

Abstract
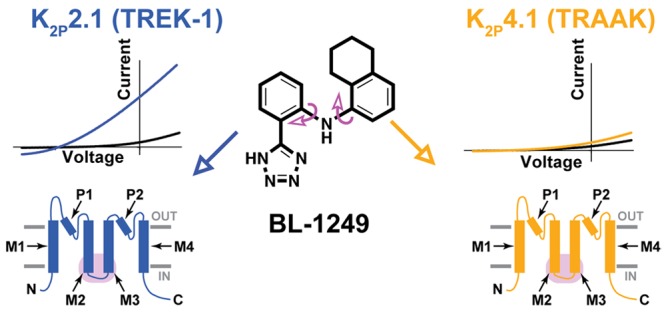
K2P potassium channels generate leak currents that stabilize the resting membrane potential of excitable cells. Various K2P channels are implicated in pain, ischemia, depression, migraine, and anesthetic responses, making this family an attractive target for small molecule modulator development efforts. BL-1249, a compound from the fenamate class of nonsteroidal anti-inflammatory drugs is known to activate K2P2.1(TREK-1), the founding member of the thermo- and mechanosensitive TREK subfamily; however, its mechanism of action and effects on other K2P channels are not well-defined. Here, we demonstrate that BL-1249 extracellular application activates all TREK subfamily members but has no effect on other K2P subfamilies. Patch clamp experiments demonstrate that, similar to the diverse range of other chemical and physical TREK subfamily gating cues, BL-1249 stimulates the selectivity filter “C-type” gate that controls K2P function. BL-1249 displays selectivity among the TREK subfamily, activating K2P2.1(TREK-1) and K2P10.1(TREK-2) ∼10-fold more potently than K2P4.1(TRAAK). Investigation of mutants and K2P2.1(TREK-1)/K2P4.1(TRAAK) chimeras highlight the key roles of the C-terminal tail in BL-1249 action and identify the M2/M3 transmembrane helix interface as a key site of BL-1249 selectivity. Synthesis and characterization of a set of BL-1249 analogs demonstrates that both the tetrazole and opposing tetralin moieties are critical for function, whereas the conformational mobility between the two ring systems impacts selectivity. Together, our findings underscore the landscape of modes by which small molecules can affect K2P channels and provide crucial information for the development of better and more selective K2P modulators of the TREK subfamily.
Keywords: K2P channel, TREK channel, electrophysiology, ion channel chemical biology
Introduction
K2P (KCNK) potassium channels are members of the voltage-gated ion channel (VGIC) superfamily, make “background” or “leak” potassium channels that are responsible for the maintenance of cellular resting potential, and play an important role in regulating cellular excitability.1−3 There are 15 K2P subtypes that form six functionally distinct subfamilies. All K2P channels comprise a dimer of subunits that each bear four transmembrane helices and two selectivity filter sequences.1,4−7 In contrast to other VGIC superfamily members, the K2P channel selectivity filter forms the principle gate that controls channel function, known as the “C-type gate”, rather than an intracellular barrier formed by the pore-lining helices.6,8−12 The activity of various K2P subtypes has been linked to a variety of physiological and pathological processes including pain,13−15 anesthetic responses,16,17 arrhythmia,18 ischemia,16,19,20 depression,21 and migraine.22,23 Yet, despite these biological links, a paucity of K2P-selective small molecule modulators has limited mechanistic and physiological studies.1,24,25 Recent advances demonstrate that it is possible to develop subtype-selective K2P small molecule modulators.6,14,26−30 Such compounds and the knowledge of how they engage K2P channels to modulate function open a path toward elaborating new K2P-specific pharmacological tools that can enlighten channel gating mechanisms and that have potential to provide new leads for issues such as pain and ischemia.25,31,32
The TREK K2P subfamily, comprising K2P2.1(TREK-1), K2P10.1(TREK-2), and K2P4.1(TRAAK), is regulated by diverse inputs that include temperature, stretch, pH, and lipids1−3 and stands out as the most structurally elucidated K2P subfamily.4,6,7,33−35 TREK subfamily structures include examples of both inhibitor–channel7 and activator–channel complexes6 that highlight two points of control that can be influenced by small molecules: the transmembrane helices7 and the K2P modulator pocket.6 Although these examples show how a small molecule can engage with the K2P channel architecture to impact function, whether other reported TREK activators14,26,28,36 act via the transmembrane domains or the K2P modulator pocket or affect other K2P channel elements remains to be elaborated.
Although not selective for K2P channels, a number of fenamates, substituted derivatives of anthranilic acid,37 activate members of the mechanosensitive TREK K2P subfamily.38,39 In particular, BL-1249, (5,6,7,8-tetrahydro-naphthalen-1-yl)-[2-(1H-tetrazol-5-yl)-phenyl]-amine, stimulates K2P2.1(TREK-1)-like currents in bladder smooth muscle cells39 and activates both K2P2.1(TREK-1)18,40 and K2P10.1(TREK-2).7 BL-1249 action is occluded by mutations that stabilize the C-type gate,40 and activation by BL-1249 has been shown to reverse the functional effects of a K2P2.1(TREK-1) genetic mutation implicated in right ventricular outflow tract (RVOT) tachycardia having a compromised ion selectivity.18 Nevertheless, how BL-1249 stimulates K2P activity and which elements of BL-1249 are crucial for its stimulatory effects have not yet been defined.
Here, we investigate the mechanism of action of BL-1249. Our studies show that this compound is a selective agonist of the TREK subfamily when applied extracellularly, having preferential action on K2P2.1(TREK-1) and K2P10.1(TREK-2) over K2P4.1(TRAAK) and establish that its mechanism of action relies on gating at the selectivity filter C-type gate. Studies of a series of K2P2.1(TREK-1)/K2P4.1(TRAAK) chimeras and mutants indicate that the M2/M3 helices are key to BL-1249 action and identify residues in M2 that contribute to subtype selectivity. These findings indicate that BL-1249 acts at a separate site from the site of action of a structurally characterized activator, ML335, that directly stimulates the C-type gate via the K2P modulator pocket.6 Investigation of the functional properties of a set of BL-1249 analogs with respect to K2P 2.1(TREK-1) and K2P 4.1(TRAAK) show that both the acidic and tetralin moieties contribute to the stimulatory action of BL-1249 and indicate that the mobility of the two aryl rings relative to each other is key to the selective action of BL-1249 on K2P 2.1(TREK-1).
Results
BL-1249 External Application Differentially and Selectively Activates Mechanosensitive K2P Channels
BL-1249 activates K2P2.1(TREK-1)18,40 and K2P10.1(TREK-2),7 but its effects on other K2P channels have not been characterized. Hence, we sought to define how BL-1249 affected the two channels most closely related to K2P2.1(TREK-1), K2P10.1(TREK-2) and K2P4.1(TRAAK), as well as representative members of the other K2P subtypes: K2P1.1(TWIK-1), K2P3.1(TASK-1), K2P5.1(TASK-2), K2P9.1(TASK-3), K2P13.1(THIK-1), and K2P18.1(TRESK) (Figure 1A,B). Two-electrode voltage-clamp (TEVC) currents measured from Xenopus oocytes expressing each of the target channels showed clear activation responses after extracellular application of 10 μM BL-1249 for only K2P2.1(TREK-1) and K2P10.1(TREK-2). Measurement of the dose–response curves for these channels (Figure 1C) revealed similar EC50 values (5.5 ± 1.2 μM and 8.0 ± 0.8 μM for K2P2.1(TREK-1) and K2P10.1(TREK-2), respectively). Ten micromolar BL-1249 weakly stimulated K2P4.1(TRAAK) in agreement with observations by Mathie and colleagues.28 Measurement of the dose–response uncovered a robust response at higher concentrations that indicated a ∼10-fold reduction in the K2P4.1(TRAAK) EC50 value relative to the other two TREK subfamily members (EC50= 48 ± 10 μM, although complete saturation of the response could not be reached due to BL-1249 solubility limits, cf. Table 1; Figure 1C). In order to test if the original 10 μM assay had missed BL-1249 effects in other K2P channels, we tested concentrations of BL-1249 up to the solubility limit (∼80 μM) against the other K2P subfamily representatives. Despite the higher concentrations of BL-1249, we failed to find evidence for activation of the other K2P subfamily representatives (Figure 1D). Thus, the data show that extracellular application of BL-1249 activates all members of the mechano- and thermosensitive TREK subfamily while sparing the other subfamilies and shows selectivity for K2P2.1(TREK-1) and K2P10.1(TREK-2) over K2P4.1(TRAAK).
