

Abstract
Background
In genomic studies, to investigate how the structure of a genetic network differs between two experiment conditions is a very interesting but challenging problem, especially in high-dimensional setting. Existing literatures mostly focus on differential network modelling for continuous data. However, in real application, we may encounter discrete data or mixed data, which urges us to propose a unified differential network modelling for various data types.
Results
We propose a unified latent Gaussian copula differential network model which provides deeper understanding of the unknown mechanism than that among the observed variables. Adaptive rank-based estimation approaches are proposed with the assumption that the true differential network is sparse. The adaptive estimation approaches do not require precision matrices to be sparse, and thus can allow the individual networks to contain hub nodes. Theoretical analysis shows that the proposed methods achieve the same parametric convergence rate for both the difference of the precision matrices estimation and differential structure recovery, which means that the extra modeling flexibility comes at almost no cost of statistical efficiency. Besides theoretical analysis, thorough numerical simulations are conducted to compare the empirical performance of the proposed methods with some other state-of-the-art methods. The result shows that the proposed methods work quite well for various data types. The proposed method is then applied on gene expression data associated with lung cancer to illustrate its empirical usefulness.
Conclusions
The proposed latent variable differential network models allows for various data-types and thus are more flexible, which also provide deeper understanding of the unknown mechanism than that among the observed variables. Theoretical analysis, numerical simulation and real application all demonstrate the great advantages of the latent differential network modelling and thus are highly recommended.
Electronic supplementary material
The online version of this article (10.1186/s12859-018-2461-2) contains supplementary material, which is available to authorized users.
Keywords: Adaptive estimation, Gaussian copula, Differential graphical model, Latent variable, Rank-based approach
Background
In genomic studies, graphical model has been an important tool to capture dependence among different genes. Particularly, Gaussian graphical model has been widely applied to infer the relationship between genes at the transcriptional level [1–4]. Under the Gaussian assumption, estimating the structure of the graphical model is equivalent to recover the support of precision matrix which is defined to be the inverse of the covariance matrix. However, in some cases, compared to focusing on a particular network, it is of greater interest to investigate how the network of connected gene pairs change from one experimental condition to another, which provides deeper insights on an underlying biological process such as identification of pathways that correspond to such a change. For instance, medical experiment usually involves two groups: the patient group and the control group. The analysis of group difference in biological networks or pathways may offer us a new insight into the underlying disease mechanism, which have extensive biomedical and clinical applications, such as identifying effective targets for drug development in a cost-effective and timely manner. Indeed, differential networking modelling has recently emerged as an important tool to analyze a set of changes in graph structure between two conditions (see, for example; [5–17]). In the context of genomic analysis, it is reasonable to assume that two genes are defined to be connected in the differential network if the magnitude of their conditional dependency relationship changes between two conditions. The precision matrix which is defined as the inverse of covariance matrix can capture the conditional dependency relationship. Thus the differential network is typically modelled as the difference of two precision matrices and this type of modelling has been widely used [7–9, 14, 15]. Figure 1a, b, c illustrate the definition of differential network. Each node represents a gene. For two groups depicted in (a) and (b), there is an edge between genes (i,j) if and only if (i,j)-th element of Ω is nonzero. For each edge, there exists a weight which is the magnitude of (i,j)-th element of Ω. Gene pair (i,j) is defined to be connected in the differential network in (c) if the magnitudes of (i,j)-th elements of two precision matrices change between two groups.
Fig. 1.
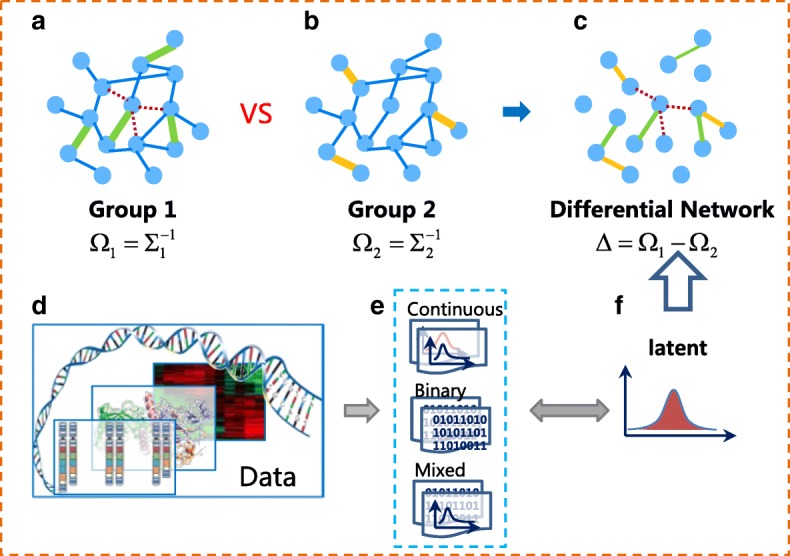
Illustration of latent differential network. a Network in group 1. b Network in group 2. c Differential network. d Data sources. e Data type. f Latent distribution
One straightforward approach to estimate the difference of two precision matrices is to separately estimate the precision matrices and then subtract the estimates. In the high dimensional setting where the dimension p is much larger than the sample size n, which is often the case for genomic study, many estimation approaches for the precision matrix have been proposed and proved to enjoy nice theoretical properties and computation advantage under the key assumption of sparsity. And this topic has been an active area of research in recent years [18–22].
Another type of approach to estimate the difference of two precision matrices is to jointly estimate the precision matrices. Guo et al. [23] penalized the joint loglikelihood with a hierarchical penalty that targets the removal of common zeros in the inverse covariance matrices across categories. Danaher et al. [24] proposed the joint graphical Lasso, which is based upon maximizing a penalized log-likelihood with generalized fused Lasso or group Lasso penalty. Motivated by the constrained ℓ1 minimization approach to precision matrix estimation of [22], Zhao et al. [7] proposed an estimation approach to directly estimate the difference of the precision matrices.
For the separately estimating methods, Liu et al. [25] proposed the nonparanormal family to relax the Gaussian assumption. While the nonparanormal family is much larger than the standard parametric Gaussian family, the independence relations among the variables are still encoded in the precision matrix. In addition, Liu et al. [26] proposed a semiparametric approach called nonparanormal SKEPTIC to estimate high dimensional undirected graphical models efficiently and robustly and proved that the nonparanormal SKEPTIC achieves the optimal parametric rates of convergency in terms of precision matrix estimation and graph recovery. Xue and Zou [27] proposed a similar regularized rank-based estimation idea for estimating nonparanormal graphical models and analyzed adaptive versions of rank-based Dantzig selector and CLIME estimators. He et al. [28] proposed a multiple testing procedure to estimate high-dimensional nonparanormal graphical model and proved that the proposed procedure can control the false discovery rate (FDR) asymptotically.
The disadvantage of Gaussian or nonparanormal graphical models lies in that they are only tailored for modeling continuous data. However, in genomic studies, we may encounter discrete data (e.g. CNV data and SNP data), continuous data (e.g. gene expression and methylation data) or data of hybrid types with both discrete and continuous variables. Besides, in some circumstances, even if the data are continuous, we still need to transform the data into discrete data to remove the heterogeneity (e.g. batch effect, outliers and population stratification). For instance, in the analysis of gene expression data collected from different platforms, to remove the unwanted variation among different experiments known as the batch effects, numerical expression data are often transformed into 0/1 binary data, where lower expression values are encoded as 0 and higher expression values are encoded as 1. In this setting, it is reasonable to assume that the discrete variable is obtained by discretizing a latent variable. Fan et al. [29] proposed a general model named the latent Gaussian copula graphical model, assuming that the observed discrete data are generated by discretizing a latent continuous variable at some unknown cutoff.
