

ABSTRACT
Background: We investigated the role of diabetes mellitus (DM) and the molecular mechanisms of antidiabetic drugs in prostate cancer (PCa).
Patients and Methods: 167 patients with both DM and PCa underwent radical prostatectomy (RPE). We divided our patient collective into “metformin” users, “insulin” users, “other antidiabetic drug” users and those with “no antidiabetic drug/diet only” (control group) and analyzed differences in PCa aggressiveness and laboratory parameters among treatment groups. In addition, we generated a tissue-micro-array (TMA) from RPE specimens for the analysis of candidate target pathways of antidiabetic drugs by immunohistochemistry (IHC).
Results: Gleason score of both biopsy and RPE, biopsy undergrading, tumor stage as well as positive resection margins did not significantly change among groups. Preoperative body mass-index, PSA, fPSA and prostate volume/weight did not change among the treatment groups. As well, CRP, GOT, GPT, yGT, LDH, amylase, hemoglobin, TSH, FT3 and FT4 did not differ. Metformin or insulin use was not associated with changes in biochemical tumor recurrence or PCa specific mortality rates. However, tissue TMA analyses by IHC showed decreased mTOR activation, as indicated by phospho-mTOR in cancer tissue of patients with metformin and also with insulin use compared to the control group. In addition, we were able to show that the androgen receptor and the epithelial-cell contact marker E-cadherin decreased upon metformin use compared to the control group.
Conclusion: We did not find a connection between antidiabetic drugs and PCa aggressiveness or progression. However, tumor biology seems to be different among patients with and without antidiabetic drugs.
Keywords: Prostate cancer, metformin, insulin, aggressiveness, mortality, molecular pathways
Introduction
Prostate cancer (PCa) is the most common non-cutaneous male cancer and the second-leading cause of cancer death among men in European countries.1 However, the risk factors for PCa development as well as the pathogenic mechanisms of initiation and progression are still under intensive investigation.
Diabetes mellitus (DM) Type 2 that accounts for 90% of DM is a metabolic disorder characterized by insulin resistance and hyperinsulinemia leading to reduced responsiveness of the skeletal muscle, liver and adipose tissue to insulin. Consequently, insulin levels increase to maintain euglycemia.2 The biguanide metformin is the most common prescribed oral antidiabetic drug used in patients with moderately elevated HbA1c levels to re-sensitize to insulin. In patients with persistent hyperglycemia despite consequent use of oral antidiabetics treatment by subcutaneous insulin injections is the standard therapy used in clinical routine.4
In the past few years, large epidemiological studies suggested a link between DM and antidiabetic drugs and several cancer entities including PCa (reviewed in4-7). Preclinical data have shown antineoplastic effects of metformin resulting in growth inhibition through activation of the AMPK pathway and downstream inhibition of the mTOR pathway, blockade of the cell cycle progression as well as attenuation of inflammation8-10. In addition, our own group recently found11 that the androgen receptor (AR), which is the key player in development, progression and therapy of PCa,12 is suppressed upon metformin treatment. However, current clinical data about metformin and PCa are very contradictious. For example, a recent large meta-analysis concluded that metformin is associated with a significant reduction in the PCa cancer risk, but not in all-cause mortality of patients with PCa.13
Previous studies have shown that insulin stimulation increases cell proliferation of PCa cells in-vitro.14 However, clinical and epidemiological reports on the impact of insulin on PCa incidence and aggressiveness are still very conflicting as summarized in a recent Cochrane analysis by Chen et al.15 Furthermore, there are conflicting literature data regarding the influence of antidiabetic drugs on cancer recurrence rates.17,18
To summarize, to date both the molecular and also the clinical effects of antidiabetic drugs in PCa are still not completely unresolved. Therefore, the aim of the present study was 1) to evaluate the impact of metformin and insulin treatment on PCa aggressiveness in the clinical setting and 2) to elucidate the molecular mechanisms of antidiabetic drugs in radical prostatectomy (RPE) specimens of drug users. As there are several studies showing preoperative biochemical standard laboratory parameters to predict PCa prognosis,19,20 we also elucidated the impact of its significance in our patient cohort.
Results
Patient characteristics
A total of 167 patients were included in the study. Among them 32 (19.2%) were insulin users, 61 (36.5%) used metformin and 27 (16.2%) other antidiabetic drugs. 47 patients (28.1%) with pre-diabetic status defined by HbA1c ≤ 6.5% (mean HbA1c 6.17%) but with no pharmacological treatment were included in the study as control group.
Metformin was administered at dosages from 500 mg to a maximum of 3000 mg per day depending on the side effects and efficacy of the substance as antidiabetic drug. The dosage was set up by the treating endocrinologist or general practitioner (GP) and not changed due to study reasons, insulin dosages were daily adapted on the present glucose levels according to food intake.
Patient characteristics are summarized in Table 1. At the time of RPE antidiabetic therapy was in line with standard therapeutic efficacy measured by HbA1c levels (Table 1).
Table 1.
Demographic Data.
| InsulinN = 32 | MetforminN = 61 | Other Antidiabetics N = 27 |
No DM-Medication N = 47 |
Total | p-value | ||
|---|---|---|---|---|---|---|---|
| Age | years | 64.52 | 63.12 | 67.12 | 63.68 | 64.19 | 0.031 |
| ± 6.00 | ± 6.28 | ± 5.23 | ± 5.72 | ± 6.02 | |||
| BMI | kg/m2 | 27.91 | 29.07 | 28.64 | 28.36 | 28.59 | 0.609 |
| ± 4.00 | ± 4.03 | ± 4.05 | ± 3.74 | ± 3.93 | |||
| PSA | ng/mL | 7.30 | 7.09 | 8.26 | 6.97 | 7.27 | 0.409 |
| ± 5.17 | ± 4.77 | ± 5.56 | ± 6.70 | ± 5.55 | |||
| fPSA | % | 13.58 | 13.24 | 14.64 | 13.29 | 13.52 | 0.575 |
| ± 4.67 | ± 4.99 | ± 5.75 | ± 7.04 | ± 5.60 | |||
| HbA1c | % | 7.87 | 6.96 | 7.31 | 6.17 | 7.05 | 0.003 |
| ± 0.68 | ± 0.95 | ± 0.46 | ± 0.59 | ± 0.92 | |||
| Prostate volume | ml | 39.78 | 45.34 | 47.14 | 36.70 | 41.80 | 0.088 |
| ± 12.67 | ± 15.07 | ± 17.06 | ± 10.98 | ± 14.39 | |||
| Prostate weight | g | 44.24 | 46.67 | 47.76 | 41.28 | 44.21 | 0.672 |
| ± 18.98 | ± 18.72 | ± 22.06 | ± 18.31 | ± 19.03 |
Mean ± Standard Deviation; p-value from Kruskal-Wallis test for comparison between groups
Biochemical parameters
As shown in Table 1 PSA (p = 0.409) and free PSA levels (p = 0.575) as well as prostate volume (p = 0.088) did not differ significantly among the patient groups. We further investigated possible differences in preoperative laboratory parameters including the inflammation parameter C-reactive protein (CRP), liver function parameters like GOT (glutamat-oxalacetat-transaminase), GPT (glutamat-pyruvat-transaminase, gamma-glutamyl-transferase (yGT), lactate-dehydrogenase (LDH) and amylase. Moreover, hemoglobin as well as the thyroid parameters TSH (thyreoidea-stimulating hormone) and free thyroxine 3 and 4 (FT3 and FT4) were assessed. However, our data did not reveal a significant difference of all investigated parameters among the treatment group as shown on Table 2.
