

Abstract
Rationale
Lymphatic vessels function to drain interstitial fluid from a variety of tissues. Although shear stress generated by fluid flow is known to trigger lymphatic expansion and remodeling, the molecular basis underlying flow-induced lymphatic growth is unknown.
Objective
We aimed to gain a better understanding of the mechanism by which laminar shear stress activates lymphatic proliferation.
Methods and Results
Primary endothelial cells from dermal blood and lymphatic vessels (BECs and LECs) were exposed to low-rate steady laminar flow. Shear stress-induced molecular and cellular responses were defined and verified using various mutant mouse models. Steady laminar flow induced the classic shear stress responses commonly in BECs and LECs. Surprisingly, however, only LECs showed enhanced cell proliferation by regulating the VEGF-A, VEGF-C, FGFR3, and p57/CDKN1C genes. As an early signal mediator, ORAI1, a pore subunit of the calcium release-activated calcium (CRAC) channel, was identified to induce the shear stress phenotypes and cell proliferation in LECs responding to the fluid flow. Mechanistically, ORAI1 induced upregulation of KLF2 and KLF4 in the flow-activated LECs and the two KLF proteins cooperate to regulate VEGF-A, VEGF-C, FGFR3 and p57 by binding to the regulatory regions of the genes. Consistently, freshly isolated LECs from Orai1 knockout embryos displayed reduced expression of KLF2, KLF4, VEGF-A, VEGF-C, and FGFR3, and elevated expression of p57. Accordingly, mouse embryos deficient of Orai1, Klf2, or Klf4 showed a significantly reduced lymphatic density and impaired lymphatic development.
Conclusions
Our study identified a molecular mechanism for laminar flow-activated LEC proliferation.
Keywords: Endothelial shear stress, KLF2, KLF4, ORAI1, lymphatic development, lymphatic capillary, endothelial cell growth, vascular endothelial growth factor
Subject Terms: Developmental Biology, Cell Signaling/Signal Transduction, Vascular Biology, Growth Factors/Cytokines
INTRODUCTION
The lymphatic system is composed of lymphatic vascular networks and lymphoid tissues and is essential for tissue fluid homeostasis, immune cell trafficking and lipid absorption and transport. Lymphatic vessels may be largely classified to two distinct compartments: capillary vs. collecting vessels. Lymphatic capillaries function to uptake tissue fluids, lipids, large molecules and cells, collectively called lymph fluid, from the interstitial space. During this lymph fluid uptake, lymphatic endothelial cells (LECs) lining the capillaries may experience interstitial fluid flow with a basal-to-apical direction at their intercellular junctions. Drained lymph fluid then flows toward downstream collecting lymphatics, imposing laminar shear stress on the luminal surface of capillary LECs. It has been recently reported that, as the embryos develop, accumulating interstitial fluid gradually increases fluid pressure and creates fluid flow that imposes shear stress on the luminal LECs in developing lymphatic capillaries 1, 2. On the other hand, the collecting lymphatic vessels function to transport the lymph fluid to the lymph nodes and back to the circulation. LECs lining the collecting vessels may more frequently experience oscillatory flows. Studies showed that this type of fluid flow serves as a key signal for development of the luminal valves, a hallmark of collecting lymphatic vessels 1, 3, 4.
It has been known that hemodynamics deliver a profound influence on vascular morphogenesis throughout development 5–7. Because interstitial fluid drainage is a primary function of lymphatic vessels, fluid flow force and pattern have been hypothesized to play key roles in lymphatic development as important non-biological stimuli 8. Indeed, previous studies reported that interstitial flow associated with functional drainage acts as a critical lymphangiogenic mediator by controlling LEC migration, VEGF-C expression, and lymphatic capillary network formation 9–11. Increased embryonic fluid drainage was demonstrated to coincide with and promote initial lymphatic development, possibly serving as an embryonic signal for lymphatic expansion 2. Here, we investigated a mechanism by which steady laminar flow can trigger lymphatic expansion. Our study revealed that low-rate steady laminar flow activates a highly selective calcium channel ORAI1 to upregulate Krüppel-Like Factors (KLF) 2 and 4, which directly regulate the genes promoting cell proliferation and survival. Our data not only offers a better understanding of shear force-induced lymphatic expansion, but also provides important insights into the mechanisms whereby endothelial cells incorporate hemodynamics signals into their biological responses.
METHODS
Cell culture, related reagents and flow application
Human primary dermal BECs and LECs were isolated from human foreskins with approval by the Institutional Review Board, University of Southern California (PI: YK Hong) and cultured in Endothelial Basal Media (EBM, Lonza)-based media as previously described 12, 13. Primary human umbilical venous endothelial cells (HUVECs) were purchased and cultured in EBM-based media (EGM Bullet Kit, Lonza). Steady laminar flow was applied using culturing media as previously reported 14 on monolayer cells for indicated times at 2 dyne/cm2 for all experiments in this study. Sources of antibodies are listed in Supplemental Method.
Animal-related work
Animal-related works were approved by the University of Southern California Institutional Animal Care and Use Committee (IACUC) (PI: YK Hong). Prox1-tdTomato 15 and Orai1 knockout (KO) 16–19 mice were previously described. Prox1-CreERT2 mouse was a gift from Dr. Taija Mäkinen (Uppsala University, Sweden) 20. Cdh5(PAC)-CreERT2 mouse was generated and provided by Dr. Ralf Adams (University of Münster, Germany) 21. Floxed Klf2 mouse 22 was kindly provided by Dr. Kristin Hogquist (University of Minnesota) and floxed Klf4 mouse (B6.129S6-Klf4tm1Khk/Mmmh Klf4) 23 was obtained from the Mutant Mouse Regional Resource Centers (MMRRC). All mutant mice were maintained outbred for the experiments.
Isolation of mouse LECs
Mouse embryonic dermal and postnatal lymph node LECs were isolated from embryos harvested at E16.5 as described in Supplemental Methods.
Statistical analyses
Error bars in all graphs represent the mean ± standard deviation (SD), unless otherwise stated. Normally distributed continuous variables between the experimental and control groups were compared by two-tailed t-test. Statistical significance between the two groups was calculated as p value using Microsoft Excel (Microsoft Office) and GraphPad PRISM6 (GraphPad Software, Inc). A P value less than 0.05 is considered to be statistically significant.
RESULTS
Steady laminar flow selectively activates proliferation of lymphatic endothelial cells
Studies have shown that steady laminar flow imposes an anti-proliferative effect to blood vessel-derived endothelial cells through mechanisms involving p21Cip1 and p53 24–30. In comparison, the impact of steady laminar flow on developing lymphatic vessels has not been fully understood. We therefore investigated the effect of steady laminar flow on proliferation of LECs and its underlying molecular basis. As the precise flow shear force levels in developing lymphatic vessels in vivo are unknown, we first evaluated the effect of different doses of shear force (0.25, 0.5, 1, 2, and 5 dyne/cm2) on cultured human dermal LECs and then searched for the effective or preferable force level, also known as the set point 31, that triggers the classic in vitro endothelial shear stress responses. Specifically, we focused on the cellular (cell elongation and alignment along the flow direction), molecular (upregulation of the key shear response regulators, KLF2 and KLF4), and biochemical (activation of intracellular calcium influx) responses 26, 27. These pilot studies revealed that elongation and alignment of LECs could be clearly triggered by steady laminar flow at 2 dyne/cm2 and above (Online Figure I A). In comparison, upregulation of KLF2 and KLF4 was detectable from the lowest shear force examined (0.25 dyne/cm2) and progressively increased as the force level increased (Online Figure I B,C). Moreover, activation of calcium uptake by the laminar flow was clearly detectable at the force levels of 2 and 5 dyne/cm2 (Online Figure I D). Based on these studies, laminar flow force at 2 dyne/cm2 was chosen for our experiments in this study. Under this shear force condition, laminar flow triggered human dermal LECs and BECs to become elongated and aligned to the direction of flow (Fig.1A). Human umbilical venous endothelial cells (HUVECs) did not show as much clear changes in their cell morphology even after 48 hr. probably because the applied shear force (2 dyne/cm2) was much lower than the reported set-point for HUVECs 31. Nonetheless, all endothelial cells upregulated the established shear stress genes, KLF2, KLF4, and eNOS, in response to this low level of shear force (Fig.1B–D). PROX1 expression in LECs was not altered by the current flow condition (Online Figure II), suggesting that the LEC identity was not compromised by this shear force condition 4. Importantly, steady laminar flow significantly stimulated LEC proliferation, while expectedly suppressing the growth of BECs and HUVECs, as determined by three independent assays measuring total cell numbers, the relative number of cells in the S-phase, and the relative percentage of BrdU-incorporated cells (Fig.1E–G). When different doses of shear force were applied from 0 to 5 dyne/cm2, a force level stronger than 1 dyne/cm2 was required to activate proliferation of cultured LECs, wherease 2 dyne/cm2 yielded the highest activation of LEC proliferation (Online Figure III A). Consistent with these data, steady laminar flow at 2 dyne/cm2 suppressed the expression of cyclin-dependent kinase inhibitor 1C (CDKN1C/p57) and, notably, this flow-induced p57 downregulation was only detectable in LECs, but not in BECs and HUVECs (Fig.1H). We also verified these results using freshly isolated mouse lymph node (LN) LECs as another source of LECs: Consistent with human LECs, mouse LN LECs displayed enhanced cell proliferation, upregulation of KLF2 and KLF4, and downregulation of p57 in response to steady laminar flow (Online Figures III B & IV A,E,F). Notably, the laminar flow suppressed cell death of LECs, as well as of BECs and HUVECs as previously reported 14, 32 (Fig.1I). Together, these results demonstrate that, although LECs, BECs and HUVECs largely display comparable classic shear stress responses in response to steady laminar flow, only LECs exhibit the unique pro-growth phenotypes.
