

Abstract
CD40/CD40L interactions play a critical role in immunity and autoimmunity. In this study, we sought to understand the requirement for CD40 signaling in the programmed cell death-1 (PD-1) checkpoint and CD28 costimulatory pathways important for maintenance of peripheral tolerance. Blocking either pathway can result in loss of self-tolerance and development of autoimmunity. We found that primary Sjögren’s syndrome (pSS) and autoimmune thyroid diseases (ATDs) that develop spontaneously in CD28-deficient IFN-γ−/− NOD.H-2h4 (CD28−/−) mice required CD40 signaling. Specifically, blockade of CD40L with the anti-CD40L mAb, MR1, inhibited autoantibody production and inflammation in thyroid and salivary gland target tissues. Unexpectedly, however, ATD and pSS in PD-1–deficient IFN-γ−/− NOD.H-2h4 (PD-1−/−) mice developed independently of CD40/CD40L interactions. Treatment with MR1 had no effect and even exacerbated disease development in pSS and ATD, respectively. Most interesting, anti-thyroglobulin and pSS-associated autoantibodies were increased following anti-CD40L treatment, even though MR1 effectively inhibited the spontaneous splenic germinal centers that form in PD-1–deficient mice. Importantly, blockade of the PD-1 pathway by administration of anti–PD-1 mAb in CD28−/− mice recapitulated the PD-1−/− phenotype, significantly impacting the ability of MR1 to suppress ATD and pSS in these mice. These results indicate that there can be different pathways and requirements to autoimmune pathogenesis depending on the availability of specific checkpoint and costimulatory receptors, and an intact PD-1 pathway is apparently required for inhibition of autoimmunity by anti-CD40L.
INTRODUCTION
NOD.H-2h4 mice provide an excellent model for studying mechanisms involved in the development and treatment of autoimmune diseases, such as autoimmune thyroid diseases (ATD) and primary Sjögren’s syndrome (pSS). Wild-type (WT) NOD.H-2h4 mice (both sexes) given NaI in the drinking water develop a relatively mild form of ATD, with infiltration of T and B cells into the thyroid, production of anti-mouse thyroglobulin (MTg) autoantibodies, and spontaneous development of germinal centers (GCs) in the spleen, thyroids, and salivary glands (1). Most female WT NOD.H-2h4 mice also develop pSS (1–4). ATD in WT NOD.H-2h4 mice does not result in hypothyroidism, the major indicator of ATD in humans. We (5, 6) generated IFN-γ−/− NOD. H-2h4 mutants that develop a more severe ATD that is characterized by extensive proliferation and hyperplasia of thyroid epithelial cells (thyrocytes) when given NaI in the drinking water. ATD in IFN-γ−/− mice is a T cell–dependent autoimmune disease in which activated autoreactive CD4+ and CD8+ T cells migrate to the thyroid where they produce TNF-α and other cytokines that act on thyrocytes to promote their proliferation (7). Because severe ATD develops slowly and in only 60–70% of IFN-γ−/− NOD.H-2h4 mice, we generated CD28-deficient IFN-γ−/− NOD.H-2h4 mutants that develop severe ATD and hypothyroid-ism, with a nearly 100% incidence at 4 mo of age (1, 8). Female CD28−/− mice also develop more severe pSS than WT NOD.H-2h4 mice (1, 8). As shown by us (4) and other investigators (2, 9, 10), pSS is also a T cell–dependent autoimmune disease with activated CD4+ and CD8+ T cells and B cells in the salivary gland infiltrates and, like ATD, proinflammatory cytokine production by T cells likely mediates the damage to the organ. In humans, ATD and pSS frequently occur in the same individuals (11), and this is also true for the NOD.H-2h4 model of autoimmune disease (2–4, 12–14). The greater severity and earlier onset of ATD and pSS in CD28−/− NOD.H-2h4 mice compared with WT NOD.H-2h4 mice provide an excellent model for examining mechanisms and devising treatment protocols for autoimmune diseases.
Interaction of CD40 with its ligand, CD40L (CD154), is important for generation of optimal immune responses to most Ags and for development of most experimentally induced and spontaneous autoimmune diseases (15–17). CD40, expressed on B cells and other APCs, interacts with CD40L, expressed on activated T cells, to facilitate T cell activation, plasma cell differentiation, and Ab production (15, 18). CD40 is also expressed on thyrocytes of NOD and NOD.H-2h4 mice (19) and humans (20). Blocking the CD40L/CD40 pathway in vivo by administration of a monoclonal anti-CD40L Ab, MR1, inhibits development of most experimentally induced autoimmune diseases (15), including experimental autoimmune thyroiditis in mice (21, 22). We previously showed that an engineered triple mutation anti-CD40L mAb that removes Fc effector function (MR1-TM) inhibits development of iodine-facilitated spontaneous autoimmune thyroiditis (I-SAT) and pSS in WT NOD.H-2h4 mice and suppresses autoimmune diabetes in NOD mice (23). MR1-TM inhibits spontaneous GC formation in the spleen and the development of tertiary lymphoid structures (TLSs) in target tissues of NOD and NOD.H-2h4 mice (23), as well as depletes GC B cells (24).
Several immune checkpoint pathways are critical for maintaining self-tolerance and preventing autoimmunity. For example, the B7/CD28 family of costimulatory molecules provides critical signals that regulate T cell activation and tolerance (25). Programmed cell death-1 (PD-1; CD279) is an immune-inhibitory receptor belonging to the B7CD28 family of costimulatory and coinhibitory molecules. PD-1 is expressed on activated T cells, B cells, and myeloid cells (26) and delivers inhibitory signals that regulate the balance of T cell activation, tolerance, and immune-mediated tissue damage (27). Blocking or elimination of the B7: CD28 or PD-1/PDL pathway can result in development of autoimmunity (27, 28). CD28−/− mice develop autoimmunity as a result of the absence of most functional regulatory T cells (Tregs) (8, 13, 29, 30), whereas autoimmunity in PD-1−/− mice develops as a result of the loss of an important inhibitory pathway (31).
The goal of this study was to examine the requirement for CD40/CD40L interactions in autoimmune mice that lack the immune checkpoint receptors CD28 or PD-1. We hypothesized that MR1 would have little or no effect on the development of ATD and pSS in CD28−/− mice, because they have been reported to lack GCs (32) that require CD40 for their development. In contrast, we assumed that MR1 would inhibit ATD and pSS in PD-1−/− mice that can develop GCs (33). Unexpectedly, however, we found that early blockade of the CD40/CD40L pathway inhibited pSS and ATD and reduced autoantibody responses in mice lacking CD28, whereas inhibition of CD40 signaling did not block development of autoimmunity in mice that lacked the PD-1 inhibitory pathway. These results indicate that CD40/CD40L interactions are essential for autoimmune disorders that develop in the absence of CD28 but are not required for the development of autoimmunity in mice lacking the PD-1 coinhibitory receptor.
MATERIALS AND METHODS
Mice
NOD.H-2h4 mice, generated as previously described, express background genes of the NOD mouse but differ at the MHC locus, expressing the K haplotype at the K and I-A loci (1). IFN-γ−/− CD28−/− NOD.H-2h4 mice (MMRRC 037141) were generated in the University of Missouri animal facility, as previously described (6, 8). IFN-γ−/− PD-1−/− NOD.H-2h4 mice (MMRRC 037145) were generated in the University of Missouri animal facility by crossing PD-1−/− NOD males (provided by Dr. A. Sharpe, Harvard Medical School) with IFN-γ−/− NOD.H-2h4 females (MMRRC 037140). The F1 offspring were crossed, and the resulting F2 mice were selected for expression of the NOD.H-2h4 MHC and absence of the NOD MHC, as described previously (6, 34). Expression of the IFN-γ and PD-1 mutations was determined by PCR analysis of tail DNA using primers described on The Jackson Laboratory Web site for IFN-γ or provided by the Sharpe laboratory for PD-1. Mice were further crossed until all offspring expressed the NOD.H-2h4 MHC and were homozygous for expression of the IFN-γ and PD-1 mutations. All animal protocols were approved by the University of Missouri Animal Care and Use Committee. All mice used in this study were IFN-γ−/− NOD.H-2h4 mutants lacking CD28 (CD28−/−) or PD-1 (PD-1−/−).
