

Abstract
The P-glycoprotein (Pgp) transporter reduces the penetration of a chemically diverse range of neurotherapeutics at the blood-brain barrier (BBB), but the molecular features of drugs and drug-Pgp interactions that drive transport remain to be clarified. In particular, the triptan neurotherapeutics, eletriptan (ETT) and sumatriptan (STT), were identified to have a greater than 10-fold difference in transport rates despite being from the same drug class. Consistent with these transport differences, ETT activated Pgp-mediated ATP hydrolysis ~2-fold, while STT slightly inhibited Pgp-mediated ATP hydrolysis by ~10%. The interactions between them were also non-competitive, suggesting that they occupy different binding sites on the transporter. Despite these differences, protein fluorescence spectroscopy revealed that the drugs have similar affinity to the transporter. NMR with Pgp and the drugs showed that they have distinct interactions with the transporter. Tertiary conformational changes probed by acrylamide quenching of Pgp tryptophan fluorescence with the drugs and a non-hydrolyzable ATP analog implied that the STT-bound Pgp must undergo larger conformational changes to hydrolyze ATP than ETT-bound Pgp. These results and previous transport studies were used to build a conformationally driven model for triptan transport with Pgp where STT presents a higher conformational barrier for ATP hydrolysis and transport than ETT.
Keywords: ABC transporters, P-glycoprotein, fluorescence, nuclear magnetic resonance (NMR)
INTRODUCTION
P-glycoprotein (Pgp) is a member of the ATP-binding cassette superfamily and acts as the gatekeeper of the blood-brain barrier (BBB) by limiting the penetration of a chemically and structurally diverse range of molecules.1,2 This function protects the brain from toxic insults, but also prevents the penetration of commercially available and potentially beneficial neurotherapeutics,3 which has made neurotherapeutic drug development a formidable challenge.4,5 One strategy to overcome this barrier has been to modulate Pgp transport with inhibitors, but so far this has been unsuccessful in the clinic.6,7 Therefore, there is a lot of interest in the pharmaceutical industry to identify molecular features of neurotherapeutics that drive Pgp transport and to determine molecular mechanisms of neurotherapeutic transport by Pgp.
Pgp is localized to the outer membrane of endothelial cells of the BBB.8,2,9 The X-ray crystal structure of mouse Pgp revealed that the transporter is a 140 kDa asymmetric monomer comprised of twelve transmembrane helices and two nucleotide-binding domains (NBDs), which lie on the cytosolic side of the membrane and bind ATP.10,11 The promiscuity of Pgp to neurotherapeutics is the result of a 6,000 Å3 cavity formed by the transmembrane helices,10,11 which allows multiple drugs to bind to the transporter simultaneously.12,13 The hydrolysis of ATP at the NBDs drive directional transport as a result of large conformational changes that bring the NBDs within contact, as seen in functionally related bacterial transporters.14,15
Triptan drugs represent a class of neurotherapeutics that abort migraines by acting on the 5-hydroxytryptamine 1B and 1D (5-HT1B/1D) receptors that are distributed throughout the brain.16–18 Several triptan drugs are substrates for Pgp and show varying degrees of brain penetration and Pgp transport,1,19–21 but it is unclear what molecular features of triptans drive transport. At one extreme is the triptan drug sumatriptan (STT, Figure 1A). It was the first clinically available triptan drug22 and it is one of the weakest Pgp substrates.e.g. 19 In vitro studies with the drug and different cell types overexpressing Pgp revealed a range of efflux ratios from 1.1 to 2.9.1,19,21 Some studies have identified STT to be a good Pgp substrate, with efflux ratios higher than other triptan drug class members,19 while other studies suggest STT is a non-transported substrate of Pgp.1 This phenomenon has also been previously noted for Pgp and the cardiovascular drug verapamil.23–25 In vivo, STT was shown to be weakly transported by Pgp at the BBB in mice.19 On the other extreme, eletriptan (ETT, Figure 1B) is a second-generation triptan drug22 and a very good Pgp substrate. In vitro studies of ETT identified the drug to have Pgp efflux ratios ranging from 1119 to 46.721. In vivo, Pgp knockout mice showed a ~50 fold increase in ETT concentration in the brain compared to wildtype.20
Figure 1.
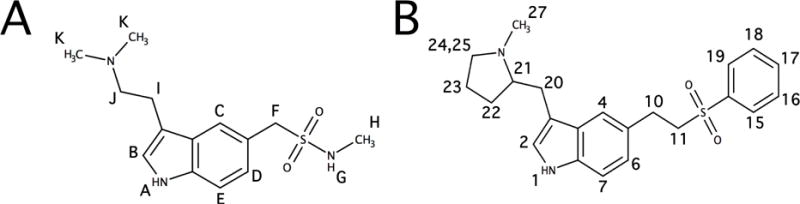
The molecular structure of (A) STT and (B) ETT with proton assignments labeled.
The molecular mechanism of transport and the specific interactions of triptan drugs with Pgp are currently unknown. The large difference in log P values of 4.1 and 0.90 for ETT and STT, respectively, may provide an explanation for the large differences in their transport rates.26 Therefore, we hypothesized that the higher transport rate of ETT by Pgp is the result of stronger interactions with Pgp because of its relatively high log P value when compared with STT. To test this hypothesis, the molecular interactions of ETT and STT with the transporter were examined. First, the effect of these drugs on Pgp-coupled ATP hydrolysis was determined. The interactions of the triptan drugs with Pgp were probed by protein fluorescence spectroscopy and the saturation transfer double difference (STDD) NMR technique. Finally, the effect of these drugs on Pgp conformation in the absence and presence of a non-hydrolyzable ATP analog was examined by acrylamide quenching of tryptophan fluorescence. These experimental results and previously published transport studies from the literature1,19–21 were used to build a conformationally gated model describing the molecular mechanism of triptan transport by Pgp.
EXPERIMENTAL
Materials
Cholesterol, Tris-HCl and disodium ATP (Na2ATP) were purchased from Amresco (Solon, OH). Escherichia coli (E. Coli) total lipid extract powder was purchased from Avanti Polar Lipids Inc. (Alabaster, AL). HEPES and acrylamide were purchased from Calbiochem (San Diego, CA). Deuterium oxide (D2O) was purchased from Cambridge Isotope Laboratories Inc. (Tewksbury, MA). Deuterated dithiothreitol (d10-DTT) was purchased from CDN Isotopes (Quebec, Canada). The detergent n-dodecyl-β-D-maltoside (DDM) was purchased from EMD Millipore Corporation (San Diego, CA). Sodium orthovanadate (Na3VO4) was purchased from Enzo Life Sciences (Farmingdale, NY). Sumatriptan was purchased from Fagron, Inc. (St. Paul, MN). Dithiothreitol (DTT) was purchased from Gold Biotechnology (Olivette, MO). Eletriptan was given as a gift from Pfizer Inc. (New York City, NY). Ammonium chloride (NH4Cl) and deuterated DMSO (DMSO-d6) was purchased from Sigma Aldrich (Milwaukee, WI). The 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) detergent was purchased from VWR International (Radnor, PA). All the remaining chemicals in this study were purchased from Thermo Fisher Scientific (Waltham, MA).