Figure 1.

External application of BL-1249 selectively activates mechanosensitive K2P channels. (A) Exemplar current traces for specified K2P channels (black) with 10 μM BL-1249 (light blue) as measured via TEVC in Xenopus oocytes. (B) K2P channel phylogenetic tree. Stars denote assayed representative K2P channels. Blue stars indicate BL-1249 responsive channels. (C) BL-1249 dose–response curves for K2P2.1(TREK-1) (blue circles), K2P10.1(TREK-2) (black triangles), and K2P4.1(TRAAK) (orange squares). EC50 = 5.5 ± 1.2 μM, 8.0 ± 0.8 μM, and 48 ± 10 μM, respectively. (D) BL-1249 responses of indicated K2P channels. Inset shows expanded view of poorly responsive K2P channels. Error bars are SEM.
Table 1. Summary of K2P Response to BL-1249a.
| background | mutant | EC50 (μM) | n (≥) |
|---|---|---|---|
| K2P2.1(TREK-1) | 5.5 ± 1.2 | 3 | |
| K2P2.1(TREK-1)GGG | 19 ± 1 | 3 | |
| K2P2.1(TREK-1)Δ322 | 26 ± 8b | 2 | |
| K2P2.1(TREK-1)Δ308 | 35 ± 8b | 2 | |
| TREK-1/AAK_T | 7.7 ± 0.6 | 2 | |
| TREK-1/AAK M4-C | 19 ± 3 | 3 | |
| TREK-1/AAK M3-C | 28 ± 2 | 3 | |
| TREK-1/AAK M2-C | 39 ± 9 | 3 | |
| TREK-1/TRAAK M2 | 26 ± 8 | 3 | |
| F172M | 15 ± 2 | 3 | |
| F185L | 27 ± 5 | 3 | |
| K2P10.1(TREK-2) | 8.0 ± 0.8 | 3 | |
| K2P4.1(TRAAK) | 48 ± 10b | 3 | |
| TRAAK/EK-1_T | 23 ± 4 | 2 | |
| TRAAK/EK-1 M4-C | 45 ± 2 | 3 | |
| TRAAK/EK-1 M3-C | 18 ± 2 | 3 | |
| TRAAK/EK-1 M2-C | 28 ± 5 | 3 | |
| TRAAK/TREK-1 M2 | 43 ± 11 | 3 | |
| M134F | 58 ± 34b | 2 | |
| L147F | 27 ± 4 | 3 |
Data derived from at least two independent experiments with each data point averaged from at least three oocytes.
Experiments where complete saturation of the response could not be reached due to BL-1249 solubility limits. For these cases, fits were imposed with an upper boundary of 15 (fold activation, I/I0) to estimate EC50 and error.
BL-1249 Activates the K2P2.1(TREK-1) C-type Gate
Diverse types of physical and chemical stimuli activate K2P2.1(TREK-1) by stabilizing the C-type gate and switching the channel into a “leak” mode that is characterized by a loss of outward rectification of the potassium current.6,8 Accordingly, we used inside-out patch clamp experiments of K2P2.1(TREK-1) expressed in HEK293 cells to test whether BL-1249 acts on the C-type gate. Application of 1 μM BL-1249 caused a clear loss of rectification similar to the effects reported for both physical and chemical activators of K2P2.1(TREK-1) (Figure 2A–E).6,8 Additionally, TEVC studies of the effects of BL-1249 on K2P2.1(TREK-1) channels bearing mutations that activate the C-type gate, G137I and W275S,6,10,41 demonstrated that these channels were insensitive to BL-1249 (Figure 2F). Together with previous single point concentration studies showing the insensitivity of K2P2.1(TREK-1) W275S in mammalian cells,40 our data provide definitive evidence that BL-1249 activates K2P2.1(TREK-1) by stimulating the C-type gate. Thus, this compound fits the mechanistic paradigm shared by varied types of activators including mechanical stretch, pH, lipids, and small molecules.6,8
Figure 2.

BL-1249 activates the K2P2.1(TREK-1) C-type gate. (A, B) Exemplar current traces for (A) K2P2.1(TREK-1) and (B) K2P2.1(TREK-1) with 1 μM BL-1249 in HEK293 inside-out patches in 150 mM K+[out]/150 mM Rb+[in]. Inset shows voltage protocol. (C, D) Current–voltage relationships for (C) K2P2.1(TREK-1) and (D) K2P2.1(TREK-1) with 1 μM BL-1249. (E) Rectification coefficients (I+100mV/I–100mV) from recordings (n ≥ 3) made in panels A–D. (F) Dose–response curves in Xenopus oocytes for K2P2.1(TREK-1) (blue circles), K2P2.1(TREK-1) G137I (purple squares), and K2P2.1(TREK-1) W275S (orange triangles). K2P2.1(TREK-1), 5.5 ± 1.2 μM; G137I and W275S, >60 μM. K2P2.1(TREK-1) data are from Figure 1C. Error bars are SEM.
K2P C-Terminal Tail Is Necessary for BL-1249 Action but Is Not the Sole Determinant of Channel Responsiveness
Gating stimuli detected by sensors in various parts of the channel converge on the K2P selectivity filter C-type gate.6,8,10,12,35 Because the C-terminal tail is the sensor for K2P2.1(TREK-1) activation by both physiological41−46 and chemical45,47 activators, we asked whether uncoupling the C-terminal tail from the channel using a triple-glycine mutant at the M4/C-terminal tail junction, K2P2.1(TREK-1)GGG,41 impacted the BL-1249 response. TEVC dose response studies of K2P2.1(TREK-1)GGG showed that the effects of BL-1249 were significantly blunted relative to wild-type channels (EC50 = 19 ± 1 μM) (Figure 3A). This result contrasts previous studies of the small molecule activator ML67-3326 for which uncoupling the C-terminal tail had no effect and suggests that unlike ML67-33, the C-terminal tail plays a role in mediating the BL-1249 response.
Figure 3.

K2P2.1(TREK-1) C-terminus affects BL-1249 response. (A) Exemplar current traces for K2P2.1(TREK-1)GGG (black) with 20 μM BL-1249 (green) and BL-1249 dose–response curves for K2P2.1(TREK-1) (blue circles) and K2P2.1(TREK-1)GGG (green circles) (EC50 = 5.5 ± 1.2 μM and 19 ± 1 μM, respectively). Green star in cartoon indicates site of GGG mutation. (B) Exemplar current traces for K2P2.1(TREK-1)μ322 (magenta) and K2P2.1(TREK-1)μ308 (purple) with 20 μM BL-1249 and BL-1249 dose–response curves for K2P2.1(TREK-1) (blue circles), K2P2.1(TREK-1)Δ322 (purple squares), and K2P2.1(TREK-1)Δ308 (magenta triangles). (EC50 = 5.5 ± 1.2 μM, 26 ± 8 μM, and 35 ± 8 μM, respectively). Magenta and purple lines in cartoon indicate sites of Δ322 and Δ308 truncations, respectively. (C) Exemplar current traces for TREK-1/AAK_T alone (black) and with 20 μM BL-1249 (cyan). BL-1249 dose–response curves for K2P2.1(TREK-1) (blue circles), K2P4.1(TRAAK) (light orange squares), and TRAAK/EK-1_T (light blue triangles). (EC50 = 5.5 ± 1.2 μM, 48 ± 10 μM, and 7.7 ± 0.6 μM, respectively). Cartoon indicates TREK-1/AAK_T channel regions from K2P2.1 (blue) and K2P4.1 (yellow). (D) Exemplar current traces for TRAAK/EK_T alone (black) and with 20 μM BL-1249 (orange). BL-1249 dose–response curves for K2P2.1(TREK-1) (blue circles), K2P4.1(TRAAK) (light orange squares), and TRAAK/EK_T (orange triangles) (EC50 = 5.5 ± 1.2 μM, 48 ± 10 μM, and 23 ± 4 μM, respectively). Cartoon indicates TRAAK/EK_T channel regions from K2P2.1 (blue) and K2P4.1 (light orange). In panels A–D, K2P2.1(TREK-1) and K2P4.1(TRAAK) data are from Figure 1C. Error bars are SEM.
K2P2.1(TREK-1) C-terminal truncations have been shown to reduce potentiation by other activating stimuli.42,48 Hence, to probe the role of the C-terminal tail in the BL-1249 response further, we examined the effects of C-terminal tail truncations at residue 322, K2P2.1(TREK-1)Δ322, equivalent to a previously described mutant called Δ89,48 and at residue 308, K2P2.1(TREK-1)Δ308. Measurement of the dose–response curves for BL-1249 showed that these changes resulted in progressively reduced responses (EC50 = 26 ± 8 μM and 35 ± 8 μM for K2P2.1(TREK-1)Δ322 and K2P2.1(TREK-1)Δ308, respectively) (Figure 3B).
