasionally in LIG4-deficient cells), the signal joint exhibits resection. In this case, the RSS would be disrupted and no longer susceptible to RAG re-cleavage (Neiditch et al., 2002). In WT cells, RAG efficiently re-cleaves RSSs once they join, and indeed, they remain bound to SEs (Figure 6B). In ATM-deficient cells, the SE complex may be destabilized, which could promote signal joint formation (Neal et al., 2016).
Figure 7. Structure of DNA Ends in Primary WT and ATM−/− Lymphocytes.

(A) SEs and CEs at the TCRα J61 segment in WT, ATM−/−, and RAG2−/− TCRβ transgenic thymocytes. The position of Jα61 is indicated above the top track and the dashed line shows the SE-CE border.
(B) Scatterplot representing the number of reads at SEs versus CEs in WT and ATM−/− thymocytes for all broken RSSs in the TCRα locus. Diagonal line represents those RSSs with equal number of reads at SEs and CEs.
(C) Examples of SE versus CE reads at Igκ V1–110 in WT pre-B cells with or without ATM inhibitor pretreatment. The position of the Igκ V1–110 gene segment is indicated above the top track and the dashed line indicates the predicted RAG cleavage site.
(D) Difference in number of reads between CEs and SEs sorted in descending order in untreated (left panel) and ATMi pre-treated (right panel) WT pre-B cells. Positive values indicate that the number of reads at the CE is higher than at the SE, whereas negative values indicate greater number of reads at the SEs.
(E) Example of a RAG off-target DSB identified by END-seq in primary thymocytes. Blue triangles represent cryptic RSSs, and the dashed red line shows the RAG cleavage site. WT (top) and ATM−/− (middle) thymocytes show two blocks of reads on both sides of the c-RSS. Sequencing track for RAG2−/− TCRβ is shown below.
(F) Left panel shows END-seq reads on plus and minus strands at the IgH locus (interval between IgH-μ in the constant region and the beginning of IgH-D) in mature WT (first two tracks) and ATM−/− (bottom two tracks) splenic B cells. Cartoon on the right illustrates a break at the IgH locus on one chromosome 12 homolog, with the centromeric fragment captured by the END-seq adaptor. The telomeric fragment is lost during replication earlier during B cell development, and as a result only one end of the original DSB is captured. Red dots denote telomeres.
Off-Target DSBs Detected in WT and ATM−/− Thymocytes
Since off-target V(D)J recombination and translocation is escalated by loss of ATM (Hu et al., 2015), we asked whether we could detect any such sites in primary thymocytes. Using the same criteria as described, we observed 27 c-RSS-associated DSBs in ATM−/− thymocytes, and seven of these sites were also observed in WT thymocytes (Table S5). These included break sites at the Trat1 gene (Figure 7E), which were found to be rearranged precisely at this same c-RSS in murine thymic lymphomas (Mijušković et al., 2015). Interestingly, the CE/SE skewing (WT SE > CE; ATM−/− CE > SE) was maintained even at off-target cryptic RSSs (Figure 7E).
Persistent One-Ended DSBs Accumulate in the Absence of ATM
We have previously suggested that RAG-dependent DSBs can persist throughout cellular division in developing ATM−/− lymphocytes (Callén et al., 2007). ATM−/− B cells that fail primary V(D)J recombination on one allele can achieve productive rearrangement on the other allele, leaving an unresolved DSB in the vicinity of the telomere on chromosome 12. Telomere-deleted chromosome 12 ends were previously detected by fluorescence in situ hybridization (FISH) analysis, suggesting that DSBs produced on the first allele in precursor cells could persist through proliferative expansion (Callén et al., 2007). An alternative hypothesis suggests that rather than long-term persistence, RAG-dependent DSB are re-joined into di-centric chromosomes, which generate new DSBs only during DNA replication after activation in culture (Hu et al., 2014). According to this scenario in resting mature B cells, chromosome ends should be stabilized and should not harbor DSBs. Since END-seq does not require cells to be in cycle, we could test whether non-activated mature ATM−/− B cells harbor DSBs at the IgH locus (Figure 7F). Indeed, whereas there were no detectable DSBs in freshly isolated WT splenic B cells, DSBs near Jh segments were spread over a region of 6.3 kb along the IgH locus in ATM−/− B cells (Figure 7F). In contrast to AsiSI and canonical RAG-dependent DSB, in which each side of the DSB is detectable, persistent RAG-dependent DSBs were “one ended” (Figure 7F). This is likely because the end containing the telomere is lost during cellular division in developing lymphocytes, but the remaining chromo-some segment containing the centromere is maintained (Callén et al., 2007). We conclude that one-ended resected DSBs persist in resting mature ATM−/− B cells.