In this paper, we consider estimating differential network for various types of biological data in a joint way. We propose a unified semi-parametric latent variable differential network model. The latent differential network model is illustrated in Fig. 1e-f. For biological data, there exist continuous data, discrete data or data of hybrid types with both continuous and discrete data. It is assumed that these data are collected by transforming latent continuous variables which are unobservable. We are interested in the differential network of the latent variables, which provide deeper understanding of the unknown mechanism than that among the observed variables. To the best of our knowledge, our work provides the first method for differential network estimation for binary or mixed data with theoretical guarantees under the high dimensional scaling. The advantages of the proposed methods lie in the following aspects: (I) Our method provides a way to infer the differential network structure among latent variables, which provides deeper understanding of the unknown mechanism than that among the observed variables. (II) Theoretical analysis shows that the proposed methods achieve the same parametric rates of convergence for both difference matrix estimation and differential graph recovery, as if the latent variables were observed. (III) The proposed methods are much more robust to outliers due to the rank-based correlation matrix estimator. (IV) The proposed approaches do not require precision matrices to be sparse, and thus can allow the individual networks to contain hub nodes. Simulation result shows that the proposed method performs much better and more robustly than several state-of-the-art methods. The proposed methods are applied on a gene expression data set associated with lung cancer. A target gene WIF1 stands out by the proposed method, which indeed is verified as a frequent target for epigenetic silencing in various human cancers [30]. The real data example illustrates the great usefulness of the current work.
Methods
In this part, we propose novel definitions of latent differential network model for various types of data. In essence, we define the differential network as the difference of two precision matrices of the latent variables, which greatly generalizes the applicability in areas such as bioinformatics, medical research and so on.
Gaussian copula differential graphical model
We first review the definition of the Gaussian copula distribution. Let f={f1,…,fp} be a set of strictly increasing univariate functions. A p dimensional random variable X=(X1,…,Xp)⊤ is said to follow the Gaussian copula distribution if and only if f(X):=(f1(X1),…,fp(Xp))⊤ :=Z∼Np(μ,Σ) and is noted as X∼NPN(μ,Σ,f), where μ=(μ1,…,μp),Σ=[Σjk] are respectively the mean vector and the correlation matrix of the Gaussian variate Z. The conditional independence structure of X is encoded by the sparsity pattern of Ω=Σ−1. Specifically, it can be shown that Xi is conditionally independent of Xj given all other variables if and only if ωij=0, where ωij is the (i,j)-th element of Ω. Therefore, the differential network of the Gaussian copula variables can be defined to be the difference between the two precision matrices, just the same as for the parametric Gaussian case.
Assume Xi=(Xi1,…,Xip)⊤ for i=1,…,nX are independent observations of the expression levels of p genes from one group denoted by X and Yi=(Yi1,…,Yip)⊤ for i=1,…,nY from the other denoted by Y, X∼NPN(μX,ΣX,fX) and Y∼NPN(μY,ΣY,fY). The differential network is defined to be the difference between the two precision matrices, denoted by Δ0=ΩY−ΩX, where ΩY and ΩX are the inverse matrices of ΣY and ΣX separately.
We propose a rank-based estimator of ΣX. It is known that if Z∼NPN(μ,Σ,f), then we have , where τjk is Kendall’s tau correlation between Zj and Zk. Thus we can estimate the unknown correlation matrix ΣX by:
| 1 |
where is the sample Kendall’s tau correlation between Xj and Xk. Similarly, we can estimate ΣY in the same way and obtain the estimator . Motivated by the direct estimation method of the difference of two precision matrices proposed by [7], one can obtain the estimator of Δ0 by solving
which is equivalent to the optimization problem:
| 2 |
where ⊗ denotes the Kronecker product, is the element-wise ℓ1 norm of the matrix Δ. Here, for a matrix A=[Ajk], |A|∞= maxjk|Ajk| and for a vector a=(aj), |a|∞= maxj|aj|.
As seen from Eq. (2), the proposed approach can directly estimate the difference matrix without implicitly estimating the individual precision matrices. Thus there is no need to assume the sparsity of (ΣY)−1 and (ΣX)−1. We only need to assume that Δ0 is sparse. Besides, compared to the sample covariance matrix, the rank-based estimators here can enjoy modelling flexibility and estimation robustness, especially when outliers exist.
Latent Gaussian copula differential graphical model for binary data
In the analysis of gene expression data, to remove the batch effects, numerical expression data are often transformed into 0/1 binary data, where lower expression values are encoded as 0 and higher expression values are encoded as 1. To estimate the underlying differential network for the binary data from two different groups, we assume that the observed discrete data are generated by discretizing a latent continuous variable at some unknown cutoff. To make the model more flexible, we assume the latent continuous variable is Gaussian copula distributed instead of Gaussian. Let B=(B1,B2,…,Bp)⊤∈{0,1}p be a p-dimensional 0/1-random vector. The 0/1-random vector B satisfies the latent Gaussian copula model (LGCM) for binary data, if there exists a p dimensional random vector X∼NPN(0,Σ,f) such that
where I(·) is the indicator function and the cutoff C=(C1,…,Cp) is a vector of constants. Then we denote B∼ LGCM(Σ,f,C). We call Σ the latent correlation matrix. The latent Gaussian copula model involves parameters (Σ,f,C). Merely based on the binary random vector B, only fj(Cj),j=1,…,p are identifiable. Denote Λ=(Λ1,…,Λp), where Λj=fj(Cj). For notational simplicity, we write LGCM(Σ,Λ) for LGCM(Σ,f,C).
Assume for i=1,…,n1 are independent observations of the binary expression levels of p genes from one group denoted by B1 and for i=1,…,n2 from the other denoted by B2, where B1∼LGCM(Σ1,Λ1) and B2∼LGCM(Σ2,Λ2). The differential network is defined to be the difference between the two precision matrices, denoted by . Motivated by Eq. (2), we should first derive estimators for Σ1 and Σ2. For ease of presentation, we only present the procedure to construct the estimator for Σ1, estimator for Σ2 can be obtained similarly. Denote the Kendall’s tau correlation between and by , it can be shown that satisfies:
where
is the cumulative distribution function of the standard bivariate normal distribution, ϕ2(x1,x2;t) is the probability density function of the standard bivariate normal distribution with correlation t. Denote by
For any fixed and , it can be shown that is a strictly monotonic increasing function on t∈(−1,1) and thus is invertible. Given and , one can estimate by . However, the cutoff values are unknown in practice. As , we can estimate by , where . Thus the Kendall’s tau rank-based correlation matrix estimator for Σ1 is a p×p matrix with element entry given by
| 3 |
Similarly, the Kendall’s tau rank-based correlation matrix estimator for Σ2 is a p×p matrix with element entry given by
| 4 |
Motivated by Eq. (2), we can obtain an estimator of by solving the following optimization problem:
| 5 |
For the binary data, we aim to infer the differential network among latent variables, which provides deeper understanding of the unknown mechanism than that among the observed binary variables. Thus, our model complements the existing work on high dimensional differential network estimation, which mostly focused on learning differential network among observed variables including, for example, the Ising model.
