

Abstract
The universally conserved Signal Recognition Particle (SRP) and SRP receptor (SR) mediate the co-translational targeting of proteins to cellular membranes. In contrast, a unique chloroplast SRP in green plants is primarily dedicated to the post-translational targeting of light harvesting chlorophyll-a/b binding (LHC) proteins. In both pathways, dimerization and activation between the SRP and SR GTPases mediate the delivery of cargo; whether and how the GTPase cycle in each system adapts to its distinct substrate proteins were unclear. Here, we show that interactions at the active site essential for GTPase activation in the chloroplast SRP and SR play key roles in the assembly of the GTPase complex. In contrast to their cytosolic homologues, GTPase activation in the chloroplast SRP-SR complex contributes marginally to the targeting of LHC proteins. These results demonstrate that complex assembly and GTPase activation are highly coupled in the chloroplast SRP and SR, and suggest that the chloroplast GTPases may forego the GTPase activation step as a key regulatory point. These features may reflect adaptations of the chloroplast SRP to the delivery of their unique substrate protein.
Keywords: Signal Recognition Particle, protein targeting, GTPases, fluorescence spectroscopy, chloroplast
Co-translational protein targeting by the Signal Recognition Particle (SRP) and the SRP receptor (SR) is a universally conserved pathway essential for the proper structure and function of the cell. Cytosolic SRP recognizes ribosomes translating SRP substrates, and via interactions with SR, delivers its cargo - the ribosome-nascent chain complexes - to the eukaryotic endoplasmic reticulum or the prokaryotic plasma membrane (1, 2). The functional core of SRP consists of a universally conserved SRP54 subunit, or Ffh in bacteria, and an SRP RNA (3). SRP54 is comprised of three domains: (i) a methionine-rich M-domain, which provides the binding site for the substrate protein and the SRP RNA (4); (ii) a GTPase G-domain that shares homology with the Ras-fold (5); and (iii) an N-terminal N-domain that interacts with the ribosome (6, 7). Together the N- and G-domains comprise a structural and functional unit called the NG-domain. The SR (FtsY in bacteria) also contains an NG-domain highly homologous to that in SRP54. The GTP-dependent interaction between the NG-domains of SRP and SR guides the delivery of cargo to protein translocation machineries on the target membrane, and subsequent GTP hydrolysis in the complex drives the dissociation of SRP and SR, recycling them for additional rounds of protein targeting (8).
A notable exception to this classic SRP pathway is provided by the chloroplast SRP (cpSRP) (9). The cpSRP pathway still uses the conserved SRP54 and SR GTPases (called cpSRP54 and cpFtsY, respectively). However, cpSRP lacks the otherwise universally conserved SRP RNA, and is instead a heterodimeric protein complex comprised of cpSRP54 and cpSRP43, a novel SRP subunit unique to the chloroplast of green plants (10-13). The most significant difference between the cytosolic and chloroplast SRP pathways lies in the nature of their substrate proteins. The cytosolic SRP must recognize its cargos within a milieu of translating ribosomes in the cytosol, based on signal sequences that differ widely in size, shape and amino acid composition. In contrast, the cpSRP is dedicated primarily to the post-translational delivery of the light-harvesting chlorophylla/b-binding (LHC) family of proteins (14, 15). LHC proteins are synthesized in the cytosol and imported into the chloroplast stroma, where they are recognized and captured by the cpSRP (10). Analogous to the cytosolic SRP pathway, the interaction of cpSRP with cpFtsY brings the LHC proteins to the Albino3 (Alb3) translocase on the thylakoid membrane, where the LHC proteins are integrated and assembled into light harvesting complexes (16).
The similarities and differences between the cytosolic and chloroplast SRP pathways raise intriguing questions: How do the targeting machineries in each pathway meet the unique challenges posed by their substrate proteins, and what are the roles of the SRP and SR GTPases in this adaptation? Extensive work on the cytosolic SRP showed that during the SRP-FtsY interaction, a series of discrete conformational changes provides multiple opportunities to exert regulation (17-21). Assembly of a stable SRP•FtsY complex requires the formation of a transient ‘early’ intermediate, which subsequently rearranges to a stable, ‘closed’ complex. GTPase activation in the complex requires yet another rearrangement, the movement of the highly conserved Insertion Box Domain (IBD) loops, which positions multiple catalytic residues adjacent to the bound GTP molecules and activates GTP hydrolysis (17). Importantly, each GTPase rearrangement allows the SRP and FtsY to sense and respond to their biological cues. A correct cargo can accelerate the assembly of the SRP•FtsY complex while delaying its GTPase activation (19). Delayed GTP hydrolysis provides an important time window for the targeting complex to search for the translocation machinery before GTP hydrolysis drives its irreversible disassembly. Once at the target membrane, the movement of the IBD loops, which mediates GTPase activation, is crucial for driving the initiation of protein translocation (22). Finally, the timing of GTP hydrolysis provides an important fidelity checkpoint: incorrect cargos, which fail to delay GTPase activation, could be more promptly rejected through premature GTP hydrolysis (19). Thus, the uncoupling of complex assembly and GTPase activation steps in the bacterial SRP and FtsY is crucial for ensuring the efficiency and fidelity of co-translational protein targeting.
On the other hand, the cpSRP handles substrate proteins of a completely different nature. The LHC family of proteins comprises 30-50% of the protein content in the thylakoid membrane and are likely the most abundant membrane proteins on earth. The sheer abundance and rapid turnover of these proteins demand a highly robust and efficient pathway for their targeting and integration. Compared to its cytosolic homologue, specific substrate selection is much easier to achieve in the cpSRP, as members of the LHC protein family are highly homologous and share a conserved sequence motif, L18, that is specifically recognized by the cpSRP. Consequently, many features have evolved in the cpSRP pathway that may represent adaptations to its unique substrate proteins. For example, cpSRP uses cpSRP43 to efficiently capture the LHC proteins (23) as well as to help localize the targeting complex to Alb3 on the thylakoid membrane (24). Here we address this issue from a different perspective: what are the similarities and differences in the GTPase cycles of the chloroplast versus cytosolic SRP and SR? Are there distinct features of the cpSRP54 and cpFtsY GTPases that may reflect their adaptation to the cpSRP pathway? Using a combination of fluorescence and mutational analyses, we dissected the molecular steps during the interaction of cpSRP54 and cpFtsY, and probed the role of the GTPase cycle in the targeting of LHCP. The results showed that, despite many similarities with their bacterial homologues, cpSRP54 and cpFtsY undergo a much more streamlined GTPase cycle in which the complex formation and GTPase activation processes are highly coupled. These differences may have evolved to maximize the efficiency of targeting for the highly abundant LHC proteins.
Materials and Methods
Mutagenesis, protein expression and purification.
The bacterial expression plasmid for cpSRP54 was constructed by inserting the coding sequence of mature cpSRP54 from Arabidopsis thaliana between the NdeI and HindIII restriction sites in pET41(a) (Novagen). cpSRP54 was over-expressed in Escherichia coli Rosetta BL21 cells (Invitrogen) at 37 °C using 0.5 mM IPTG (EMD Biosciences). cpSRP54 was purified by cation exchange chromatography in buffer A (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5% Glycerol), first using SP-Sepharose FF beads (GE Healthcare), followed by a MonoS HP column (GE Healthcare), both using a linear gradient of 150 - 600 mM NaCl.