DISCUSSION
The last decades have seen major advances in our understanding of how DNA breaks are detected, signaled to cell-cycle checkpoints, and repaired (Jackson and Bartek, 2009). However, a more complete understanding of how the genomic landscape influences DSB generation and repair is needed. By offering a global view of DSBs at a given time in a population of cells, END-seq provides a means to investigate interfaces between chromatin state and the DNA damage response. Cellular dilution experiments reveal that peak heights across the genome are proportional to the number of cells carrying the break, and that the sensitivity of the method is at least 1 in 10,000. This is at least an order of magnitude more sensitive than previous methods that assess DNA breakage (Tsai and Joung, 2016). In contrast to technologies that utilize viral transduction, transfection, or translocation, END-seq provides a direct snapshot of DNA ends and is therefore well-suited for in vivo applications.
Global Spectrum of DSBs In Vivo
Previous methods that assess DSB formation or translocation have been limited to tissue culture. To begin to study the spectrum of DSBs in vivo, we monitored site-specific DSBs in freshly isolated thymocytes. Although the post-selection diversity of the T cell repertoire has been previously scrutinized, we are not aware of any global representation of the pre-selection DSB landscape. Several biological insights emerged: (1) the DSB repertoire is remarkably reproducible; (2) a wide range of RSSs are utilized (e.g., all Jα segments annotated to be productive are broken), although there is an overrepresentation of some RSSs over others; (3) the ATM-deficient repertoire is distinct from WT, and resected CEs accumulate; (4) off-target RAGDSBs are detectable in both primary WT and ATM−/− thymocytes; and (5) persistent IgH DSBs are detectable in freshly isolated peripheral B cells from ATM−/− mice. Based on these observations, and the requirement for relatively few cells (two to ten million), we believe that END-seq should be amenable to study DSB and resection dynamics over a broad spectrum of cellular models derived from diverse tissues and organisms.
Applications
Recently, CRISPR-associated Cas9 nuclease has been used to edit disease genes in adult mice (Swiech et al., 2015; Xue et al., 2014), and ZFNs have been utilized for gene therapy in HIV infection in humans (Tebas et al., 2014). To translate somatic genome engineering approaches into safe and effective tools for clinical applications, it is critical to identify Cas9 off-target sites genome-wide. The most sensitive method to date detects off-target sites with frequencies of 1 in 1,000 in a population of cells but is not suitable for in vivo applications (Tsai et al., 2015). As an alternative strategy, END-seq could be used to evaluate faithful targeting as well as off-target cleavage sites of therapeutic nucleases directly in the tissue of interest.
Some anti-cancer agents produce recurrent DNA damage. For example, toposimerase inhibitors preferentially cause DSBs in lineage-specific and transcriptionally active genes (Baranello et al., 2014, 2016). The resulting mutations could be a source of genetic variation both for tumor cells that survive the treatment and for primary cells. Indeed, secondary leukemias can arise from topoisomerase II inhibitor chemotherapy, which is frequently associated with mixed-lineage leukemia (MLL) gene rearrangements. END-seq could potentially be used to map DSBs produced by uncharacterized genotoxic agents in drug development and to interrogate structural variation during tumor evolution.
