

Abstract
Premise of the Study
Simple sequence repeat (SSR) or microsatellite markers have been used in a broad range of studies mostly scoring alleles on the basis of amplicon size as a proxy for the number of repeat units of an SSR motif. However, additional sources of variation within the SSR or in the flanking regions have largely remained undetected.
Methods
In this study, we implemented a next‐generation sequencing–based genotyping approach in a newly characterized set of 18 nuclear SSR markers for the carob tree, Ceratonia siliqua. Our aim was to evaluate the effect of three different methods of scoring molecular variation present within microsatellite markers on the genetic diversity and structure results.
Results
The analysis of the sequences of 77 multilocus genotypes from four populations revealed SSR variation and additional sources of polymorphism in 87% of the loci analyzed (42 single‐nucleotide polymorphisms and five insertion/deletion polymorphisms), as well as divergent paralog copies in two loci. Ignoring sequence variation under standard amplicon size genotyping resulted in incorrect identification of 69% of the alleles, with important effects on the genetic diversity and structure estimates.
Discussion
Next‐generation sequencing allows the detection and scoring of SSRs, single‐nucleotide polymorphisms, and insertion/deletion polymorphisms to increase the resolution of population genetic studies.
Keywords: carob tree, genetic diversity, homoplasy, MicNeSs, next‐generation sequencing, simple sequence repeat (SSR)
Microsatellite markers or simple sequence repeats (SSRs) have been broadly used in molecular studies due to their high polymorphism rates, codominant nature, and frequent occurrence throughout genomes (Chistiakov et al., 2006). Among these advantages, their high mutation rate per generation is particularly useful to document scenarios involving recent population demographic changes (Guichoux et al., 2011; Aimé and Austerlitz, 2017), especially in plant species characterized by low genetic diversity (e.g., Zehdi‐Azouzi et al., 2015). These markers will likely continue to be widely used because budgets are affordable once a set of SSRs is characterized for a taxon (Jennings et al., 2011). Although several studies in the past decade have focused on SSR isolation and characterization strategies (Zane et al., 2002; Viruel et al., 2010, 2015; Meglécz et al., 2014; Merritt et al., 2015), few improvements on the genotyping procedures of microsatellite markers can be found in the literature (e.g., Suez et al., 2016).
Usually SSR genotyping is based on amplicon size variation, detected through capillary gel electrophoresis, as a proxy for the number of repeat units of an SSR motif. However, two alleles scored as identical in size under standard SSR genotyping procedures can be different in sequence due to variation in their flanking regions or within the SSR motif itself (Brinkmann et al., 1998; Rossetto et al., 2002). The difference in sequence but not in amplicon size, frequently referred to as size homoplasy, can be revealed through sequencing (Estoup et al., 2002). The term size homoplasy is rightly applied when using standard SSR genotyping because, as in phylogenetics (where the term originates), similarity that is not due to common ancestry is revealed after the analysis. When genotyping SSRs by sequencing, the use of size homoplasy is not ideal because, in contrast to standard SSR procedures, it is determined from the onset that two alleles are not identical. That is why we have here avoided using size homoplasy. Although the advent of next‐generation sequencing (NGS) in molecular ecology and conservation genetics has provided the tools to refine the scoring of SSRs, few studies have tackled the specific challenge of sequencing SSR amplicons to integrate the additional variation detected in sequences. For example, MicNeSs software (Suez et al., 2016) automatically estimates SSR genotypes from NGS data (originally optimized for Roche 454 sequencing). MicNeSs estimates the true alleles for each individual and locus from the observed distribution of SSR lengths aiming to correct the artifactual insertions or deletions produced by PCR amplification. Regarding the detection of distinct alleles that could be taken as identical by standard genotyping approaches, the main improvement of MicNeSs is the inclusion of substitutions during genotype scoring, allowing point mutations to occur within the SSR motif. However, it does not consider the potential single‐nucleotide polymorphisms (SNPs) and insertion/deletion polymorphisms (indels) in the flanking regions. Vartia et al. (2016) found a high infraspecific incidence of SNPs and indels in flanking regions of SSR loci. However, these authors did not explore the influence of this information in the estimation of genetic diversity and structure. The potential of NGS to significantly improve SSR genotyping stems from increasing data quality by correctly identifying alleles, facilitating data comparisons among laboratories and studies (Guichoux et al., 2011), and allowing a better understanding of the molecular evolution of SSR loci by discerning variation due to the number of units of the SSRs, indels, and SNPs (Putman and Carbone, 2014).
In the context of a project dealing with the evolutionary history of carob (Ceratonia siliqua L.), a Mediterranean fruit tree, we aimed at improving SSR genotyping using NGS and comparing different scoring methods. Sequence variation within newly characterized microsatellite markers was investigated in carob populations.
MATERIALS AND METHODS
A diagram chart of the pipeline followed in the present study to genotype SSR regions using NGS is shown in Figure 1.
Figure 1.
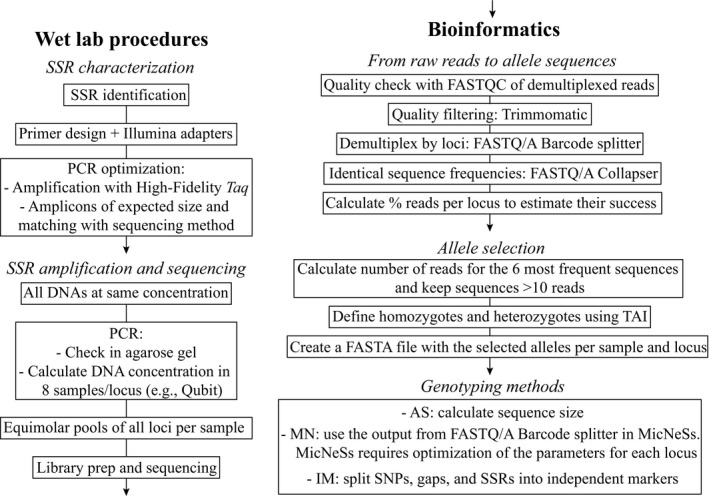
Diagram chart of the pipeline followed in this study to genotype SSR regions using next‐generation sequencing. See Materials and Methods for details. AS = amplicon size scoring; MN = MicNeSs scoring; IM = independent marker scoring; TAI = true allele index.