Methods
Purification and reconstitution of Pgp
The his-tagged wildtype mouse Pgp (Abcb1a, MDR3) was purified from Pichia pastoris in two stages with nickel-nitrilotriacetic acid (Ni-NTA) (Thermo Fisher Scientific, Waltham, MA) and Whatman DE52 cellulose resin (Thermo Fisher Scientific, Waltham, MA) as previously described.27,28 Detergent solubilized Pgp was directionally integrated into liposomes with the NBDs on the outside using the procedure described in.23,24 These liposomes were composed of 80% w/v Avanti total Escherichia (E.) coli Total Lipid Extract (Avanti Polar Lipids, Alabaster, AL) and 20% w/v cholesterol with a lipid to protein ratio of 0.16 mg ml−1.23,24 The Pgp orientation in the liposomes was determined by permeabilizing the liposomes to ATP with CHAPS detergent so that any NBDs located on the inside of the liposome would hydrolyze ATP. There was no increase in the apparent ATPase activity as a result of NBDs located on the inside of the liposomes (data not shown) demonstrating that the NBDs are essentially on the external side of the liposome.23 Proteoliposome aliquots were stored at −80°C in HE PES buffer (20 mM HEPES, 100 mM NaCl2, 5 mM MgCl2, 2 mM DTT pH 7.4). The concentration of protein was determined with the extinction coefficient of 1.28 mg mg−1 cm−1 and the DC Protein Assay Kit II (Bio-Rad, Hercules, CA).23,24
ATPase activity measurements
The ATPase activity of Pgp was measured using the Chifflet method on a FlexStation 3 spectrometer (Molecular Devices, Sunnyvale, CA), as previously described.29,23,24 The concentration of free inorganic phosphate (Pi) after ATP hydrolysis was estimated by the absorbance signal at 850 nm from the formation of a Pi-molybdenum complex. The ATPase activity of ETT and STT was measured in the presence of 50 nM Pgp reconstituted in liposomes in Chifflet buffer (150 mM NH4Cl, 5 mM MgSO4, 0.02% w/v NaN3, 50 mM Tris-HCl, pH 7.4).29,23,24
Simple enzyme kinetics is traditionally analyzed using linear transformations such as Lineweaver-Burk, Hans-Woolf and Eadie-Hofstee plots.30,31 Unfortunately, these transformations suffer from lack of variable independence across the axes and they have the tendency to bias the data points.32−34 In contrast, non-linear regression is considerably more accurate than linear transformation, and is easily performed with modern computers.33 Therefore, the Pgp-coupled ATP hydrolysis kinetics was fit by non-linear regression with Igor Pro 6.2 (Wavemetrics, Tigard, OR) and the following equations. Hyperbolic Pgp-coupled ATP hydrolysis kinetics of the drugs were fit to the modified Michaelis-Menten equation (Eq. 1):30
| (1) |
where Vmax is the maximum ATP hydrolysis, Km is the Michaelis-Menten constant, [L] is the concentration of ligand and vbasal is the basal ATPase activity in the absence of drugs. For drug interactions that are competitive, the ATP hydrolysis curves were fit to Eq. 1 and an apparent Michaelis-Menten constant (Km,app) shown in Eq. 2:30
| (2) |
where [I] is the inhibitor and KI is the inhibitory constant. For non-competitive inhibition, the apparent Vmax (Vmax,app) can be calculated with the following equation (Eq. 3):30
| (3) |
This equation assumes that the enzyme velocity will approach 0 at saturating [I]. In cases where there is only partial inhibition of the enzyme at saturating [I], the Vmax,app will approach a new Vmax (Vmax,uninhibited). In this case, Eq. 3 can be modified to Eq. 4:
| (4) |
where Vmax,inhibited represents the maximum inhibitable enzyme velocity at saturating [I].
Fluorescence Spectroscopy
Quenching of protein fluorescence has been used to measure the dissociation constants (KDs) of a wide range of ligands to Pgp.35,36,23,24 This technique was used to measure the affinity of ETT and STT to Pgp. An Olis DM 45 spectrofluorimeter (Olis Corp, Bogart, GA) was used to measure the drug-induced quenching of 1 μM Pgp reconstituted in liposomes in 100 mM potassium phosphate (KPi) and 2 mM DTT (pH 7.4) to prevent cysteine disulfide linkage between Pgp monomers, as previously described.23,24 Pgp protein fluorescence emission was observed between 300 and 500 nm with a fluorescence emission maximum at ~330 nm, when the protein was excited at wavelengths between 260 and 295 nm.37 The ETT and STT molecules also emit fluorescence in this region, which could potentially complicate Pgp protein fluorescence measurements. Fortunately, the drug fluorescence only partially overlaps with the Pgp protein fluorescence (Figure S1), even at the highest concentrations used in the study. For drug concentrations less than 400 μM, the drug fluorescence contributions could be effectively subtracted from the overall fluorescence signal. The drug fluorescence contribution was further minimized for ETT and STT by exciting Pgp at 280 and 295 nm, respectively. The drug induced fluorescence quenching was corrected (Fcorrected) for background fluorescence and inner filter effects with the following equation (Eq. 5):37,23
| (5) |
where F is the measured protein fluorescence at 330 nm, B is the background, [Q] is the quenching ligand concentration, ε is the extinction coefficients for the excitation (εex) and emission (εem) and b is the pathlength for the excitation (bex) and emission (bem). The εex at 280 nm and εem at 330 nm for ETT was 2.13 mM−1 cm−1 and 0.432 mM−1 cm−1, respectively. For STT, the εex at 295 nm was determined to be 0.808 mM−1 cm−1 and the εem at 330 nm was 0.344 mM−1 cm−1
Protein fluorescence quenching can occur by static or dynamic mechanisms. Static quenching occurs from ligand forming a complex with the protein and is directly correlated to the ligand’s affinity.37 In contrast, dynamic or collisional quenching is the result of random collisions between a ligand and protein.37 To differentiate between different quenching mechanisms, protein fluorescence quenching titrations were measured at different temperatures as described.37,23,24 The corrected fluorescence (Fcorrected) of a monophasic fluorescent quenching curve was fit to the following rearrangement of the Stern-Volmer equation (Eq. 6):37,23
| (6) |
where Fcorrected,0 is fluorescence in the absence of a quenching ligand, Funquenched is an offset related to the unquenched fluorescence, KSV is the Stern-Volmer constant, and [Q] is the concentration of quenching ligand. Biphasic fluorescence quenching curves were fit to the following equation (Eq. 7):37
| (7) |
where F0,L and F0,H are the fluorescence amplitudes at low and high quenching ligand concentrations, respectively. The KSV,L and KSV,H are the Stern-Volmer constants at low and high quenching ligand concentrations, respectively.
The collisional quenching of Pgp by acrylamide has been used to probe changes in exposure to solvent accessible tryptophan residues to determine conformational changes of Pgp upon ligand binding.e.g. 38,39,24 Acrylamide is an uncharged polar compound that has minimal penetration into lipids and the hydrophobic core of proteins.40 Fluorescence emission of 1 μM Pgp reconstituted in liposomes was monitored at 330 nm after excitation at 295 nm in the presence of saturating concentrations of ETT and STT in 100 mM KPi and 2 mM DTT (pH 7.4). The fluorescence emission was corrected for inner filter effects and background fluorescence with Eq. 5. To compare the results with the ATP hydrolysis experiments, the fluorescence quenching was monitored in the presence of 3.2 mM of the non-hydrolyzable ATP analog adenosine 5′-(β,γ-imido)triophsophate (AMPPNP). N-acetyl-L-tryptophanamide (NATA) is an analog of solvent accessible tryptophans that was used to determine the maximum possible tryptophan quenching.39 The Stern-Volmer plots were created by plotting the Fcorrected,0/Fcorrected as a function of acrylamide concentrations. The KSV value is a quantitative measure of exposure to solvent accessible tryptophans, which was estimated from the slopes of the Stern-Volmer plots as described previously.e.g. 37,23
Saturation transfer double difference NMR technique
All NMR experiments were performed at 25°C on a Varian INOVA 600 MHz spectrometer with a 5mm z-gradient 1H{13C/15N} cryoprobe and analyzed in iNMR software (http://ww.inmr.net) and Igor Pro 6.2, as described previously.23,24 The 1H NMR peaks of 100 mM STT and ETT in DMSO-d6 were assigned using standard 1H 1D NMR techniques. The 1H NMR peak assignments for STT and ETT are shown in Figure 1. This labeling scheme was used for the assigning 1H NMR peaks.