Influence of Chromatin Structure on Gene Targeting
Stage and cell-type-specific regulation of VDJ recombination is achieved through specific targeting of RAG to accessible Ig and TCR (Yancopoulos and Alt, 1985). Consistent with this, we saw no correlation between the DSB repertoire and the recombination potential of associated RSSs. END-seq offers the most direct measure of RAG-mediated cleavage in vivo and could substantially improve our understanding of the various molecular mechanisms that control V(D)J gene segment usage in developing lymphocytes (Gopalakrishnan et al., 2013). We also monitored the spectrum of DSBs produced by the AsiSI restriction enzyme. Unlike RAG, but similar to Cas9, AsiSI does not normally interact with eukaryotic chromatin but rather phage or plasmid DNA in its physiological setting. We found that AsiSI activity was preferentially targeted to demethylated, open, and transcriptionally active chromatin, and that off-target ZFN DSBs were also associated with transcription. A recent study leveraging large genetic screens suggests that nucleosomes also provide a strong barrier to Cas9 binding and activity (Horl-beck et al., 2016). We therefore hypothesize that the relative strength of Cas9-mediated breakage across the genome would similarly be dependent on chromatin accessibly. Whether or not common sets of rules govern the targeting of various genome-editing enzymes, we suggest that END-seq will provide a rigorous test for future algorithms that predict cleavage sites based both on sequence and chromatin features.
Limitations
Peaks are not detectable by END-seq unless DNA breakage is recurrent. Random damage, such as occurs after γ-irradiation, would produce “background noise” across the genome, which could potentially overwhelm low-level recurrent breakage. Even spontaneous DNA breaks that occur during replication can contribute to END-seq signals. Indeed, when we examined the background levels of DNA breaks in v-abl transformed cells lines, we observed a 1.2- to 2-fold increase in background in the dividing versus G1-arrested cultures. Thus, END-seq is most sensitive when DNA breaks are site specific and requires at least 2,000 cells containing DSBs to work reliably.
Although we observed resection tracks of up to 13 kb, we do not know if there is a limit in the size of ssDNA that can be detected by END-seq. Because the method blunts DNA ends, information about the exact structure of the original overhang is lost. Recent studies indicate that deficiency in 53BP1 promotes not only 3′ ssDNA, but also long 5′ overhangs (Dorsett et al., 2014). The resulting non-uniform distribution of 3′ overhangs would nevertheless produce the same blunted DNA end, and the first base sequenced would be identical. Finally, END-seq may not capture the full spectrum of DNA end structures (e.g., hairpinned intermediates or covalent complexes between proteins and DNA ends). Nevertheless, END-seq could be adapted to remove such structures prior to end ligation.
In summary, we have developed a new resource that provides a simple and robust strategy to measure DNA breaks in vivo. END-seq may therefore extend the range of applications that rely on measuring the location and frequency of low-level DNA breaks. This opens up the possibility for better understanding the causes and consequences of genomic instability.