Plant material
This study is part of a wider phylogeographic project focused on the carob tree (Ceratonia siliqua), in which a total of 1135 leaf samples were collected from populations throughout the Mediterranean Basin. SSR isolation and characterization were performed using DNA of one individual from Èze (Alpes Maritimes, France). To optimize the PCR amplification and select the polymorphic loci, we chose 77 samples from carob trees sampled in four wild populations. Leaves were collected from individual trees and dried using silica gel. These populations were selected focusing on the eastern and western parts of the Mediterranean Basin aiming to cover an adequate representation of the genetic differentiation within this species. The populations sampled were ESGRA (Sierra de Grazalema, Spain; 36.75605°N, 5.41916°W), GRLOU (Loutro, Crete, Greece; 35.198983°N, 24.076279°E), LIENF (Saydit el Nourieh, Anfeh, Lebanon; 34.30194°N, 35.68203°E), and MAIMO (Imouzzer des Ida‐Outanane, Morocco; 30.6557°N, 9.4956°W).
SSR characterization
Total DNA of one individual was extracted from leaves stored with silica gel using the NucleoSpin Plant II Kit (Macherey‐Nagel Sarl, Hoerdt, France). Size‐selected fragments from genomic DNA were enriched for SSR content using magnetic streptavidin beads and biotin‐labeled GATA and GTAT repeat oligonucleotides (Dynabeads M‐280; Thermo Fisher Scientific, Waltham, Massachusetts, USA). The SSR‐enriched library was sequenced in a paired‐end MiSeq 250 × 250 Nano V2 (Illumina, San Diego, California, USA), and the paired‐end reads were merged with FLASH 1.2.9 (Magoč and Salzberg, 2011). A de novo assembly was then performed with the merged paired‐end reads using MIRA 4.0.1 software (Chevreux et al., 2004). Primers were designed with MSATCOMMANDER 0.8.2.0 (Faircloth, 2008); duplicated reads and those containing more than one SSR array were removed. Primers fulfilling the following criteria were selected (Viruel et al., 2015): optimal size 20–25 bp, not directly flanking the SSR motif, lacking ambiguous bases, low self‐ and pair product complementary parameters, amplicon expected size <390 bp, and melting temperature (T m) difference <1.5°C.
SSR genotyping optimization with NGS
Total DNA of 77 individuals was quantified using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific), and concentration was normalized to 5 ng/μL. PCR amplifications were performed in a total volume of 25 μL and contained 4 μL of dNTPs (1.25 mM), 1 μL of each primer (10 mM), 2 mM of MgCl2, 0.2 μL of GoTaq DNA Polymerase (5 U/μL; Promega Corporation, Madison, Wisconsin, USA), and approximately 5 ng of DNA. The PCR program consisted of an initial denaturation of 4 min at 95°C; followed by 35 cycles of 30 s at 95°C, 30 s at 56°C, and 45 s at 72°C; and a final extension step of 7 min at 72°C. PCR programs were further optimized for four loci: for C18 and C20 the annealing temperature (T a) was 54°C, for C19 and C30 T a was 58°C. Amplicons of the expected size were verified in 3% agarose gel.
Illumina universal adapter sequences 5′‐AAGACTCGGCAGCATCTCCA‐3′ and 5′‐GCGATCGTCACTGTTCTCCA‐3′ were then added to the 5′ or 3′ end of the locus‐specific forward and reverse primers, respectively. We also included five pairs of primers for expressed sequence tag (EST)‐SSR regions previously described for C. siliqua (La Malfa et al., 2014) and a plastid region (rpl32‐trnL spacer). The primers for rpl32‐trnL were specifically designed for C. siliqua for this study using the plastome sequence available in GenBank (KJ468096). The set of primers with the Illumina adapters (Appendix S1) was amplified in a total volume of 15 μL containing 0.3 μL of dNTPs (10 mM), 0.6 μL of each primer (10 μM), 0.15 μL of Q5 High‐Fidelity DNA Polymerase (5 U/μL; New England Biolabs, Ipswich, Massachusetts, USA), and approximately 5 ng of DNA. The PCR program consisted of a pre‐melt of 30 s at 98°C; followed by a touch‐up from 56–59°C (16 cycles of 10 s of denaturation at 98°C, 30 s of annealing with touch‐up temperature increase of 0.2°C per cycle, and 20 s of extension at 72°C); plus 19 cycles of 10 s at 98°C, 30 s of annealing at 59°C, and 20 s of extension at 72°C; followed by 7 min of final extension at 72°C.
The PCRs were automated with an epMotion 5075 TMX robot (Eppendorf, Hamburg, Germany) to promote uniformity. PCR success was verified in agarose gels, and amplicon quantification was then performed using a Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific).
MiSeq sequencing
After characterizing new microsatellite markers for C. siliqua and evaluating their performance in a MiSeq run, 18 microsatellite markers were selected (see Results, Appendix S1). SSR performance and optimization in an NGS platform as well as genotyping reproducibility were then evaluated through independent runs containing 96, 192, and 384 pooled samples (Appendix S2). Sets of 96, 192, and 384 samples were pooled separately by combining Nextera and TruSeq universal barcodes (Illumina) and sequenced in a paired‐end MiSeq 250 × 250 standard V2 (Illumina). The 77 selected samples from four wild populations were demultiplexed and extracted for the subsequent analyses.
Demultiplexed raw FastQ reads were evaluated using FastQC (Andrews, 2010), and quality filtering was applied using Trimmomatic version 0.35 (Bolger et al., 2014). Paired‐end sequencing allowed us to guarantee that both reads retrieved the same sequence. Reads were demultiplexed by loci and sample with FASTQ/A Barcode splitter, and identical sequence frequencies were calculated with FASTQ/A Collapser using FASTX‐Toolkit (Gordon and Hannon, 2010).
Genotyping from sequences
We defined an index to identify the true alleles for each sample and locus (true allele index [TAI]) by calculating the percentage of reads obtained for each sequence variant (i.e., putative alleles). Homozygotes were identified when ≥30% of the reads were retrieved by one sequence variant, and the subsequent sequence variants represented ≤10% of the total reads. Heterozygotes were identified when the two most frequent sequence variants retrieved ≥10% of the reads, and the difference between them was not more than 50% (Appendix S3). In all cases, the frequencies of the remaining sequence variants were less than 5% of the total reads and were discarded. The selected sequence variants (alleles) per sample were compiled in FASTA files per locus.
Reproducibility was estimated using technical and biological replicates over the whole project. The amplification and sequencing process was repeated twice for 16 samples (technical replicates). We also included in the analysis 10 trees from Sicily that had been grafted with scions from the cultivar Tantillo. These samples from the same cultivar are comparable to branches of the same tree and therefore they are expected to have the same genotype (biological replicates); differences between these 10 trees would be considered as genotyping error.