The saturation transfer difference (STD) NMR technique is used to identify drug functional groups that interact with a protein receptor by selectively saturating the receptor and monitoring the saturation transfer from the receptor to the drug.41−43 This observed degree of saturation transfer is directly correlated to the drug’s interaction with the receptor.41,42 For membrane proteins reconstituted into a lipid system, the lipid-drug interactions can lead to nonspecific interactions and interference in the 1H STD NMR spectrum. The saturation transfer double difference (STDD) NMR technique was developed to extract specific NMR signals corresponding to drug-protein interactions from samples with lipid membranes.43,23,24 The STDD NMR spectrum is created by subtracting the STD NMR spectrum of the drug in the presence of lipid from the STD NMR spectrum of a drug in the presence of a protein embedded in the same lipid.44,45,43,24 This technique was used to probe the ETT and STT binding interactions with Pgp reconstituted in liposomes. NMR samples contained 1 μM reconstituted Pgp in 80% deuterated 100 mM KPi, pD 7.4 and saturating concentrations of triptans, 250 μM. Control liposome samples were prepared identically to reconstituted Pgp samples. A train of 50 ms gaussian shaped pulses selectively saturated reconstituted Pgp for a total saturation of 2 s. The water suppression by gradient tailored excitation (WATERGATE) pulse sequence was added to suppress the water signal.23 The STD NMR spectra were produced by phase cycling and alternating the saturation frequency between -1.5 and 40 ppm during 512 scans.23,24 To create the 1H STDD NMR spectrum of the drugs with Pgp, the 1H STD NMR spectrum of the control liposome sample with drug was subtracted from the 1H STD NMR spectrum of the drug with reconstituted Pgp. This difference (ΔI) correlates to the molecular interactions between the drug and the transporter. This was quantitated using STDD amplification factors calculated from the following equation (Eq. 8):23
| (8) |
where [P] is the protein concentration, [L] is the concentration of ligand and I0 is the amplitude of the 1H NMR peaks in the spectrum without radio frequency saturation.
RESULTS
The effect of ETT and STT on Pgp ATP activation
Figure 2 shows the Pgp-coupled ATPase activity in the presence of ETT and STT. In the absence of drugs, the Pgp-coupled ATP hydrolysis activity was 560 ± 40 nmol min−1 mg−1 (Figure 2A), which is similar to previous estimates23,24. The ATP hydrolysis activity was almost completely abolished in the presence of 200 μM of the Pgp inhibitor Na3VO4 (data not shown). In Figure 2A (closed squares), the Pgp-coupled ATP hydrolysis activity was measured with a range of ETT concentrations. The ATP hydrolysis kinetics was monophasic and reached a maximum at ~1000 nmol min−1 mg−1. Fitting the curve to the modified Michaelis-Menten equation (Eq. 1) generated a VMAX and KM value of 508 ± 27 nmol min−1 mg−1 and 14.8 ± 3.1 μM, respectively. Figure 2A (open circles) also shows the Pgp-coupled ATP hydrolysis kinetics in the presence of STT. In contrast to ETT, STT weakly inhibits Pgp-coupled ATP hydrolysis kinetics by < 10%. This effect is not inconsistent with STT being transported by Pgp. Several known Pgp transported substrates, colchicine, doxorubicin and chloramphenicol, have little impact on Pgp-coupled ATP hydrolysis kinetics46−48 and the fluorescent probe substrate Hoechst 33342, in fact, inhibits ATP hydrolysis by Pgp, but is still transported.49
Figure 2. The effect of ETT and STT on Pgp-mediated ATP hydrolysis.
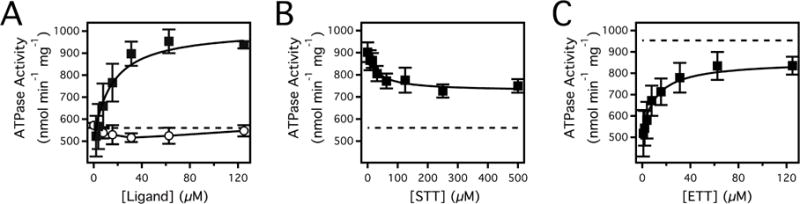
(A) Pgp-coupled ATPase activity as a function of basal ATPase activity (dashed line), ETT (closed squares) and STT (open circles) concentrations. (B) Pgp-coupled ATPase activity in the presence of 125 μM ETT and a range of STT concentrations. For comparison, the level for basal ATPase activity is shown as a dotted line in the panel. (C) Pgp-coupled ATPase activity in the presence of 500 μM STT and a range of ETT concentrations. The dashed line in the panel represents the maximum Pgp-coupled ATP hydrolysis of ETT. Error bars represent the standard deviation and the points represent the average of at least three independent experiments.
The fact that Pgp-mediated ATP hydrolysis kinetics in the presence of ETT and STT were distinct in Figure 2A allowed us to probe the competition of these drugs to Pgp. Classically, competitive inhibition will lead to an increase in Km as a result of drug displacement from Pgp, while non-competitive inhibition will lead to a decrease in Vmax, but will have little effect on the Km.30 Non-competitive inhibition also implies simultaneous binding of both drugs.30 Figure 2B shows the effect of a range of STT concentrations on the activation of Pgp-coupled ATP hydrolysis by 125 μM ETT. Increasing STT concentrations only decreased the apparent ATP hydrolysis rate about 30%. The fact that the ATP hydrolysis is only partially inhibited implies that both drugs are bound simultaneously and that the interactions are non-competitive. Fitting the ATP hydrolysis kinetics in Figure 2B with Eqs. 1 and 4 gives a KI, Vmax,inhibited and Vmax,uninhibited values of 30.2 ± 12.1 μM, 201 ± 21 nmol min−1 mg−1 and 187 ± 17 nmol min−1 mg−1, respectively. To demonstrate that ETT and STT interactions were in fact non-competitive, an ETT titration of Pgp was performed in the presence of 500 μM STT in Figure 2C. Fitting the curve to Eq. 1 reveals that the activated Vmax decreases 30% to 341 ± 20 nmol min−1 mg−1 and the extracted Km is 11.1 ± 2.8 μM. The decrease in Vmax and essentially no change from the previous Km value of 14.8 ± 3.1 μM (Figure 2A, closed squares) demonstrate that the interactions between ETT and STT are indeed non-competitive. The KI for STT estimated by fitting Figure 2C with Eqs. 1 and 4 was 48.3 ± 18.0 μM, which is similar to the KI obtained from the curve fit in Figure 2B.
Probing the interactions of ETT and STT with Pgp by intrinsic protein fluorescence
Equilibrium constants (i.e. Km, KI) derived from fitting Pgp-mediated ATP hydrolysis kinetics curves do not necessarily correlate directly to ETT and STT binding affinity, since the fitting equations assume specific enzyme mechanisms.30 One might anticipate that there could be significant differences in the affinities of ETT and STT to Pgp because of large differences in their log P values50 and their apparent transport rates.e.g.20 Ligand affinities to Pgp have been directly estimated with fluorescent or radiolabeled ligands,51,52,12 fluorescence quenching of labeled Pgp53,54 and quenching of intrinsic Pgp tryptophan fluorescence.35,23,24 Directly measuring drug binding with fluorescently labeled Pgp may be problematic due to interference from the label.53,54 Therefore, the affinities of ETT and STT were estimated by measuring their impact on Pgp protein fluorescence.35,23,24
Figure 3 shows the protein fluorescence and analysis of Pgp in the presence of ETT and STT. Figure 3A shows the Pgp fluorescence emission spectra in the presence of ETT excited at 280 nm with the background fluorescence subtracted. The protein fluorescence produces a fluorescence maximum at 330 nm (thin line) that decreases ~50% with saturating ETT (thick line). After adjustment for inner filter effects with Eq. 5, the amplitude at 330 nm was monitored as a function of ETT concentration (Figure 3B). Quenching of Pgp by ETT was biphasic with low and high concentration phases. Fitting the quenching curve to Eq. 7 gave KSV,L and KSV,H of 7.15 ± 1.14 μM−1 and 8.69 × 10−3 ± 0.74 × 10−3 μM−1, respectively. The titration was repeated at 37°C to determine the quenching mechanism for each phase as done previously23,24. The Stern-Volmer constants, KSV,L and KSV,H, increased and decreased, respectively (data not shown). Therefore, the low concentration-quenching phase associated with KSV,L quenches by a dynamic mechanism, and the high quenching concentration phase associated with KSV,H quenches by a static mechanism. Thus, the KA for ETT was 8.69 × 10−3 ± 0.74 × 10−3 μM−1 and the corresponding dissociation constant (KD) was 115 ± 10 μM. Figure 3C shows the effects of STT on Pgp fluorescence after excitation at 295 nm with the fluorescence background subtracted. Saturating STT quenched Pgp fluorescence about 20%. In Figure 3D, Pgp fluorescence emission corrected for inner filter effects was monitored at 330 nm as a function of STT concentration. The fluorescence quenching curve was monophasic and fitting the curve to Eq. 6 gave a KSV value of 10.8 × 10−3 ± 2.9 × 10−3 μM−1. The KSV value increased when this titration was repeated at 10°C showing that the quenching was the result of a static mechanism (data not shown) and the KD at 25°C was 92.6 ± 25 μM. The surprising similarities between the KDs between ETT and STT reveal their binding is not driven by drug hydrophobicity. Instead, the binding may be driven by other factors such as the number and strength of hydrogen bonds as suggested by a SAR study.55
Figure 3.