Table 2.
Biochemical Parameters.
| InsulinN = 32 | MetforminN = 61 | Other Antidiabetics N = 27 |
No DM-Medication N = 47 |
Total | p-value | ||
|---|---|---|---|---|---|---|---|
| CRP | mg/dL | 0.40 | 0.28 | 0.51 | 0.29 | 0.34 | 0.930 |
| ± 0.47 | ± 0.31 | ± 1.14 | ± 0.32 | ± 0.56 | |||
| GOT | U/L | 24.53 | 25.47 | 22.47 | 24.04 | 24.44 | 0.790 |
| ± 10.45 | ± 11.16 | ± 9.22 | ± 8.98 | ± 10.15 | |||
| GPT | U/L | 23.73 | 28.78 | 22.53 | 27.54 | 26.60 | 0.348 |
| ± 9.74 | ± 14.87 | ± 13.10 | ± 12.87 | ± 13.51 | |||
| γGT | 50.35 | 46.62 | 43.58 | 67.70 | 51.34 | 0.351 | |
| U/L | ± 50.74 | ± 29.51 | ± 29.65 | ± 57.24 | ± 41.31 | ||
| LDH | 246.67 | 168.63 | 171.69 | 185.90 | 186.22 | 0.289 | |
| U/L | ± 255.36 | ± 35.37 | ± 35.80 | ± 38.45 | ± 111.16 | ||
| Amylase | 43.50 | 43.50 | 56.00 | 56.50 | 49.88 | 0.839 | |
| U/L | ± 7.78 | ± 7.78 | ± 25.46 | ± 16.26 | ± 13.93 | ||
| Hemoglobin | g/L | 149.53 | 146.87 | 141.84 | 151.70 | 147.61 | 0.178 |
| ± 12.91 | ± 12.68 | ± 17.81 | ± 11.24 | ± 13.63 | |||
| TSH | mU/L | 1.93 | 1.92 | 1.52 | 1.84 | 1.82 | 0.865 |
| ± 1.13 | ± 0.79 | ± 0.64 | ± 0.91 | ||||
| fT4 | ng/L | 19.60 | 18.33 | 19.50 | 14.40 | 18.11 | 0.313 |
| ± 1.36 | ± 1.99 | ||||||
| fT3 | ng/L | 5.27 | 5.06 | 4.24 | 3.20 | 4.64 | 0.299 |
| ± 0.40 | ± 0.83 |
Mean ± Standard Deviation; p-value from Kruskal-Wallis test for comparison between groups
Histopathology of tumor samples
Next we investigated, based on histopathological characteristics, if antidiabetic drugs are associated with PCa aggressiveness in both the biopsy and RPE specimen. Gleason scores of both biopsy and RPE specimen are shown in Table 3 respectively. Analyzing differences in PCa aggressiveness we found no difference concerning Gleason score of both biopsy (p = 0.932) and RPE specimen (p = 0.212) among patients with antidiabetic drugs and the control group (Table 3). In addition, biopsy under-grading or over-grading did not differ among the treatment groups (p = 0.251, Table 3). Moreover, we investigated the pathological states of RPE specimens and divided our patient cohort into two groups ≤ pT2c (organ confined) versus ≥ pT3a (non-organ confined) (Table 3). In line with the Gleason scores also pT stages of the RPE specimens were not significantly different among the treatment groups (p = 0.696). Also the surgical margins of the RPE specimens did not differ among the treatment groups (p = 0.603) (Table 3).
Table 3.
Histology of Biopsy and RPE.
