

Abstract
Alzheimer's disease (AD) is the most frequent age-associated dementia with no treatments that can prevent or slow its progression. Since age is by far the major risk factor for AD, there is a strong rationale for an alternative approach to drug discovery based upon the biology of aging. Phenotypic screening assays that reflect multiple, age-associated neurotoxicity pathways rather than single molecular targets can identify compounds that have therapeutic efficacy by targeting aspects of aging that contribute to AD pathology. And, while the suitability of any single assay can be questioned, a combination of assays can make reliable predictions about the neuroprotective effects of compounds in vivo. Therefore, our lab has developed a combination of phenotypic screening assays that are ideally suited not only to identify novel neuroprotective compounds for the treatment of AD but also their target pathways, thereby potentially providing new therapeutic targets for disease treatment. Using these assays, we screened a large library of extracts from plants with identified pharmacological uses. Analysis of one of these extracts from the plant Yerba santa (Eriodictyon californicum) identified the flavanone sterubin as the active component and further studies showed it to be a potent neuroprotective and anti-inflammatory compound.
Keywords: Oxidative stress, Ferroptosis, Glutathione, Inflammation, Nrf2, ATF4
Graphical abstract

Highlights
-
•
Phenotypic screening of a curated library of plant extracts identifies Yerba santa.
-
•
The flavonoid sterubin is the main active component of Yerba santa.
-
•
Sterubin is very neuroprotective against multiple toxicities of the aging brain.
-
•
Sterubin has potent anti-inflammatory activity that is dependent on Nrf2 induction.
-
•
Sterubin is also an iron chelator which could enhance its neuroprotective activity.
1. Introduction
Since the 1990's, the combination of molecular and structural biology, combinatorial chemistry and high throughput screening has dominated drug discovery [1]. This approach provides a rapid process for the discovery of drug candidates with high selectivity and affinity for a specific molecular target. However, it has not produced the successes that were initially expected, especially with respect to complex neurodegenerative diseases such as Alzheimer's disease (AD).
Prior to the development of this target-based drug discovery paradigm, new drugs were discovered by evaluating chemicals against observable characteristics or phenotypes in biological systems such as cells or animals. This method is known as phenotypic screening. While this approach has fallen out of favor with the pharmaceutical industry, surprisingly a recent study showed that it still continues to be more successful than target-based approaches for the identification of first in class small molecule drugs [2]. It has been argued that this is because target-based discovery is based on a priori assumptions that do not take into account the complexities of biological systems [2], [3]. Moreover, phenotypic screening against human disease models can lead to the identification of new, disease-related molecular pathways and targets.
The ideal phenotypic drug screening paradigm would employ the ultimate end user-humans; and this is how most of the natural product-based, first in class drugs were originally discovered. However, for obvious reasons, this strategy is no longer ethically viable. Laboratory animals, primarily disease models in mice, are currently used for preclinical testing. However, using them for the initial screening of drug candidates is impractical due to cost and time constraints as well as the drive to reduce animal use in research. A reasonable alternative is to create cell-based assays based on toxicity pathways relevant to age-associated neurodegeneration and use these assays to identify novel drug candidates. In this way, the screening paradigms have disease relevance, reproducibility and reasonable throughput.
Many arguments can be made against the relevance of any single cellular screening assay, based on the cell type or the nature of the toxic insult. Thus, to account for individual weaknesses, our phenotypic screening approach combines multiple assays. This enables the identification of potent, disease-modifying compounds for preclinical testing in animal models of neurodegenerative diseases. In general, these assays involve primary neurons, neuron-like cell lines or microglial cell lines that are subjected to toxic insults that have been observed to occur in the aging brain and to a larger extent in AD.
In this report, we describe the use of these assays to screen a commercial library of extracts from plants with identified pharmacological uses and the identification of a previously uncharacterized neuroprotective flavonoid. All plant extracts were first tested in the oxytosis assay in HT22 mouse hippocampal nerve cells. Extracts that were positive in this assay were then screened in additional assays including: protection against energy depletion in HT22 hippocampal nerve cells, intracellular amyloid toxicity in MC65 human nerve cells, inhibition of inflammation mediated by microglial activation using BV-2 mouse microglial cells and differentiation of rat PC12 cells. These assays reflect multiple, age-associated neurotoxicity/survival pathways directly relevant to AD, such as increased oxidative stress and glutathione (GSH) depletion, reduced energy metabolism, accumulation of misfolded, aggregated proteins, loss of neurotrophic support and inflammation [3]. In addition, these particular models were selected to provide a replicable, cost- and time-effective screening approach.
2. Materials and methods
All reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise stated. The plant extract library was obtained from Caithness Biotechnologies (Leicester, UK). Eriodictyol and homoeriodictyol were purchased from Indofine Chemical Company (Hillsborough, NJ, USA). Sterubin was a gift from Jakob Ley at Symrise AG (Holzminden, Germany).
2.1. Phenotypic screening assays
2.1.1. Oxytosis (HT22 cells)
This assay, also called oxidative glutamate toxicity, tests the ability of compounds to rescue cells from oxidative stress-induced programmed cell death caused by GSH depletion after treatment with glutamate [4]. A reduction in GSH is seen in the aging brain and is accelerated in AD [5]. The depletion of GSH from cells leads to lipoxygenase activation, reactive oxygen species production and calcium influx which initiates a form of programmed cell death with features similar to those implicated in the nerve cell damage seen in AD [6]. Because of the generality of the toxicity pathway in oxytosis and its mechanistic association with aging and AD [7], [8], it is used as our primary screen. In this assay, 5 × 103 HT22 mouse hippocampal nerve cells, grown in high-glucose Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (FCS) (Hyclone, Logan, UT, USA), were plated in 96 well plates. After 24 h of culture, the medium was exchanged with fresh medium and 5 mM glutamate and the indicated concentrations/dilutions of extracts/fractions/compounds were added. After 24 h of treatment, viability was measured by the 3-(4, 5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) assay as previously described [9]. Results were confirmed by visual inspection of the wells.
2.1.2. Anti-inflammatory activity (BV-2 cells)
Inflammation is a major feature of AD [10]. Microglia are the resident immune cell population of the CNS and activated, pro-inflammatory microglia are implicated in the pathogenesis of AD [10]. Activated microglia produce a wide array of pro-inflammatory and cytotoxic factors including cytokines, free radicals, excitatory neurotransmitters and eicosanoids that may work in concert to promote neurodegeneration [10]. Thus, inhibiting the generation of activated pro-inflammatory microglia is another important therapeutic target for AD. Briefly, mouse BV2 microglial cells were grown in low glucose DMEM supplemented with 10% FCS. For the assay the cells were plated at 5 × 105 cells in 35 mm tissue culture dishes. After growth overnight, the cells were treated with 50 µg/ml bacterial lipopolysaccharide (LPS) alone or in the presence of the extracts/compounds. After 24 h the medium was removed, spun briefly to remove floating cells and 100 µl assayed for nitrite using 100 µl of the Griess Reagent in a 96 well plate. After incubation for 10 min at room temperature the absorbance at 550 nm was read on a microplate reader, as described previously [11]. The levels of IL-6, IL1β and TNF-α in the supernatants were determined using ELISAs (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instruction.
2.1.3. Neurotrophic activity (PC12 cell differentiation)
Connections between nerve cells are altered in AD. Thus, compounds that can promote the regeneration of these connections might be of particular benefit, thereby promoting the recovery of higher neuronal function. As a model for this property, we used neurite outgrowth in rat PC12 cells, a well-studied model system of neuronal differentiation. In response to neurotrophic factors such as nerve growth factor (NGF), PC12 cells undergo a series of physiological changes culminating in a phenotype resembling that of sympathetic neurons [12]. In this assay, PC12 cells (originally obtained from Greene [13]) grown in high-glucose DMEM supplemented with 10% FCS and 5% horse serum were plated in 35 mm tissue culture dishes. After 3 days of growth, the medium was replaced with serum-free N2 medium (Invitrogen) and the cells were treated with the extracts/compounds. After 24 h the cells were scored for the presence of neurites. PC12 cells produce neurites much more rapidly when treated in N2 medium than when treated in regular growth medium. For each treatment, 100 cells in each of three separate fields were counted. Cells were scored positive if one or more neurites longer than one cell body diameter in length were observed [14].