Latent Gaussian copula differential graphical model for mixed data
In the analysis of biological data, there also exists the case where some biological data are discrete while some others are continuous. For instance, multi-level omics data integrative analysis involves gene mutation, expression, methylation, metabolome and phenome data. In this case, mixed data appear naturally. We start with the definition of the latent Gaussian copula model for mixed data. Assume that M=(M1,M2), where M1 represents the p1-dimensional binary variables and M2 represents the p2-dimensional continuous variables. The random vector M satisfies the latent Gaussian copula model for mixed data, if there exists a p1 dimensional random vector X1 such that X=(X1,M2)∼ NPN(0,Σ,f) and
where is a vector of constants. Then we denote M∼ LGCM(0,Σ,f,C), and call Σ the latent correlation matrix. In the latent Gaussian copula regression model, the binary components M1 are generated by a latent continuous random vector X1 truncated at C, and combining with the continuous components M2, X=(X1,M2) satisfies the Gaussian copula model. For the binary data M1, only Λj=fj(Cj),j=1,…,p1 are identifiable. For the continuous components M2, the marginal transformations fj(·),j=p1+1,…,p are identifiable.
Assume for i=1,…,n1 are independent observations of the expression levels of p genes from one group denoted by M1 and for i=1,…,n2 from the other denoted by M2, where M1∼LGCM(Σ1,Λ1) and M2∼LGCM(Σ2,Λ2). The differential network is defined to be the difference between the two precision matrices, denoted by . Similar to the discussions in the last sections, we first need to construct estimators for Σ1 and Σ2. For ease of presentation, we only present the procedure to construct the estimator for Σ1, estimator for Σ2 can be obtained similarly. For discrete components , as what we have discussed in the last subsection with a slight change of notation, we can estimate by:
| 6 |
For continuous components , as what we have discussed, we can estimate by:
| 7 |
where is defined as follows:
We still need to consider the mixed case. Without loss of generality, we assume that is binary and is continuous. In this case, the Kendall’s tau correlation can be expressed by
The population version of Kendall’s tau correlation can be expressed by , where
Moreover, for fixed , is an invertible function of t. The parameter could be estimated by , where . Thus when is binary and is continuous, can be estimated by the Kendall’ tau rank-based estimator:
| 8 |
where is the inverse function of for fixed . Thus the Kendall’s tau rank-based correlation matrix estimator for Σ1 is a p×p matrix with corresponding element entry given by Eqs. (6), (7), and (8) respectively. Similarly, we can obtain estimator for Σ2. Motivated by Eq. (2), we can obtain an estimator of Δ0 by solving the following optimization problem:
| 9 |
We show that the rank-based covariance matrix estimators achieve the same parametric rate of convergence for both difference matrix estimation and differential graph recovery in the Additional file 1. Thus the extra modelling flexibility comes at almost no cost of statistical efficiency. Besides, for the binary data or data of hybrid types with both binary and continuous variables, the differential network among latent variables can be well estimated, which provides deeper understanding of the unknown mechanism than that among the observed variables.
Implementation
In this section we will present how to solve the optimization problems in Eqs. (2), (5), and (9). For ease of presentation, we only present the procedure to obtain the solution to optimization problem in Eq. (2) and solutions to optimization problems in Eqs. (5) and (9) can be obtained in the similar way.
Recall that in Eq. (2), the optimization problem is
Let Δ= [δjk]1≤j,k≤p and define θ to be the p(p+1)/2×1 vector with θ=(δjk)1≤j≤k≤p. Estimating a symmetric Δ is thus equivalent to estimating θ, which alleviates the computation burden especially when p is large. Define the p2×p(p+1)/2 matrix Γ with columns indexed by 1≤j≤k≤p and with rows indexed by l=1,…,p and m=1,…,p, so that each entry is labeled by Γlm,jk. For j≤k, let Γjk,jk=Γkj,jk=1 and set all other entries of Γ equal to zero. With these notations, one may consider the following optimization problem:
| 10 |
where , and for a p(p+1)/2×1 vector c, |c|O∞ denotes the sup-norm of the entries of c corresponding to the off diagonal elements of its matrix form, while |c|D∞ denotes the sup-norm of the entries of c corresponding to the diagonal elements. The matrix form of will be denoted by in the following sections. The optimization problem in Eq. (10) can be solved by the alternating direction method of multipliers (ADMM), for a thorough discussion, we refer to [31]. For the optimization problem in Eq. (10), to apply the ADMM algorithm, we rewrite it as:
where the function g(·) is defined by
The augmented Lagrangian can be written as
| 11 |
where u is the Lagrange multiplier and ρ is a positive penalty parameter which can be specified by users. The ADMM algorithm is based on minimizing the augmented Lagrangian in (11) over θ and z and then applying a dual variable update to the Lagrange multiplier u, which yields the updates
for iterations t=0,1,2…. As for the tuning parameter λn in (10), it can be chosen by an approximate Akaike information criterion (AIC). λn is chosen to minimize
where k is the effective degrees of freedom that can be approximated by and L(λn) represents the loss function either L∞ or LF which are defined by
In this paper we focus on the loss functions with the supremum and Frobenius norms for further theoretical development. One may also use other matrix norms, such as spectral norm:
Similarly, for the latent Gaussian copula model for binary data, one can solve the following optimization problem:
| 12 |
where , . The matrix form of will be denoted by in the following sections. For the latent Gaussian copula model for mixed data, one can solve the following optimization problem:
| 13 |
where , . The matrix form of will be denoted by in the following sections. Besides, corresponding Akaike information criterion can be proposed to choose the tuning parameter λn.
Simulation
Simulation for Gaussian copula differential graphical model In this part, we conduct simulation study for differential network estimation under Gaussian copula model. We mainly focus on the graphs that contain hub nodes. First we generate the edge set EX for the group X. We partition p features into 5 equally-sized and non-overlapping sets: C1∪C2⋯∪C5={1,…,p}, |Ck|=p/5, Ci∩Cj=∅. For the smallest i∈Ck, we set (i,j)∈Ck for all {j≠i:j∈Ck}. The non-zero entries of ΩX is then determined by the edge set EX, where ΩX=(ΣX)−1. Next, the value of each nonzero entry of ΩX was generated from a uniform distribution with support [−0.75,−0.25]∪[0.25,0.75]. To ensure positive definiteness of ΩX, let ΩX=ΩX+(0.2+|λmin(ΩX)|)I. At last the ΩX is rescaled such that ΣX is a correlation matrix. Then we proceed to generate the differential network Δ0. We randomly select two hub nodes from the 5 equally-sized and non-overlapping sets. The differential network Δ0 is generated such that the connections of these two hub nodes change sign between ΩX and ΩY. The correlation matrix ΣX and ΣY are generated by (ΩX)−1 and (ΩY)−1 respectively. Finally we generate nX i.i.d observations of ZX from the N(0,ΣX) distribution and nY i.i.d observations of ZY from the N(0,ΣY) distribution. Next we sample nX i.i.d samples from the nonparanormal distribution NPN(0,ΣX,fX) and nY i.i.d samples from the nonparanormal distribution NPN(0,ΣY,fY). For simplicity, we use the same univariate transformations on each dimension: and fX=fY. To sample data from the nonparanormal distribution, we also need g:=f−1. We consider the Gaussian CDF Transformation of g which is used in [26].