| Insulin | Metformin | Other Antidiabetics | No DM-Medication | Total | ||
|---|---|---|---|---|---|---|
| p = 0.932 (Fisher’s exact test for comparison between groups) | ||||||
| Biopsy GS ≤ 6 |
n | 17 | 33 | 14 | 24 | 88 |
| % | 63.0% | 61.1% | 70.0% | 58.5% | 62.0% | |
| Biopsy GS 7 |
n | 6 | 16 | 5 | 13 | 40 |
| % | 22.2% | 29.6% | 25.0% | 31.7% | 28.2% | |
| Biopsy GS ≥ 8 |
n | 4 | 5 | 1 | 4 | 14 |
| % | 14.8% | 9.3% | 5.0% | 9.8% | 9.8% | |
| Total | n | 27 | 54 | 20 | 41 | 142 |
| % | 100.0% | 100.0% | 100.0% | 100.0% | 100.0% | |
| p = 0.212 (Fisher’s exact test for comparison between groups) | ||||||
| RPE GS ≤ 6 |
n | 13 | 11 | 8 | 16 | 48 |
| % | 40.6% | 18.0% | 29.6% | 34.7% | 28.9% | |
| RPE GS 7 |
n | 15 | 41 | 14 | 21 | 91 |
| % | 46.9% | 67.2% | 51.9% | 45.7% | 54.8% | |
| RPE GS ≥ 8 |
n | 4 | 9 | 5 | 9 | 27 |
| % | 12.5% | 14.8% | 18.5% | 19.6% | 16.3% | |
| Total | n | 32 | 61 | 27 | 46 | 166 |
| % | 100.0% | 100.0% | 100.0% | 100.0% | 100.0% | |
| p = 0.251 (Fisher’s exact test for comparison between groups) | ||||||
| Same Histology | n | 13 | 20 | 4 | 12 | 49 |
| % | 48.1% | 37.0% | 20.0% | 30.0% | 34.8% | |
| Under-grading | n | 11 | 30 | 14 | 20 | 75 |
| % | 40.7% | 55.6% | 70.0% | 50.0% | 53.1% | |
| Over- grading |
n | 3 | 4 | 2 | 8 | 17 |
| % | 11.2% | 7.4% | 10.0% | 20.0% | 12.1% | |
| Total | n | 27 | 54 | 20 | 40 | 141 |
| % | 100.0% | 100.0% | 100.0% | 100.0% | 100.0% | |
| p = 0.696 (Fisher’s exact test for comparison between groups) | ||||||
| ≤ pT2c | n | 21 | 43 | 18 | 28 | 110 |
| % | 65.6% | 70.5% | 66.7% | 59.6% | 65.9% | |
| ≥ pT3a | n | 11 | 18 | 9 | 19 | 57 |
| % | 34.4% | 29.5% | 33.3% | 40.4% | 34.1% | |
| Total | n | 32 | 61 | 27 | 47 | 167 |
| % | 100.0% | 100.0% | 100.0% | 100.0% | 100.0% | |
| p = 0.603 (Fisher’s exact test for comparison between groups; Rx excluded) | ||||||
| R0 | n | 19 | 42 | 18 | 23 | 102 |
| % | 59.4% | 68.8% | 66.7% | 48.9% | 61.1% | |
| R1 | n | 8 | 17 | 7 | 16 | 48 |
| % | 25.0% | 27.9% | 25.9% | 34.1% | 28.7% | |
| Rx | n | 5 | 2 | 2 | 8 | 17 |
| % | 15.6% | 3.3% | 7.4% | 17.0% | 10.2% | |
| Total | n | 32 | 61 | 27 | 41 | 167 |
| % | 100.0% | 100.0% | 100.0% | 100.0% | 100.0% | |
Recurrence rates and pca specific mortality rates
Moreover, we aimed to analyze the long-term clinical courses of patients after RPE. Among 147 patients (follow up of 20 patients missing) 39 patients experienced a biochemical recurrence (BCR). 19.4% of insulin users, 23.1% of metformin users and 22.7% of patients who consumed other antidiabetic drugs had a BCR, while in the group of patients with no medication 38.1% of patients had a BCR (p = 0.274). Analyzing the time to BCR we found no significant differences among patients using antidiabetic drugs compared to the control group. However, this could be due to a lack of statistical power since the cumulative recurrence-free survival is slightly worse in the no medication group compared to the treated patients from a mere descriptive point of view (Figure 1).
Figure 1.
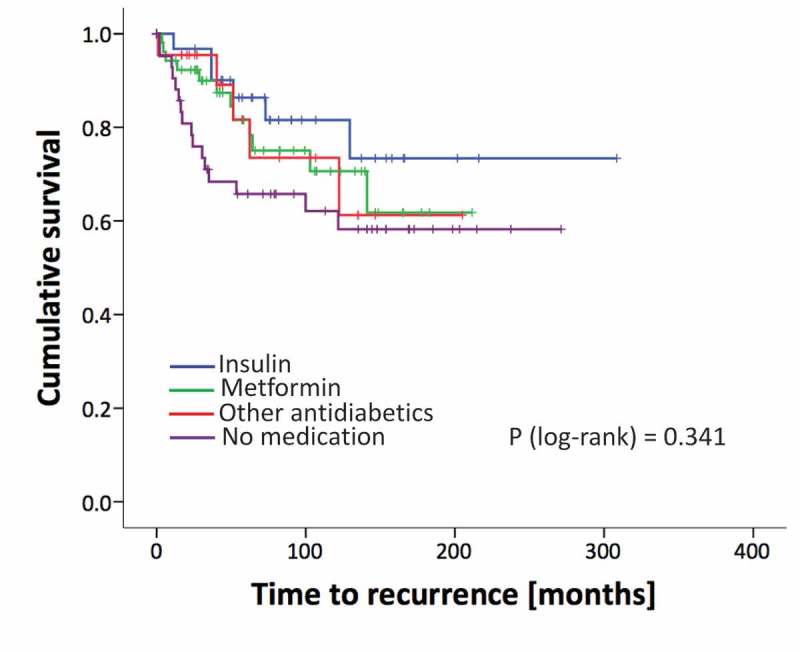
Kaplan Meier curves of recurrence rates (BCR) of patients analyzed for different treatment groups. p-value from the log-rank test.
Long-term follow up revealed a BCR in 39 patients who then underwent salvage radiation therapy. Of these patients 17 experienced a second recurrence and received hormonal therapy subsequently. 10/17 patients of them progressed to a metastatic castration resistant stage and were treated with docetaxel chemotherapy. Out of these patients, 8 died from PCa. PCa specific survival time is shown in Figure 2. There were no statistically significant differences among groups, which again might be causal due to the small patient number.
Figure 2.
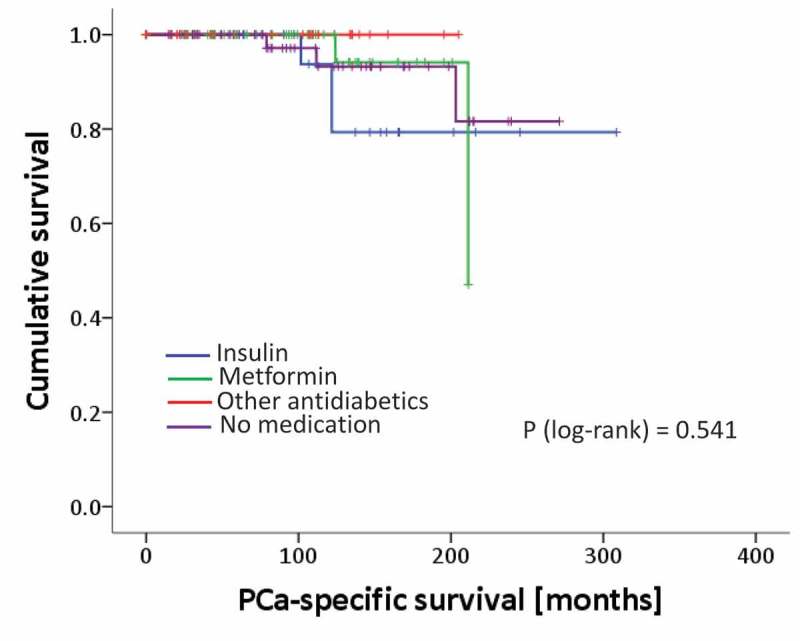
Kaplan Meier curves of patients´ prostate cancer specific mortality rates analyzed for different treatment groups. p-value from the log-rank test.