Three different methods were applied to obtain genotypes:
Amplicon size scoring (AS): This reproduces the standard scoring method of microsatellite markers, which uses amplicon size to identify alleles and re‐identifies allelic size to be multiple of the SSR repeat pattern. This method assumes that variation in each locus is exclusively due to changes in the number of repeat units of the same SSR motif.
MicNeSs scoring (MN): Substitutions within the SSR motif were accounted for in addition to AS by using MicNeSs (Suez et al., 2016), which identifies alleles using as input the FASTA files obtained after FASTQ/A Barcode splitter and then converted from FASTQ to FASTA.
Independent marker scoring (IM): We coded independently variation resulting from the number of SSR units vs. SNPs or indels either within the SSR or the flanking regions. We used a custom script in R to build a new FASTA file containing the different alleles (Appendix S4). We used MUSCLE (Edgar, 2004) with default settings to align these alleles and edited manually in MEGA7 software (Kumar et al., 2016). This alignment was used to score SSR and sequence variations.
Molecular evolution of microsatellite markers
To explain the molecular variation and its consequences on allele identification, we used TCS Software (Clement et al., 2000) to construct a network that was modified to reflect the evolution of SSRs and SNPs as suggested by Barthe et al. (2012). This was done for all loci but is shown here only for locus C08 (Fig. 2) because it contains a higher frequency of sequence polymorphisms in the shortest SSR motifs and a reduced frequency of the longest SSR alleles (see Discussion for details).
Figure 2.
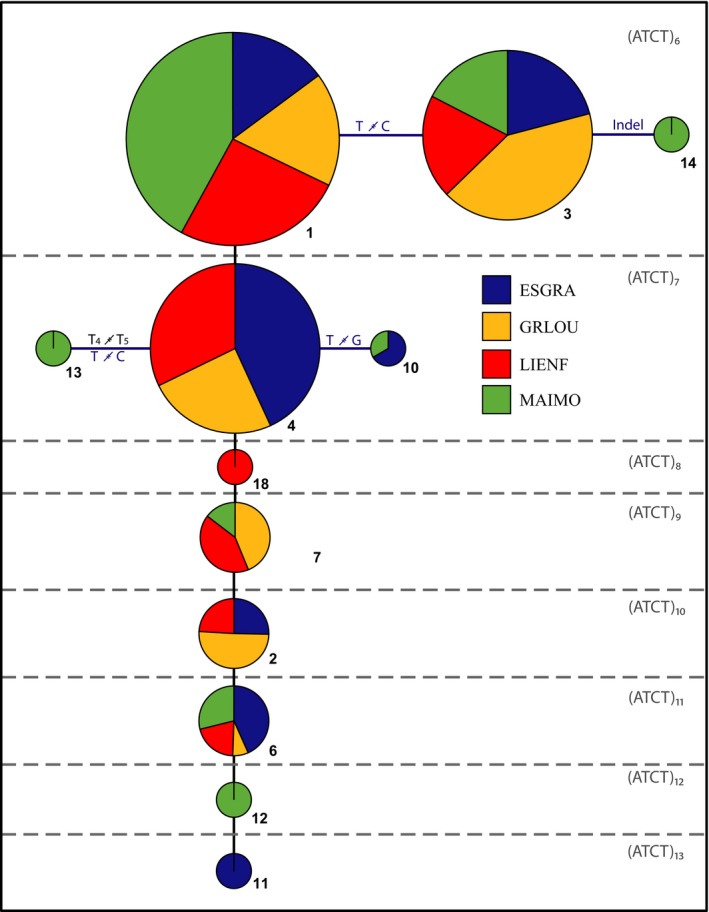
Network showing the relatedness of the 12 alleles found in the SSR locus C08 in four populations of Ceratonia siliqua. The size of the SSR allele (number of repeat units) is shown at the top right of each section (separated by dashed lines), which groups alleles of the same repeat number. Circles represent an allele and its frequencies per population. Pie chart size is proportional to the abundance of each allele: 1 (41), 2 (8), 3 (29), 4 (33), 6 (14), 7 (14), 10 (3), 11 (2), 12 (4), 13 (3), 14 (2), 18 (1). Alleles connected by horizontal lines differ by mutations in the sequence due to indels, SNPs, or mononucleotide SSR polymorphisms.
Genetic diversity and structure analyses
Allele frequencies and genetic diversity indices were calculated in GenAlEx 6.5 (Peakall and Smouse, 2012). Deviations from Hardy–Weinberg equilibrium (HWE) were estimated using GENEPOP 4.0 (Rousset, 2008) using 10,000 permutations. Genetic differentiation between populations was analyzed by calculating pairwise F ST values in GenAlEx to investigate the genetic structure under different population groupings and the three scoring methods. Finally, Bayesian clustering using STRUCTURE 2.3.4 (Pritchard et al., 2000) was applied to infer population genetic structure under an admixture ancestry model for K genetic clusters from 1 to 5, for 10 replicates of 7 × 105 generations of burn‐in and 7 × 106 iterations of Markov chain Monte Carlo (MCMC) run length. A correlated allele frequency model without priors on population origins was used. The most probable number of clusters was calculated using Evanno et al. (2005) criterion.
RESULTS
Primer design and optimization
The total number of reads obtained from the SSR‐enriched library was 11,130, had an average size of 387 bp, and 2900 of them contained SSR motifs (26%). Primers were designed for 505 reads, and 40 pairs of primers with the best parameter values were selected (see Materials and Methods, Appendix S1). Thirty‐eight out of the 40 pairs of primers produced good amplifications (one clear, bright band). After adding the Illumina adapter sequences to the primers, the amplification was successful for 30 loci (Appendix S1). Concentration values showed a broad range from 2.25 to 39.00 ng/μL and, in accordance with the expected size for each locus, nanomolar concentrations ranged between 11 and 191 nM (Appendix S1). We divided all loci into two sets of 18 markers depending on their concentration, below (Set A) or above (Set B) 90 nM. All PCR products from each sample were mixed under equimolarity conditions to 90 nM in Set A and to 11 nM in Set B (Appendix S1).