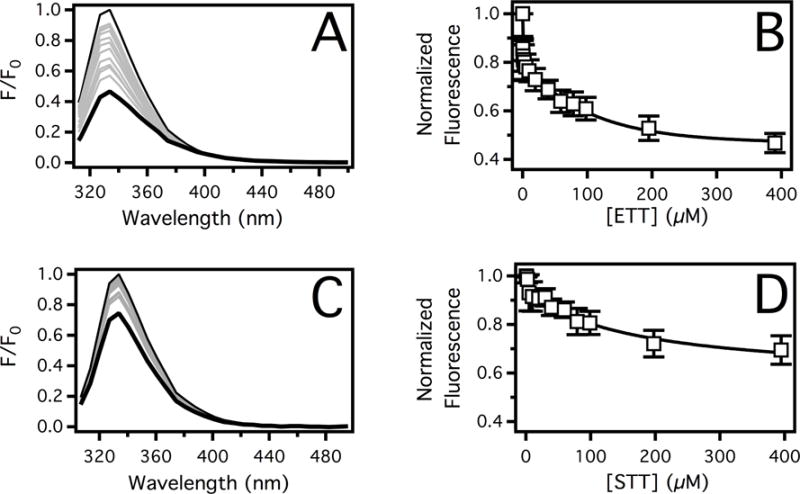
The affinity of ETT and STT to Pgp deduced by protein fluorescence spectroscopy. Pgp fluorescence emission spectra in the presence of a range of (A) ETT and (C) STT concentrations. The fluorescence emission spectrum at 0 μM and 400 μM triptan drugs are shown as thin and thick black lines, respectively. Spectra at intermediate concentrations are shown in gray. The corrected fluorescence emission amplitude at 330 nm was plotted as a function of (B) ETT and (D) STT concentration. The points and error bars represent the average and standard deviations, respectively, of at least three independent experiments.
Functional groups of ETT and STT involved in molecular recognition by Pgp
Although ETT and STT affinities are similar, the effects of these drugs on Pgp-coupled ATP hydrolysis were distinct and the interactions between the two drugs on the transporter were non-competitive. Therefore, their interactions with the transporter were probed by the STDD NMR technique. The degree of drug interaction with the transporter is reflected in the amplitude of the 1H STDD NMR peaks and quantitatively by 1H STDD NMR amplification factors.24
Figure 4 shows the STDD NMR spectra, STDD amplification factors and %ΔSTDD amplification factors of ETT and STT in the presence of Pgp. The figure follows the labeling scheme of the STT and ETT molecular structures in Figure 1. The 1H STDD NMR spectrum of 125 μM ETT in the presence of 1 μM Pgp is illustrated in Figure 4A. 1H STDD NMR peaks with significant saturation transfer are labeled. Protons emanating from the liposomes and exchange broadening prevented us from reliably measuring the amplitudes of several alkyl and an amine 1H NMR peaks of ETT labeled 1, 10–11 and 21–25 (see Figure 1B). From the STDD amplitudes in Figure 4A, the STDD amplification factors are estimated in Figure 4B using Eq. 8. The average STDD amplification factor for ETT to Pgp was 15. The strongest STDD amplification factor of 20 was observed for aromatic phenyl protons labeled 15,19, while the weakest STDD amplification factor of 10 came from the methyl labeled 27 of the pyrrolidine functional group. To exemplify differences in the STDD amplification factors, they were normalized against protons of the pyrrolidine methyl in Figure 4C. The phenyl group (protons 15–19, black) of ETT had STDD amplification factors that were 72% larger than the pyrrolidine methyl. The indole ring (protons 2–7, gray) of ETT had amplification factors that were 34% larger than the pyrrolidine methyl. These results imply that the phenyl of ETT plays the largest role in molecular recognition of Pgp.
Figure 4. ETT and STT interactions with Pgp investigated by the saturation transfer double difference (STDD) NMR technique.
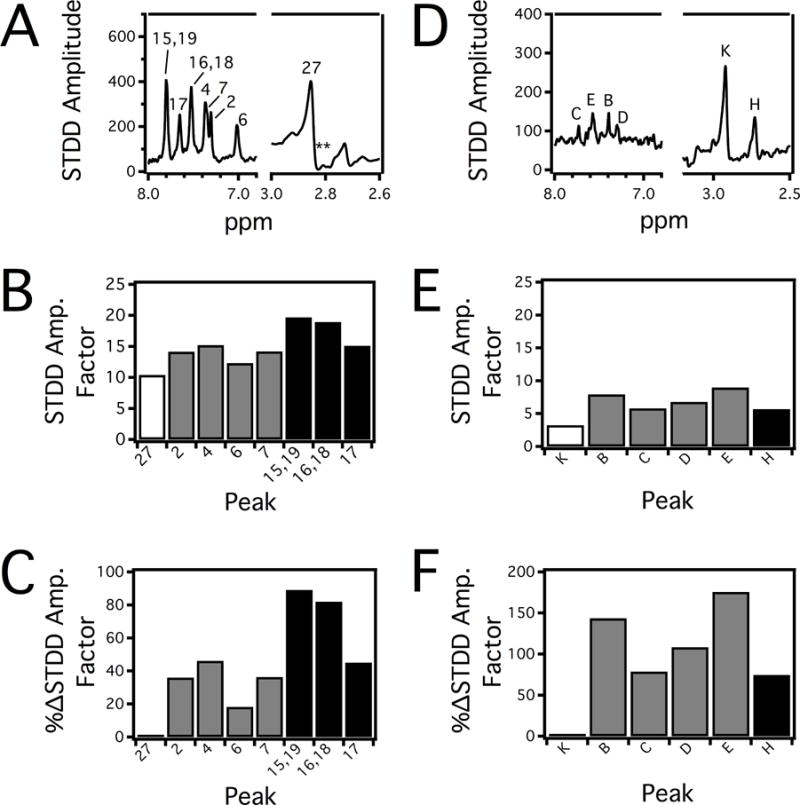
The 1H STDD NMR spectra with the peaks labeled of 250 μM (A) ETT and (D) STT in the presence of 1 μM Pgp. STDD amplification factors calculated for (B) ETT and (E) STT from the 1H STDD NMR spectra. For clarity, STDD amplification factors from the indole protons are colored gray and protons from flanking functional groups are shown in white and black. To emphasize differences in the STDD amplification factors, they were normalized against the left most (weakest) STDD amplification factor as Δ%STDD for (C) ETT and (F) STT.
Figure 4D shows the 1H STDD NMR spectrum of 250 μM STT in the presence of 1 μM Pgp. Several 1H STDD NMR peaks of STT were identified and labeled in the figure. Like ETT, reliable 1H STDD NMR measurements were not possible for several alkyl protons (I and J) and amine protons (A and G) due to overlap with proton NMR signals from the liposome and exchange broadening of amines from STT (see Figure 1A). The STDD amplification factors from Figure 4D were calculated using Eq. 8 and are shown in Figure 4E. The average STDD amplification factor for STT was 6, which is less than half of ETT. This difference does not necessarily correspond to weaker interaction of STT with the transporter than ETT, since fluorescence spectroscopy with these drugs and Pgp in Figure 3 has already demonstrated that they have similar KDs. Instead, lower STDD amplification factors by STT can result from lower saturation transfer efficiency41,56 because of how STT is distinctly sequestered by the transporter. For STT, the strongest STDD amplification factor of 9 was observed with proton E of the indole ring, while the weakest STDD amplification factor of 3 was observed for the methyl protons labeled K. Interestingly, the analogous methyl protons on ETT also had the weakest STDD amplification factor. To contrast the differences in STDD amplification factors of STT, they were normalized against the methyl protons labeled K in Figure 4F. The indole ring had 130% higher STDD amplification factors (gray) than the methyl protons. The N-linked methyl near the sulfone labeled H had STDD amplification factors (black) that were 75% of the methyl protons. These results imply that the indole functional group of STT plays the largest role in molecular recognition of the transporter.