DM drug effects in prostate tissue
To evaluate effects of DM drugs on benign and malignant prostate cells a tissue microarray (TMA) including benign and cancerous tissue with either metformin or insulin consumption as well as of patients without antidiabetic medication was employed (n = 95).
Thereby we found that the tumor proliferation rate, measured by KI67 staining, was not different in prostate tissue of patients using antidiabetic drugs compared to the control group (Figure 3A).
Figure 3.
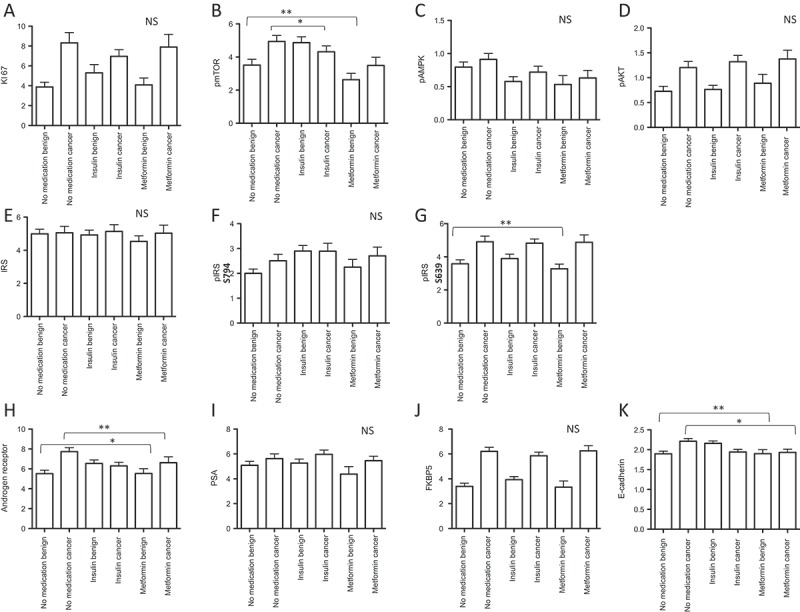
Immunohistochemistry results of radical prostatectomy specimens (benign and cancer cores) stained for different antibodies (A-K). Data are presented as mean values± standard deviations, p-values < 0.05 were considered significant (*p < 0.05; **p < 0.01).
In general, the mTOR pathway has an important impact on tumor growth and metastasis. Previous findings suggest that the antineoplastic effects of metformin include growth inhibition by attenuation of mTOR activity with subsequent blockade of cell cycle progression.21 In contrast, insulin can activate the mTOR pathway via the PI3K-Akt survival pathway. Thus we evaluated the levels of active AMPK – a key primary target of metformin – estimated as phospho-AMPK (Thr172), pAMPK and mTOR, estimated as phospho-mTOR-Ser2448 (pmTOR) in patients’ tissue samples by immunohistochemistry (IHC). Thereby we found that pmTOR was downregulated in the benign prostate tissue of metformin users in comparison to the control group (p = 0.006) (Figure 3B, Supplementary Figure 1A). Interestingly, also insulin use significantly decreased active mTOR (pmTOR) in the cancer tissue of patients (p = 0.024) and showed the same, but statistically insignificant trend in benign tissue (Figure 3B, Supplementary Figure 1A). However, we did not find any differences in pAMPK levels between treatment groups (Figure 3C). Furthermore, we stained for active AKT (as phosphor-pAKT(Ser473), pAkt) on TMA but did not find differences in pAKT levels among treatment groups suggesting an AMPK and AKT independent regulation of the mTOR pathway. In line with previous studies we observed that PCa tissue harbors higher levels of pAKT compared to benign prostate tissue (Figure 3D).
In addition, we measured both total and phosphorylated insulin receptor substrat-1 protein levels (IRS). In general, the insulin signaling pathway is activated when insulin or IGF-1 bind to the insulin receptor, activate the insulin receptor kinase and auto-phosphorylate the insulin receptor which binds and phosphorylates IRS proteins thereby creating docking sites for SH2 domain-containing proteins that bring the signal to the effectors. Thus, we used two different antibodies (pIRS- S639 and pIRS- S794) as IRS-1 contains more than 30 potential serine/threonine phosphorylation sites among them the mTOR pathway mediates phosphorylation of IRS-1 at Ser636/639. In contrast, phosphorylation of IRS-1 at S794 mediates the control of various cellular processes by insulin. When phosphorylated by the insulin receptor it binds specifically to proteins containing SH2 domains such as phosphatidylinositol 3-kinase p85 subunit. In addition, it activates phosphatidylinositol 3-kinase when bound to the regulatory p85 subunit. Interestingly analyzing our patients samples we found that only pIRS- S639 is significantly decreased in benign cores of metformin users compared to the control (p = 0.002) indicating an activated mTOR pathway (Figure 3 E-G, Supplementary Figure 1B).
Our previous in-vitro data demonstrated an attenuation of metformin on the AR and its activity in PCa cells.11 In line with these data, AR immunoreactivity was significantly decreased in tumor tissue of metformin users compared to the control group (p = 0.01). An opposite effect of metformin was observed in the benign tissue cores of metformin users (p = 0.03) (Figure 3H, Supplementary Figure 1C). However, we did not observe significant changes in AR target genes PSA (Figure 3 I) or FKBP5 (Figure 3 J) expression in metformin users compared to the control indicating that metformin is not able to influence AR response mechanisms. In contrast to metformin, insulin use had no significant effect on AR expression.
A prerequisite of metastasis is the ability of tumor cells to migrate and invade surrounding tissue. Epithelial-to-mesenchymal transition (EMT) transforms epithelial tumor cells to motile mesenchymal-like cells with enhanced metastasizing capacity.22,23 A characteristic marker for EMT is the loss of E-cadherin expression. E-cadherin immunostaining revealed that metformin has the capacity to reduce this epithelial phenotype cell marker in cancer tissue (p = 0.04) and to increase it in the benign tissue (p = 0.003) (Figure 3K, Supplementary Figure 1D). Insulin use again was not associated with a difference compared to no medication.