Given the importance of the C-terminal tail for the BL-1249 response and the fact that the C-terminal tails of K2P2.1(TREK-1) and K2P4.1(TRAAK) vary substantially (17.6% sequence similarity, 13.5% identity), we wondered if these differences could contribute to the different potencies observed in K2P2.1(TREK-1) and K2P4.1(TRAAK) BL-1249 responses. To test this possibility, we made chimeras that swapped the C-terminal tail between K2P2.1(TREK-1) and K2P4.1(TRAAK), TREK-1/AAK_T and TRAAK/EK_T. The TREK-1/AAK_T chimera yielded channels having responses similar to K2P2.1(TREK-1) (EC50 = 7.7 ± 0.6 μM and 5.5 ± 1.2 μM for TREK-1/AAK_T and K2P2.1(TREK-1), respectively; Figure 3C). By contrast, swapping the K2P2.1(TREK-1) C-terminal tail onto K2P4.1(TRAAK), TRAAK/EK-1_T, increased the sensitivity of K2P4.1(TRAAK) core to BL-1249 by ∼2-fold (EC50 = 23 ± 4 μM and 48 ± 10 μM, for TRAAK/EK-1_T and K2P4.1(TRAAK) respectively) (Figure 3D). Despite this modest change, it is clear that the C-terminal tail alone is not sufficient to endow the K2P4.1(TRAAK) core with a K2P2.1(TREK-1)-like BL-1249 response. Although the C-terminal tail is not a major locus for the selective actions of BL-1249, the strong impact that uncoupling the C-terminal tail from the core channel and C-terminal tail truncations has on the K2P2.1(TREK-1) response to BL-1249 indicate that this channel element is an important factor that allows channel activation by BL-1249.
Multiple Transmembrane Regions Contribute to BL-1249 Activation
To look for other elements that might contribute to BL-1249 responses, we constructed a set of K2P2.1(TREK-1)/K2P4.1(TRAAK) chimeras in which an increasing amount of one channel was spliced with the other. For the purposes of nomenclature, the channels are named using the parent N-terminal portion and the C-terminal chimera junction even though the chimera set forms a continuum spanning the two wild-type channels (Figure 4A). For example, TREK-1/AAK M4-C bears the K2P2.1(TREK-1) sequence up to the junction with M4, whereas TRAAK/EK-1 M2-C bears K2P4.1(TRAAK) up to the junction with M2 even though the largest portion of both of these channels comes from K2P2.1(TREK-1).
Figure 4.

BL-1249 responses of TREK-1/TRAAK chimeras (A) Exemplar current traces for of BL-1249 responses for K2P2.1(TREK-1) (blue), K2P4.1(TRAAK) (light orange), and chimeras TREK-1/AAK M4-C (light blue), TREK-1/AAK M3-C (green), TREK-1/AAK M2-C (light green), TRAAK/EK-1 M4-C (light green), TRAAK/EK-1 M3-C (green), and TRAAK/EK-1 M2-C (light blue). Black and colored traces show basal and currents with 20 μM BL-1249, respectively. Cartoon schematics show channel portions from K2P2.1(TREK-1) (blue) and K2P4.1(TRAAK) (light orange). (B, C) Dose–response curves for K2P2.1(TREK-1), K2P4.1(TRAAK), and the indicated chimeras. (EC50 = 5.5 ± 1.2 μM, 48 ± 10 μM, 19 ± 3 μM, 28 ± 3 μM, 39 ± 9 μM, 45 ± 2 μM, 18 ± 2 μM, and 28 ± 5 μM for K2P2.1(TREK-1), K2P4.1(TRAAK), TREK-1/AAK M4-C, TREK-1/AAK M3-C, TREK-1/AAK M2-C, TRAAK/EK-1 M4-C, TRAAK/EK-1 M3-C, and TRAAK/EK-1 M2-C, respectively). K2P2.1(TREK-1) and K2P4.1(TRAAK) data are from Figure 1C. Error bars are SEM.
TEVC experiments showed that all of the chimeras formed functional channels (Figures S1 and S2). To test the ability of the chimeras to report on channel determinants for compound action, we examined the responses of the chimeras to two previously characterized activators, ML335, a compound that selectively activates K2P2.1(TREK-1) but not K2P4.1(TRAAK),6 and ML67-33, an activator showing no clear preference for either channel.26 The chimeras showed an essentially binary response to ML335 that was entirely dependent on the presence or absence of a lysine on the extracellular end of M4 that forms a cation−π interaction with ML335.6 Only constructs bearing a lysine at position equivalent to K2P2.1(TREK-1) residue 271 (K2P2.1(TREK-1), TRAAK/EK-1 M2-C, TRAAK/EK-1 M3-C, and TREK-1/AAK M4-C) robustly responded to ML335 (Figure S1A–C,G). In contrast to these results, we found no major changes with respect to the responses of the various chimeras to ML67-33 (Figure S1D–F,H). These findings are consistent with the inability of ML67-33 to discriminate between K2P2.1(TREK-1) and K2P4.1(TRAAK).26 Together, these studies show that this chimera set can identify selectivity determinants for activator compounds within the TREK subfamily.
We next examined how this panel of chimeras responded to BL-1249. We found that the character of the donor channel with respect to BL-1249 response became progressively prevalent as larger portions were swapped into the recipient channel, contrasting the binary changes seen for ML335 responses (Figures 4A–C and S1G, Table 1). Further, the patterns of changes in the BL-1249 responses were not equivalent with respect to the direction of the substitution. Substituting K2P4.1(TRAAK) sequence into K2P2.1(TREK-1) from the C-terminal direction caused stepwise changes in EC50 as the construct became dominated by the K2P4.1(TRAAK) sequence (Figure 4A,C, clockwise in Figure 4A from K2P2.1(TREK-1), EC50 = 5.5 ± 1.2, 19 ± 3, 28 ± 2, and 39 ± 9 μM for K2P2.1(TREK-1), TREK-1/AAK M4-C, TREK-1/AAK M3-C, and TREK-1/AAK M2-C, respectively). By contrast, substitution of K2P4.1(TRAAK) sequence into K2P2.1(TREK-1) from the N-terminal direction caused a loss, mild recovery, and then further loss of BL-1249 response (Figure 4A,B, counterclockwise in Figure 4A from K2P2.1(TREK-1), EC50 = 5.5 ± 1.2, 28 ± 5, 18 ± 2, 45 ± 2, and 48 ± 10 μM, for K2P2.1(TREK-1), TRAAK/EK-1 M2-C, TRAAK/EK-1 M3-C, TRAAK/EK-1 M4-C, and K2P4.1(TRAAK)). The complexity of the EC50 changes displayed by the chimeras with respect to BL-1249 contrasted with how these chimeras responded for the case in which there is a single site responsible for compound selectivity (Figure S1G). Such a contrast suggests that multiple parts of the channel make contributions that influence BL-1249 selectivity rather than just a single site.
K2P2.1(TREK-1) M2 Residues Contribute to BL-1249 Selectivity
In the course of converting K2P2.1(TREK-1) to K2P4.1(TRAAK) and vice versa by chimeras, the M2/M3 region stood out as a point where we found both gradual changes in EC50 (i.e., TREK-1/AAK M3-C → TREK-1/AAK M2-C) and stepwise changes that reversed the general EC50 trend (i.e., TRAAK/EK-1 M3-C → TRAAK/EK-1 M2-C). To investigate this issue further, we generated two chimeras in which only the M2 helix was exchanged between K2P2.1(TREK-1) and K2P4.1(TRAAK), TREK-1/TRAAK M2 and TRAAK/TREK-1 M2. The substitution of the K2P2.1(TREK-1) M2 helix into K2P4.1(TRAAK) had little effect on the BL-1249 response, yielding a channel having a response indistinguishable from K2P4.1(TRAAK) (EC50 = 43 ± 11 μM and 48 ± 10 μM for TRAAK/TREK-1 M2 and K2P4.1(TRAAK), respectively) (Figure 5A). By contrast, the substitution of the K2P4.1(TRAAK) M2 helix into K2P2.1(TREK-1) caused a substantial loss in BL-1249 response (EC50 = 26 ± 8 μM and 5.5 ± 1.2 μM for TREK-1/TRAAK M2 and K2P2.1(TREK-1), respectively), indicating that elements from M2 contribute to the K2P2.1(TREK-1) response to BL-1249.