Drug-induced conformational changes of Pgp in the presence of ETT and STT
Both X-ray crystal structures of analogous bacterial transporters with non-hydrolyzable ATP analogs14,15 and studies with Pgpe.g. 57,58 have suggested that conformational changes are involved in Pgp-mediated ATP hydrolysis. The Pgp conformations were probed by estimating the relative exposure of solvent accessible tryptophan residues (e.g. residues 158, 799, 1104) through acrylamide quenching as we did previously.23,24 Figure 5 shows the Stern-Volmer plots in the presence of drugs and the non-hydrolyzable ATP analog AMPPNP, where the slope is equivalent to the KSV value. In all cases, the curves in the Stern-Volmer plots were linear with respect to acrylamide concentration, which shows that acrylamide does not directly bind to Pgp or affect Pgp’s conformation.37
Figure 5. ETT and STT induced conformational changes of Pgp in the absence and presence of an ATP analog AMPPNP inferred by fluorescence spectroscopy.
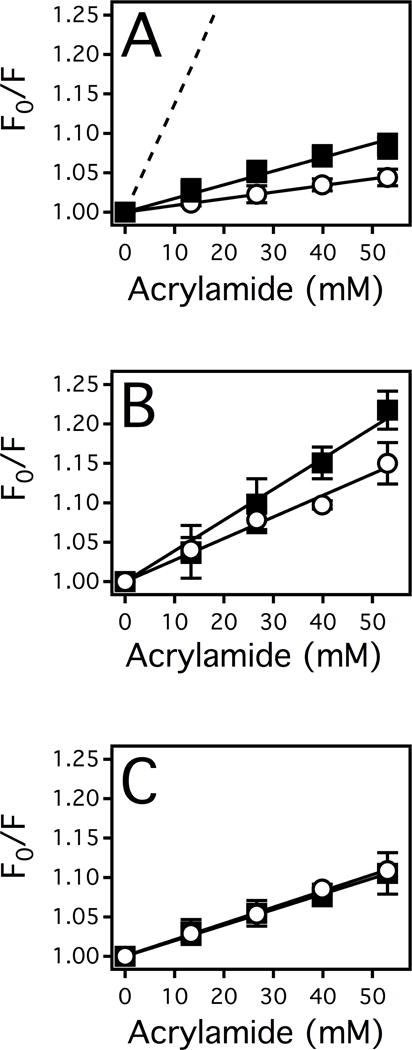
(A) The Stern-Volmer plots of NATA (dashed line), Pgp in the absence of drugs (closed squares) and in the presence of AMPPNP (open circles). (B) The Stern-Volmer plot of Pgp in the presence of STT (closed squares) and STT with AMPPNP (open circles). (C) The Stern-Volmer plot of Pgp in the presence of ETT (closed squares) and ETT with AMPPNP (open circles). The points represent the average and the error bars represent the standard deviation of at least three independent experiments.
In the absence of drugs, the Stern-Volmer plot of Pgp in Figure 5A (closed squares) had a KSV value of 1.52 ± 0.20 M−1 with 34% of the total tryptophan fluorescence quenched at 1 M acrylamide. This degree of quenching correlates well to the ratio of fully solvent accessible tryptophans (3) versus the total number of tryptophan residues (11) deduced from the X-ray crystal structure of Pgp.59,60 As a positive control, NATA was used to assess the maximum tryptophan quenching. NATA was quenched 93% at 1M acrylamide and had a KSV value of 13.4 ± 0.30 M−1 (Figure 5A, dashed line).
The addition of the ATP analog AMPPNP (open circles) to Pgp reduced the KSV value to 0.84 ± 0.20 M−1, resulting in a ΔKSV of 0.68 ± 0.20 M−1. The change in tryptophan accessibility or the ΔKSV value is consistent with nucleotide-induced conformational shift observed in previous crystallography studies.e.g. 14,15
The effect of STT on the solvent accessibility of tryptophan residues from Pgp is shown in Figure 5B (closed squares). The STT-bound complex has a significantly higher KSV value of 4.13 ± 0.20 M−1 and in the presence of AMPPNP (open circles), the KSV value decreases to 2.69 ± 0.20 M−1, resulting in a ΔKSV value of 1.44 ± 0.20 M−1. This ΔKSV value is considerably larger than the ΔKSV value in the absence of drugs, implying that the STT-bound complex undergoes a relatively large conformational change in the presence of the ATP analog. In the presence of ETT, the KSV value increased modestly to 1.96 ± 0.07 M−1 (Figure 5C, closed squares). Furthermore, the KSV value of the ETT-bound complex remains essentially unchanged by the addition of AMPPNP to 2.06 ± 0.05 M−1 (Figure 5C, open circles) with a ΔKSV value of −0.10 ± 0.09 M−1. These results imply that the nucleotide-induced Pgp conformational transitions are more modest in the presence of ETT than STT.
DISCUSSION
A conformationally gated model of triptan transport is presented in Figure 6 to explain the molecular mechanism driving ETT and STT transport by Pgp. The model is based on our results, the conformational changes that Pgp undergoes in the presence of nucleotides and ligands61−64,24 and the observed differences in triptan transport.e.g. 1,19−21 By analogy with the bacterial transporters,15 Pgp can assume a wide range of conformations. For simplicity, we define three conformations of Pgp: “open”, “intermediate” and “closed”, as we did previously.23,24 In the “open” conformation, the NBDs are separated with the binding cavity exposed to the cytosol. The NBDs are together in the “closed” conformation with the binding cavity exposed to the extracellular space. The “intermediate” conformation is between the “open” and “closed” Pgp conformations with equivalent exposure to the extracellular and cytosolic sides of the membrane.
Figure 6. Conformationally gated model of triptan transport by Pgp.
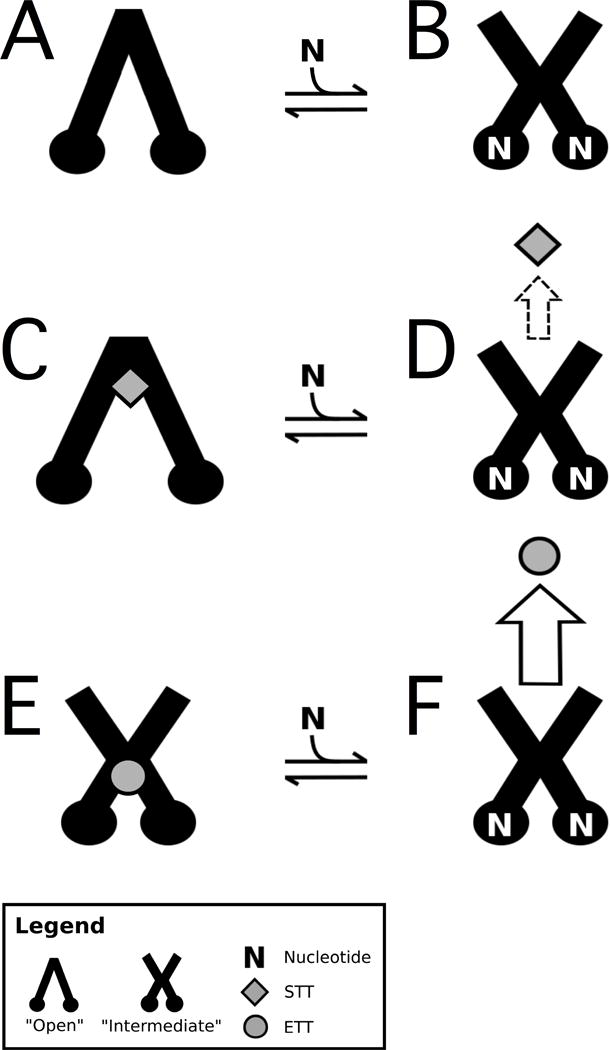
Pgp is depicted as a cartoon image of the “open” and “intermediate” states in the (A,B) absence of drug, the presence of (C,D) STT and (E,F) ETT. STT are shown as gray diamonds, ETT are gray circles and N is ATP. The horizontal arrows represent equilibrium between the nucleotide-bound conformational states of Pgp. The sizes of the vertical arrows represent the degree of transport. The dashed arrow reflects the fact that STT has been observed to be both transported19 and not transported by Pgp1 depending on the cell type.