Discussion
In the recent years many studies explored the impact of DM and antidiabetic drugs on PCa. A large number of studies found that the antidiabetic drug metformin reduces the risk of developing PCa.24,25 However, less data is available addressing the question if metformin influences prognosis of PCa patients suffering from concurrent DM. For example, in 2013, Spratt et al published the first clinical retrospective data indicating that metformin use may improve progression free survival and PCa mortality.26
In the present study we demonstrate no significant differences concerning pathological stage and Gleason score of different diabetic drug users in comparison to the control group. Moreover, there was no significant difference in biopsy tumor over- or under-grading. These findings are in line with previous reports also demonstrating no pathological changes upon metformin use.19,27,28
Considering the hypothesized biological mechanisms of metformin and insulin, we investigated their impact on cancer progression however, failed to show any significant beneficial or worsening effects of antidiabetic drugs with respect to PCa pathological stage, PCa specific mortality as well as BCR, albeit both metformin and insulin users showed a statistical trend towards a lower recurrence rate.
Implicating the relatively low patient number in this and all other studies investigating this issue, our data encourage for further elucidating the role of metformin, but also insulin with regard to recurrence rates after RPE. Several previous studies also found no substantial changes of BCR rates after RPE.19,28,29 In contrast, a study of Patel et al. demonstrated increased BCR after RPE in metformin users29 whereas Danzig et al. found metformin use in combination with statins significantly associated with a reduced risk of BCR (hazard ratio (HR) = 0.190; 95% CI 0.040–0.905; p = 0.037).20
Given the fact, that only a small number of patients included in our study died from PCa and the observational time frame was relatively short, we did not find an effect of antidiabetic drugs on PCa mortality, although a statistical under-powering cannot be excluded. Nevertheless our finding is in line with results of a recent meta-analysis involving 334.532 participants and reporting no association between metformin and PCa mortality.30 On the other hand, two other large studies found a reduced mortality of PCa patients upon metformin use.26,31
Although the anti-tumor effect of metformin has been observed in different types of cancers a clear mechanism of action remained elusive. To our best knowledge the present study is the first analyzing proposed molecular pathways of antidiabetic drugs in the prostate tissue of patients suffering from both PCa and DM. In line with the clinical data we did not find any differences in cell proliferation in the prostate tumor tissue of different DM drug users. This finding is in contrast to a recent experimental study on human PC3 cells (PCa bone metastasis derived cell line) showing that metformin reduces cell proliferation in vitro and in vivo.32
As mentioned a downstream effector of metformin is the mTOR pathway. Indeed, we observed that pmTOR was downregulated in the benign prostate tissue of metformin users in comparison to the control group (p = 0.006).This finding is in line with previous studies.33,34
In contrast to other clinical studies we investigated not only the effect of metformin, but also of insulin on PCa aggressiveness. Insulin and insulin-like growth factors (IGF) are key regulators of cellular growth and metabolism. A large number of experimental studies including ours have shown that insulin, IGFs and their receptors are overexpressed in PCa.35,36 Despite the substantial effects in preclinical models insulin use had no impact on tumor histopathology or on recurrence rates after RPE in PCa patients with concurrent DM. AMPK is a prime target of metformin. Interestingly, we did not find any differences in pAMPK levels of patients treated with metformin or insulin or those without medication. This is in line with other studies presenting that AMPK is dispensable for metformin’s beneficial effects.37–39 For example, metformin restricts the nuclear pore complex (NPC) and thus attenuates RagC activation of mTOR signaling independent of AMPK.40
Concerning AKT a survival kinase upstream of mTOR, we also did not find differences of the pAKT levels among patients with antidiabetic drugs (metformin and insulin) or those without antidiabetic medication. This finding might support the hypothesis of an AKT-independent regulation of mTOR by mitogen-responsive pathways as described by Memmott et al.41
As we observed an AMPK and AKT independent mTOR regulation, we speculated abn involvement of the IRS-1 and found that pIRS- S639 is significantly decreased in benign cores of metformin users compared to the control (p = 0.002) indicating an activated mTOR pathway. Both metformin and insulin use reduced pmTOR levels in both benign and cancer areas compared to no medication and thereby may exert antitumor effects. Whereas this is expected for metformin it is contradictory to most published studies describing a tumor promoting effect of insulin partly regulated via activation of the mTOR pathway. We speculate that this may be due to the heterogeneous mode of action of insulin. For example, in a previous study we stimulated both diverse prostate cancer cells and benign cells with insulin and or IGF (insulin like growth factor) and found that benign cells do not react with an increase in cell proliferation upon insulin and/or IGF stimulation and activate basal to luminal differentiation upon insulin and/or IGF stimulation instead.35 In addition, others and we observed negative feedback loops when cells were stimulated with higher insulin concentrations.35,42 As the present study is a real-life study and insulin was administered according to the glucose levels and insulin sensitivity of the hyperglycemic patients. We do not have information about the exact dosage of insulin, which can consequently vary significantly. Therefore, an induction of a negative feedback-loop cannot be excluded and may result in the observed reduced pmTOR levels.
Recent work from our research group found that metformin disrupts the AR translational MID1 regulator complex leading to downregulation of the AR protein.11 In agreement with that finding metformin use decreased the AR in cancerous tissue and increased it in the non-cancerous tissue of RPE specimens. This novel metformin effect might be confirmed in a recently started prospective clinical study investigating the impact of addition of metformin to the AR modulating agent abiraterone for the treatment of metastatic castration resistant PCa patients (NCT01677897). However, further analyses revealed that the AR target genes PSA and FKBP5 are not affected upon metformin or insulin treatment.
Metformin was also suggested to attenuate metastasis formation by inhibiting EMT.43 However, E-cadherin expression, downregulation of which is a consequence of EMT, displayed an incoherent picture, which does not support this hypothesis. It was moderately upregulated in the non-malignant but downregulated in the malignant tissue of metformin users in comparison to the patients without DM drugs.
A clear limitation of our study is the fact that we do not have any information about the duration of antidiabetic drug use. Our control group included pre-diabetic patients with a mean HbA1c of 6.17% (manifest DM HbA1c ≥ 6.5%). As this study presents a “real-life” situation we did not attempt to adapt/optimize the corresponding antidiabetic therapy. From clinical practice we know that adherence and compliance to anti-diabetic drug treatment vary significantly and for sure this is one limitation of the study protocol.44,45 For future studies, especially prospective studies working on this topic, for sure, a second control group of patients without DM is inevitable. Moreover, a major limitation of the study is the retrospective single center character and the relatively small patient number.
