

Summary
CellMinerCDB provides a web-based resource (https://discover.nci.nih.gov/cellminercdb/) for integrating multiple forms of pharmacological and genomic analyses, and unifying the richest cancer cell line datasets (the NCI-60, NCI-SCLC, Sanger/MGH GDSC, and Broad CCLE/CTRP). CellMinerCDB enables data queries for genomics and gene regulatory network analyses, and exploration of pharmacogenomic determinants and drug signatures. It leverages overlaps of cell lines and drugs across databases to examine reproducibility and expand pathway analyses. We illustrate the value of CellMinerCDB for elucidating gene expression determinants, such as DNA methylation and copy number variations, and highlight complexities in assessing mutational burden. We demonstrate the value of CellMinerCDB in selecting drugs with reproducible activity, expand on the dominant role of SLFN11 for drug response, and present novel response determinants and genomic signatures for topoisomerase inhibitors and schweinfurthins. We also introduce LIX1L as a gene associated with mesenchymal signature and regulation of cellular migration and invasiveness.
Subject Areas: Genomics, Bioinformatics, Biological Database, Cancer Systems Biology
Graphical Abstract
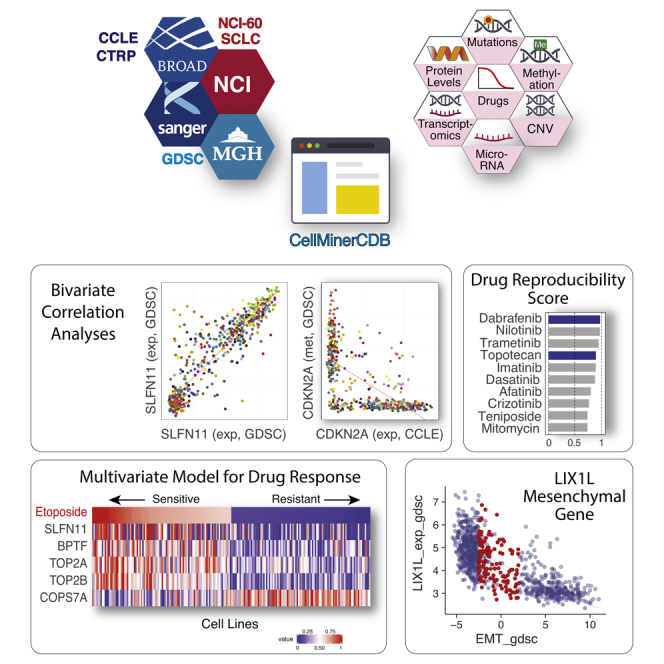
Highlights
-
•
CellMinerCDB integrates pharmacogenomic data of the major cancer cell line databases
-
•
It seamlessly enables genomic and drug data exploration within and across databases
-
•
It tests genomic data reproducibility and proposes drug response determinants
-
•
We expand the GDSC drug panel and advance LIX1L as a novel mesenchymal gene
Genomics; Bioinformatics; Biological Database; Cancer Systems Biology
Introduction
A critical aim of precision medicine is to match drugs with genomic determinants of response. Identifying tumor molecular features that affect response to specific drug treatments is especially challenging because of the typically encountered diversity of patient experiences, incomplete knowledge of the multiple molecular determinants of response and resistance factors downstream of the primary drug targets, and tumor heterogeneity. In this setting, the relative homogeneity of cell lines is advantageous, making them model systems for resolving and establishing cellular intrinsic drug response mechanisms. These features motivated the development of cancer cell line pharmacogenomic databases.
Building on the NCI-60 paradigm (Abaan et al., 2013, Reinhold et al., 2012, Reinhold et al., 2015, Reinhold et al., 2017, Zoppoli et al., 2012), pharmacogenomic data portals such as the Genomics of Drug Sensitivity in Cancer (GDSC) (Garnett et al., 2012, Iorio et al., 2016), the Cancer Cell Line Encyclopedia (CCLE) (Barretina et al., 2012, Cancer Cell Line Encyclopedia Consortium and Genomics of Drug Sensitivity in Cancer Consortium, 2015), and the Cancer Therapeutics Response Portal (CTRP) (Rees et al., 2016) have expanded to span ∼1,400 cancer cell lines. Each database provides a readily available resource for translational research, and proposals have been advanced to further enrich them to over 10,000 cancer cell lines for better coverage of tumor type diversity (Boehm and Golub, 2015). The NCI-60 dataset includes drug activity data for over 21,000 compounds, together with a wide range of molecular profiling data (gene expression, mutations, copy number, methylation, and protein expression). The GDSC and CCLE collections focus on drug activity data for clinically relevant drugs over larger cell line sets, together with an array of molecular profiling data that match the NCI-60 and clinical genomic analyses. The CTRP provides independent drug activity data for nearly 500 compounds over cell lines spanning most of the CCLE and GDSC collections. Each source-specific portal allows deep exploration of its associated datasets, but does not allow immediate cross-database analyses. Yet, substantial overlaps in both cell lines and drugs have the potential to empower integrative analyses, building on the complementarity of the cancer cell line datasets. However, data complexity and mundane (but significant) sources of friction, such as differences in entity naming (cell lines, drugs) and data preparation, have until now made working across databases challenging, even for those with informatics training.
To enable integrative analyses within and across data sources, we are introducing CellMinerCDB (https://discover.nci.nih.gov/cellminercdb/), a web application allowing immediate, interactive exploration of the richest cancer cell line genomic and pharmacogenomic databases (Figure 1). In CellMinerCDB, named entities are transparently matched across sources, allowing cell line molecular features and drug responses to be readily compared using bivariate scatterplots and correlation analyses. Multivariate models of drug response or any genomic cell line attribute can also be assessed. Analyses can be restricted to tissues of origin, with cell lines across all sources mapped to a uniform tissue type hierarchy. Gene pathway annotations allow assessment and filtering of analysis results. CellMinerCDB is built using the publicly available rcellminer R/Bioconductor package, which provides analyses and a standard data representation format (Luna et al., 2016). The latter also allows CellMinerCDB to be readily updated to include additional data. Although the rcellminer package (Luna et al., 2016) is available for bioinformaticists, it requires knowledge of the R programming language to install, configure, and conduct analyses. CellMinerCDB, by contrast, is accessible via a web-based interface meant for direct, general use. Furthermore, CellMinerCDB is enhanced with new data sources and analyses, including a wide range of fully interoperable pharmacogenomics datasets, as well as multivariate analyses that can be used to explore the biological complexity of these data. The accessibility of these analyses and breadth of available data make CellMinerCDB a unique resource for cancer cell line pharmacogenomic data exploration and hypothesis generation.
Figure 1.

CellMinerCDB Overview
(A) CellMinerCDB integrates cancer cell line information from principal resources and provides powerful, user-friendly analysis tools.
(B) Summary of molecular and drug activity data for the five data sources currently included in CellMinerCDB. For molecular data types, the numbers indicate the number of genes with a particular data type. GDSC gene-level mutation and methylation data (numbers in red) were prepared from raw data as part of the development of CellMinerCDB. Asterisks indicate molecular data under development, but not publicly available. Protein expression was determined by reverse-phase protein array.
(C) Cell line and drug overlaps between data sources.
(D) Drug overlaps between data sources.
(E) Small cell lung cancer (SCLC) cell line overlaps between data sources.
(F) SCLC cell line-tested drug overlaps between data sources.