2.1.4. Protection against energy loss (HT22 cells)
A breakdown in neuronal energy production leading to adenosine triphosphate (ATP) loss is associated with nerve cell damage and death in AD [15]. In order to induce ATP loss, we used the compound iodoacetic acid (IAA) in combination with the HT22 hippocampal nerve cells. IAA has been used in a number of other studies to cause a rapid loss of ATP [16]. HT22 cells were seeded onto 96 well plates as described in the oxytosis assay. The medium was exchanged 24 h later with fresh medium and the cells were treated with 20 µM IAA alone (which results in 90–95% cell death) or in the presence of extracts/compounds. After 2 h the medium in each well was aspirated and replaced with fresh medium without IAA but containing the extracts/compounds. After 24 h of treatment, viability was measured by the MTT assay. Results were confirmed by visual inspection of the wells.
2.1.5. Intracellular abeta toxicity (MC65 cells)
Accumulation of intracellular amyloid beta peptide (Aβ) is considered by many as being a primary toxic event in AD [17]. The human nerve cell line MC65 conditionally expresses the C99 fragment of the amyloid precursor protein (APP) leading to the accumulation of intracellular Aβ. The MC65 cells are routinely grown in the presence of tetracycline and, following its removal, the expression of C99 is induced and the cells die within 4 days because of the accumulation of intracellular, toxic protein aggregates [18], [19]. Briefly, MC65 cells were regularly grown in high glucose DMEM supplemented with 10% FCS. For the assay, cells were dissociated and plated at 4 × 105 cells per 35 mm tissue culture dishes in Opti-minimal essential media (Opti-MEM, Invitrogen) in the presence (no induction) or absence (APP-C99 induced) of 1 μg/ml tetracycline in the presence or absence of the indicated extracts/compounds. At day 4, the control cells in the absence of tetracycline were dead, and cell viability was determined by the MTT assay, as previously described [19].
2.2. HPLC fractionation
Initially, the plant extract (1 µl of original DMSO extract) was diluted with 150 µl of 50% aqueous acetonitrile and injected onto a Vydac C-18 column (4.6 × 250 mm). A linear gradient was run over 30 min, beginning at 9% acetonitrile in 0.1% trifluoroacetic acid and increasing to 90% acetonitrile with a flow rate of 1 ml/min. Absorbance was monitored at 330 nm. One milliliter fractions were collected automatically for the entire gradient. Fractions were dried in vacuo employing a Savant Speed-Vac concentrator and fractions were re-dissolved in DMSO before testing in the oxytosis assay described above. Once the active fraction was identified, a shallower HPLC gradient was employed (40 min, beginning at 18% acetonitrile in 0.1% trifluoroacetic acid increasing to 54% acetonitrile with a flow rate of 1 ml/min) and fractions surrounding the active peak were collected manually based on absorbance at 330 nm. An aliquot of these fractions was dried in vacuo and assayed in the oxytosis assay. The active fraction was then subjected to mass spectrometric analysis.
2.3. Mass spectrometry
The fraction eluting at 19.4 min under the HPLC conditions described above was diluted 1:100 with 1% formic acid in methanol/water. The analyte was directly infused into a ThermoFisher Q Exactive mass spectrometer (Waltham, MA, USA) via a micro-flow source. A major signal was detected at m/z = 303.09 (singly charged). The signal was isolated and subjected to HCD fragmentation at a normalized collision energy setting of 45. MS/MS spectra were recorded and averaged for 2 min. After the analysis of the natural compound, spectra were also recorded for the synthetic compounds (homoeriodictyol, hesperitin, and sterubin), all of which possess a predicted precursor mass of MH+ = 303.09. Only the MS/MS spectrum of pure sterubin exhibited a match with that of the isolated natural product.
2.4. Measurement of total glutathione (tGSH)
For measurement of tGSH, 3 × 105 HT22 cells were plated in 60 mm dishes. After 24 h of culture, the medium was exchanged with fresh medium and the indicated concentrations of glutamate and the compounds were added. The cells were treated for 2–24 h and then scraped into ice-cold PBS and 10% sulfosalicylic acid was added at a final concentration of 3.3%. GSH was determined by the recycling assay based on the reduction of 5,5-dithiobis(2-nitrobenzoic acid) with glutathione reductase and NADPH [20] and normalized to protein recovered from the acid-precipitated pellet by treatment with 0.2 N NaOH at 37 °C overnight and measured by the bicinchoninic acid assay (Pierce, Rockford, IL, USA).
2.5. Reactive oxygen species (ROS) measurement
HT22 cells were seeded onto 96-well black walled microtiter plates at a density of 5 × 103 cells per well. The next day, the cells were treated with the 2.5–10 µM sterubin, eriodictyol or homoeridodictyol alone or in the presence of 5 mM glutamate for 7 h. The medium was then replaced with 100 µl loading medium (phenol red-free Hank's balanced salt solution containing 10 µM CM-H2DCFDA). After 30 min in CM-H2DCFDA (C6827, Invitrogen), the fluorescence (λ excitation = 495 nm, λ emission = 525 nM) was determined using a Molecular Devices microplate reader (San Jose, CA, USA). Each treatment was done in sextuplicate. DCFDA fluorescence was normalized to control cells not exposed to metals or glutamate.
2.6. Iron binding
Ferrous iron binding was measured using ferrozine as described [21]. Briefly, different dilutions of sterubin, eriodicyol and homoeridictyol were mixed with an equal volume of 5 µM FeCl2 in 50 mM HEPES, pH 7.5 in a 96 well plate. After 2 min, 50 µl of 5 mM ferrozine was added and the absorbance at 562 nm measured on a Molecular Devices microplate reader. The amount of remaining iron was calculated from the difference in absorbance between the sample with ferrozine and the same sample without ferrozine divided by the difference between the control sample (iron plus ferrozine) and its blank (iron alone).
2.7. Western blots
2.7.1. Sample preparation
For Western blotting, 3 × 105 HT22 cells per 60 mm dish were grown for 24 h prior to the indicated treatments. For nuclear extracts, cells were rinsed twice in ice-cold Tris-buffered saline (TBS), scraped into ice-cold nuclear fractionation buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 1 mM Na3VO4, 1× protease inhibitor cocktail and 1× phosphatase inhibitor cocktail) and incubated on ice for 15 min. Then NP40 at a final concentration of 0.6% was added, cells were vortexed and the nuclei pelleted by centrifugation. Nuclear proteins were extracted by sonication of the nuclear pellet in nuclear fractionation buffer and the extracts were cleared by additional centrifugation. Total protein extracts were prepared by rinsing the cells twice with ice-cold phosphate-buffered saline. The cells were scraped into lysis buffer (50 mM HEPES, pH 7.5, 50 mM NaCl, 50 mM NaF, 10 mM Na pyrophosphate, 5 mM EDTA, 1% Triton X-100, 1 mM Na3VO4, 1× protease inhibitor cocktail, 1× phosphatase inhibitor cocktail) and incubated on ice for 30 min. Extracts were sonicated and cleared by centrifugation. The supernatants were stored at − 70 °C until analysis. Protein concentrations were quantified by the bicinchoninic acid method (Pierce) and adjusted to equal concentrations. 5× Western blot sample buffer (74 mM Tris-HCl, pH 8.0, 6.25% SDS, 10% β-mercaptoethanol, 20% glycerol) was added to a final concentration of 2.5× and samples were boiled for 5 min.