Currently no clear evidence exists about dosing of antidiabetic drugs for their anticancer efficacy. To investigate this in detail and set-up separate dose finding studies would be needed (also in patients without diabetes, who were not included in this study). Furthermore, best to our knowledge there is no known biomarker to monitor anticancer-efficacy of antidiabetic drugs although it would be of high medical need. As mentioned, we included only patients with DM in the study, therefore the study cannot be generalized to a whole patient population. For sure, studies on metformin in non-diabetic and euglycemic patients would deliver important information on general efficacy of antidiabetic drugs as therapeutic options in prostate cancer. Several studies using metformin in addition to approved PCa drugs are currently running.
To summarize, this study elucidates for the first time preclinical identified anti-tumorous molecular mechanisms of metformin in patient tissue samples. Moreover, we provide tissue expression data indicating attenuation of the metabolic- and proliferative mTOR pathway independently of pAMPK and pAKT. With view on clinical outcome after RPE, we failed to show beneficial or worsening effects of antidiabetic drugs use by PCa patients.
Patients and methods
Ethical statement
The study was approved by the Ethical Committee of the Medical University Innsbruck (study number AN2014-0145 336/4.24) and written informed consent to participate in research studies was obtained from all patients.
Patients and data acquisition
We retrospectively analyzed 167 patients diagnosed with both DM and PCa who underwent an open retropubic or robotic assisted (Da Vinci) RPE at our department. We divided our patient cohort into four groups: “metformin” users, “insulin” users, “other antidiabetic drug” users (glitazones, sulfonyl urea and α-glucosidase inhibitors) as well as those with “no antidiabetic drug- diet only” (pre-diabetic situation, control group) and scrutinized for differences in PCa aggressiveness.
For monitoring the efficacy of antidiabetic drugs we measured HbA1c at one-time point before RPE (the day before surgery) and we did not include long term follow up measurements or investigation of long term antidiabetic drug adherence.
Moreover, we assessed potential changes in serum biochemical parameters C-reactive protein (CRP), liver function parameters like GOT (glutamat-oxalacetat-transaminase), GPT (glutamat-pyruvat-transaminase, gamma-glutamyl-transferase (yGT), lactate-dehydrogenase (LDH) and amylase, hemoglobin as well as thyroid parameters TSH (thyreoidea-stimulating hormone) and free thyroxine 3 and 4 (FT3), which were assessed in a blood sample routinely drawn the day before RPE.
In addition, we performed a long-term follow-up analysis of patients (until 02/2015) including PCa specific mortality rates of the local tumor registry. If patients did not have their oncological follow-up at the Department of Urology, Medical University Innsbruck, their treating outpatient urologists where contacted to communicate the oncological follow up information.
Statistical evaluation
All demographic and baseline characteristics as well as histopathological and biochemical parameters were analyzed descriptively (absolute and relative frequency for qualitative data and mean and standard deviation [SD] for quantitative data), stratified by medication group (metformin/insulin/other antidiabetics/no medication). Fisher’s exact test and Kruskal-Wallis test (since assumption of normality was violated for most parameters) were performed for group comparisons. Kaplan Meier product-limit estimation curves for time to recurrence of PCa and PCa-specific survival were produced and groups were compared with the log-rank test. A significance level of α = 0.05 (two-tailed) was applied. Statistical analyses were conducted in SPSS, version 22.0 (IBM Corp).
Tissue microarray (TMA) and immunohistochemistry (IHC)
To investigate potential changes of candidate target pathways in patients a TMA of patients´ RPE specimens was constructed. Cylindrical samples including three cancer areas and three benign areas were re-located from formalin-fixed, paraffin-embedded tissue blocks to the TMA block. In total, 570 tissue samples from 95 patients were collected. Embedded hepatic cells as well as prostate cells (LNCaP and PC3) were used as controls.
The TMA was assembled using a manual tissue arrayer (Beecher Instruments). H&E and p63/α-methylacyl-CoA racemase IHC double staining was performed to confirm the histological diagnosis. IHC was performed on a Discovery-XT staining device (Ventana). The following antibodies were used: anti-E-cadherin (M3612, Dako), AR (EPR 1535, Abcam); phospho-mTOR (Ser2448, Cell Signaling Technologies), KI67 (M7240, Dako), phospho-AMPKα (Thr172, Cell Signaling Technologies), phospho-AKT (Ser473, Cell Signaling Technologies), PSA (PSA/KLK3 (D11E1) XP, Cell Signaling Technologies), FKBP5 (A301-430A-M, Bethyl), IRS1 (ab40777, Abcam), phospho-IRS-S639 (ab47404, Abcam) and phospho-IRS-S794 (S794, Abcam).
Microscope images were taken with a Zeiss Imager Z2 microscope (Zeiss, Vienna) equipped with a Pixelink PL-B622-CU camera (Canimpex Enterprises Ltd). The IHC evaluation was supervised by an experienced uro-pathologist (G.S.) AR, pmTOR, evaluated using quick score (the proportion of positively stained cells (0–10%: 1 point, 10–50%: 2 points, 50–75%: 3 points, 75–100%: 4 points) and the average staining intensity (light: 1 point, medium: 2 points, strong: 3 points)). KI67 was assessed by the number of KI67-positively stained cells per 100 prostate gland cells. E-cadherin, pAMPK and pAKT, PSA, FKBP5, and pIRS were scored by staining intensity (very week: 0 point, light: 1 point, medium: 2 points, strong: 3 points). T- test was used to compare the differences between the different treatment groups.
supplemental data
Supplemental data for this article can be accessed on the publisher’s website.
