

ABSTRACT
Complex tissue communication networks function throughout an organism's lifespan to maintain tissue homeostasis. Using the genetic model Drosophila melanogaster, we have defined a network of immune responses that are activated following the induction of muscle stresses, including hypercontraction, detachment and oxidative stress. Of these stressors, loss of the genes that cause muscle detachment produced the strongest levels of JAK-STAT activation. In one of these mutants, fondue (fon), we also observe hemocyte recruitment and the accumulation of melanin at muscle attachment sites (MASs), indicating a broad involvement of innate immune responses upon muscle detachment. Loss of fon results in pathogen-independent Toll signaling in the fat body and increased expression of the Toll-dependent antimicrobial peptide Drosomycin. Interestingly, genetic interactions between fon and various Toll pathway components enhance muscle detachment. Finally, we show that JAK-STAT and Toll signaling are capable of reciprocal activation in larval tissues. We propose a model of tissue communication for the integration of immune responses at the local and systemic level in response to altered muscle physiology.
KEY WORDS: Drosophila, Innate immunity, Tissue communication, Muscle
Summary: Activation of Drosophila innate immune responses is coordinated at local and systemic levels through tissue crosstalk following disruptions to muscle homeostasis.
INTRODUCTION
In insects, the detection of foreign molecules activates a robust immune signaling cascade coupled to the production of biological outputs and cellular activities that minimize damage to the host. Innate immunity can be broken down into the humoral arm, which uses signaling pathways for antimicrobial peptide (AMP) and target gene expression, and the cellular arm, which regulates the mobilization of hemocytes and melanin production (Royet et al., 2003). Drosophila possesses three types of blood cells: hemocytes, migratory plasmatocytes similar to macrophages; lamellocytes for pathogen encapsulation; and crystal cells, which are crucial for releasing molecules essential for melanization (Evans and Wood, 2014; Wang et al., 2014). Depending on the type of infection, signaling during pathogen challenge proceeds through the canonical Toll (Tl) and immune deficiency (Imd) pathways (Lemaitre et al., 1997). Both of these signal transduction pathways require binding of an extracellular ligand to transmembrane receptors to activate a series of intracellular events that lead to the nuclear translocation of NF-κB transcription factors, Dorsal (Dl) and/or Dif, or Relish (Rel), respectively (Bergmann et al., 1996; De Gregorio et al., 2002; Hetru and Hoffmann, 2009; Reach et al., 1996; Stöven et al., 2000). Once in the nucleus, these transcription factors induce the expression of AMPs and other immune-responsive genes. How these individual aspects of the innate immune response are initiated and integrated to preserve the function of tissues remains an important question in animal physiology.
Recently, work in model organisms has highlighted novel relationships between muscle tissue and activation of the innate immune response. During parasitoid wasp infections, JAK-STAT signaling originating in larval muscle tissue is essential for the encapsulation of wasp eggs (Yang et al., 2015). Drosophila indirect flight muscles (IFMs) also act as an immune-responsive tissue essential for surviving bacterial challenges during adulthood (Chatterjee et al., 2016). During an infection, AMP production occurs in the adult IFMs. Importantly, reduction of AMPs through knocking down Toll or Imd pathway components, or through compromising IFM structural integrity, limits an individual's survival. Similarly, immune responsiveness through TLR-mediated signaling was observed in zebrafish muscles following infection (Chatterjee et al., 2016). These experiments establish muscle as a key immune-responsive tissue in the defense against infection in both invertebrates and vertebrates. Further supporting a shared connection between muscle maintenance and innate immunity, we recently identified fondue (fon) as essential for maintaining the extracellular matrix (ECM) of the larval muscle attachment site (MAS) (Green et al., 2016). The common suite of secreted hemolymph proteins that accumulate and function at the MAS were also found to act with Fon in its previously described role in coagulation (Bajzek et al., 2012; Lindgren et al., 2008; Scherfer et al., 2006).
The invasion of immune cells and inflammation following muscle injury is a standard strategy for cellular repair in vertebrates (Tidball and Villalta, 2010). However, individuals with myopathies and muscular dystrophies often experience persistent immune responses in damaged muscle, which may contribute to disease progression (Madaro and Bouché, 2014; Nitahara-Kasahara et al., 2016; Rosenberg et al., 2015; Villalta et al., 2015). Gene expression profiles obtained from Drosophila mutants with hypercontraction-induced myopathy (Montana and Littleton, 2006) and human muscular dystrophy patients (Chien et al., 1991; Haslett et al., 2002; Hathout et al., 2014) reveal an upregulation of genes involved in actin-dependent remodeling and chaperone transcripts, as well as a downregulation of metabolic and mitochondrial genes characteristic of metabolic stress in dystrophic muscle. Additional increases in classes of immune-responsive genes upon muscle damage suggests that the capacity for immune responses to act not only in response to, but to potentially drive tissue damage, emphasizes the necessity for understanding the fundamental mechanisms regulating immune and tissue physiology.
Here, we have uncovered a tissue communication network linking muscle maintenance and innate immune signaling. While characterizing the muscle phenotypes of fon mutants (Green et al., 2016), we noted several immune responses resulting from damaged muscle tissue, including the deposition of melanin at MASs, hemocyte recruitment to detached and torn muscles, and the activation of both JAK-STAT and Toll signaling. Importantly, we also show that JAK-STAT and Toll have the ability to establish a reciprocal signaling network between tissues that sense and respond to cellular stress. Therefore, we conclude that muscle tissue maintenance is coordinated with innate immunity in a multi-organ response mediated through local JAK-STAT signaling and systemic Toll activation.
RESULTS
JAK-STAT signaling occurs in response to specific classes of muscle stress
The role of JAK-STAT signaling in tissue stress has been well documented (Myllymäki and Rämet, 2014). More recently, a novel and essential role for JAK-STAT activity within muscle tissue has been described in the Drosophila response to parasitoid wasp infections (Yang and Hultmark, 2016; Yang et al., 2015). Furthermore, JAK-STAT signaling is upregulated at the borders of epithelial wound sites and in the closely associated muscle layer of third instar (L3) larvae (Lee et al., 2017). Based on these observations, we wanted to determine whether JAK-STAT signaling could act as a local response to muscle tissue stress using the JAK-STAT reporter, 10xSTAT92E-GFP (STAT-GFP). Bach et al. (2007) genetically engineered flies containing a reporter construct consisting of STAT92E binding sites fused to GFP. When JAK-STAT signaling is active, the transcription factor STAT92E translocates into the nucleus. With STAT in the nucleus, the transcription factor associates with the tandem STAT-binding sites present in the reporter construct and leads to the production of GFP that can be seen in both the nucleus and cytoplasm of cells (Fig. 1A).
Fig. 1.

JAK-STAT is activated by specific classes of muscle stress. (A) Schematic of the JAK-STAT reporter, 10xSTAT92E-GFP (STAT-GFP), which contains STAT binding sites enabling GFP transcription upon STAT activity in the nucleus. Activation of JAK-STAT signaling causes increased expression of the STAT-GFP reporter in both the cytoplasm and nucleus. (B–G) Muscle phenotypes (F-actin, red) and nuclear positioning (DAPI, blue) in select RNAi knockdowns via muscle-specific GAL4/UAS inducing different classes of cellular stress. (B′–G′) STAT expression in L3 larval muscles as measured with the STAT-GFP reporter (GFP, green). (B,B′) STAT levels are low or nearly undetectable in the cytoplasm and nucleus of unstressed larval muscles. (C,C′) RNAi knockdown of Tig in muscles leads to weakened tendon cell anchoring, but muscle attachments are maintained across hemisegments. Partial detachment is associated with cytoplasmic and nuclear increases in STAT expression (arrowheads). (D,D′) RNAi knockdown of stck, which encodes the integrin-associated PINCH protein, enhances STAT-GFP expression in muscles. Although knockdown of stck can cause muscle detachment, an attached muscle was selected for analysis to simplify interpretation. (E,E′) Perturbations to oxidative stress induced through knockdown of Sod1 causes subtle disruptions to the sarcomeric spacing of muscles (see regions marked by yellow line in E) and STAT-GFP levels to weakly increase. (F,F′) Mitochondrial stress caused through knockdown of the mitochondrial protein, TFAM, causes similar disruptions to muscle sarcomeres (yellow line in F) and activates JAK-STAT signaling. (G,G′) Myofibrillar unbundling and muscle degeneration resulting from tn knockdown is not capable of activating JAK-STAT signaling. (H–M) Line plot analysis of STAT-GFP fluorescence intensity throughout the cytoplasm and nucleus (white lines) in panels B′–G′. Green dots on each graph correspond to arrowheads marking nuclei in each image panel. GFP intensity from STAT-GFP control muscle in panel H is overlaid on line plots as a gray-filled profile in panels I–M. Additional examples of stresses can be found in Fig. S1. Scale bars: 100 µm.
We devised a reporter screen using RNAi stocks to determine which classes of muscle stresses could activate JAK-STAT in muscle tissue. In healthy muscle, JAK-STAT signaling is held at low levels (Fig. 1B,B′,H). Using the STAT-GFP reporter in combination with the muscle-specific mef2-GAL4 driver, we could selectively knock down genes known to induce tissue damage and assay for increased JAK-STAT activation in muscles. We identified representative genes where knockdown resulted in well-defined muscle phenotypes including degeneration, detachment, oxidative stress and mitochondrial stress to assess the ability of each type of tissue stress to activate JAK-STAT.
One of the most damaging phenotypes to tissue is muscle detachment, which can occur as either a complete detachment of muscle from the ECM or a partial detachment compromising the connection of the actin cytoskeleton to molecules in the extracellular space. The knockdown of Tiggrin (Tig), a component of the larval ECM, causes muscles to round up and detach at indirect MASs (Bunch et al., 1998). The Drosophila gene steamer duck (stck) encodes PINCH, which is an adaptor protein associated with integrin complexes whose loss leads to a detachment of the internal actin cytoskeleton (Clark et al., 2003). RNAi knockdown of either Tig or stck results in muscle morphology defects and increased STAT-GFP expression (Fig. 1C-D′,I,J). Based on these observations, it is not surprising that muscles intentionally torn with forceps during live larval dissections quickly induce a local JAK-STAT response in comparison to wild-type larvae that have been heat-killed prior to dissection (Fig. S1). We also examined the effect of an external pinch wound, which preserves cuticle integrity while damaging the underlying epithelial and muscle layers (Burra et al., 2013). Larvae with pinch wounds show only a muted response to subtle muscle damage (Fig. S1), which may reflect the small degree of damage inflicted by this technique.