In the simulation study, we let p= 50,80,100,120 and nX=nY=100. The simulation result is based on 100 replications. For each simulated data set, we apply three estimation methods. That is, the direct differential network estimator (DDN) in [7], the rank-based differential network estimator (RDN) and the direct differential network estimator based on the latent variable Z and Pearson correlation (ZP-DDN). In ZP-DDN, we assume that ZX and ZY are observed and the Pearson correlation estimator of cov(ZX) and cov(ZY) are plugged into the direct estimation procedure. While ZP-DDN are often not available in real applications, we use ZP-DDN as benchmarks for quantifying the information loss of the remaining estimators.
We evaluate the performance of the estimation methods from two aspects: support recovery and estimation error. The support recovery results are evaluated by true positive rate (TPR) and true negative rate (TNR) along a range of tuning parameter λ. Suppose the true difference matrix Δ0 has the support and its estimator has the support set . TPR and TNR are defined as follows:
where TP and TN are the numbers of true positives and true negatives respectively, which are defined as
To evaluate the support recovery performance, we use the true discovery rate, which is defined as TD =TP. As for the estimation error, we calculate the element-wise L∞ norm and Frobenius norm of .
Simulation for latent Gaussian copula differential graphical model In this part, we conduct simulation study for differential network estimation under Latent Gaussian copula model. We assume that the cutoff vector C∼Unif [0,1] and let Σ1 and Σ2 be generated in the same way as ΣX and ΣY described in the last subsection. We consider the following three Scenarios:
∙Scenario 1 Generate data , where and X∼NPN(0,Σ1,f1); Generate data , where and Y∼NPN(0,Σ2,f2). The transformation functions f1 and f2 are Gaussian CDF transformation.
∙Scenario 2 Generate data , where , X∼NPN(0,Σ1,f1) and ;
Generate data , where , and Y∼NPN(0,Σ2,f2) and . The transformation functions f1 and f2 are Gaussian CDF transformation.
∙Scenario 3 Generate data , where and Z1∼N(0,Σ1), where 10 entries in each Z1 is randomly sampled and replaced by -5 or 5;
Generate data , where and Z2∼N(0,Σ2), where 10 entries in each Z2 is randomly sampled and replaced by -5 or 5.
In Scenario 1 and Scenario 3, we generate binary data. Scenario 1 corresponds to the latent Gaussian copula model and Scenario 3 corresponds to the setting where the binary data can be misclassified due to the outliers of the latent Gaussian variable. Scenario 3 is designed to investigate the robustness of the proposed approach. Scenario 2 corresponds to the mixed data generated from the latent Gaussian copula model.
Application to gene expression data sets related to lung cancer
In this section we consider the differential network estimation for a gene expression data set related to lung cancer. The data set is publicly available from the Gene Expression Omnibus at accession number GDS2771 and was studied in [24]. It includes 22,283 microarray-derived gene expression measurements from large airway epithelial cells sampled from 97 patients with lung cancer and 90 controls in the data set. It is of interest to investigate how the structure of the gene co-expression network differs between the group of patients with lung cancer and the control group. It may shed light on underlying lung cancer mechanisms. In this real example study, we limited our analysis to the 122 genes in the Wnt signaling pathway. The Wnt signaling pathway has recently emerged as a critical pathway in lung carcinogenesis as already demonstrated in many cancers and particularly in colorectal cancer [32]. The Gene expression levels were analyzed on a logarithmic scale. Each gene feature was standardized to have mean zero and standard deviation 1 within the cancer samples and the controls separately.
Results
Simulation results for Gaussian copula differential graphical model
The receiver operating characteristic (ROC) curves of the three estimation methods are depicted in Fig. 2. It shows that the proposed method RDN compares favourably with the benchmark method ZP-DDN, which means that the information loss is negligible. Besides, Fig. 2 also shows that DDN performs pretty bad in the non-Gaussian case.
Fig. 2.
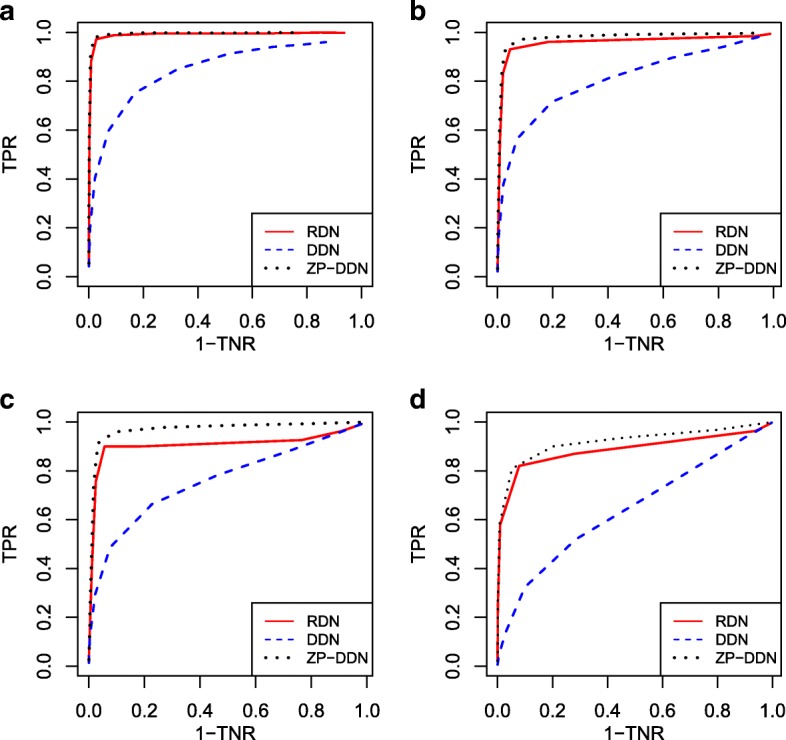
Receiver operating characteristic curves under Gaussian copula model with dimensionality varying from 50 to 120. The red line represents the proposed RDN method, the black dotted represents the benchmark method ZP-DDN, the blue dotted line represents DDN method. a Scenario 3, p = 50. b Scenario 3, p = 80. c Scenario 3, p = 100. d Scenario 3, p = 120
Table 1 gives the true discovery rates with different loss functions. The results also show the method RDN compares favourably with the benchmark method ZP-DDN. For all the methods, tuning using the LF gives better true discovery rates than tuning using the L∞. Table 1 depicts the elementwise L∞ norm estimation accuracies of the thresholded estimators tuned using the loss functions L∞ and LF. From Table 1, we can see that the LF loss function gives slightly better results than the L∞ loss function. For all the methods, the elementwise L∞ norm estimation accuracy are comparable. We point out that it is possible for RDN to simultaneously give better support recovery but similar estimation than DDN. The reason is that estimation error depends on the magnitudes of the estimated entries, while support recovery depends only on whether the entries are nonzero. Besides, RDN has comparable performance with the benchmark method ZP-DDN in terms of both support recovery and estimation accuracy, which indicates that the information loss of the estimator RDN is negligible.
Table 1.