Figure 1. Low-rate steady laminar flow selectively activates proliferation of LECs.


(A) Steady laminar flow (LF, 2 dyne/cm2) induced elongated cell morphology and alignment in LECs and BECs, with marginal changes in HUVECs. Scale bars: 50 μm. (B,C) Western blot assays showing upregulation of KLF2 (B) and KLF4 (C) in LECs, BECs and HUVECs in response to steady laminar flow (2 dyne/cm2) for indicated time. (D) Quantitative RT-PCR (qRT-PCR) showing upregulation of eNOS in LECs, BECs and HUVECs by steady laminar flow (2 dyne/cm2) for 8 and 16 hr. (E) Total cell number increase after static culturing or laminar flow exposure. Equal number of LECs, BECs, and HUBECs were plated and subjected or not to laminar flow (2 dyne/cm2). After 24 hr., total cell number was counted and the percent increase from the initial cell numbers was graphed. (F) LECs, BECs, and HUBECs were cultured under the static or flow condition (2 dyne/cm2) and the relative percentage of cells in the S-phase was determined using flow cytometry. (G) BrdU-incorporation assays showing the percent of BrdU-positive LECs under the static or flow condition for 48 hr. Top: Fluorescent images showing BrdU-incorporated cells (Green) and total nuclei (Blue). Bottom: Bar graph representing the percent of the BrdU-positive cells. (H) Western blot assays showing LEC-specific downregulation of p57 by laminar flow (2 dyne/cm2). (I) ELISA-based cell death assays showing that laminar flow (2 dyne/cm2) commonly reduced cell death in all cell types. Error bars in the graphs represent the standard deviation (SD) of the mean. Laminar flow was steadily applied at 2 dyne/cm2 as previously described 14. Using two-tailed t-test, statistical significance was calculated between the static vs. laminar flow conditions, and the significance level was expressed as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Molecular players in the laminar flow-induced lymphatic cell proliferation
We next set out to identify the molecular players in laminar flow-activated LEC proliferation and found that laminar flow at 2 dyne/cm2 commonly upregulated VEGF-A in LECs, BECs and, as reported14, 34, in HUVECs 14, 33 (Fig.2A). Interestingly, however, the flow force induced the expression of VEGF-C and FGFR3 selectively in LECs, not in BECs and HUVECs (Fig.2B,C). A progressive upregulation of VEGF-A, VEGF-C and FGFR3, as well as a gradual downregulation of p57, were detectable in LECs by the increasing laminar flow forces (Online Figure I E–H). This flow-induced regulation of VEGF-A, VEGF-C, and FGFR3 was also confirmed in freshly isolated mouse LECs (Online Figure IV B–D). Moreover, steady laminar flow increased phosphorylation of VEGFR2 and VEGFR3 in LECs without changing the total protein levels (Fig.2D,E). Notably, previous studies reported ligand-independent activation of VEGFR2 34 and VEGFR3 31 by fluid shear stress to induce eNOS activation and arterial remodeling, respectively. We therefore investigated whether the laminar flow-induced phosphorylation of these VEGFRs was dependent on the presence of their ligands. Pre-treatment of LECs with anti-VEGF-A antibody and/or soluble VEGFR3 protein significantly reduced the flow-induced phosphorylation of both VEGFR2 and VEGFR3 (Fig.2F). These data suggest that VEGF-A and VEGF-C, which are upregulated by laminar flow (Fig.2A,B), are necessary for the flow-induced phosphorylation of VEGFR2 and VEGFR3 in LECs. Moreover, small chemical-based inhibition of VEGFR2, VEGFR3, and FGFR3, but not CXCR2 (chosen as a negative control), reduced the flow-activated LEC proliferation (Fig.2G). To exclude potential off-target effects of these chemical inhibitors, non-chemical blocking reagents were also used: An anti-VEGF-A neutralizing antibody and a soluble VEGFR3 protein, individually or together, profoundly inhibited the laminar flow-induced LEC proliferation (Fig.2H). Similarly, siRNA-mediated knockdown of FGFR3 reduced the flow-activated LEC growth (Fig.2I), confirming a significant contribution of FGFR3 to the flow-activated LEC proliferation. Together, our studies demonstrate that low-rate steady laminar flow upregulates VEGF-A, VEGF-C, and FGFR3 in LECs, which together play important roles in the flow-activated proliferation of LECs.
Figure 2. Molecular players in laminar flow-induced LEC proliferation.


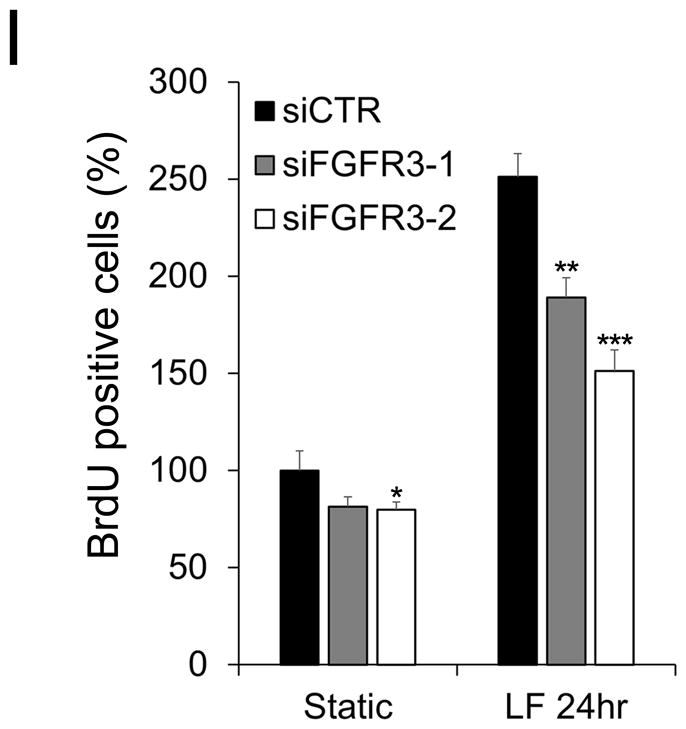
(A–C) Steady laminar flow (LF) upregulated VEGF-A (A) in LECs, BECs, and HUVECs, while activing the expression of VEGF-C (B) and FGFR3 (C) specifically in LECs, determined by ELISA (A,B) and western blot assays (C). (D,E) Laminar flow increased phosphorylation of VEGFR2 (D) and VEGFR3 (E) in LECs. LECs were exposed to laminar flow and then subjected to immunoprecipitation (IP) for VEGFR2 or VEGFR3, followed by immunoblotting (IB) for phosphorylated tyrosine (pY). As controls, LECs under the static condition were treated with VEGF-A (20 ng/ml) or VEGF-C (20 ng/ml) for 15 minutes before cell harvest. (F) Western blot assays showing the ligand-dependency of the flow-induced phosphorylation of VEGFR2 and VEGFR3 in LECs. LECs were pre-incubated for 10 min. with a VEGF-A neutralizing antibody (α-VEGF-A, 20 ng/mL) and/or soluble VEGFR3 receptor (sVEGFR3, 20 ng/mL), followed by static culturing or laminar flow for 24hr. Western blots were performed using antibodies against phospho-VEGFR2, whole VEGFR2, phospho-VEGFR3, whole VEGFR3 and β-actin. As controls, LECs under the static condition were treated with VEGF-A (20 ng/ml) or VEGF-C (20 ng/ml) for 15 min. before cell harvest. (G–I) BrdU-incorporation assays were performed to estimate the roles of VEGFRs and FGFR3 in the laminar flow-induced LEC proliferation. (G) LECs were pre-treated for 10 min. with chemical inhibitors of FGFR3 (FGFRi, 50 μM of PD 166866), VEGFR2 (Ki, 50 μM of Ki8751), VEGFR3 (MAZ, 50 μM of MAZ51), or CXCR2 (SB, 50 μM of SB225002), followed by static culturing or laminar flow exposure for 24 hr. before BrdU assays. (H) LECs were pre-incubated for 10 min. with a VEGF-A neutralizing antibody (α-VEGF-A, 20 ng/mL) and/or soluble VEGFR3 receptor (sVEGFR3, 20 ng/mL), followed by 24-hr. exposure to static culturing or laminar flow and then subjected to the BrdU assays. (I) LECs were transfected with two different siRNAs for FGFR3 (siFGFR3-1, siFGFR3-2), or control siRNA (siCTR), overnight prior to static culturing or laminar flow for 24 hr. and then subjected to BrdU assay. Laminar flow was applied at 2 dyne/cm2 as previously described 14. Error bars indicate the standard deviations (SD) of the mean. Using two-tailed t-test, statistical significance was calculated between the static vs. laminar flow conditions (A,B) or between the control vs. treated groups (G–I). Statistical values: **, p < 0.01; ***, p < 0.001.