Skeletal muscle aging and several pathological conditions are associated with a loss of metabolic homeostasis from mitochondrial dysfunction and increasing levels of reactive oxygen species (ROS) (Fabio et al., 2013). Tissue balance of ROS is maintained by a series of enzymes including Superoxide dismutase (Sod) and Catalase (Cat). Both overexpression and knockdown of Sod1 have been reported to increase levels of ROS (Buettner et al., 2006; Cabreiro et al., 2011). Disruption of ROS balance through the reduction (Fig. 1E,E′,K) or overexpression (Fig. S1) of the Sod1 enzyme results in active JAK-STAT signaling. Similarly, altering levels of Cat causes increased STAT-GFP in larval musculature (Fig. S1). Inducing mitochondrial stress through knockdown of mitochondrial transcription factor a (TFAM) (Fig. 1F,F′,L) or parkin (park) (Fig. S1) yielded the lowest levels of JAK-STAT activation from the chosen classes to screen (Greene et al., 2003; Larsson et al., 1998). Surprisingly, the myofibrillar unbundling phenotype present in thin (tn) RNAi fillets is not capable of activating JAK-STAT signaling (Fig. 1G,G′,M), suggesting that activation of this pathway is subject to different cellular or molecular stresses.
JAK-STAT signaling is activated locally in response to muscle damage
Our results thus far show that knockdown of two genes required for muscle attachment (Tig and stck) activate local JAK-STAT signaling in muscle. We previously identified Fon as a novel ECM organizer in preserving larval MASs (Green et al., 2016). Owing to its strong detachment phenotype and the ability to bypass the embryonic lethality that complicates the analysis of similar mutants (i.e. integrin subunits), we next examined the muscle-specific activation of the JAK-STAT pathway in fon knockout mutants. Indeed, removal of fon promotes widespread muscle detachment and elicits dramatic increases compared with wild type in STAT-GFP reporter expression in both the cytoplasm and nucleus of muscles (Fig. 2 A,A′,F,C,C′,H). This exceeds the increased expression of STAT-GFP that occurs in response to activation of the temperature-sensitive, constitutively active JAK allele, hopTum-l (Fig. 2B,B′,G). We noted in our reporter screen that basal levels of JAK-STAT signaling are present in muscle, cuticle and epithelial cells in wild-type larvae, although there was a clear increase in muscle-based JAK-STAT signaling from knockout of genes in the muscle detachment class (Fig. 1; Fig. S2). The majority of homozygous fon mutants (∼80%) share this expression pattern, although there were occasional instances (∼20%) where STAT-GFP in the epithelial layer underneath muscles appeared to experience activated JAK-STAT signaling as well (Fig. S2).
Fig. 2.

JAK-STAT signaling is a local response to muscle damage via detachment. (A–E′) Expression of STAT-GFP reporter (green) in L3 larval muscle stained with phalloidin (F-actin, red) and DAPI (blue). (A,A′) In normal muscle, STAT-GFP expression is at low levels. (B,B′) Activation of JAK-STAT signaling using the constitutively active JAK allele, hopTum-l, increases STAT-GFP levels in both the cytoplasm and nucleus (arrowheads). (C,C′) Loss of fon causes dramatic increases in STAT-GFP both in the cytoplasm and the nucleus. For clarity and consistency, a muscle that remained attached was imaged. Lower magnification images of STAT-GFP expression in fon mutants can be found in Fig. S2. (D–E′) In the hypercontractile mutants, MhcS1 and BrkdJ29, STAT-GFP expression is increased throughout muscle tissue and concentrates in the nucleus (arrowheads). (F–J) Line plot analysis of STAT-GFP fluorescence intensity along white lines drawn through muscles in panels A′–E′. Green dots on each graph correspond to arrows marking nuclei in each image panel. GFP intensity from STAT-GFP control muscle in panel F is overlaid on line plots as a gray-filled profile in panels G–J. Scale bars: 100 µm.
Genetic mutations characterized in Caenorhabditis elegans and Drosophila show that hypercontraction can act as a precursor to muscle detachment (Bessou et al., 1998; Grisoni et al., 2002; Myers et al., 1996; Nongthomba et al., 2007; Nongthomba et al., 2004; Raghavan et al., 2000). Furthermore, expression profiling of the dominant MhcS1 allele in Drosophila adults revealed that immune signaling was upregulated in response to hypercontraction-induced myopathy (Montana and Littleton, 2006). We wanted to examine whether immune activation occurred at steps preceding muscle detachment (i.e. prolonged hypercontraction) and confirm that immune activation was responsive to muscle damage in larval stages. We tested two temperature-sensitive hypercontractile mutants, MhcS1 and BrkdJ29, which contain hypercontracted muscle as larvae and exhibit indented thorax phenotypes in adults as IFMs degenerate (Montana and Littleton, 2004). At restrictive temperatures, both MhcS1 and BrkdJ29 exhibit hypercontraction and active JAK-STAT signaling (Fig. 2D–E′,I,J), suggesting that JAK-STAT acts as a local mediator in the progression towards muscle detachment.
Muscle stress induces a broad suite of immune responses
During our analysis of fon-induced muscle attachment, we noted the appearance of several phenotypes attributed to innate immunity. In wild-type larvae, unchallenged individuals are free of melanization (Fig. 3A) (Binggeli et al., 2014; Tang, 2009; Tang et al., 2006). It was known that mutations in fon result in diffuse melanization at wound sites as well as the presence of melanotic tumors throughout the hemocoel (Fig. S3) (Scherfer et al., 2006). A small percentage of fon mutants (∼5%) present a unique melanization phenotype where melanin spontaneously accumulates at sites of muscle attachment (Fig. 3D; Fig. S3).
Fig. 3.
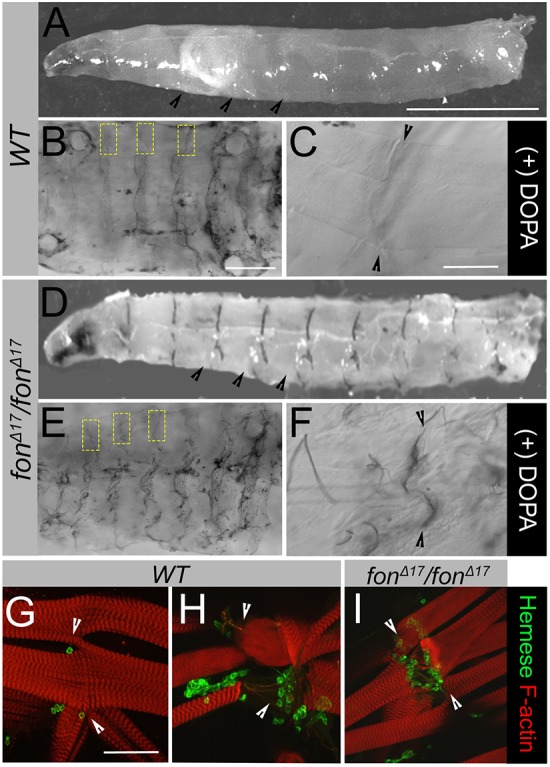
Loss of Fon activates innate immune processes. (A–F) Presence of melanin in wild-type (WT) versus fonΔ17 homozygous mutants visualized in exterior views (A,D) and dissected muscle fillets (B,C,E,F). (A) Wild-type larvae lack a visible melanization response throughout the body cavity or MASs (black arrowheads). (B) Addition of the phenoloxidase substrate, L-DOPA, allows for conversion into melanin which collects non-specifically throughout the cuticle of wild-type larvae. Areas containing MASs along the hemisegmental borders are marked with yellow dashed boxes. (C) Wild-type muscle attachments (black arrowheads) imaged at higher magnification are free of melanin. (D) Melanin is spontaneously deposited at MASs (black arrowheads) in low percentages (∼5%) of fonΔ17 mutant larvae. (E) Melanization at MASs (yellow boxes) can be induced by providing excess L-DOPA substrate to dissected fillets. (F) High magnification of melanin deposits at muscle attachments of fonΔ17 larvae observed upon addition of L-DOPA (black arrowheads). (G–I) Distribution of hemocytes at MASs in filleted larvae. Low magnification images can be found in Fig. S3. (G) Hemocytes (detected through labeling for Hemese) are found at low levels near intact MASs. (H,I) In wild-type muscles that have been mechanically damaged during dissection (H) or upon fon-mediated muscle detachment (I), hemocytes are recruited to sites of muscle attachment and/or damaged muscles. Arrowheads denote the MAS in panels G–I. Scale bars: 1 mm in A,D; 500 µm in B,E; 100 µm in C,F–I.
We utilized two methods to increase the frequency of melanization in fon mutants for further study: 1) uniform crush wounding to exacerbate tissue stress; and 2) dissection of larvae in excess L-DOPA substrate to increase phenoloxidase (PO) enzymatic activity (see Materials and Methods for details). A uniform crush-based trauma was administered to fon mutant larvae through gently compressing the dorsal surface along the body axis. This additional stress resulted in melanization of the dorsal vessel, particularly the pericardial cells, and occasionally localized melanin accumulation at MASs (Fig. S3). Next, fillets were dissected and incubated in the presence of the PO substrate L-DOPA. In wild-type fillets free of muscle damage, melanin does not accumulate at MASs but may be found along dissection sites and within the cuticle (Fig. 3B,C). However, fon mutant fillets in the presence of L-DOPA accumulate melanin that phenocopies spontaneously-occurring patterns at nearly 100% of MASs (Fig. 3D–F; Fig. S3). If tissue-based melanization is a result of muscle damage and not simply a consequence of fon removal, we would expect to see melanin accumulating in wild-type muscles intentionally damaged during dissection or by trauma. Indeed, when muscles are damaged during dissection and incubated in L-DOPA, melanin strongly accumulates along the borders of damage sites (Fig. S3). Additionally, melanin accumulation is obvious at MASs upon tear-induced muscle detachment and at the site of forceps pinching in conjunction with damage to the underlying muscle hemisegments (Fig. S3). These data, taken together, show that muscle damage, whether through genetic manipulation or methods of artificial muscle stress, is sufficient to trigger melanization-induced immune responses.