EXPERIMENTAL PROCEDURES
END-Seq
Single-cell suspensions of thymocytes (70 million), pre-B cells (40 million), or B cells (15 million) were washed in PBS, resuspended in cell suspension buffer, embedded in agarose, and transferred into plug molds (Bio-Rad CHEF Mammalian Genomic DNA plug kit). Plugs were allowed to solidify at 4°C and were then incubated with Proteinase K solution (Puregene, QIAGEN) for 1 hr at 50°C and then for 7 hr at 37°C, followed by consecutive washes in a wash buffer containing 10 mM Tris (pH 8.0) and 50 mM EDTA (Wash Buffer) and then in a buffer containing 10 mM Tris (pH 8.0) and 1 mM EDTA (TE Buffer). Washed plugs were subsequently treated with RNaseA (Puregene, QIAGEN), washed again in Wash Buffer, and stored at 4°C (up to 2 weeks). Blunting, A-tailing, and finally ligation to biotinylated hairpin adaptor 1 (ENDseq-adaptor-1, 5′-Phos-GATCGGAAGAGCGTCGTGTAGGGAAAGAGTGUU[Biotin-dT]U[Biotin-dT]UUACACTCTTT CCCTACACGACGCTCTTCCGATC*T-3′ [*phosphorothioate bond]) were performed in the plug (as detailed in Figure 1A) to minimize externally produced DNA damage. For in vitro digestion of AsiSI samples, plugs were equilibrated in NEB CutSmart buffer and incubated with 100 U AsiSI in a volume of 500 μL for 4 hr at 37°C. Following ligation of adaptor 1, DNA was recovered by first melting the agarose plugs and then digesting the agarose with GELase (Epicenter), using manufacturer-recommended protocols. DNA recovered from melted plugs was sheared to a length between 150 and 200 bp by sonication (Covaris), and biotinylated DNA fragments were purified using streptavidin beads (MyOne C1, Invitrogen). Following streptavidin capture, the newly generated ends were end repaired using T4 DNA polymerase (15 U), Klenow fragment (5 U), and T4 polynucleotide kinase (15 U); A-tailed with Klenow exo-fragment (15U); andfinally ligated to hairpin adaptor 2 using theNEB Quick ligation kit(Figure 1A; ENDseq-adaptor-2, 5′-Phos-GATCGGAAGAGCACACGTCUUUUUUUUAGACGTGTGCTCTTCCGATC*T-3′ [*phosphorothioate bond]).
After the second adaptor ligation, libraries were prepared by first digesting the hairpins on both adapters with USER enzyme (NEB) and PCR amplified for 16 cycles using TruSeq index adapters. Control input libraries from thymocytes, pre-B cells, or B cells were generated using 5 ng sheared DNA and processed as detailed above in the second adaptor processing step, with the exception of the streptavidin purification and USER digestion steps. Control libraries were prepared using Illumina TruSeq universal adapters and 15 cycles of PCR. All libraries were quantified using Picogreen or qPCR. Sequencing was performed either on the Illumina Hiseq2500 (50 bp single-end reads) or on the Illumina Nextseq500 (75 bp single-end reads).
Cell Lines, Immunofluorescence, and Mice
Supplemental Experimental Procedures provides an in-depth description of the Abelson-transformed pre-B cell lines, DSB induction conditions, and mice. Animal experiments were approved by the Animal Care and Use Committee of NCI-Bethesda.
ChIP-Seq, RNA-Seq, and BLESS
RPA ChIP-seq was performed in parallel with END-seq as described (Yamane et al., 2013). H3K4me3 and H3K27Ac ChIP-seq data were derived from Lane et al. (2014) (GEO: GSE48555), ATAC-seq in pre-B cells from Mandal et al. (2015) (GEO: GSE63302), methylC-seq in pre-B cells from Benner et al. (2015) (GEO: GSM1867947), H3K9me2 in pre-B cells from Choukrallah et al. (2015) (GEO: GSM1463436), RAG1 ChIP-seq in WT thymocytes from Teng et al. (2015) (GEO: GSE69478), and RAG2 ChIP-seq in WT thymocytes from Ji et al. (2010) (GEO: GSE21207). For RNA-seq, total RNA was isolated from RAG1−/− pre-B cells, mRNA libraries were prepared using the TruSeq Stranded mRNA HT Sample Prep Kit (Illumina), and RNA was sequenced on an Illumina HiSeq2500. For comparing END-seq and BLESS, we induced AsiSI in G1-arrested LIG4−/− pre-B cells and divided the cell pellets (40 million cells each) for parallel processing by END-seq and BLESS following the published protocol (Crosetto et al., 2013).
Sequence Analysis
Unprocessed END-seq single-end reads were aligned to the GRCm38/mm10 assembly of the mouse genome using either Novoalign (Novocraft) or Bowtie2 (Langmead and Salzberg, 2012), and alignment files were generated using SAMtools (Li et al., 2009) and BEDtools (Quinlan and Hall, 2010). The UCSC Genome Browser was used for data visualization (Kent et al., 2002). AsiSI sites were identified by scanning the mouse genome for its target sequence 5′-GCGATCGC-3′. For BLESS, only reads containing the proximal adaptor barcode were used and evaluated against the same number of reads derived from END-seq.