The construct expressing mature cpFtsY fused to thioredoxin is a generous gift from R. Henry (25). Thioredoxin-fused cpFtsY was over-expressed in Escherichia coli BL21-DE3· cells (Invitrogen) at 37 °C using 0.5 mM IPTG (EMD Biosciences). cpFtsY was first purified over Talon resin (Clonetech) in buffer B (50 mM KHEPES, pH 7.5, 150 mM NaCl, 1 mM PMSF) following manufacturer’s instructions. Following thrombin digestion to remove the thioredoxin tag, cpFtsY was further purified by anion exchange chromatography over a MonoQ column (GE healthcare) in Buffer C (50 mM Tris, pH 7.5, 50 mM NaCl, 1 mM EDTA, 2 mM DTT) using a linear gradient of 50 - 300 mM NaCl, as previously described (26).
cpSRP54 and cpFtsY mutants were constructed using the QuikChange protocol (Stratagene), and were expressed and purified using the same procedures as those for the wildtype cpSRP54, with the following exceptions. Cys-less and single cysteine mutants of cpFtsY were expressed in Escherichia coli BL21-DE3* cells (Invitrogen). Inclusion bodies containing mutant cpFtsY were solubilized using 8M urea. Solubilized cpFtsY was refolded into the native structure by dialyzing in refolding buffer (100 mM Tris-HCl pH 8.0, 400 mM L-Arginine, 5 mM reduced Glutathione, 0.5 mM Oxidized Glutathione, Complete EDTA free protease inhibitor cocktail tablet). The refolded proteins were dialyzed in Buffer A and purified by affinity chromatography using Ni-NTA (Qiagen) followed by anion exchange chromatography using MonoQ as described above for wildtype cpFtsY.
Fluorescence labeling.
For FRET experiments, single cysteine mutants were labeled with maleimide derivatives of coumarin N-(7-dimethylamino-4-methylcoumarin-3-yl)maleimide (DACM) and BODIPY-Fluorescein-N-(2-aminoethyl)maleimide (BODIPY-FL) (Invitrogen). Proteins were dialyzed in labeling buffer (50 mM KHEPES (7.5), 300 mM NaCl, 2 mM EDTA and 10% Glycerol) and treated with 2 mM TCEP at RT to reduce disulfide bonds. The labeling reaction was carried out with a 30-fold excess of dye over protein for 2 hrs at 4 °C and stopped by addition of 2 mM DTT. Acrylodan labeling was done similarly except that the labeling reaction was carried out for >12 hrs at 4 °C. The excess dye was removed by gel filtration using Sephadex G25 resin (Sigma-Aldrich). The absorbance of DACM, BODIPY-FL and acrylodan (ε363 = 27,000 M−1 cm−1, ε504 = 79,000 M−1 cm−1, ε391 = 20,000 M−1 cm−1, respectively) were used to determine the concentration of labeled protein. The labeling efficiency was typically over 80% for all the probes and the background labeling estimated from cys-less or cys-lite constructs was less than 10%.
Fluorescence measurement.
All measurements were carried out at 25°C in assay buffer (50mM KHEPES pH 7.5, 150 mM KOAc, 2 mM Mg(OAc)2, 0.01% Nikkol, 10% glycerol) on a Fluorolog 3-22 spectrofluorometer (Jobin Yvon). For formation of the GTP-bound cpSRP54•cpFtsY complex, 2 mM GTP (Sigma-Aldrich) was used to ensure that both proteins were predominantly GTP-bound. The amount of GDP generated during the course of the experiment was minimal, as estimated from the GTPase rate constants. For complex formation with 5’-guanylylimido-diphosphate (GMPPNP), 200 μM GMPPNP (Sigma-Aldrich) was used. For equilibrium or kinetic measurements using FRET, an excitation wavelength of 380 nm was used and the donor fluorescence emission was monitored at 450 nm. The FRET efficiency was calculated as described (18). For measurements using acrylodan-labeled cpSRP54, an excitation wavelength of 370 nm and an emission wavelength of 495 nm were used (19).
Equilibrium titrations were carried out using a constant concentration of labeled protein and varying concentrations of the binding partner. The data were fit to Eq. 1 or 2,
| (1) |
| (2) |
where Fobs is the observed fluorescence at a particular protein concentration, F1 is the fluorescence with saturating protein, and Kd is the equilibrium dissociation constant of the complex.
The association rate constant (kon) for the cpSRP54•cpFtsY complex was measured using the stop-flow apparatus as described in (18). For FRET, 0.5 μM DACM-labeled cpFtsY was mixed with 1-50 μM BODIPY-FL-labeled cpSRP54 in the presence of 2 mM GTP. For measurements based on acrylodan fluorescence, 0.5 μM acrylodan-labeled cpSRP54 was mixed with 1-50 μM wildtype cpFtsY. The observed rate constants (kobs) for each reaction was plotted against cpSRP54 or cpFtsY concentration, respectively, and fitted to a linear (Eq. 3) or hyperbolic function (Eq. 4).
| (3) |
| (4) |
in which kobs is observed rate of association at a particular protein concentration, kon (slope) is the association rate constant and koff, app (y-intercept) is the apparent dissociation rate constant, and k1 and Kd are defined in Figure 2E.
Figure 2.
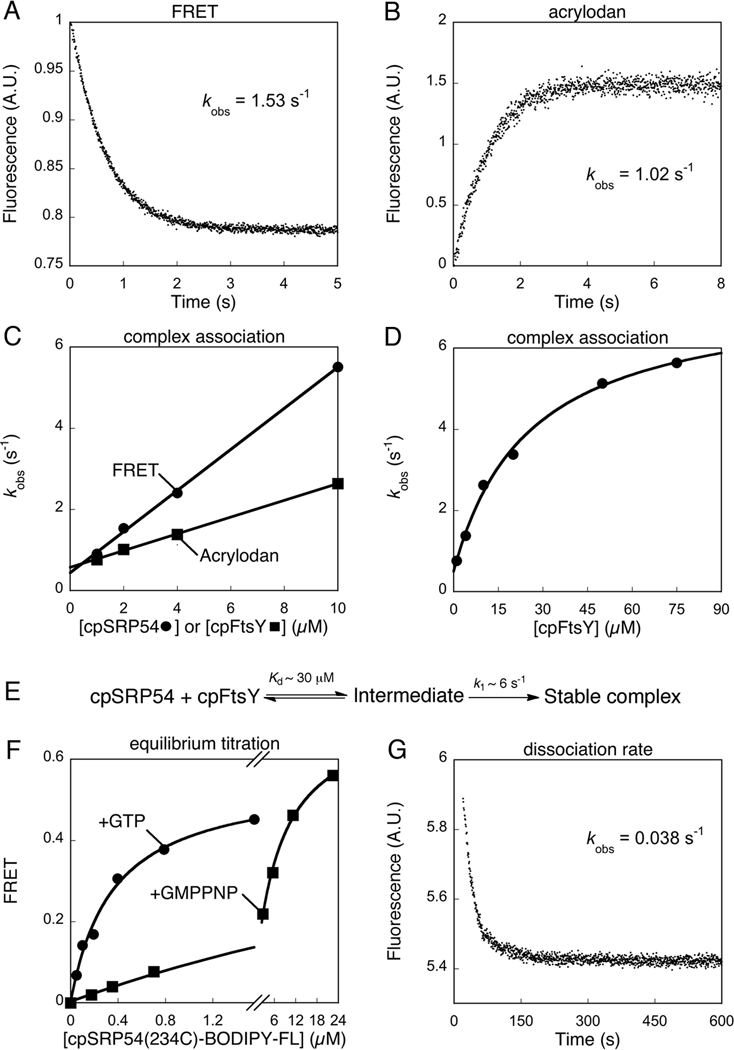
Thermodynamic and kinetics for formation of the cpSRP54•cpFtsY complex. (A) Complex assembly between 0.5 μM cpFtsY(321C)-DACM and 2 μM cpSRP54(234C)-BODIPY-FL, measured in a stopped-flow apparatus as described in Methods. Single exponential fit of the data gave a kobs value of 1.53 s-1. (B) Complex assembly between 0.5 μM cpSRP54(234C)-acrylodan and 2 μM cpFtsY, measured in a stopped-flow apparatus as described in Methods. Single exponential fit of the data gave a kobs value of 1.02 s-1. (C) Association rate constants for cpSRP54-cpFtsY complex formation with GTP measured by FRET (●) and acrylodan fluorescence (■). Linear fits of the data gave complex assembly rate constants (kon) of 5 × 105 and 1.57 × 105 M−1 s−1 with FRET and acrylodan fluorescence, respectively. (D) A hyperbolic dependence of complex assembly rate constants on cpFtsY concentration. The data were fit to Eq. 4 in the Methods, which gave a Kd value of 30 μM and a rate constant of 6 s−1 at saturating cpFtsY. (E) A two-step schematic of cpSRP54-cpFtsY complex assembly. (F) Equilibrium titration of the cpSRP54•cpFtsY complex formed with GTP (●) or GMPPNP (■) measured by FRET. Complex formation with GTP was carried out using mutant cpFtsY(A168W) to minimize GTP hydrolysis. The data were fit to Eq. 2, which gave Kd values of 0.35 μM with GTP and 7 μM with GMPPNP. (G) Dissociation kinetics of the cpSRP54(234C, A142W)•cpFtsY complex, measured as described in the Methods. Single exponential fit of the data gave an apparent dissociation rate constant of 0.038 s-1. After subtracting the GTP hydrolysis rate from this complex (0.008 s−1), the corrected dissociation rate constant was 0.030 s-1.