Here we present CellMinerCDB (https://discover.nci.nih.gov/cellminercdb/), highlighting key features of molecular and drug data reproducibility, and complementarity across sources. We provide examples illustrating cancer biology explorations and drug response determinants. We propose the potential repurposing of oxyphenisatin acetate (acetalax; NSC59687) as an anticancer agent for triple-negative breast cancer. We demonstrate multivariate analyses for the exploration of genomic response determinants for topoisomerase inhibitors and schweinfurthins, a class of National Cancer Institute (NCI)-developed compounds derived from natural products. CellMinerCDB also provides phenotypic genomic signatures for cancer cell lines, including a gene-expression-based measure of epithelial-to-mesenchymal (EMT) transition status. We demonstrate the use of the latter to assess EMT stratification within specific tissues of origin, leading to the identification of a novel EMT gene, LIX1L. Detailed use of CellMinerCDB is described in a video tutorial (https://youtu.be/XljXazRGkQ8).
Results
Data Source Comparisons
CellMinerCDB integrates four prominent cancer cell line data sources: the CellMiner NCI-60 (Abaan et al., 2013, Luna et al., 2016, Reinhold et al., 2015, Reinhold et al., 2017), Sanger/Massachusetts General Hospital GDSC (Garnett et al., 2012), the Broad/Novartis CCLE, the Broad CTRP (Barretina et al., 2012, Rees et al., 2016), and a tissue-specific dataset encompassing 66 small cell lung cancer lines (NCI-SCLC) (Polley et al., 2016) (Figure 1). Collectively, these databases provide drug activity and molecular profiling data for approximately 1,400 distinct cancer cell lines (Figure 1B, Supplemental Information). Each source has particular strengths. The NCI-60 is unmatched with respect to the breadth of molecular profiling data, as well as the number of tested drugs, compounds, and natural products (>20,000). It also includes replicate data readily accessible via the established CellMiner data portal (Reinhold et al., 2015). The GDSC, CCLE, and CTRP sources feature much larger numbers of cell lines, spanning tissues of origin not included in the NCI-60. The range of tested compounds in these expanded cell line panels is narrow relative to the NCI-60, although the GDSC and CTRP focus on a wide range of clinically relevant anticancer drugs. The CTRP provides data for 170 US Food and Drug Administration (FDA)-approved or investigational anticancer drugs and 196 other compounds with mechanism of action information. The CTRP molecular data in CellMinerCDB are from the CCLE (Figure 1B).
Despite ongoing data acquisition and processing efforts, gaps exist with respect to genomic profiling data (Figure 1B, dark gray table entries). For the GDSC gene mutation and methylation data, we took advantage of processing pipelines developed for the NCI-60 (Reinhold et al., 2014, Reinhold et al., 2017) to compute gene-level summary data from publicly available raw data. Remaining source-specific molecular profiling data gaps can be filled within CellMinerCDB by effectively extending data provided by one source to another. This is possible because of extensive overlaps between tested cell lines and drugs (Figure 1). For example, gene-level methylation data are not publicly available for the CCLE, but GDSC methylation data are available for the matching 671 CCLE lines and 597 CTRP lines (Figure 1C). CellMinerCDB automatically matches synonymous cell line and drug names (https://discover.nci.nih.gov/cellminercdb/), freeing users from a mundane but time-consuming impediment to work across data sources.
Molecular Data Reproducibility
Integrative analyses presuppose data concordance across sources. Such analyses can be readily performed with CellMinerCDB because of the extensive overlaps across the cancer cell line databases: 55 of the NCI-60 lines are in GDSC and 44 are in CCLE, 671 lines (∼60%) are shared between CCLE and GDSC (Figure 1C), 40 of the 67 NCI-SCLC lines are in GDSC and 36 are in CCLE (Figure 1E), 74 drugs are in both GDSC and CTRP, and 63 drugs are in both NCI-60 and CTRP (Figure 1D).
For the genomic data, we assessed concordance by computing Pearson's correlations between gene-specific molecular profiles over matched cell lines for all pairs of sources and comparable data types. The distributions of expression, copy number, and methylation data correlations indicate highly significant concordance across sources (Figure 2A). Concordance was also evident based on non-parametric Spearman's rank correlations (Figure S1, related to Figure 2A). For these analyses, gene-level transcript expression and methylation patterns with uniformly low values across matched cell lines were excluded due to their lack of meaningful pattern (Transparent Methods). The median correlations exceed 0.7 in all cases (Figure 2A). The striking concordance between NCI-60 and GDSC methylation data (median R = 0.97, median n = 52) may derive in part from the use of same technology platform (Reinhold et al., 2017) and gene-level data summarization approach (Transparent Methods). Examples for specific genes are displayed in Figure S2 (related to Figure 2A), demonstrating the high data reproducibility for SLFN11 (Schlafen 11) expression in the NCI-60 versus GDSC, CDH1 (E-cadherin) expression in GDSC versus CCLE, SLFN11 methylation in the GDSC versus NCI-60, and CDKN2A (p16INK4/p19ARF) copy number in NCI-60 versus CCLE. Readers are invited to explore their own queries at https://discover.nci.nih.gov/cellminercdb/ by selecting a genomic feature for any given gene in two different datasets of their choice.
Figure 2.

Molecular Data Reproducibility across Sources
Comparison of the available genomic features of the cell lines shared between the CellMinerCDB data sources. Bar plots indicate the median and inter-quartile range.
(A) Pearson's correlation distributions for comparable expression (exp), DNA copy number (cop), and DNA methylation (met) data.
(B) Jaccard coefficient distributions for comparable binary mutation (mut) data. The Jaccard coefficient for a pair of gene-specific mutation profiles is the ratio of the number of mutated cell lines reported by both sources to the number of mutated lines reported by either source.
(C and D) Overlaps of function-impacting mutations as predicted using SIFT/PolyPhen2 for selected tumor suppressor genes and oncogenes. Matched cell line mutation data were binarized by assigning a value of 1 to lines with a homozygous mutation probability greater than a threshold, which was set to 0.3 for (B) and for oncogenes in (C) and to 0.7 for tumor suppressor genes in (D).
Gene-level mutation values in CellMinerCDB indicate the probability that an observed mutation is homozygous and is function impacting. For genes with multiple deleterious mutations in a given cell line, values are converted to cumulative probability values (Reinhold et al., 2014), and are available in graphical and tabular forms at https://discover.nci.nih.gov/cellminercdb/. To compare mutation profiles across sources, we binarized the matched cell line data by assigning a value of 1 to lines with an aforementioned probability value greater than 0.3. This value was selected to be below the formally expected value of 0.5 for a heterozygous mutation to allow for technical variability.
Entirely matched mutation profiles across sources should have a Jaccard index value of 1. As such, the similarity index distributions indicate greater discordance for the mutation data (Figure 2B) than for the other types of genomic data (Figure 2A). The similarity distribution values are higher for the NCI-60 (NCI-60/GDSC median J = 0.5, n = 55; NCI-60/CCLE median J = 0.71, n = 39) than for the GDSC/CCLE comparison (median J = 0.38, n = 593). One caveat, however, is that the large cell line database comparisons entail far larger numbers of matched cell lines. Indeed, the Jaccard similarity values approaching 1 with the NCI-60 comparisons often derive from just one or two matched mutant cell lines. We used similar processing steps to derive gene-level mutation data from variant call data for the NCI-60, GDSC, and CCLE (Transparent Methods). Still, inconsistencies were notable.