2.7.2. Western blotting
For SDS-PAGE, equal amounts of cellular protein, typically 10–20 µg per lane, were used. All samples were separated using 10% Criterion XT Precast Bis-Tris Gels (Biorad, Hercules, CA). Proteins were transferred to nitrocellulose membranes and the quality of protein measurement, electrophoresis and transfer checked by staining with Ponceau S. Membranes were blocked with 5% skim milk in TBS-T (20 mM Tris buffer pH 7.5, 0.5 M NaCl, 0.1% Tween 20) for 1 h at room temperature and incubated overnight at 4 °C in the primary antibody diluted in 5% BSA in TBS/0.05% Tween 20. The primary antibodies used were: HRP-conjugated rabbit anti-actin (#5125, 1/20,000) from Cell Signaling (Danvers, MA, USA); rabbit anti-ATF4 (#sc-200, 1/500), rabbit anti-Nrf2 (#sc-13032, 1/500), rabbit anti-GCLC (#sc-22755, 1/1000), rabbit anti-GCLM (#sc-22754, 1/1000) from Santa Cruz Biotechnology (Dallas, TX, USA); rabbit anti-heme oxygenase 1 (#SPA-896, 1/5000) from Stressgen (Victoria, BC, Canada), mouse anti-COX2 (#610203, 1/1000) and mouse anti-iNOS (#610431, 1/1000) from BD Transduction Labs (San Jose, CA, USA) and guinea pig anti-p62 (#03-GP62-C, 1/1000) from American Research Products (Belmont, MA, USA). Subsequently, blots were washed in TBS/0.05% Tween 20 and incubated for 1 h at room temperature in horseradish peroxidase-goat anti-rabbit or goat anti-mouse (Biorad) diluted 1/5000 in 5% skim milk in TBS/0.1% Tween 20. After additional washing, protein bands were detected by chemiluminescence using the Super Signal West Pico Substrate (Pierce). For all antibodies, the same membrane was re-probed for actin or an antiserum reacting with the total protein. Autoradiographs were scanned using a Biorad GS800 scanner. Band density was measured using the manufacturer's software. Relative protein expression was normalized to actin band density. Each Western blot was repeated at least three times with independent protein samples.
2.8. Transfection
For siRNA transfection, HT22 cells were plated in 60 mm dishes at 5 × 105 cells/dish and 20 pmol ATF4 siRNA (#sc-35113), Nrf2 siRNA (#sc-37049) or control siRNA (#sc-37007), all from Santa Cruz Biotechnology were used along with RNAi max (Invitrogen) according to the manufacturer's instructions.
2.9. Determination of the Trolox equivalent activity concentration (TEAC)
TEAC values were determined as previously described [22].
2.10. Statistical analysis
Data from at least three independent experiments were normalized, pooled and analyzed using Graph Pad Prism 4 software followed by the statistical tests indicated in the figure legends. P < 0.05 was taken as significant.
3. Results
3.1. Phenotypic screening of plant extracts
We used a commercial plant extract library from Caithness Biotechnologies (http://caithnessbiotechnologies.com/) as the starting point for this study. This library was designed to maximize the potential for hit finding against diverse molecular targets while minimizing the content of potentially toxic compounds by focusing only on plants with a history of use as traditional medicines or a history of inverse association with disease risk. Moreover, this library has been carefully sized to optimally balance high molecular diversity and potential for lead finding by smaller research groups. Furthermore, the library contains only extracts from plants that are commercially available thereby negating any potential issues with recollection, sustainability or sovereignty of the discovery.
The library contains a total of 400 different plant extracts with two pre-fractions per sample, one polar and one non-polar. For this study, we focused on the non-polar extracts which were screened at a final dilution of 1/10,000 in the oxytosis assay using HT22 hippocampal nerve cells plated in 96 well plates [3]. Because of the generality of the toxicity pathway in oxytosis and its mechanistic association with both aging and AD, it is a good assay to quickly and simply screen the different extracts. Using this approach, we identified 9 extracts that provided significant protection from glutamate at the dilution used (Table 1). Further analysis of these extracts for neurotrophic activity (PC12 differentiation) and anti-inflammatory activity (BV2 cells) as well as direct toxicity (Table 1) identified two extracts, C and G, that not only were highly protective in the oxytosis assay but also had strong anti-inflammatory and neurotrophic activity and showed < 10% cell growth inhibition at the protective doses. Here we report our studies on C whose source, Yerba santa (Eriodictyon californicum), is a shrub native to California and thus of significant local interest.
Table 1.
Biological activities of the plant extracts.
| Plant extract | Oxytosis EC50(µg/ml) | BV-2 cells EC50(µg/ml) | PC12 cell differentiation | % Decrease in cell number at EC50 |
|---|---|---|---|---|
| A | 16.5 | 50 | yes | 10 |
| B | 53 | 85 | no | 0 |
| C | 20 | < 25 | yes | 9 |
| D | 14 | 103 | no | 1.5 |
| E | 43 | 179 | no | 4 |
| F | 18 | < 25 | yes | 17 |
| G | 3 | < 25 | yes | 8 |
| H | 8.3 | 22.5 | yes | 25 |
| I | 32 | 62.5 | no | 20 |
The results for the 9 most potent plant extracts in the primary screen (oxytosis assay using HT22 cells) as well as the BV-2 cell assay for anti-inflammatory activity and the PC12 cell differentiation assay. The effects of the extracts on the HT22 cell number was also determined. EC50 = half maximal effective concentration.
3.2. HPLC fractionation and screening in the oxytosis assay
Given the complex composition of compounds in a crude extract, C was fractionated by HPLC (Fig. 1A) and each fraction was then prepared identically and tested at the same dilution in the oxytosis assay (Fig. 1B). Fraction 17 was identified as the one that protected the most with ~ 50% cell survival (Fig. 1B). No other fractions showed any significant protection. Therefore, this fraction was selected for further study.
Fig. 1.

Purification and Identification of sterubin. (A) Fractionation of Yerba Santa extract by reversed phase HPLC. Conditions were similar to the ones described in Methods but a gradient from 9% to 90% acetonitrile within 30 min was employed. One minute fractions were collected. (B) Survival assay of HPLC fractions. HPLC fractions were dried in vacuo and reconstituted in DMSO before being tested in the oxytosis assay. (C) MS spectrum of active fraction. A major signal was detected at m/z = 303.09. Signals which were also present in the blank sample are labeled by an asterisk (*). (D) MS/MS spectrum of the isolated m/z = 303.09 signal. (E) MS/MS spectrum of pure sterubin recorded under the same conditions as for the natural material. The fragmentation pattern is essentially identical to one of the natural isolate. (F) MS/MS spectrum of synthetic homoeriodictyol recorded under the same conditions as for sterubin.
3.3. Identification and purification of the flavonoid sterubin
In order to determine the composition of fraction 17 and possibly identify individual biologically active compounds, the fraction was analyzed by mass spectrometry. A strong peak was identified with a molecular weight (MW) identical to two flavonoids known to be present in Yerba santa, homoeriodictyol and sterubin (MW = 302) (Fig. 1C) [23]. The mass fragmentation pattern was also determined to serve as a fingerprint to identify the compound in the active fraction (Fig. 1D). Homoeriodictyol and sterubin were obtained from commercial sources and their HPLC elution times and mass fragmentation patterns determined. While both characteristics of homoeriodictyol were distinct from those of the active fraction, those of commercially sourced sterubin (Fig. 1E) were almost identical strongly suggesting that the active compound of this fraction was sterubin.