Average true discovery rates (%) and average estimation errors over 100 simulations
| ZP-DDN | RDN | DDN | ||||
|---|---|---|---|---|---|---|
| p | L ∞ | L F | L ∞ | L F | L ∞ | L F |
| Average true discovery rates | ||||||
| 50 | 74.0 (13.6) | 83.2 (10.9) | 75.6 (14.0) | 89.1 (11.3) | 45.9 (24.7) | 27.8 (17.3) |
| 80 | 91.4 (16.4) | 99.6 (4.3) | 95.2 (14.2) | 100.0 (0.0) | 44.9 (34.6) | 51.0 (42.8) |
| 100 | 96.3 (14.1) | 100.0(0.0) | 99.5 (5.2) | 100.0 (0.0) | 39.3 (40.3) | 50.0 (49.1) |
| 120 | 78.8 (16.8) | 100.0(0.0) | 79.0 (18.2) | 100.0 (0.0) | 23.4 (41.3) | 30.0 (46.3) |
| Average estimation errors in the elementwise L∞ norm | ||||||
| 50 | 3.26 (0.41) | 2.91 (0.33) | 3.08 (0.32) | 2.59 (0.35) | 2.27 (0.12) | 2.41 (0.21) |
| 80 | 2.06 (0.28) | 1.92 (0.06) | 1.98 (0.21) | 1.91 (0.00) | 1.97 (0.09) | 1.94 (0.08) |
| 100 | 1.86 (0.15) | 1.82 (0.00) | 1.82 (0.04) | 1.82 (0.00) | 1.87 (0.10) | 1.83 (0.04) |
| 120 | 1.12 (0.17) | 0.87 (0.00) | 1.12 (0.18) | 0.87 (0.00) | 0.89 (0.07) | 0.87 (0.00) |
Simulation results for Latent Gaussian copula differential graphical model
The ROC curves for Scenario 1 and Scenario 2 with different dimensionality p (varying from 50 to 120) is presented in Fig. 3. Table 2 give the true discovery rates with different loss functions and the elementwise L∞ norm estimation accuracies of the thresholded estimators tuned using the loss functions L∞ and LF, respectively. For method ZR-RDN, we assume that the latent Gaussian copula variables are observed. In particular, the rank-based correlation matrix estimator of the latent Gaussian copula variables are plugged into the direct estimation procedure. With a slight abuse of notation, the RDN method here refers to either the rank-based method for binary data or for mixed data. The ROC curves in Fig. 3 show that the rank-based methods RDN proposed for latent Gaussian copula model (binary and mixed) perform pretty well even when the dimensionality is larger than the sample size.
Fig. 3.

Receiver operating characteristic curves for Scenario 1 and Scenario 2 under latent Gaussian copula model, with dimensionality varying from 50 to 120. a Scenario 1. b Scenario 2
Table 2.
Simulation results over 100 replications for Scenario 1 and Scenario 2
| Scenario 1 | Scenario 2 | |||
|---|---|---|---|---|
| p | L ∞ | L F | L ∞ | L F |
| Average true discovery rates(%) | ||||
| 50 | 78.8 (15.2) | 98.4 (5.9) | 79.6 (13.8) | 40.8 (25.6) |
| 80 | 76.4 (23.1) | 100.0(0.0) | 83.4 (17.0) | 88.2 (17.6) |
| 100 | 89.5 (22.1) | 100.0(0.0) | 84.8 (20.0) | 99.3 (3.9) |
| 120 | 76.5 (31.0) | 94.0(24.0) | 82.4 (15.2) | 100.0 (0.0) |
| Average estimation errors in the elementwise L∞ norm | ||||
| 50 | 2.66 (0.26) | 2.21 (0.15) | 3.23 (0.40) | 3.85 (0.55) |
| 80 | 2.10 (0.20) | 1.91 (0.00) | 2.29 (0.35) | 2.14 (0.32) |
| 100 | 1.88 (0.13) | 1.82 (0.00) | 2.03 (0.28) | 1.83 (0.08) |
| 120 | 1.00 (0.16) | 0.87 (0.00) | 1.17 (0.16) | 0.88 (0.07) |
By the ROC curves in Fig. 4, we can find that RDN is more robust to the data misclassification than the benchmark estimator ZP-DDN. The robustness of RDN to outliers illustrates the advantage of the dichotomization method. In the absence of misclassification, it is seen that the ROC curves of RDN and ZR-RDN are similar, which indicates little information loss for differential network recovery due to the dichotomization procedure. Table 3 gives the true discovery rates with different different loss functions for Scenario 3 and presents the elementwise L∞ norm estimation accuracies of the thresholded estimators tuned using the loss functions L∞ and LF for Scenario 3. From Table 3, we can see that the LF loss function gives slightly better results than the L∞ loss function. Besides, we can see that the elementwise L∞ norm estimation accuracy are comparable. This is also true for Scenario 1 and Scenario 2.
Fig. 4.

Receiver operating characteristic curves for Scenario 3 under latent Gaussian copula model, with dimensionality varying from 50 to 120. The red line represents the proposed RDN method, the black dotted represents the benchmark method ZP-DDN, the blue dotted line represents DDN method. a Scenario 3, p = 50. b Scenario 3, p = 80. c Scenario 3, p = 100. d Scenario 3, p = 120
Table 3.
Simulation results over 100 replications for Scenario 3
| ZP-DDN | RDN | ZR-RDN | ||||
|---|---|---|---|---|---|---|
| p | L ∞ | L F | L ∞ | L F | L ∞ | L F |
| Average true discovery rates(%) | ||||||
| 50 | 39.8 (39.5) | 46.1 (47.3) | 87.6 (14.7) | 97.3 (7.4) | 88.0 (11.0) | 90.0 (12.4) |
| 80 | 32.4 (41.9) | 35.5 (47.9) | 80.7 (14.8) | 99.8 (2.5) | 89.5 (8.7) | 95.4 (7.2) |
| 100 | 23.5 (40.2) | 31.7 (46.9) | 75.6 (20.3) | 100.0(0.0) | 84.0 (12.0) | 99.1 (4.2) |
| 120 | 16.0 (37.0) | 16.0 (37.0) | 52.9 (44.6) | 68.0(47.1) | 70.4 (26.8) | 93.0 (24.8) |
| Average estimation errors in the elementwise L∞ norm | ||||||
| 50 | 2.15 (0.03) | 2.16 (0.01) | 2.05 (0.15) | 2.12 (0.08) | 2.05 (0.17) | 2.02 (0.15) |
| 80 | 1.91 (0.02) | 1.91 (0.01) | 1.91 (0.12) | 1.92 (0.04) | 1.92 (0.12) | 1.91 (0.08) |
| 100 | 1.82 (0.02) | 1.82 (0.00) | 1.88 (0.12) | 1.82 (0.00) | 1.90 (0.12) | 1.83 (0.04) |
| 120 | 0.87 (0.00) | 0.87 (0.00) | 0.91 (0.09) | 0.87 (0.00) | 0.97 (0.11) | 0.88 (0.05) |
Theoretical results
The estimators , and , after an additional threshold step, are shown to be able to recover not only the support of the true Δ0 but also the signs of its nonzero entries as long as those entries are sufficiently large. Besides, under mild conditions, the estimation errors bounds in terms of matrix Frobenius norm and elementwise ℓ∞ norm both achieve the parametric rate , see details in Additional file 1. It indicates that the extra modeling flexibility and robustness come at almost no cost of statistical efficiency and it seems as if the latent variable can be observed. Thus these new estimators can be used as a safe replacement of Gaussian estimators even when the data are truly Gaussian. Compared to the separate and joint approaches to estimating differential networks (e.g. [22, 23],) which require sparsity on each Σ−1, the proposed direction estimation methods for different types of data only require the sparsity of the difference matrix Δ0. The detailed theorems and proofs are in the Additional file 1 available online.