Abs
The anti-CD154 mAb, MR1, was a triple mutation (TM) mAb generated and provided by MedImmune. It was engineered to contain the Fc region of human IgG1 with TM in the Ch2 region to prevent interactions with the Fc receptor (24). Other Abs included anti-CD25 (PC61; Leinco Technologies, St. Louis, MO) and anti–PD-1 (RMP1–4; Bio X Cell, New Lebanon, NH). Human IgG1 TM was used as isotype control for MR1-TM, and normal rat IgG was used as isotype control for anti-CD25 and anti–PD-1.
Experimental design for induction of ATD and pSS
Mice were given 0.08% (w/v) NaI in their drinking water at 5–7 wk of age, and thyroids and submandibular salivary glands were removed 7–12 wk later, as indicated for individual experiments. In all experiments, mice in different treatment groups were cohoused. Individual groups within an experiment were litter-mates, and most groups consisted of more than one litter of comparable age (±2 wk). NaI supplementation of the drinking water is essential for early development and a high incidence of severe ATD (1) and has no effect on the incidence or severity of pSS (H. Braley-Mullen, unpublished observations). MR1-TM (400 μg) was administered i.v. at 4–6 wk of age, and mice were given NaI-supplemented water for 1–7 d, unless indicated otherwise. In some experiments, Tregs were transiently depleted by i.p. administration of 0.5 mg of anti-CD25 (PC61) or rat IgG, as previously described (12). Most peripheral Foxp3+ CD4+ T cells are depleted for 7–10 d, after which they gradually repopulate and are present in relatively normal numbers 3 wk later (12). Anti-CD25 or rat IgG was administered to 5–7-wk-old mice, followed by anti-CD40L (MR1-TM) or isotype control 5–7 d later. For experiments using anti–PD-1 to block the PD-1–inhibitory pathway, CD28−/− mice were given anti–PD-1 or rat IgG i.p. (300 μg per mouse), 5–7 d before injection of MR1-TM or isotype control. Injections were repeated 1 d before and 4–5 d after MR1-TM or isotype control treatment.
Serum T4
Blood was collected from the retro-orbital plexus immediately before collecting tissue for histology. Serum T4 was measured by ELISA using a kit from Leinco Technologies, according to the manufacturer’s protocol. As previously determined, mice were considered hypothyroid at T4 < 3 μg/dl of serum (35). Serum T4 levels are highly correlated with ATD severity scores (35). Mice with few or no residual thyroid follicles (severity scores of 4–5+) always have low serum T4 levels (8, 35).
Autoantibody determination
Anti-MTg autoantibodies were determined by ELISA, as described in detail previously (36). Sera from individual mice were diluted 1/50 or 1/100 and plated in duplicate on MTg-coated ELISA plates. Anti-Ro and anti-La Abs were quantified by ELISA. Briefly, plates were coated overnight with 5 μg/ml anti-Ro Ab (Sigma-Aldrich, St. Louis, MO) or anti-La Ab (Abcam, Cambridge, MA) diluted in PBS. After washing with PBS with 0.5% Tween and blocking with 0.2% BSA in PBS, serum was diluted and incubated on plates overnight. Bound Ig was detected with goat anti-mouse IgG–alkaline phosphatase (0.2 μg/ml; Southern-Biotech, Birmingham, AL) diluted in blocking buffer. Plates were developed with SIGMAFAST p-Nitrophenyl phosphate Tablets (Sigma-Aldrich), and specific absorbance was measured at 405 nm using a Molecular Devices SpectraMax micro-plate reader.
Scoring of ATD
Thyroid lobes were fixed in formalin, sectioned, and stained with H&E. Thyroids were scored for the extent of thyroid follicular cell hyperplasia/proliferation and inflammatory cell infiltration using a scale of 0–5+, as previously described (1, 5). Briefly, a score of 0 indicates no changes compared with thyroids from unmanipulated mice, and 0+ indicates mild follicular changes with very few inflammatory cells. Cellular infiltrates with ≥125 cells and hyperplastic changes sufficient to replace several follicles are scored 1+. 2+ indicates 10–20 areas of inflammatory cell infiltration and hyperplastic changes or destruction up to one fourth of the gland, 3+ indicates that one fourth to one half of the gland has inflammatory cells and hyperplastic changes, 4+ indicates that more than one half of the gland is hyperplastic and infiltrated, and 5+ indicates that thyroids have few or no remaining normal follicles. IFN-γ−/− mice with severe (4–5+) ATD have widespread clusters of proliferating thyrocytes, with lymphocyte infiltration and areas of proliferating thyrocytes surrounded by collagen (fibrosis). All scoring was done in a blinded manner.
Scoring of salivary gland infiltration
Submandibular salivary glands were removed when thyroids were collected, fixed in formalin, sectioned, and stained using H&E. Salivary glands were scored in a blinded manner by counting the number of foci containing >50 lymphocytes, referred to as focus scores (14, 37), and results are expressed as the number of foci in both submandibular gland lobes. Many salivary glands were also scored using a Zeiss Axiovert 200M microscope and MetaMorph software, as previously described (8). The focus scores obtained in this manner were defined as the number of foci with >50 lymphocytes per four square millimeters (8).
Immunohistochemical staining
Salivary glands and spleens were removed at the indicated ages. Tissues were frozen in OCT, and blocks were stored at −70°C until sectioning. Frozen tissues were sectioned (5 μm thickness), fixed in acetone, and stained with the following primary Abs: anti-IgM–Texas Red (SouthernBiotech), anti-B220, anti-CD4–FITC, anti-CD8–FITC, anti-GL7–FITC, anti–peanut agglutinin (PNA)–FITC, and biotinylated PNA (Vector Laboratories, Burlingame, CA). Anti-rat–Oregon Green and streptavidin–Oregon Green were used as secondary Abs. All Abs were purchased from Life Technologies, unless otherwise indicated. The number of splenic GCs was determined by dividing the total number of GCs by the total number of B cell follicles, to normalize for area in each cryosection. Two or three cryosections per mouse were evaluated. Expression of CD3 and B220 in thyroids and salivary glands was examined using formaldehyde-fixed paraffin sections, as previously described (8). All fixed tissue was stained by IDEXX RADIL (Columbia, MO).
Statistics
GraphPad Prism 4.0 was used with the nonparametric Mann–Whitney U test for determining the significance of differences in severity of ATD and pSS. The Student t test was used for all other statistical analyses. A p value < 0.05 was considered statistically significant.