ORAI1 mediates the laminar flow-induced calcium influx and KLF2/4 regulation in LECs
We next investigated a mechanism that may contribute to LEC-specific activation of cell proliferation by laminar flow. Because intracellular calcium increase is known to be an immediate response of endothelial cells upon the onset of laminar flow 7, through which shear stress upregulates KLF2 35, we investigated whether the calcium signaling may play a role in orchestrating the laminar flow-induced LEC phenotypes. Time-lapse calcium imaging using a protein calcium reporter GCaMP3 36 showed that low-rate steady laminar flow activated the calcium influx in LECs within the first minute of the flow onset (Online Figure V A,B) 37, consistent with previous studies 37, 38. Moreover, this calcium influx was efficiently blocked by a low concentration of SKF-96365 39, a chemical inhibitor of the store-operated Ca2+ entry (SOCE) 40. In addition, SKF-96365 prevented the laminar flow-induced cell elongation in both LECs and BECs (Fig.3A). To be more specific, we next knocked-down the expression of ORAI1, a pore subunit of the highly selective calcium release-activated calcium (CRAC) channel on the plasma membrane 16–19. Importantly, siRNA-mediated ORAI1 inhibition significantly blocked or delayed the laminar flow-induced cellular elongation of both cell types (Fig.3A). Moreover, ORAI1 knockdown significantly reversed the flow-induced upregulation of KLF2 and KLF4 in LECs with much less impact to BECs (Fig.3B,C). Using freshly isolated mouse embryonic LECs, we also confirmed that SKF-96365 treatment reversed the flow-induced regulation of KLF2 and KLF4 (Online Figure IV G–I). Together, our studies identified ORAI1 as an essential calcium channel for the laminar flow-induced calcium uptake, cell morphology change, and regulation of KLF2 and KLF4 in LECs.
Figure 3. ORAI1 mediates the laminar flow-induced upregulation of KLF2 and KLF4 in LECs.

(A) Inhibition of ORAI1 impaired the flow-induced cellular elongation and alignment of LECs and BECs. LECs or BECs were exposed to laminar flow (2 dyne/cm2) using culture media in the absence (CTR) or presence of SKF (SKF-96365, 10 μM). Alternatively, the cells were transfected with non-specific siRNA (siCTR) or ORAI1 siRNA (siORAI1-1) for 24 hr. before the onset of laminar flow (2 dyne/cm2). Scale bars: 50 μm. (B,C) qRT-PCR analyses showing the expression of KLF4, KLF2 and ORAI1 in LECs (B) and BECs (C), which were transfected with non-specific siRNA (siCTR) or ORAI1 siRNA (siORAI1) for 24 hr. before the onset of laminar flow (2 dyne/cm2). Another set of ORAI1 siRNA (siOrai1-2) was used and comparable results were obtained (Online Figure IV G–I). Error bars indicate the standard deviations (SD) of the mean. Using two-tailed t-test, statistical significance was calculated between the siCTR vs. siORAI1 groups. Statistical values: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
ORAI1 plays a key role in the laminar flow-induced LEC proliferation
In addition, ORAI1 knockdown in LECs strongly reduced the flow-induced expression of VEGF-C and FGFR3, with a marginal suppression of VEGF-A only at 12 hr. (Fig.4A, Online Figure IV J–L). In comparison, ORAI1 knockdown in BECs mainly reversed the VEGF-A upregulation without much impact to the expression of VEGF-C, and FGFR3 (Fig.4B). When calcium influx in LECs was chemically blocked by Bapta-AM or SKF-96365, or genetically inhibited by ORAI1 knockdown, the laminar flow-induced p57 downregulation was significantly abolished (Fig.4C,D, Online Figure IV M). Similarly, inhibition of calcium influx in freshly isolated mouse LECs with SKF-96365 reversed the laminar flow-induced regulation of VEGF-A, VEGF-C, FGFR3, and p57 (Online Figure IV A–D). Consistent with these molecular phenotypes, ORAI1 knockdown also prevented the laminar flow-activated LEC proliferation (Fig.4E). In comparison, however, ORAI1 knockdown did not reverse the flow-mediated suppression of BEC growth. Moreover, freshly isolated mouse embryonic LECs from wild type vs. Orai1 KO embryos displayed differential responses to laminar flow: genetic deletion of Orai1 largely abrogated the above-observed flow-induced regulation of KLF2, KLF4, VEGF-A, VEGF-C, FGFR3 and p57 (Online Figure VI), further verifying an essential role of ORAI1 in the flow-induced regulation of these genes. Therefore, our studies show that ORAI1 is responsible for the shear stress-induced intracellular calcium influx in LECs and plays an essential role in the laminar flow-activated LEC proliferation.
Figure 4. ORAI1 is essential for the laminar flow-response phenotypes of LECs.