Differences between the underlying sequencing technologies and initial data preparation methods are likely to account for the observed discrepancies between the gene mutation data across the datasets. For example, the CCLE mutation data were obtained for a selected set of 1,667 cancer-associated genes subject to high-depth exome capture sequencing (Barretina et al., 2012). They consistently yielded the largest numbers of cell lines with function-impacting mutations. The greater number of mutations found for KRAS, PTEN, BRAF, NRAS, or MSH6 in CCLE relative to the GDSC or NCI-60 databases (evaluated by global exome sequencing; Figure S3, related to Figures 2C and 2D) reflects the importance of sequencing depth for accurate assessment of mutations.
For a more focused and translational assessment of mutation data concordance, we examined the overlap between sources for established oncogenes and tumor suppressor genes (Figures 2C and 2D, Table S1). For the tumor suppressors, we binarized the data using a probability threshold of 0.7 (to account for the recessive nature of such mutations), whereas for the oncogenes, a 0.3 threshold was used (to account for the dominance of oncogene-activating mutations). These values were set below the formally expected values of 1 and 0.5 for homozygous and heterozygous mutations, respectively, to allow for technical variability. As expected, the most frequently mutated genes were TP53, KRAS, BRAF, APC, RB1, NF1, PTEN, SMARCA4, and MLH1 (Table S1, related to Figure 2). BRAF mutation profiles showed the expected overlap (J > 0.7) across datasets, as was the case for the TP53 gene across the GDSC and CCLE (J = 0.69). On the other hand, PIK3CA, BRCA2, BRCA1, MLH1, MSH6, and MSH2 mutation comparisons were largely divergent. These discrepancies reflect the ongoing challenges and trade-offs with mutation profiling technologies and mutation calling procedures. The ability of CellMinerCDB to compare and integrate data across sources highlights the fundamental research efforts and technological standards still required for the accurate identification of mutations. As a practical matter, CellMinerCDB users can readily compare cell line mutation calls across sources for any given gene of interest. For follow-up studies, they can then select either cell lines that are consistently identified as mutant across sources or the larger set of mutant lines (according to one or more sources).
Drug Activity Data Reproducibility and Enrichment
Independent studies have examined drug data reproducibility, noting potential sources of data divergence such as assay type and duration of drug treatments (Cancer Cell Line Encyclopedia Consortium and Genomics of Drug Sensitivity in Cancer Consortium, 2015, Haibe-Kains et al., 2013, Haverty et al., 2016). To explore the reproducibility and the ability of CellMinerCDB to identify genomic signatures over a larger number of cell lines from different tissues of origin, we tested a selected set of NCI-60-screened compounds in the larger GDSC panel (Table S2, related to Figure 3). Noting that the GDSC and the NCI/Developmental Therapeutics Program (DTP) used different assays to determine their IC50 values (Cell Titer Glo measurements of ATP at 72 hr post-treatment versus sulforhodamine B measurement of total protein at 48 hr post-treatment, with additional differences in cell seeding densities and drug dose ranges), we tested in parallel 19 drugs referenced by their NSCs (National Service Center identifiers) and associated with a range of mechanisms of action.
Figure 3.

Drug Activity Data Reproducibility
(A and B) GDSC versus NCI-60 drug activity data in matched cell lines for (A) oxyphenisatin acetate (acetalax; NSC59687) and (B) MJ-III-65 (LMP744; NSC706744). Each point represents a matched cell line. Red points in (A) indicate triple-negative breast cancer cell lines.
(C–H) (C and D) A total of 38 drugs were tested in the NCI-60, GDSC, and CTRP. CCLE was excluded because of its small drug dataset (24 drugs), which is largely included in CTRP. For each of the three inter-source comparisons, drugs were ranked by activity correlation strength (q-value), with ranks scaled between 0 (lowest) and 1 (highest). Specifically active compounds, such as the BRAF inhibitor dabrafenib, show strong correlations based on the response of melanoma lines shown in red (E and F), whereas broadly active compounds, such as the topoisomerase I inhibitor topotecan, show strong correlations based on broad response patterns (G and H). The NCI-60-matched data in (F) and (H) capture the pattern observed with matched data between the larger GDSC and CTRP collections. The full data table excerpted in (D) is shown in Figure S6.
Two drugs with the strongest correlations were oxyphenisatin acetate (acetalax) and bisacodyl (Figure 3A, R = 0.84, p = 8.6 × 10−13, N = 44 and R = 0.80, N = 43, p = 1.0 × 10−10, respectively). These FDA-approved laxatives were included in our comparative analysis based on their range of antiproliferative activity in the NCI-60 (further corroborated by NCI-60 activity data for several derivatives), unique pattern of activity compared with the FDA-approved anticancer drugs, outstanding activity in two of the three NCI-60 triple-negative breast cancer cell lines, and lack of pre-existing data in the CTRP, CCLE, or GDSC. The GDSC results confirmed that oxyphenisatin acetate (acetalax) elicits a broad range of cytotoxic responses in the expanded GDSC cell line collection. Extending our NCI-60 observations, it is more active than any of the 15 tested oncologic drugs by a significant margin (p < 7 × 10−10) in the 22 GDSC triple-negative breast cancer lines (Table S3, related to Figure 3).
Overall, 16 of the 19 newly tested compounds across the NCI-60 and GDSC gave significant correlations (Table S2, related to Figure 3). Technical discrepancies were evident for three drugs. Dacarbazine, an alkylating agent related to temozolomide, and vincristine, an anti-tubulin, both had poor reproducibility even within DTP assay replicates. Fulvestrant appeared to be out of the proper concentration range in the DTP assay (Figure S4, related to Figure 3). The non-camptothecin indenoisoquinoline-based topoisomerase I inhibitor in clinical trial, LMP744 (NSC 706744; MJ-III-65) (Burton et al., 2018), was also included in our 19-compound test set to assess the similarity of its activity profile with that of topotecan over a larger cell line collection and to enrich the genomic signature associated with its activity (see section Exploring Drug Response Determinants). Consistent with its activity as a topoisomerase I inhibitor (Antony et al., 2003, Burton et al., 2018), LMP744 is highly correlated with topotecan in the GDSC testing (R = 0.83, p = 4.2 × 10−187, N = 715) (Figure S5, related to Figure 3), and exhibits significant activity data concordance between NCI-60 and GDSC (R = 0.66, p = 9.8 × 10−7, N = 44) (Figure 3B).
Further focusing on drug activity data reproducibility, we analyzed the 38 drugs previously tested in each of the three databases with larger numbers of tested drugs (NCI-60, GDSC, and CTRP) (Figure 3C). For each of the three inter-source comparisons, drugs were ranked by activity correlation strength (q-value, scaled between 0 [lowest] and 1 [highest]). The drugs were then ordered by the average of the three inter-source comparison rank scores (Figures 3D and S6, related to Figure 3). As noted in earlier studies of drug activity data reproducibility (Haverty et al., 2016), strong activity correlations were observed for specifically active compounds (Figures 3E and 3F), such as the BRAF inhibitor dabrafenib, wherein outstanding response occurs in cell lines with the activated kinase target. Notably, we also observed high correlations for broadly active drugs, such as the topoisomerase I inhibitor topotecan (Figures 3G and 3H), indicating that the cancer cell line responses are reproducible across databases and assays and are not limited to protein kinase inhibitors. Still, for many of the 38 assessed drugs (see lower half of Figure S6, related to Figure 3), there were discordant activity data between one or more pairs of sources. The inter-source activity data comparisons enabled by CellMinerCDB allow individual researchers to identify drugs with concordant data, so they can pursue reliable molecular pharmacology and translational genomic analyses (see below and Figures 5 and 6).
Figure 5.