RESULTS
CD40/CD40L interactions are required for development of ATD and pSS in CD28−/− NOD.H-2h4 mice
CD28−/− mice reportedly do not develop GCs (32), but CD28−/− NOD.H-2h4 mice develop accelerated ATD and pSS due to a lack of functional Tregs (8). We hypothesized that autoimmune disorders that develop in the absence of CD28 might not require CD40L signaling, because our previous study showed that MR1 suppressed autoimmune diseases in WT NOD.H-2h4 mice by eliminating the spontaneous development of splenic GCs (23). To test this hypothesis, CD28−/− NOD.H-2h4 female and male mice, 5–6 wk of age, were given a single injection of MR1-TM. The next day, they were given NaI in their drinking water to promote optimal development of severe ATD (8). Thyroids and submandibular salivary glands were removed 7–8 wk later. Unexpectedly, a single injection of anti-CD40L significantly prevented the development of severe ATD in male and female CD28−/− NOD.H-2h4 mice (Fig. 1A, 1B). All but one female mouse given isotype control had severe ATD (4–5+ score), whereas most mice given anti-CD40L had minimal or no ATD (Fig. 1A). Blocking CD40/CD40L interactions also resulted in normalization of thyroid function (serum T4 >3 μg/dl) in all but one MR1-treated mouse, whereas most mice given isotype control were hypothyroid, with low serum T4 levels (Fig. 1C). Anti-CD40L had no effect on ATD if it was administered 2 wk after mice were given NaI-supplemented water, when thyroid lesions were beginning to develop (Supplemental Fig. 1), indicating that CD40L signaling is important at early time points for the initiation of ATD.
FIGURE 1. MR1-TM inhibits the development of severe ATD and pSS in CD28−/− NOD.H-2h4 mice.

(A) Mice (both sexes) were given 400 μg of MR1-TM or isotype control at 5–6 wk of age. The next day, they were given NaI-supplemented water, and thyroids were removed 7–8 wk later. Each point represents an individual mouse given isotype control or MR1. Female: n = 16, male: n = 10. ****p < 0.0001. (B) Representative H&E-stained sections of thyroids from a mouse given isotype control or MR1 (original magnification ×100). Scale bar, 10 μM. (C) Serum T4 levels in some mice from (A). Values less than or equal to three indicate low thyroid function (see Materials and Methods). ****p < 0.0001. (D) Salivary gland infiltration of some mice in (A). Each point represents the focus score of an individual mouse (n = 11 for each group). **p < 0.001 Iso versus MR1, for females and males. Females have significantly higher focus scores than males (*p < 0.05). (E) Representative H&E-stained and immunohistochemistry-stained sections (B cells, red; T cells, green) of a salivary gland from a female mouse given isotype control and a female mouse given MR1. Mice are 4 mo old. Original magnification ×100 (immunohistochemistry). Scale bar (H&E), 1 mm.
Female CD28−/− NOD.H-2h4 mice develop pSS at 4 mo of age (8). Evaluation of salivary gland infiltration of the mice in Fig. 1A indicated that mice given MR1-TM had greatly reduced inflammatory cell infiltrates compared with salivary glands of isotype control–treated mice. Salivary gland focus scores of MR1-treated mice (both sexes) were significantly reduced compared with isotype controls (Fig. 1D, 1E). As expected, female mice had higher focus scores than males. These results indicate that development of pSS in CD28-deficient mice is highly dependent on CD40/CD40L signaling.
GC formation is largely absent in CD28−/− NOD.H-2h4 mice
As reported previously (23), anti-CD40L suppresses autoimmunity in WT NOD.H-2h4 mice by eliminating splenic GCs and preventing TLS neogenesis. If CD28−/− mice do not develop GCs, as reported by other investigators (32, 38, 39), suppression of autoimmunity in CD28−/− mice must develop by a GC-independent mechanism. To determine whether CD28−/− NOD. H-2h4 mice spontaneously develop splenic GCs, spleens were obtained from 3–6-mo-old CD28−/−female mice. Consistent with earlier reports, splenic GCs do not develop spontaneously in CD28−/−female NOD.H-2h4 mice up to 6 mo of age, whereas they are present in female age-matched CD28-sufficient WT NOD.H-2h4 mice (Fig. 2). Surprisingly, GC-like PNA+ structures were present in salivary gland–associated lymph nodes of 3 of 24 mice examined (data not shown). Because all female mice had pSS, the fact that GC-like structures were detected in lymph nodes of only a few mice suggests that they are not critical for the development of pSS in this strain. Salivary glands of all female CD28−/− mice with pSS had clusters of CD4+ T cells and B cells that were not GL-7+, and they were not detected in salivary glands of MR1-treated mice (Fig. 1E). These results indicate that pSS in CD28−/− mice develops independently of GCs but requires CD40/CD40L interactions.
FIGURE 2. Splenic GCs are absent in CD28−/− NOD.H-2h4 mice.
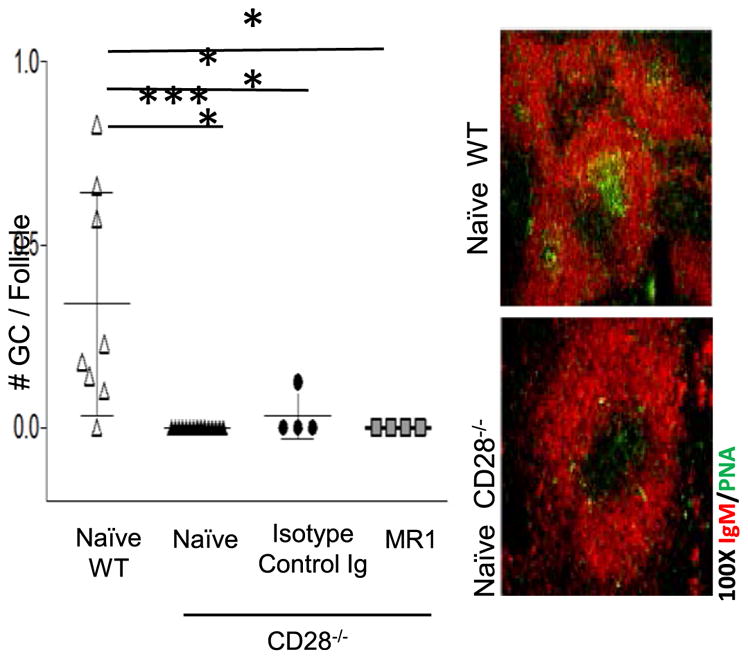
WT or CD28−/− NOD.H-2h4 females were left untreated (naive; n = 8) or were given MR1 (n = 4) or isotype-control (n = 4) mAb, as indicated, at 7 wk of age. Spleens were removed at 14 wk of age, and frozen sections were stained for expression of B cells (IgM; red) and GCs (PNA; green). *p < 0.05, ***p < 0.001.
CD40/CD40L interaction blockade accelerates ATD and pSS in PD-1–deficient− NOD.H-2h4 mice
The PD-1/PDL pathway is a negative regulatory pathway critical for terminating immune responses. The blocking or absence of PD-1 results in breakdown of tolerance and development of autoimmunity (31, 40). PD-1−/− NOD.H-2h4 mice (IFN-γ sufficient and IFN-γ deficient) develop severe ATD and pSS earlier and with a higher frequency compared with their PD-1–sufficient counterparts, although both diseases develop more slowly and with a lower incidence than in CD28−/− IFN-γ−/− mice (H. Braley-Mullen, unpublished observations). We hypothesized that blocking CD40/CD40L interactions would suppress the development of pSS and ATD in IFN-γ−/− PD-1−/− NOD.H-2h4 mice that should develop spontaneous GCs. To test this, PD-1−/− NOD.H-2h4 mice were given a single injection of MR1-TM at 5–6 wk of age and were given NaI-supplemented water for 12 wk before tissues were collected. Surprisingly, female and male mice given anti-CD40L had a significantly higher incidence of severe ATD compared with the corresponding mice given isotype-control mAb (Fig. 3A, 3B). We next examined the effect of CD40/CD40L blockade on the development of pSS. Salivary gland infiltration was generally lower in PD-1−/− mice than in female CD28−/− mice and was greater in females than in males. In contrast to the results with CD28−/− mice given anti-CD40L, a single injection of anti-CD40L did not inhibit pSS in PD-1–deficient mice (Fig. 3C). Representative images of thyroid and salivary gland infiltration in isotype-control– versus MR1-treated PD-1−/− mice are shown in Fig. 3B and 3D.