(A,B) qRT-PCR analyses showing the expression of VEGF-A, VEGF-C and FGFR3 in LECs (A) and BECs (B) that were transfected with non-specific siRNA (siCTR) or ORAI1 siRNA (siORAI1-1) for 24 hr. and exposed to laminar flow (2 dyne/cm2) for 0, 12 or 24 hr. Another set of ORAI1 siRNA (siOrai1-2) was used and comparable results were obtained (Online Figure IV J-L). (C) Western blot analyses (left) showing the expression of p57 in LECs that were pre-treated with vehicle (DMSO), Bapta-AM (3 μM), or SKF-96365 (SKF, 10 μM) for 30 min. and exposed to laminar flow (2 dyne/cm2) for 0 (Static), 6, 12, or 24 hr. Expression of p57 protein was quantified and normalized against β-actin in the graph (right). (D) Western blot analyses (top) showing the expression of p57 in LECs that were transfected with non-specific siRNA (siCTR) or ORAI1 siRNA (siORAI1) for 24 hr. and exposed to laminar flow (2 dyne/cm2) for 0 (Static), 12, and 24 hr. p57 expression was quantified and normalized against β-actin in the graph (bottom). Another set of ORAI1 siRNA (siOrai1-2) was used and comparable results were obtained (Online Figure IV M). (E) BrdU-incorporation assay showing the relative percent of cells in the S-phase. LECs or BECs were transfected with control siRNA (siCTR) or ORAI1 siRNA (siORAI1) for 24 hr. and cultured under the static or flow condition (2 dyne/cm2) for 24 hr. before BrdU-incorporation assay. Error bars: the standard deviations (SD) of the mean. Statistical significance was calculated using two-tailed t-test between the control vs. treated groups (C), between the siCTR vs. siORAI1 groups (D), or between the siCTR/Static vs. other groups (E). Statistical values: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
ORAI1 deletion reduces lymphatic vessel density during development
We next investigated lymphatic phenotypes in Orai1 KO mice to validate our in vitro findings. Heterozygote Orai1 KO (Orai1 +/−) mice 16, 17 were intercrossed with the Prox1-tdTomato lymphatic reporter mice 15, which allows a convenient visualization of lymphatic vessels due to the expression of the tdTomato reporter under the direction of the Prox1 promoter. From this genetic cross, we obtained Prox1-tdTomato/Orai1 KO embryos (Prox1-tdTomato; Orai1−/−) along with their control heterozygote embryos (Prox1-tdTomato; Orai1+/−) at E14.5. Indeed, Orai1 KO significantly inhibited embryonic lymphatic development with a notable reduction in lymphatic vessel area (Fig.5A–D, Online Figure VII A). Moreover, Orai1 deletion also profoundly reduced lymphatic sprouting, which was documented in detail in a separate study 37. The reduced number of LECs was also detectable in the trachea of rarely-surviving postnatal Orai1 KO mice, where the tracheal lymphatics were significantly smaller in diameter, compared to those in the wild type littermates (Fig.5E–H, Online Figure VII B). In order to confirm the ORAI1-regulated gene expression phenotypes, we determined the expression levels of the flow-regulated molecular players in LECs and BECs freshly isolated from the back skins of control and Orai1 KO embryos. Indeed, genetic deletion of Orai1 caused a reduced expression of VEGF-A, VEGF-C, KLF2, KLF4, and FGFR3 as well as upregulation of p57 in Orai1 KO LECs (Online Figure VIII). These in vivo expression signatures are consistent with the in vitro gene expression profiles seen in the ORAI1-depleted cultured LECs (Figs.3 & 4). In BECs, by comparison, Orai1 KO altered expression of VEGF-A and KLF4 only. Together, our studies suggest that Orai1 deletion prevents the flow-enhanced LEC proliferation and thus impairs lymphatic development due to dysregulation of the molecular players involved in the flow-induced LEC proliferation.
Figure 5. ORAI1 is required for embryonic lymphatic development.

(A–D) Developing dermal lymphatic vessels were visualized in Orai1 heterozygote (+/−) and homozygote (−/−) KO embryos (E14.5) using the Prox1-tdTomato lymphatic reporter 15. Compared to lymphatic vessels of Orai1 heterozygote embryos (A,C, n=5), those of homozygote KO embryos (B,D, n=4) displayed a significantly reduced number of LECs and impaired lymphatic vessel formation in the dorsolateral (A,B) and dorsal midline (C,D) areas. (E–H) Lymphatic vessels in the trachea of rarely surviving 3-week old Orai1 KO mouse (F,H, n=3) were abnormally thinner, compared to those of heterozygote littermates (E,G, n=6). Boxed areas in panels E and F are enlarged in panels G and H, respectively. Scale bars; 100 μm (A–D, G,H), 500 μm (E,F). Relative lymphatic vascular areas were shown in Online Figure VII A,B.
KLF2 and KLF4 directly regulate the molecular players in flow-induced LEC proliferation
Previous studies have genetically placed KLF proteins upstream of the VEGF signaling in the shear-exposed vascular endothelial cells 14, 41, 42. We therefore aimed to establish the genetic relationships among these molecular players. Individual knockdown of KLF2 or KLF4 using two different siRNA complexes markedly altered the flow-induced regulation of VEGF-A, VEGF-C, FGFR3 and p57 (Fig.6A, Online Figure IX A–D). Consistent with this regulation, when mouse LECs freshly isolated from Klf2 KO embryos were subjected to the laminar flow, they displayed defective regulation of these genes (Online Figure IX E). Combined knockdown of KLF2 and KLF4 caused largely additive effects on the flow-induced regulation of the genes (Fig.6A, Online Figure IX F).
Figure 6. KLF2 and KLF4 regulate molecular players in flow-induced LEC proliferation.



(A) qRT-PCR data showing the effects of KLF2, KLF4 or combined knockdown on the expression of VEGF-A, VEGF-C, FGFR3 and p57 in LECs exposed to laminar flow. Knockdown was performed for 24hr. prior to the onset of laminar flow (2 dyne/cm2) for 12 or 24hr. Data were obtained by using 2 independent siRNA for KLF2 (siKLF2-1, siKLF2-2) and KLF4 (siKLF4-1, siKLF4-2), individually and together, and displayed here and Online Figure IX A–D. Expression levels of KLF2 and KLF4 after individual or combined knockdowns are shown in Online Figure IX F. (B,C) qRT-PCR assays showing the expression of these genes in LECs that were infected for 48 hr. with adenovirus expressing KLF2 (Ade-KLF2) (B), or KLF4 (Ade-KLF4) (C). Ade-CTR, control adenovirus. (D) ChIP assays showing association of KLF2 and KLF4 proteins to the regulatory regions of the VEGF-C, FGFR3, and p57 genes. LECs were exposed to laminar flow (2 dyne/cm2) for 6 hr. and ChIP assays were performed using normal IgG, anti-KLF2 or anti-KLF4 antibody and the primers against the indicated upstream sequences (UPS) regions. Detailed locations of these regions can be found in Online Figure X. (E,F) Effects of the individual or combined knockdown of KLF2 and/or KLF4 on the percent of cells in the S phase (E) and on the cell death (F) of LECs. LECs were transfected with control siRNA (siCTR), KLF2 siRNA (siKLF2) and/or KLF4 siRNA (siKLF4) for 24 hr. under the static condition, followed by flow cytometry-based measurement of the S phase cell population (E) or by ELISA-based measurement of cell death (F). Statistical significance was calculated using two-tailed t-test between the siCTR vs. siKLF2/siKLF4 groups (A,E,F), or between the Ade-CTR vs. Ade-KLF2/KLF4 groups (B,C). Statistical values: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Conversely, adenoviral overexpression of KLF2 or KLF4 in LECs significantly upregulated VEGF-A, VEGF-C, and FGFR3, while downregulating p57 (Fig.6B,C). Chromatin immunoprecipitation (ChIP) assays were performed to investigate whether KLF2 and/or KLF4 proteins directly bind to the regulatory sequences of these genes. Many KLF proteins have been reported to bind to similar DNA sequences, known as the KLF consensus binding motif (CACCC), presumably due to a high homology in their zinc finger domains 41. Because KLF2 and KLF4 proteins were previously reported to bind to the VEGF-A gene and regulate its expression 42–44, we focused our study on the other three genes, VEGF-C, FGFR3 and p57. In order to find the functional enhancer regions of the VEGF-C and FGFR3 genes, we took advantage of the epigenetic signatures reported by the Encyclopedia of DNA Elements (ENCODE) Consortium, and identified several regions with the enhancer histone marks (high H3K4Me1 and H3K27Ac, low H3K4Me3) upstream the VEGF-C and FGFR3 genes (Online Figure X). We then investigated whether KLF2 and/or KLF4 proteins are physically associated with these putative enhancer regions by ChIP assays. Indeed, both KLF2 and KLF4 were found to bind to the 210-kb upstream area of the VEGF-C gene and these bindings were profoundly increased by laminar flow (Fig.6D, Online Figure X). Notably, neither protein bound to the130-kb and 50-kb upstream regions. Similarly, when LECs were exposed to steady laminar flow, KLF2 and KLF4 were recruited to a putative enhancer region present 34-kb upstream the FGFR3 coding sequence (Fig.6D, Online Figure X). In comparison, both KLF proteins occupy the proximal promoter of p57 under the static condition, and the laminar flow only slightly increased their binding to the region (Fig.6D).