The first MiSeq run including 48 pooled samples organized in two sets of 17 markers (MiSeq96) produced a total of 9,832,485 paired reads. FastQC quality analysis indicated that our results were within standards; a threshold quality score of 20 and a minimum length of 110 were applied in subsequent steps. These results allowed selection of a final set of 18 polymorphic SSR loci, which were sequenced in two additional MiSeq runs with 192 (MiSeq192) and 384 (MiSeq384) pooled samples obtaining 9,561,116 and 9,174,599 total paired reads, respectively. Similar FastQC quality patterns were observed and the same threshold values were applied. The average number of paired reads per sample was 110,477 ± 16,223 in MiSeq96, 54,017 ± 15,315 in MiSeq192, and 26,748 ± 6771 in MiSeq384. After filtering paired reads with average quality reads below 35 (AVGQUAL:35), 77% ± 11% reads per sample and per locus were retained in MiSeq96 run, 96% ± 3% in MiSeq192, and 96% ± 3% in MiSeq384.
A positive association between the nanomolar concentration of loci and the number of reads retrieved per locus was observed (Appendices S2, S5). Seven out of 34 loci retrieved less than 0.6% of the total reads and were therefore discarded (Cesi187, Cesi976, C07, C32, rpl32‐trnL, Cesi21, Cesi74). These loci had concentrations below 50 nM except for Cesi187, which had a concentration of 91 nM (Appendix S1). PCR failure was observed in locus C28 (48 nM) as 33 samples obtained less than 13 reads by amplicon and therefore this locus was also discarded. Loci Cesi17, C16, C19, C27, C30, and C38 contained either highly divergent sequences or mononucleotide (C16) or dinucleotide (C19) motifs. These six loci were discarded because the TAI (see Materials and Methods) failed at identifying one or two alleles per sample. Two additional loci were discarded because of their low polymorphism rates: only two alleles were found for locus C26 and locus C40 was monomorphic in all samples. Finally, we kept 18 polymorphic loci suitable for genotyping through NGS.
Comparative analysis of genotyping methods
A complete matrix was generated under each scoring method for the 77 genotypes coming from four populations; this matrix included few missing data for loci C10, C11, C22, C23, and C31 that were due to PCR or sequencing failures. Two highly divergent sequences, representing paralogs, were detected in two loci (C6 and C17), and for both loci one of the copies was discarded due to a lack of polymorphisms.
Identification of alleles
Only three out of the 18 selected markers (loci C10, C29, and C31) showed polymorphisms exclusively due to the number of repeat units of the same SSR motif: 15 markers contained variability that could only be retrieved through sequencing. For these loci, 69.4% of total alleles would have been incorrectly identified under standard procedures (Appendix S1). This erroneous identification of alleles under the AS method ranged from 25.2% to 99.2% across loci.
A total of 42 SNPs (nine within the SSR and 33 in the flanking regions) and nine indels (five within the SSR and four in the flanking regions) were found. Seven and five loci had SNPs and indels within the SSR, respectively. Fourteen had variations in the flanking regions, and two loci that exhibited two divergent sequences turned out to represent two paralogs.
The networks built to represent the evolution of the SSR alleles showed that a trend to accumulate SNPs and indels variation occurs more frequently in the most common alleles, which usually were the smallest SSR alleles (Fig. 2). For example, of eight SSR alleles found in the marker C08 in the four carob populations ranging from (ATCT)6 to (ATCT)13, only the two shortest—(ATCT)6 and (ATCT)7—contained SNPs or indels.
We did not find alleles that were equal in length, but had different numbers of repeats due to indels, probably because most are tetra‐ or trinucleotides. Only locus C17 has a dinucleotide motif, and only one SNP was scored. Two loci contain mononucleotide SSRs (C08 and C21), and in both cases these were biallelic and did not match with the amplicon size of a different allele.
When comparing the 16 samples that were amplified and sequenced twice to test reproducibility (technical replicates), an error rate of 2.85% was found when accounting for missing data. In each case, the difference between the two repeated genotypes concerned only one allele in the genotype. In the second test, which involved expected clones coming from 10 scions of the cultivar Tantillo, nine differences were counted, leading to an error rate of 2.5%; eight out of nine cases were due to failure to detect the second allele in a heterozygous genotype.
Implications of scoring method for genetic diversity
Globally, the average number of alleles per locus in the four studied populations of C. siliqua was similar (P = 0.27; 10,000 permutations) for the AS scoring (3.57 ± 0.11) and the MN scoring (3.44 ± 0.12). The average number of sequence variants per population was higher when both SSR and sequence polymorphisms were considered (3.87 ± 0.15). Under the IM scoring method, where the different sources of variation were separated, the global number of alleles per marker decreased to 2.89 ± 0.07 per locus. When considering only the number of repeat units (SSR), the average number of alleles was 3.157 ± 0.098, compared to 2.52 ± 0.10 when considering only SNPs and indels. This pattern holds at the population level (Table 1). The scoring method radically affects the average number of private alleles per locus (Table 1). For example, population LIENF showed an average of 0.11 private alleles per locus under AS, 0.06 under MN, and 0.03 under IM. This marked difference is explained by the fact that LIENF has private SSR alleles but no private variation in the flanking regions (Table 1). The MAIMO population contained the highest proportion of private alleles both when considering only SNPs (0.53) and when considering only number of repeat units (0.40) in IM scoring. Altogether, inbreeding coefficient (F IS) global values were close to HWE in all matrices, but a deviation toward heterozygote excess was found under MN and AS genotyping (Table 1).
Table 1.