For peak calling, control input libraries were generated in parallel using the same sheared DNA of END-seq samples, but without streptavidin purification, to have a quantitative representation of the DNA that was available for END-seq adaptor ligation and purification. Input libraries were used by the peak calling algorithm to estimate the background distribution and identify regions of the genome significantly enriched in the samples above background. Peak calling for AsiSI-expressing samples was performed using the FindPeaks function (parameters were as follows: ‒region, size ‒150, ‒minDist 500, min reads 35) in HOMER v4.8.2 (Heinz et al., 2010). Peak calling for RAG-expressing samples was performed using SICER v1.1 (Zang et al., 2009) by first comparing END-seq reads to a Poissonian distribution, followed by comparison with control input library (parameters were as follows: window size 300, e-value 100, gap size 0, minimum summit height 20). The peaks identified with the set of parameters described above were used as bona fide peaks for downstream analysis using SAMtools (Li et al., 2009) and BEDtools (Quinlan and Hall, 2010). Peak calling for RAG off-targets and RIC scores is described in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
END-seq provides a high-resolution view of DNA breaks and end resection
END-seq detects at least one DSB among 10,000 cells not harboring DSBs
END-seq maps DSBs during antigen receptor rearrangements and genome editing
END-seq can be used to study DSB formation/repair in various tissues and organisms
ACKNOWLEDGMENTS
We thank Avinash Bhandoola, Ferenc Livak, Sam John, and Nicola Crosetto for stimulating discussions, and Robert L. Walker, Marbin Pineda, Gustavo Gutierrez-Cruz, Stefania Dell’Orso, and Jαck Zhu for help on sequencing and data handling. This work was supported by the Intramural Research Program of the NIH, the National Cancer Institute, and by the Center for Cancer Research FLEX Program. B.P.S. was supported by NIH grant R01 AI074953.
Footnotes
ACCESSION NUMBERS
The accession number for the sequencing data reported in this paper is SRA: PRJNA326246.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and five tables and can be found with the article online at http://dx.doi.org/10.1016/j.molcel.2016.06.034.
REFERENCES
- Baranello L, Kouzine F, Wojtowicz D, Cui K, Przytycka TM, Zhao K, and Levens D (2014). DNA break mapping reveals topoisomerase II activity genome-wide. Int. J. Mol. Sci 15, 13111–13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranello L, Wojtowicz D, Cui K, Devaiah BN, Chung HJ, Chan-Salis KY, Guha R, Wilson K, Zhang X, Zhang H, et al. (2016). RNA polymerase II regulates topoisomerase 1 activity to favor efficient transcription. Cell 165, 357–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow JH, Faryabi RB, Callén E, Wong N, Malhowski A, Chen HT, Gutierrez-Cruz G, Sun HW, McKinnon P, Wright G, et al. (2013). Identification of early replicating fragile sites that contribute to genome instability. Cell 152, 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner C, Isoda T, and Murre C (2015). New roles for DNA cytosine modification, eRNA, anchors, and superanchors in developing B cell progenitors. Proc. Natl. Acad. Sci. USA 112, 12776–12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, and Sleckman BP (2006). ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 442, 466–470. [DOI] [PubMed] [Google Scholar]
- Bunch H, Lawney BP, Lin YF, Asaithamby A, Murshid A, Wang YE, Chen BP, and Calderwood SK (2015). Transcriptional elongation requires DNA break-induced signalling. Nat. Commun 6, 10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, and Nussenzweig A (2013). End-joining, translocations and cancer. Nat. Rev. Cancer 13, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callén E, Jαnkovic M, Difilippantonio S, Daniel JA, Chen HT, Celeste A, Pellegrini M, McBride K, Wangsa D, Bredemeyer AL, et al. (2007). ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell 130, 63–75. [DOI] [PubMed] [Google Scholar]
- Carico Z, and Krangel MS (2015). Chromatin dynamics and the development of the TCRα and TCRδ repertoires. Adv. Immunol 128, 307–361. [DOI] [PubMed] [Google Scholar]
- Cathomen T, and Joung JK (2008). Zinc-finger nucleases: the next generation emerges. Mol. Ther 16, 1200–1207. [DOI] [PubMed] [Google Scholar]