The dissociation rate constant (koff) was determined by a pulse-chase experiment. 2 μM wildtype cpFtsY was incubated with 0.5 μM acrylodan-labeled cpSRP54(234C, A142W) for 10 min to form the GTP-bound cpSRP54•cpFtsY complex, and mixed with 200 mM EDTA or a 20-fold excess of unlabeled cpSRP54 to drive disassociation of the complex. The time course for decrease in acrylodan fluorescence was fit to a single exponential function to obtain the dissociation rate constant. Both the complex association and dissociation kinetics were measured on a Kintek stopped-flow apparatus.
GTPase assays.
All GTPase assays were performed at 25 °C in assay buffer as described previously (26). GTP hydrolysis reactions were followed and analyzed as described in (27). The reciprocally stimulated GTPase reaction between cpFtsY and cpSRP was measured in multiple-turnover experiments ([GTP] > [E]) with a small fixed amount of cpSRP54 (100 nM), varying concentrations of wildtype or mutant cpFtsY, and 100 μM GTP. The cpFtsY concentration dependence of the observed rate (kobs) was fit to Eq. 5,
| (5) |
in which kcat is the maximal rate constant with saturating cpFtsY and Km is the concentration of cpFtsY required to reach half saturation. Analogous set-ups were used when cpSRP54 mutants were tested, with the concentration of cpSRP54 being varied instead of cpFtsY.
The affinity of mutant cpFtsY for cpSRP54 was determined using an inhibition assay that measures the ability of mutant cpFtsY to compete with wildtype cpFtsY and inhibit its interaction with cpSRP54, as described in Shan et al (17). The data were fit to Eq. 6,
| (6) |
in which Ki is the inhibition constant, k0 is the rate constant of GTP hydrolysis in the absence of the inhibitor, and k1 is the rate constant of GTP hydrolysis from the cpSRP54•cpFtsY(mt) complex. At subsaturating concentrations of the wildtype cpFtsY ( < Km), the value of Ki equals Kd, the dissociation constant of the cpSRP54•cpFtsY(mt) complex. Analogous set-ups and analyses were used when cpSRP54 mutants were tested.
Gel filtration.
Complex formation was carried out in column buffer [50 mM KHEPES (pH 7.5), 200 mM NaCl, 2 mM Mg(OAc)2, 2 mM DTT]. 10 μM of cpSRP54 was mixed with 10 μM wildtype or mutant cpFtsY in the presence of 450 μM GMPPNP and incubated on ice for 10 minutes before loading onto Superdex 200 (GE Healthcare). Reference runs of the individual proteins confirmed the identities of the peaks.
LHCP integration assay.
The thylakoids were collected from chloroplasts of 9-12 day-old pea seedlings (Laxton Progressive 9) hypotonically lysed in lysis buffer (10 mM KHEPES, pH 8.0, 10 mM MgCl2) for 10 minutes as described by Yuan et al (28). The stromal extract was removed, and the thylakoid pellet was resuspended in lysis buffer and washed twice in Import Buffer (50 mM KHEPES, pH 8.0, 330 mM sorbitol) containing 1M KOAc to remove residual cpFtsY associated with the membrane. Thylakoids were resuspended in Import Buffer to a concentration of 1 mg chlorophyll/mL (1X). Each 150 μL light-harvesting chlorophylla/b-binging protein (LHCP) targeting/integration reaction contained 10 μL in vitro translated 35S-methionine-labeled LHCP, 50 μL 1X salt-washed thylakoid, 50 mM GTP, 50 mM ATP, 0.5 μM cpSRP54 and varying concentrations of cpFtsY. Analogous set-ups were used when cpSRP54 mutants were tested. The reactions were incubated at 25°C for 10 minutes before being quenched on ice. The reaction mixtures were thermolysin-treated for 40 minutes and centrifuged to remove any non-integrated LHCP in the supernatant. The resulting pellets were resuspended in 2X SDS, and analyzed by SDS-PAGE. The two lower bands that represent the protease-protected fragments of the integrated LHCP (25-kDa and 18.5-kDa) were quantified using a Molecular Dynamics Storm 840 and the ImageQuant software (GE healthcare).
RESULTS
Fluorescence assays to monitor the cpSRP54-cpFtsY interaction
To directly visualize the interaction between cpSRP54 and cpFtsY in real-time, we developed fluorescence-based assays, which have been used in the bacterial SRP and other systems to elucidate key features of protein interaction mechanisms. To this end, we constructed cys-lite and cys-less versions of cpSRP54 and cpFtsY, respectively. cpSRP54 has a solvent exposed cysteine 198 which can be mutated to serine to obtain cys-lite cpSRP54 without disrupting its interaction with cpFtsY (Figure S1A of Supporting Information); the remaining two cysteines in cpSRP54 are likely buried inside the folded protein based on homology modeling with Ffh, and did not react significantly with fluorescent dyes in control experiments (Figure S1B of Supporting Information). cpFtsY contains five native cysteines, all of which were replaced with serines. Cys-less cpFtsY was purified from inclusion bodies and refolded into the native structure. Refolded cys-less cpFtsY interacted with and stimulated cpSRP54’s GTPase activity with efficiencies within two-fold of that of wildtype cpFtsY (Figure S1C of Supporting Information).
As the crystal structure of cpSRP54 or its complex with cpFtsY is not available, we constructed a homology model of the cpSRP54•cpFtsY complex based on superposition of the crystal structure of apo-cpFtsY onto that of T. aquaticus FtsY in complex with Ffh (Figure 1A). Based on this model, single cysteines were introduced at solvent exposed positions and labeled with fluorescent dyes using thio-specific chemistry. In FRET experiments, a cysteine was engineered at residue 321 of cys-less cpFtsY and labeled with DACM as the donor fluorophore, and a cysteine was introduced at residue 234 of cys-lite cpSRP54 and labeled with BODIPY-FL as the acceptor dye (Figure 1A). Both probes are located at the N-G domain interface of the respective GTPases and are ~30 Å apart as estimated from the homology model. Significant FRET was observed upon assembly of the cpSRP54•cpFtsY complex in the presence of GTP (Figure 1B). At saturating protein concentrations and when complications from GTP hydrolysis were minimized (see below), the FRET efficiency in the cpSRP54•cpFtsY complex was ~0.60 (Figure 2F below). In addition, the cysteine at residue 234 of cys-lite cpSRP54 was labeled with an environmentally sensitive dye, acrylodan. Formation of the cpSRP54•cpFtsY complex with GTP induced a blue shift and a ~30% increase in the fluorescence intensity of this dye (Figure 1C), providing an additional measurement of the cpSRP54-cpFtsY interaction. Fluorescently labeled cpSRP54 and cpFtsY interacted with and activated each other’s GTPase activity with rate constants within two fold of the wild-type proteins (Figures S1D, E of Supporting Information). Further, both the FRET and fluorescence change of cpSRP54(234C)-acrylodan upon complex formation could be competed away by EDTA or unlabeled protein (Figures S2A, B of Supporting Information). Thus these fluorescence assays faithfully report on the kinetics and stability of the cpSRP54-cpFtsY interaction.
Figure 1.