Genetic diversity indices for Ceratonia siliqua sampled at four sites based on sequencing 18 SSR loci and comparing three different genotyping procedures: AS (amplicon size scoring), MN (MicNeSs scoring), and IM (independent marker scoring).a
| Populationb | N | AS | MN | IM | IM (only SSR)c | IM (only SNPs and indels)c | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | Priv. | H o | H e | F IS | A | Priv. | H o | H e | F IS | A | Priv. | H o | H e | F IS | A | Priv. | H o | H e | F IS | A | Priv. | H o | H e | F IS | ||
| ESGRA | 19 | 3.667 (0.229) | 0.389 (0.143) | 0.597 (0.048) | 0.515 (0.033) | −0.196 (0.062) | 3.556 (0.294) | 0.333 (0.114) | 0.638 (0.069) | 0.518 (0.044) | −0.250 (0.078) | 3.000 (0.188) | 0.114 (0.055) | 0.421 (0.041) | 0.383 (0.035) | −0.129 (0.036) | 3.350 (0.244) | 0.100 (0.069) | 0.477 (0.051) | 0.430 (0.042) | −0.139 (0.047) | 2.533 (0.256) | 0.133 (0.091) | 0.348 (0.065) | 0.321 (0.058) | −0.115 (0.056) |
| GRLOU | 19 | 3.111 (0.322) | 0.000 (0.000) | 0.447 (0.071) | 0.439 (0.059) | −0.042 (0.070) | 3.167 (0.305) | 0.000 (0.000) | 0.535 (0.067) | 0.479 (0.055) | −0.141 (0.067) | 2.600 (0.197) | 0.000 (0.000) | 0.373 (0.042) | 0.384 (0.042) | −0.027 (0.040) | 2.850 (0.254) | 0.000 (0.000) | 0.413 (0.056) | 0.439 (0.056) | 0.004 (0.056) | 2.267 (0.300) | 0.000 (0.000) | 0.319 (0.062) | 0.310 (0.059) | −0.078 (0.053) |
| LIENF | 20 | 3.333 (0.302) | 0.722 (0.311) | 0.526 (0.048) | 0.527 (0.045) | −0.039 (0.048) | 3.222 (0.329) | 0.111 (0.076) | 0.556 (0.058) | 0.512 (0.047) | −0.111 (0.070) | 2.629 (0.201) | 0.029 (0.029) | 0.386 (0.039) | 0.417 (0.041) | 0.027 (0.034) | 2.800 (0.277) | 0.050 (0.050) | 0.406 (0.048) | 0.446 (0.053) | 0.033 (0.038) | 2.400 (0.289) | 0.000 (0.000) | 0.359 (0.067) | 0.378 (0.064) | 0.019 (0.065) |
| MAIMO | 19 | 4.056 (0.366) | 0.722 (0.311) | 0.553 (0.049) | 0.562 (0.033) | −0.015 (0.061) | 3.722 (0.321) | 0.444 (0.166) | 0.614 (0.073) | 0.524 (0.051) | −0.173 (0.076) | 3.457 (0.189) | 0.457 (0.125) | 0.440 (0.041) | 0.469 (0.034) | 0.051 (0.059) | 3.650 (0.244) | 0.400 (0.184) | 0.505 (0.049) | 0.534 (0.036) | 0.038 (0.072) | 3.200 (0.296) | 0.533 (0.165) | 0.353 (0.066) | 0.383 (0.057) | 0.068 (0.100) |
| Total | 77 | 3.542 (0.157) | 0.530 (0.028) | 0.510 (0.022) | −0.074 (0.031) | 3.417 (0.155) | 0.586 (0.033) | 0.508 (0.024) | −0.166 (0.027) | 2.886 (0.073) | 0.411 (0.015) | 0.417 (0.014) | −0.018 (0.015) | 3.157 (0.098) | 0.457 (0.019) | 0.466 (0.017) | −0.020 (0.020) | 2.524 (0.100) | 0.349 (0.023) | 0.353 (0.022) | −0.016 (0.025) | |||||
A = number of alleles; F IS = inbreeding coefficient; H e = unbiased expected heterozygosity; H o = observed heterozygosity; N = population size; Priv. = number of private alleles.
Values presented are mean (SE).
Population codes correspond to the information provided in the Materials and Methods.
For IM scoring, SSRs and SNPs were split in independent matrices and genetic diversity indices were also calculated.
Implications of scoring method in the genetic structure estimation
Regarding the optimal number of clusters (Fig. 3), the AS genotypes showed a higher ΔK for K = 2 (ΔK = 159.8) than for K = 4 (ΔK = 111.6); the optimal partition under MN was K = 4 (ΔK = 1403.0). For the IM scoring, K = 2 (ΔK = 17.6) was the most likely partition followed by K = 4 (ΔK = 9.3). The genetic groups defined for K = 2 were largely coincident across all genotyping methods, but clear differences appeared in the groups inferred for K = 4. Individual assignment resolution was higher for the IM scoring (i.e., lowest admixture; Fig. 3). Population pairwise F ST values were also influenced by the scoring method as significant differences were found for interpopulation differentiation (Table 2). For instance, the AS and MN scoring methods estimated 7.6% and 8.0% F ST values for the ESGRA and MAOUM populations, respectively, whereas the IM scoring method estimated a higher value (10.9%). Between the GRLOU and LIENF populations, the AS and MN scoring methods estimated an F ST of 7.5% and 10.4%, respectively, whereas the IM scoring method revealed a lower differentiation of 5.4%.
Figure 3.
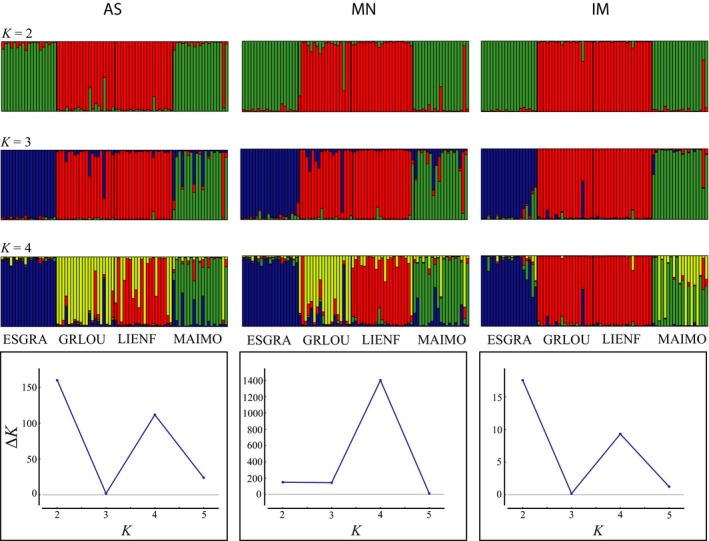
Comparative results of the Bayesian analysis of the genetic structure of four populations of Ceratonia siliqua based on 18 SSR loci sequenced and genotyped under three different approaches (see Materials and Methods): AS (amplicon size scoring), MN (MicNeSs scoring), and IM (independent marker scoring). The probabilities of membership of each sample to the genetic clusters K = 2, 3, and 4 are shown. The most likely number of genetic clusters (K) determined according to Evanno et al. (2005) is shown for each scoring method, as well as the FST values calculated by STRUCTURE (Pritchard et al., 2000) for each predefined population and K.
Table 2.