Because melanization requires the activity of PO, which is expressed only in a subset of hemocytes known as crystal cells, we wanted to determine whether hemocytes were localized to damaged muscle tissue. During a wounding event, hemocytes are recruited to the sites of damage to aid in repair through release of the PO enzyme and other secreted proteins (Krautz et al., 2014). In L3 larvae, hemocytes can be circulating throughout the hemolymph, reside as sessile populations along the dorsal vessel, or in hematopoietic pockets between body wall muscles and epithelia (Holz et al., 2003; Makhijani et al., 2011; Márkus et al., 2005; Stofanko et al., 2008; Zaidman-Rémy et al., 2012). Hemocytes are dispersed in these patterns along muscle hemisegments in unwounded larvae, but do not specifically localize to MASs (Fig. 3G; Fig. S3). In wild-type fillets with mechanical damage to muscles, hemocytes are recruited to sites of tissue damage (Fig. 3H; Fig. S3). Similarly, hemocytes are targeted to detached muscles and attachment sites where muscles have begun to pull away from one another upon genetic loss of fon (Fig. 3I; Fig. S3). Taken together, these data indicate that loss of muscle maintenance is linked to the initiation of stress-based signaling and cellular immune activities.
Muscle damage caused by loss of fon activates pathogen-independent, systemic Toll signaling
In previous studies that examined the role of Fon in immunity, fon mutants constitutively expressed the AMP Drosomycin (Drs), and fon itself was identified as a target of Toll signaling (Scherfer et al., 2006). The muscle morphology of dissected wild-type larval fillets features repeating hemisegments of broad, rectangular muscles stably anchored at MASs (Fig. 4B). In fon mutants, unstable muscle attachments lead to muscle detachment that generates extensive tissue damage (Fig. 4D). In the absence of pathogens, muscle morphology is maintained in wild-type fillets while still being disrupted in fon mutants (Fig. 4C,E).
Fig. 4.

Toll signaling is activated in fon mutants. (A) Schematic showing Dl localization during Toll signaling. Dl is primarily cytoplasmic when Toll signaling is turned off in L3 fat body cells. Following Toll activation, Dl moves into the nucleus to induce gene expression. (B–E) Muscle fillets of dissected L3 wild-type and fonΔ17 mutants raised in either normal or axenic conditions and visualized with phalloidin (F-actin, red). (B′–E′) Dl localization (green) in larval fat body tissue. A single fat body cell is outlined in white for each panel. (B,C) Wild-type muscles are rectangular and firmly anchored to adjacent muscles and tendon cells in both the presence and absence of pathogens (axenic). (B′,C′) Dl is localized to the cytoplasm of fat body cells regardless of the presence or absence of pathogens. (D,E) Muscles round up and detach (arrowheads) upon loss of fon, independent of the presence of pathogens. (D′,E′) Dl is enriched in the nucleus of fon mutant fat body cells in both normal and axenic conditions. (F) Mean±s.e.m relative transcript levels of Drs RNA collected from the pooled fat bodies of wild-type larvae, larvae expressing fon alleles (Δ17/Df and Δ24/Df), and larvae demonstrating fat-body induced Toll (Tl) overexpression, Cg>Tl10B. Scale bars: 500 µm in B–E; 50 µm in B′–E′.
We utilized the subcellular localization of the NF-κB transcription factor, Dl, to evaluate Toll pathway activation upon loss of fon. In wild-type larvae without infection or damage, Toll signaling is inactive and Dl is localized throughout the cytoplasm of the cell (Fig. 4A–B′). Upon Toll activation, Dl translocates into the fat body nucleus to initiate transcription of Toll-responsive genes (Fig. 4A). Dl staining is concentrated in the nuclei of fon null mutants, indicating that a loss of Fon activates Toll signaling (Fig. 4D′). We also observe activation of Toll in the fat bodies of wild-type larvae several hours after the muscle and underlying epithelium is damaged via pinch wounding (Fig. S3). This nuclear localization of Dl is absent in muscle tissue of both wild-type and fon mutant larvae, narrowing Toll activation to a systemic rather than local response (Fig. S4). Previously, Drs expression in unchallenged fon mutants was detected using a Drs–GFP reporter (Scherfer et al., 2006). Complementary to these studies and our observations that fon mutants activate Toll in the fat body, Drs transcripts are dramatically increased in isolated fat bodies lacking fon (Fig. 4F).
To determine whether microbes are important for Toll activation in fon mutants, these same experiments were performed under axenic, or germ-free, conditions. The appearance of muscle detachment and the translocation of Dl into the nucleus indicate that the consequences of loss of fon are pathogen-independent (Fig. 4D–E′). We also tested whether the other major immune response pathway, Imd, and its NF-κB transcription factor, Rel, were activated in response to fon-mediated muscle damage. Similar to Dl, Rel is restricted to the cytoplasm when Imd signaling is inactive and then translocates into the nucleus upon signal transduction to begin immune-related gene expression (Stöven et al., 2000). We found that Rel is retained in the cytoplasm of wild-type and fon mutant fat bodies and transcription of the AMP diptericin (dpt) is not upregulated in response to muscle stress (Fig. S4). Therefore, Toll signaling is activated systemically and selectively in response to muscle damage owing to loss of fon.
fon interacts with genes necessary for the activation of Toll signaling
Because knockdown of Fon induces systemic Toll activation, we wanted to determine whether canonical components of Toll signaling are required. We implemented a fon-sensitized background (fonΔ24/+; da-GAL4) screen using the GAL4/UAS system to target genes at regulatory points in the Toll signal transduction cascade (Brand and Perrimon, 1993). In both wild-type larvae and fon heterozygotes, rarely is muscle detachment observed (Fig. 5A,E,I). However, when crossed to lines with candidate genes that interact with fon, genetic interactions would lead to an enhancement of muscle detachment greater than that observed in lines where the candidate gene alone was knocked down or overexpressed. Candidate lines were chosen to simulate the genetic activation of Toll signaling, i.e., either the overexpression of genes (UAS-SPE, UAS-dl, US-Dif, UAS-Toll10B) at key activation steps or the knockdown of inhibitors (UAS-cact RNAi) present in the pathway.
Fig. 5.

Genetic interactions between fon and Toll pathway components enhance muscle detachment. (A–H) Two hemisegments of muscle fillets stained with phalloidin (F-actin, green) in a wild-type control, candidate RNAi lines alone (B–D) or in the fon-sensitized genetic background, fonΔ24/+; da-GAL4 (E–H). (A,E) Morphological defects are absent in the muscles of wild-type and fonΔ24/+; da-GAL4 larval fillets. (B) RNAi knockdown of the NFκB inhibitor, cact, alone does not disrupt muscle attachment. (C,D) Overexpression of Dl or SPE has no obvious consequences for muscle attachment stability. (E–H) In comparison to heterozygous fonΔ24/+ alone (E), loss of cact (F) or the overexpression of dl (G) or SPE (H) in a fon-sensitized background enhances muscle detachment (arrowheads). (I) Mean±s.e.m quantification of muscle detachment of select genotypes (fonΔ24; da>yw, n=16; da>cact RNAi, n=12; fonΔ24; da>cact RNAi, n=21; da>UAS-SPE, n=20, fonΔ24; da>UAS-SPE, n=19; da>UAS-dl, n=10; fonΔ24; da>UAS-dl, n=13; da>UAS-spz(FL) #1, n=20; fonΔ24; da>UAS-spz(FL) #1, n=12); da>UAS-spz(FL) #2, n=17; fonΔ24; da>UAS-spz(FL) #2, n=10; da>UAS-Dif, n=16). Lethality of UAS-Dif and UAS-Toll10B combinations at and above 18°C prevented a similar larval analysis. Dots in each plot indicate results for individual larval fillet samples. (J) Mean±s.e.m effectiveness of RNAi knockdown of cact transcripts through cact RNAi #3 determined using qPCR. RNAi phenotypes of additional cact RNAi lines tested can be found in Fig. S5. P-values determined via Kruskal–Wallis statistical test; ***P<0.001; ****P<0.0001; n.s., not significant. P-values for comparisons to the sensitized background (fonΔ24; da>yw) alone are placed above each RNAi line. P-values of comparisons between RNAi alone and in combination with the sensitized background are denoted in parentheses. Scale bars: 500 µm.
We first tested the effects of knocking down cactus (cact), as elongated pupal phenotypes present in cact mutants are similar to those observed in fon mutants (Green et al., 2016; Letsou et al., 1991). The NF-κB inhibitor, cact, is responsible for sequestering the transcription factors, Dif and Dl, in the cytoplasm until Toll signal transduction proceeds. When cact is ubiquitously knocked down using RNAi, muscles remain intact (Fig. 5B,I). Knocking down cact in the fon-sensitized background causes dramatic, widespread muscle detachment denoting a genetic interaction between fon and cact (Fig. 5F,I). We note that only small decreases in cact transcripts (Fig. 5J) are necessary to induce muscle detachment as knockdown of cact at 29°C in the fon-sensitized background results in lethality. As verification that this effect was due to knockdown of cact transcripts and not off-target effects, we observed the expected melanization and Toll activation phenotypes upon tissue-specific RNAi knockdown (Fig. S5) (Lemaitre et al., 1995; Qiu et al., 1998). Overexpression of Dl in the fon-sensitized background results in low levels of muscle detachment, although this trend is not statistically significant (Fig. 5G,I). Dif and Dl have been shown to play redundant roles in larval immune tissues that could account for the minimal effect exerted by overexpression of Dl (Lemaitre et al., 1995; Rutschmann et al., 2000). However, genetic interactions between fon and two important pathway members, Dif and Tl, could not be determined due to lethality of these crosses at temperatures as low as 18°C (Fig. 5I).