We next investigated the cellular phenotypes of LECs after the individual or combined knockdown of KLF2 and/or KLF4. Single knockdown of each gene did not clearly alter the flow-induced activation of cell cycle progression of LECs or BECs (Fig.6E). However, simultaneous knockdown of both KLF proteins decreased the S-phase population in LECs, but not in BECs. On the other hand, the flow-enhanced cell survival was reversed in both cell types by combined inhibition of KLF2 and KLF4 (Fig.6F). We next studied whether overexpression of KLF2 and KLF4 could reverse the dysregulation of VEGF-A, VEGF-C, FGFR3 and p57 in ORAI1-depleted LECs. To address this, ORAI1 was knocked-down first and then KLF2 and KLF4 were adenovirally expressed in LECs. These LECs were then subjected to laminar flow, or cultured under the static condition. As expected, laminar flow upregulated VEGF-A, VEGF-C, and FGFR3, and suppressed p57 expression (Online Figure XI A–D) and this flow-induced gene regulation was abrogated by ORAI1 knockdown as seen above (Figs.3 & 4). Importantly, when KLF2 and KLF4 were ectopically expressed in the ORAI1-depleted, flow-exposed LECs, the flow-mediated regulations of VEGF-A, VEGF-C, FGFR3, and p57 were significantly restored. The expected expression levels of ORAI1, KLF2, and KLF4 were also verified (Online Figure XI E–G). Together, these data demonstrate that KLF2 and KLF4, which are downstream targets of ORAI1, directly regulate VEGF-A, VEGF-C, FGFR3, and p57, and play key roles in the laminar flow-induced activation of LEC proliferation.
Abnormal lymphatic development by tissue-specific deletion of Klf2 or Klf4
We next studied the in vivo roles of KLF2 and KLF4 in developing lymphatic vessels through targeted deletion. Mice harboring the floxed Klf2 alleles (Klf2 fl/fl) 45 were crossed with the Cdh5(PAC)-CreERT2 mice (also known as VE-CadCreERT2) expressing the tamoxifen-responsive Cre in endothelial cells 21. Resulting pregnant females were i.p. injected with tamoxifen at E11.5 and E13.5 to induce endothelial-specific deletion of Klf2 (Klf2 ECKO), and the embryos were harvested at E15.5 for vascular analyses. Indeed, the Klf2 ECKO embryos revealed defective lymphatic network formation, characterized with reduced lymphatic branching, irregular vessel thickness, and round-end sprouts, without similar defects in blood vessel development (Fig.7A–D, Online Figure XII A) 37. Image analyses revealed that the relative lymphatic area is significantly reduced in Klf2 ECKO embryos, compared to their control litter embryos (Online Figure VII C).
Figure 7. Defective lymphatic development by targeted deletion of Klf2 or Klf4.


(A–D) Developing dermal lymphatic and blood vessels were stained using anti-Lyve1 (A,C) and anti-Cd31 (B,D) antibodies, respectively, in the control embryos (Klf2 +/+; Cdh5(PAC)-CreERT2) or endothelial-specific inducible Klf2 KO embryos (Klf2 fl/fl;Cdh5(PAC)-CreERT2). Tamoxifen-responsive Cre was activated by intraperitoneal injection of tamoxifen (1.5 mg) into pregnant females at E11.5 and 13.5, and their embryos were harvest at E15.5 for vascular analyses. Relative vascular areas (%) are shown in Online Figure VII C. More than 6 embryos were analyzed per genotype. (E–J) Dermal lymphatic and blood vessels were visualized in the control embryos (Klf4 +/+; Prox1-CreERT2; Prox1-tdTomato) or lymphatic-specific Klf4 KO embryos (Klf4 fl/fl; Prox1-CreERT2; Prox1-tdTomato) at E15.5. Tamoxifen-responsive Cre was activated in the same way as for the Klf2 deletion described above. Lymphatic vessels were visualized using the tdTomato reporter. Enlarged images of the boxed regions are shown in the specified panels. Relative vascular areas (%) are shown in Online Figure VII D. More than 6 embryos were analyzed per genotype. (K,L) The ear lymphatics of wild type (WT) or Cdh5-KLF4 transgenic adult mice 46 were stained with anti-Lyve1 antibody (K). Relative lymphatic vascular area (%) in wild type and Cdh5-KLF4 transgenic mice (n >3) were quantitated (L). Error bars display the standard deviations (SD) of the mean. Statistical values: *, p < 0.05. Scale bars: 500 μm (A–N, S), 100 μm (O–R).
Similarly, the Klf4 gene was deleted selectively in developing lymphatic vessels in embryos using the Prox1-CreERT2 driver line 20. Compared to those in the control litter embryos, the back skins of Klf4 ECKO embryos displayed profound defects in lymphatic branching morphogenesis and hierarchical network formation with a decreased lymphatic density (Fig.7E–J, Online Figure VII D). Cd31-positive blood vessels were unaffected as expected due to the Prox1-CreERT2 –driven lymphatic deletion (Online Figure XII B). Finally, we asked whether overexpression of KLF4 could increase lymphatic density using endothelial-specific Klf4 transgenic mouse (Klf4 EC-Tg) 46, where KLF4 is ectopically expressed under the direction of the Cdh5/VE-Cad promoter. Indeed, lymphatic vessels were significantly enlarged in the ear skins of the Cdh5-Klf4 mouse compared to those in their control litter mates (Fig.7K,L), indicating that ectopic KLF4 expression may increase lymphatic density presumably by activating LEC proliferation. Together, the outcome of these animal-based studies was consistent with our in vitro studies as described above and further demonstrated the important roles of KLF2 and KLF4 in lymphatic development.
DISCUSSION
As oxygen delivery is a major function of blood vessels, oxygen deficiency serves as a strong non-biological stimulus for blood vessel growth. Similarly, as lymphatic vessels function to drain tissue fluid, fluid flow generated by interstitial fluid drainage triggers lymphatic vessel expansion and remodeling 1. Because of their distinct physiological roles, blood and lymphatic vessels are expected to differentially respond to various patterns and forces of fluid flow. Studies have shown that laminar flow suppresses proliferation of blood vessel-derived cells through mechanisms involving p21Cip1 and p53 24–29. However, the effects of laminar flow on lymphatic development and function need to be better understood. In this study, we investigated whether and how low-rate steady laminar flow triggers lymphatic expansion and remodeling, particularly focusing on proliferation and survival of LECs. Based on the outcome of our study, we built a working model for a molecular mechanism underlying the laminar flow-induced LEC proliferation and survival (Online Figure XIII). In this model, the CRAC calcium channel ORAI1 is an early and essential mediator of the laminar flow-induced LEC proliferation. In response to laminar flow, ORAI1 activates intracellular calcium influx and upregulates KLF2 and KLF4. These two KLF proteins together promote the cell cycle progression of LECs through upregulation of VEGF-A, VEGF-C, and FGFR3, and concurrent downregulation of the cell cycle inhibitor, p57. Secreted VEGF-A and VEGF-C may deliver their activities through autocrine and/or paracrine manners, especially onto those present immediately downstream. In comparison, upregulated FGFR3 may make the cells more sensitive to its limited ligands, such as FGF2. We therefore conclude that the interplay of ORAI1 and KLF2/4 proteins may direct the laminar flow-induced lymphatic expansion and remodeling by activating the proliferation and survival of LECs.
The precise shear force level in developing lymphatic networks is not known and it will be technically challenging to determine the force level. Previously, several reports estimated shear levels in certain postnatal lymphatics. The shear force level in a collecting lymphatic vessel was found to be ~0.64 dyne/cm2 under the normal physiological condition 47. Shear levels for mouse tail capillaries 48 and human skin capillaries 49 were about ~0.001 dyne/cm2 and ~0.003 dyne/cm2, respectively. Compared to these quiescent mature postnatal lymphatics, developing lymphatics in the rapidly expanding embryos are likely to experience significantly elevated levels of shear force in order to deal with the overwhelming amount of embryonic tissue fluid. In fact, an elegant study by Planas-Paz et al 2 demonstrated the presence of functional fluid drainage and flow as early as embryonic day 11.5 by showing the interrelationship among embryonic fluid accumulation, fluid pressure increase, stretching of LECs, VEGFR-3 phosphorylation, proliferation of LECs, and functional fluid drainage. In comparison, an anti-proliferative effect on BECs and HUVECs, which was previously seen by the higher, physiologically more relevant shear levels (10~30 dyne/cm2) 24–30, was also detected by our low-rate shear force (2 dyne/cm2).