FIGURE 3. MR1-TM does not inhibit the development of ATD and pSS in mice lacking the PD-1 inhibitory molecule.

(A) The experimental design is identical to that in Fig. 1A. Mice were 17–20 wk old when thyroids were removed. Isotype control (n = 18) versus MR1 (n = 19): **p < 0.001 for females, isotype control (n = 18) versus MR1 (n = 19). *p < 0.05 for males. (B) Representative H&E-stained sections of thyroids from a mouse given isotype control or MR1. ATD severity scores are 4+, isotype control; 5+, MR1. Original magnification ×100. (C) Salivary gland focus scores (foci per four square millimeters) of some mice from (A). There are no significant differences between isotype controls and MR1-treated mice. Female mice have more severe pSS than males. (D) Representative H&E-stained sections of salivary glands of a female mouse given isotype control or MR1. Scale bars, 1 mm.
The above results indicate that CD40L blockade has opposing effects on the development of autoimmunity depending on the presence of specific costimulatory or checkpoint-inhibitory receptors. CD28−/− mutants develop very severe ATD and pSS that are highly CD40/CD40L dependent. In contrast, blocking CD40/CD40L interactions promotes, rather than inhibits, the development of severe ATD and has no effect on pSS in PD-1−/− mutants; PD-1−/− mutants have a lower incidence of severe ATD and pSS than CD28−/− mutants (Figs. 1, 3), perhaps because CD28−/− mice have few functional Tregs (8), and PD-1−/− mice should have normal numbers of functional Tregs (41, 42). To determine whether the reduced incidence of severe ATD and pSS in PD-1−/− mice compared with CD28−/− mutants is due, at least in part, to increased Treg activity, PD-1−/− mice were transiently depleted of Tregs, as previously described (12). Treg depletion greatly increased the incidence of severe ATD and pSS, such that most mice (both sexes) had severe disease at 4–5 mo of age (Supplemental Fig. 2), comparable to CD28−/− mice that permanently lack most functional Tregs (Fig. 1A). The effects of blocking CD40L were not changed when Tregs were depleted; neither autoimmune disease was suppressed by anti-CD40L in Treg-depleted PD-1−/− mice (Supplemental Fig. 2).
Anti-CD40L dissolves GCs in PD-1−/− NOD.H-2h4 mice
Next, we addressed whether PD-1−/− NOD.H-2h4 mice develop spontaneous GCs and whether their GCs are dissolved by anti-CD40L. PD-1−/− mice of both sexes spontaneously developed splenic GCs (Fig. 4A) that were not detected in MR1-treated mice (Fig. 4B). Therefore, MR1 dissolves splenic GCs in PD-1−/− mice as it does in WT NOD.H-2h4 mice (23), but this does not result in suppression of their autoimmune disease. Treg depletion promoted splenic GC development in PD-1−/− mice, and this was also independent of CD40/CD40L interactions (data not shown). These results suggest that, in contrast to WT NOD.H-2h4 mice, autoimmunity in PD-1−/− and CD28−/− mutants develops independently of GCs.
FIGURE 4. PD-1 deficiency promotes the development of splenic GCs in male mice, and MR1 dissolves GCs in both genders.

Frozen sections of spleens from 4–5-mo-old WT (n = 4) and PD-1−/− NOD.H-2h4 mice (n = 4 females, n = 8 males) were stained for expression of B cells (IgM, red; PNA, green). Original magnification ×100. Results are expressed as the number of GCs per follicle. (A) Untreated versus WT NOD.H-2h4 mice. *p < 0.05. (B) PD-1−/− mice given MR1 or isotype control (n = 5 females, n = 4 males). (B) **p < 0.001, ***p < 0.0001.
MR1-TM has opposing effects on autoantibody responses in CD28−/− and PD-1−/− mutants
NOD.H-2h4 mice with ATD produce anti-MTg autoantibodies, the levels of which generally correlate with ATD severity scores (1, 5). To begin to address the mechanisms underlying the opposing effects of blocking CD40/CD40L signaling on ATD and pSS development in CD28−/− and PD-1−/− mice, anti-MTg autoantibody levels were determined. As previously reported (8), CD28−/− mice produce anti-MTg autoantibodies, and autoantibody levels correlate with ATD severity scores (data not shown). Anti-MTg autoantibody responses in PD-1−/− mice also correlated with ATD severity scores (Fig. 5B, top). Anti-MTg autoantibody responses in CD28−/− mice were reduced when mice were given anti-CD40L (Fig. 5A), whereas anti-MTg autoantibody responses in anti-CD40L-treated PD-1−/− mice with 3–5+ ATD severity scores were significantly higher than those in mice with comparable severity scores that were given isotype control (Fig. 5B). Therefore, in contrast to CD28−/− mice, blocking CD40/CD40L interactions in PD-1−/− mice increased, rather than inhibited, anti-MTg autoantibody responses, indicating that the opposing requirements for CD40/CD40L interactions in the two mutants are comparable for the development of autoimmune inflammation and the production of anti-MTg autoantibodies.
FIGURE 5. MR1 inhibits anti-MTg autoantibody responses in CD28−/− NOD.H-2h4 mice but increases autoantibody responses in PD-1−/− NOD. H-2h4 mice.

(A) Anti-MTg autoantibodies were measured in individual CD28−/− mice from Fig. 1A (n = 15). (B) Anti-MTg autoantibodies [isotype control (0–2) n = 20, MR1 (0–2) n = 7, isotype control (3–5) n = 18, MR1 (3–5) n = 23] were measured in individual 16–20-wk-old PD-1−/− NOD.H-2h4 mice from Fig. 3A. Mice were separated into two groups based on their ATD severity scores (0–2+ or 3–5+). (C) Anti-La autoantibodies (WT n = 7, isotype control n = 10, MR1 n = 12) and anti-Ro autoantibodies (WT n = 6, isotype control n = 9, MR1 = 9) were measured in some 18–20-wk-old PD-1−/− female mice from Fig. 3A. *p < 0.05, **p < 0.001, ***p < 0.001.
Anti-SSA (anti-Ro) and anti-SSB (anti-La) are anti-nuclear autoantibodies associated with some autoimmune diseases, such as systemic lupus erythematosus and pSS (2, 43, 44). Consistent with our previous results (8), these autoantibodies were very low and often undetectable in serum of CD28−/− NOD.H-2h4 females with severe salivary gland inflammation (data not shown), perhaps because the mice used for these experiments were 12–14 wk old, and our earlier studies indicated that Ro and La autoantibody levels in WT NOD.H-2h4 mice were also very low at this age (23). PD-1−/− mice (16–18 wk old) produced Ro and La autoantibodies at levels comparable to those in age-matched WT NOD.H-2h4 mice (Fig. 5C). Surprisingly however, PD-1−/− females treated with anti-CD40L had significantly higher levels of both autoantibodies (Fig. 5C), whereas production of anti-Ro and anti-La was reduced by treatment of WT NOD.H-2h4 mice with MR1 (23). These results indicate that autoantibody responses are differentially regulated by CD40/CD40L interactions in CD28−/− and PD-1−/− NOD.H-2h4 mice.
Blocking PD-1 in CD28−/− mice partially reverses MR1-induced suppression of autoimmunity
The results presented above show different requirements for CD40/CD40L interactions for the development of autoimmunity that are dependent on which costimulatory or checkpoint inhibitors are present. Previously, two studies (44, 45) reported that an intact PD-1/PD-L1 pathway was required for anti-CD40L to prolong graft survival and induce transplantation tolerance. Therefore, we asked whether blocking the PD-1 inhibitory pathway in CD28−/− mice using anti–PD-1 would influence the ability of anti-CD40L to suppress their autoimmune diseases. CD28−/− mice were given anti–PD-1, as indicated in Materials and Methods, and thyroids or salivary glands were removed 7 wk later (Fig. 6). Anti–PD-1 significantly reduced the ability of anti-CD40L to suppress ATD and pSS in CD28−/− mice. These results suggest that the requirement for CD40/CD40L interactions for the development of autoimmunity is greatly reduced when the PD-1 checkpoint–inhibitory pathway is absent (PD-1−/− mice) or blocked (by anti–PD-1).