Exploring Drug Response Determinants
(A and B) Response to the pre-mRNA splicing inhibitor indisulam versus expression of its target complex component DCAF15 in the CTRP. Drug response in (B) is measured by the activity area above the dose-response curve, with higher values indicating relative drug sensitivity. A report of increased indisulam sensitivity in hematopoietic cell lines (shown in red) with high DCAF15 expression is readily verified (Han et al., 2017).
(C) Response to the aurora kinase inhibitor alisertib is associated with increased MYC expression in small cell lung cancer lines (Mollaoglu et al., 2017).
(D) Heatmap indicating etoposide drug activity and candidate determinant gene expression in the 100 most sensitive and resistant CTRP cell lines.
(E) Scatterplots of etoposide activity versus candidate determinant gene expression in CTRP cell lines, with hematopoietic cell lines shown in red.
(F) A statistical summary of a multivariate linear model of etoposide response in the CTRP.
(G) A mechanistic scheme indicating how the selected determinants may influence etoposide drug response.
Figure 6.

A Multivariate Model of Schweinfurthin A Drug Activity
(A) Reproducibility of the data for the two schweinfurthin derivatives tested in the GDSC.
(B) Heatmap indicating Schweinfurthin A drug activity and candidate determinant gene expression in the 100 most sensitive and resistant non-hematopoietic GDSC cell lines.
(C) A statistical summary of a multivariate linear model of Schweinfurthin A response in the GDSC.
(D) Scheme of the proposed molecular pharmacology of the schweinfurthins. Schweinfurthins have been shown to inhibit PI3K/AKT signaling and cell survival by binding oxysterol-binding-protein-related proteins (ORPs) to disrupt trans-Golgi network trafficking required for robust pathway activity (Bao et al., 2015). Together with the ORPs OSBP, OSBPL3, and OSBPL10, the other candidate determinants, PLEKHO1 and THEM4, have also been implicated in PI3K/AKT signaling (Liu et al., 2013, Tokuda et al., 2007).
Plots and analyses in panels B–D are based on non-hematopoietic GDSC cell lines.
Exploring Gene Regulatory Determinants
Cancer-specific gene expression is known to be affected by DNA copy number variations (CNVs) and epigenetic alterations such as promoter methylation. CellMinerCDB makes it easy to explore these and other potential gene regulatory determinants. For example, in the NCI-60, reduced expression of the tumor suppressor gene CDKN2A (p16INK4a) is associated with both DNA copy loss (Figure 4A) and promoter hypermethylation (Figure 4B) across tissue types. Notably, Figure 4C shows that approximately 25% of NCI-60 cell lines show both alterations, consistent with biallelic, “two-hit” suppression of CDKN2A expression. Integration of matched cell line GDSC methylation and CCLE copy number data illustrates the same CDKN2A regulatory relationships in a larger cell line collection (Figures 4D–4F). Table S4 (related to Figure 4) shows that CDKN2A stands out with respect to the high proportion of cell lines showing co-occurrence of promoter methylation and DNA copy loss. Conversely, the impact of copy gain on increased oncogene expression can be similarly assessed with CellMinerCDB. Figure 4G shows that a subset of MYC-driven CCLE small cell lung cancer lines (red dots) exhibits both MYC copy gain and increased MYC gene expression. KRAS activation, typically regarded as mutation driven, can also occur by copy gain, as evident in a subset of CCLE lines (Figure 4H), consistent with clinical studies (Wagner et al., 2011).
Figure 4.

Exploring Gene Expression Determinants
Reduced mRNA expression (xai, average log2 intensity) of the cell cycle inhibitor and tumor suppressor CDKN2A (p16) is associated with DNA copy loss (cop) (A) and promoter methylation (met) (B) in the NCI-60 lines. In a subset of NCI-60 lines, enclosed in the red box, (C), DNA copy loss accompanies higher levels of promoter methylation. DNA copy number and promoter methylation data from the CCLE and GDSC, respectively, can be also be visualized over matched cell lines to verify a similar pattern in larger cell line collections (D–F). Note that the corroboration of the NCI-60 regulatory relationships in a far larger and more diverse cell line set is uniquely enabled by CellMinerCDB, which allows gene-level methylation data only available in the GDSC to complement gene-level DNA copy number data only available in the CCLE (for automatically matched cell lines). DNA copy number gain is associated with increased expression (exp, Z score microarray log2 intensity data) of the oncogenes MYC (G) and KRAS (H) in selected CCLE cell lines. In (G), small cell lung cancer lines are indicated in red to highlight a subset potentially derived from MYC-driven tumors (within red box).
Exploring Drug Response Determinants
CellMinerCDB allows correlation analyses and scatterplots for testing and visualizing potential response-determinant relationships (univariate analyses) as well as multivariate linear regression methods for integrating multiple determinants (multivariate analyses; see Figures 5D and 6B). CellMinerCDB also enables the discovery of candidate genomic determinants of drug response as well as drug-drug correlations (“Compare Pattern” tab in the “Univariate analyses” tool; https://discover.nci.nih.gov/cellminercdb/). This method led to the discovery of Schlafen 11 (SLFN11) expression as a causal determinant of response to widely used DNA-targeted anticancer agents, including topoisomerase inhibitors, platinum derivatives, PARP inhibitors, and antimetabolites (Barretina et al., 2012, Murai et al., 2016, Murai et al., 2018, Zoppoli et al., 2012). Starting with target expression profiles, CellMinerCDB correlation analyses can identify compounds with matching activity profiles. For example, CellMinerCDB can be used to demonstrate that epidermal growth factor receptor (EGFR) expression is significantly correlated with the activity of erlotinib and afatinib in all major cell line databases, as well as with the activity of other established EGFR inhibitors available in one or more data sources. CellMinerCDB correlation analyses also allow direct evaluation of drug resistance determinants. For example, potential substrates of drug efflux ABC transporters can be recovered because strong negative activity correlates with the expression of ABC drug transporters, as in the case of paclitaxel and ABCB1 in the GDSC (r = −0.33; p value = 5.7 × 10−12).
CellMinerCDB also allows users to assess on the spot the generality of results presented in the literature, and iteratively explore evidence for multifactorial mechanistic models. Figure 5A shows an example for indisulam, which targets the splicing factor RBM39 for proteasomal degradation by forming a ternary complex with RBM39 and the E3 ubiquitin ligase receptor DCAF15. A report of increased indisulam sensitivity in hematopoietic cell lines with high DCAF15 expression is readily verified with CellMinerCDB (Figure 5B, red dots) (Han et al., 2017). CellMinerCDB also corroborates a report of MYC-driven small cell lung cancer exhibiting vulnerability to aurora kinase inhibition (Mollaoglu et al., 2017) (Figure 5C).
As determinants of drug responses are multifactorial, CellMinerCDB includes a multivariate analysis tool under the “Regression Models” tab. Figures 5D–5G illustrate its use for the topoisomerase II inhibitor etoposide. Starting with expression of the drug target (TOP2A and TOP2B) (Pommier et al., 2016) and SLFN11 (Zoppoli et al., 2012), users can select additional determinants based on biological knowledge. Determinant selection can be further guided by pathway annotations, as well as partial correlation analyses, which measure the capacity of additional features to improve the current model (Figure 5G). Additional determinants can be found using the “LASSO” tool in the “Algorithm” dropdown menu of the “Regression Models” tab of the CellMinerCDB website. The use of the multivariate modeling tools included in the “Regression Models” tab is outlined in the video tutorial (https://youtu.be/XljXazRGkQ8) and will be exemplified below in the A Multivariate Model of Schweinfurthin A Drug Activity section.