3.4. Validation of the biological activity of sterubin in screening assays
In order to determine whether the potent biological activity of C could be explained by the presence of sterubin, the biological activity of the purified compound was tested in all of the different screening assays (Table 2). For comparison, the EC50 values for homoeriodictyol and eriodictyol, two other flavonoids which are also present in Yerba santa and have been much more widely studied, are also shown. In addition, the EC50 values for fisetin are included. Fisetin is a neuroprotective and cognition-enhancing molecule which our laboratory has identified using some of the same screening assays [14], [24] and that we have recently shown to protect in in vivo models of dementia and AD [25], [26]. The results show that sterubin is indeed a highly effective compound in all of the assays. In particular, sterubin is very effective at inhibiting intracellular Abeta toxicity with an EC50 less than 1 µM. While eriodictyol has weaker but still significant activity in the same assays, homoeriodictyol is completely inactive at concentrations up to 10 µM. Importantly, the EC50 values of sterubin were substantially better than those of fisetin.
Table 2.
Biological activities of sterubin, eriodictyol, homoeriodictyol and fisetin in the five phenotypic screening assays.
| Compound | Protection against oxytosis EC50(µM) | Protection against energy loss EC50(µM) | Anti-inflammatory activity EC50(µM) | Neurotrophic activity EC50(µM) | Intracellular abeta toxicity EC50(µM) | TEAC |
|---|---|---|---|---|---|---|
| Sterubin | 0.8 | 0.9 | 1.4 | 5 | 0.8 | 0.6 |
| Eriodictyol | 2.9 | 1 | 4.1 | > 10 | 2.0 | 0.6 |
| Homoeriodictyol | > 10 | > 10 | > 10 | > 10 | > 10 | 0.08 |
| Fisetin | 3 | 3 | 2.5 | 5 | 3.3 | 3.0 |
The EC50s for the three flavonoids found in Yerba santa (sterubin, eriodictyol, homoeriodictyol) along with fisetin as a positive control in the 5 different phenotypic screening assays as well as their TEAC values, a measure of direct anti-oxidant activity, were determined as described in Section 2.
3.5. Mechanisms underlying the beneficial activities of sterubin
The steps involved in oxytosis have been well characterized. The first step involves the loss of the major endogenous intracellular antioxidant glutathione (GSH). Thus, we first asked if sterubin could preserve GSH levels. As shown in Fig. 2, sterubin dose-dependently increased GSH levels in control cells (Fig. 2A, B) and maintained the levels of GSH in cells treated with glutamate to induce oxytosis (Fig. 2A, B). Eriodictyol was less effective at both increasing control GSH levels and maintaining GSH in the presence of glutamate (Fig. 2B) while homoeriodictyol was completely ineffective (Fig. 2B). Consistent with the ability of sterubin to maintain GSH levels, it also provided excellent, dose-dependent protection against two inducers of ferroptosis, a cell death pathway closely related, if not identical, to oxytosis [7]. Erastin, similar to glutamate, blocks system xc- activity while RSL3 is an inhibitor of glutathione peroxidase 4 that is downstream of GSH loss and plays a key role in the cell death process in both oxytosis and ferroptosis [7] (Fig. 2C).
Fig. 2.

Neuroprotective properties of sterubin. (A) Dose dependent effects of sterubin on HT22 cell total GSH levels in the absence or presence of 5 mM glutamate. GSH levels were measured after 24 h with a chemical assay. Results are the average of 3 independent experiments. *** p < 0.001 relative to control. (B) Comparison of the effects of 10 µM sterubin, eriodictyol and homoeriodictyol on basal GSH levels and GSH levels in the presence of 5 mM glutamate. GSH levels were measured after 24 h with a chemical assay. Results are the average of 3 independent experiments. *** p < 0.001 relative to control; # p < 0.05 relative to glutamate alone; ### p < 0.001 relative to glutamate alone. (C) Dose dependent effects of sterubin on HT22 cell survival in the presence of 500 nM erastin or 100 nM RSL3. Cell survival was measured after 24 h with the MTT assay. Results are the average of 3 independent experiments. (D) Dose dependent effects of sterubin and eriodictyol on basal and glutamate-induced ROS levels. Cells were treated with 5 mM glutamate alone or in the presence of the indicated concentrations of the compounds. After 7 h, ROS levels were determined using CM-H2DCFDA as described in Experimental Procedures. The treatments were done in sextuplicate and the results are the average of 4 independent experiments. (E) Dose dependent effects of sterubin on HT22 cell survival in the presence of 5 µM tBOOH or 750 µM H2O2. Cell survival was measured after 24 h with the MTT assay. (F) Iron binding of sterubin, eriodictyol and homoeridictyol. Iron (Fe+2) binding by the flavanones was measured using the ferrozine assay at flavanone:iron ratios of 10:1, 5:1, 2:1 and 1:1. Results are presented as percent of Fe+2 chelated relative to controls with no flavonoid and are the average of 3 independent experiments.
Downstream of GSH loss in oxytosis is an increase in ROS levels. As shown in Fig. 2D, sterubin strongly and dose-dependently reduced glutamate-induced ROS production. Eriodictyol was less effective (Fig. 2D) and homoeriodictyol had no effect (not shown). Interestingly, sterubin also significantly reduced the basal levels of ROS (Fig. 2D) prompting an examination of the effects of sterubin against two direct pro-oxidants, H2O2 and t-butyl peroxide (tBOOH), both of which can also promote cell death. As shown in Fig. 2E, sterubin was surprisingly effective against cell death induced by either H2O2 or tBOOH with EC50s of ~ 1 µM. These results prompted the measurement of the sterubin's TEAC value which was quite low (Table 2) suggesting that it is unlikely to act as a direct antioxidant.
Iron can promote cell death through its ability to generate ROS. Since ferroptosis is described as an iron dependent form of cell death [27] and iron chelators also protect from oxytosis [7], we next asked if sterubin can chelate iron, which might contribute to its antioxidant actions. To test its iron binding, we used the ferrozine assay [21] which looks at the ability of a compound to prevent iron binding to ferrozine. As shown in Fig. 2F, sterubin is quite an effective iron chelator even at ratios of 1:1 flavonoid:iron. While eriodictyol showed similar activity, homoeriodictyol demonstrated very little iron binding even at a ratio of 10:1 flavonoid:iron.
Two transcription factors contribute to the upregulation of GSH, Nrf2 and ATF4 [5]. Thus, it was next asked if sterubin can alter the levels of these proteins in the HT22 cells. As shown in Fig. 3A, sterubin dose-dependently increased nuclear Nrf2 levels to a maximum of 6-fold at 5 µM. This increase was seen within 30 min of sterubin treatment and remained constant for up to 6 h (not shown). Eriodictyol also induced Nrf2 with similar efficacy as sterubin (Fig. 3B). In contrast, homoeriodictyol had a much lesser effect on Nrf2 levels (not shown). Since we have shown that ATF4 plays an important role in the maintenance of GSH levels in nerve cells [28], we also examined the effects of the three flavonoids on ATF4 levels using the same nuclear extracts. As shown in Fig. 3C, sterubin dose-dependently increased nuclear ATF4 levels to a maximum of 3-fold at 5 µM and eriodictyol had a similar effect. As with Nrf2, the increase in nuclear ATF4 was seen within 30 min of treatment and remained constant for up to 6 h (not shown).
Fig. 3.

Nrf2 induction by sterubin. HT22 cells were treated with increasing concentrations of sterubin or eriodictyol for 4 h. Nuclei were prepared and examined by Western blotting for Nrf2 and ATF4. Representative blots are shown (A). (B) and (C) Quantification of the results from 3 independent experiments as shown in (A). * p < 0.05; ** p < 0.01; *** p < 0.001. (D) HT22 cells were treated with 10 µM sterubin for 2–8 h and whole cell extracts examined for the indicated proteins by Western blotting. Similar results were obtained in 3 independent experiments.