Abbreviation lists
| AMP-Kinase | AMPK |
| Androgen receptor | AR |
| Biochemical recurrence | BCR |
| Body mass index | BMI |
| C-reactive protein | CRP |
| Diabetes Mellitus | DM |
| Epithelial-to-mesenchymal transition | EMT |
| Gamma-glutamyl-transferase | yGT |
| General practitioner | GP |
| Gleason Score | GS |
| Glutamat-oxalacetat-transaminase | GOT |
| Glutamat-pyruvat-transaminase | GPT |
| Glycated hemoglobin A1C | HbA1C |
| Insulin-like growth factor | IGF |
| Insulin receptor subatrate | IRS |
| Immunohistochemistry | IHC |
| Lactate-dehydrogenase | LDH |
| Mammalian target of rapamycin | mTOR |
| Prostate cancer | PCa |
| Prostate specific antigen | PSA |
| Protein kinase B/AKT | AKT |
| Radical prostatectomy | RPE |
| Tissue Microarray | TMA |
| Thyreoidea-stimulating hormone | TSH |
Declarations of interest
The study was approved by the Ethical Committee of the Medical University Innsbruck (study number AN2014-0145 336/4.24) and written informed consent to participate in research studies was obtained from all patients. Data and material are available. There are no competing interests or funding. We thank Irma Sottsas and Sarah Peer for helping with TMA generation. Author contributions: Heidegger: Ethics, Study design, data acquisition, data interpreting, manuscript, revision, Zieher: Data analysis, TMA construction and analysis, Pircher: TMA analysis, Revision, Eigentler: IHC preparation, Schäfer: TMA analysis, Fritz: Statistics, Pichler: Data acquisition (recurrence rates, mortality rates), Puhr: TMA picture acquisition, Steiner: Statistics, Horninger: Providing data, principal investigator, Klocker: General idea, supervision, manuscript, revision.
References
- 1.Jemal A. Global burden of cancer: opportunities for prevention. Lancet. 2012. November 24;380(9856):1797–1799. doi: 10.1016/S0140-6736(12)61688-2. [DOI] [PubMed] [Google Scholar]
- 2.Vijan S. Type 2 diabetes. Ann Intern Med. 2010. March 2;152(5):ITC31–15. doi: 10.7326/0003-4819-152-5-201003020-01003. [DOI] [PubMed] [Google Scholar]
- 3.Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996. February 29;334(9):574–579. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 4.Cannata D, Fierz Y, Vijayakumar A, LeRoith D. Type 2 diabetes and cancer: what is the connection? Mt Sinai J Med. 2010. March;77(2):197–213. doi: 10.1002/msj.20167. [DOI] [PubMed] [Google Scholar]
- 5.Vallianou NG, Evangelopoulos A, Kazazis C. Metformin and cancer. Rev Diabet Stud. 2013;10(4):228–235. doi: 10.1900/RDS.2013.10.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, Gandini S. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila). 2010. November;3(11):1451–1461. doi: 10.1158/1940-6207.CAPR-10-0157. [DOI] [PubMed] [Google Scholar]
- 7.Clements A, Gao B, Yeap SH, Wong MK, Ali SS, Gurney H. Metformin in prostate cancer: two for the price of one. Ann Oncol. 2011. December;22(12):2556–2560. doi: 10.1093/annonc/mdr037. [DOI] [PubMed] [Google Scholar]
- 8.Sahra IB, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, Le Marchand-Brustel Y, Giorgetti-Peraldi S, Cormont M, Bertolotto C, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010. March 15;70(6):2465–2475. doi: 10.1158/0008-5472.CAN-09-2782. [DOI] [PubMed] [Google Scholar]
- 9.Sahra IB, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008. June 5;27(25):3576–3586. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 10.Flavin R, Zadra G, Loda M. Metabolic alterations and targeted therapies in prostate cancer. J Pathol. 2011. January;223(2):283–294. doi: 10.1002/path.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demir U, Koehler A, Schneider R, Schweiger S, Klocker H. Metformin anti-tumor effect via disruption of the MID1 translational regulator complex and AR downregulation in prostate cancer cells. BMC Cancer. 2014;14:52. doi: 10.1186/1471-2407-14-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong YN, Ferraldeschi R, Attard G, De BJ. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat Rev Clin Oncol. 2014. May 20;11:365–376. doi: 10.1038/nrclinonc.2014.72. [DOI] [PubMed] [Google Scholar]
- 13.Yu H, Yin L, Jiang X, Sun X, Wu J, Tian H, Gao X, He X, Medeiros R. Effect of metformin on cancer risk and treatment outcome of prostate cancer: a meta-analysis of epidemiological observational studies. PLoS One. 2014;9(12):e116327. doi: 10.1371/journal.pone.0116327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heidegger I, Ofer P, Doppler W, Rotter V, Klocker H, Massoner P. Diverse functions of IGF/insulin signaling in malignant and noncancerous prostate cells: proliferation in cancer cells and differentiation in noncancerous cells. Endocrinology. 2012. October;153(10):4633–4643. doi: 10.1210/en.2012-1348. [DOI] [PubMed] [Google Scholar]
- 15.Chen YB, Chen Q, Wang Z, Zhou J. Insulin therapy and risk of prostate cancer: a systematic review and meta-analysis of observational studies. PLoS One. 2013;8(11):e81594. doi: 10.1371/journal.pone.0081594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rieken M, Kluth LA, Xylinas E, Fajkovic H, Becker A, Karakiewicz PI, et al. Association of diabetes mellitus and metformin use with biochemical recurrence in patients treated with radical prostatectomy for prostate cancer. World J Urol. 2014. August;32(4):999–1005. doi: 10.1007/s00345-013-1171-7. [DOI] [PubMed] [Google Scholar]
- 17.Danzig MR, Kotamarti S, Ghandour RA, Rothberg MB, Dubow BP, Benson MC, Badani KK, McKiernan JM. Synergism between metformin and statins in modifying the risk of biochemical recurrence following radical prostatectomy in men with diabetes. Prostate Cancer Prostatic Dis. 2015. March;18(1):63–68. doi: 10.1038/pcan.2014.47. [DOI] [PubMed] [Google Scholar]
- 18.Rocha P, Morgan CJ, Templeton AJ, Pond GR, Naik G, Sonpavde G. Prognostic impact of C-reactive protein in metastatic prostate cancer: a systematic review and meta-analysis. Oncol Res Treat. 2014;37(12):772–776. doi: 10.1159/000369545. [DOI] [PubMed] [Google Scholar]
- 19.Matsuyama H, Shimabukuro T, Hara I, Kohjimoto Y, Suzuki K, Koike H, Uemura H, Hayashi T, Ueno M, Kodaira K, et al. Combination of hemoglobin, alkaline phosphatase, and age predicts optimal docetaxel regimen for patients with castration-resistant prostate cancer. Int J Clin Oncol. 2014. October;19(5):946–954. doi: 10.1007/s10147-013-0638-2. [DOI] [PubMed] [Google Scholar]