Benefits of Analyses over Multiple Data Sources
The uniform data representation, accessibility, and interoperability provided by CellMinerCDB allows direct exploratory analyses across the datasets (NCI-60, CCLE, GDSC, CTRP, NCI/DTP-SCLC). This is critically important to identify molecular and drug response determinants with consistent data across sources for specific analyses. The automatic management of cell line overlaps also enables comprehensive analyses encompassing all databases, using complementary data to supplement source-associated gaps in molecular and drug activity data (see Figure 1, and prior section). These application features are highlighted in the examples presented below.
The main ABC transporter, ABCB1 (PgP), is a dominant factor conferring resistance to multiple classes of clinically relevant drugs. Because CellMinerCDB integrates different databases with different drugs tested in each database, it can reliably test the relationship between drug resistance and ABCB1 expression. The first step (https://discover.nci.nih.gov/cellminercdb/) is to ensure that ABCB1 expression exhibits a high dynamic range (i.e., cells with and without expression and high expression in the positive cells) and that ABCB1 expression is highly correlated across databases. Figure S7A (expression of ABCB1 across GDSC and CCLE) shows very high correlation between the two databases (r = 0.91; p value = 1.4 × 10−238). The next step is to use the “Compare Patterns” tool of CellMinerCDB by entering “ABCB1” as the “x-Axis Data Type” for each of the databases (selected via the “x-Axis Cell Line Set”). Table S5 (related to Figure 5) shows the top 50 drugs with activity negatively correlated with ABCB1 for the three datasets: GDSC, CTRP (CCLE), and NCI-60. Overlapping and established drugs effluxed by ABCB1 in each dataset are highlighted in yellow. In addition, each dataset includes many additional drugs. Therefore, if a drug is not in one dataset, it may be found in others. Figure S8 (related to Figure 5) shows that adding ABCB1 to SLFN11 enhances the prediction of doxorubicin activity. This analysis is readily done using the “Regression Models” tool of CellMinerCDB. Finally, Figure S7 (related to Figure 4) shows that ABCB1 is epigenetically regulated by promoter hypermethylation Figure S7B rather than by copy number alteration Figure S7C.
Figure S9 (related to Figure 5) presents an example of cross-database exploration to identify cyclin D1 (CCND1) as a potential determinant of response to the HDAC inhibitor belinostat, together with evidence of CCND1 expression regulation by DNA copy number and promoter methylation.
A Multivariate Model of Schweinfurthin A Drug Activity
Schweinfurthin A was discovered by the NCI natural products initiative (Thornburg et al., 2018) to identify compounds with distinctive NCI-60 activity profiles indicative of novel targets (via COMPARE analysis; Paull et al., 1989). Its wide activity range with notable potency in leukemia and CNS lines (<10 nmol/L) motivated the synthesis of a series of derivatives (Kodet et al., 2014). Because the development of schweinfuthins has been hampered by limited understanding of their molecular pharmacology, we tested Schweinfurthin A and 5’-methylschweinfurthin G (NSC 746620) in the GDSC panel and applied the various features of CellMinerCDB to reveal the molecular pathways for response. After confirming that the activities of both compounds were highly correlated (R = 0.87, p = 8.8 × 10−182, N = 585, Figure 6A), we explored the genomic correlates with activity for the ≈700 GDSC cell lines tested. The CellMinerCDB Univariate Analysis tool (“Compare Patterns” tab) indicates that the leading molecular correlate (by lowest p value) of schweinfurthin activity is the expression of PLEKHO1, a negative regulator of phosphatidylinositol 3-kinase (PI3K)/AKT signaling (R = 0.47, p = 1.95 × 10−33, N = 582) (Tokuda et al., 2007). This result is consistent with a recent study showing that schweinfurthins inhibit mammalian target of rapamycin (mTOR)/AKT signaling by interfering with trans-Golgi network (TGN) trafficking (Bao et al., 2015). In particular, schweinfurthins bind to oxysterol-binding proteins, which regulate TGN trafficking (Burgett et al., 2011, Mesmin et al., 2017), thereby arresting lipid-raft-mediated PI3K activation and functional mTOR/RheB complex formation.
Next, using the multivariate analysis feature of CellMinerCDB (“Regression Models” tab; https://discover.nci.nih.gov/cellminercdb/), we developed a linear predictive model for schweinfurthin response integrating the expressions of PLEKHO1, THEM4, a positive regulator of AKT signaling (Liu et al., 2013), and the genes encoding the oxysterol-binding protein family members OSBP, OSBPL3, and OSBP10 (Figures 6C and 6D). Increased expression of the oxysterol-binding protein family members conceivably sustains TGN trafficking and PI3K/AKT signaling, in keeping with their negative regression coefficient weights as resistance determinants in the model. The negative weighting of THEM4 expression and positive weighting of PLEKHO1 expression are similarly consistent with their respective roles in activating and suppressing PI3K/AKT signaling. These analyses give molecular insight into the cholesterol trafficking and intracellular membrane pathways as the targets of schweinfurthins and open new opportunities for testing the potential activity of schweinfurthins with genomic and molecular signatures.
Relating EMT Status with Gene Expression to Identify LIX1L Expression and Schweinfurthin Activity in Mesenchymal Cells
EMT is a fundamental process in development, wound healing, and cancer progression, characterized by the loss of cell-cell adhesion and the acquisition of motile and invasive properties (Figure 7A) (Kalluri and Weinberg, 2009, Lamouille et al., 2014). EMT is driven by dominant transcription factors, including ZEB1/2, SNAI1/2, and TWIST1/2, and is reversible through a continuum of states from epithelial to mesenchymal. These attributes have motivated the development of gene-expression-based EMT signatures to identify cell line state and understand drug resistance.
Figure 7.

Relating Epithelial Mesenchymal Transition (EMT) Status with Gene Expression to Identify LIX1L as a Novel EMT Gene
(A) A 37-gene EMT signature developed in (Kohn et al., 2014) was used to derive a numerical index of EMT status as a weighted sum of cell-line-specific EMT gene expression values (see Transparent Methods for details). Epithelial and mesenchymal statuses are associated with positive and negative index values, respectively.
(B) For 821 non-hematopoietic cell lines in the GDSC collection, the EMT index values show a bimodal distribution, which can be modeled as a normal mixture. Cell lines with EMT index values less than (greater than) 1 standard deviation above (below) the putative mesenchymal (epithelial) group mean are annotated as mesenchymal (epithelial).
(C) EMT stratification by tissue of origin.
(D and E) Expression of LIX1L, a novel mesenchymal gene, is strongly correlated with the EMT index signature. “Epithelial-mesenchymal” lines with intermediate EMT index values are indicated in red. Mesenchymal lines are in blue at the left, and epithelial are in blue at the right.
(F) Western blot showing the efficient knockdown of LIX1L in MDA-MB231 cells.
(G) Representative image showing increased migration and invasion after LIX1L knockdown.
(H) Quantitation of the increased migration and invasion of cells after LIX1L knockdown. Individual experiments are shown as dots. Error bars indicate the standard error of the mean.
We applied a 37-gene EMT signature initially developed in the NCI-60 (Kohn et al., 2014) to derive a numerical index of EMT status as a weighted sum of cell-line-specific EMT expression values (Transparent Methods). Figure 7B shows the bimodal distribution for the EMT index values across the 821 non-hematopoietic GDSC cell lines, allowing cell line stratification into epithelial, mesenchymal, and epithelial-mesenchymal categories. EMT stratification within particular tissues of origin also shows a substantial proportion of intermediate epithelial-mesenchymal lines in liver, ovary, and lung cancer cell lines (Figure 7C). The numerical EMT index is available for all CellMinerCDB-integrated data sources as the variable KOHN_EMT_PC1 (“Metadata”), allowing its correlation with any chosen molecular or drug response feature.