Average pairwise differentiation F ST values between four populations of wild Ceratonia siliqua (ESGRA, GRLOU, LIENF, and MAOUM) based on scoring the variability of SSR amplicons with three different methods: AS (amplicon size scoring), MN (MicNeSs scoring), and IM (independent marker scoring).a
| Scoring | AS | MN | IM | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Site | MAOUM | ESGRA | GRLOU | MAOUM | ESGRA | GRLOU | MAOUM | ESGRA | GRLOU |
| ESGRA | 0.076 | — | — | 0.08 | — | — | 0.109 | — | — |
| GRLOU | 0.108 | 0.113 | — | 0.130 | 0.108 | — | 0.114 | 0.111 | — |
| LIENF | 0.116 | 0.132 | 0.075 | 0.152 | 0.157 | 0.104 | 0.14 | 0.136 | 0.054 |
See Materials and Methods for details about the scoring methods used.
DISCUSSION
SSR variation has been the most widely used molecular marker for population genetics and molecular ecology since the 1990s (Guichoux et al., 2011). Due to their high mutation rate and their potential to screen hundreds to thousands of individuals, microsatellite markers have been recently used in several studies focusing on the evolutionary history of fruit trees (Besnard et al., 2013; Cornille et al., 2014; Diez et al., 2015; Pollegioni et al., 2017). In this study, we developed 18 new polymorphic SSR markers in C. siliqua. Previous studies on genetic diversity in carob populations found a moderately low genetic diversity (La Malfa et al., 2014). Therefore, the carob tree constitutes a suitable model to investigate whether NGS could increase the resolution of SSR markers. Our results provide new insights on the consequences of scoring the variation found within microsatellite markers compared to scoring only the amplicon size.
Toward an improved identification of SSR loci variation
The occurrence of hidden mutations in the SSR amplicon has usually been attributed to divergence among species, and it was usually identified when transferring microsatellite markers between species or jointly analyzing SSR data for divergent taxa (van Oppen et al., 2000; Henriques et al., 2016; Germain‐Aubrey et al., 2016). However, according to our study, the incidence of alleles containing hidden variation is also likely to occur in microsatellite markers specifically designed for a single species. Our analyses revealed that 15 out of 18 microsatellite markers contained SNPs or indels in their sequences. SNPs and indels were found in both the flanking regions and within the SSR motif and occur at higher frequencies in the most common alleles, which were usually those with the lowest number of repeats of the SSR motif (Fig. 2). For these markers characterized by several sources of sequence variation, scoring SSRs by amplicon size led to incorrect allele identification for 69.44% of the alleles (see Results). These values are similar to those found by Vartia et al. (2016), who genotyped microsatellites using genotyping by sequencing (GBS) procedures. They concluded that 38 out of 40 SSR loci showed variation of SNPs and/or indels and that 32% of the alleles that were considered identical by size were truly non‐identical. Therefore, we strongly recommend GBS and analyses of sequences to score the variation of SSR loci in future studies.
NGS‐based pipeline to score SSR amplicons
The NGS‐based approach proposed in this study (Fig. 1) to genotype SSRs offers perspectives to improve the precision in the detection of the alleles compared to amplicon size scoring. We have optimized the allele scoring by proposing the true allele index (TAI, see Materials and Methods; Appendix S3) to detect and differentiate the noisy sequences obtained during the processes of PCR amplification (significantly reduced by using a High‐Fidelity DNA Polymerase) and sequencing. Some improvements have been proposed for producing SSR data based on NGS, such as MEGASAT (Zhan et al., 2016) and SSR_PIPELINE (Miller et al., 2013). These methods allow SSR detection and allele genotyping based on NGS‐produced sequences. An improved allele identification in this approach is only focused on discarding amplification artifacts (stutter products) identified by sequence depth. However, in contrast to our approach, the final genotype is based solely on variation in the number of repeat units, whereas the existence of other polymorphisms within the SSR or flanking regions is not investigated. Suez et al. (2016) produced an innovative software (MicNeSs) capable of recognizing repeat motifs within NGS‐produced sequences and, additionally, following up the scoring of an SSR pattern stopped by single mutations. We compared our results with those obtained by MicNeSs and found that this software significantly overestimates heterozygosity indices, as its inner algorithm for detecting the true alleles in the data set is exclusively based on the frequency of the observed distributions of the SSR patterns (see Suez et al., 2016 and the software manual for details).
In addition to improving the accuracy of genotyping, our approach offers more information on molecular variation. Depending on the objective, working on the sequence of microsatellite markers allows SSR and SNP alleles to be scored separately. Working with different types of markers that exhibit different rates of molecular evolution was recently recommended by Aimé and Austerlitz (2017) to get complementary insights on demographic history.
Integrating sequence and SSR polymorphisms in genetic diversity and structure studies
Depending on whether divergent but equally sized SSR alleles are treated as different, or additional sequence variation is recorded, and how this variation is treated, one would expect that different scoring methods led to differences in genetic diversity analyses. Sequence‐based SSR genotyping allows a better estimate of population divergence. By scoring both types of molecular variation independently (i.e., IM), the clusters detected through Bayesian inference of the genetic structure, as well as the pairwise F ST, reduced the admixture inferred for individual assignments (Fig. 3). This potential is also well illustrated here by private allele richness, a classical indicator of evolutionary uniqueness and long‐term persistence, which differed markedly depending on the scoring method (Table 1). Moreover, IM also purged the excess of heterozygotes that other scoring methods artificially generated. An increased deviation toward heterozygotes was observed in F IS values in both the MN and AS scoring methods (Table 1). Such deviation could be due to AS and MN being sensitive to paralog copies that generate false heterozygote genotypes or to the method used in MicNeSs (MN) software to select true alleles (Suez et al., 2016). In addition, the AS method can also misinterpret alleles by scoring amplicons containing gaps and inferring allele identity based on amplicon sizes multiple to the repetition motif. This excess of heterozygotes was corrected for when splitting the different types of molecular variation of each locus into separate data sets (IM), in which all populations showed F IS values close to HWE. We also estimated the F IS index for both SNPs and SSRs independently (Table 1), and they did not significantly deviate from HWE. Our analyses thus allow us to conclude that the traditional size‐based microsatellite markers may constitute chimerical genotypes that combine genetic variability evolving under different mutation rates, which may lead to disparate conclusions at the population level.
Due to the emergence of NGS techniques, a renovated SSR genotyping definition should combine all the sources of DNA sequence variation, corresponding to markers evolving at different mutation rates that potentially could capture recent demographic events, such as the last post‐glacial expansion, while keeping imprints of older events of vicariance or admixture.