ORAI1 is a pore component of a calcium-selective ion channel on the plasma membrane and activates the SOCE process 40. Although it was initially discovered from studying defective Ca2+ entry in T cells that is associated with severe combined immune deficiency (SCID), ORAI1 has been found to be expressed in a number of different cell types including arterial, venous and capillary endothelial cells, and to play essential roles in various molecular and cellular responses toward physiological stimuli and pathological insults 50, 51. A recent study convincingly demonstrated that the intracellular calcium dynamics in cultured LECs depends on the magnitude of the shear stress and also that blockage of CRAC channels significantly reduced the calcium mobilization 38. Consistent with this study, we identified ORAI1 as an important CRAC channel responsible for the SOCE process activated by laminar flow in LECs. When ORAI1 was chemically or genetically inhibited in cultured LECs, laminar flow could no longer activate the classic shear stress responses. Moreover, the ORAI1 inhibition in LECs abolished the flow-induced regulation of VEGF-A, VEGF-C, FGFR3 and p57, and efficiently reversed the cell proliferation activated by laminar flow. Consistent with these cellular phenotypes, Orai1 KO embryos displayed significant defects in lymphatic development with reduced numbers of LECs. Our data suggest that KLF2 and KLF4 are downstream effectors of ORAI1 for the laminar flow-induced lymphatic phenotypes.
KLF2 and KLF4 transcription factors have been shown to act as critical regulators of endothelial homeostasis. Because of their closely related structures, functions, and expressions 52–55, they are believed to play shared and/or overlapping roles in vascular development and maintenance. Notably, numerous previous studies show that KLF2 and KLF4 negatively regulate vasculogenesis and angiogenesis 41. KLF2 inhibits VEGF-mediated angiogenesis 56 and laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3 57. KLF2 induces a gene expression pattern that can be seen in functionally quiescent endothelial cells 58 and suppresses angiogenesis of liver endothelial cells through ERK1/2 59. KLF2 decreased the hypoxia-induced VEGF protein level in HUVECs 42. Loss of epigenetic KLF4-mediated transcriptional suppression was found to be crucial for upregulation of VEGF-A in breast cancer cells 44. However, their positive roles in vascular development have also been documented. KLF2 was shown to activate VEGF/VEGFR-2 signaling and survival of HUVECs in response to laminar flow 14. This study, however, did not report any mitogenic activity of the flow-activated VEGF/VEGFR-2 signaling in HUVECs 14. KLF2 cooperates with a ETS family protein ERG to activate Flk1/VEGFR2 expression during vascular development 60. KLF2 and KLF4 genetically interact to maintain endothelial integrity in mouse embryonic vasculogenesis 53. Considering these debated roles of KLF2 and KLF4 in vascular development, it is quite unexpected and unique to find that KLF2 and KLF4 concertedly activated the cell cycle progression of LECs by regulating the expression of VEGF-C, FGFR3 and p57 in response to laminar flow. Especially, the two KLF proteins bind to the enhancer areas present as far as ~210-kb and ~34-kb from the coding sequences of VEGF-C and FGFR3, respectively. In comparison, the two KLF proteins bind to the proximal promoter of p57 to suppress its expression, suggesting that differential transcriptional programs regulate VEGF-C/ FGFR3 vs. p57 in response to the laminar flow. Together, KLF2 and KLF4 proteins may, individually and concertedly, regulate vascular development and maintenance in different manners depending on the physiological and pathological settings of the cells.
One important question in our study was why the low-rate steady laminar flow delivers distinct cell proliferative effects to LECs and BECs. Although different types of endothelial cells may prefer different levels of flow rates, or set points 31, to initiate their remodeling program, our data showed that LECs, BECs and HUVECs commonly displayed the molecular signatures of shear stress responses, most clearly the upregulation of KLF2, KLF4 and eNOS, in response to the low-rate laminar flow condition. In addition, higher rate laminar flows, which are comparable to blood flow, instead suppress proliferation of vascular endothelial cells 24–30. Therefore, we speculate that the flow-induced cell proliferation program is unique to LECs, but absent in BECs, and that distinct pathways may operate to trigger the seemingly opposing proliferative responses between LECs and BECs. It seems that the flow-responsive cell proliferation program in LECs employs the ORAI1-KLF pathway to regulate VEGF-A, VEGF-C, FGFR3 and p57 and to activate the cell cycle progression of LECs. Accordingly, ORAI1 inhibition abrogated the flow-induced cell cycle progression of LECs, while not affecting the flow-induced growth suppression of BECs.
In summary, we demonstrated that low-rate laminar flow activates proliferation of LECs. We also identified the important molecular mediators and players involved in the laminar flow-induced LEC proliferation. We propose that this phenotype and the underlying mechanism are unique to LECs, as proliferation of blood vessel-derived endothelial cells was not stimulated by the same condition, despite the comparable upregulation of KLF2 and KLF4. These findings are consistent with the function and physiology of lymphatic vessels, as LECs would experience an extensive shear stress during functional interstitial fluid drainage. Recent studies showed that the pore-forming subunit of a mechanosensitive ion channel is required for vascular development and plays a key role in integrating vascular architecture with physiological force 61, 62. It will be interesting to define the flow sensing mechanism in LECs, and to study how the flow sensors activate ORAI1 and downstream genes.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Fluid flow profoundly influences lymphatic development and remodeling.
Interstitial pressure-driven lymphatic drainage may activate lymphatic expansion.
What New Information Does This Article Contribute?
Our study revealed that steady laminar flow selectively enhances endothelial cells from lymphatic vessels, but not from blood vessels.
We defined a molecular mechanism by which steady laminar flow activates lymphatic endothelial cell proliferation.
A major function of lymphatic vessel is to drain tissue fluid. While lymphatic vessels are essential for fluid flow, fluid flow also controls lymphatic vessel development. This reciprocal feedback control mechanism appears to be critical for tissue fluid balance and distribution in our body. Here, we investigated this reciprocal feedback control, specifically by deciphering a molecular mechanism explaining how physical force generated by fluid flow can be translated to biological phenomenon, namely cell proliferation. Our study uncovered that fluid flow enhances the intracellular calcium level and thus activates key transcriptional regulators. These regulators induce the expression of genes involved in endothelial growth. Our study provides a mechanistic understanding on flow-induced lymphatic growth.
Acknowledgments
We thank Taija Mäkinen (Uppsala University) for sharing Prox1-CreERT2 mice. We also thank Drs. Guillermo Garcia-Cardena (Harvard Medical School) and Chunming Liu (University of Kentucky College of Medicine) for their kind sharing of KLF2 and KLF4 adenoviruses, respectively.
SOURCE OF FUNDING
This study was supported by NIH grants (HL121036 (YH), HL119583 (YH), EY026260 (YH)), American Heart Association Grant-In-Aid (13GRNT17060131 (YH)) and the L.K. Whittier Foundation (YH, AW). The project was also supported in part by an award (P30CA014089) from the National Cancer Institute.
Nonstandard Abbreviations and Acronyms
- BECs
Blood vascular Endothelial Cells
- LECs
Lymphatic Endothelial Cells
- CRAC
Calcium Release-Activated Calcium
- KLF
Krüppel-Like Factors
- HUVECs
Human Umbilical Venous Endothelial Cells
- SOCE
Store-Operated Ca2+ Entry
Footnotes
AUTHOR CONTRIBUTIONS
DC, EP, EJ, YS, MH, SL, JB, and GG performed experiments and collected data.
SS, YG, CK, EB, AH provided the resources.
JP, AW, YH designed and supervised the research.
DISCLOSURES
The authors declare no conflict of interest with this study.