We also investigated genes involved in upstream activation events that take place in the extracellular space for ligand-receptor binding. Spatzle-processing enzyme (SPE) is a protease required for cleavage of the proenzyme, pro-Spatzle, into its active form (Spz), which binds to Toll to initiate signal transduction (Mulinari et al., 2006). Overexpression of SPE alone is not sufficient to cause muscle detachment (Fig. 5D,I). However, SPE overexpression in a heterozygous fon background disrupts muscle detachment (Fig. 5H,I). These results show that fon functions with members of the Toll pathway to mitigate muscle damage via detachment.
The major output of Toll signaling is the expression of AMPs, which eliminate pathogens through mechanisms that are presently not understood (Lemaitre and Hoffmann, 2007). AMP overexpression has recently been shown to be a driving force in Drosophila neurodegeneration (Cao et al., 2013). We reasoned that a potential mechanism for disrupting muscle tissue could come from the constitutive expression of AMPs as a result of systemic Toll activation. Using the same sensitized background approach, we examined the effects of overexpressing specific AMPs on muscle architecture. We tested a subset of AMPs including Drs, Metchnikowin (Metch, also known as Mtk), and Drosocin (Dro), selected to represent expression that encompasses Toll and/or Imd signaling. Although the ubiquitous overexpression of these AMPs did not significantly enhance muscle detachment, we did see the appearance of hypercontracted regions within the muscle when AMPs were overexpressed in a fon-sensitized background (Fig. S6). We then overexpressed two of these AMPs in either neural (C155-GAL4) or muscle (mef2-GAL4) tissue during larval stages. This tissue-specific overexpression of AMPs was not sufficient to significantly cause muscle hypercontraction, although there were populations of analyzed muscle fillets that contained large amounts of hypercontracted muscles (Fig. S6). Although these data suggest that AMPs could contribute to muscle defects, overexpression of AMPs alone is not sufficient to drive muscle damage.
JAK-STAT and Toll signaling are coordinated during muscle stress
Having identified a breadth of immune responses at both the local and systemic level, we wanted to understand how muscle and fat body cells could communicate to coordinate an efficient overall response to muscle stress. To determine the potential for a reciprocal network between fat body and muscle cells, we chose to ectopically activate JAK-STAT signaling using genetic tools. Constitutively active forms of JAK (hopTum-l) or the Tl receptor (Tl10B and Tl3) are available as both dominant alleles and UAS constructs for ubiquitous or tissue-specific expression, respectively. Using previously described assays to report JAK-STAT (STAT-GFP) or Toll (nuclear Dl translocation) activation, we induced each of these pathways in healthy tissue and examined either fat body or muscle tissue to determine mechanisms of tissue crosstalk (Fig. 6A).
Fig. 6.

JAK-STAT and Toll pathways are activated in a reciprocal signaling network. (A) Schematic showing experimental design used to determine the relationship between Toll and JAK-STAT signaling. When Toll signaling is activated in the fat body, Dl staining concentrates in the nucleus. Activation of JAK-STAT results in increased STAT-GFP expression in both the cytoplasm and nucleus of muscles. (B–E) Impact of JAK-STAT signaling in L3 muscle on systemic Toll signaling. (B) Two hemisegments of wild-type muscle (F-actin, red) with stable attachment sites. (C) The NFκB transcription factor, Dl (Dorsal, green), localizes to the cytoplasm of fat body cells (outline, inset). Nuclei are stained with DAPI (blue). (D) Constitutively active hopTum-l mutants have muscles with no visible defects. (E) Dl translocates into the nucleus of fat body cells following JAK-STAT activation via the hopTum-l mutation (outline, inset). For tissue-specific expression of hopTum-l see Fig. S6. (F–J′) Analysis of JAK-STAT signaling following Toll activation. (F,F′) In wild-type muscles, expression levels of the STAT reporter STAT-GFP (green) are low. (G,G′) Overall activation of the Drosophila JAK allele, hopTum-l, causes STAT-GFP levels to increase in both the cytoplasm and nuclei (arrowheads) of muscle tissue. (H–J′) Constitutively active Toll signaling using the activated Toll allele Tl3 (H,H′) or UAS-Tl10B expressed using a fat body- and salivary gland-specific driver (I,I′) or fat body- and hemocyte-specific driver (J,J′) increases STAT levels in larval muscle. (K–O) Line plot analysis of STAT-GFP fluorescence intensities measured from white lines drawn in panels F′–J′. Green dots on each graph correspond to arrowheads marking nuclei in each image panel. GFP intensity from STAT-GFP control muscle in panel K is overlaid on line plots as a gray-filled profile in panels L–O. Scale bars: 500 µm in B,D; 50 µm in C,E; 100 µm in F–J′.
In hopTum-l larvae, muscles appear similar to wild-type (Fig. 6B,D). When JAK-STAT signaling is activated in all tissues, we observe Dl redistributing from the cytoplasm as seen in wild-type fat bodies (Fig. 6C) into the nucleus coincident with active Toll signaling (Fig. 6E). Next, we wanted to determine whether tissue-specific activation of JAK-STAT could induce fat body-based Toll signaling. Activation of JAK-STAT in fat body, muscle, tendon or hemocytes alone was not sufficient to induce activation of systemic Toll signaling (Fig. S7). Conversely, when Toll signaling is constitutively activated in all larval tissues with the Tl3 allele, STAT-GFP expression increases in damaged muscle in comparison to undamaged muscle and is similar to ubiquitous hopTum-l activation (Fig. 6F–H′,K–M). This effect persists when UAS-Tl10B is expressed using two fat body drivers (Fig. 6I–J′,N,O). These data suggest that JAK-STAT and Toll function in a signaling network involving multiple tissues in response to muscle stress and damage.
DISCUSSION
We have made the unanticipated discovery that innate immune activation occurs upon muscle stress at both the local and systemic levels. First, we observe increases in JAK-STAT signaling upon loss of muscle homeostasis, which has been described in many tissue stresses to act as a local mediator of immune induction and gene expression (Myllymäki and Rämet, 2014; Srinivasan et al., 2016). Our data also shows that specific muscle stresses activate JAK-STAT signaling (Figs 1, 2), further emphasizing that immune pathways are responsive to the physiological states of tissues. We show that muscle detachment caused by loss of fon specifically activates Toll signaling and that this signaling is restricted to the fat body (Fig. 4; Fig. S4). Moreover, our experiments indicate that JAK-STAT signaling is capable of activating Toll in the fat body to drive the systemic immune response and vice versa (Fig. 6). We have compiled these data into a proposed model (Fig. 7) that outlines how JAK-STAT signaling and Toll signaling are used to sense and respond to damage in a complex network involving tissue coordination.
Fig. 7.

Model of damage-based tissue communication. Schematic representation of the tissue communication network that is activated following disruptions to muscle homeostasis. Muscle health depends on muscle integrity and the strong attachment of muscle (red) to tendon (green, asterisk) via ECM interactions. Muscle hypercontraction (Damage 1) and weakened MASs that progress to detachment (Damage 2) generate stress responses that activate local and systemic immune responses. Muscle damage prompts a series of cellular immune responses including hemocyte recruitment and melanization. Locally, JAK-STAT signaling is activated in muscle tissue through the binding of Upd ligands to the Domeless (Dome) receptor to induce expression of immune-responsive genes. Toll signaling is activated in conjunction with the JAK-STAT pathway in a reciprocal network. Upon Toll signal transduction, Dl moves into the nuclei of fat body cells to activate Toll-responsive genes such as the AMP, drosomycin. Other forms of cellular stress in muscle tissue or possibly in coordinating tissues such as epithelium (Damage 3) may coincide with destabilization of muscle attachment and contribute to further loss of muscle maintenance.
Muscle damage results in a broad array of humoral and cellular events, including the recruitment of hemocytes, which are known to secrete a variety of bioactive molecules at wound sites (Krautz et al., 2014). A subset of hemocytes called crystal cells are responsible for secreting PO to activate melanization, a critical step in hardening and stabilizing the clot when melanin polymerizes around the soft clot structure. Hemocyte localization to damaged MASs and the strengthening role of melanization provides solid rationale for the involvement of hemocytes in a stabilizing response at damaged MASs (Fig. 7). Mutations in genes important for hemocyte function or misregulation of PO activity result in the formation of melanotic tumors (Minakhina and Steward, 2006). Interestingly, constitutively active mutants of both Tl and hopscotch (hop) produce the accumulation of melanotic tumors in Drosophila larvae and adults, exposing relationships between the melanotic cascade and two major immune signaling pathways, Toll and JAK-STAT (Harrison et al., 1995; Lemaitre et al., 1995). Furthermore, Toll signaling is required for both hemocyte recruitment and melanization (Schmid et al., 2014). These observations suggest an intricate orchestration of the cellular and humoral systems during the immune response, the details of which remain to be elucidated.
Interesting future directions suggested by our model include dissecting the extracellular events that respond to damage-based signals, identifying secreted ligands, and determining the contribution of hemocytes that link JAK-STAT and Toll signaling following muscle damage. It has previously been shown that the Toll ligand Spz is produced and secreted primarily from hemocytes (Irving et al., 2005). Upd ligands (Upd1–Upd3) necessary for JAK-STAT activation are also secreted by hemocytes in specific circumstances (Yang and Hultmark, 2016; Yang et al., 2015), making these mobile cells a prime candidate for sensing and responding to both muscle and fat body cues through ligand expression (Fig. 7). Skeletal muscle has also been shown to produce Upd ligands during homeostatic communication and may act as a source for long-range secretion (Zhao and Karpac, 2017). At present, our efforts are concentrated on understanding how multiple tissues function in concert to modulate signaling events during immune responses induced by muscle damage.
In a normal infection, the dramatic expression of AMPs targets pathogens, eliminating and deactivating foreign molecules through a variety of destructive mechanisms. When AMPs lack explicit pathogenic targets, the action of AMPs on healthy tissue creates a paradox for innate immune activation during sterile tissue damage (Cao et al., 2013). In the context of muscle damage, AMPs may act on stressed muscle tissue to exacerbate damage, which we see in the appearance of hypercontraction as a result of AMP overexpression (Fig. S6). In Drosophila, excessive levels of AMPs have already been shown to induce neurodegeneration when activated through neural bacterial infections or artificial neuronal-specific expression (Cao et al., 2013). The fact that several key regulatory points in Toll signaling, and possibly the activity of AMPs, intensify muscle detachment in a fon-sensitized background seems to suggest that prolonged periods of systemic immune signaling can act as an additional source of cellular stress in already damaged tissue (Damage 3 in Fig. 7).