Nrf2 regulates the expression of multiple enzymes involved in GSH metabolism and protection against oxidative stress [for review see [29]]. Thus, we examined the levels of some of these proteins by Western blotting. As observed in Fig. 3D, 10 µM sterubin increased the levels of one of the glutamate cysteine ligase (GCL) subunits (GCLM) within 6 h of treatment (1.37-fold). In addition to GCLM, sterubin increased the levels of p62 (22.7-fold) and heme oxygenase 1 (HO-1) (4.1-fold), two proteins involved in protection against oxidative stress and known to be regulated by Nrf2 [30], [31] with a similar time course (Fig. 3D) further demonstrating that sterubin induces a variety of anti-oxidant proteins.
Yerba santa has long been known for its anti-inflammatory properties [32] and in our phenotypic screens we found that sterubin had potent anti-inflammatory activity in BV-2 microglial cells whereas eriodictyol was less effective and homoeridodicytol had no effect at concentrations up to 10 µM (Table 2). To expand upon this observation, the BV-2 cells were treated with different doses of sterubin or eriodictyol in the presence of LPS and the production of NO, IL-6, IL-1β and TNFα were determined on culture supernatants and the levels of iNOS and COX2 assessed in cell extracts. Sterubin strongly and dose-dependently reduced the production of NO, IL-6 and IL1β (Fig. 4A–C) as well as the levels of both iNOS and COX2 (Fig. 5A, B). A lesser effect was seen on the induction of TNFα (Fig. 4D). Eriodictyol was much less effective (Fig. 4A–D) and homoeriodictyol, even at 5–10 µM, had little or no effect on any of these parameters (not shown). Sterubin also dose-dependently increased nuclear Nrf2 and ATF4 levels in the BV-2 cells (Fig. 5C–E) suggesting that either or both of these transcription factors might be a common target for both its neuroprotective and anti-inflammatory effects.
Fig. 4.

Sterubin reduces microglial activation. Dose dependent effects of sterubin on the production of NO and pro-inflammatory cytokines by LPS-treated BV-2 microglial cells. BV-2 cells were treated overnight with 50 ng/ml LPS alone or in the presence of sterubin or eriodictyol. Cell culture supernatants were cleared and assayed for NO by the Griess assay or pro-inflammatory cytokines using specific ELISAs. Results are presented as the percent (%) of the value obtained with LPS alone which was set at 100%. (A) NO; (B) IL6; (C) IL-1β; (D) TNFα. Results represent the average of 3–4 independent experiments. Sterubin is significantly more effective as compared with eriodictyol at all of the concentrations tested in all of the assays.
Fig. 5.

Effects of sterubin on pro-inflammatory proteins. (A) Dose dependent effects of sterubin on iNOS and COX2 production in LPS-treated BV-2 microglia. Cells were treated overnight with LPS alone or in the presence of the indicated concentrations of sterubin. Whole cell extracts were prepared for Western blotting. (B) Quantitation of the results shown in (A) ** p < 0.01 relative to LPS treated cells. (C) Sterubin induces Nrf2 and ATF4 in BV-2 microglial cells. Cells were treated for 4 h with the indicated concentrations of sterubin or eriodictyol or 10 µM homoeriodictyol (D and E) Quantitation of the results shown in (C). All results are the average of 3 independent experiments. ** p < 0.01, *** p < 0.001 relative to untreated cells.
To test this idea, we first downregulated Nrf2 in the HT22 nerve cells and the BV-2 microglia using specific siRNA. In the HT22 cells, the Nrf2 levels were decreased by ~ 90% (Fig. 6A) while in the BV-2 cells the Nrf2 levels were decreased by ~ 80% (Fig. 7A). In the HT22 nerve cells, knockdown of Nrf2 significantly shifted the dose-response curve for neuroprotection by sterubin in the oxytosis assay (Fig. 6B). In contrast, Nrf2 knockdown had little or no effect on the protective effects of sterubin against H2O2 or tBOOH (Fig. 6B, C) suggesting that these actions were mediated by as yet unidentified pathways. In the BV-2 cells, the effects of Nrf2 knockdown were much more dramatic with the EC50s for inhibition of NO, IL-6 and IL-1β production shifted from 1–2 µM to greater than 10 µM (Fig. 7B–D). Thus, Nrf2 appears to play a key role in the anti-inflammatory effects of sterubin and contribute to its neuroprotective activity in the HT22 cells.
Fig. 6.

Nrf2 siRNA but not ATF4 siRNA reduces sterubin-induced increases in cell survival. HT22 cells were transfected with control siRNA, ATF4 siRNA or Nrf2 siRNA 24 h before seeding. (A) Nuclear extracts from untreated and sterubin-treated (4 h, 10 µM) cells were analyzed by Western blotting for Nrf2 or ATF4 with actin as a loading control. (B–D) Transfected HT22 cells in 96-well plates were treated with 5 mM glutamate (B), 10 µM tBOOH (C) or 750 µM H2O2(D) and the indicated concentrations of sterubin. MTT reduction was measured after 24 h. Results in (B)–(D) are the average of three independent experiments. ** p < 0.01 relative to control siRNA transfection.
Fig. 7.

Nrf2 siRNA but not ATF4 siRNA reduces the anti-inflammatory effects of sterubin. BV-2 cells were transfected with control siRNA, ATF4 siRNA or Nrf2 siRNA 24 h before seeding. (A) Nuclear extracts from untreated and sterubin-treated (4 h, 10 µM) cells were analyzed by Western blotting for Nrf2 or ATF4 with actin as a loading control. (B–D) Transfected BV2 cells were treated overnight with 50 ng/ml LPS and the indicated concentrations of sterubin. Cell culture supernatants were cleared and assayed for NO by the Griess assay or pro-inflammatory cytokines using specific ELISAs. Results are presented as the percent (%) of the value obtained with LPS alone which was set at 100%. NO (B); IL6 (C) or IL-1β (D). Results in (B)–(D) are the average of three independent experiments. * p < 0.05 relative to control siRNA transfection; ** p < 0.01 relative to control siRNA transfection; *** p < 0.001 relative to control siRNA transfection.
Since sterubin also induces ATF4 (Fig. 3), we looked at the effects of ATF4 knockdown on the activities of sterubin in both BV2 microglia and HT22 nerve cells. In contrast to the results with Nrf2 knockdown, loss of ATF4 by 80–85% in the HT22 cells (Fig. 6A) had little or no effect on neuroprotection against glutamate (Fig. 6) and actually enhanced the neuroprotective effects of sterubin against H2O2 or tBOOH (Fig. 6B, C). These results further support the idea that these actions by sterubin are mediated by pathways distinct from those involving ATF4 or Nrf2. Similarly, loss of ATF4 by ~ 80% (Fig. 7A) had no effect on the anti-inflammatory activity of sterubin (Fig. 7B–D).
4. Discussion
In this manuscript, we show for the first time that the most significant neuroprotective flavonoid in Yerba santa is the flavanone sterubin. Although sterubin has long been known to be present in Yerba santa [23], it has remained remarkably unstudied with most reports focusing on the other Yerba santa flavonoids, eriodictyol or homoeriodicytol. Moreover, studies which have identified sterubin in various screens have often ignored it based on the incorrect assumption that it was already well studied [eg 33]. While eriodictyol has some of the same properties as sterubin, albeit with significantly higher EC50s, homoeriodictyol is essentially inactive in most of our assays at concentrations up to 10 µM.
Yerba santa has long been known to have medicinal properties. For example, a treatise published in 1902 on plants used by the native people in Mendocino County, California [34] states that “No plant is more highly valued as a medicine by all the tribes of Mendocino County.” Historically, it was used to treat a variety of indications including various respiratory conditions and fever as well as bruises, infections and pain including headaches [32], [34]. Thus, its anti-inflammatory properties are well documented, consistent with the potent anti-inflammatory effects of sterubin in our in vitro assay.