References
- 1.Schwartz MA, Simons M. Lymphatics thrive on stress: mechanical force in lymphatic development. EMBO J. 2012;31:781–782. doi: 10.1038/emboj.2011.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Planas-Paz L, Strilić B, Goedecke A, Breier G, Fässler R, Lammert E, Strilic B. Mechanoinduction of lymph vessel expansion. The EMBO journal. 2012;31:788–804. doi: 10.1038/emboj.2011.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sabine A, Agalarov Y, Maby-El Hajjami H, Jaquet M, Hagerling R, Pollmann C, Bebber D, Pfenniger A, Miura N, Dormond O, Calmes JM, Adams RH, Makinen T, Kiefer F, Kwak BR, Petrova TV. Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic-valve formation. Dev Cell. 2012;22:430–445. doi: 10.1016/j.devcel.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 4.Sweet DT, Jimenez JM, Chang J, Hess PR, Mericko-Ishizuka P, Fu J, Xia L, Davies PF, Kahn ML. Lymph flow regulates collecting lymphatic vessel maturation in vivo. J Clin Invest. 2015;125:2995–3007. doi: 10.1172/JCI79386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dewey CF, Jr, Bussolari SR, Gimbrone MA, Jr, Davies PF. The dynamic response of vascular endothelial cells to fluid shear stress. J Biomech Eng. 1981;103:177–185. doi: 10.1115/1.3138276. [DOI] [PubMed] [Google Scholar]
- 6.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies PF. Hemodynamics in the Determination of Endothelial Phenotype and Flow Mechanotransduction. In: Aird WC, editor. Endothelial Biomedicine. Cambridge University Press; 2007. pp. 230–245. [Google Scholar]
- 8.Swartz MA, Boardman KC., Jr The role of interstitial stress in lymphatic function and lymphangiogenesis. Ann N Y Acad Sci. 2002;979:197–210. doi: 10.1111/j.1749-6632.2002.tb04880.x. discussion 229–134. [DOI] [PubMed] [Google Scholar]
- 9.Ng CP, Helm CL, Swartz MA. Interstitial flow differentially stimulates blood and lymphatic endothelial cell morphogenesis in vitro. Microvasc Res. 2004;68:258–264. doi: 10.1016/j.mvr.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Boardman KC, Swartz MA. Interstitial flow as a guide for lymphangiogenesis. Circ Res. 2003;92:801–808. doi: 10.1161/01.RES.0000065621.69843.49. [DOI] [PubMed] [Google Scholar]
- 11.Breslin JW, Kurtz KM. Lymphatic endothelial cells adapt their barrier function in response to changes in shear stress. Lymphat Res Biol. 2009;7:229–237. doi: 10.1089/lrb.2009.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin JW, Min M, Larrieu-Lahargue F, Canron X, Kunstfeld R, Nguyen L, Henderson JE, Bikfalvi A, Detmar M, Hong YK. Prox1 promotes lineage-specific expression of fibroblast growth factor (FGF) receptor-3 in lymphatic endothelium: a role for FGF signaling in lymphangiogenesis. Mol Biol Cell. 2006;17:576–584. doi: 10.1091/mbc.E05-04-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S, Kang J, Yoo J, Ganesan SK, Cook SC, Aguilar B, Ramu S, Lee J, Hong YK. Prox1 physically and functionally interacts with COUP-TFII to specify lymphatic endothelial cell fate. Blood. 2009;113:1856–1859. doi: 10.1182/blood-2008-03-145789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.dela Paz NG, Walshe TE, Leach LL, Saint-Geniez M, D’Amore PA. Role of shear-stress-induced VEGF expression in endothelial cell survival. J Cell Sci. 2012;125:831–843. doi: 10.1242/jcs.084301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong M, Jung E, Yang S, Jung W, Seong YJ, Park E, Bramos A, Kim KE, Lee S, Daghlian G, Seo JI, Choi I, Choi IS, Koh CJ, Kobielak A, Ying QL, Johnson M, Gardner D, Wong AK, Choi D, Hong YK. Efficient Assessment of Developmental, Surgical and Pathological Lymphangiogenesis Using a Lymphatic Reporter Mouse and Its Embryonic Stem Cells. PLoS ONE. 2016;11:e0157126. doi: 10.1371/journal.pone.0157126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gwack Y, Srikanth S, Oh-Hora M, Hogan PG, Lamperti ED, Yamashita M, Gelinas C, Neems DS, Sasaki Y, Feske S, Prakriya M, Rajewsky K, Rao A. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol. 2008;28:5209–5222. doi: 10.1128/MCB.00360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim KD, Srikanth S, Yee MK, Mock DC, Lawson GW, Gwack Y. ORAI1 deficiency impairs activated T cell death and enhances T cell survival. J Immunol. 2011;187:3620–3630. doi: 10.4049/jimmunol.1100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 19.Liao Y, Erxleben C, Yildirim E, Abramowitz J, Armstrong DL, Birnbaumer L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci U S A. 2007;104:4682–4687. doi: 10.1073/pnas.0611692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bazigou E, Lyons OT, Smith A, Venn GE, Cope C, Brown NA, Makinen T. Genes regulating lymphangiogenesis control venous valve formation and maintenance in mice. J Clin Invest. 2011;121:2984–2992. doi: 10.1172/JCI58050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD, Adams RH. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 22.Weinreich MA, Takada K, Skon C, Reiner SL, Jameson SC, Hogquist KA. KLF2 transcription-factor deficiency in T cells results in unrestrained cytokine production and upregulation of bystander chemokine receptors. Immunity. 2009;31:122–130. doi: 10.1016/j.immuni.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katz JP, Perreault N, Goldstein BG, Lee CS, Labosky PA, Yang VW, Kaestner KH. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development. 2002;129:2619–2628. doi: 10.1242/dev.129.11.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levesque MJ, Nerem RM, Sprague EA. Vascular endothelial cell proliferation in culture and the influence of flow. Biomaterials. 1990;11:702–707. doi: 10.1016/0142-9612(90)90031-k. [DOI] [PubMed] [Google Scholar]
- 25.Dimmeler S, Haendeler J, Rippmann V, Nehls M, Zeiher AM. Shear stress inhibits apoptosis of human endothelial cells. FEBS Lett. 1996;399:71–74. doi: 10.1016/s0014-5793(96)01289-6. [DOI] [PubMed] [Google Scholar]
- 26.Dardik A, Chen L, Frattini J, Asada H, Aziz F, Kudo FA, Sumpio BE. Differential effects of orbital and laminar shear stress on endothelial cells. J Vasc Surg. 2005;41:869–880. doi: 10.1016/j.jvs.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 27.Akimoto S, Mitsumata M, Sasaguri T, Yoshida Y. Laminar shear stress inhibits vascular endothelial cell proliferation by inducing cyclin-dependent kinase inhibitor p21(Sdi1/Cip1/Waf1) Circ Res. 2000;86:185–190. doi: 10.1161/01.res.86.2.185. [DOI] [PubMed] [Google Scholar]
- 28.Lin K, Hsu PP, Chen BP, Yuan S, Usami S, Shyy JY, Li YS, Chien S. Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc Natl Acad Sci U S A. 2000;97:9385–9389. doi: 10.1073/pnas.170282597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abaci HE, Shen YI, Tan S, Gerecht S. Recapitulating physiological and pathological shear stress and oxygen to model vasculature in health and disease. Sci Rep. 2014;4:4951. doi: 10.1038/srep04951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kadohama T, Nishimura K, Hoshino Y, Sasajima T, Sumpio BE. Effects of different types of fluid shear stress on endothelial cell proliferation and survival. J Cell Physiol. 2007;212:244–251. doi: 10.1002/jcp.21024. [DOI] [PubMed] [Google Scholar]
- 31.Baeyens N, Nicoli S, Coon BG, Ross TD, Van den Dries K, Han J, Lauridsen HM, Mejean CO, Eichmann A, Thomas JL, Humphrey JD, Schwartz MA. Vascular remodeling is governed by a VEGFR3-dependent fluid shear stress set point. Elife. 2015:4. doi: 10.7554/eLife.04645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boon RA, Horrevoets AJ. Key transcriptional regulators of the vasoprotective effects of shear stress. Hamostaseologie. 2009;29:39–40. 41–33. [PubMed] [Google Scholar]