Changes to tryptophan accessibility of Pgp by ETT, STT and the non-hydrolyzable analog AMPPNP were deduced by acrylamide fluorescence quenching in Figure 5. The KSV and ΔKSV values were used to correlate the exposure of solvent accessible tryptophan residues to Pgp conformational changes. An increase in the KSV value implies a shift to a more “open” conformation, while a decrease in KSV value suggests a shift to a “closed” conformation. Moreover, the ΔKSV value infers the degree of this conformational change. However, tryptophan accessibility and the corresponding KSV values cannot be used to assign specific ligand-bound conformations, since it does not correlate directly to a specific Pgp conformation. We know from X-ray crystallography, cryo-em, cross-linking and mutagenesis studies with Pgp and the analogous bacterial transporters that the interaction of the NBDs is essential for ATP hydrolysis.65−68,57,69 Therefore, Pgp-mediated ATP hydrolysis rates, relative changes in tryptophan accessibility from Figure 5 and previous Pgp conformational assignments23,24 were used to direct conformational assignment of Pgp in the presence of nucleotides, ETT and STT. In general, when the ligand-induced activation of Pgp-mediated ATP hydrolysis is relatively high, we assume that Pgp is shifted toward the “closed” conformation. When there is ligand-induced inhibition or no inhibition in Pgp-mediated ATP hydrolysis, we assume that Pgp is shifted toward or in an “open” conformation.
We know from X-ray crystallography of Pgp in the absence of ligands that the NBDs are relatively far apart in the “open” conformation.e.g. 10 Therefore, Pgp is depicted in the “open” conformation in the absence of ligands in Figure 6A. Figure 5A shows that tryptophan accessibility decreases in the presence of AMPPNP (ΔKSV = 0.68 M−1) suggesting a shift to the “closed” conformation. However, the baseline P-gp mediated ATP hydrolysis rate hovers around 600 nmol min−1 mg−1. Therefore, we propose that Pgp in the presence of ATP, but in the absence of ligands, is in an “intermediate” conformation in Figure 6B as we hypothesized previously24.
Figure 5B shows that STT increases the KSV value of Pgp by 2.61 M−1, while STT decreases the Pgp-mediated ATP hydrolysis rate by ~10% in Figure 2. These results imply a ligand-induced separation of the NBDs and a shift to a more “open” conformation. This is consistent with X-ray crystallographic studies showing Pgp in an “open” conformation, when complexed with inhibitors.10,70 Therefore, we propose that the STT-bound Pgp structure is in an “open” conformation and bound near the extracellular side of the transporter in Figure 6C like inhibitors in the Pgp X-ray crystal structures.10,70 In the presence of AMPPNP, the KSV value of the STT-bound Pgp decreases by 1.44 M−1, implying a large conformational shift toward the “closed” conformation. A conformational barrier to ATP hydrolysis and transport would explain the 10% reduction in Pgp-mediated ATP hydrolysis in the presence of STT (Figure 2A) and the significantly lower Pgp-mediated transport of STT versus ETT19. Since Pgp-mediated ATP hydrolysis in the presence of STT is similar to basal activity, the STT-nucleotide-bound complex is hypothesized to be in an “intermediate” conformation in Figure 6D. In vitro transport studies with STT with Pgp have demonstrated that Pgp-mediated STT transport is cell type dependent.1,19−21 Therefore, STT transport or lack there of is represented as a dashed arrow in Figure 6.
ETT induces ~2-fold activation of Pgp-mediated ATP hydrolysis, which is similar to digoxin-induced activation but is half of the verapamil-induced activation of Pgp.23 Digoxin was hypothesized to shift Pgp into an “intermediate” conformation, while verapamil shifted the transporter to the “closed” conformation. Therefore, we propose that Pgp in the presence of ETT is in an “intermediate” conformation in Figure 6E like the digoxin-bound complex.23 The Stern-Volmer plot in Figure 5C shows that the KSV value for acrylamide quenching of Pgp in the presence of ETT changes little with saturating AMPPNP, implying that the conformation is changed little by the non-hydrolyzable ATP analog. Therefore, we propose that Pgp in the presence of ETT and ATP is also in an “intermediate” conformation in Figure 6F. With the ETT-bound Pgp being in the same conformation in the presence and absence of ATP, the conformational barrier for ATP hydrolysis is minimized when compared to STT. This reduction in the conformational barrier correlates well to the 5–20 fold higher efflux ratios observed for ETT than STT.1,19 In addition, the interactions of STT and ETT on Pgp probed by Pgp-mediated ATP hydrolysis (Figure 2B and 2C) were non-competitive suggesting that they occupy distinct sites on the transporter. Since ETT induces significant activation of Pgp-mediated ATP hydrolysis, we place ETT far from the hypothetical STT binding site near the NBDs in Figure 6E.
Our results in this study imply that triptan transport is not merely driven by chemical properties of STT and ETT. Triptans have distinct interactions with the transporter and induce conformational changes that activate or hinder Pgp-mediated ATP hydrolysis.1,19−21 These studies are the first to provide direct insight into the molecular mechanism of triptan transport by Pgp. Although this study is only focused on two neurotherapeutics, our simplified transport model may apply to a range of neurotherapeutics.
Supplementary Material
Acknowledgments
We would like to give special thanks to Dr. Ina L. Urbatsch of Texas Tech University Health Sciences Center for her generous gift of Pichia (P.) pastoris with the wild-type mouse Pgp transporter gene. We would also like to thank her postdoc Dr. Douglas J. Swartz for sending us the materials and providing protocols for genetically manipulating and purifying Pgp from P. pastoris for our laboratory. Without their contribution and generosity, this research would not be possible. We would also like to thank Pfizer, Inc. for generously providing us eletriptan for our research. Without this kind gift, we would not have been able to conduct these experiments.
FUNDING
This work was supported by the American Heart Association Grant (14GRNT20450044) and the National Institute of Health R15 Area Grant (1R15GM10793-01A1).
ABBREVIATIONS
- Amp
amplitude
- AMPPNP
adenosine 5′-(β,γ-imido)triophsophate
- BBB
blood-brain barrier
- CHAPS
3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate
- DDM
n-dodecyl-β-D-maltoside
- DEAE
diethylaminoethyl cellulose
- ETT
eletriptan
- KD
Dissociation constant
- KPi
potassium phosphate
- KSV
Stern-Volmer quenching constant
- n
number of binding sites
- NATA
N-acetyl-L-tryptophanamide
- NBD
nucleotide-binding domain
- Ni-NTA
nickel-nitrilotriacetic acid
- NMR
nuclear magnetic resonance
- Pgp
P-glycoprotein
- Pi
inorganic phosphate
- SAR
structure-activity relationship
- STDD
saturation transfer double difference
- STT
sumatriptan
- WATERGATE
water suppression by gradient tailored excitation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
AGR and LAW conceived and coordinated the study and wrote the paper. AGR and LAW designed all experiments in the study. LAW analyzed the experiments for all the figures and prepared the figures. DN collected fluorescence data for Figure 5 and LAW collected data for the remaining figures. All the authors approved the final version of the manuscript.