Figure 5.

M2 residues contribute to BL-1249 selectivity between K2P2.1(TREK-1) and K2P4.1(TRAAK). (A) Exemplar current traces for TREK-1/TRAAK M2 (light blue) and TRAAK/TREK-1 M2 (orange) with 20 μM BL-1249 (left). Insets depict M2 helix swap. BL-1249 dose–response curves (right) for K2P2.1(TREK-1) (blue circles), TREK-1/TRAAK M2 (light blue squares), K2P4.1(TRAAK) (light orange circles), and TRAAK/TREK-1 M2 (orange triangles). (EC50 = 5.5 ± 1.2 μM, 26 ± 8 μM, 48 ± 10 μM, and 43 ± 11 μM, respectively). (B) Alignment of K2P2.1(TREK-1) and K2P4.1(TRAAK) M2 sequences. Nonconserved residues are highlighted in yellow. (C) K2P2.1(TREK-1) (PDB 6CQ6)6 structure. Residues that differ from K2P4.1(TRAAK) are highlighted yellow. Panel insets show the environment surrounding the highlighted M2 residues. (D) Exemplar current traces for TREK-1/F172M (pink) and TREK-1/F185L (light blue), with 20 μM BL-1249. BL-1249 dose–response curves for K2P2.1(TREK-1) (blue circles), TREK-1/F172M (pink circles), K2P2.1 (F185L) (light blue squares), and K2P4.1(TRAAK) (light orange circles). (EC50 = 5.5 ± 1.2 μM, 15 ± 2 μM, 27 ± 5 μM, and 48 ± 10 μM, respectively). (E) Exemplar current traces for TRAAK/M134F (orange triangles) and TRAAK/L147F (olive green squares) with 20 μM BL-1249. BL-1249 dose–response curves for K2P2.1(TREK-1) (blue circles), TRAAK/M134F (orange triangles), TRAAK/L147F (olive green squares), and K2P4.1(TRAAK) (light orange circles). (EC50 = 5.5 ± 1.2 μM, 58 ± 34 μM, 27 ± 4 μM, and 48 ± 10 μM, respectively). K2P2.1(TREK-1) and K2P4.1(TRAAK) data are from Figure 1C. Inset compares responses at 35 μM BL-1249: *** indicates p < 0.001 for a one-way ANOVA test; ns indicates no significant difference. Error bars are SEM.
To identify K2P2.1(TREK-1) M2 residues that might participate in the BL-1249 response, we mapped the residues that differ between K2P2.1(TREK-1) and K2P4.1(TRAAK) in the context of the K2P2.1(TREK-1) structure6 (Figure 5B,C). Two K2P2.1(TREK-1) M2 residues stood out as candidates that could explain the reduction in BL-1249 response when the entire M2 helix was replaced with M2 from K2P4.1(TRAAK). One is at the M2/M4 interface and occurs between Phe172 and Arg297 via a π–cation interaction that would be lost when Phe172 is replaced with the equivalent K2P4.1(TRAAK) residue Met134 (Figure 5C, right inset). The second is an intrasubunit π–π interaction between Phe185 from M2, a site whose equivalent in K2P10.1(TREK-2) (Phe215) has a role in membrane stretch responses,7 and Phe214 from M3 that would be disrupted by the replacement with the equivalent K2P4.1(TRAAK) residue Leu147 (Figure 5C, left inset). To assess the importance of these interactions in the context of BL-1249 response, we made the K2P2.1(TREK-1) mutants F172M and F185L and measured their responses to BL-1249 (Figure 5D). Both changes reduced the BL-1249 response (EC50 = 15 ± 2 μM and 27 ± 5 μM for F172M and F185L, respectively) to levels similar to the M2 helix swap. Notably, the M2/M3 interface substitution, F185L had a larger impact on the EC50, whereas the M2/M4 change F172M caused a substantial reduction in the extent to which the channel could be activated by BL-1249 (Figure 5D). Unlike the case for K2P2.1(TREK-1), the two corresponding inverse mutations in K2P4.1(TRAAK), M134F at the M2/M4 interface and L147F at the M2/M3 interface did not cause similar outcomes. The mutation at the M2/M4 interface had no impact on BL-1249 response (EC50, M134F 58 ± 34 μM), whereas the change in the M2/M3 interface conferred a modest improvement in the BL-1249 response (EC50, L147F EC50 = 27 ± 4 μM; p < 0.001 at 35 μM (n = 7)) (Figure 5E). Taken together, these data highlight the importance of the M2 helix in BL-1249 activation. The observation that amino acid swaps in the M2/M3 interface are able to blunt the response of K2P2.1(TREK-1) but enhance the response of K2P4.1(TRAAK) points to the M2/M3 interface as a key element in the differential effects of BL-1249 on TREK subfamily members.
The BL-1249 Acidic Group and Tetralin Are Critical for Potency and Selectivity
Fenamates are weak K2P modulators38,39,49 and their structure–activity relationships (SAR) with respect to K2P channels are poorly defined. Hence, we synthesized a set of BL-1249 derivatives in order to probe which portions of the small molecule were important for channel activation in the context of the differential responses of K2P2.1(TREK-1) and K2P 4.1(TRAAK). BL-1249 has two ring systems, one bearing a tetrazole and a second bearing a tetralin moiety. Replacement of the tetrazole by other similar functionalities resulted in compounds having poorer potency than BL-1249 against K2P2.1(TREK-1) (EC50 = 22 ± 8 μM and 44 ± 10 μM for BL-1249-amide and BL-1249-acid, respectively; Figure 6A,B, Table 2). Notably, even though BL-1249-acid was slightly less potent than BL-1249-amide (∼2-fold), it had a stronger stimulatory effect on the current than either BL-1249 or BL-1249-amide, suggesting that the acidic nature of the side chain is important for BL-1249 function (Figure 6B). Curiously, unlike what we observe for the TREK subfamily, for K2P18.1(TRESK) the change from BL-1249 to BL-1249-acid has been reported to switch the functional effects of the compound from an activator to an inhibitor.49 Both BL-1249-amide and BL-1249-acid retained selectively for K2P2.1(TREK-1) over K2P4.1(TRAAK) (Figure 6C,D) indicating that this moiety is not the key determinant of selectivity.
Figure 6.

Structure–activity relationships of BL-1249 analogues. (A) Chemical structures of BL-1249 and analogues. “I” and “II” indicate the substitution sites. (B) Dose–response of K2P2.1(TREK-1) for BL-1249 (black) (from Figure 1C), BL-1249-amide (blue), BL-1249-acid (light blue), and BL-1249-Ph (red) (EC50= 5.5 ± 1.2 μM, 22 ± 8 μM, 44 ± 10 μM, and >100 μM, respectively). K2P2.1(TREK-1) and K2P4.1(TRAAK) data are from Figure 1C. (C–E) Exemplar current traces for K2P2.1(TREK-1) with 25 μM BL-1249-amide (blue), BL-1249-acid (light blue), and BL-1249-Ph (red), respectively (top), chemical structures of BL-1249 analogues (middle), and dose response of K2P2.1(TREK-1) (blue) and K2P4.1(TRAAK) (light orange) (bottom). (F) Chemical structure of BL-1249-tricycle. (G) Exemplar current traces for K2P2.1(TREK-1) and K2P4.1(TRAAK) with 25 μM BL-1249-tricycle (purple). (H) Dose–response of K2P2.1(TREK-1) (blue) and K2P4.1(TRAAK) (light orange) for BL-1249-tricycle (EC50= 34 ± 6 μM and 42 ± 9 μM, respectively). Error bars are SEM.
Table 2. Summary of BL-1249 Analogue Activation of K2P2.1(TREK-1) and K2P4.1(TRAAK)a.
| compound | K2P2.1(TREK-1) EC50 (μM) | n (≥) | K2P4.1(TRAAK) EC50 (μM) | n (≥) |
|---|---|---|---|---|
| BL-1249 | 5.5 ± 1.2 | 3 | 48 ± 10b | 3 |
| BL-1249-amide | 22 ± 8 | 3 | >200 | 3 |
| BL-1249-acid | 44 ± 10c | 4 | >200 | 2 |
| BL-1249-Ph | >200 | 3 | >200 | 3 |
| BL-1249-tricycle | 34 ± 6 | 2 | 42 ± 9 | 3 |
Data derived from at least two independent experiments with each data point averaged from at least three oocytes.
EC50 estimated imposing an upper boundary of 15 (fold activation, I/I0).