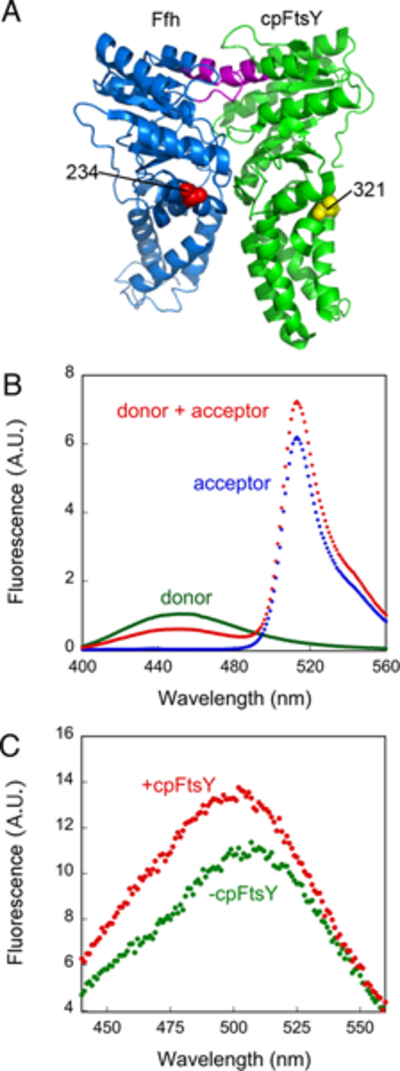
Fluorescence assays to report on complex assembly between cpSRP54 and cpFtsY. (A) The positions of the FRET donor (yellow sphere) and acceptor (red sphere) probes in cpFtsY and cpSRP54, respectively, mapped onto a homology model of the complex generated by superimposing the crystal structure of cpFtsY (2OG2) onto that of the T. aquaticus Ffh•FtsY NG domain complex (1RJ9). The same residue in cpSRP54 was also used for labeling with acrylodan. The IBD loops in cpSRP54 and cpFtsY are highlighted in magenta. (B) Fluorescence emission spectra of donor labeled cpFtsY (0.5 μM, green), acceptor labeled cpSRP54 (2 μM, blue), and their complex formed with 2 mM GTP (red). (C) Fluorescence emission spectra of acrylodan-labeled cpSRP54(234C) in the absence (green) and presence (red) of cpFtsY (2 μM).
Two-step complex assembly
Using the fluorescence assays, we characterized the kinetics and stability of the interaction between cpSRP54 and cpFtsY. These analyses, however, were complicated by the hydrolysis of GTP, which occurs quickly in the cpSRP54•cpFtsY complex and drives rapid disassembly of the GTPase complex. In the bacterial SRP and FtsY GTPases, this problem can be overcome by using the non-hydrolyzable GTP analogue GMPPNP, which provides a good mimic for GTP to support efficient assembly of a stable SRP•FtsY complex (18). However as shown below (Figure 2F), GMPPNP does not provide an adequate mimic of GTP to support stable complex assembly between cpSRP54 and cpFtsY. To overcome this problem, we used the mutant GTPases, cpSRP54(A142W) or cpFtsY(A168W). The corresponding mutations in bacterial SRP and FtsY, Ffh(A144W) and FtsY(A335W) respectively, specifically disrupted GTPase activation in the Ffh•FtsY complex without affecting rapid and stable complex assembly (17). Similarly, both mutants in cpSRP54 and cpFtsY allowed a stable cpSRP54•cpFtsY complex to be efficiently assembled (Figures 3B and 4B below) but specifically blocked GTP hydrolysis in the complex, and thus provided a reasonable estimate for the kinetic and thermodynamic stabilities of the wildtype cpSRP54•cpFtsY complex.
Figure 3.
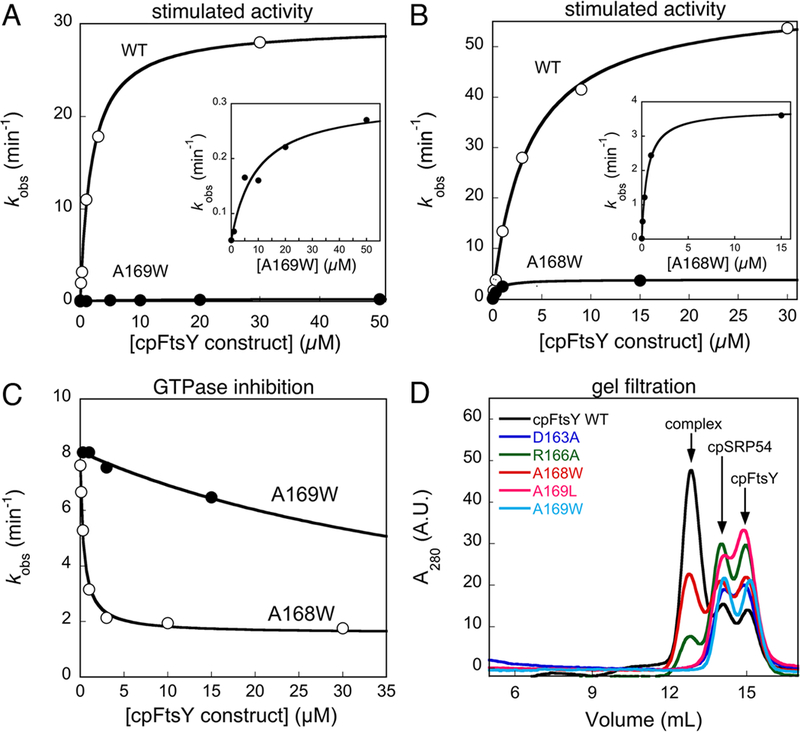
Effects of cpFtsY mutations on its stimulated GTPase reaction with cpSRP54. (A, B) The stimulated GTPase reactions of wildtype cpFtsY (○), and mutants cpFtsY(A169W) (part A, ● and inset) and cpFtsY(A168W) (part B, ● and inset). (C) Inhibition assays for determining the affinities of mutants cpFtsY(A168W) (○) and cpFtsY(A169W) (●) for cpSRP54. The figures show representative data, and Table 1 summarizes the average values from two or more measurements. (D) Gel filtration analyses of stable complex formation of cpSRP54 with wildtype cpFtsY (black) and mutants cpFtsY D163A (blue), R166A (green), A168W (red), A169L (magenta) and A169W (cyan).
Figure 4.