- 33.Goettsch W, Gryczka C, Korff T, Ernst E, Goettsch C, Seebach J, Schnittler HJ, Augustin HG, Morawietz H. Flow-dependent regulation of angiopoietin-2. J Cell Physiol. 2008;214:491–503. doi: 10.1002/jcp.21229. [DOI] [PubMed] [Google Scholar]
- 34.Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res. 2003;93:354–363. doi: 10.1161/01.RES.0000089257.94002.96. [DOI] [PubMed] [Google Scholar]
- 35.Wang W, Ha CH, Jhun BS, Wong C, Jain MK, Jin ZG. Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood. 2010;115:2971–2979. doi: 10.1182/blood-2009-05-224824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi D, Park E, Jung E, Seong YJ, Yoo J, Lee S, Hong M, Lee S, Ishida H, Burford J, Peti-Peterdi J, Adams RH, Srikanth S, Gwack Y, Chen CS, Vogel H, Koh CJ, Wong AK, Hong YK. Notch Downregulation by Laminar Flow Promotes Lymphatic Sprouting. J Clin Invest. 2017 doi: 10.1172/JCI87442. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jafarnejad M, Cromer WE, Kaunas RR, Zhang SL, Zawieja DC, Moore JE., Jr Measurement of shear stress-mediated intracellular calcium dynamics in human dermal lymphatic endothelial cells. Am J Physiol Heart Circ Physiol. 2015;308:H697–706. doi: 10.1152/ajpheart.00744.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15:124–134. doi: 10.1016/j.ccr.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 40.Putney JW. Capacitative calcium entry: from concept to molecules. Immunol Rev. 2009;231:10–22. doi: 10.1111/j.1600-065X.2009.00810.x. [DOI] [PubMed] [Google Scholar]
- 41.Atkins GB, Jain MK. Role of Kruppel-like transcription factors in endothelial biology. Circ Res. 2007;100:1686–1695. doi: 10.1161/01.RES.0000267856.00713.0a. [DOI] [PubMed] [Google Scholar]
- 42.Kawanami D, Mahabeleshwar GH, Lin Z, Atkins GB, Hamik A, Haldar SM, Maemura K, Lamanna JC, Jain MK. Kruppel-like factor 2 inhibits hypoxia-inducible factor 1alpha expression and function in the endothelium. J Biol Chem. 2009;284:20522–20530. doi: 10.1074/jbc.M109.025346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Yang C, Gu Q, Sims M, Gu W, Pfeffer LM, Yue J. KLF4 Promotes Angiogenesis by Activating VEGF Signaling in Human Retinal Microvascular Endothelial Cells. PLoS ONE. 2015;10:e0130341. doi: 10.1371/journal.pone.0130341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ray A, Alalem M, Ray BK. Loss of epigenetic Kruppel-like factor 4 histone deacetylase (KLF-4-HDAC)-mediated transcriptional suppression is crucial in increasing vascular endothelial growth factor (VEGF) expression in breast cancer. J Biol Chem. 2013;288:27232–27242. doi: 10.1074/jbc.M113.481184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Katz JP, Perreault N, Goldstein BG, Actman L, McNally SR, Silberg DG, Furth EE, Kaestner KH. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology. 2005;128:935–945. doi: 10.1053/j.gastro.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 46.Hale AT, Tian H, Anih E, Recio FO, 3rd, Shatat MA, Johnson T, Liao X, Ramirez-Bergeron DL, Proweller A, Ishikawa M, Hamik A. Endothelial Kruppel-like factor 4 regulates angiogenesis and the Notch signaling pathway. J Biol Chem. 2014;289:12016–12028. doi: 10.1074/jbc.M113.530956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dixon JB, Greiner ST, Gashev AA, Cote GL, Moore JE, Zawieja DC. Lymph flow, shear stress, and lymphocyte velocity in rat mesenteric prenodal lymphatics. Microcirculation. 2006;13:597–610. doi: 10.1080/10739680600893909. [DOI] [PubMed] [Google Scholar]
- 48.Berk DA, Swartz MA, Leu AJ, Jain RK. Transport in lymphatic capillaries. II. Microscopic velocity measurement with fluorescence photobleaching. Am J Physiol. 1996;270:H330–337. doi: 10.1152/ajpheart.1996.270.1.H330. [DOI] [PubMed] [Google Scholar]
- 49.Fischer M, Franzeck UK, Herrig I, Costanzo U, Wen S, Schiesser M, Hoffmann U, Bollinger A. Flow velocity of single lymphatic capillaries in human skin. Am J Physiol. 1996;270:H358–363. doi: 10.1152/ajpheart.1996.270.1.H358. [DOI] [PubMed] [Google Scholar]
- 50.Beech DJ. Orai1 calcium channels in the vasculature. Pflugers Arch. 2012;463:635–647. doi: 10.1007/s00424-012-1090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruhle B, Trebak M. Emerging roles for native Orai Ca2+ channels in cardiovascular disease. Curr Top Membr. 2013;71:209–235. doi: 10.1016/B978-0-12-407870-3.00009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suzuki T, Aizawa K, Matsumura T, Nagai R. Vascular implications of the Kruppel-like family of transcription factors. Arterioscler Thromb Vasc Biol. 2005;25:1135–1141. doi: 10.1161/01.ATV.0000165656.65359.23. [DOI] [PubMed] [Google Scholar]
- 53.Chiplunkar AR, Curtis BC, Eades GL, Kane MS, Fox SJ, Haar JL, Lloyd JA. The Kruppel-like factor 2 and Kruppel-like factor 4 genes interact to maintain endothelial integrity in mouse embryonic vasculogenesis. BMC Dev Biol. 2013;13:40. doi: 10.1186/1471-213X-13-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Villarreal G, Jr, Zhang Y, Larman HB, Gracia-Sancho J, Koo A, Garcia-Cardena G. Defining the regulation of KLF4 expression and its downstream transcriptional targets in vascular endothelial cells. Biochem Biophys Res Commun. 2010;391:984–989. doi: 10.1016/j.bbrc.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Z, Tang AT, Wong WY, Bamezai S, Goddard LM, Shenkar R, Zhou S, Yang J, Wright AC, Foley M, Arthur JS, Whitehead KJ, Awad IA, Li DY, Zheng X, Kahn ML. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature. 2016;532:122–126. doi: 10.1038/nature17178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bhattacharya R, Senbanerjee S, Lin Z, Mir S, Hamik A, Wang P, Mukherjee P, Mukhopadhyay D, Jain MK. Inhibition of vascular permeability factor/vascular endothelial growth factor-mediated angiogenesis by the Kruppel-like factor KLF2. J Biol Chem. 2005;280:28848–28851. doi: 10.1074/jbc.C500200200. [DOI] [PubMed] [Google Scholar]
- 57.Doddaballapur A, Michalik KM, Manavski Y, Lucas T, Houtkooper RH, You X, Chen W, Zeiher AM, Potente M, Dimmeler S, Boon RA. Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler Thromb Vasc Biol. 2015;35:137–145. doi: 10.1161/ATVBAHA.114.304277. [DOI] [PubMed] [Google Scholar]
- 58.Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, Meijers JC, Voorberg J, Pannekoek H, Horrevoets AJ. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. 2006;107:4354–4363. doi: 10.1182/blood-2005-08-3465. [DOI] [PubMed] [Google Scholar]
- 59.Zeng XQ, Li N, Pan DY, Miao Q, Ma GF, Liu YM, Tseng YJ, Li F, Xu LL, Chen SY. Kruppel-like factor 2 inhibit the angiogenesis of cultured human liver sinusoidal endothelial cells through the ERK1/2 signaling pathway. Biochem Biophys Res Commun. 2015;464:1241–1247. doi: 10.1016/j.bbrc.2015.07.113. [DOI] [PubMed] [Google Scholar]
- 60.Meadows SM, Salanga MC, Krieg PA. Krüppel-like factor 2 cooperates with the ETS family protein ERG to activate Flk1 expression during vascular development. Development. 2009;136:1115–1125. doi: 10.1242/dev.029538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, Sedo A, Hyman AJ, McKeown L, Young RS, Yuldasheva NY, Majeed Y, Wilson LA, Rode B, Bailey MA, Kim HR, Fu Z, Carter DA, Bilton J, Imrie H, Ajuh P, Dear TN, Cubbon RM, Kearney MT, Prasad KR, Evans PC, Ainscough JF, Beech DJ. Piezo1 integration of vascular architecture with physiological force. Nature. 2014;515:279–282. doi: 10.1038/nature13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ranade SS, Qiu Z, Woo SH, Hur SS, Murthy SE, Cahalan SM, Xu J, Mathur J, Bandell M, Coste B, Li YS, Chien S, Patapoutian A. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci U S A. 2014;111:10347–10352. doi: 10.1073/pnas.1409233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.