EC50 estimated imposing an upper boundary of 20 (fold activation, I/I0)
To test the importance of the tetralin moiety, we synthesized a BL-1249 derivative in which this entity was replaced by a simple phenyl ring (BL-1249-Ph) (Figure 6A). This substitution proved very detrimental to activity and yielded a compound that had only a small amount of stimulatory effect against K2P2.1(TREK-1) (EC50 > 200 μM) and showed a similar profile against K2P4.1(TRAAK) revealing the importance of the bicyclic tetralin ring for BL-1249 function (Figure 6B,E). Finally, we tested whether the conformation of the two aryl rings with respect to each other was important for the potency and selectivity of BL-1249. We made a BL-1249 derivative in which the tetralin structure was fused into a tricyclic scaffold to constrain the available conformations between the two aryl rings (BL-1249-tricycle) (Figure 6F). BL-1249-tricycle showed poorer activity against K2P2.1(TREK-1) relative to BL-1249 (Figure 6G,H, EC50 = 34 ± 6 μM versus 5.5 ± 1.1 μM for BL-1249-tricycle and BL-1249, respectively) but, surprisingly, retained essentially the same activity against K2P4.1(TRAAK) (Figure 6G,H, EC50 = 42 ± 9 μM versus 48 ± 10 μM for BL-1249-tricycle and BL-1249, respectively). This loss in selectivity between K2P2.1(TREK-1) and K2P4.1(TRAAK) indicates that the ability of the aryl and tetralin rings to adopt non-coplanar conformations is key to the preferential action of BL-1249 on K2P2.1(TREK-1). We also observed that BL-1249-tricycle showed a small but robust “mode switch” behavior versus K2P2.1(TREK-1) manifested as inhibition between 0.1 and 10 μM followed by activation at higher concentrations. This behavior was not evident against K2P4.1(TRAAK) and further indicates the importance of the mobility between the two ring systems for the stimulatory action of BL-1249 on K2P2.1(TREK-1). Notably, other tricyclic compounds have been reported to inhibit K2P2.1(TREK-1) but not K2P4.1(TRAAK)50 similar to the properties of BL-1249-tricycle. Together, our studies demonstrate that both the acidic and tetralin moieties are important contributors to the stimulatory action of BL-1249 against K2P2.1(TREK-1) and indicate that the mobility of the two aryl rings relative to each other is key to its selective effects on K2P2.1(TREK-1) over K2P 4.1(TRAAK).
Discussion
Addressing the relatively poor chemical biology surrounding the K2P family is an important goal that has the potential to provide tool compounds that can remove the current barriers to understanding how diverse inputs modulate K2P function, as well as the physiological roles of K2P channels in various tissues.1,24,25 Our studies show that the fenamic acid derivative BL-1249,39 previously shown to activate K2P2.1(TREK-1)18,40 and K2P10.1(TREK-2),7 potently and selectively activates all three members of the mechanosensitive TREK K2P subfamily, K2P2.1(TREK-1), K2P10.1(TREK-2), and K2P4.1(TRAAK), by potentiating the potassium currents with EC50 values in the low micromolar range when applied extracellularly. Similar to many K2P activators,6,8,10,26,41 BL-1249 enhances TREK subfamily currents by stimulating the selectivity filter C-type gate. This mode of action provides further evidence for the central role of this C-type gate in the control of K2P function.
There are currently two structurally characterized examples for how small molecules can engage members of the K2P family. The cocrystal structure of K2P10.1(TREK-2) with inhibitor norfluoxetine shows that this inhibitor binds in a pocket underneath the P2 helix of the selectivity filter at a site that is framed by the M2, M3, and M4 transmembrane helices and that becomes accessible when the pore lining M4 helix adopts a “down” conformation.7,51 Although the binding site is clearly demarcated, how state-dependent binding of norfluoxetine inhibits is still unclear. Interestingly, besides inhibiting the movement of the M4 helix, the structural data indicates that the primary amine of norfluoxtine is near to the lower side of the selectivity filter where it could impact ion conduction by interfering with the electrostatic environment of the pore. The other structural example shows that a pair of related molecules, ML335 and ML402, bind to a cryptic binding site, the K2P modulator pocket, situated behind the selectivity filter and sandwiched at the interface between the P1 pore helix and top of the M4 transmembrane helix of K2P2.1(TREK-1).6 These activators act as wedges that stabilize the mobility of the P1/M4 interface, a site also impacted by gain-of-function mutations,6,10,35,41 and directly activate the C-type gate.6 Understanding the extent to which other K2P modulators, such as BL-1249 and ML67-33, act at the norfluoxetine site, the K2P modulator pocket, or elsewhere on the channel is important for outlining the landscape of druggable sites for the K2P potassium channel class.
Our studies of mutants and chimeras of K2P2.1(TREK-1) and K2P4.1(TRAAK) indicate that multiple channel elements that include C-terminal tail and the M2, M3, and M4 transmembrane helices contribute to BL-1249 responses. The integrity of the C-terminal tail is essential for BL-1249 stimulation (Figure 3), placing BL-1249 within a diverse class of TREK subfamily activators that are functionally dependent on this channel element including lipids,42 arachidonic acid,45 intracellular acidosis,46 chloroform,45 and temperature.41 This dependence on the C-terminal tail is notably not shared by another tetrazole containing small molecule activator, ML67-33.26 Despite the importance of the C-terminal tail in the BL-1249 response, our data indicate that this channel element has a limited role in mediating the selective action of BL-1249 on K2P2.1(TREK-1) over K2P4.1(TRAAK). By contrast, we do find evidence that multiple transmembrane domains contribute to BL-1249 selectivity. Although in the context of K2P2.1(TREK-1)/K2P4.1(TRAAK) chimeras no single transmembrane domain emerged as the predominant contributor, we were able to identify a site in the M2/M3 interface where exchange of a single amino acid between K2P2.1(TREK-1) and K2P4.1(TRAAK), F185L and L147F, respectively, was able to shift the BL-1249 phenotype in the direction of the donor channel, impairing the K2P2.1(TREK-1) response while enhancing the K2P4.1(TRAAK) response. Interestingly, the M2/M3 interface is also important for TREK subfamily responses to temperature35 and membrane stretch.52 Taken together, our findings suggest that BL-1249 does not act in the K2P modulator pocket but affects a site that is a composite of elements from multiple transmembrane helices. Although this general characteristic is shared with the norfluoxetine site, the role of the M2/M3 interface in BL-1249 selectivity suggests that BL-1249 may act outside of both structurally defined small molecule sites.
Our studies of a small set of BL-1249 derivatives show that the two defining moieties of BL-1249, the tetrazole and the tetralin groups, contribute to the stimulatory effect of BL-1249 TREK subfamily channels. The acidic nature at the tetrazole site and the hydrophobicity of the tetralin ring are both crucial for the potency of BL-1249 (Figure 6). Whether or not the two rings are constrained is key for the compound to discriminate between K2P2.1(TREK-1) and K2P4.1(TRAAK) as demonstrated by the properties of BL-1249-tricycle. This dependence on the ability of the ring systems to adopt non-coplanar conformations in order to achieve selectivity within the TREK subfamily suggests that further exploration of strategies to modify the conformational preferences between the two ring systems might be a means to achieve better subtype discrimination. Interestingly, the importance of the tetrazole is a property shared by BL-1249 and ML67-33,26 and both compounds share the general architecture of a hydrophobic ring system linked to the tetrazole. However, ML67-33 is not selective within the TREK subfamily and, rather than having hydrophobic moieties that can adopt a non-coplanar conformation, has an acridine ring system that is constrained, not unlike BL-1249-tricycle. This commonality between ML67-33 and BL-1249-tricycle lends further support to the idea that constrained versus conformationally adaptable hydrophobic ring systems are an important property for the tetrazole-bearing class of TREK subfamily activators. These shared features suggest that further optimization of hydrophobic scaffolds bearing tetrazoles or other acidic groups could provide a path toward the development of other TREK family modulators.
The actions of multiple diverse physical and chemical activators of the TREK subfamily converge at the C-type gate.6,8−10,12,26,41 Our observation that BL-1249 also stimulates the C-type gate fits this paradigm. Our data support the idea that the BL-1249 site of action is not in the K2P modulator pocket, the chemical modulator site closest to the C-type gate, but appears to reside among the transmembrane helices and supports the notion that changes in the channel architecture distant from the selectivity filter can impact the C-type gate.6,35
Elaboration of small molecule modulators for the TREK subfamily provides essential chemical biology tools for unraveling channel function and may offer new paths for treating issues such as pain14,25 and arrhythmia.18 Our study, together with recent structural work,6,7 paints a complex landscape in which there are multiple points for small molecules to intervene in K2P function. Given the growing and diverse list of small molecules that influence various K2P channels,14,26−28,36,50,53−57 further definition of the types of sites with which small molecules can bind and impact K2P function through combined efforts of structural, functional, and computational studies will be crucial for defining how precise chemical control of K2P activity can be achieved.