FIGURE 6. Blocking the PD-1 pathway in CD28−/− NOD.H-2h4 mice recapitulates the PD-1 deficiency phenotype for development of CD40L-independent autoimmunity.

(A) ATD severity score (isotype control n = 10, MR1 n = 13, isotype control + anti-PD-1 n = 9, MR1 + anti–PD-1 n = 14). (B) pSS (isotype control n = 8, MR1 n = 11, isotype control + anti–PD-1 n = 7, MR1 + anti–PD-1 n = 11) focus scores (total foci per gland) were measured in CD28−/− NOD.H-2h4 mice treated with isotype control, MR1, and anti–PD-1, as indicated in (A). Anti–PD-1 or rat IgG was administered three times, as indicated in Materials and Methods. *p = 0.015, MR1 versus MR1 + anti–PD-1. **p < 0.001, isotype control versus MR1. ***p = 0.0008, isotype control versus MR1. ****p = 0.00001, MR1 versus MR1 + anti–PD-1.
DISCUSSION
Blocking CD40/CD40L interactions inhibits the development of experimentally induced (21, 22, 46) and spontaneous autoimmune diseases, such as type I diabetes in NOD mice and ATD and pSS in NOD.H-2h4 mice (16, 23). In our previous studies, a single injection of anti-CD40L (CD154) prevented the development of diabetes, pSS, and thyroiditis in NOD and NOD.H-2h4 mice by eliminating the spontaneous development of splenic GCs (23). Blocking CD40/CD40L interactions interferes with T/B cell interactions, resulting in reduced T cell activation and autoantibody production by self-reactive B cells (15). The goal of this study was to examine the requirement for CD40/CD40L interactions for the development of autoimmunity in NOD.H-2h4 mutants lacking particular checkpoint or costimulatory receptors (PD-1 or CD28) important for maintenance of self-tolerance. A single injection of anti-CD40L profoundly inhibited the development of ATD and pSS (Fig. 1) and resulted in reduced anti-MTg autoantibody responses in CD28−/− NOD.H-2h4 mice (Fig. 5). CD28−/− mice did not develop splenic GCs (Fig. 2), suggesting that their autoimmunity develops via a GC-independent pathway. Although salivary glands in CD28−/− mice with pSS had many infiltrating B cells, the B cells did not express GC markers. Salivary gland–associated lymph nodes with GC markers were detected in only a few mice, whereas all mice developed autoimmune disease, indicating that suppression of autoimmunity by MR1 in CD28−/− mice likely indicates a requirement for CD40/CD40L interactions for autoreactive T and B cell activation that is independent of its ability to dissolve GCs. Because CD28−/− mice lack most functional Tregs (13), suppression of autoimmunity by blocking CD40/CD40L interactions is apparently not due to effects on Tregs.
More interesting and unexpected results were obtained using PD-1−/− NOD.H-2h4 mice. Although PD-1−/− mice have a higher incidence of severe ATD and pSS than PD-1+ mice, the incidence of severe autoimmunity at 4 mo of age is much lower than in CD28−/− mice (H. Braley-Mullen, unpublished observations). This is due, in part, to Treg activity in PD-1−/− mice, because transient depletion of Tregs resulted in a higher incidence of ATD and pSS, comparable to that of CD28−/− mice (Supplemental Fig. 2). The unexpectedly strong Treg-mediated suppression of autoimmunity in PD-1−/− mice suggests that PD-1–deficient Tregs or T follicular regulatory cells (Tfrs) may be more potent than WT Tregs and Tfrs, as suggested by other investigators (47). Unlike CD28−/− mice that lack splenic GCs, PD-1−/− mice spontaneously develop splenic GCs (Fig. 4). Importantly, in WT NOD.H-2h4 mice, spontaneous GCs develop in young female, but not male, mice (23). In this case, loss of PD-1 in male mice was associated with the development of spontaneous GCs and increased salivary gland inflammation, suggesting a connection between early-life splenic GCs and the development of glandular inflammation, as we previously described for female WT mice (21). Unexpectedly, anti-CD40L effectively eliminated splenic GCs in PD-1−/− mice (Fig. 4), but the development of pSS was unaffected, and there was a higher incidence of severe ATD (Fig. 3). In addition, WT (23) and CD28−/− mice given anti-CD40L had reduced anti-MTg autoantibody responses compared with mice given isotype control, whereas autoantibody responses were higher in PD-1−/− mice given anti-CD40L compared with their respective controls (Fig. 5). These results indicate that activation of autoreactive B cells in PD-1−/− mice, in contrast to most other strains of mice, is CD40/CD40L and GC independent. Autoantibody responses to some self-Ags and foreign Ags, as in autoimmune-prone MRL/lpr mice, develop extrafollicularly and independently of GCs (48–54). However, those extrafollicular Ab responses require CD40/CD40L interactions (48, 50–52). PD-1 regulates GC B cell survival and development of follicular helper T cells (Tfhs) and Tfrs that are important for GC responses (47), but GCs, Tfhs, and Tfrs develop in PD-1−/− mice (33, 55–57), and PD-1−/− mice have normal Ab responses to most Ags (31). The inability of anti-CD40L to suppress autoantibody responses in PD-1−/− mice may be due, in part, to the fact that PD-1 expression by B cells can be required for suppression of autoantibody production (58). To our knowledge, others have not determined the effect of blocking CD40/CD40L interactions on Ab responses or autoimmune diseases in PD-1−/− mice.
As shown in this article, the requirements for CD40/CD40L interactions and splenic GCs for development of autoimmunity differ in PD-1−/− mice compared with WT and CD28−/− NOD.H-2h4 mice, as summarized in Fig. 7. ATD and pSS in WT and mutant NOD.H-2h4 mice are mediated by activated T cells (CD4+ and CD8+) that target the thyroid (1, 8) or salivary gland (4, 9, 10). Interactions between CD4+ T cells and B cells or other APCs are required for activation of most CD4+ T cells. Anti-CD40L blocks these interactions, resulting in reduced activation of T and B cells (15, 17). In WT and CD28−/− mice, autoreactive T and B cell activation is effectively inhibited by anti-CD40L, as evidenced by suppression of autoimmunity and autoantibody responses. However, in PD-1−/− mice, activation of autoreactive T and B cells is apparently not diminished, because autoimmunity and autoantibody responses are increased after anti-CD40L, even though GCs are effectively depleted. B cells function as important APCs for the development of ATD and pSS (59), suggesting that anti-CD40L does not effectively inhibit T cell/APC and/or B cell activation in PD-1−/− mice. Effective inhibition by anti-CD40L apparently requires an intact PD-1/PDL pathway, as shown for CD28−/− mice given anti–PD-1 (Fig. 6). Experiments using cell-specific deletion of PD-1 in B cells, CD4+ T cells, and dendritic cells would be required to determine which specific cells have to express PD-1 for anti-CD40L to suppress immune responses. Because the effects of MR1 on ATD and pSS are similar when functional Tregs are or are not depleted (Supplemental Fig. 2), the effects of MR1 on autoimmunity are presumably not focused on interactions between Tregs and other autoreactive T or B cells. Because anti-CD40L effectively dissolved spontaneous splenic GCs and GCs induced by immunization in PD-1−/− mice (data not shown), the mechanism underlying the development of ATD and pSS apparently differs among PD-1−/−, CD28−/−, and WT NOD.H-2h4 mice, because intact GCs are required for the development of autoimmunity in WT mice but not in PD-1−/−or CD28−/− mice (Fig. 7). The development of autoimmunity in CD28−/− and WT NOD.H-2h4 mice is highly dependent on CD40/CD40L interactions, but autoimmunity can develop independently of this pathway when the inhibitory PD-1 pathway is absent or blocked.