Results of application
In the real application part, we compare three estimation methods. The first method is the Gaussian copula RDN method, which we denote as C-RDN. The second method is the latent Gaussian copula RDN method, which we denote as B-RDN. In specific, we first apply the adaptive dichotomization method implemented by the ArrayBin package in R to remove the batch effect in the gene expression data. The adaptive dichotomization method transforms the numerical gene expression data into 0/1 binary data. The genes with high expression level are encoded as 1 and the genes with lower expression level are encoded as 0. Then we apply the B-RDN to the 0/1 binary data. The third method is the direct differential network estimation method proposed by [7] with Gaussian assumption, which we denote as DDN.
We conduct Shapiro-Wilk test on the gene data set and 63% of the genes reject the normality null hypothesis. Therefore, the Gaussian assumption of DDN method is violated in this real data example. Thus we expect that C-RDN which relaxes the Gaussian assumption may provide a more reliable result. The deficiency of the C-RDN method lies in that it does not take the batch effect of the genes expression data from different platforms into consideration. For the B-RDN method, it removes the batch effect.
Figure 5 depicts the differential network estimated by the three methods. Table 4 gives the hub genes selected out by different estimation methods. For method C-RDN, the tuning parameter λ is selected by the AIC criterion with the elementwise ℓ1 norm loss function. To ensure a fair comparison, the tuning parameter λ for method B-RDN and DDN are selected such that the number of edges in the estimated differential graphs by all three methods are almost the same. The number of edges selected by the three methods are 56, 59 and 52, respectively. From Fig. 5, we can see that B-RDN identifies an obvious hub gene WIF1 that is an extracellular antagonist of WNT. WIF1 is a frequent target for epigenetic silencing in various human cancers [30]. WIF1 promoter is frequently methylated in non-small cell lung cancer (NSCLC) cells to down-regulate its mRNA expression [33]. Both C-RDN and B-RDN select out a common hub gene APC. APC expression in lung cancer are associated with survival time and is also related to cancer metastasis [34]. Both C-RDN and DDN select out a common hub gene, MAPK8, which plays a significant role in the promotion of lung inflammation and tumorigenesis subsequent to tobacco smoke exposure [35]. The expression level of DVL2 was reported significantly higher in lung adenocarcinomas than in squamous carcinomas, and was associated with poor tumor differentiation [36]. Winn et al. [37] reported that the restoration of FZD9 signaling inhibited both cell proliferation and anchorageindependent growth, promoted cellular differentiation, and reversed the transformed phenotype in NSCLC. The overexpression of MMP7 was associated with tumor proliferation, and a poor prognosis in NSCLC [38]. RAC1 generally plays an important role in cancer progression and metastasis [39].
Fig. 5.

Differential network estimated by different methods. Orange edges show an increase in conditional dependency from control group to lung cancer patient group; grey edges show a decrease. Red points stand for hub genes which have edges with more than 3 other genes. a C-RDN. b B-RDN. c DDN
Table 4.
Hub genes selected by different methods
| DDN | PRKACA | MAPK8 | CACYBP | CAMK2B | SFRP1 | CSNK2A2 | TCF7 |
| BTRC | RUVBL1 | ||||||
| C-RDN | PLCB2 | DVL2 | MAPK8 | PLCB1 | APC | WNT2 | FZD9 |
| WNT11 | DKK1 | SFRP4 | |||||
| B-RDN | WIF1 | MMP7 | RAC1 | LEF1 | APC | PRKACA | WNT8B |
| BAMBI |
By comparing (a) and (b) in Fig. 5, we can see that the estimated differential network can be very different with/without considering the batch effect. Although it is inevitable to result in information loss in the discretization procedure for method B-RDN, [40] argued that this procedure can potentially improve the accuracy of the statistical analysis. In real data example, we recommend to use the B-RDN method to remove the batch effect despite the little information loss. At last we argue that statistical comparison of group difference in this biological network or pathway can provide new insight into the underlying lung cancer mechanism, which may further offer more effective targets for drug development.
To further interpret the underlying biological implications of the identified hub genes, we conducted Gene Ontology (GO) enrichment analysis. Table 5 shows the common GO terms enriched by C-RDN, B-RDN and DDN. The GO enrichment analysis is performed using R package “clusterProfiler” with the P-value adjusted by Benjamini-Hochberg method. It shows that our methods (C-RDN, B-RDN) have smaller P-value than DDN. The common molecular function and cellular component suggest that the change of frizzled binding, Wnt-protein binding and beta-catenin destruction complex are important in the etiology of lung cancer. These predictions are supported by the literatures [41–43], which indicates that the proposed differential network model can provide biological meaningful underlying signals.
Table 5.
Gene Ontology (GO) enrichment analysis result
| ID | Functional term | Ontology | Adjust P-value | ||
|---|---|---|---|---|---|
| C-RDN | B-RDN | DDN | |||
| GO:0016055 | Wnt signaling pathway | BP | 1.69×10−11 | 2.96×10−6 | 0.0022 |
| GO:0198738 | cell-cell signaling by wnt | BP | 1.69×10−11 | 2.96×10−6 | 0.0022 |
| GO:0060828 | regulation of canonical Wnt signaling pathway | BP | 1.49×10−9 | 0.0012 | 0.0027 |
| GO:0060070 | canonical Wnt signaling pathway | BP | 4.78×10−9 | 0.0012 | 0.0027 |
| GO:0030111 | regulation of Wnt signaling pathway | BP | 6.67×10−9 | 0.0058 | 0.0091 |
| GO:0005109 | frizzled binding | MF | 5.28×10−5 | 0.0058 | 0.0091 |
| GO:0007369 | gastrulation | BP | 0.0024 | 0.0058 | 0.0276 |
| GO:0017147 | Wnt-protein binding | MF | 0.0025 | 0.0073 | 0.0286 |
| GO:0060562 | epithelial tube morphogenesis | BP | 0.0068 | 0.0073 | 0.0290 |
| GO:0003002 | regionalization | BP | 0.0074 | 0.0080 | 0.0331 |
| GO:0035239 | tube morphogenesis | BP | 0.0082 | 0.0090 | 0.0332 |
| GO:0001503 | ossification | BP | 0.0093 | 0.0131 | 0.0341 |
| GO:0007389 | pattern specification process | BP | 0.0113 | 0.0131 | 0.0357 |
| GO:0043393 | regulation of protein binding | BP | 0.0202 | 0.0175 | 0.0377 |
| GO:0034329 | cell junction assembly | BP | 0.0205 | 0.0178 | 0.0382 |
| GO:0030877 | beta-catenin destruction complex | CC | 0.0223 | 0.0377 | 0.0382 |
| GO:0045216 | cell-cell junction organization | BP | 0.0229 | 0.0409 | 0.0402 |
| GO:0034330 | cell junction organization | BP | 0.0259 | 0.0411 | 0.0408 |
| GO:0071496 | cellular response to external stimulus | BP | 0.0281 | 0.0411 | 0.0418 |
| GO:0071214 | cellular response to abiotic stimulus | BP | 0.0290 | 0.0421 | 0.0448 |
| GO:0104004 | cellular response to environmental stimulus | BP | 0.0290 | 0.0450 | 0.0453 |
| GO:0051098 | regulation of binding | BP | 0.0330 | 0.0474 | 0.0478 |
| GO:0045992 | negative regulation of embryonic development | BP | 0.0341 | 0.0479 | 0.0495 |
| GO:1903829 | positive regulation of cellular protein localization | BP | 0.0397 | 0.0489 | 0.0495 |
| GO:1901990 | regulation of mitotic cell cycle phase transition | BP | 0.0409 | 0.0489 | 0.0498 |
BP: biological process; MF: molecular function; CC: cellular component
Discussion
A complex disease phenotype (e.g. diabetes, cancer) often reflects various pathobiological processes that interact in a network rather than the abnormality of a single gene. Such interactions are not static processes, instead they are dynamic in response to changing genetic, epigenetic and environmental factors, which further entails the analysis of differential network. In this paper, we propose adaptive estimation approaches for latent variable differential network model with the assumption that the true differential network is sparse, which do not require precision matrices to be sparse. The latent variable differential network model is fundamentally different from the existing ones in the literature in the sense that the differential structure in the unobserved latent variables are of primary interest. Theoretical analysis shows that the proposed methods achieve the same parametric convergence rate for both the difference of the precision matrices estimation and differential structure recovery, which means that the extra modelling flexibility comes at almost no cost of statistical efficiency. The unified latent variable differential network model provides deeper understanding of the unknown genomic mechanism than that among the observed variables.