Materials and Methods
Molecular Biology
Constructs for murine K2P channels, including K2P2.1(TREK-1) (Gene ID 16526), K2P10.1(TREK-2) (Gene ID 72258), K2P4.1(TRAAK) (Gene ID 16528), K2P5.1(TASK-2) (Gene ID 16529), K2P3.1(TASK-1) (Gene ID 16527), K2P9.1(TASK-3) (Gene ID 223604), and K2P18.1(TRESK) (Gene ID 332396) in pGEMHE/pMO, were used for Xenopus oocyte experiments as previously described.10,26 Murine K2P2.1(TREK-1) (Gene ID 16526) was expressed in HEK-293 cells using a pIRES-EGFP construct as previously described.26 Murine K2P13.1(THIK-1) (Gene ID 217826) and K2P1.1(TWIK-1) (Gene ID 16525) were cloned into pGEMHE/pMO for use in Xenopus oocytes. Chimeras were designed using EMBOSS Needle pairwise sequence alignment tool58 to match homologous helices in K2P2.1(TREK-1) and K2P4.1(TRAAK) and were assembled using the Gibson assembly method.59 Chimera boundaries are Thr152 (TREK-1/AAK M2-C), Trp199 (TREK-1/AAK M3-C), Y272 (TREK-1/AAK M4-C), Thr114 (TRAAK/EK-1 M2-C), Trp161 (TRAAK/EK-1 M3-C), and Tyr234 K2P4.1(TRAAK). All sequences were verified using DNA sequencing.
Patch Clamp Electrophysiology
Mouse K2P2.1 was expressed from a previously described pIRES2-EGFP vector in HEK293T cells (ATCC CRL-1573). Cells at 70% confluence were transfected (in 35 mm diameter wells) using LipofectAMINE 2000 (Invitrogen) for 6 h and plated onto coverslips coated with Matrigel (BD Biosciences).
Voltage-dependent activation of K2P2.1 was recorded on excised patches in inside-out configuration (50 kHz sampling) in the absence and presence of 1 μM BL-1249. Pipette solution contained the following: 150 mM KCl, 3.6 mM CaCl2, 10 mM HEPES (pH 7.4 with KOH). Bath solution contained the following: 150 mM RbCl, 2 mM EGTA, and 10 mM HEPES (pH 7.4 with RbOH), and it was continuously perfused at 200 mL/h during the experiment. TREK-1 currents were elicited by a 10 mV voltage step protocol from −100 mV to +100 mV, from a −80 mV holding potential. Data were analyzed using Clampfit 9 and Origin 7.
Two-Electrode Voltage-Clamp (TEVC) Electrophysiology
Xenopus laevis oocytes were harvested in accordance with UCSF IACUC protocol AN129690 and digested in calcium-free ND-96 (96 mM NaCl, 2 mM KCl, 3.8 mM MgCl2) immediately following harvest, as previously described.6,10 Digested oocytes were maintained in standard ND96 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 2 mM MgCl2) with antibiotics (100 units mL–1 penicillin, 100 μg mL–1 streptomycin, 50 μg mL–1 gentimycin). Defolliculated stage V–VI oocytes were injected with 0.2–6.0 ng of mRNA in 50 nL, and currents were recorded 24–48 h after injection. mRNA was synthesized from plasmid DNA using mMessage mMachine Kit (T7 promoter, Ambion, Life Technologies) and purified using RNEasy Kit (Qiagen). Injected oocytes were impaled with two standard microelectrodes (0.2–1.0 MΩ) filled with 3 M KCl and subjected to constant perfusion of standard ND96 during recording. Currents were amplified using the GeneClamp 500B (MDS Analytical Technologies) amplifier controlled by the pClamp software (Molecular Devices). Data were digitized at 1 kHz using Digidata 1332A (MDS Analytical Technologies). For all experiments with small molecules, basal currents were evoked using 1 s long ramps from −150 to +50 mV under constant perfusion of ND96. Once stabilized basal currents were recorded, compounds were perfused at various concentrations in standard ND96 and currents were allowed to increase to stabilization before recording final current. Fold activation upon compound application is expressed as I/I0 (0 mV), derived from the current at 0 mV in the presence of compound divided by the basal current at 0 mV in standard ND96 without compound. Data were analyzed and plotted using Graphpad Prism Version 5 (GraphPad Software, San Diego California USA, www.graphpad.com). In cases where saturation could not be reached due to BL-1249 solubility limits, EC50 was estimated using an upper bound of I/I0 was set to 15 for the fits. In the case of BL-1249-acid EC50 estimation, upper bound of I/I0 was set to 20 to account for the strong stimulation of BL-1249-acid.
BL-1249 Analogue Chemical Synthesis
Complete methods for the synthesis of BL-1249 analogs, BL-1249-acid, BL-1249-amide, BL-1249-Ph, and BL-1249-tricycle are found in the Supporting Information.
Acknowledgments
We thank P. Riegelhaupt for input in the early stages of this project and K. Brejc for comments on the manuscript. This work was supported by Grant NIH-R01-MH093603 to D.L.M. and an American Heart Foundation Postdoctoral fellowship to C.A.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.8b00337.
Responses of TREK-1/TRAAK chimeras to ML355 and ML-67-33 and methods and schemes for synthesis of compounds (PDF)
Author Contributions
L.P., C.A., H.L., and D.L.M. conceived the study and designed the experiments. L.P. and H.L. performed two-electrode voltage-clamp electrophysiology experiments. C.A. performed patch clamp electrophysiology experiments. L.P. and C.A. analyzed the electrophysiology data. L.P. and H.L. performed molecular biology experiments. C.B. and A.G.-G. synthesized and purified the compounds. C.B., A.G.-G., and A.R.R. designed the synthetic routes. D.L.M. analyzed data and provided guidance and support. L.P., C.A., C.B., A.R.R., and D.L.M. wrote the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Feliciangeli S.; Chatelain F. C.; Bichet D.; Lesage F. (2015) The family of K channels: salient structural and functional properties. J. Physiol. 593, 2587. 10.1113/jphysiol.2014.287268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedi P.; Czirjak G. (2010) Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol. Rev. 90, 559–605. 10.1152/physrev.00029.2009. [DOI] [PubMed] [Google Scholar]
- Renigunta V.; Schlichthorl G.; Daut J. (2015) Much more than a leak: structure and function of K(2)p-channels. Pfluegers Arch. 467, 867–894. 10.1007/s00424-015-1703-7. [DOI] [PubMed] [Google Scholar]
- Brohawn S. G.; del Marmol J.; MacKinnon R. (2012) Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science 335, 436–441. 10.1126/science.1213808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A. N.; Long S. B. (2012) Crystal structure of the human two-pore domain potassium channel K2P1. Science 335, 432–436. 10.1126/science.1213274. [DOI] [PubMed] [Google Scholar]
- Lolicato M.; Arrigoni C.; Mori T.; Sekioka Y.; Bryant C.; Clark K. A.; Minor D. L. Jr. (2017) K2P2.1 (TREK-1)-activator complexes reveal a cryptic selectivity filter binding site. Nature 547, 364–368. 10.1038/nature22988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y. Y.; Pike A. C.; Mackenzie A.; McClenaghan C.; Aryal P.; Dong L.; Quigley A.; Grieben M.; Goubin S.; Mukhopadhyay S.; Ruda G. F.; Clausen M. V.; Cao L.; Brennan P. E.; Burgess-Brown N. A.; Sansom M. S.; Tucker S. J.; Carpenter E. P. (2015) K2P channel gating mechanisms revealed by structures of TREK-2 and a complex with Prozac. Science 347, 1256–1259. 10.1126/science.1261512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schewe M.; Nematian-Ardestani E.; Sun H.; Musinszki M.; Cordeiro S.; Bucci G.; de Groot B. L.; Tucker S. J.; Rapedius M.; Baukrowitz T. (2016) A Non-canonical Voltage-Sensing Mechanism Controls Gating in K2P K(+) Channels. Cell 164, 937–949. 10.1016/j.cell.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberberg N.; Ilan N.; Goldstein S. A. (2001) KCNKO: opening and closing the 2-P-domain potassium leak channel entails “C-type” gating of the outer pore. Neuron 32, 635–648. 10.1016/S0896-6273(01)00503-7. [DOI] [PubMed] [Google Scholar]