The EMT index identified LIX1L as a novel mesenchymal gene whose expression is highly correlated with the EMT index signature (Kohn et al., 2014) across multiple cancer types (Figure 7D, R = −0.75, p = 8.9 × 10−179, N = 823). LIX1L is also broadly expressed in TCGA tumor samples (Figure S10A, related to Figure 7). Knockdown analyses in the breast cancer MDA-MB-231 cell line suggest that LIX1L expression reduces cell migration and invasiveness (Figures 7F–7H, S10C, and S10D).
We also correlated the EMT index with the activity profiles of the 297 compounds in the GDSC database, including the 19 additional compounds tested for the current study (Figure S11, related to Figure 7). Schweinfurthin A is the strongest negative correlate, indicating its selective antiproliferative activity in mesenchymal cancer cell lines, such as those derived from bone or soft tissue (Figure S11C). The second strongest negative correlate with the EMT index is the RHO-associated kinase 1 inhibitor GSK269962A (Figure S11C), whose target regulates actin dynamics and cell motility associated with EMT (Kalluri and Weinberg, 2009, Lamouille et al., 2014). On the opposite side, the drug most highly correlated with epithelial cell line status was acetalax (oxyphenisatin), with its two independent samples (NSC59687 and 614826) at the top of the list above afatinib and lapatinib (Figure S11C), consistent with its potential activity in epithelial breast cancer cells (Morrison et al., 2013).
Discussion
CellMinerCDB (https://discover.nci.nih.gov/cellminercdb/) allows researchers to interact directly with an unparalleled breadth of cancer cell line genomic and pharmacologic data. The examples described here, spanning data assessment, integration, and discovery, demonstrate the value of working within and across data sources.
The CellMinerCDB analyses support the maturity and essential reproducibility of most molecular profiling technology platforms, such as gene transcript expression and DNA CNV. Mutation data are prominently featured in translational studies, and CellMinerCDB exposes the issue of discrepancies between matched cell line mutation profiles across sources. This provides a foundation for understanding and mitigating sources of variability, which reflect the ongoing technical challenges and trade-offs with the acquisition and interpretation of genome sequencing data. Somatic variant calling in cancer cell lines is inherently challenging because of the absence of matched normal tissue for comparison, as well as the potentially higher mutation burden in cell lines relative to primary tumor tissue. One approach for excluding potentially cell-line-specific passenger mutations is to filter variants based on frequency in patient populations (Iorio et al., 2016). Variability in cell line mutation data across sources may also arise from differences in variant calling algorithms, as well as data sources used for filtering likely germline variants. Indeed, the reproducibility of matched cell line gene expression, DNA copy number, and methylation signatures across databases (see Figures 2A, S1, and S2) indicates that the mutation data inconsistencies are likely technical. An existing strategy for acquiring more robust mutation data is to pursue higher-depth targeted sequencing of a restricted gene set. Indeed, we noted that the CCLE data, derived from the latter approach, consistently identified more mutant cell lines for prominent oncogenes and tumor suppressor genes than genome-wide exome sequencing (as in the NCI-60 and GDSC databases). CellMinerCDB can directly integrate mutation data across sources to identify cell lines with consistent mutation calls for a gene of interest, together with other potentially impacted lines. Given the primarily technical basis for the mutation data discrepancies, the best course for users remains this sort of comparison of data across sources. CellMinerCDB enables researchers to focus on consistently mutation-impacted cell lines for further bioinformatic analyses and experimental use (including targeted high-depth sequencing for specific genes of interest).
Regarding drug reproducibility, assays across the major cancer cell line data sources measure different biochemical features at different time points. Still, CellMinerCDB demonstrates significant concordance between drug activity data generated at the NCI (NCI-60 and NCI/DTP-SCLC), at the Broad Institute (CCLE, CTRP), and at the Sanger Institute and Massachusetts General Hospital (GDSC), including data for widely used anticancer drugs. The cross-database comparison features of CellMinerCDB allow researchers to explore potential drug reproducibility issues and focus on drugs with reliable data. Scatterplots of matched cell line activity data can highlight problem areas with particular assays, such as inappropriate concentration ranges, as illustrated for fulvestrant in the NCI/DTP assay.
In addition, among the 19 NCI-60 drugs tested for reproducibility and expansion of genomic correlates, we found significant consistency for 16 drugs, including the novel non-camptothecin topoisomerase I inhibitor LMP744 (Burton et al., 2018), and identified the FDA-approved laxatives oxyphenisatin acetate (acetalax) and bisacodyl as potential novel anticancer drug candidates. CellMinerCDB shows (https://discover.nci.nih.gov/cellminercdb/) that oxyphenisatin exhibits a wide range of concordant antiproliferative activity across the NCI-60 cell lines and across the 710 GDSC cell lines tested, being substantially more active in triple-negative breast cancer lines relative to other cancer drugs tested on the GDSC panel. These findings suggest the potential of oxyphenisatin derivatives for repurposing as anticancer drugs.
With both drug activity reproducibility and broader associations between molecular features, such as CDKN2A expression and gene copy/methylation, we noted that the NCI-60 could effectively capture relationships evident in larger cell line sets. The latter better reflect tissue type diversity and context-specific molecular features. Still, for dominant associations, such as SLFN11 expression and DNA-targeted drug responses, representative cell line sets such as the NCI-60 are often sufficient (Zoppoli et al., 2012). In addition, the NCI-60 provides drug responses for over 21,000 individual agents, making it an unmatched resource for the discovery of new chemotypes based on correlations with genomic data and response patterns for drugs with known targets. CellMinerCDB makes correlation-based COMPARE analyses readily accessible for drug discovery (https://dtp.cancer.gov/databases_tools/compare.htm [Paull et al., 1989]). It also enables a direct visualization of activity data for compounds retrieved in the analysis together with data for the queried entity, using the “Univariate Analysis” tool (https://discover.nci.nih.gov/cellminercdb/). The NCI databases are also a tractable starting point for molecular data expansion with leading-edge technologies. RNA sequencing data with isoform-specific transcript expression and SWATH mass spectrometry-based protein expression data have been generated for the NCI-60, and will be made available within CellMinerCDB (Shao et al., 2015). We are committed to sustain development of CellMinerCDB as an ongoing resource in the mold of the existing NCI CellMiner data portal, which has steadily integrated new data and analyses since its inception (Reinhold et al., 2012, Reinhold et al., 2014, Reinhold et al., 2015, Reinhold et al., 2017). These developments will expand the current features of CellMinerCDB. For example, as existing and emerging data sources provide novel proteomic and isoform-specific transcript expression data, we are planning to integrate these with regular updates of the same website.
CellMinerCDB (https://discover.nci.nih.gov/cellminercdb/) ultimately aims to provide a seamless platform for data exploration and hypothesis generation, integrating previously isolated data sources and enhancing their interpretation through the intuition and expertise of experimental scientists and clinicians. The present publication provides only a sample of the potential uses of CellMinerCDB. CellMinerCDB uniquely complements existing data portals that provide detailed information on their associated data, together with specialized analyses. By empowering researchers to easily build on the strengths of individual databases and pursue their own questions, CellMinerCDB aims to advance the potential of cancer cell line pharmacogenomic data to lay the foundation, validate, and focus experimental and, ultimately, clinical drug development and precision medicine.