FIGURE 7. (A) The requirement for CD40/CD40L interactions for the development of autoimmune diseases differs depending on the presence of specific immune checkpoint pathways.

Development of autoimmunity is dependent on CD40/CD40L interaction in CD28 deficient mice, while this pathway is dispensable for development of ATD and pSS when the PD-1 pathway is absent or blocked. (B) Summary of the results described in the article.
All of our experiments used IFN-γ–deficient CD28−/− and PD-1−/− NOD.H-2h4 mutants. IFN-γ+ PD-1−/− and CD28−/− NOD.H-2h4 mutants, like their IFN-γ−/− counterparts, develop more severe ATD and pSS than WT NOD.H-2h4 mice (13) (H. Braley-Mullen, unpublished observations). With the experimental design used in this study, MR1 suppressed the development of ATD and pSS in IFN-γ−/− and IFN-γ+ CD28−/− NOD.H-2h4 mice (H. Braley-Mullen, unpublished observations). We have not used anti-CD40L in PD-1–deficient IFN-γ+ mice, but we would predict that the effects on ATD and pSS in PD-1−/− mice should be comparable, irrespective of whether they express IFN-γ. Although the effects of IFN-γ signaling in immune responses are very complex (60), tumors that are resistant to suppression by checkpoint blockade or other modes of suppression can be more responsive to immunotherapy when type I or type II IFNs are absent (61).
Why is an intact PD-1 pathway apparently required for CD40/CD40L interactions for the development of autoimmune pathology and autoantibody production? In agreement with our results, the ability of anti-CD40L to prolong graft survival was lost after PD-1 blockade (45), and tolerance induction by allogeneic bone marrow transplantation requires an intact PD-1 inhibitory pathway (44, 62). T cell exhaustion, mediated by the PD-1 inhibitory pathway, is reversed after blocking PD-1 or PD-L1. An intact CD40/CD40L pathway is required to rescue exhausted T cells (63, 64), suggesting that there could be potentially important cross-talk between these pathways. Blocking or absence of the PD-1 pathway results in increased immune responses and autoimmune pathology, but why should this bypass the requirement for CD40/CD40L interactions? Absence of PD-1 creates a proinflammatory environment, and immune responses developing in proinflammatory environments can be CD40L independent (65). We hypothesize that increased production of proinflammatory cytokines in PD-1−/− mice creates an environment in which autoimmunity can develop in the absence of CD40/CD40L interactions. Further studies are required to test the validity of this hypothesis.
The results of this study have important implications for therapies that use checkpoint inhibitors for treating patients with tumors or chronic viral infections if patients also have an autoimmune disease or have had an organ transplant. Development of autoimmunity and transplant rejection can be a consequence of such treatments, as has been reported in animal models (66–68) and in humans (69, 70). Patients with an autoimmune disease or who have had organ transplants can have adverse effects after checkpoint inhibitor therapy (71–73). The adverse effects can be more serious after anti–PD-1 therapy compared with anti–CTLA-4 therapy (74), treatments that inhibit tumors by distinct mechanisms (75). As shown in this article, some therapies that are used to treat autoimmunity or promote allograft tolerance might be ineffective or harmful when used in combination with checkpoint inhibitors that block the PD-1 pathway, whereas blocking other pathways, such as CD28, do not have these effects. In fact, diabetes in PD-1−/− NOD mice was refractory to anti-CD3–induced immunotherapy or induction of Ag-specific tolerance (67). As shown in this article, autoimmunity in PD-1−/− mice is significantly reduced in the presence of endogenous Tregs and Tfrs that have potent suppressive activity in PD-1–deficient mice (47) (Supplemental Fig. 2). Treg activity was not reduced after PD-1/PD-L1 blockade (67), and PD-1 and Foxp3 function independently to maintain tolerance (76). Therefore, therapies that increase Treg numbers and/or function might be useful for suppressing autoimmunity in patients treated with checkpoint inhibitors. In any event, these results demonstrate unexpected effects that can interfere with suppression of autoimmune diseases when certain therapies are combined with checkpoint inhibitors that target the PD-1 pathway.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health Grant AI 074857 (to H.B.-M.) and by the Lottie Caroline Hardy Trust.
We thank Dr. Arlene Sharpe for providing NOD PD-1−/− mice and for critically reading the manuscript and the Pathology Department at Medimmune for technical help with experiments.
Abbreviations used in this article
- ATD
autoimmune thyroid disease
- GC
germinal center
- MTg
mouse thyroglobulin
- PD-1
programmed cell death-1
- PNA
peanut agglutinin
- pSS
primary Sjögren’s syndrome
- Tfh
follicular helper T cell
- Tfr
T follicular regulatory cell
- TLS
tertiary lymphoid structure
- TM
triple mutation
- Treg
regulatory T cell
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
DISCLOSURES
E.V., T.M., and R.E. are full-time employees and shareholders of Medimmune/AstraZeneca. The other authors have no financial conflicts of interest.
References
- 1.Braley-Mullen H, Yu S. NOD.H-2h4 mice: an important and underutilized animal model of autoimmune thyroiditis and Sjögren’s syndrome. Adv Immunol. 2015;126:1–43. doi: 10.1016/bs.ai.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Delaleu N, Nguyen CQ, Peck AB, Jonsson R. Sjögren’s syndrome: studying the disease in mice. Arthritis Res Ther. 2011;13:217–233. doi: 10.1186/ar3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ciháková D, Talor MV, Barin JG, Baldeviano GC, Fairweather D, Rose NR, Burek CL. Sex differences in a murine model of Sjögren’s syndrome. Ann N Y Acad Sci. 2009;1173:378–383. doi: 10.1111/j.1749-6632.2009.04760.x. [DOI] [PubMed] [Google Scholar]
- 4.Karnell JL, Mahmoud TI, Herbst R, Ettinger R. Discerning the kinetics of autoimmune manifestations in a model of Sjögren’s syndrome. Mol Immunol. 2014;62:277–282. doi: 10.1016/j.molimm.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Yu S, Sharp GC, Braley-Mullen H. Thyroid epithelial cell hyperplasia in IFN-gamma deficient NOD.H-2h4 mice. Clin Immunol. 2006;118:92–100. doi: 10.1016/j.clim.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 6.Yu S, Sharp GC, Braley-Mullen H. Dual roles for IFN-gamma, but not for IL-4, in spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J Immunol. 2002;169:3999–4007. doi: 10.4049/jimmunol.169.7.3999. [DOI] [PubMed] [Google Scholar]
- 7.Yu S, Fang Y, Sharav T, Sharp GC, Braley-Mullen H. CD8+ T cells induce thyroid epithelial cell hyperplasia and fibrosis. J Immunol. 2011;186:2655–2662. doi: 10.4049/jimmunol.1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kayes TD, Weisman GA, Camden JM, Woods LT, Bredehoeft C, Downey EF, Cole J, Braley-Mullen H. New murine model of early onset autoimmune thyroid disease/hypothyroidism and autoimmune exocrinopathy of the salivary gland. J Immunol. 2016;197:2119–2130. doi: 10.4049/jimmunol.1600133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karabiyik A, Peck AB, Nguyen CQ. The important role of T cells and receptor expression in Sjögren’s syndrome. Scand J Immunol. 2013;78:157–166. doi: 10.1111/sji.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barr JY, Wang X, Meyerholz DK, Lieberman SM. CD8 T cells contribute to lacrimal gland pathology in the nonobese diabetic mouse model of Sjögren syndrome. Immunol Cell Biol. 2017;95:684–694. doi: 10.1038/icb.2017.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jara LJ, Navarro C, Brito-Zerón MP, García-Carrasco M, Escárcega RO, Ramos-Casals M. Thyroid disease in Sjögren’s syndrome. Clin Rheumatol. 2007;26:1601–1606. doi: 10.1007/s10067-007-0638-6. [DOI] [PubMed] [Google Scholar]