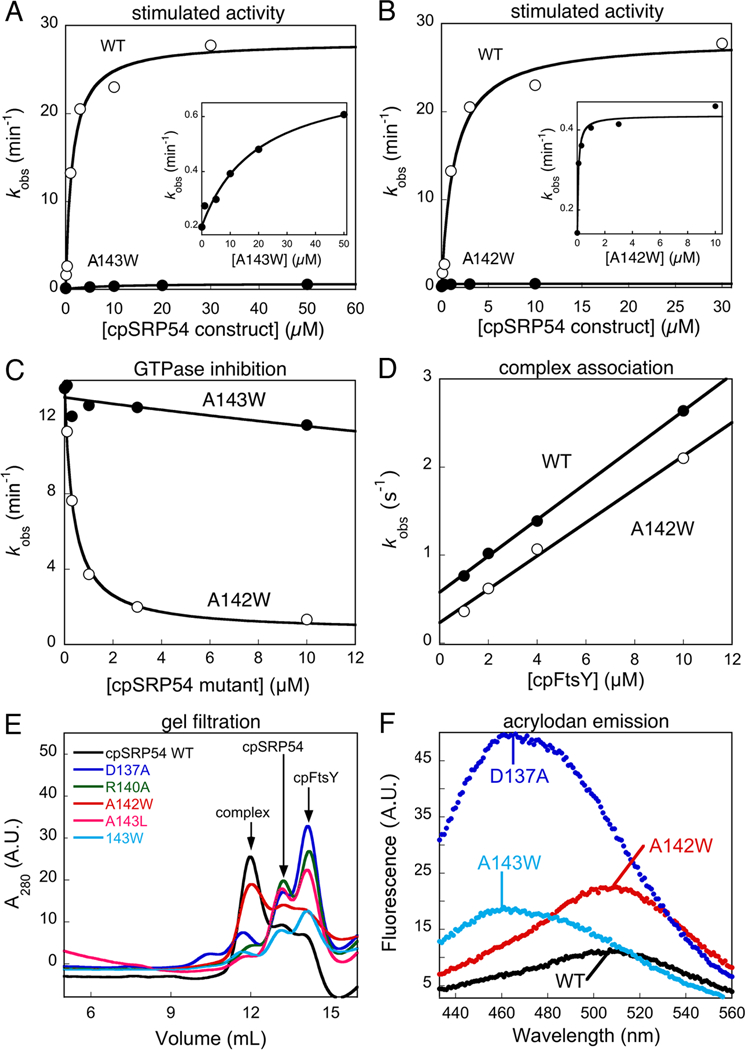
Effects of cpSRP54 mutations on its stimulated GTPase reaction with cpFtsY. (A, B) The stimulated GTPase reactions of cpFtsY with wildtype cpSRP54 (○), and mutants cpSRP54(A143W) (part A, ● and inset) and cpSRP54(A142W) (part B, ● and inset). (C) Inhibition assays for determining the affinities of mutants cpSRP54(A142W) (○) and cpSRP54(A143W) (●) for cpFtsY. The figures show representative data, and Table 1 summarizes the average values from two or more measurements. (D) Mutant cpSRP54(A142W) (○) exhibits the same GTP-dependent complex assembly kinetics as wildtype cpSRP54 (●), measured using acrylodan labeled cpSRP54(234C) as described in the Methods. Linear fits of data gave complex formation rate constants of 1.9 and 2.3 × 105 M−1s−1 for mutant and wildtype cpSRP54, respectively. (E) Gel filtration analyses of stable complex formation of cpFtsY with wildtype cpSRP54 (black), and mutants cpSRP54 D137A (blue), R140A (green), A142W (red), A143L (magenta) and A143W (cyan). (F) Fluorescence emission spectra of the acrylodan labeled at cpSRP54(234C) in the wildtype protein (black) compared with mutants cpSRP54 D137A (blue), A142W (red) and A143W (cyan).
We determined the kinetics of complex assembly in the presence of GTP by either following the gain of FRET (Figure 2A) or the increase in fluorescence of acrylodan-labeled cpSRP54 (Figure 2B). Surprisingly, the complex assembly rate constant (kon) measured using FRET was over three-fold faster than that determined using cpSRP54(234C)-acrylodan (Figure 2C). This difference was not caused by a larger deleterious effect of acrylodan labeling at cpSRP54(234C) on complex assembly, as cpSRP54(234C)-acrylodan exhibited comparable activity in the stimulated GTPase reaction than the cpSRP54 and cpFtsY labeled with the FRET dyes (compare Figures S1D and E of Supporting Information, ●). Instead, we reasoned that the difference in the observed complex assembly rates arises from the fact that the acrylodan probe reports on a local conformational change surrounding residue 234 that accompanies complex assembly, whereas FRET directly reports on approximation of distance between cpSRP54 and cpFtsY as soon as a complex is formed. This raised the possibility that assembly of the stable cpSRP54-cpFtsY complex occurs in two steps, with the initial formation of an intermediate detected by FRET followed by conformational rearrangement to form a more stable, final complex detected specifically by cpSRP54(234C)-acrylodan.
To provide additional evidence for this model, we analyzed the concentration dependence of the observed complex assembly rates using cpSRP54(234C)-acrylodan. If formation of a stable complex occurred in a single bimolecular association, then the observed complex assembly rate constants should increase linearly with increasing protein concentration. In contrast, if additional steps were required for stable complex assembly, deviations from linearity would be expected. Indeed, the observed complex assembly rate constant exhibited a hyperbolic dependence on cpFtsY concentration and plateaued at 6 s−1 with saturating cpFtsY (Figure 2D). Control experiments showed that this plateau was unlikely to be caused by protein aggregation or inactivation at high concentrations (Figure S1E of Supporting Information). These results are consistent with the formation of a transient intermediate with a Kd value of 30 μM during complex assembly (Figure 2E), such that complex formation is rate-limited by the bimolecular cpSRP54-cpFtsY association at low protein concentrations but becomes rate-limited by a unimolecular rearrangement from this intermediate at saturating protein concentrations. Together, these results strongly suggest that assembly of the cpSRP54•cpFtsY complex requires at least two steps.
We further determined the kinetic and thermodynamic stabilities of the cpSRP54•cpFtsY complex. The affinity of the cpSRP54•cpFtsY complex was measured by equilibrium titrations using mutant cpSRP54(A142W) or cpFtsY(A168W), as rapid GTP hydrolysis from the wildtype complex will artificially raise the observed equilibrium dissociation constant (Kd) (Figures 2F vs S3A of Supporting Information, ●). These analyses yielded a Kd value of 300-500 nM using both the FRET assay and acrylodan-labeled cpSRP54 (Figures 2F and S3B,C of Supporting Information). In addition, pulse-chase experiments gave a dissociation rate constant of 0.03 s−1 for the cpSRP54(A142W)•cpFtsY complex (Figure 2G). In conjunction with the association rate constant measured above (Figure 2C), this yielded a Kd value of 200 nM for this complex, consistent with the value determined from equilibrium titrations.
IBD loops play essential roles in both complex assembly and GTPase activation
To probe the molecular determinants essential for the interaction between cpSRP54 and cpFtsY, we generated a collection of site-directed mutant GTPases that map to the putative interaction surface of cpSRP54 and cpFtsY based on structural homology, with an emphasis on the universally conserved IBD loops (Figure 1A, magenta and Tables S1 and S2 of Supporting Information). Control experiments showed that the basal GTP binding and hydrolysis activity (Tables S3 and S4 of Supporting Information, respectively) of the individual cpFtsY and cpSRP54 mutants were comparable to that of the wildtype proteins, ensuring that defects did not arise from disruption of the global structure of the mutant proteins. We then screened the mutants by monitoring the reciprocally stimulated GTPase reaction between cpSRP54 and cpFtsY (Figures 3A, B and 4A, B). As demonstrated above, the complex assembly rate constants measured directly using the fluorescence assays agreed well with the value of kcat/Km (~3 × 105 M−1s−1) in the stimulated GTPase reaction; further, dissociation of the GTP•cpSRP54•cpFtsY•GTP complex (0.030 s−1) is at least 20-fold slower than GTP hydrolysis from this complex (~0.7 s−1). Both observations indicate that in the stimulated GTPase reaction, the value of kcat/Km is rate-limited by, and hence reports on, the rate of assembly of a stable cpSRP54-cpFtsY complex, whereas the maximal rate constant kcat reports on either GTP hydrolysis from the complex or a rate-limiting rearrangement that activates the chemical step.
The vast majority of mutants exhibited defects in this reaction (Tables 1 and S1, S2 of Supporting Information). Among them, perturbations of the IBD loops produced the most deleterious effect on the reciprocally stimulated GTPase reaction between cpSRP54 and cpFtsY (Table 1), consistent with their high evolutionary conservation. Inspection of the concentration dependence of the stimulated GTPase reactions further suggested that the majority of these mutants have defects in both the complex assembly and GTP hydrolysis steps. For example, the cpFtsY(A169W) and the corresponding cpSRP54(A143W) mutations not only reduced the GTPase rate from the complex by over 50-fold (Figure 3A and4A, and Table 1, kcat), but a significantly higher concentration of mutant proteins were required to reach saturation (Figures 3A and4A, insets and Table 1, Km). Only two mutations, cpFtsY(A168W) and cpSRP54(A142W), were exceptions: both mutants reduced the maximal rate of GTP hydrolysis by 15 - 50-fold (Figures 3B and4B and Table 1, kcat), but saturation in GTPase rate could be reached at low protein concentrations, suggesting that efficient complex assembly could occur in these mutants (Figures 3B and4B, insets and Table 1, Km).
Table 1.