The current work could be extended in the following two aspects. First, in this paper, we consider the following optimization problem to directly estimate the difference matrix Δ:
where and denote the rank-based estimators of the covariance matrices. The D-trace loss function [15, 44] can also be applied to to directly estimate the precision matrix difference. Thus, we may also consider the D-trace loss function to estimate the Gaussian copula and latent Gaussian copula differential graphical models. In specific, the difference matrix Δ could be eatimated by:
where λ>0 is a regularization parameter and is a decomposable non-convex penalty function which has the form , such as smoothly clipped absolute deviation (SCAD) penalty [45]. The theoretical guarantees are still needed to be investigated, but we expect that the empirical performance could be comparable.
Second, for the latent Gaussian copula differential graphical model, we focus on the binary data. In fact, the methods can be extended to the discrete data with more than two categories. The properties of this procedure are left for future investigation as there are a lot of work still needed to be done.
Conclusions
The proposed latent variable differential network models are very flexible and provide deeper understanding of the unknown biological mechanism. It is demonstrated latent differential network models enjoy great advantages over existing models and thus are highly recommended in real application.
Additional file
Contains the theoretical guarantee of of the proposed methods and proofs. (PDF 284 kb)
Acknowledgments
Funding
This work was supported by grants from the National Natural Science Foundation of China (grant number 81803336, 11801316, 11571080 and 81573259) and Natural Science Foundation of Shandong Province (ZR2018BH033). Publication of this article was sponsored by 81803336 grant. The funding body played no role in the design, writing or decision to publish this manuscript.
Availability of data and materials
The gene expression data set related to lung cancer is publicly available from the Gene Expression Omnibus at accession number GDS2771.
About this supplement
This article has been published as part of BMC Bioinformatics Volume 19 Supplement 17, 2018: Selected articles from the International Conference on Intelligent Biology and Medicine (ICIBM) 2018: bioinformatics. The full contents of the supplement are available online at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-19-supplement-17.
Abbreviations
- CNV
Copy Number Variation
- FDR
False Discovery Rate
- GO
Gene Ontology
- ROC
Receiver Operating Characteristic
- SNP
Single Nucleotide Polymorphisms
- TDR
True Discovery Rate
- TNR
True Negative Rate
- TPR
True Positive Rate
Authors’ contributions
YH and JJ contributed to the study design, analytical preparation and the writing of the manuscript. YH and JJ performed the simulation studies. JJ analyzed the data, LX, XZ and FX wrote and revised the manuscript. All authors read and approved this version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yong He, Email: heyong@sdufe.edu.cn.
Jiadong Ji, Email: jiadong@sdufe.edu.cn.
Lei Xie, Email: lxie@iscb.org.
Xinsheng Zhang, Email: xszhang@fudan.edu.cn.
Fuzhong Xue, Email: xuefzh@sdu.edu.cn.
References
- 1.Li H, Gui J. Gradient directed regularization for sparse gaussian concentration graphs, with applications to inference of genetic networks. Biostatistics. 2006;7(2):302–17. doi: 10.1093/biostatistics/kxj008. [DOI] [PubMed] [Google Scholar]
- 2.Segal E, Friedman N, Kaminski N, Regev A, Koller D. From signatures to models: understanding cancer using microarrays. Nat Genet. 2005;37:38–45. doi: 10.1038/ng1561. [DOI] [PubMed] [Google Scholar]
- 3.Peng J, Wang P, Zhou N, Zhu J. Partial correlation estimation by joint sparse regression models. J Am Stat Assoc. 2009;104(486):735. doi: 10.1198/jasa.2009.0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai T, Li H, Liu W, Xie J. Covariate-adjusted precision matrix estimation with an application in genetical genomics. Biometrika. 2013;100(1):139–56. doi: 10.1093/biomet/ass058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuente ADL. From differential expression to differential networking–identification of dysfunctional regulatory networks in diseases. Trends Genet. 2010;26(7):326–33. doi: 10.1016/j.tig.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Ideker T, Krogan NJ. Differential network biology. Mol Syst Biol. 2012;8(1):565. doi: 10.1038/msb.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao SD, Cai T, Li H. Direct estimation of differential networks. Biometrika. 2014;101(2):253–68. doi: 10.1093/biomet/asu009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tian D, Gu Q, Jian M. Identifying gene regulatory network rewiring using latent differential graphical models. Nucleic Acids Res. 2016;44(17):140. doi: 10.1093/nar/gkw581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia Y, Cai T, Cai T. Testing differential networks with applications to detecting gene-by-gene interactions. Biometrika. 2015;102(2):247–66. doi: 10.1093/biomet/asu074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ji J, Yuan Z, Zhang X, Li F, Xu J, Liu Y, Li H, Wang J, Xue F. Detection for pathway effect contributing to disease in systems epidemiology with a case-control design. Bmj Open. 2015;5(1):006721. doi: 10.1136/bmjopen-2014-006721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ji J, Yuan Z, Zhang X, Xue F. A powerful score-based statistical test for group difference in weighted biological networks. BMC Bioinformatics. 2016;17(1):86. doi: 10.1186/s12859-016-0916-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan Z, Ji J, Zhang T, Liu Y, Zhang X, Chen W, Xue F. A novel chi-square statistic for detecting group differences between pathways in systems epidemiology. Stat Med. 2016;35(29):5512–24. doi: 10.1002/sim.7094. [DOI] [PubMed] [Google Scholar]
- 13.Yuan Z, Ji J, Zhang X, Xu J, Ma D, Xue F. A powerful weighted statistic for detecting group differences of directed biological networks. Sci Rep. 2016;6:34159. doi: 10.1038/srep34159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu W. Structural similarity and difference testing on multiple sparse gaussian graphical models. Ann Stat. 2017;45(6):2680–2707. [Google Scholar]