- 12.Ellis JS, Braley-Mullen H. Regulatory T cells in B-cell-deficient and wild-type mice differ functionally and in expression of cell surface markers. Immunology. 2015;144:598–610. doi: 10.1111/imm.12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellis JS, Hong SH, Zaghouani H, Braley-Mullen H. Reduced effectiveness of CD4+Foxp3+ regulatory T cells in CD28-deficient NOD.H-2h4 mice leads to increased severity of spontaneous autoimmune thyroiditis. J Immunol. 2013;191:4940–4949. doi: 10.4049/jimmunol.1301253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellis JS, Wan X, Braley-Mullen H. Transient depletion of CD4+ CD25+ regulatory T cells results in multiple autoimmune diseases in wild-type and B-cell-deficient NOD mice. Immunology. 2013;139:179–186. doi: 10.1111/imm.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152–172. doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balasa B, Krahl T, Patstone G, Lee J, Tisch R, McDevitt HO, Sarvetnick N. CD40 ligand-CD40 interactions are necessary for the initiation of insulitis and diabetes in nonobese diabetic mice. J Immunol. 1997;159:4620–4627. [PubMed] [Google Scholar]
- 17.Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 18.Klaus SJ, Pinchuk LM, Ochs HD, Law CL, Fanslow WC, Armitage RJ, Clark EA. Costimulation through CD28 enhances T cell-dependent B cell activation via CD40-CD40L interaction. J Immunol. 1994;152:5643–5652. [PubMed] [Google Scholar]
- 19.Kayes T, Fang Y, Yu S, Downey E, Wang S, Braley-Mullen H. Agonistic anti-CD40 induces thyrocyte proliferation and promotes thyroid autoimmunity by increasing CD40 expression on thyroid epithelial cells. J Immunol. 2013;190:3928–3938. doi: 10.4049/jimmunol.1202929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huber AK, Finkelman FD, Li CW, Concepcion E, Smith E, Jacobson E, Latif R, Keddache M, Zhang W, Tomer Y. Genetically driven target tissue overexpression of CD40: a novel mechanism in autoimmune disease. J Immunol. 2012;189:3043–3053. doi: 10.4049/jimmunol.1200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peterson KE, Braley-Mullen H. CD40L is necessary for the priming of effector cells for lymphocytic and granulomatous experimental autoimmune thyroiditis. J Autoimmun. 1999;12:1–12. doi: 10.1006/jaut.1998.0256. [DOI] [PubMed] [Google Scholar]
- 22.Carayanniotis G, Masters SR, Noelle RJ. Suppression of murine thyroiditis via blockade of the CD40–CD40L interaction. Immunology. 1997;90:421–426. doi: 10.1111/j.1365-2567.1997.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahmoud TI, Wang J, Karnell JL, Wang Q, Wang S, Naiman B, Gross P, Brohawn PZ, Morehouse C, Aoyama J, et al. Autoimmune manifestations in aged mice arise from early-life immune dysregulation. Sci Translat Med. 2016;8:361ra137. doi: 10.1126/scitranslmed.aag0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yusuf I, Stern J, McCaughtry TM, Gallagher S, Sun H, Gao C, Tedder T, Carlesso G, Carter L, Herbst R, Wang Y. Germinal center B cell depletion diminishes CD4+ follicular T helper cells in autoimmune mice. PLoS One. 2014;9:e102791. doi: 10.1371/journal.pone.0102791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 26.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 27.Francisco LM, V, Salinas H, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bour-Jordan H, Salomon BL, Thompson HL, Szot GL, Bernhard MR, Bluestone JA. Costimulation controls diabetes by altering the balance of pathogenic and regulatory T cells. J Clin Invest. 2004;114:979–987. doi: 10.1172/JCI20483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bour-Jordan H, Bluestone JA. CD28 function: a balance of costimulatory and regulatory signals. J Clin Immunol. 2002;22:1–7. doi: 10.1023/a:1014256417651. [DOI] [PubMed] [Google Scholar]
- 30.Bour-Jordan H, Bluestone JA. Regulating the regulators: costimulatory signals control the homeostasis and function of regulatory T cells. Immunol Rev. 2009;229:41–66. doi: 10.1111/j.1600-065X.2009.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–245. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 32.Ferguson SE, Han S, Kelsoe G, Thompson CB. CD28 is required for germinal center formation. J Immunol. 1996;156:4576–4581. [PubMed] [Google Scholar]
- 33.Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat Immunol. 2010;11:535–542. doi: 10.1038/ni.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braley-Mullen H, Yu S. Early requirement for B cells for development of spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J Immunol. 2000;165:7262–7269. doi: 10.4049/jimmunol.165.12.7262. [DOI] [PubMed] [Google Scholar]
- 35.Yu S, Downey EF, Braley-Mullen H. Agonistic anti-CD40 promotes early development and increases the incidence of severe thyroid epithelial cell hyperplasia (TEC H/P) in CD4−/− mice. Immun Inflamm Dis. 2013;1:14–25. doi: 10.1002/iid3.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu S, Dunn R, Kehry MR, Braley-Mullen H. B cell depletion inhibits spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J Immunol. 2008;180:7706–7713. doi: 10.4049/jimmunol.180.11.7706. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen CQ, Yin H, Lee BH, Chiorini JA, Peck AB. IL17: potential therapeutic target in Sjögren’s syndrome using adenovirus-mediated gene transfer. Lab Invest. 2011;91:54–62. doi: 10.1038/labinvest.2010.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walker LS, Gulbranson-Judge A, Flynn S, Brocker T, Raykundalia C, Goodall M, Förster R, Lipp M, Lane P. Compromised OX40 function in CD28-deficient mice is linked with failure to develop CXC chemokine receptor 5-positive CD4 cells and germinal centers. J Exp Med. 1999;190:1115–1122. doi: 10.1084/jem.190.8.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Linterman MA, Denton AE, Divekar DP, Zvetkova I, Kane L, Ferreira C, Veldhoen M, Clare S, Dougan G, Espéli M, Smith KG. CD28 expression is required after T cell priming for helper T cell responses and protective immunity to infection. ELife. 2014 doi: 10.7554/eLife.03180. doi:10.7554.eLife03180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X, Fosco D, Kline DE, Meng L, Nishi S, Savage PA, Kline J. PD-1 regulates extrathymic regulatory T-cell differentiation. Eur J Immunol. 2014;44:2603–2616. doi: 10.1002/eji.201344423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ellestad KK, Thangavelu G, Ewen CL, Boon L, Anderson CC. PD-1 is not required for natural or peripherally induced regulatory T cells: severe autoimmunity despite normal production of regulatory T cells. Eur J Immunol. 2014;44:3560–3572. doi: 10.1002/eji.201444688. [DOI] [PubMed] [Google Scholar]
- 43.Chiorini JA, Cihakova D, Ouellette CE, Caturegli P. Sjögren syndrome: advances in the pathogenesis from animal models. J Autoimmun. 2009;33:190–196. doi: 10.1016/j.jaut.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lucas CL, Workman CJ, Beyaz S, LoCascio S, Zhao G, Vignali DA, Sykes M. LAG-3, TGF-β, and cell-intrinsic PD-1 inhibitory pathways contribute to CD8 but not CD4 T-cell tolerance induced by allogeneic BMT with anti-CD40L. Blood. 2011;117:5532–5540. doi: 10.1182/blood-2010-11-318675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mai G, del Rio ML, Tian J, Ramirez P, Buhler L, Rodriguez-Barbosa JI. Blockade of the PD-1/PD-1L pathway reverses the protective effect of anti-CD40L therapy in a rat to mouse concordant islet xenotransplantation model. Xenotransplantation. 2007;14:243–248. doi: 10.1111/j.1399-3089.2007.00402.x. [DOI] [PubMed] [Google Scholar]