Limitations of the Study
We see CellMinerCDB as primarily a data exploration and hypothesis generation tool. Its selection of analyses reflects what is practically manageable in this context, both computationally and conceptually. For example, we do not provide analyses with extended runtimes that are less suitable for interactive data exploration. We do, however, make all data integrated within the application easily downloadable, to expedite more specialized or computationally intensive analyses. CellMinerCDB still provides interactive access to the most fundamental methods, including regression-based predictive models, which have been prominently featured in highly cited studies of cancer cell line pharmacogenomic data.
We have also attempted to minimize the conceptual barrier for basic exploratory analyses by making reasonable default choices for this setting. In keeping with existing CellMiner tools (http://discover.nci.nih.gov/cellminer) (Reinhold et al., 2012, Reinhold et al., 2014, Reinhold et al., 2015, Reinhold et al., 2017) and leading studies, we use Pearson's correlations to measure association between molecular or drug response variables. We do note for users that statistical significance results for these correlations presuppose approximately multivariate normal data; substantial deviations from this assumption can be readily noted through the provided scatterplots. Still, a comparative study of CCLE and GDSC data favored Pearson correlations over non-parametric Spearman correlations, showing that the latter often failed to detect patterns in which responses are restricted to a relatively small fraction of cell lines (as will often be the case for pathway-targeted drugs) (Cancer Cell Line Encyclopedia Consortium and Genomics of Drug Sensitivity in Cancer Consortium, 2015).
In keeping with the exploratory focus of CellMinerCDB, we do not enforce formal adjustments for multiple hypothesis testing (although our pattern comparison and related results do include tabulation of q-values to allow false-discovery-rate-based filtering). The strictest adjustment for multiple testing would require inaccessible knowledge of all analyses conducted by a user toward a particular aim. This sort of application of statistical filters would likely exclude many experimentally established relationships that involve more than one determinant.
Our goal is to strike a balance between providing statistical measures (with reasonable caveats), and allowing scientific experts to apply their judgment when exploring data. We finally note that it is possible to consider additional levels of inter-source data integration. For example, pooling molecular or drug data for distinct cell lines across sources (as compared to strictly overlapping cell lines) could increase the power of statistical analyses. This approach, although potentially valuable, would require careful assessment and adjustment for source-specific effects and is outside the scope of the current study.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to thank Dr. David Goldstein of the NCI Office of Science and Technology Resources for supporting the purchase of software required to enable efficient, multi-user access to the CellMinerCDB site. The work was supported by the Center for Cancer Research, Intramural Program of the National Cancer Institute (Z01 BC006150 to Y.P.), Ruth L. Kirschstein National Research Service Award (F32 CA192901 to A.L.), and the National Resource for Network Biology (NRNB) from the National Institute of General Medical Sciences (NIGMS) (P41 GM103504 to C.S.). M.J.G was funded by the Wellcome Trust (086375 and 102696). This study was also supported by fellowships from the Japanese Society of Clinical Pharmacology and Therapeutics and the Japan Society for the Promotion of Science (to M.Y).
Author Contributions
Conception, design, and development, V.N.R., A.L., F.E., L.L., W.C.R., and Y.P.; acquisition and preparation of data, V.N.R., S.V., M.S., F.I., C.H.B, M.J.G., and W.C.R.; analysis and interpretation of data, development of results, V.N.R., A.L., M.Y., S.V., F.G.S., M.I.A., A.T., K.W.K., C.H.B., M.G., W.C.R., and Y.P.; experimental validation studies, M.Y., K.W.K, and Y.P.; writing, review, and revision of the manuscript, V.N.R., A.L., M.Y., F.I., F.G.S., M.I.A., A.T., C.S., C.H.B., M.G., W.C.R., and Y.P.; study supervision, W.C.R.and Y.P.
Declaration of Interests
The authors declare no competing interests.
Published: December 12, 2018
Footnotes
Supplemental Information includes Transparent Methods, 11 figures, and 5 tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.11.029.
Contributor Information
Vinodh N. Rajapakse, Email: vinodh.rajapakse@nih.gov.
Augustin Luna, Email: augustin_luna@hms.harvard.edu.
Yves Pommier, Email: pommier@nih.gov.
Supplemental Information
Oncogene and tumor suppressor gene mutation call frequencies and overlaps across data sources.
Comparison of drug activities as measured by the GDSC and the NCI/DTP.
Activity of bisacodyl and oxyphenisatin acetate (acetalax) in GDSC triple-negative breast cancer lines.
Co-occurrence of gene promoter methylation and DNA copy loss.
Top 50 drugs with respect to negative activity correlation with ABCB1 expression for the GDSC, CTRP (CCLE), and NCI-60 datasets.
References
- Abaan O.D., Polley E.C., Davis S.R., Zhu Y.J., Bilke S., Walker R.L., Pineda M., Gindin Y., Jiang Y., Reinhold W.C. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013;73:4372–4382. doi: 10.1158/0008-5472.CAN-12-3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony S., Jayaraman M., Laco G., Kohlhagen G., Kohn K.W., Cushman M., Pommier Y. Differential induction of topoisomerase I-DNA cleavage complexes by the indenoisoquinoline MJ-III-65 (NSC 706744) and camptothecin: base sequence analysis and activity against camptothecin-resistant topoisomerases I. Cancer Res. 2003;63:7428–7435. [PubMed] [Google Scholar]
- Bao X., Zheng W., Hata Sugi N., Agarwala K.L., Xu Q., Wang Z., Tendyke K., Lee W., Parent L., Li W. Small molecule schweinfurthins selectively inhibit cancer cell proliferation and mTOR/AKT signaling by interfering with trans-Golgi-network trafficking. Cancer Biol. Ther. 2015;16:589–601. doi: 10.1080/15384047.2015.1019184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm J.S., Golub T.R. An ecosystem of cancer cell line factories to support a cancer dependency map. Nat. Rev. Genet. 2015;16:373–374. doi: 10.1038/nrg3967. [DOI] [PubMed] [Google Scholar]
- Burgett A.W., Poulsen T.B., Wangkanont K., Anderson D.R., Kikuchi C., Shimada K., Okubo S., Fortner K.C., Mimaki Y., Kuroda M. Natural products reveal cancer cell dependence on oxysterol-binding proteins. Nat. Chem. Biol. 2011;7:639–647. doi: 10.1038/nchembio.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton J.H., Mazcko C.N., LeBlanc A.K., Covey J.M., Ji J.J., Kinders R.J., Parchment R.E., Khanna C., Paoloni M., Lana S.E. NCI comparative oncology program testing of non-camptothecin indenoisoquinoline topoisomerase i inhibitors in naturally occurring canine lymphoma. Clin. Cancer Res. 2018 doi: 10.1158/1078-0432.CCR-18-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Cell Line Encyclopedia Consortium. Genomics of Drug Sensitivity in Cancer Consortium Pharmacogenomic agreement between two cancer cell line data sets. Nature. 2015;528:84–87. doi: 10.1038/nature15736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett M.J., Edelman E.J., Heidorn S.J., Greenman C.D., Dastur A., Lau K.W., Greninger P., Thompson I.R., Luo X., Soares J. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haibe-Kains B., El-Hachem N., Birkbak N.J., Jin A.C., Beck A.H., Aerts H.J.W.L., Quackenbush J. Inconsistency in large pharmacogenomic studies. Nature. 2013;504:389–393. doi: 10.1038/nature12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T., Goralski M., Gaskill N., Capota E., Kim J., Ting T.C., Xie Y., Williams N.S., Nijhawan D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. 2017;356 doi: 10.1126/science.aal3755. [DOI] [PubMed] [Google Scholar]