Compared to traditional size‐based SSR genotyping, our study identifies additional sources of variation within microsatellite markers. By scoring sequence polymorphisms independently, the IM method described here improves genetic diversity estimates, correcting for HWE deviations in the traditional genotyping. Not accounting for the null alleles resulting from PCR failure remains a problem. However, our renovated microsatellite marker genotyping could help address this problem in future studies by considering mutation rates in flanking regions.
DATA ACCESSIBILITY
DNA sequences are available from GenBank (accessions KY913123–KY913162 [SSR characterization, see Appendix S1] and KY913163–KY913279 [SSR alleles]).
AUTHOR CONTRIBUTIONS
J.V., G.N.F., and A.B. wrote the manuscript. J.V., A.B., and A.H. developed the molecular methods. M.J. and F.M. extracted DNA. J.V. performed PCR, controls, and amplicon dilutions. D.B. set the amplicon sequencing libraries and ran the samples in the MiSeq. J.V. set the bioinformatics pipeline. J.V., G.N.F., and A.B. did the genetic diversity and structure analyses. M.B.K., L.O., G.N.F., and F.M. participated in field sampling. M.B.K. and S.L.M. contributed to the manuscript. H.S. coordinated the funding and devised the project DYNAMIC. All authors reviewed the manuscript.
Supporting information
Appendix S1. Characteristics of the SSR loci developed for Ceratonia siliqua.
Appendix S2. Number of reads per locus and sample obtained in three MiSeq 250 × 250 runs in which 48 samples were pooled for two sets of 17 loci (A), 192 samples were pooled for the final selected 18 loci (B), and 384 samples were pooled for the final selection of 18 loci (C).
Appendix S3. Frequencies of the selected alleles in homozygous and heterozygous loci according to the true allele index (TAI), see Materials and Methods.
Appendix S4. Custom R script to convert an input file in FASTA format into a list of genotypes.
Appendix S5. Representation of the number of sequences obtained for a locus vs. the number of different sequences obtained when genotyping SSRs through next‐generation sequencing.
ACKNOWLEDGMENTS
This work benefited from equipment and services from the molecular biology lab facility at the Institut Méditerranéen de Biodiversité et d'Ecologie marine et continentale (IMBE) and from the genotyping and sequencing core facility (iGenSeq) at ICM (Hôpital Pitié Salpêtrière, Paris, France). This research is part of the DYNAMIC project funded by the French National Research Agency (ANR‐14‐CE02‐0016). J.V. benefited from a postdoctoral fellowship funded by DYNAMIC (ANR‐14‐CE02‐0016) and a Marie Skłodowska‐Curie Individual Fellowship (704464‐YAMNOMICS‐MSCA‐IF‐EF‐ST).
Viruel, J. , Haguenauer A., Juin M., Mirleau F., Bouteiller D., Boudagher‐Kharrat M., Ouahmane L., La Malfa S., Médail F., Sanguin H., Nieto Feliner G., and Baumel A.. 2018. Advances in genotyping microsatellite markers through sequencing and consequences of scoring methods for Ceratonia siliqua (Leguminosae). Applications in Plant Sciences 6(12): e1201.
LITERATURE CITED
- Aimé, C. , and Austerlitz F.. 2017. Different kinds of genetic markers permit inference of Paleolithic and Neolithic expansions in humans. European Journal of Human Genetics 25: 360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S. 2010. FastQC: A quality control tool for high throughput sequence data. Website http://www.bioinformatics.babraham.ac.uk/projects/fastqc [accessed 13 November 2018].
- Barthe, S. , Gugerli F., Barckley N. A., Maggia L., Cardi C., and Scotti I.. 2012. Always look on both sides: Phylogenetic information conveyed by Simple Sequence Repeat allele sequences. PLoS ONE 7: e40699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard, G. , El Bakkali A., Haouane H., Baali‐Cherif D., Moukhli A., and Khadari B.. 2013. Population genetics of Mediterranean and Saharan olives: Geographic patterns of differentiation and evidence for early generations of admixture. Annals of Botany 112: 1293–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse M., and Usadel B.. 2014. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann, B. , Klintschar M., Neuhuber F., Hühne J., and Rolf B.. 1998. Mutation rate in human microsatellites: Influence of the structure and length of the tandem repeat. American Journal of Human Genetics 62: 1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevreux, B. , Pfisterer T., Drescher B., Driesel A. J., Müller W. E. G., Wetter T., and Suhai S.. 2004. Using the miraEST Assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Research 14: 1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakov, D. A. , Hellemans B., and Volckaert F. A. M.. 2006. Microsatellites and their genomic distribution, evolution, function and applications: A review with special reference to fish genetics. Aquaculture 255: 1–29. [Google Scholar]
- Clement, M. , Posada D., and Crandall K. A.. 2000. TCS: A computer program to estimate gene genealogies. Molecular Ecology 9: 1657–1659. [DOI] [PubMed] [Google Scholar]
- Cornille, A. , Giraud T., Smulders M. J., Roldán‐Ruiz I., and Gladieux P.. 2014. The domestication and evolutionary ecology of apples. Trends in Genetics 30: 57–65. [DOI] [PubMed] [Google Scholar]
- Diez, C. M. , Trujillo I., Martinez‐Urdiroz N., Barranco D., Rallo L., Marfil P., and Gaut B. S.. 2015. Olive domestication and diversification in the Mediterranean Basin. New Phytologist 206: 436–447. [DOI] [PubMed] [Google Scholar]
- Edgar, R. C. 2004. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estoup, A. , Jarne P., and Cournet J.‐M.. 2002. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Molecular Ecology 11: 1591–1604. [DOI] [PubMed] [Google Scholar]
- Evanno, G. , Regnaut S., and Goudet J.. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Molecular Ecology 14: 2611–2620. [DOI] [PubMed] [Google Scholar]
- Faircloth, B. C. 2008. MSATCOMMANDER: Detection of microsatellite repeat arrays and automated, locus‐specific primer design. Molecular Ecology Resources 8: 92–94. [DOI] [PubMed] [Google Scholar]
- Germain‐Aubrey, C. C. , Nelson C., Soltis D. E., Soltis P. S., and Gitzendanner M. A.. 2016. Are microsatellite fragment lengths useful for population‐level studies? The case of Polygala lewtonii (Polygalaceae). Applications in Plant Sciences 4: 1500115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, A. , and Hannon G. J.. 2010. FASTX‐Toolkit. FASTQ/A short‐reads preprocessing tools (unpublished). Website http://hannonlab.cshl.edu/fastx_toolkit/ [accessed 13 November 2018].