- 15.Yuan H, Xi R, Chen C, Deng M. Differential network analysis via the lasso penalized d-trace loss. Biometrika. 2017;104:755–70. [Google Scholar]
- 16.He Y, Zhang X, Ji J, Liu B. Joint estimation of multiple high-dimensional gaussian copula graphical models. Aust N Z J Stat. 2017;59:289–310. [Google Scholar]
- 17.Ji J, He D, Feng Y, He Y, Xue F, Xie L. Jdinac: joint density-based non-parametric differential interaction network analysis and classification using high-dimensional sparse omics data. Bioinformatics. 2017;33(19):3080–87. doi: 10.1093/bioinformatics/btx360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meinshausen N, Bühlmann P. High-dimensional graphs and variable selection with the lasso. Ann Stat. 2006;34:1436–62. [Google Scholar]
- 19.Yuan M, Lin Y. Model selection and estimation in the gaussian graphical model. Biometrika. 2007;94(1):19–35. [Google Scholar]
- 20.Friedman J, Hastie T, Tibshirani R. Sparse inverse covariance estimation with the graphical lasso. Biostatistics. 2008;9(3):432–41. doi: 10.1093/biostatistics/kxm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan M. High dimensional inverse covariance matrix estimation via linear programming. J Mach Learn Res. 2010;11(12):2261–86. [Google Scholar]
- 22.Cai T, Liu W, Luo X. A constrained ℓ1 minimization approach to sparse precision matrix estimation. J Am Stat Assoc. 2011;106(494):594–607. [Google Scholar]
- 23.Guo J, Levina E, Michailidis G, Zhu J. Joint estimation of multiple graphical models. Biometrika. 2011;98(1):1–15. doi: 10.1093/biomet/asq060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Danaher P, Wang P, Witten DM. J R Stat Soc Ser B (Stat Methodol). 2014; 76(2):373–97. [DOI] [PMC free article] [PubMed]
- 25.Liu H, Lafferty J, Wasserman L. The nonparanormal: Semiparametric estimation of high dimensional undirected graphs. J Mach Learn Res. 2009;10(3):2295–328. [Google Scholar]
- 26.Liu H, Han F, Yuan M, Lafferty J, Wasserman L. High-dimensional semiparametric gaussian copula graphical models. Ann Stat. 2012;40(4):2293–326. [Google Scholar]
- 27.Xue L, Zou H. Regularized rank-based estimation of high-dimensional nonparanormal graphical models. Ann Stat. 2012;40(5):2541–71. [Google Scholar]
- 28.He Y, Zhang X, Wang P, Zhang L. High dimensional Gaussian copula graphical model with FDR control. Comput Stat Data Anal. 2017;113:457–74. [Google Scholar]
- 29.Fan J, Liu H, Ning Y, Zou H. High dimensional semiparametric latent graphical model for mixed data. J R Stat Soc. 2017;79(2):405–21. [Google Scholar]
- 30.Ying Y, Tao Q. Epigenetic disruption of the wnt/ß-catenin signaling pathway in human cancers. Epigenetics. 2009;4(5):307–12. doi: 10.4161/epi.4.5.9371. [DOI] [PubMed] [Google Scholar]
- 31.Boyd S, Parikh N, Chu E, Peleato B, Eckstein J. Distributed optimization and statistical learning via the alternating direction method of multipliers. Found Trends Mach Learn. 2011;3(1):1–122. [Google Scholar]
- 32.Mazieres J, He B, You L, Xu Z, Jablons DM. Wnt signaling in lung cancer. Cancer Lett. 2005;222(1):1–10. doi: 10.1016/j.canlet.2004.08.040. [DOI] [PubMed] [Google Scholar]
- 33.Lee SM, Park J, Kim DS. Wif1 hypermethylation as unfavorable prognosis of non-small cell lung cancers with egfr mutation. Mol Cells. 2013;36(1):69–73. doi: 10.1007/s10059-013-0060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brabender J, Usadel H, Danenberg KD, Metzger R, Schneider PM, Lord RV, Wickramasinghe K, Lum CE, Park J, Salonga D, et al. Adenomatous polyposis coli gene promoter hypermethylation in non-small cell lung cancer is associated with survival. Oncogene. 2001;20(27):3528–32. doi: 10.1038/sj.onc.1204455. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering ikkbeta- and jnk1-dependent inflammation. Cancer Cell. 2010;17(1):89. doi: 10.1016/j.ccr.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei Q, Zhao Y, Yang ZQ, Dong QZ, Dong XJ, Han Y, Zhao C, Wang EH. Dishevelled family proteins are expressed in non-small cell lung cancer and function differentially on tumor progression. Lung Cancer. 2008;62(2):181–92. doi: 10.1016/j.lungcan.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 37.Winn RA, Marek L, Han SY, Rodriguez K, Rodriguez N, Hammond M, Scoyk MV, Acosta H, Mirus J, Barry N. Restoration of wnt-7a expression reverses non-small cell lung cancer cellular transformation through frizzled-9-mediated growth inhibition and promotion of cell differentiation. J Biol Chem. 2005;280(20):19625. doi: 10.1074/jbc.M409392200. [DOI] [PubMed] [Google Scholar]
- 38.Liu D, Nakano J, Ishikawa S, Yokomise H, Ueno M, Kadota K, Urushihara M, Huang CL. Overexpression of matrix metalloproteinase-7 (mmp-7) correlates with tumor proliferation, and a poor prognosis in non-small cell lung cancer. Lung Cancer. 2007;58(3):384–91. doi: 10.1016/j.lungcan.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 39.Kaneto N, Yokoyama S, Hayakawa Y, Kato S, Sakurai H, Saiki I. Rac1 inhibition as a therapeutic target for gefitinib-resistant non-small-cell lung cancer. Cancer Sci. 2014;105(7):788–94. doi: 10.1111/cas.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCall MN, Irizarry RA. Thawing frozen robust multi-array analysis (frma) BMC Bioinformatics. 2011;12(1):1. doi: 10.1186/1471-2105-12-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst. 2014;106(1):356. doi: 10.1093/jnci/djt356. [DOI] [PubMed] [Google Scholar]
- 42.Nakayama S, Sng N, Carretero J, Welner R, Hayashi Y, Yamamoto M, Tan AJ, Yamaguchi N, Yasuda H, Li D. β-catenin contributes to lung tumor development induced by egfr mutations. Cancer Res. 2014;74(20):5891–902. doi: 10.1158/0008-5472.CAN-14-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rapp J, Jaromi L, Kvell K, Miskei G, Pongracz JE. Wnt signaling-lung cancer is no exception. Respir Res. 2017;18(1):167. doi: 10.1186/s12931-017-0650-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wadsworth JL, Tawn JA. Sparse precision matrix estimation via lasso penalized d-trace loss. Biometrika. 2014;1(1):103–20. [Google Scholar]
- 45.Fan J, Li R. Variable selection via nonconvave penalized likelihood and its oracle properties. J Am Stat Assoc. 2001;96(456):1348–60. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Contains the theoretical guarantee of of the proposed methods and proofs. (PDF 284 kb)
Data Availability Statement
The gene expression data set related to lung cancer is publicly available from the Gene Expression Omnibus at accession number GDS2771.