- Bagriantsev S. N.; Peyronnet R.; Clark K. A.; Honore E.; Minor D. L. Jr. (2011) Multiple modalities converge on a common gate to control K2P channel function. EMBO J. 30, 3594–3606. 10.1038/emboj.2011.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A.; Ben-Abu Y.; Hen S.; Zilberberg N. (2008) A novel mechanism for human K2P2.1 channel gating. Facilitation of C-type gating by protonation of extracellular histidine residues. J. Biol. Chem. 283, 19448–19455. 10.1074/jbc.M801273200. [DOI] [PubMed] [Google Scholar]
- Piechotta P. L.; Rapedius M.; Stansfeld P. J.; Bollepalli M. K.; Erhlich G.; Andres-Enguix I.; Fritzenschaft H.; Decher N.; Sansom M. S.; Tucker S. J.; Baukrowitz T. (2011) The pore structure and gating mechanism of K2P channels. EMBO J. 30, 3607–3619. 10.1038/emboj.2011.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devilliers M.; Busserolles J.; Lolignier S.; Deval E.; Pereira V.; Alloui A.; Christin M.; Mazet B.; Delmas P.; Noel J.; Lazdunski M.; Eschalier A. (2013) Activation of TREK-1 by morphine results in analgesia without adverse side effects. Nat. Commun. 4, 2941. 10.1038/ncomms3941. [DOI] [PubMed] [Google Scholar]
- Vivier D.; Soussia I. B.; Rodrigues N.; Lolignier S.; Devilliers M.; Chatelain F. C.; Prival L.; Chapuy E.; Bourdier G.; Bennis K.; Lesage F.; Eschalier A.; Busserolles J.; Ducki S. (2017) Development of the first Two-Pore Domain Potassium Channel TREK-1 (TWIK-Related K+ Channel 1)-selective agonist possessing in vivo anti-nociceptive activity. J. Med. Chem. 60, 1076. 10.1021/acs.jmedchem.6b01285. [DOI] [PubMed] [Google Scholar]
- Alloui A.; Zimmermann K.; Mamet J.; Duprat F.; Noel J.; Chemin J.; Guy N.; Blondeau N.; Voilley N.; Rubat-Coudert C.; Borsotto M.; Romey G.; Heurteaux C.; Reeh P.; Eschalier A.; Lazdunski M. (2006) TREK-1, a K+ channel involved in polymodal pain perception. EMBO J. 25, 2368–2376. 10.1038/sj.emboj.7601116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurteaux C.; Guy N.; Laigle C.; Blondeau N.; Duprat F.; Mazzuca M.; Lang-Lazdunski L.; Widmann C.; Zanzouri M.; Romey G.; Lazdunski M. (2004) TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J. 23, 2684–2695. 10.1038/sj.emboj.7600234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarenko R. M.; Fortuna M. G.; Shi Y.; Mulkey D. K.; Takakura A. C.; Moreira T. S.; Guyenet P. G.; Bayliss D. A. (2010) Anesthetic activation of central respiratory chemoreceptor neurons involves inhibition of a THIK-1-like background K(+) current. J. Neurosci. 30, 9324–9334. 10.1523/JNEUROSCI.1956-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decher N.; Ortiz-Bonnin B.; Friedrich C.; Schewe M.; Kiper A. K.; Rinne S.; Seemann G.; Peyronnet R.; Zumhagen S.; Bustos D.; Kockskamper J.; Kohl P.; Just S.; Gonzalez W.; Baukrowitz T.; Stallmeyer B.; Schulze-Bahr E. (2017) Sodium permeable and “hypersensitive” TREK-1 channels cause ventricular tachycardia. EMBO Mol. Med. 9, 403–414. 10.15252/emmm.201606690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laigle C.; Confort-Gouny S.; Le Fur Y.; Cozzone P. J.; Viola A. (2012) Deletion of TRAAK potassium channel affects brain metabolism and protects against ischemia. PLoS One 7, e53266. 10.1371/journal.pone.0053266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Liu Y.; Chen X.; Sun Q.; Tang R.; Wang W.; Yu Z.; Xie M. (2013) Involvement of TREK-1 activity in astrocyte function and neuroprotection under simulated ischemia conditions. J. Mol. Neurosci. 49, 499–506. 10.1007/s12031-012-9875-5. [DOI] [PubMed] [Google Scholar]
- Heurteaux C.; Lucas G.; Guy N.; El Yacoubi M.; Thummler S.; Peng X. D.; Noble F.; Blondeau N.; Widmann C.; Borsotto M.; Gobbi G.; Vaugeois J. M.; Debonnel G.; Lazdunski M. (2006) Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nat. Neurosci. 9, 1134–1141. 10.1038/nn1749. [DOI] [PubMed] [Google Scholar]
- Andres-Enguix I.; Shang L.; Stansfeld P. J.; Morahan J. M.; Sansom M. S.; Lafreniere R. G.; Roy B.; Griffiths L. R.; Rouleau G. A.; Ebers G. C.; Cader Z. M.; Tucker S. J. (2012) Functional analysis of missense variants in the TRESK (KCNK18) K channel. Sci. Rep. 2, 237. 10.1038/srep00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafreniere R. G.; Cader M. Z.; Poulin J. F.; Andres-Enguix I.; Simoneau M.; Gupta N.; Boisvert K.; Lafreniere F.; McLaughlan S.; Dube M. P.; Marcinkiewicz M. M.; Ramagopalan S.; Ansorge O.; Brais B.; Sequeiros J.; Pereira-Monteiro J. M.; Griffiths L. R.; Tucker S. J.; Ebers G.; Rouleau G. A. (2010) A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat. Med. 16, 1157–1160. 10.1038/nm.2216. [DOI] [PubMed] [Google Scholar]
- Mathie A.; Veale E. L. (2015) Two-pore domain potassium channels: potential therapeutic targets for the treatment of pain. Pfluegers Arch. 467, 931–943. 10.1007/s00424-014-1655-3. [DOI] [PubMed] [Google Scholar]
- Vivier D.; Bennis K.; Lesage F.; Ducki S. (2016) Perspectives on the Two-Pore Domain Potassium Channel TREK-1 (TWIK-Related K(+) Channel 1). A Novel Therapeutic Target?. J. Med. Chem. 59, 5149–5157. 10.1021/acs.jmedchem.5b00671. [DOI] [PubMed] [Google Scholar]
- Bagriantsev S. N.; Ang K. H.; Gallardo-Godoy A.; Clark K. A.; Arkin M. R.; Renslo A. R.; Minor D. L. Jr. (2013) A high-throughput functional screen identifies small molecule regulators of temperature- and mechano-sensitive K2P channels. ACS Chem. Biol. 8, 1841–1851. 10.1021/cb400289x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z. W.; Brown E. C.; Wang W. W.; MacKinnon R. (2016) Novel cell-free high-throughput screening method for pharmacological tools targeting K+ channels. Proc. Natl. Acad. Sci. U. S. A. 113, 5748–5753. 10.1073/pnas.1602815113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loucif A. J. C.; Saintot P. P.; Liu J.; Antonio B. M.; Zellmer S. G.; Yoger K.; Veale E. L.; Wilbrey A.; Omoto K.; Cao L.; Gutteridge A.; Castle N. A.; Stevens E. B.; Mathie A. (2018) GI-530159, a novel, selective, mechanosensitive two-pore-domain potassium (K2P) channel opener, reduces rat dorsal root ganglion neuron excitability. British journal of pharmacology 175, 2272. 10.1111/bph.14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn C. A.; Luo Y.; Cui M.; Wang J.; Soll R.; Dong J.; Hu B.; Lyon M. A.; Santarelli V. P.; Kraus R. L.; Gregan Y.; Wang Y.; Fox S. V.; Binns J.; Doran S. M.; Reiss D. R.; Tannenbaum P. L.; Gotter A. L.; Meinke P. T.; Renger J. J. (2012) Discovery of a pharmacologically active antagonist of the two-pore-domain potassium channel K2P9.1 (TASK-3). ChemMedChem 7, 123–133. 10.1002/cmdc.201100351. [DOI] [PubMed] [Google Scholar]
- Rodrigues N.; Bennis K.; Vivier D.; Pereira V.; Chapuy E.; Deokar H.; Busserolles J.; Lesage F.; Eschalier A.; Ducki S.; Chatelain F. C. (2014) Synthesis and structure-activity relationship study of substituted caffeate esters as antinociceptive agents modulating the TREK-1 channel. Eur. J. Med. Chem. 75, 391–402. 10.1016/j.ejmech.2014.01.049. [DOI] [PubMed] [Google Scholar]