Although sterubin has not previously been studied in the context of neuroprotection, there are a number of reports on eriodictyol. However, consistent with our results, eriodictyol was previously shown not to be effective either against oxytosis in HT22 cells or LPS-induced nitrite production in BV-2 microglial cells [35]. Nevertheless, other studies have found that eriodictyol could reduce both oxidative stress and inflammation in both cell culture and animal models [36], [37], [38], [39]. The in vitro neuroprotective effects were at least partially dependent on Nrf2 [36], [37] but required relatively high concentrations of the flavonoid (> 20 µM). This is in contrast to the strong effects seen in this study with sterubin even at 1–2 µM. Although it has recently been reported that homoeriodictyol can also induce Nrf2 and protect against oxidative stress in endothelial cells [40], we did not observe a similar effect in the HT22 or BV-2 cells.
Previously, we showed that the flavonol fisetin could upregulate both Nrf2 and ATF4 [41] and, at the time, this appeared to be the first molecule described which could do so. As shown in this manuscript, sterubin, as well as eriodictyol, exhibit a similar property. However, in contrast to fisetin [41], Nrf2 rather than ATF4 plays a role in the neuroprotective effects of sterubin. Nrf2 is also critical to the anti-inflammatory actions of sterubin. Nrf2 has long been known to have both cytoprotective [31] and anti-inflammatory activity [42]. The cytoprotective activity is related to its ability to simultaneously upregulate the transcription of genes encoding a variety of antioxidant and detoxification proteins including those that regulate intracellular GSH homeostasis, major components of the thioredoxin system which modulates protein oxidation, and proteins involved in heme and iron metabolism. A number of these proteins, such as HO-1, also contribute to the anti-inflammatory effects of Nrf2 [42]. Nrf2 can also directly suppress the transcription of some pro-inflammatory cytokines including IL-6 and IL-1β [43] consistent with our data in the BV-2 cells.
In addition, we show that sterubin is an effective iron chelator. Since iron can contribute to nerve cell damage in aging and neurodegenerative diseases [8], this property is likely to contribute to the neuroprotective activity of sterubin both in vitro and potentially in vivo. Importantly, we recently showed that iron can potentiate GSH loss and cell death [20] in response to compounds that promote GSH loss but not in response to other oxidants. This finding might help to explain at least some of the protective activity of sterubin that is not dependent on Nrf2 in the glutamate-treated HT22 cells.
We also show that sterubin is effective against multiple inducers of cell death that activate distinct cell death pathways. Importantly, not only is sterubin very effective against oxytosis induced by glutamate treatment but also against inducers of the very closely related ferroptotic pathway [7] including erastin which has the same target as glutamate and RSL3 which inhibits glutathione peroxidase 4, a downstream target of this pathway [7]. This finding indicates that not only does sterubin protect against oxytosis/ferroptosis by maintaining GSH levels under conditions of stress but it also acts downstream of GSH loss. In addition, sterubin prevents cell death induced by both H2O2 and tBOOH, although in contrast to the protection against oxytosis, Nrf2 does not seem to be involved in sterubin-mediated protection against these two agents. This protection may involve the iron chelating activity of sterubin as iron chelators can protect against H2O2 and tBOOH toxicity (not shown). Importantly, sterubin is also very effective at protecting the MC65 nerve cells against the toxic accumulation of intracellular amyloid beta peptide which is considered by many to be a primary toxic event in AD [17].
An important property that distinguishes sterubin from either eriodictyol or homoeriodictyol is its ability to induce PC12 cell differentiation (Table 2). We [3] and others [44] have suggested that the ability to activate neurotrophic signaling pathways is an important characteristic of potential neuroprotective compounds. Indeed, treatments that only activate anti-oxidant systems may not be adequate to address the numerous changes that occur in the aging brain and that are exacerbated in AD. Thus, a compound such as sterubin that not only activates global anti-oxidant systems via Nrf2 but also mimics neurotrophic signaling may be particularly effective for the treatment of neurodegenerative diseases.
By combining the analyses of the neuroprotective activities of sterubin, eriodictyol, homoeriodictyol, and fisetin in our phenotypic screening assays (Table 2) with their physicochemical properties (Table 3), we were able to elaborate a plausible structure-activity relationship (SAR). The catechol B-ring with phenolic hydroxyls on sterubin is essential for its neuroprotective activities. Comparing their chemical structures, sterubin, eriodictyol, and fisetin contain a 3′,4′-dihydroxy on the catechol B-ring and they are all active in our cell-based assays with EC50 values less than 5 µM. In contrast, homoeriodictyol, which contains a 3′-methoxy 4′-hydroxy on the B-ring, is inactive (EC50s > 10 µM). For flavonoids, free phenolic hydroxyl groups on the catechol ring provide hydrogen bonds important for molecular recognition. They are key structural features known to be responsible for the radical scavenging, anti-oxidant, anti-inflammatory and anti-protein aggregation activities [45], [46]. Because our phenotypic screens in cell culture models mimic several aspects of the changes seen in the aging brain including increases in oxidative stress, energy loss, neuroinflammation, and intracellular Aβ aggregation, it is logical that we identified sterubin, eriodictyol, and fisetin as being active in these assays. Interestingly, sterubin has a relatively low TEAC value supporting the idea that it protects from oxidative stress-induced death via an indirect anti-oxidant mechanism. In addition, the ortho-dihydroxyls on the B-ring play a role in chelation of oxidizing metal ions [45]. This was confirmed in the present study which showed that sterubin and eriodictyol effectively bind Fe2+ in a dose-dependent manner while homoeriodictyol does not (Fig. 2F). On the other hand, the hydroxyl groups in the A-ring of sterubin are not critical for the neuroprotection but could play a modulating role. Sterubin is a derivative of eriodictyol where the only difference is the 7-methoxy group on the A-ring of sterubin. Such O-methylation on the A-ring increases the overall lipophilicity of sterubin (ClogP, 2.37) compared to eriodictyol (ClogP, 1.85). It also decreases the total polar surface area (tPSA) from 107 to 96 Å and reduces the number of hydrogen bond donors (HBD) to 3 (Table 3). It is conceivable that such changes in its physiochemical properties increase, at least in part, the passive membrane permeability of sterubin for better target engagement in cells, which in turn contributes to the approximately 3-fold increase of neuroprotective potency in comparison with eriodictyol (Table 2). In addition, methylation of flavonoids has been reported to improve their bioavailability and metabolic stability [47].
Table 3.
Structures and physiochemical properties of sterubin, eriodictyol, homoeriodictyol and fisetin.
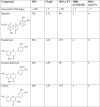 |
Successful CNS drugs should present molecular weights (MW) ≤ 400, CLogP ≤ 5, total polar surface area (tPSA) ≤ 90, hydrogen bond donors (HBD) ≤ 3 and hydrogen bond acceptors (HBA) ≤ 7 to improve their penetration of the BBB [53], [54]. The physicochemical properties were predicted using ChemBioDraw software and Molinspiration Chemoinformatics (http://www.molinspiration.com/cgi-bin/properties).
Sterubin is also about 4-fold more potent in the phenotypic screening assays than fisetin, an AD drug candidate [25], [26]. Despite their structural differences in the A- and C-rings, sterubin has a higher ClogP, a smaller tPSA, and one less HBD compared to fisetin. Again, in support of our hypothesis, this may result in sterubin being more cell membrane permeable and thereby more potent than fisetin. Touil et al. [48] previously reported that geraldol, a 3′-O-methylated derivative of fisetin, is an active metabolite in mice and shows a higher potency of cytotoxic and antiangiogenic activities in vitro. Although those compounds were tested for anticancer rather than neuroprotective activities, this provides additional evidence that methylation of flavonoids can modulate their physiochemical properties and potency and the methylation position may play a distinct roles in modulating different biochemical. Strikingly, sterubin meets the desirable physicochemical criteria to be a viable CNS drug candidate (Table 3), and it has a higher score of CNS druglikeness than fisetin [49]. However, it will be important to investigate the sterubin metabolites and pharmacokinetics in vivo in order to provide a better understanding of the pharmacological behavior of sterubin.