It is consistent that broad-scale injuries induced during live dissection or caused by intentional wounding produced a robust increase in STAT-GFP expression similar to muscle detachment (Fig. 1; Fig. S1). Although a less invasive and aggressive form of wounding via an external pinch wound produced only low levels of JAK-STAT signaling or hemocyte recruitment (Fig. S3), this type of injury sufficiently activated Toll signaling (Fig. S3), which suggests differential mechanisms for Toll activation. This is supported by work demonstrating activation of the larval humoral response following a gentler method of pinch wounding, which was shown to be independent of hemocytes (Kenmoku et al., 2017). It should be noted that the overall robustness of immune responses following muscle stress are attenuated in comparison to those observed during infection. One explanation for this difference could be the urgency required to eradicate invading pathogens, which is not as imperative in sterile tissue damage.
It was surprising that muscle detachment elicited such a strong increase in JAK-STAT signaling, as muscle degeneration had no detectable effect on reporter expression (Fig. 1). Distinct differences in the pathology underlying each type of muscle stress or damage may be related to the degree to which tissue integrity is disrupted and/or the nature of molecules released into the extracellular environment. There is evidence that the manner of cell death (necrosis or apoptosis) releases different subsets of cellular signals (Kolb et al., 2017). Subtle details in these differences may help to explain why programmed cell death fails to activate immunity during development, while injuries and subsequent tissue stress later in life induce robust immune responses (Pradeu and Cooper, 2012).
Models for initiating innate immune responses have been proposed through either pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs). Examples of ‘damage-based’ innate immune activation can be found throughout the invertebrate and vertebrate literature (for reviews see Chen and Nuñez, 2010; Kono et al., 2014; Shaukat et al., 2015). We show that both muscle detachment and systemic immune activation are pathogen-independent, which could implicate the release of a DAMP as part of this tissue network. More recently, efforts to understand DAMP-based immune activation have focused on the identification of molecules capable of initiating immune signaling upon tissue damage. Examples of identified DAMPs include intracellular components such as chromatin, nucleotides (ATP), ROS, cytoskeletal components (Mhc, actin), or fragments of the ECM or basement membrane that are released and recognized as foreign to activate immune pathways, often through Toll-like receptor (TLR) signaling (Kono et al., 2014). Complementary studies in Drosophila and vertebrates showed that F-actin released during tissue damage is sufficient to elicit an immune response (Ahrens et al., 2012; Srinivasan et al., 2016). Torn and leaky membranes characteristic of muscle damage in chronic diseases present opportunities for investigations that integrate DAMP release and the immune phenotypes noted in clinical descriptions. The large arsenal of genetic and molecular tools in Drosophila will continue to prove useful in the identification of novel conserved DAMPs.
Precedence for mechanical damage activating NF-κB pathways was previously reported in the mdx mouse model of Duchenne muscular dystrophy (DMD) and in patients with a variety of muscle diseases including DMD, skeletal muscle atrophy and cachexia-induced muscle wasting (Acharyya et al., 2007; Evans et al., 2009; Kumar and Boriek, 2003; Li et al., 2008; Messina et al., 2011; Monici et al., 2003; Mourkioti and Rosenthal, 2008; Peterson and Guttridge, 2008; Peterson et al., 2011). In addition to Toll acting as the main signaling pathway in many types of infections in invertebrates, mitigating vertebrate TLR signaling implicated in the pathology of myositis and inflammatory myopathies is being explored as a therapeutic intervention (Rayavarapu et al., 2013; Tournadre and Miossec, 2013). Drosophila has yielded many insights into vertebrate TLR immune response, which garnered a Nobel Prize in 2011 (Lemaitre et al., 1996). Because of the largely conserved nature of Toll signaling between Drosophila and humans (Buchon et al., 2014; Valanne et al., 2011), determining the relationship between tissue stress and Toll signaling could have benefits in defining new methods of immune activation and in providing novel perspectives on pathological conditions.
MATERIALS AND METHODS
Fly genetics
Flies were raised on standard cornmeal medium at 25°C unless otherwise specified. The control stock used in all experiments was w1118. Two fon null alleles, fonΔ17 and fonΔ24 (Bajzek et al., 2012) were used to remove fon and paired with the deficiency stock, Df(2L)Exel6043. Other mutant alleles used in experiments were BrkdJ29 (a gift from Troy Littleton, Massachusetts Institute of Biology, Cambridge, MA, USA), hopTum-l (Bloomington Drosophila Stock Center, BL-8492), MhcS1 (a gift from Troy Littleton), and Tl3 (BL-3338). The following GAL4 lines were used to direct tissue-specific expression: da-GAL4 (originally BL-37291 outcrossed ten times to w1118 to remove background lethals), C155-GAL4 (BL-458), Cg-GAL4 (BL-7011), mef2-GAL4 (BL-27390), sr-GAL4, UAS-CD8-GFP/TM6 (a gift from Talila Volk, Weizmann Institute of Science, Rehovot, Israel), hml-GAL4, UAS-2xEGFP (BL-30140) and ppl-GAL4 (a gift from Len Dobens, University of Missouri-Kansas City, Kansas City, MO, USA). Stocks analyzed in screens include UAS-cat (BL-24621), UAS-Dif (BL-22201), UAS-dl (BL-9319), UAS-Dro (a gift from David Wassarman, University of Wisconsin-Madison, Madison, WI, USA), UAS-Drs (a gift from David Wassarman), UAS-hopTum-l (a gift from Michelle Starz-Gaiano, University of Maryland-Baltimore County, Baltimore, MD, USA), UAS-Metch (a gift from David Wassarman), UAS-Sod1 (BL-33605), UAS-SPE [a gift from Won-Jae Lee; (Jang et al., 2006)], UAS-spz(FL) #1 (a gift from Tony Ip, University of Massachusetts Medical School, Worcester, MA, USA), UAS-spz(FL) #2 (a gift from Tony Ip), UAS-Toll10B (BL-58987), UAS-cact RNAi #1 (BL-31713), UAS-cact RNAi #2 (BL-34775), UAS-cact RNAi #3 (BL-37484), UAS-cat RNAi (BL-34020), UAS-Sod1 RNAi (BL-22491), UAS-stck RNAi (BL-31536), UAS-Tfam RNAi (BL-17644), UAS-Tig RNAi (BL-31570; RNAi validation in Green et al., 2016), UAS-tn RNAi (BL-31588; RNAi validation in Brooks et al., 2016), and UAS-park RNAi (BL-38333; RNAi validation in Brooks et al., 2016). The reporter stock used in these experiments was 10xSTAT92E-GFP (BL-26197).
Mutant alleles and genetic constructs were maintained over the appropriate balancer chromosome: FM7C (I), Cyo-Act-GFP or Cyo, Tb (II), or TM6, Tb (III). Individuals were chosen by selection against the Tb or GFP marker for second and third chromosome crosses or gender in first chromosome balanced alleles. All temperature-dependent crosses were performed at 29°C with the following exceptions: (1) crosses involving temperature-sensitive alleles BrkdJ29 and MhcS1 were raised at 29°C and heat shocked for 1 h at 37°C immediately before dissection; (2) to bypass embryonic development, hopTum-l and UAS- hopTum-l crosses were shifted from 18°C to the permissive temperature at 29°C following embryogenesis; (3) crosses for the muscle-specific RNAi knockdown of stck were shifted from 18°C to 29°C at L1 to overcome lethality in embryogenesis; (4) experiments using cact RNAi #3 were performed at 25°C to avoid early lethality in combination with the fon-sensitized background. Other cact RNAi stocks (#1 and #2) were analyzed at 29°C.
Immunostaining and microscopy
Wandering L3 larvae were dissected to either retain fat body tissue or isolate muscle fillets, and the tissue fixed in 4% formaldehyde. All dissections were performed on larvae which were heat-killed, chilled and dissected in ice-cold PBS, except for larvae where the effects of mechanical damage on muscle were examined or to test the effect of live dissection on STAT-GFP levels (Fig. S1). To induce mechanical damage to muscle tissue, larvae were dissected live in HL3 buffer and forceps were used to tear muscles along the middle of a hemisegment on both halves of the fillet, being careful to avoid damage to other tissues. Dissections were left unfixed with all tissues intact for 15–30 min before resuming standard fixation and staining protocols. Extraneous tissues were removed after fixation for the visualization of muscles. Crush-based trauma was performed by gently compressing and rolling L3 larvae between two microscope slides on a level surface to create uniform stress to the body cavity, pinch wounding was performed using forceps (Burra et al., 2013) with care taken to preserve cuticle integrity but to encompass a large enough area to induce damage to the muscle layer. Larvae were dissected 2 h after pinch wounding and 4–6 h post-wound to assess STAT-GFP activation.