Summary of Equilibrium and Kinetic Properties of cpSRP54 and cpFtsY IBD-loop mutants.
| Mutations | kcat (min−1) | Km (μM) | kcat/Km (μM−1min−1) | Ki (μM) | Kd (μM) | |||
|---|---|---|---|---|---|---|---|---|
| WT | 27 ± 3 | (1)1 | 1.6 ± 0.3 | (1) | 18 ± 3 | (1) | 0.802 | |
| A142W cpSRP54 | 0.50 ± 0.34 | (0.02) | 0.24 ± 0.17 | (0.2) | 2.8 ± 1.7 | (0.2) | 0.11 ± 0.09 | 0.443, 0.164 |
| A168W cpFtsY | 1.8 ± 0.1 | (0.07) | 0.27 ± 0.02 | (0.2) | 6.8 ± 0.6 | (0.4) | 0.23 ± 0.15 | 0.523 |
| D137A cpSRP54 | 1.0 ± 0.5 | (0.04) | 5.1 ± 0.1 | (3) | 0.19 ± 0.09 | (0.01) | 2.0 ± 1.3 | |
| D163A cpFtsY | 1.5 ± 0.4 | (0.05) | 5.0 ± 0.1 | (3) | 0.34 ± 0.06 | (0.02) | ND | |
| R140A cpSRP54 | 3.4 ± 0.2 | (0.1) | 6.3 ± 1.2 | (4) | 0.54 ± 0.08 | (0.03) | 0.51 ± 0.20 | |
| R166A cpFtsY | 4.3 ± 0.8 | (0.2) | 5.1 ± 1.7 | (3) | 0.89 ± 0.18 | (0.05) | 3.2 ± 1.4 | |
| A143L cpSRP54 | 0.23 ± 0.05 | (0.008) | 20 ± 1 | (13) | 0.012 ± 0.003 | (0.001) | > 40 | |
| A143W cpSRP54 | 0.52 ± 0.07 | (0.02) | 15 ± 7 | (10) | 0.037 ± 0.012 | (0.002) | > 40 | |
| A169L cpFtsY | 0.32 ± 0.10 | (0.01) | 15 ± 2 | (10) | 0.031 ± 0.019 | (0.002) | > 40 | |
| A169W cpFtsY | 0.36 ± 0.15 | (0.01) | 13 ± 5 | (8) | 0.028 ± 0.001 | (0.002) | > 40 | |
| F165A cpFtsY | 28 ± 3 | (1) | 0.8 ± 0.3 | (0.5) | 38 ± 10 | (2) | ND | |
| A167W cpFtsY | 11 | (0.4) | 7.4 | (5) | 1.5 | (0.1) | ND | |
Values in parenthesis are relative to that of the wildtype proteins, which is normalized to 1. ND, not determined.
Apparent Kd values were obtained from equilibrium titrations using FRET1 or acrylodan2-labeled cpSRP54 in Figures 2F and S3 of the Supporting Information.
Kd obtained from koff/kon.
To further dissect the contribution of each residue to complex assembly and/or GTPase activation, we used a well-established inhibition assay (Figures 3C, 4C) (17). For example, if a mutant cpFtsY could bind cpSRP54 but failed to efficiently hydrolyze GTP, then it would compete with wildtype cpFtsY in binding and inhibit its stimulated GTPase reaction with cpSRP54. Under subsaturating concentrations of the wildtype cpSRP54 and cpFtsY, the inhibition constant Ki obtained from this assay represents the dissociation constant between the mutant and its partner GTPase. In agreement with their kinetic parameters from the stimulated GTPase reactions, cpFtsY(A168W) and cpSRP54(A142W) exhibited strong competitive binding to its partner GTPase, with inhibition constants below 0.2 μM (Figure 3C and4C, open circles and Table 1, Ki). In contrast, all the other deleterious mutations in the IBD loop severely impaired complex formation (Table 1). For example, cpFtsY(A169W) and cpSRP54(A143W) could not act as competitive inhibitors in the inhibition assay, and exhibited Ki values over 50 μM (Figure 3C and4C, ●).
To independently corroborate the results from the inhibition assay, we used gel filtration and fluorescence analyses to independently evaluate the mutational effects on the stability and/or conformational changes of the complex. In gel filtration analyses, which qualitatively assess the ability of the mutant proteins to form a kinetically stable complex (29, 30), cpSRP54(A142W) and cpFtsY(A168W) assembled complexes with efficiencies within two fold of the wildtype proteins (Figure 3D & 4E, red vs. black). On the other hand, all the other deleterious mutations in the IBD loops (residues D163, R166, A169 of cpFtsY and D137, R140, A143 of cpSRP54) showed no or little detectable complex formation (Figures 3D and4E). Similarly, fluorescence assays showed that mutant cpSRP54(A142W) exhibited the same complex assembly rate constant as wildtype cpSPR54 (Figure 4D), and both cpSRP54(A142W) and cpFtsY(A168W) assembled stable complexes with their binding partners (Figures S3B, C of Supporting Information). In contrast, complex formation could not be detected for mutants cpSRP54(D137A) and cpSRP54(A169W) using the fluorescence assay (data not shown). Together, these results strongly suggest that the IBD loops, which provide key catalytic motifs for GTPase activation, are also intimately involved in the assembly of the cpSRP54-cpFtsY complex.
Two additional lines of evidence support this notion and showed that in the cpSRP54•cpFtsY complex, interactions at the catalytic active site are tightly coupled to assembly of the GTPase complex. First, several mutations in the IBD loop of cpSRP54 caused extensive blue shift and increase in fluorescence intensity of the acrylodan labeled at cpSRP54(234C) compared to that of wildtype cpSRP54 (Figure 4F). This indicates that perturbation of the IBD loop effects a change in the local environment at the NG-domain interface of cpSRP54, a region critical for efficient complex assembly (Table S1 of Supporting Information and (30)). Second, replacement of the β,γ-bridging oxygen of GTP strongly reduced both the rate (data not shown) and the stability of the complex (Figure 2F), and GMPPNP could not induce the change in the fluorescence of acrylodan-labeled cpSRP54(234C) upon complex formation (data not shown). Thus interactions at sites critical for the chemical reaction are also integrally involved in the complex assembly process.
Defects in complex formation and GTPase activation block LHCP targeting
To assess the contribution of the individual steps in cpSRP54 and cpFtsY’s GTPase cycle to the protein targeting reaction, we tested the effects of the mutant cpFtsY and cpSRP54 GTPases on the targeting and translocation of LHCP. The overall efficiency of LHCP targeting and integration was analyzed based on protease protection of LHCP upon its proper integration into salt-washed thylakoid membranes (see Methods). In vitro translocation reached completion after 10 minutes and the reaction saturated at cpFtsY concentrations above 150 nM (Figures S5A and B of Supporting Information, respectively). Based on these observations, a concentration of 500 nM and a time point of 10 minutes were used to test the effect of mutant proteins on the efficiency of the targeting reaction.
In general, a significant defect in LHCP integration was observed only with a >10-fold reduction in the individual steps of cpSRP54 and cpFtsY’s GTPase cycle (complex formation or GTPase activation). This is analogous to observations in the co-translational protein targeting reaction carried out by bacterial SRP and FtsY (31), and suggests that the targeting of LHCP by cpSRP and cpFtsY is not the major rate-limiting step in the translocation/integration assay. Nevertheless, this assay revealed moderate to strong defects in LHCP integration for most of the mutant GTPases (Figures 5A, B and S5C of Supporting Information). The two mutants that specifically block GTPase activation, cpSRP54(A142W) and cpFtsY(A168W), reduced translocation efficiency ~two-fold (Figure 5, A and B), suggesting that activated GTP hydrolysis in the cpSRP54•cpFtsY complex is not crucial but does modulate the efficiency of the targeting reaction. In comparison, mutant GTPases that also impair complex assembly, such as cpFtsY(A169W) and cpSRP54(A143W), showed stronger defects in LHCP targeting and translocation (Figure 5, A and B). The reduction in translocation efficiency of the various GTPase mutants correlated with their values of kcat/Km in the GTPase assay, an indicator for the efficiency of complex assembly (Figure 5, C and D). Collectively, these results demonstrate that efficient assembly of the cpSRP54-cpFtsY complex is crucial for the targeting and integration of LHCP, whereas GTPase activation and/or GTP hydrolysis plays a modulatory role to help enhance the efficiency of targeting.