- Guichoux, E. , Lagache L., Wagner S., Chaumeil P., Léger P., Lepais O., Lepoittevin C., et al. 2011. Current trends in microsatellite genotyping. Molecular Ecology Resources 11: 591–611. [DOI] [PubMed] [Google Scholar]
- Henriques, R. , von der Heyden S., and Matthe C. A.. 2016. When homoplasy mimics hybridization: A case study of Cape hakes (Merluccius capensis and M. paradoxus). PeerJ 4: e1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings, T. N. , Knaus B. J., Mullins T. D., Haig S. M., and Cronn R. C.. 2011. Multiplexed microsatellite recovery using massively parallel sequencing. Molecular Ecology Resources 11: 1060–1067. [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Stecher G., and Tamura K.. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology and Evolution 33: 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Malfa, S. , Currò S., Bugeja Douglas A., Brugaletta M., Caruso M., and Gentile A.. 2014. Genetic diversity revealed by EST‐SSR markers in carob tree (Ceratonia siliqua L.). Biochemical Systematics and Ecology 55: 205–211. [Google Scholar]
- Magoč, T. , and Salzberg S. L.. 2011. FLASH: Fast Length Adjustment of SHort reads to improve genome assemblies. Bioinformatics 27: 2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meglécz, E. , Pech N., Gilles A., Dubut V., Hingamp P., Trilles A., Grenier R., and Martin J. F.. 2014. QDD version 3.1: A user friendly computer program for microsatellite selection and primer design revisited: Experimental validation of variables determining genotyping success rate. Molecular Ecology Resources 14: 1302–1313. [DOI] [PubMed] [Google Scholar]
- Merritt, B. J. , Culley T. M., Avanesyan A., Stokes R., and Brzyski J.. 2015. An empirical review: Characteristics of plant microsatellite markers that confer higher levels of genetic variation. Applications in Plant Sciences 3: 1500025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M. P. , Knaus B. J., Mullins T. D., and Haig S. M.. 2013. SSR_pipeline: A bioinformatic infrastructure for identifying microsatellites from paired‐end Illumina high‐throughput DNA sequencing data. Computer Note 104: 881–885. [DOI] [PubMed] [Google Scholar]
- van Oppen, M. J. H. , Rico C., Turner G. F., and Hewitt G. M.. 2000. Extensive homoplasy, nonstepwise mutations and shared ancestral polymorphism at a complex microsatellite locus in Lake Malawi cichlids. Molecular Biology and Evolution 17: 489–498. [DOI] [PubMed] [Google Scholar]
- Peakall, R. , and Smouse P. E.. 2012. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research–an update. Bioinformatics 28: 2537–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollegioni, P. , Woeste K., Chiocchini F., Del Lungo S., Ciolfi M., Olimpieri I., Tortolano V., et al. 2017. Rethinking the history of common walnut (Juglans regia L.) in Europe: Its origins and human interactions. PLoS ONE 12: e0172541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K. , Stephens M., and Donnelly P.. 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putman, A. I. , and Carbone I.. 2014. Challenges in analysis and interpretation of microsatellite data for population genetic studies. Ecology and Evolution 4: 4399–4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetto, M. , McNally J., and Henry R. J.. 2002. Evaluating the potential of SSR flanking regions for examining taxonomic relationships in the Vitaceae. Theoretical and Applied Genetics 104: 61–66. [DOI] [PubMed] [Google Scholar]
- Rousset, F. 2008. GENEPOP'007: A complete reimplementation of the GENEPOP software for Windows and Linux. Molecular Ecology Resources 8: 103–106. [DOI] [PubMed] [Google Scholar]
- Suez, M. , Behdenna A., Brouillet S., Graça P., Higuet D., and Achaz G.. 2016. MicNeSs: Genotyping microsatellite loci from a collection of (NGS) reads. Molecular Ecology Resources 16: 524–533. [DOI] [PubMed] [Google Scholar]
- Vartia, S. , Villanueva‐Cañas J. L., Finarelli J., Farrell E. D., Collins P. C., Hughes G. M., Carlsson J. E. L., et al. 2016. A novel method of microsatellite genotyping‐by‐sequencing using individual combinatorial barcoding. Royal Society Open Science 3: 150565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viruel, J. , Catalán P., and Segarra‐Moragues J. G.. 2010. New microsatellite loci in the dwarf yams Dioscorea group Epipetrum (Dioscoreaceae). American Journal of Botany 97: e121–e123. [DOI] [PubMed] [Google Scholar]
- Viruel, J. , Ortiz P. L., Arista M., and Talavera M.. 2015. Characterization of nuclear microsatellite markers for Rumex bucephalophorus (Polygonaceae) using 454 sequencing. Applications in Plant Sciences 3: 1500088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zane, L. , Bargelloni L., and Patarnello T.. 2002. Strategies for microsatellite isolation: A review. Molecular Ecology 11: 1–16. [DOI] [PubMed] [Google Scholar]
- Zehdi‐Azouzi, S. , Cherif E., Moussouni S., Gros‐Balthazard M., Abbas Naqvi S., Ludeña B., Castillo K., et al. 2015. Genetic structure of the date palm (Phoenix dactylifera) in the Old World reveals a strong differentiation between eastern and western populations. Annals of Botany 116: 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan, L. , Paterson I. G., Fraser B. A., Watson B., Bradbury I. R., Nadukkalam Ravindran P., Reznick D., et al. 2016. MEGASAT: Automated inference of microsatellite genotypes from sequence data. Molecular Ecology Resources 17: 247–256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Characteristics of the SSR loci developed for Ceratonia siliqua.
Appendix S2. Number of reads per locus and sample obtained in three MiSeq 250 × 250 runs in which 48 samples were pooled for two sets of 17 loci (A), 192 samples were pooled for the final selected 18 loci (B), and 384 samples were pooled for the final selection of 18 loci (C).
Appendix S3. Frequencies of the selected alleles in homozygous and heterozygous loci according to the true allele index (TAI), see Materials and Methods.
Appendix S4. Custom R script to convert an input file in FASTA format into a list of genotypes.
Appendix S5. Representation of the number of sequences obtained for a locus vs. the number of different sequences obtained when genotyping SSRs through next‐generation sequencing.
Data Availability Statement
DNA sequences are available from GenBank (accessions KY913123–KY913162 [SSR characterization, see Appendix S1] and KY913163–KY913279 [SSR alleles]).