Tissues were stained with the following primary antibodies: mouse anti-Dl [1:200; 7A4, Developmental Studies Hybridoma Bank (DSHB)], rabbit anti-Hemese (1:1000; a gift from Dan Hultmark, Umeå University, Umeå, Sweden), and mouse anti-Relish-C (1:400; 21F3, DSHB). Fluorescence was detected using the following secondary antibodies: Alexa Fluor anti-mouse 488 and Alexa Fluor anti-rabbit 488 (1:400; A-11001, A-11034, Molecular Probes). F-actin was labeled with phalloidin 488, 594 or 647 (1:400; A12379, A22283, A22287, Molecular Probes). Images were captured using a Zeiss 700 confocal microscope. Image processing and analysis was performed using a combination of Zen Black (Zeiss), ImageJ (NIH) and Adobe Photoshop.
qPCR analysis
Transcript levels were assessed using quantitative PCR (qPCR) to verify RNAi knockdown and to compare gene expression amongst genotypes. Total RNA was collected in triplicate from individual wandering L3 larvae using the RNeasy Mini Kit (Qiagen). Fat body-specific RNA was obtained by isolating the fat bodies of five L3 larvae and homogenizing isolated tissues in ice-cold RLT buffer (Qiagen). Synthesis of cDNA from 125 ng RNA was performed using the qScript XLT cDNA Supermix kit (Quantabio). Dilutions of cDNA were optimized according to each primer set and combined with PowerUp SYBR Green Master Mix (Thermo Fisher). The following primers and cDNA dilutions were used: rp49 forward 5′-GCCCAAGGGTATCGACAACA-3′, reverse 3′-GCGCTTGTTCGATCCGTAAC-5′ (1:50) (generated via FlyPrimer Bank; Hu et al., 2013); Drs forward 5′-CCCTCTTCGCTGTCCTGA-3′, reverse 3′-GCGTCCCTCCTCCTTGC-5′ (1:50) (Deng et al., 2009); cact forward 5′-CTCACTAGCCACTAGCGGTAA-3′, reverse 3′-CCCGAATCACTGGTTTCGTTT-5′ (1:50) (Wang et al., 2015); and dpt forward 5′-ATTGGACTGAATGGAGGATATGG-3′, reverse 3′-CGGAAATCTGTAGGTGTAGGT-5′ (Chatterjee et al., 2016). All primers were synthesized at Integrated DNA Technologies (IDT). Quantitative transcript levels were obtained using the 2-ΔΔCt method and graphed as mean±s.e.m. using GraphPad Prism 6.0.
Phenotypic quantification and statistical analysis
Detachment
Images were quantified as described (Green et al., 2016). Muscles were considered detached if they had rounded up following detachment or were beginning to strip away from the attachment site. Percent detachment was calculated by dividing the number of hemisegments containing one or more detached muscles by the total number of hemisegments within the fillet. These percentages were compiled in GraphPad Prism 6.0 and graphically represented as a dot plot.
Hypercontraction
Muscles containing differentially compressed regions of sarcomeres were scored as ‘hypercontracted’ as previously defined (Montana and Littleton, 2004; Montana and Littleton, 2006). Percent hypercontraction was calculated in the same manner as percent detachment, input into GraphPad Prism 6.0 and graphed as mean±s.d. dot plots.
Line plot intensities
All images within a particular analysis were taken using the same power and gain configurations. Fluorescence intensities of both STAT-GFP and phalloidin were analyzed along a single line across the length of ventral longitudinal muscles using the plot profile feature in ImageJ. Line plots of hypercontracted muscles were taken to illustrate the spacing of sarcomeres that correspond to the peaks and valleys of phalloidin staining. For STAT-GFP expression comparisons, axis range was set to best fit the genotype plot with the most signal intensity. Control plot profiles were traced and overlaid on experiment plots to highlight differences in plot intensities. Note that control STAT-GFP muscles in both Figs 1 and 2 contain two copies of the STAT-GFP reporter as well as the fonΔ17 allele (Fig. 2; Fig. S2), which was recombined with the STAT-GFP reporter and analyzed as a homozygote. Larvae with the following genetic manipulations contain a single copy of the STAT-GFP reporter: RNAi lines (Fig. 1; Fig. S1), dominant alleles (Figs 2, 6), and overexpression constructs (Fig. 1; Figs S1, S6).
Statistical analysis
Statistical analyses were performed in GraphPad Prism 6.0 using the Kruskal–Wallis test to analyze non-Gaussian distributions of three or more unmatched genotypes. Significance values are listed for each quantification within the figure legend.
DOPA incubation
Larvae were live dissected and washed in cold PBS. Muscle fillets were then incubated in L-DOPA (Cayman Chemical) solution (60 mM dissolved in PBS) for 1 h at 25°C in the dark to allow melanization to proceed. Fillets were then washed and fixed in 4% formaldehyde and stained with phalloidin (1:400, Molecular Probes) to determine muscle morphology at points of melanization. Images were taken on a Nikon 80i and processed using ImageJ (NIH) and Adobe Photoshop.
Axenic conditions
Axenic larvae were generated using the protocol generated by Sabat et al. (Sabat et al., 2015). In a sterilized hood, embryos were dechorionated and sterilized using bleach and 70% ethanol solutions. Sterilized embryos were transferred to autoclaved food vials until individuals matured to the larval stage. Larvae were then collected and dissected as described above. The microbe status of axenic lines was analyzed by means of growing overnight cultures on Luria broth (LB) containing larval lysates from either normal or axenic conditions. No bacterial growth was observed in axenic lines compared to an LB only control, whereas larval lysates not sterile and grown on normal food showed obvious bacterial growth.
Supplementary Material
Acknowledgements
We thank Len Dobens, Dan Hultmark, Tony Ip, Won-Jae Lee, Troy Littleton, Michelle Starz-Gaiano, Talila Volk and David Wassarman for contributing reagents; David Brooks for reviewing of the manuscript; and Michael Kanost, Maureen Gorman and Neal Dittmer for valuable consultation on immunity experiments. We also thank the Bloomington Drosophila Stock Center for stocks used in this study and the Developmental Studies Hybridoma Bank, created by the National Institute of Child Health and Human Development of the National Institutes of Health (NIH) and maintained at the University of Iowa for monoclonal antibodies.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: N.G., E.R.G.; Methodology: N.G., E.R.G.; Validation: N.G.; Formal analysis: N.G.; Investigation: N.G., J.W., A.B., M.Z.; Resources: E.R.G.; Writing - original draft: N.G., E.R.G.; Writing - review & editing: N.G., E.R.G.; Visualization: N.G., E.R.G.; Supervision: E.R.G.; Project administration: E.R.G.; Funding acquisition: E.R.G.
Funding
We acknowledge funding from the National Science Foundation for a GK-12 award to N.G. (0841414) and from the National Institutes of Health for RO1 (AR060788) and K-INBRE (GM103418) grants to E.R.G. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.217943.supplemental
References
- Acharyya S., Villalta S. A., Bakkar N. Bupha-Intr T., Janssen P. M. L., Carathers M., Li Z.-W., Beg A. A., Ghosh S., Sahenk Z. et al. (2007). Interplay of IKK/ NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J. Clin. Invest. 117, 889-901. 10.1172/JCI30556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrens S., Zelenay S., Sancho D., Hanč P., Kjær S., Feest C., Fletcher G., Durkin C., Postigo A., Skehel M. et al. (2012). F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 36, 635-645. 10.1016/j.immuni.2012.03.008 [DOI] [PubMed] [Google Scholar]
- Bach E. A., Ekas L. A., Ayala-Camargo A., Flaherty M. S., Lee H., Perrimon N. and Baeg G.-H. (2007). GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene Expr. Patterns 7, 323-331. 10.1016/j.modgep.2006.08.003 [DOI] [PubMed] [Google Scholar]
- Bajzek C., Rice A. M., Andreazza S. and Dushay M. S. (2012). Coagulation and survival in Drosophila melanogaster fondue mutants. J. Insect Physiol. 58, 1376-1381. 10.1016/j.jinsphys.2012.07.013 [DOI] [PubMed] [Google Scholar]
- Bergmann A., Stein D., Geisler R., Hagenmaier S., Schmid B., Fernandez N., Schnell B. and Nüsslein-Volhard C. (1996). A gradient of cytoplasmic Cactus degradation establishes the nuclear localization gradient of the dorsal morphogen in Drosophila. Mech. Dev. 60, 109-123. 10.1016/S0925-4773(96)00607-7 [DOI] [PubMed] [Google Scholar]
- Bessou C., Giugia J.-B., Franks C. J., Holden-Dye L. and Ségalat L. (1998). Mutations in the Caenorhabditis elegans dystrophin-like gene dys-1 lead to hyperactivity and suggest a link with cholinergic transmission. Neurogenetics 2, 61-72. 10.1007/s100480050053 [DOI] [PubMed] [Google Scholar]
- Binggeli O., Neyen C., Poidevin M. and Lemaitre B. (2014). Prophenoloxidase activation is required for survival to microbial infections in Drosophila. PLoS Path. 10, e1004067 10.1371/journal.ppat.1004067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A. H. and Perrimon N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401-415. [DOI] [PubMed] [Google Scholar]
- Brooks D. S., Vishal K., Kawakami J., Bouyain S. and Geisbrecht E. R. (2016). Optimization of wrMTrck to monitor Drosophila larval locomotor activity. J. Insect. Physiol. 93-94, 11-17. 10.1016/j.jinsphys.2016.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchon N., Silverman N. and Cherry S. (2014). Immunity in Drosophila melanogaster – from microbial recognition to whole-organism physiology. Nat. Rev. Immunol. 14, 796-810. 10.1038/nri3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner G. R., Ng C. F., Wang M., Rodgers V. G. J. and Schafer F. Q. (2006). A new paradigm: manganese superoxide dismutase influences the production of H2O2 in cells and thereby their biological state. Free Radic. Biol. Med. 41, 1338-1350. 10.1016/j.freeradbiomed.2006.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunch T. A., Graner M. W., Fessler L. I., Fessler J. H., Schneider K. D., Kerschen A., Choy L. P., Burgess B. W. and Brower D. L. (1998). The PS2 integrin ligand tiggrin is required for proper muscle function in Drosophila. Development 125, 1679-1689. [DOI] [PubMed] [Google Scholar]