Figure 5.
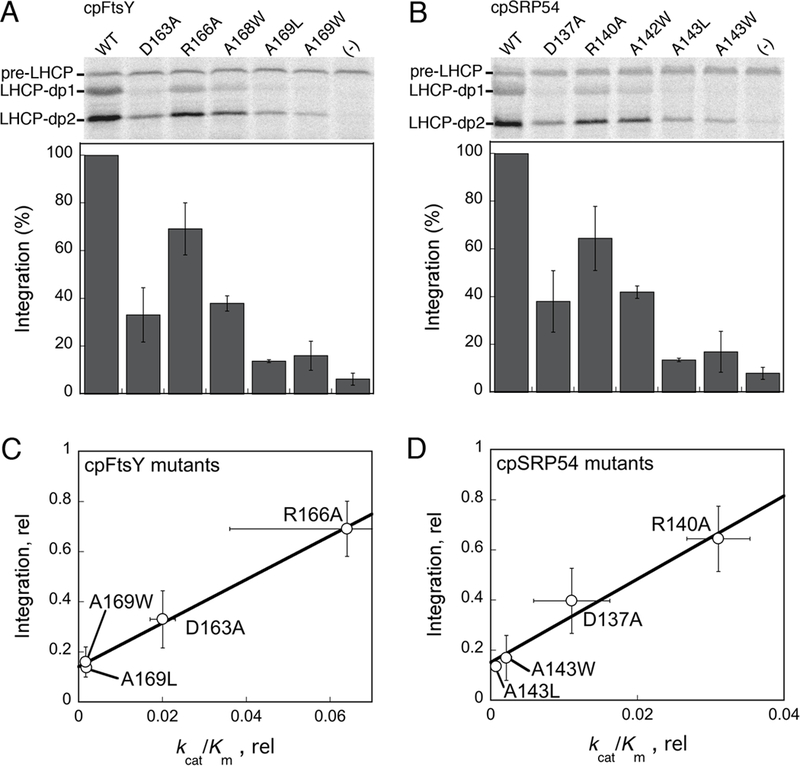
Effect of mutant GTPases on the targeting and integration of LHCP into thylakoid membranes. LHCP-dp1 and -dp2 denote the two 18.5 kDa and 21 kDa protease-protected fragments that represent LHCP successfully targeted and integrated into the thylakoid membrane. pre-LHCP was added to the reaction after the protease treatment and served as a loading control. (A, B) LHCP integration efficiency by the individual cpFtsY and cpSRP54 mutants, respectively. The top panels show representative data, and the lower panels show quantification of two or more measurements. All the data were normalized to that of the wildtype protein, which was set to 100%. (C, D) Correlation of the translocation defect of cpFtsY (part C) and cpSRP54 (part D) mutants with their kcat/Km values.
DISCUSSION
The interaction between the SRP and SR GTPases delivers cargo proteins to a target membrane, and hence plays a crucial role in the proper localization of membrane proteins. During the interaction of the bacterial SRP and SR, formation of a stable complex is a two-step process that requires initial formation of a transient ‘early’ intermediate, followed by a slow rearrangement of this intermediate to a stable complex (Figure 6, black line, steps 1 and 2) (18). Here, real-time fluorescence analyses strongly suggest that a two-step assembly process also occurs during the interaction between cpSRP54 and cpFtsY. First, the complex assembly rate constant measured by acrylodan, which detects a local rearrangement at the NG-domain interface accompanying complex formation, is significantly slower than that reported by the FRET probes, which are less sensitive to the conformational state of the complex. Further, the observed complex assembly rate constant exhibits a hyperbolic, instead of linear dependence on protein concentration. Both observations are indicative of the presence of an intermediate during complex assembly (Figures 2E and6, red lines). Compared to the bacterial SRP and FtsY, the assembly intermediate formed by the chloroplast GTPases is less stable (Kd ~ 30 μM compared to 4-8 μM in the bacterial complex; (18)) but rearranges to the stable complex much faster (k1 ~ 6 s−1 compared to 0.6-1 s−1 for the bacterial complex; (18)) (Figure 6, red vs. black lines, step 2). These observations suggest that the transient intermediate assembled by the chloroplast GTPases is more productive, and possibly requires less extensive rearrangements to attain the final complex than their bacterial homologues. This is consistent with our previous observation that cpFtsY is pre-organized into a conformation more conducive to stable complex assembly than the bacterial FtsY (26, 32).
Figure 6.
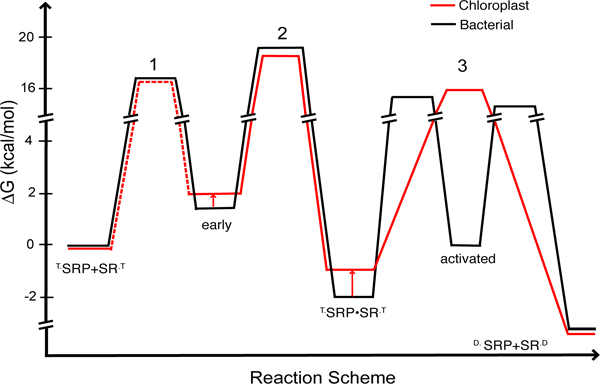
Free-Energy profile for the GTP-dependent binding and activation cycles between the SRP and SR GTPases from bacteria (black) or chloroplast (red). The values for the E. coli GTPases were obtained from references (19, 27). A standard state of 1 μM was used. The activation free energies were calculated from the observed association and dissociation rate constants using ΔG‡ = -RT ln(kh/kBT), where R = 1.987 cal K−1 mol−1, Planck constant h = 1.58 × 10−37 kcal s−1, Boltzmann constant kB = 3.3 × 10−27 kcal s, and T = 298K. Equilibrium stabilities of complexes were calculated using ΔG = ΔG° - RT ln(K/K°).
An important feature of the bacterial SRP system is that movement of the IBD loops, which activates GTP hydrolysis, can be conceptually and experimentally uncoupled from the rearrangements in the rest of the protein that mediate stable complex assembly (17, 22). Numerous mutations in the IBD loops result in specific inhibition of GTPase activation, without significantly disrupting formation of the complex (17). Given these observations, it was surprising to find that the vast majority of mutations in the IBD loops of cpSRP54 and cpFtsY severely compromised assembly of the GTPase complex. This raises the possibility that in the cpSRP54•cpFtsY complex, these two steps are more tightly coupled, as the catalytic active sites are also intimately involved in the assembly of the complex. Supporting this notion is the observation that conservative perturbations at the site of chemical transformation, such as replacement of the β, γ-bridging oxygen of GTP with -NH-, severely disrupted complex stability and assembly rate, in contrast to the bacterial SRP with which GMPPNP specifically inhibits GTP hydrolysis (31). Further, mutations of the IBD loops induced large changes in the fluorescence of an acrylodan dye over 30Å away at the NG-domain interface, suggesting that the GTPase active sites are intimately linked to sites crucial for complex assembly. Taken together, these observations suggest that during the interaction between cpSRP54 and cpFtsY, the complex formation and GTPase activation steps are highly coupled, in contrast to the cytosolic SRP•FtsY complex in which these processes occur in two distinct molecular steps (Figure 6, step 3, black vs. red lines).
What gives rise to this difference? To address this question, one might begin by reflecting on the role of the multiple conformational steps during the assembly and activation of the cytosolic SRP•FtsY complex. Uncoupling complex formation and GTPase activation allows each of these steps to provide an independent fidelity checkpoint, thus providing the SRP multiple opportunities to reject the incorrect cargos (19). This is crucial for the bacterial SRP to achieve a high fidelity of substrate selection, as it has to recognize highly divergent signal sequences and to distinguish between the correct and incorrect cargos based on subtle variations (20). We speculate that the absence of a similar challenge in attaining specific substrate selection may underlie the different behavior of the chloroplast GTPases. In contrast to the cytosolic SRP, the cpSRP is primarily dedicated to a highly conserved LHC family of proteins, and the cpSRP43 subunit can provide highly specific recognition of these substrates (14, 15, 23). Although cpSRP54 also participates in the co-translational targeting of several membrane proteins (such as D1 protein) (33), the number and diversity of these substrates are much more limited than those handled by the cytosolic SRP. It could therefore be envisioned that the chloroplast SRP system can afford to forego the GTPase activation step as an additional regulatory point.