- 46.Howard LM, Miga AJ, Vanderlugt CL, Dal Canto MC, Laman JD, Noelle RJ, Miller SD. Mechanisms of immunotherapeutic intervention by anti-CD40L (CD154) antibody in an animal model of multiple sclerosis. J Clin Invest. 1999;103:281–290. doi: 10.1172/JCI5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sage PT, Ron-Harel N, Juneja VR, Sen DR, Maleri S, Sungnak W, Kuchroo VK, Haining WN, Chevrier N, Haigis M, Sharpe AH. Suppression by TFR cells leads to durable and selective inhibition of B cell effector function. Nat Immunol. 2016;17:1436–1446. doi: 10.1038/ni.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med. 2012;209:597–606. doi: 10.1084/jem.20111696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takemori T, Kaji T, Takahashi Y, Shimoda M, Rajewsky K. Generation of memory B cells inside and outside germinal centers. Eur J Immunol. 2014;44:1258–1264. doi: 10.1002/eji.201343716. [DOI] [PubMed] [Google Scholar]
- 50.Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol. 2009;9:845–857. doi: 10.1038/nri2637. [DOI] [PubMed] [Google Scholar]
- 51.Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, Flavell RA, Craft J. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med. 2008;205:2873–2886. doi: 10.1084/jem.20080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 2002;297:2066–2070. doi: 10.1126/science.1073924. [DOI] [PubMed] [Google Scholar]
- 53.Sang A, Niu H, Cullen J, Choi SC, Zheng YY, Wang H, Shlomchik MJ, Morel L. Activation of rheumatoid factor-specific B cells is antigen dependent and occurs preferentially outside of germinal centers in the lupus-prone NZM2410 mouse model. J Immunol. 2014;193:1609–1621. doi: 10.4049/jimmunol.1303000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fu YX, Storb U. Immunology. Autoreactive B cells migrate into T cell territory. Science. 2002;297:2006–2008. doi: 10.1126/science.1077011. [DOI] [PubMed] [Google Scholar]
- 55.Hams E, McCarron MJ, Amu S, Yagita H, Azuma M, Chen L, Fallon PG. Blockade of B7-H1 (programmed death ligand 1) enhances humoral immunity by positively regulating the generation of T follicular helper cells. J Immunol. 2011;186:5648–5655. doi: 10.4049/jimmunol.1003161. [DOI] [PubMed] [Google Scholar]
- 56.Sage PT, Sharpe AH. T follicular regulatory cells. Immunol Rev. 2016;271:246–259. doi: 10.1111/imr.12411. [DOI] [PubMed] [Google Scholar]
- 57.Zhu Y, Zou L, Liu YC. T follicular helper cells, T follicular regulatory cells and autoimmunity. Int Immunol. 2016;28:173–179. doi: 10.1093/intimm/dxv079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gotot J, Gottschalk C, Leopold S, Knolle PA, Yagita H, Kurts C, Ludwig-Portugall I. Regulatory T cells use programmed death 1 ligands to directly suppress autoreactive B cells in vivo. Proc Natl Acad Sci USA. 2012;109:10468–10473. doi: 10.1073/pnas.1201131109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ellis JS, Braley-Mullen H. Mechanisms by which B cells and regulatory T cells influence development of murine organ-specific autoimmune diseases. J Clin Med. 2017;6:13. doi: 10.3390/jcm6020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Minn AJ, Wherry EJ. Combination cancer therapies with immune checkpoint blockade: convergence on interferon signaling. Cell. 2016;165:272–275. doi: 10.1016/j.cell.2016.03.031. [DOI] [PubMed] [Google Scholar]
- 61.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 167:1540–1554.e12. doi: 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lucas CL, Sykes M. Layers of regulation in induction of mixed chimerism by anti-CD40L. Chimerism. 2011;2:111–113. doi: 10.4161/chim.2.4.19053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhadra R, Gigley JP, Khan IA. Cutting edge: CD40-CD40 ligand pathway plays a critical CD8-intrinsic and -extrinsic role during rescue of exhausted CD8 T cells. J Immunol. 2011;187:4421–4425. doi: 10.4049/jimmunol.1102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhadra R, Cobb DA, Khan IA. CD40 signaling to the rescue: a CD8 exhaustion perspective in chronic infectious diseases. Crit Rev Immunol. 2013;33:361–378. doi: 10.1615/critrevimmunol.2013007444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shugart JA, Bambina S, Alice AF, Montler R, Bahjat KS. A self-help program for memory CD8+ T cells: positive feedback via CD40-CD40L signaling as a critical determinant of secondary expansion. PLoS One. 2013;8:e64878. doi: 10.1371/journal.pone.0064878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, Yamazaki T, Azuma M, Iwai H, Khoury SJ, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med. 2003;198:63–69. doi: 10.1084/jem.20022125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fife BT, Guleria I, Gubbels Bupp M, Eagar TN, Tang Q, Bour-Jordan H, Yagita H, Azuma M, Sayegh MH, Bluestone JA. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J Exp Med. 2006;203:2737–2747. doi: 10.1084/jem.20061577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kochupurakkal NM, Kruger AJ, Tripathi S, Zhu B, Adams LT, Rainbow DB, Rossini A, Greiner DL, Sayegh MH, Wicker LS, Guleria I. Blockade of the programmed death-1 (PD1) pathway undermines potent genetic protection from type 1 diabetes. PLoS One. 2014;9:e89561. doi: 10.1371/journal.pone.0089561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Byun DJ, Wolchok JD, Rosenberg LM, Girotra M. Cancer immunotherapy - immune checkpoint blockade and associated endocrinopathies. Nat Rev Endocrinol. 2017;13:195–207. doi: 10.1038/nrendo.2016.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van der Vlist M, Kuball J, Radstake TR, Meyaard L. Immune checkpoints and rheumatic diseases: what can cancer immunotherapy teach us? Nat Rev Rheumatol. 2016;12:593–604. doi: 10.1038/nrrheum.2016.131. [DOI] [PubMed] [Google Scholar]
- 71.Calabrese C, Kirchner E, Kontzias K, Velcheti V, Calabrese LH. Rheumatic immune-related adverse events of checkpoint therapy for cancer: case series of a new nosological entity. [Published erratum appears in 2017 RMD Open 3: e000412corr1.] RMD Open. 2017;3:e000412. doi: 10.1136/rmdopen-2016-000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cappelli LC, Shah AA, Bingham CO., III Immune-related adverse effects of cancer immunotherapy-implications for rheumatology. Rheum Dis Clin North Am. 2017;43:65–78. doi: 10.1016/j.rdc.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, Hicks M, Puzanov I, Alexander MR, Bloomer TL, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375:1749–1755. doi: 10.1056/NEJMoa1609214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wanchoo R, Riella LV, Uppal NN, Lopez CA, Nair V, Devoe C, Jhaveri KD. Immune checkpoint inhibitors in the cancer patient with an organ transplant. J Onco-Nephrol. 1:42–48. [Google Scholar]
- 75.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, Allison JP. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell. 2017;170:1120–1133.e17. doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang B, Chikuma S, Hori S, Fagarasan S, Honjo T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc Natl Acad Sci USA. 2016;113:8490–8495. doi: 10.1073/pnas.1608873113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.