- Bayliss D. A.; Barrett P. Q. (2008) Emerging roles for two-pore-domain potassium channels and their potential therapeutic impact. Trends Pharmacol. Sci. 29, 566–575. 10.1016/j.tips.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekkirala A. S.; Roberson D. P.; Bean B. P.; Woolf C. J. (2017) Breaking barriers to novel analgesic drug development. Nat. Rev. Drug Discovery 16, 545. 10.1038/nrd.2017.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohawn S. G.; Campbell E. B.; MacKinnon R. (2013) Domain-swapped chain connectivity and gated membrane access in a Fab-mediated crystal of the human TRAAK K+ channel. Proc. Natl. Acad. Sci. U. S. A. 110, 2129–2134. 10.1073/pnas.1218950110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohawn S. G.; Campbell E. B.; MacKinnon R. (2014) Physical mechanism for gating and mechanosensitivity of the human TRAAK K+ channel. Nature 516, 126–130. 10.1038/nature14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolicato M.; Riegelhaupt P. M.; Arrigoni C.; Clark K. A.; Minor D. L. Jr. (2014) Transmembrane helix straightening and buckling underlies activation of mechanosensitive and thermosensitive K(2P) channels. Neuron 84, 1198–1212. 10.1016/j.neuron.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadi P. K.; Vierra N. C.; Days E. L.; Dickerson M.; Vinson P. N.; Weaver C. D.; Jacobson D. A. (2017) Selective small molecule activators of TREK-2 channels stimulate DRG c-fiber nociceptor K2P currents and limit calcium influx. ACS Chem. Neurosci. 8, 558. 10.1021/acschemneuro.6b00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham G. G. (2016) Fenamates, in Compendium of Inflammatory Diseases (Parnham M. J., Ed.), pp 477–482, Springer Basel, Basel. [Google Scholar]
- Takahira M.; Sakurai M.; Sakurada N.; Sugiyama K. (2005) Fenamates and diltiazem modulate lipid-sensitive mechano-gated 2P domain K(+) channels. Pfluegers Arch. 451, 474–478. 10.1007/s00424-005-1492-5. [DOI] [PubMed] [Google Scholar]
- Tertyshnikova S.; Knox R. J.; Plym M. J.; Thalody G.; Griffin C.; Neelands T.; Harden D. G.; Signor L.; Weaver D.; Myers R. A.; Lodge N. J. (2005) BL-1249 [(5,6,7,8-tetrahydro-naphthalen-1-yl)-[2-(1H-tetrazol-5-yl)-phenyl]-amine]: a putative potassium channel opener with bladder-relaxant properties. J. Pharmacol. Exp. Ther. 313, 250–259. 10.1124/jpet.104.078592. [DOI] [PubMed] [Google Scholar]
- Veale E. L.; Al-Moubarak E.; Bajaria N.; Omoto K.; Cao L.; Tucker S. J.; Stevens E. B.; Mathie A. (2014) Influence of the N terminus on the biophysical properties and pharmacology of TREK1 potassium channels. Mol. Pharmacol. 85, 671–681. 10.1124/mol.113.091199. [DOI] [PubMed] [Google Scholar]
- Bagriantsev S. N.; Clark K. A.; Minor D. L. Jr. (2012) Metabolic and thermal stimuli control K(2P)2.1 (TREK-1) through modular sensory and gating domains. EMBO J. 31, 3297–3308. 10.1038/emboj.2012.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J.; Patel A. J.; Duprat F.; Lauritzen I.; Lazdunski M.; Honore E. (2005) A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J. 24, 44–53. 10.1038/sj.emboj.7600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J.; Patel A. J.; Duprat F.; Sachs F.; Lazdunski M.; Honore E. (2007) Up- and down-regulation of the mechano-gated K(2P) channel TREK-1 by PIP (2) and other membrane phospholipids. Pfluegers Arch. 455, 97–103. 10.1007/s00424-007-0250-2. [DOI] [PubMed] [Google Scholar]
- Murbartian J.; Lei Q.; Sando J. J.; Bayliss D. A. (2005) Sequential phosphorylation mediates receptor- and kinase-induced inhibition of TREK-1 background potassium channels. J. Biol. Chem. 280, 30175–30184. 10.1074/jbc.M503862200. [DOI] [PubMed] [Google Scholar]
- Patel A. J.; Honore E.; Maingret F.; Lesage F.; Fink M.; Duprat F.; Lazdunski M. (1998) A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 17, 4283–4290. 10.1093/emboj/17.15.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore E.; Maingret F.; Lazdunski M.; Patel A. J. (2002) An intracellular proton sensor commands lipid- and mechano-gating of the K(+) channel TREK-1. EMBO J. 21, 2968–2976. 10.1093/emboj/cdf288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotshaw D. P. (2007) Biophysical, pharmacological, and functional characteristics of cloned and native mammalian two-pore domain K+ channels. Cell Biochem. Biophys. 47, 209–256. 10.1007/s12013-007-0007-8. [DOI] [PubMed] [Google Scholar]
- Maingret F.; Honore E.; Lazdunski M.; Patel A. J. (2002) Molecular basis of the voltage-dependent gating of TREK-1, a mechano-sensitive K(+) channel. Biochem. Biophys. Res. Commun. 292, 339–346. 10.1006/bbrc.2002.6674. [DOI] [PubMed] [Google Scholar]
- Monteillier A.; Loucif A.; Omoto K.; Stevens E. B.; Lainez S.; Saintot P. P.; Cao L.; Pryde D. C. (2016) Investigation of the structure activity relationship of flufenamic acid derivatives at the human TRESK channel K2P18.1. Bioorg. Med. Chem. Lett. 26, 4919–4924. 10.1016/j.bmcl.2016.09.020. [DOI] [PubMed] [Google Scholar]
- Thummler S.; Duprat F.; Lazdunski M. (2007) Antipsychotics inhibit TREK but not TRAAK channels. Biochem. Biophys. Res. Commun. 354, 284–289. 10.1016/j.bbrc.2006.12.199. [DOI] [PubMed] [Google Scholar]
- McClenaghan C.; Schewe M.; Aryal P.; Carpenter E. P.; Baukrowitz T.; Tucker S. J. (2016) Polymodal activation of the TREK-2 K2P channel produces structurally distinct open states. J. Gen. Physiol. 147, 497–505. 10.1085/jgp.201611601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryal P.; Jarerattanachat V.; Clausen M. V.; Schewe M.; McClenaghan C.; Argent L.; Conrad L. J.; Dong Y. Y.; Pike A. C. W.; Carpenter E. P.; Baukrowitz T.; Sansom M. S. P.; Tucker S. J. (2017) Bilayer-Mediated Structural Transitions Control Mechanosensitivity of the TREK-2 K2P Channel. Structure 25, 708–718e702. 10.1016/j.str.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun G.; Lengyel M.; Enyedi P.; Czirjak G. (2015) Differential sensitivity of TREK-1, TREK-2 and TRAAK background potassium channels to the polycationic dye ruthenium red. British journal of pharmacology 172, 1728–1738. 10.1111/bph.13019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chokshi R. H.; Larsen A. T.; Bhayana B.; Cotten J. F. (2015) Breathing Stimulant Compounds Inhibit TASK-3 Potassium Channel Function Likely by Binding at a Common Site in the Channel Pore. Mol. Pharmacol. 88, 926–934. 10.1124/mol.115.100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luethy A.; Boghosian J. D.; Srikantha R.; Cotten J. F. (2017) Halogenated Ether, Alcohol, and Alkane Anesthetics Activate TASK-3 Tandem Pore Potassium Channels Likely through a Common Mechanism. Mol. Pharmacol. 91, 620–629. 10.1124/mol.117.108290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennard L. E.; Chumbley J. R.; Ranatunga K. M.; Armstrong S. J.; Veale E. L.; Mathie A. (2005) Inhibition of the human two-pore domain potassium channel, TREK-1, by fluoxetine and its metabolite norfluoxetine. Br. J. Pharmacol. 144, 821–829. 10.1038/sj.bjp.0706068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A. J.; Honore E.; Lesage F.; Fink M.; Romey G.; Lazdunski M. (1999) Inhalational anesthetics activate two-pore-domain background K+ channels. Nat. Neurosci. 2, 422–426. 10.1038/8084. [DOI] [PubMed] [Google Scholar]
- Rice P.; Longden I.; Bleasby A. (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277. 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Gibson D. G.; Young L.; Chuang R. Y.; Venter J. C.; Hutchison C. A. 3rd; Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.