- Chen HT, Bhandoola A, Difilippantonio MJ, Zhu J, Brown MJ, Tai X, Rogakou EP, Brotz TM, Bonner WM, Ried T, and Nussenzweig A (2000). Response to RAG-mediated VDJ cleavage by NBS1 and gamma-H2AX. Science 290, 1962–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choukrallah MA, Song S, Rolink AG, Burger L, and Matthias P (2015). Enhancer repertoires are reshaped independently of early priming and heterochromatin dynamics during B cell differentiation. Nat. Commun 6, 8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell LG, Davila M, Kepler TB, and Kelsoe G (2002). Identification and utilization of arbitrary correlations in models of recombination signal sequences. Genome Biol. 3, H0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosetto N, Mitra A, Silva MJ, Bienko M, Dojer N, Wang Q, Karaca E, Chiarle R, Skrzypczak M, Ginalski K, et al. (2013). Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 10, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett Y, Zhou Y, Tubbs AT, Chen BR, Purman C, Lee BS, George R, Bredemeyer AL, Zhao JY, Sodergen E, et al. (2014). HCoDES reveals chromosomal DNA end structures with single-nucleotide resolution. Mol. Cell 56, 808–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, and Alt FW (2015). Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol 33, 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugmann SD, Lee AI, Shockett PE, Villey IJ, and Schatz DG (2000). The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu. Rev. Immunol 18, 495–527. [DOI] [PubMed] [Google Scholar]
- Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, Nowrouzi A, Bartholomae CC, Wang J, Friedman G, et al. (2011). An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol 29, 816–823. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan S, Majumder K, Predeus A, Huang Y, Koues OI, Verma-Gaur J, Loguercio S, Su AI, Feeney AJ, Artyomov MN, and Oltz EM (2013). Unifying model for molecular determinants of the preselection Vb repertoire. Proc. Natl. Acad. Sci. USA 110, E3206–E3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, Isaacs WB, Bova GS, Liu W, Xu J, et al. (2010). Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet 42, 668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, and Sleckman BP (2012). The response to and repair of RAG-mediated DNA double-strand breaks. Annu. Rev. Immunol 30, 175–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck MA, Witkowsky LB, Guglielmi B, Replogle JM, Gilbert LA, Villalta JE, Torigoe SE, Tjian R, and Weissman JS (2016). Nucleosomes impede Cas9 access to DNA in vivo and in vitro. eLife 5, e12677, 10.7554/eLife.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Tepsuporn S, Meyers RM, Gostissa M, and Alt FW (2014). Developmental propagation of V(D)J recombination-associated DNA breaks and translocations in mature B cells via dicentric chromosomes. Proc. Natl. Acad. Sci. USA 111, 10269–10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Zhang Y, Zhao L, Frock RL, Du Z, Meyers RM, Meng FL, Schatz DG, and Alt FW (2015). Chromosomal loop domains direct the recombination of antigen receptor genes. Cell 163, 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Meyers RM, Dong J, Panchakshari RA, Alt FW, and Frock RL (2016). Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat. Protoc 11, 853–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, and Legube G (2010). High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 29, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Pellicioli A, BalijJα A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, et al. (2004). DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431, 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jαckson SP, and Bartek J (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Resch W, Corbett E, Yamane A, Casellas R, and Schatz DG (2010). The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell 141, 419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, and Rosenfeld MG (2006). A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 312, 1798–1802. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AA, Chapuy B, Lin CY, Tivey T, Li H, Townsend EC, van Bodegom D, Day TA, Wu SC, Liu H, et al. (2014). Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat. Genet 46, 618–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SM, Agard E, Suh S, and Czyzyk L (1997). Cryptic signals and the fidelity of V(D)J joining. Mol. Cell. Biol 17, 3125–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao PC, et al. (2015). Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161, 1592–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M, Hamel KM, Maienschein-Cline M, Tanaka A, Teng G, TuteJα JH, Bunker JJ, Bahroos N, Eppig JJ, Schatz DG, and Clark MR (2015). Histone reader BRWD1 targets and restricts recombination to the Igκ locus. Nat. Immunol 16, 1094–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijušković M, Chou YF, Gigi V, Lindsay CR, Shestova O, Lewis SM, and Roth DB (2015). Off-target V(D)J recombination drives lymphomagenesis and is escalated by loss of the Rag2 C terminus. Cell Rep. 12, 1842–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal JA, Xu Y, Abe M, Hendrickson E, and Meek K (2016). Restoration of ATM expression in DNA-PKcs-deficient cells inhibits signal end joining.J. Immunol 196, 3032–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiditch MB, Lee GS, Huye LE, Brandt VL, and Roth DB (2002). The V(D)J recombinase efficiently cleaves and transposes signal joints. Mol. Cell 9, 871–878. [DOI] [PubMed] [Google Scholar]
- Pannunzio NR, Li S, Watanabe G, and Lieber MR (2014). Non-homologous end joining often uses microhomology: implications for alternative end joining. DNA Repair (Amst.) 17, 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, Alexandrov LB, Van Loo P, Cooke SL, Marshall J, et al. (2014). RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet 46, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross WE, and Shipley N (1980). Relationship between DNA damage and survival in formaldehyde-treated mouse cells. Mutat. Res 79, 277–283. [DOI] [PubMed] [Google Scholar]
- Roth DB, Nakajima PB, Menetski JP, Bosma MJ, and Gellert M (1992). V(D)J recombination in mouse thymocytes: double-strand breaks near T cell receptor delta rearrangement signals. Cell 69, 41–53. [DOI] [PubMed] [Google Scholar]
- Schatz DG, and Ji Y (2011). Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol 11, 251–263. [DOI] [PubMed] [Google Scholar]
- Schatz DG, and Swanson PC (2011). V(D)J recombination: mechanisms of initiation. Annu. Rev. Genet 45, 167–202. [DOI] [PubMed] [Google Scholar]
- Schlissel M, Constantinescu A, Morrow T, Baxter M, and Peng A (1993). Double-strand signal sequence breaks in V(D)J recombination are blunt, 5′-phosphorylated, RAG-dependent, and cell cycle regulated. Genes Dev. 7 (12B), 2520–2532. [DOI] [PubMed] [Google Scholar]
- Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y, Trombetta J, Sur M, and Zhang F (2015). In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol 33, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, et al. (2014). Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med 370, 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng G, and Schatz DG (2015). Regulation and evolution of the RAG recombinase. Adv. Immunol 128, 1–39. [DOI] [PubMed] [Google Scholar]
- Teng G, Maman Y, Resch W, Kim M, Yamane A, Qian J, Kieffer-Kwon KR, Mandal M, Ji Y, Meffre E, et al. (2015). RAG represents a widespread threat to the lymphocyte genome. Cell 162, 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, and Joung JK (2016). Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat. Rev. Genet 17, 300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol 33, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson LM, and Lees-Miller SP (2011). Estrogen receptor α-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis 32, 279–285. [DOI] [PubMed] [Google Scholar]
- Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, Cai W, Yang G, Bronson R, Crowley DG, et al. (2014). CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane A, Robbiani DF, Resch W, Bothmer A, Nakahashi H, Oliveira T, Rommel PC, Brown EJ, Nussenzweig A, Nussenzweig MC, and Casellas R (2013). RPA accumulation during class switch recombination represents 5′−3′ DNA-end resection during the S-G2/M phase of the cell cycle. Cell Rep. 3, 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancopoulos GD, and Alt FW (1985). Developmentally controlled and tissue-specific expression of unrearranged VH gene segments. Cell 40, 271–281. [DOI] [PubMed] [Google Scholar]
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, and Peng W (2009). A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25, 1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.