- 20.Zakikhani M, Dowling RJ, Sonenberg N, Pollak MN. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res (Phila). 2008. October;1(5):369–375. doi: 10.1158/1940-6207.CAPR-08-0081. [DOI] [PubMed] [Google Scholar]
- 21.Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013. November;19(11):1438–1449. doi: 10.1038/nm.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nauseef JT, Henry MD. Epithelial-to-mesenchymal transition in prostate cancer: paradigm or puzzle? Nat Rev Urol 2011;Aug(8):428–439. doi: 10.1038/nrurol.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lehman DM, Lorenzo C, Hernandez J, Wang CP. Statin use as a moderator of metformin effect on risk for prostate cancer among type 2 diabetic patients. Diabetes Care. 2012. May;35(5):1002–1007. doi: 10.2337/dc11-1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Preston MA, Riis AH, Ehrenstein V, Breau RH, Batista JL, Olumi AF, Mucci LA, Adami H-O, Sørensen HT. Metformin use and prostate cancer risk. Eur Urol. 2014. December;66(6):1012–1020. doi: 10.1016/j.eururo.2014.04.027. [DOI] [PubMed] [Google Scholar]
- 25.Spratt DE, Zhang C, Zumsteg ZS, Pei X, Zhang Z, Zelefsky MJ. Metformin and prostate cancer: reduced development of castration-resistant disease and prostate cancer mortality. Eur Urol. 2013. April;63(4):709–716. doi: 10.1016/j.eururo.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaushik D, Karnes RJ, Eisenberg MS, Rangel LJ, Carlson RE, Bergstralh EJ. Effect of metformin on prostate cancer outcomes after radical prostatectomy. Urol Oncol. 2014. January;32(1):43–47. doi: 10.1016/j.urolonc.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allott EH, Abern MR, Gerber L, Keto CJ, Aronson WJ, Terris MK, Kane CJ, Amling CL, Cooperberg MR, Moorman PG, et al. Metformin does not affect risk of biochemical recurrence following radical prostatectomy: results from the SEARCH database. Prostate Cancer Prostatic Dis. 2013. December;16(4):391–397. doi: 10.1038/pcan.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel T, Hruby G, Badani K, Abate-Shen C, McKiernan JM. Clinical outcomes after radical prostatectomy in diabetic patients treated with metformin. Urology. 2010. November;76(5):1240–1244. doi: 10.1016/j.urology.2010.03.059. [DOI] [PubMed] [Google Scholar]
- 29.Deng D, Yang Y, Tang X, Skrip L, Qiu J, Wang Y, Zhang F.. Association between metformin therapy and incidence, recurrence and mortality of prostate cancer: evidence from a meta-analysis. Diabetes Metab Res Rev. 2015. February 23;31:595–602. doi: 10.1002/dmrr.v31.6. [DOI] [PubMed] [Google Scholar]
- 30.Margel D, Urbach DR, Lipscombe LL, Bell CM, Kulkarni G, Austin PC, Fleshner N. Metformin use and all-cause and prostate cancer-specific mortality among men with diabetes. J Clin Oncol. 2013. September 1;31(25):3069–3075. doi: 10.1200/JCO.2012.46.7043. [DOI] [PubMed] [Google Scholar]
- 31.Kato H, Sekine Y, Furuya Y, Miyazawa Y, Koike H, Suzuki K. Metformin inhibits the proliferation of human prostate cancer PC-3 cells via the downregulation of insulin-like growth factor 1 receptor. Biochem Biophys Res Commun. 2015. May 22;461(1):115–121. doi: 10.1016/j.bbrc.2015.03.178. [DOI] [PubMed] [Google Scholar]
- 32.Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, Tanti J-F, Giorgetti-Peraldi S, Bost F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011. July 1;71(13):4366–4372. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- 33.Li HX1, Gao JM1, Liang JQ1, Xi JM1, Fu M1, Wu YJ1. Vitamin D3 potentiates the growth inhibitory effects of metformin in DU145 human prostate cancer cells mediated by AMPK/mTOR signalling pathway. Clin Exp Pharmacol Physiol. 2015. June;42(6):711–717. doi: 10.1111/1440-1681.12409. [DOI] [PubMed] [Google Scholar]
- 34.Heidegger I, Kern J, Ofer P, Klocker H, Massoner P. Oncogenic functions of IGF1R and INSR in prostate cancer include enhanced tumor growth, cell migration and angiogenesis. Oncotarget. 2014. April 2;5. doi: 10.18632/oncotarget.v5i9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012. March;12(3):159–169. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- 36.Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010. July;120(7):2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Griss T, Vincent EE, Egnatchik R, Chen J, Ma EH, Faubert B, Viollet B, DeBerardinis RJ, Jones RG, Green DR. Metformin antagonizes cancer cell proliferation by suppressing mitochondrial-dependent biosynthesis. PLoS Biol. 2015. December 1;13(12):e1002309. doi: 10.1371/journal.pbio.1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalender A, Selvaraj A, Kim SY, Gulati P, Brûlé S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010. May 5;11(5):390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu L, Zhou B, Oshiro-Rapley N, Li M, Paulo JA, Webster CM, Mou F, Kacergis MC, Talkowski ME, Carr CE, et al. An ancient, unified mechanism for metformin growth inhibition in C. elegans and cancer. Cell. 2016. December 15;167(7):1705–1718.e13. doi: 10.1016/j.cell.2016.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Memmott RM, Dennis PA. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal. 2009. May;21(5):656–664. doi: 10.1016/j.cellsig.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi R, Berkel HJ, Yu H. Insulin-like growth factor-I and prostate cancer: a meta-analysis. Br J Cancer. 2001. September 28;85(7):991–996. doi: 10.1038/sj.bjc.6691961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Shen C, Wang L, Ma Q, Xia P, Qi M, Yang M, Han B. Metformin inhibits epithelial-mesenchymal transition in prostate cancer cells: involvement of the tumor suppressor miR30a and its target gene SOX4. Biochem Biophys Res Commun. 2014. September 26;452(3):746–752. doi: 10.1016/j.bbrc.2014.08.154. [DOI] [PubMed] [Google Scholar]
- 43.García-Pérez LE1, Alvarez M, Dilla T, Gil-Guillén V, Orozco-Beltrán D. Adherence to therapies in patients with type 2 diabetes. Diabetes Ther. 2013. December 4;4(2):175–194. doi: 10.1007/s13300-013-0034-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahmad NS, Ramli A, Islahudin F, Paraidathathu T. Medication adherence in patients with type 2 diabetes mellitus treated at primary health clinics in Malaysia. Patient Prefer Adher. 2013. June 17;7:525–530. doi: 10.2147/PPA.S44698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.