- Haverty P.M., Lin E., Tan J., Yu Y., Lam B., Lianoglou S., Neve R.M., Martin S., Settleman J., Yauch R.L. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature. 2016;533:333–337. doi: 10.1038/nature17987. [DOI] [PubMed] [Google Scholar]
- Iorio F., Knijnenburg T.A., Vis D.J., Bignell G.R., Menden M.P., Schubert M., Aben N., Gonçalves E., Barthorpe S., Lightfoot H. A landscape of pharmacogenomic interactions in cancer. Cell. 2016;166:740–754. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R., Weinberg R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodet J.G., Beutler J.A., Wiemer D.F. Synthesis and structure activity relationships of schweinfurthin indoles. Bioorg. Med. Chem. 2014;22:2542–2552. doi: 10.1016/j.bmc.2014.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn K.W., Zeeberg B.M., Reinhold W.C., Pommier Y. Gene expression correlations in human cancer cell lines define molecular interaction networks for epithelial phenotype. PLoS One. 2014;9:e99269. doi: 10.1371/journal.pone.0099269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.-P., Liao W.-C., Ger L.-P., Chen J.-C., Hsu T.-I., Lee Y.-C., Chang H.-T., Chen Y.-C., Jan Y.-H., Lee K.-H. Carboxyl-terminal modulator protein positively regulates Akt phosphorylation and acts as an oncogenic driver in breast cancer. Cancer Res. 2013;73:6194–6205. doi: 10.1158/0008-5472.CAN-13-0518. [DOI] [PubMed] [Google Scholar]
- Luna A., Rajapakse V.N., Sousa F.G., Gao J., Schultz N., Varma S., Reinhold W., Sander C., Pommier Y. rcellminer: exploring molecular profiles and drug response of the NCI-60 cell lines in R. Bioinformatics. 2016;32:1272–1274. doi: 10.1093/bioinformatics/btv701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B., Bigay J., Polidori J., Jamecna D., Lacas-Gervais S., Antonny B. Sterol transfer, PI4P consumption, and control of membrane lipid order by endogenous OSBP. EMBO J. 2017;36:3156–3174. doi: 10.15252/embj.201796687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollaoglu G., Guthrie M.R., Böhm S., Brägelmann J., Can I., Ballieu P.M., Marx A., George J., Heinen C., Chalishazar M.D. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell. 2017;31:270–285. doi: 10.1016/j.ccell.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison B.L., Mullendore M.E., Stockwin L.H., Borgel S., Hollingshead M.G., Newton D.L. Oxyphenisatin acetate (NSC 59687) triggers a cell starvation response leading to autophagy, mitochondrial dysfunction, and autocrine TNFalpha-mediated apoptosis. Cancer Med. 2013;2:687–700. doi: 10.1002/cam4.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J., Feng Y., Yu G.K., Ru Y., Tang S.-W., Shen Y., Pommier Y. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget. 2016;7:76534–76550. doi: 10.18632/oncotarget.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J., Tang S.-W., Leo E., Baechler S.A., Redon C.E., Zhang H., Al Abo M., Rajapakse V.N., Nakamura E., Jenkins L.M.M. SLFN11 blocks stressed replication forks independently of ATR. Mol. Cell. 2018;69:371–384.e6. doi: 10.1016/j.molcel.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull K.D., Shoemaker R.H., Hodes L., Monks A., Scudiero D.A., Rubinstein L., Plowman J., Boyd M. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of a mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- Polley E., Kunkel M., Evans D., Silvers T., Delosh R., Laudeman J., Ogle C., Reinhart R., Selby M., Connelly J. Small cell lung cancer screen of oncology drugs, investigational agents, and gene and microRNA expression. J. Natl. Cancer Inst. 2016;108 doi: 10.1093/jnci/djw122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y., Sun Y., Huang S.N., Nitiss J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016;17:703–721. doi: 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees M.G., Seashore-Ludlow B., Cheah J.H., Adams D.J., Price E.V., Gill S., Javaid S., Coletti M.E., Jones V.L., Bodycombe N.E. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016;12:109–116. doi: 10.1038/nchembio.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold W.C., Sunshine M., Liu H., Varma S., Kohn K.W., Morris J., Doroshow J., Pommier Y. CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2012;72:3499–3511. doi: 10.1158/0008-5472.CAN-12-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold W.C., Sunshine M., Varma S., Doroshow J.H., Pommier Y. Using cellminer 1.6 for systems pharmacology and genomic analysis of the NCI-60. Clin. Cancer Res. 2015;21:3841–3852. doi: 10.1158/1078-0432.CCR-15-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold W.C., Varma S., Sousa F., Sunshine M., Abaan O.D., Davis S.R., Reinhold S.W., Kohn K.W., Morris J., Meltzer P.S. NCI-60 whole exome sequencing and pharmacological CellMiner analyses. PLoS One. 2014;9:e101670. doi: 10.1371/journal.pone.0101670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold W.C., Varma S., Sunshine M., Rajapakse V., Luna A., Kohn K.W., Stevenson H., Wang Y., Heyn H., Nogales V. The NCI-60 methylome and its integration into cellminer. Cancer Res. 2017;77:601–612. doi: 10.1158/0008-5472.CAN-16-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S., Koh C.C., Gillessen S., Joerger M., Jochum W., Aebersold R. Minimal sample requirement for highly multiplexed protein quantification in cell lines and tissues by PCT-SWATH mass spectrometry. Proteomics. 2015;5:3711–3721. doi: 10.1002/pmic.201500161. [DOI] [PubMed] [Google Scholar]
- Thornburg C.C., Britt J.R., Evans J.R., Akee R.K., Whitt J.A., Trinh S.K., Harris M.J., Thompson J.R., Ewing T.L., Shipley S.M. NCI program for natural product discovery: a publicly-accessible library of natural product fractions for high-throughput screening. ACS Chem. Biol. 2018;13:2484–2497. doi: 10.1021/acschembio.8b00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuda E., Fujita N., Oh-hara T., Sato S., Kurata A., Katayama R., Itoh T., Takenawa T., Miyazono K., Tsuruo T. Casein kinase 2–interacting protein-1, a novel Akt pleckstrin homology domain-interacting protein, down-regulates PI3K/Akt signaling and suppresses tumor growth in vivo. Cancer Res. 2007;67:9666–9676. doi: 10.1158/0008-5472.CAN-07-1050. [DOI] [PubMed] [Google Scholar]
- Wagner P.L., Stiedl A.-C., Wilbertz T., Petersen K., Scheble V., Menon R., Reischl M., Mikut R., Rubin M.A., Fend F. Frequency and clinicopathologic correlates of KRAS amplification in non-small cell lung carcinoma. Lung Cancer. 2011;74:118–123. doi: 10.1016/j.lungcan.2011.01.029. [DOI] [PubMed] [Google Scholar]
- Zoppoli G., Regairaz M., Leo E., Reinhold W.C., Varma S., Ballestrero A., Doroshow J.H., Pommier Y. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc. Natl. Acad. Sci. U S A. 2012;109:15030–15035. doi: 10.1073/pnas.1205943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oncogene and tumor suppressor gene mutation call frequencies and overlaps across data sources.
Comparison of drug activities as measured by the GDSC and the NCI/DTP.
Activity of bisacodyl and oxyphenisatin acetate (acetalax) in GDSC triple-negative breast cancer lines.
Co-occurrence of gene promoter methylation and DNA copy loss.
Top 50 drugs with respect to negative activity correlation with ABCB1 expression for the GDSC, CTRP (CCLE), and NCI-60 datasets.