Finally, one of the known properties of sterubin that overlaps with those of eriodictyol and homoeriodictyol is its bitter masking property [23]. Studies have shown that all three flavonoids are antagonists of bitter taste receptors (T2Rs) [50]. While bitter receptors are best known and studied for their role in taste, recent studies have shown that these receptors are also located in multiple regions of the brain [51] where their roles are presently unknown. However, there is evidence for the upregulation of some of these receptors in AD as well as other neurodegenerative diseases [52]. Thus, a compound such as sterubin which can act as a T2R receptor antagonist might have additional, and as yet undescribed, beneficial effects in vivo.
Together these studies provide further support for the use of our set of phenotypic screening assays to identify more potent neuroprotective molecules. In this report, we used a curated library of plant extracts and found that 9/400 (2.25%) extracts were highly active. Using the oxytosis assay as our primary screen, we identified extracts that are extremely effective against a form of cell death that is associated with a variety of age-associated diseases including AD [7]. The subsequent identification of sterubin in one of the most active extracts shows that even compounds which are generally thought of as well studied [33] can provide surprising results. Thus, based on its potent Nrf2-inducing as well as neurotrophic, anti-inflammatory and iron chelating properties, we believe that sterubin deserves further examination in the context of AD, as well as other neurodegenerative diseases.
Acknowledgements
We would like to thank Jakob Ley from Symrise AG for the sterubin.
Acknowledgments
Funding sources
This work was supported by funding from NIH (RO1 AG046153 and RF1 AG054714), the Edward N. & Della Thome Memorial Foundation and the Paul F. Glenn Center for Aging Research at the Salk Institute.
References
- 1.Hao H., Zheng X., Wang G. Insights into drug discovery from natural medicines using reverse pharmacokinetics. Trends Pharmacol. Sci. 2014;35:168–177. doi: 10.1016/j.tips.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Swinney D.C. Phenotypic vs. target-based drug discovery for first-in-class medicines. Clin. Pharm. Ther. 2013;93:299–301. doi: 10.1038/clpt.2012.236. [DOI] [PubMed] [Google Scholar]
- 3.Prior M., Chiruta C., Currais A., Goldberg J., Ramsey J., Dargusch R., Maher P.A., Schubert D. Back to the future with phenotypic screening. ACS Chem. Neurosci. 2014;5:503–513. doi: 10.1021/cn500051h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan S., Schubert D., Maher P. Oxytosis: a novel form of programmed cell death. Curr. Top. Med. Chem. 2001;1:497–506. doi: 10.2174/1568026013394741. [DOI] [PubMed] [Google Scholar]
- 5.Currais A., Maher P. Functional consequences of age-dependent changes in glutathione status in the brain. Antioxid. Redox Signal. 2013;19:813–822. doi: 10.1089/ars.2012.4996. [DOI] [PubMed] [Google Scholar]
- 6.Sonnen J.A., C B.J., Lovell M.A., Markesbery W.R., Quinn J.F., Montine T.J. Free radical-mediated damage to brain in Alzheimer's disease and its transgenic mouse models. Free Radic. Biol. Med. 2008;45:219–230. doi: 10.1016/j.freeradbiomed.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewerenz J., Ates G., Methner A., Conrad M., Maher P. Oxytosis/ferroptosis-(re-)emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases on the central nervous system. Front. Neurosci. 2018;12:214. doi: 10.3389/fnins.2018.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lane D.J.R., Ayton S., Bush A.I. Iron and Alzheimer's disease: an update on emerging mechanisms. J. Alzheimer's Dis. 2018;64:S379–S395. doi: 10.3233/JAD-179944. [DOI] [PubMed] [Google Scholar]
- 9.Davis J.B., Maher P. Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell lines. Brain Res. 1994;652:169–173. doi: 10.1016/0006-8993(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 10.Wyss-Coray T., Rogers J. Inflammation in Alzheimer's disease – a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012;2:a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiruta C., Schubert D., Dargusch R., Maher P. Chemical modification of the multi-target neuroprotective compound fisetin. J. Med. Chem. 2012;55:378–389. doi: 10.1021/jm2012563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keegan K., Halegoua S. Signal transduction pathways in neuronal differentiation. Curr. Opin. Neurobiol. 1993;3:14–19. doi: 10.1016/0959-4388(93)90029-x. [DOI] [PubMed] [Google Scholar]
- 13.Greene L., Tischler A.S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sagara Y., Vahnnasy J., Maher P. Induction of PC12 cell differentiation by flavonoids is dependent upon extracellular signal-regulated kinase activation. J. Neurochem. 2004;90:1144–1155. doi: 10.1111/j.1471-4159.2004.02563.x. [DOI] [PubMed] [Google Scholar]
- 15.Saxena U. Bioenergetics failure in neurodegenerative diseases: back to the future. Expert Opin. Ther. Targets. 2012;16:351–354. doi: 10.1517/14728222.2012.664135. [DOI] [PubMed] [Google Scholar]
- 16.Maher P., Salgado K.F., Zivin J.A., Lapchak P.A. A novel approach to screening for new neuroprotective compounds for the treatment of stroke. Brain Res. 2007;1173:117–125. doi: 10.1016/j.brainres.2007.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LaFerla F.M., Green K.N., Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat. Rev. Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 18.Sopher B.L., Fukuchi K., Smith A.C., Leppig K.A., Furlong C.E., Martin G.M. Cytotoxicity mediated by conditional expression of a carboxyl-terminal derivative of the beta-amyloid precursor protein. Brain Res. Mol. Brain Res. 1994;26:207–217. doi: 10.1016/0169-328x(94)90092-2. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y., Dargusch R., Maher P., Schubert D. A broadly neuroprotective derivative of curcumin. J. Neurochem. 2008;105:1336–1345. doi: 10.1111/j.1471-4159.2008.05236.x. [DOI] [PubMed] [Google Scholar]
- 20.Maher P. Potentiation of glutathione loss and nerve cell death by the transition metals iron and copper: implications for age-related neurodegenerative diseases. Free Rad. Biol. Med. 2017;115:92–104. doi: 10.1016/j.freeradbiomed.2017.11.015. [DOI] [PubMed] [Google Scholar]
- 21.Mladenka P., Macakova K., Filipsky T., Zatloukalova L., Jahodar L., Bovicelli P., Silvestri I.P., Hrdina R., Saso L. In vitro analysis of iron chelating activity of flavonoids. J. Inorg. Biochem. 2011;105:693–701. doi: 10.1016/j.jinorgbio.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Maher P. A comparison of the neurotrophic activities of the flavonoid fisetin and some of its derivatives. Free Radic. Res. 2006;40:1105–1111. doi: 10.1080/10715760600672509. [DOI] [PubMed] [Google Scholar]
- 23.Ley J.P., Krammer G., Reinders G., Gatfield I.L., Bertram H.J. Evaluation of bitter masking flavanones from Herba Santa (Eriodictyon californicum (H. and A.) Torr., Hydrophyllaceae) J. Agric. Food Chem. 2005;53:6061–6066. doi: 10.1021/jf0505170. [DOI] [PubMed] [Google Scholar]