- Burra S., Wang Y., Brock A. R. and Galko M. J. (2013). Using Drosophila larvae to study epidermal wound closure and inflammation. Methods Mol. Biol. (Clifton, N.J.) 2013, 449-461. 10.1007/978-1-62703-505-7_26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabreiro F., Ackerman D., Doonan R., Araiz C., Back P., Papp D., Braeckman B. P. and Gems D. (2011). Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radic. Biol. Med. 51, 1575-1582. 10.1016/j.freeradbiomed.2011.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., Chtarbanova S., Petersen A. J. and Ganetzky B. (2013). Dnr1 mutations cause neurodegeneration in Drosophila by activating the innate immune response in the brain. Proc. Natl. Acad. Sci. USA 110, E1752 10.1073/pnas.1306220110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A., Roy D., Patnaik E. and Nongthomba U. (2016). Muscles provide protection during microbial infection by activating innate immune response pathways in Drosophila and zebrafish. Dis. Model. Mech. 9, 697-705. 10.1242/dmm.022665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G. Y. and Nuñez G. (2010). Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826-837. 10.1038/nri2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien K. R., Knowlton K. U., Zhu H. and Chien S. (1991). Regulation of cardiac gene expression during myocardial growth and hypertrophy: molecular studies of an adaptive physiologic response. FASEB J. 5, 3037-3046. 10.1096/fasebj.5.15.1835945 [DOI] [PubMed] [Google Scholar]
- Clark K. A., McGrail M. and Beckerle M. C. (2003). Analysis of PINCH function in Drosophila demonstrates its requirement in integrin-dependent cellular processes. Development 130, 2611-2621. 10.1242/dev.00492 [DOI] [PubMed] [Google Scholar]
- De Gregorio E., Spellman P. T., Tzou P., Rubin G. M. and Lemaitre B. (2002). The Toll and Imd pathways are the major regulators of the immune response in Drosophila. EMBO J. 21, 2568-2579. 10.1093/emboj/21.11.2568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.-J., Yang W.-Y., Huang Y.-D., Cao Y., Wen S.-Y., Xia Q.-Y. and Xu P. (2009). Gene expression divergence and evolutionary analysis of the Drosomycin gene family in Drosophila melanogaster. J. Biomed. Biotechnol. 2009, 1-9. 10.1155/2009/315423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans I. R. and Wood W. (2014). Drosophila blood cell chemotaxis. Curr. Opin. Cell Biol. 30, 1-8. 10.1016/j.ceb.2014.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans N. P., Misyak S. A., Robertson J. L., Bassaganya-Riera J. and Grange R. W. (2009). Immune-mediated mechanisms potentially regulate the disease time-course of Duchenne muscular dystrophy and provide targets for therapeutic intervention. PM&R 1, 755-768. 10.1016/j.pmrj.2009.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabio D., Rosanna P., Alfred L. G. and Norbert P. (2013). Mechanisms of skeletal muscle aging: insights from Drosophila and mammalian models. Dis. Model. Mech. 6, 1339-1352. 10.1242/dmm.012559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green N., Odell N., Zych M., Clark C., Wang Z.-H., Biersmith B., Bajzek C., Cook K. R., Dushay M. S. and Geisbrecht E. R. (2016). A common suite of coagulation proteins function in Drosophila muscle attachment. Genetics 204, 1075-1087. 10.1534/genetics.116.189787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene J. C., Whitworth A. J., Kuo I., Andrews L. A., Feany M. B. and Pallanck L. J. (2003). Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 100, 4078-4083. 10.1073/pnas.0737556100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisoni K., Martin E., Gieseler K., Mariol M.-C. and Ségalat L. (2002). Genetic evidence for a dystrophin-glycoprotein complex (DGC) in Caenorhabditis elegans. Gene 294, 77-86. 10.1016/S0378-1119(02)00762-X [DOI] [PubMed] [Google Scholar]
- Harrison D. A., Binari R., Nahreini T. S., Gilman M. and Perrimon N. (1995). Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J. 14, 2857-2865. 10.1002/j.1460-2075.1995.tb07285.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslett J. N., Sanoudou D., Kho A. T., Bennett R. R., Greenberg S. A., Kohane I. S., Beggs A. H. and Kunkel L. M. (2002). Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc. Natl. Acad. Sci. USA 99, 15000-15005. 10.1073/pnas.192571199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hathout Y., Marathi R. L., Rayavarapu S., Zhang A., Brown K. J., Seol H., Gordish-Dressman H., Cirak S., Bello L., Nagaraju K. et al. (2014). Discovery of serum protein biomarkers in the mdx mouse model and cross-species comparison to Duchenne muscular dystrophy patients. Hum. Mol. Genet. 23, 6458-6469. 10.1093/hmg/ddu366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetru C. and Hoffmann J. A. (2009). NF- kappaB in the immune response of Drosophila. Cold Spring Harb. Perspect. Biol. 1, a000232 10.1101/cshperspect.a000232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz A., Bossinger B., Strasser T., Janning W. and Klapper R. (2003). The two origins of hemocytes in Drosophila. Development 130, 4955-4962. 10.1242/dev.00702 [DOI] [PubMed] [Google Scholar]
- Hu Y., Sopko R., Foos M., Kelley C., Flockhart I., Ammeux N., Wang X., Perkins L., Perrimon N. and Mohr S. E. (2013). FlyPrimerBank: an online database for Drosophila melanogaster gene expression analysis and knockdown evaluation of RNAi reagents. G3 (Bethesda) 3, 1607-1616. 10.1534/g3.113.007021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving P., Ubeda J.-M., Doucet D., Troxler L., Lagueux M., Zachary D., Hoffmann J. A., Hetru C. and Meister M. (2005). New insights into Drosophila larval haemocyte functions through genome-wide analysis. Cell. Microbiol. 7, 335-350. 10.1111/j.1462-5822.2004.00462.x [DOI] [PubMed] [Google Scholar]
- Jang I.-H., Chosa N., Kim S.-H., Nam H.-J., Lemaitre B., Ochiai M., Kambris Z., Brun S., Hashimoto C., Ashida M. et al. (2006). A spätzle-processing enzyme required for toll signaling activation in Drosophila Innate Immunity. Dev. Cell 10, 45-55. 10.1016/j.devcel.2005.11.013 [DOI] [PubMed] [Google Scholar]
- Kenmoku H., Hori A., Kuraishi T. and Kurata S. (2017). A novel mode of induction of the humoral innate immune response in larvae. Dis. Model. Mech. 10, 271 10.1242/dmm.027102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb J. P., Oguin T. H., Oberst A. and Martinez J. (2017). Programmed cell death and inflammation: winter is coming. Trends Immunol. 38, 705-718. 10.1016/j.it.2017.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H., Onda A. and Yanagida T. (2014). Molecular determinants of sterile inflammation. Curr. Opin. Immunol. 26, 147-156. 10.1016/j.coi.2013.12.004 [DOI] [PubMed] [Google Scholar]
- Krautz R., Arefin B. and Theopold U. (2014). Damage signals in the insect immune response. Front. Plant Sci. 5, 342 10.3389/fpls.2014.00342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A. and Boriek A. M. (2003). Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: a possible role in Duchenne muscular dystrophy. FASEB J. 17, 386-396. 10.1096/fj.02-0542com [DOI] [PubMed] [Google Scholar]
- Larsson N.-G., Wang J., Wilhelmsson H., Oldfors A., Rustin P., Lewandoski M., Barsh G. S. and Clayton D. A. (1998). Mitochondrial transcription factor A is necessary for mtDNA maintance and embryogenesis in mice. Nat. Genet. 18, 231-236. 10.1038/ng0398-231 [DOI] [PubMed] [Google Scholar]
- Lee J.-H., Lee C.-W., Park S.-H. and Choe K.-M. (2017). Spatiotemporal regulation of cell fusion by JNK and JAK/STAT signaling during wound healing. J. Cell Sci. 130, 1917 10.1242/jcs.187658 [DOI] [PubMed] [Google Scholar]
- Lemaitre B. and Hoffmann J. (2007). The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 25, 697-743. 10.1146/annurev.immunol.25.022106.141615 [DOI] [PubMed] [Google Scholar]
- Lemaitre B., Meister M., Govind S., Georgel P., Steward R., Reichhart J. M. and Hoffmann J. A. (1995). Functional analysis and regulation of nuclear import of dorsal during the immune response in Drosophila. EMBO J. 14, 536-545. 10.1002/j.1460-2075.1995.tb07029.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre B., Nicolas E., Michaut L., Reichhart J.-M. and Hoffmann J. A. (1996). The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86, 973-983. 10.1016/S0092-8674(00)80172-5 [DOI] [PubMed] [Google Scholar]
- Lemaitre B., Reichhart J.-M. and Hoffmann J. (1997). Drosophila host defense: differential induction of antimicrobial peptide genes after infection by various classes of microorganisms. Proc. Natl. Acad. Sci. USA 94, 14614-14619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letsou A., Alexander S., Orth K. and Wasserman S. A. (1991). Genetic and molecular characterization of tube, a Drosophila gene maternally required for embryonic dorsoventral polarity. Proc. Natl. Acad. Sci. USA 88, 810-814. 10.1073/pnas.88.3.810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Malhotra S. and Kumar A. (2008). Nuclear factor-kappa B signaling in skeletal muscle atrophy. J. Mol. Med. 86, 1113-1126. 10.1007/s00109-008-0373-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren M., Riazi R., Lesch C., Wilhelmsson C., Theopold U. and Dushay M. S. (2008). Fondue and transglutaminase in the Drosophila larval clot. J. Insect. Physiol. 54, 586-592. 10.1016/j.jinsphys.2007.12.008 [DOI] [PubMed] [Google Scholar]
- Madaro L. and Bouché M. (2014). From innate to adaptive immune response in muscular dystrophies and skeletal muscle regeneration: the role of lymphocytes. BioMed. Res. Int. 2014, 1-12. 10.1155/2014/438675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhijani K., Alexander B., Tanaka T., Rulifson E. and Brückner K. (2011). The peripheral nervous system supports blood cell homing and survival in the Drosophila larva. Development 138, 5379-5391. 10.1242/dev.067322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Márkus R., Kurucz É., Rus F. and Andó I. (2005). Sterile wounding is a minimal and sufficient trigger for a cellular immune response in Drosophila melanogaster. Immunol. Lett. 101, 108-111. 10.1016/j.imlet.2005.03.021 [DOI] [PubMed] [Google Scholar]