References
- 1.Mahar Doan KM, et al. Passive permeability and p-glycoprotein-mediated efflux differentiate central nervous system (cns) and non-cns marketed drugs. J Pharmacol Exp Ther. 2002;303(3):1029–1037. doi: 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- 2.Ramakrishnan P. The role of p-glycoprotein in the blood-brain barrier. Einstein Q J Biol Med. 2003;19:160–165. [Google Scholar]
- 3.Jose M, Thomas SV. Role of multidrug transporters in neurotherapeutics. Ann Indian Acad Neurol. 2009;12(2):89–98. doi: 10.4103/0972-2327.53076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cutler NR, et al. The impending crisis in cns drug development. Oxford, UK: Wiley-Blackwell; 2010. [Google Scholar]
- 5.Kaitin KI, Milne CP. A dearth of new meds. Sci Am. 2011;305(2):16. doi: 10.1038/scientificamerican0811-16. [DOI] [PubMed] [Google Scholar]
- 6.Binkhathlan Z, Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr Cancer Drug Targets. 2013;13(3):326–346. doi: 10.2174/15680096113139990076. [DOI] [PubMed] [Google Scholar]
- 7.Callaghan R, et al. Inhibition of the multidrug resistance p-glycoprotein: Time for a change of strategy? Drug Metab Dispos. 2014;42(4):623–631. doi: 10.1124/dmd.113.056176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bendayan R, et al. Functional expression and localization of p-glycoprotein at the blood brain barrier. Microsc Res Tech. 2002;57(5):365–380. doi: 10.1002/jemt.10090. [DOI] [PubMed] [Google Scholar]
- 9.Lee G, Bendayan R. Functional expression and localization of p-glycoprotein in the central nervous system: Relevance to the pathogenesis and treatment of neurological disorders. Pharm Res. 2004;21(8):1313–1330. doi: 10.1023/b:pham.0000036905.82914.8e. [DOI] [PubMed] [Google Scholar]
- 10.Aller SG, et al. Structure of p-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323(5922):1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, et al. Refined structures of mouse p-glycoprotein. Protein Sci. 2014;23(1):34–46. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin C, et al. Communication between multiple drug binding sites on p-glycoprotein. Mol Pharmacol. 2000;58(3):624–632. doi: 10.1124/mol.58.3.624. [DOI] [PubMed] [Google Scholar]
- 13.Safa AR. Identification and characterization of the binding sites of p-glycoprotein for multidrug resistance-related drugs and modulators. Curr Med Chem Anticancer Agents. 2004;4(1):1–17. doi: 10.2174/1568011043482142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawson RJ, et al. Uptake or extrusion: Crystal structures of full abc transporters suggest a common mechanism. Mol Microbiol. 2007;65(2):250–257. doi: 10.1111/j.1365-2958.2007.05792.x. [DOI] [PubMed] [Google Scholar]
- 15.Ward A, et al. Flexibility in the abc transporter msba: Alternating access with a twist. Proc Natl Acad Sci U S A. 2007;104(48):19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barnes NM, Sharp T. A review of central 5-ht receptors and their function. Neuropharmacology. 1999;38(8):1083–1152. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- 17.Napier C, et al. Characterisation of the 5-ht receptor binding profile of eletriptan and kinetics of [3h] eletriptan binding at human 5-ht 1b and 5-ht 1d receptors. Eur J Pharmacol. 1999;368(2):259–268. doi: 10.1016/s0014-2999(99)00026-6. [DOI] [PubMed] [Google Scholar]
- 18.Tepper SJ, et al. Mechanisms of action of the 5-ht1b/1d receptor agonists. Arch Neurol. 2002;59(7):1084–1088. doi: 10.1001/archneur.59.7.1084. [DOI] [PubMed] [Google Scholar]
- 19.Evans DC, et al. Eletriptan metabolism by human hepatic cyp450 enzymes and transport by human p-glycoprotein. Drug Metab Disposition. 2003;31(7):861–869. doi: 10.1124/dmd.31.7.861. [DOI] [PubMed] [Google Scholar]
- 20.Kalvass JC, et al. Use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs: Comparison of unbound concentration ratios to in vivo p-glycoprotein efflux ratios. Drug Metab Disposition. 2007;35(4):660–666. doi: 10.1124/dmd.106.012294. [DOI] [PubMed] [Google Scholar]
- 21.Kikuchi R, et al. In vitro p-glycoprotein efflux ratio can predict the in vivo brain penetration regardless of biopharmaceutics drug disposition classification system class. Drug Metab Dispos. 2013;41(12):2012–2017. doi: 10.1124/dmd.113.053868. [DOI] [PubMed] [Google Scholar]
- 22.Humphrey PP. The discovery and development of the triptans, a major therapeutic breakthrough. Headache. 2008;48(5):685–687. doi: 10.1111/j.1526-4610.2008.01097.x. [DOI] [PubMed] [Google Scholar]
- 23.Ledwitch KV, et al. Unravelling the complex drug-drug interactions of the cardiovascular drugs, verapamil and digoxin, with p-glycoprotein. Biosci Rep. 2016;36(2) doi: 10.1042/BSR20150317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ledwitch KV, et al. Cooperativity between verapamil and atp bound to the efflux transporter p-glycoprotein. Biochem Pharmacol. 2016 doi: 10.1016/j.bcp.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ledwitch KV, Roberts AG. Cardiovascular ion channel inhibitor drug-drug interactions with p-glycoprotein. The AAPS Journal. 2016:1–12. doi: 10.1208/s12248-016-0023-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S, et al. Pubchem substance and compound databases. Nucleic Acids Res. 2015:gkv951. doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lerner-Marmarosh N, et al. Large scale purification of detergent-soluble p-glycoprotein frompichia pastoris cells and characterization of nucleotide binding properties of wild-type, walker a, and walker b mutant proteins. J Biol Chem. 1999;274(49):34711–34718. doi: 10.1074/jbc.274.49.34711. [DOI] [PubMed] [Google Scholar]
- 28.Bai JP, et al. A gene optimization strategy that enhances production of fully functional p-glycoprotein in pichia pastoris. PLoS One. 2011;6(8):e22577. doi: 10.1371/journal.pone.0022577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chifflet S, et al. A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: Application to lens atpases. Anal Biochem. 1988;168(1):1–4. doi: 10.1016/0003-2697(88)90002-4. [DOI] [PubMed] [Google Scholar]
- 30.Segel IH. Enzyme kinetics : Behavior and analysis of rapid equilibrium and steady state enzyme systems. New York: Wiley; 1975. [Google Scholar]
- 31.Cook PF, Cleland WW. Enzyme kinetics and mechanism. London; New York: Garland Science; 2007. [Google Scholar]
- 32.Leatherbarrow RJ. Using linear and non-linear regression to fit biochemical data. Trends Biochem Sci. 1990;15(12):455–458. doi: 10.1016/0968-0004(90)90295-m. [DOI] [PubMed] [Google Scholar]
- 33.Martin RB. Disadvantages of double reciprocal plots. J Chem Educ. 1997;74(10):1238. [Google Scholar]
- 34.Ranaldi F, et al. What students must know about the determination of enzyme kinetic parameters. Biochemical Education. 1999;27(2):87–91. [Google Scholar]
- 35.Liu R, et al. Intrinsic fluorescence of the p-glycoprotein multidrug transporter: Sensitivity of tryptophan residues to binding of drugs and nucleotides. Biochemistry. 2000;39(48):14927–14938. doi: 10.1021/bi0018786. [DOI] [PubMed] [Google Scholar]
- 36.Sharom FJ, et al. Fluorescence techniques for studying membrane transport proteins: The p-glycoprotein multidrug transporter. Methods Mol Biol. 2003;227:109–128. doi: 10.1385/1-59259-387-9:109. [DOI] [PubMed] [Google Scholar]
- 37.Lakowicz JR. Principles of fluorescence spectroscopy. New York: Kluwer Academic/Plenum; 1999. [Google Scholar]
- 38.Sonveaux N, et al. Ligand-mediated tertiary structure changes of reconstituted p-glycoprotein. A tryptophan fluorescence quenching analysis. J Biol Chem. 1999;274(25):17649–17654. doi: 10.1074/jbc.274.25.17649. [DOI] [PubMed] [Google Scholar]