- 24.Ishige K., Schubert D., Sagara Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic. Biol. Med. 2001;30:433–446. doi: 10.1016/s0891-5849(00)00498-6. [DOI] [PubMed] [Google Scholar]
- 25.Currais A., Prior M., Dargusch R., Armando A., Ehren J., Schubert D., Quehenberger O., Maher P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer's disease transgenic mice. Aging Cell. 2014;13:379–390. doi: 10.1111/acel.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Currais A., Farrokhi C., Dargusch R., Armando A., Quehenberger O., Schubert D., Maher P. Fisetin reduces the impact of aging on behavior and physiology in the rapidly aging SAMP8 mouse. J. Gerentol. A Biol. Sci. Med. Sci. 2018;73:299–307. doi: 10.1093/gerona/glx104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., Morrison B., Stockwell B.R. Ferroptosis: an iron-dependent form of non-apoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewerenz J., Maher P. Basal levels of eIF2α phosphorylation determine cellular antioxidant status by regulating ATF4 and xCT expression. J. Biol. Chem. 2009;284:1106–1115. doi: 10.1074/jbc.M807325200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kensler T.W., Wakabayashi N., Biswal S. Cell Survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 30.Jain A., Lamark T., Sjottem E., Larsen K.B., Awuh J.A., Overvatn A., McMahon M., Hayes J.D., Johansen T. p62/SQSTM1 is a target gene for transcription factor Nrf2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayes J.D., Dinkova-Kostova A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Moore M.J. Red Crane Books; Santa Fe: 1993. Medicinal Plants of the Pacific West. [Google Scholar]
- 33.Yang J., Liang Q., Wang M., Jeffries C., Smithson D., Tu Y.-C., Boulos N., Jacob M.R., Shelat A.A., Wu Y., Ravu R.R., Gilbertson R., Avery M.A., Khan I.A., Walker L.A., Guy R.K., Li X.C. UPLC-MS-ELSD-PDA as a powerful dereplication tool to facilitate compound identification from small-molecule natural product libraries. J. Nat. Prod. 2014;77:902–909. doi: 10.1021/np4009706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chestnut V.K. US Government Printing Office; California: 1902. Plants used by the Indians of Mendocino County. [Google Scholar]
- 35.Cho N., Choi J.H., Yang H., Jeong E.J., Lee K.Y., Kim Y.C., Sung S.H. Neuroprotective and anti-inflammatory effects of flavonoids isolated from Rhus verniciflua in neuronal HT22 and microglial BV2 cell lines. Food Chem. Toxicol. 2012;50:1940–1945. doi: 10.1016/j.fct.2012.03.052. [DOI] [PubMed] [Google Scholar]
- 36.Lou H., Jing X., Ren D., Wei X., Zhang X. Eriodictyol protects against H2O2-induced neuron-like PC12 cell death through activation of Nrf2/ARE signaling pathway. Neurochem. Int. 2012;61:251–257. doi: 10.1016/j.neuint.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 37.Jing X., Shi H., Zhu X., Wei X., Ren M., Han M.Y., Ren D., Lou H. Eriodictyol attenuates b-amyloid 25–35 peptide-induced oxidative death in primary cultured neurons by activation of Nrf2. Neurochem. Res. 2015;40:1463–1471. doi: 10.1007/s11064-015-1616-z. [DOI] [PubMed] [Google Scholar]
- 38.Ferreira P.S., Spolidorio L.C., Manthey J.A., Cesar T.B. Citrus flavanones prevent systemic inflammation and ameliorate oxidative stress in C57BL/6J mice fed high-fat diet. Food Funct. 2016;7:2675–2681. doi: 10.1039/c5fo01541c. [DOI] [PubMed] [Google Scholar]
- 39.Ferreira E.O., Fernandes M.Y., Lima N.M., Neves K.R., Carmo M.R., Lima F.A., Fonteles A.A., Menezes A.P., Andrade G.M. Neuroinflammatory response to experimental stroke is inhibited by eriodictyol. Behav. Brain Res. 2016;312:321–332. doi: 10.1016/j.bbr.2016.06.046. [DOI] [PubMed] [Google Scholar]
- 40.Shen T., Li H.Z., Li A.L., Li Y.R., Wang X.N., Ren D.M. Homoeriodictyol protects human endothelial cells against oxidative insults through activation of Nrf2 and inhibition of mitochondrial dysfunction. Vasc. Pharmacol. 2018;1537:30443. doi: 10.1016/j.vph.2018.06.007. [DOI] [PubMed] [Google Scholar]
- 41.Ehren J.L., Maher P. Concurrent regulation of the transcription factors Nrf2 and ATF4 mediates the enhancement of glutathione levels by the flavonoid fisetin. Biochem. Pharmacol. 2013;85:1816–1826. doi: 10.1016/j.bcp.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 42.Ahmed S.M.U., Luo L., Namani A., Wang X.J., Tang X. Nrf2 signaling pathway: Pivotal role in inflammation. Biochim. Biophys. Acta. 1863;2017:585–597. doi: 10.1016/j.bbadis.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi E.H., Suzuki T., Funayama R., Nagashima T., Hayashi M., Sekine H., Tanaka N., Moriguchi T., Motohashi H., Nakayama K., Yamamoto A. Nrf2 suppresses macrofage inflammatory response by blocking pro-inflammatory cytokine transcription. Nat. Commun. 2016;7:11624. doi: 10.1038/ncomms11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murphy K.E., Park J.J. Can co-activation of Nrf2 and neurotrophic signaling pathway slow Alzheimer's disease? Int. J. Mol. Sci. 2017;18:1168. doi: 10.3390/ijms18061168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verma A.K., Pratrap R. The biological potential of flavones. Nat. Prod. Rep. 2010;27:1571–1593. doi: 10.1039/c004698c. [DOI] [PubMed] [Google Scholar]
- 46.Dhouafli Z., Cuanalo-Contreras K., Hayouni E.A., Mays C.E., Soto C., Moreno-Gonzalez I. Inhibition of protein misfolding and aggregation by natural phenolic compounds. Cell. Mol. Life Sci. 2018;75:3521–3538. doi: 10.1007/s00018-018-2872-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walle T. Methylation of dietary flavones greatly improves their hepatic metabolic stability and intestinal absorption. Mol. Pharmacol. 2007;4:826–832. doi: 10.1021/mp700071d. [DOI] [PubMed] [Google Scholar]
- 48.Touil Y.S., Auzeil N., Boulinguez F., Saighi H., Regazzetti A., Scherman D., Chabot G.G. Fisetin deposition and metabolism in mice: identification of geraldol as an active metabolite. Biochem. Pharmacol. 2011;82:1731–1739. doi: 10.1016/j.bcp.2011.07.097. [DOI] [PubMed] [Google Scholar]
- 49.Wager T.T., Hou X.Y., Verhoest P.R., Villalobos A. Central nervous system multiparameter optimization desirability: application in drug discovery. ACS Chem. Neurosci. 2016;7:767–775. doi: 10.1021/acschemneuro.6b00029. [DOI] [PubMed] [Google Scholar]
- 50.Pluskal T., Weng J.-K. Natural product modulators of human sensations and mood: molecular mechanisms and therapeutic potential. Chem. Soc. Rev. 2018;47:1592. doi: 10.1039/c7cs00411g. [DOI] [PubMed] [Google Scholar]
- 51.Singh N., Vrontakis M., Parkinson F., Chelikani P. Functional bitter taste receptors are expressed in brain cells. Biochem. Biophys. Res. Commun. 2011;406:146–151. doi: 10.1016/j.bbrc.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 52.Ferrer I., Garcia-Esparcia P., Carmona M., Carro E., Aronica E., Kovacs G.G., FGrison A., Gustincich S. Olfactory receptors in non-chemosensory organs: the nervous system in health and disease. Front. Aging Neurosci. 2016;8:163. doi: 10.3389/fnagi.2016.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pajouhesh H., Lenz G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hitchcock S.A., Pennington L.D. Structure-brain exposure relationships. J. Med. Chem. 2006;49:7559–7583. doi: 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]