- Messina S., Vita G. L., Aguennouz M., Sframeli M., Romeo S., Rodolico C. and Vita G. (2011). Activation of NF- kappaB pathway in Duchenne muscular dystrophy: relation to age. Acta Myol. 30, 16-23. [PMC free article] [PubMed] [Google Scholar]
- Minakhina S. and Steward R. (2006). Melanotic mutants in Drosophila: pathways and phenotypes. Genetics 174, 253-263. 10.1534/genetics.106.061978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monici M. C., Aguennouz M., Mazzeo A., Messina C. and Vita G. (2003). Activation of nuclear factor-κB in inflammatory myopathies and Duchenne muscular dystrophy. Neurology 60, 993-997. 10.1212/01.WNL.0000049913.27181.51 [DOI] [PubMed] [Google Scholar]
- Montana E. S. and Littleton J. T. (2004). Characterization of a hypercontraction-induced myopathy in Drosophila caused by mutations in Mhc. J. Cell Biol. 164, 1045-1054. 10.1083/jcb.200308158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montana E. S. and Littleton J. T. (2006). Expression profiling of a hypercontraction-induced myopathy in Drosophila suggests a compensatory cytoskeletal remodeling response. J. Biol. Chem. 281, 8100-8109. 10.1074/jbc.M512468200 [DOI] [PubMed] [Google Scholar]
- Mourkioti F. and Rosenthal N. (2008). NF-κB signaling in skeletal muscle: prospects for intervention in muscle diseases. J. Mol. Med. 86, 747-759. 10.1007/s00109-008-0308-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulinari S., Häcker U. and Castillejo - López C. (2006). Expression and regulation of Spätzle-processing enzyme in Drosophila. FEBS Lett. 580, 5406-5410. 10.1016/j.febslet.2006.09.009 [DOI] [PubMed] [Google Scholar]
- Myers C. D., Goh P.-Y., Allen T. S., Bucher E. A. and Bogaert T. (1996). Developmental genetic analysis of troponin T mutations in striated and nonstriated muscle cells of Caenorhabditis elegans. J. Cell Biol. 132, 1061-1077. 10.1083/jcb.132.6.1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myllymäki H. and Rämet M. (2014). JAK/STAT Pathway in Drosophila Immunity. Scand. J. Immunol. 79, 377-385. 10.1111/sji.12170 [DOI] [PubMed] [Google Scholar]
- Nitahara-Kasahara Y., Takeda S. and Okada T. (2016). Inflammatory predisposition predicts disease phenotypes in muscular dystrophy. Inflamm. Regen. 36, 14 10.1186/s41232-016-0019-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nongthomba U., Clark S., Cummins M., Ansari M., Stark M. and Sparrow J. C. (2004). Troponin I is required for myofibrillogenesis and sarcomere formation in Drosophila flight muscle. J. Cell Sci. 117, 1795-1805. 10.1242/jcs.01024 [DOI] [PubMed] [Google Scholar]
- Nongthomba U., Ansari M., Thimmaiya D., Stark M. and Sparrow J. (2007). Aberrant splicing of an alternative exon in the Drosophila troponin-T gene affects flight muscle development. Genetics 177, 295-306. 10.1534/genetics.106.056812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson J. M. and Guttridge D. C. (2008). Skeletal muscle diseases, inflammation, and NF-kappa B signaling: Insights and opportunities for therapeutic intervention. Int. Rev. Immunol. 27, 375-387. 10.1080/08830180802302389 [DOI] [PubMed] [Google Scholar]
- Peterson J. M., Bakkar N. and Guttridge D. C. (2011). NF-κB signaling in skeletal muscle health and disease. Curr. Top. Dev. Biol. 96, 85-119. 10.1016/B978-0-12-385940-2.00004-8 [DOI] [PubMed] [Google Scholar]
- Pradeu T. and Cooper E. L. (2012). The danger theory: twenty years later. Front. Immunol. 3, 287 10.3389/fimmu.2012.00287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P., Pan P. C. and Govind S. (1998). A role for the Drosophila Toll/ Cactus pathway in larval hematopoiesis. Development 125, 1909-1920. [DOI] [PubMed] [Google Scholar]
- Raghavan S., Williams I., Aslam H., Thomas D., Szöőr B., Morgan G., Gross S., Turner J., Fernandes J., Vijayraghavan K. et al. (2000). Protein phosphatase 1β is required for the maintenance of muscle attachments. Curr. Biol. 10, 269-272. 10.1016/S0960-9822(00)00364-X [DOI] [PubMed] [Google Scholar]
- Rayavarapu S., Coley W., Kinder T. B. and Nagaraju K. (2013). Idiopathic inflammatory myopathies: pathogenic mechanisms of muscle weakness. Skeletal Muscle 3, 13 10.1186/2044-5040-3-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reach M., Galindo R. L., Towb P., Allen J. L., Karin M. and Wasserman S. A. (1996). A gradient of Cactus protein degradation establishes dorsoventral polarity in the Drosophila embryo. Dev. Biol. 180, 353-364. 10.1006/dbio.1996.0308 [DOI] [PubMed] [Google Scholar]
- Rosenberg A. S., Puig M., Nagaraju K., Hoffman E. P., Villalta S. A., Rao V. A., Wakefield L. M. and Woodcock J. (2015). Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 7, 299rv4 10.1126/scitranslmed.aaa7322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royet J., Meister M. and Ferrandon D. (2003). Humoral and cellular responses in Drosophila innate immunity. In Innate Immun. (ed. Ezekowitz R. A. B. and Hoffmann J. A.), pp. 137-153. Totowa, NJ: Humana Press. [Google Scholar]
- Rutschmann S., Jung A. C., Hetru C., Reichhart J.-M., Hoffmann J. A. and Ferrandon D. (2000). The Rel protein DIF mediates the antifungal but not the antibacterial host defense in Drosophila. Immunity 12, 569-580. 10.1016/S1074-7613(00)80208-3 [DOI] [PubMed] [Google Scholar]
- Sabat D., Johnson E. M., Abhinay A., Jayabalan R. and Mishra M. (2015). A protocol to generate germ free Drosophila for microbial interaction studies. Adv. Tech. Biol. Med. S1, 001 10.4172/2379-1764.S1-001 [DOI] [Google Scholar]
- Scherfer C., Qazi M. R., Takahashi K., Ueda R., Dushay M. S., Theopold U. and Lemaitre B. (2006). The Toll immune-regulated Drosophila protein Fondue is involved in hemolymph clotting and puparium formation. Dev. Biol. 295, 156-163. 10.1016/j.ydbio.2006.03.019 [DOI] [PubMed] [Google Scholar]
- Schmid M. R., Anderl I., Vesala L., Vanha-aho L.-M., Deng X.-J., Ramet M. and Hultmark D. (2014). Control of Drosophila blood cell activation via toll signaling in the fat body. PLoS One 9, e102568 10.1371/journal.pone.0102568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaukat Z., Liu D. and Gregory S. (2015). Sterile inflammation in Drosophila. Mediators Inflamm. 2015, 369286 10.1155/2015/369286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan N., Gordon O., Ahrens S., Franz A., Deddouche S., Chakravarty P., Phillips D., Yunus A. A., Rosen M. K., Valente R. S. et al. (2016). Actin is an evolutionarily-conserved damage- associated molecular pattern that signals tissue injury in Drosophila melanogaster. eLife 22, e19662 10.7554/eLife.19662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stofanko M., Kwon S. Y. and Badenhorst P. (2008). A misexpression screen to identify regulators of Drosophila larval hemocyte development. Genetics 180, 253-267. 10.1534/genetics.108.089094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöven S., Ando I., Kadalayil L., Engström Y. and Hultmark D. (2000). Activation of the Drosophila NF-κB factor Relish by rapid endoproteolytic cleavage. EMBO Rep. 1, 347-352. 10.1093/embo-reports/kvd072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H. (2009). Regulation and function of the melanization reaction in Drosophila. Fly 3, 105-111. 10.4161/fly.3.1.7747 [DOI] [PubMed] [Google Scholar]
- Tang H., Kambris Z., Lemaitre B. and Hashimoto C. (2006). Two proteases defining a melanization cascade in the immune system of Drosophila. J. Biol. Chem. 281, 28097-28104. 10.1074/jbc.M601642200 [DOI] [PubMed] [Google Scholar]
- Tidball J. and Villalta S. A. (2010). Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1173-R1187. 10.1152/ajpregu.00735.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournadre A. and Miossec P. (2013). A critical role for immature muscle precursors in myositis. Nat. Rev. Rheumatol. 9, 438-442. 10.1038/nrrheum.2013.26 [DOI] [PubMed] [Google Scholar]
- Valanne S., Wang J.-H. and Ramet M. (2011). The Drosophila toll signaling pathway. J. Immunol. 186, 649-656. 10.4049/jimmunol.1002302 [DOI] [PubMed] [Google Scholar]
- Villalta S. A., Rosenberg A. S. and Bluestone J. A. (2015). The immune system in Duchenne muscular dystrophy: friend or foe. Rare Dis. 23, e1010966 10.1080/21675511.2015.1010966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Kounatidis I. and Ligoxygakis P. (2014). Drosophila as a model to study the role of blood cells in inflammation, innate immunity and cancer. Front. Cell Infect. Microbiol. 3, 113 10.3389/fcimb.2013.00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Wu D., Liu Y., Xia X., Gong W., Qiu Y., Yang J., Zheng Y., Li J., Wang Y.-F. et al. (2015). Drosophila Dicer-2 has an RNA interference-independent function that modulates Toll immune signaling. Sci. Adv. 1, e1500228 10.1126/sciadv.1500228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. and Hultmark D. (2016). Tissue communication in a systemic immune response of Drosophila. Fly 10, 115-122. 10.1080/19336934.2016.1182269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Kronhamn J., Ekström J.-O., Korkut G. G. and Hultmark D. (2015). JAK/ STAT signaling in Drosophila muscles controls the cellular immune response against parasitoid infection. EMBO Reps. 16, 1664-1672. 10.15252/embr.201540277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidman-Rémy A., Regan J. C., Brandão A. S. and Jacinto A. (2012). The Drosophila larva as a tool to study gut-associated macrophages: PI3K regulates a discrete hemocyte population at the proventriculus. Dev. Comp. Immunol. 36, 638-647. 10.1016/j.dci.2011.10.013 [DOI] [PubMed] [Google Scholar]
- Zhao X. and Karpac J. (2017). Muscle directs diurnal energy homeostasis through a myokine-dependent hormone module in Drosophila. Curr. Biol. 27, 1941-1955.e6. 10.1016/j.cub.2017.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.