- 39.Russell PL, Sharom FJ. Conformational and functional characterization of trapped complexes of the p-glycoprotein multidrug transporter. Biochem J. 2006;399(2):315–323. doi: 10.1042/BJ20060015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eckford PD, Sharom FJ. Functional characterization of escherichia coli msba: Interaction with nucleotides and substrates. J Biol Chem. 2008;283(19):12840–12850. doi: 10.1074/jbc.M708274200. [DOI] [PubMed] [Google Scholar]
- 41.Mayer M, Meyer B. Group epitope mapping by saturation transfer difference nmr to identify segments of a ligand in direct contact with a protein receptor. Journal of American Chemical Society. 2001;123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 42.Viegas A, et al. Saturation-transfer difference (std) nmr: A simple and fast method for ligand screening and characterization of protein binding. J Chem Educ. 2011;88(7):990–994. [Google Scholar]
- 43.Venkitakrishnan RP, et al. Use of nmr saturation transfer difference spectroscopy to study ligand binding to membrane proteins. Membrane Protein Structure and Dynamics. Methods and Protocols. 2012:47–63. doi: 10.1007/978-1-62703-023-6_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Claasen B, et al. Direct observation of ligand binding to membrane proteins in living cells by a saturation transfer double difference (stdd) nmr spectroscopy method shows a significantly higher affinity of integrin αiibβ3 in native platelets than in liposomes. J Am Chem Soc. 2005;127(3):916–919. doi: 10.1021/ja044434w. [DOI] [PubMed] [Google Scholar]
- 45.Haselhorst T, et al. Direct detection of ligand binding to sepharose-immobilised protein using saturation transfer double difference (stdd) nmr spectroscopy. Biochem Biophys Res Commun. 2007;359(4):866–870. doi: 10.1016/j.bbrc.2007.05.204. [DOI] [PubMed] [Google Scholar]
- 46.Borgnia MJ, et al. Competition of hydrophobic peptides, cytotoxic drugs, and chemosensitizers on a common p-glycoprotein pharmacophore as revealed by its atpase activity. J Biol Chem. 1996;271(6):3163–3171. doi: 10.1074/jbc.271.6.3163. [DOI] [PubMed] [Google Scholar]
- 47.Litman T, et al. Structure-activity relationships of p-glycoprotein interacting drugs: Kinetic characterization of their effects on atpase activity. Biochimica Et Biophysica Acta-Molecular Basis of Disease. 1997;1361(2):159–168. doi: 10.1016/s0925-4439(97)00026-4. [DOI] [PubMed] [Google Scholar]
- 48.von Richter O, et al. A novel screening strategy to identify abcb1 substrates and inhibitors. Naunyn Schmiedebergs Arch Pharmacol. 2009;379(1):11–26. doi: 10.1007/s00210-008-0345-0. [DOI] [PubMed] [Google Scholar]
- 49.Clay AT, Sharom FJ. Lipid bilayer properties control membrane partitioning, binding, and transport of p-glycoprotein substrates. Biochemistry. 2013;52(2):343–354. doi: 10.1021/bi301532c. [DOI] [PubMed] [Google Scholar]
- 50.Wishart DS, et al. Drugbank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34(suppl 1):D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Busche R, et al. Equilibrium, kinetic and photoaffinity labeling studies of daunomycin binding to p-glycoprotein-containing membranes of multidrug-resistant chinese hamster ovary cells. Eur J Biochem. 1989;183(1):189–197. doi: 10.1111/j.1432-1033.1989.tb14912.x. [DOI] [PubMed] [Google Scholar]
- 52.Doppenschmitt S, et al. Radioligand-binding assay employing p-glycoprotein-overexpressing cells: Testing drug affinities to the secretory intestinal multidrug transporter. Pharm Res. 1998;15(7):1001–1006. doi: 10.1023/a:1011965707998. [DOI] [PubMed] [Google Scholar]
- 53.Liu R, Sharom FJ. Site-directed fluorescence labeling of p-glycoprotein on cysteine residues in the nucleotide binding domains. Biochemistry. 1996;35(36):11865–11873. doi: 10.1021/bi960823u. [DOI] [PubMed] [Google Scholar]
- 54.Romsicki Y, Sharom FJ. The membrane lipid environment modulates drug interactions with the p-glycoprotein multidrug transporter. Biochemistry. 1999;38(21):6887–6896. doi: 10.1021/bi990064q. [DOI] [PubMed] [Google Scholar]
- 55.Seelig A, Landwojtowicz E. Structure-activity relationship of p-glycoprotein substrates and modifiers. Eur J Pharm Sci. 2000;12(1):31–40. doi: 10.1016/s0928-0987(00)00177-9. [DOI] [PubMed] [Google Scholar]
- 56.Angulo J, et al. Ligand-receptor binding affinities from saturation transfer difference (std) nmr spectroscopy: The binding isotherm of std initial growth rates. Chemistry (Easton) 2010;16(26):7803–7812. doi: 10.1002/chem.200903528. [DOI] [PubMed] [Google Scholar]
- 57.Loo TW, et al. The atpase activity of the p-glycoprotein drug pump is highly activated when the n-terminal and central regions of the nucleotide-binding domains are linked closely together. J Biol Chem. 2012;287(32):26806–26816. doi: 10.1074/jbc.M112.376202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scian M, et al. Reaction dynamics of atp hydrolysis catalyzed by p-glycoprotein. Biochemistry. 2014;53(6):991–1000. doi: 10.1021/bi401280v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Swartz DJ, et al. P-glycoprotein is fully active after multiple tryptophan substitutions. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2013;1828(3):1159–1168. doi: 10.1016/j.bbamem.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Esser L, et al. Structures of the multidrug transporter p-glycoprotein reveal asymmetric atp binding and the mechanism of polyspecificity. J Biol Chem. 2016 doi: 10.1074/jbc.M116.755884. jbc. M116. 755884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loo TW, et al. Drug binding in human p-glycoprotein causes conformational changes in both nucleotide-binding domains. J Biol Chem. 2003;278(3):1575–1578. doi: 10.1074/jbc.M211307200. [DOI] [PubMed] [Google Scholar]
- 62.Loo TW, et al. Substrate-induced conformational changes in the transmembrane segments of human p-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J Biol Chem. 2003;278(16):13603–13606. doi: 10.1074/jbc.C300073200. [DOI] [PubMed] [Google Scholar]
- 63.Verhalen B, et al. Dynamic ligand-induced conformational rearrangements in p-glycoprotein as probed by fluorescence resonance energy transfer spectroscopy. J Biol Chem. 2012;287(2):1112–1127. doi: 10.1074/jbc.M111.301192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Mara ML, Mark AE. Structural characterization of two metastable atp-bound states of p-glycoprotein. PLoS One. 2014;9(3):e91916. doi: 10.1371/journal.pone.0091916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beaudet L, Gros P. Functional dissection of p-glycoprotein nucleotide-binding domains in chimeric and mutant proteins. Modulation of drug resistance profiles. J Biol Chem. 1995;270(29):17159–17170. doi: 10.1074/jbc.270.29.17159. [DOI] [PubMed] [Google Scholar]
- 66.Urbatsch IL, et al. Both p-glycoprotein nucleotide-binding sites are catalytically active. J Biol Chem. 1995;270(45):26956–26961. doi: 10.1074/jbc.270.45.26956. [DOI] [PubMed] [Google Scholar]
- 67.Hrycyna CA, et al. Both atp sites of human p-glycoprotein are essential but not symmetric. Biochemistry. 1999;38(42):13887–13899. doi: 10.1021/bi991115m. [DOI] [PubMed] [Google Scholar]
- 68.Lawson J, et al. Structure-based interpretation of the mutagenesis database for the nucleotide binding domains of p-glycoprotein. Biochim Biophys Acta. 2008;1778(2):376–391. doi: 10.1016/j.bbamem.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 69.Frank GA, et al. Cryo-em analysis of the conformational landscape of human p-glycoprotein (abcb1) during its catalytic cycle. Mol Pharmacol. 2016;90(1):35–41. doi: 10.1124/mol.116.104190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nicklisch SC, et al. Global marine pollutants inhibit p-glycoprotein: Environmental levels, inhibitory effects, and cocrystal structure. Sci Adv. 2016;2(4):e1600001. doi: 10.1126/sciadv.1600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.