Consistent with this notion, GTPase activation plays a less essential role in protein targeting by the cpSRP than the cytosolic SRP pathway. In the cytosolic SRP pathway, mutant GTPases that specifically block the activation of GTP hydrolysis severely inhibit protein targeting at late stages (31). Thus the molecular rearrangements that lead to GTPase activation, notably the movement of the IBD loops, play an essential role in the unloading of cargo from the SRP and the initiation of protein translocation. In contrast, mutations that specifically inhibit GTPase activation in the cpSRP54•cpFtsY complex resulted in only a two-fold reduction in the targeting of LHCP. Although in previous reports, the observation that GMPPNP inhibited LHCP targeting has implicated a crucial role of GTP hydrolysis for LHCP targeting and integration (24, 25), our findings here suggest that these defects could instead arise from the failure of GMPPNP to support efficient and stable cpSRP54-cpFtsY complex assembly. Indeed, mutant GTPases that impair complex assembly between cpSRP54 and cpFtsY led to much larger deleterious effects on LHCP targeting, and the defects in their targeting efficiency correlated with their defects in complex assembly. Thus GTPase activation or GTP hydrolysis plays a modulatory role in enhancing the targeting efficiency of LHCP, but is not as crucial as is the case with the cytosolic SRP. The ability of cpSRP43 to directly interact with the Alb3 translocase and thus regulate substrate binding and release (24, 34) might allow the cpSRP pathway to bypass the use of GTPase activation as a critical mechanism to drive the unloading of cargo from the cpSRP; this possibility remains to be tested.
Collectively, these results suggest a more streamlined cpSRP54-cpFtsY interaction cycle compared to their bacterial homologues (Figure 6), which might be a consequence of their adaptation to targeting a different set of substrate proteins. This pair of GTPases is primed to efficiently form a complex with one another and to quickly turn over the complex (through rapid GTP hydrolysis), bypassing conformational steps that serve as important fidelity checkpoints in the bacterial SRP pathway. These features could allow the cpSRP pathway to cater to the LHC family of proteins, whose sequence conservation allows specific substrate selection to be more easily achieved, but whose high abundance demands a highly efficient targeting pathway with rapid turnover. In this light, one might further speculate that the complex series of dynamic conformational changes observed for the bacterial SRP•FtsY GTPase complex could be fine-tuned to allow efficient targeting only in response to the correct signal sequences while minimizing the targeting of empty ribosomes and incorrect cargo proteins. The divergent properties of the bacterial and chloroplast SRP and FtsY GTPases might reflect different mechanisms to achieve the balance between efficiency and selectivity as the two pathways adapt to distinct challenges posed by their substrate proteins.
Supplementary Material
Acknowledgment.
We thank X. Zhang, N. Pierce and members of the Shan lab for helpful comments on the manuscript.
This work was supported by NIH grant GM078024 to S.S. S.S. was supported by the Beckman Young Investigator award, the Packard and Lucile award in science and engineering, and the Henry Dreyfus teacher-scholar award.
Abbreviations:
- SRP
Signal recognition particle
- SR
Signal recognition particle receptor
- cpSRP
chloroplast SRP
- LHCP
light-harvesting chlorophylla/b binding protein
- GTP
guanosine-5’-triphosphate
- GMPPNP
5’-guanylylimido-diphosphate
References
- 1.Koch HG, Moser M, and Muller M (2003) Signal recognition particle-dependent protein targeting, universal to all kingdoms of life, Re. Physiol. Biochem. Pharmacol. 146, 55–94. [DOI] [PubMed] [Google Scholar]
- 2.Walter P, and Johnson AE (1994) Signal sequence recognition and protein targeting to the endoplasmic reticulum membrane, Ann. Rev. Cell Biol. 10, 87–119. [DOI] [PubMed] [Google Scholar]
- 3.Keenan RJ, Freymann DM, Stroud RM, and Walter P (2001) The signal recognition particle, Annu. Rev. Biochem. 70, 755–775. [DOI] [PubMed] [Google Scholar]
- 4.Zopf D, Bernstein HD, Johnson AE, and Walter P (1990) The methionine-rich domain of the 54 kd protein subunit of the signal recognition particle contains an RNA binding site and can be crosslinked to a signal sequence, EMBO J. 9, 4511–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zopf D, Bernstein HD, and Walter P (1993) GTPase Domain of the 54-kD subunit of the mammalian signal recognition particle is required for protein translocation but not for signal sequence binding, J. Cell Biol. 120, 1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halic M, Blau M, Becker T, Mielke T, Poll MR, Wild K, Sinning I, and Beckmann R (2006) Following the signal sequence from ribosomal tunnel exit to signal recognition particle, Nature 444, 507–511. [DOI] [PubMed] [Google Scholar]
- 7.Schaffitzel C, Oswald M, Berger I, Ishikawa T, Abrahams JP, Koerten HK, Koning RI, and Ban N (2006) Structure of the E. coli signal recognition particle bound to a translating ribosome, Nature 444, 503–506. [DOI] [PubMed] [Google Scholar]
- 8.Bourne HR, Sanders DA, McCormick F (1991) The GTPase superfamily: Conserved structure and molecular mechanism, Nature 349, 117–127. [DOI] [PubMed] [Google Scholar]
- 9.Eichacker LA, and Henry R (2001) Function of a chloroplast SRP in thylakoid protein export, Biochim. et Biophys. Acta 1541, 120–134. [DOI] [PubMed] [Google Scholar]
- 10.Schunemann D, Gupta S, Persello-Cartieaux F, Klimyuk VI, Jones JDG, Nussaume L, and Hoffman NE (1998) A novel signal recognition particle targets light-harvesting proteins to the thylakoid membranes, Proc. Natl. Acad. Sci. U. S. A. 95, 10312–10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Funke S, Knechten T, Ollesch J, and Schunemann D (2005) A unique sequence motif in the 54-kDa subunit of the chloroplast signal recognition particle mediates binding to the 43-kDa subunit, J. Biol. Chem. 280, 8912–8917. [DOI] [PubMed] [Google Scholar]
- 12.Richter CV, Trager C, and Schunemann D (2008) Evolutionary substitution of two amino acids in chloroplast SRP54 of higher plants cause its inability to bind SRP RNA, FEBS Lett. 582, 3223–3229. [DOI] [PubMed] [Google Scholar]
- 13.Franklin KE, and Hoffman NE (1993) Characterization of a chloroplast homologue of the 54-kDa subunit of the signal recognition particle, J. Biol. Chem. 268, 22175–22180. [PubMed] [Google Scholar]
- 14.Delille J, Peterson EC, Johnson T, Morre M, Kight A, and Henry R (2000) A novel precursor recognition element facilitates posttranslational binding to the signal recognition particle in chloroplasts, Proc. Natl. Acad. Sci. 97, 1926–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tu CJ, Peterson EC, Henry R, and Hoffman NE (2000) The L18 domain of light-harvesting chlorophyll proteins binds to chloroplast signal recognition particle 43, J. Biol. Chem. 275, 13187–13190. [DOI] [PubMed] [Google Scholar]
- 16.Tu C-J, Schuenemann D, and Hoffman NE (1999) Chloroplast FtsY, Chloroplast Signal Recognition Particle, and GTP are required to reconstitute the soluble phase of light-harvesting chlorophyll protein transport into thylakoid membranes, J. Biol. Chem. 274, 27219–27224. [DOI] [PubMed] [Google Scholar]
- 17.Shan S, Stroud R, and Walter P (2004) Mechanism of association and reciprocal activation of two GTPases, Plos Biology 2, e320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Kung S, and Shan S (2008) Demonstration of a two-step mechanism for assembly of the SRP-SRP receptor complex: implications for the catalytic role of SRP RNA, J. Mol. Biol. 381 581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Schaffitzel C, Ban N, and Shan S (2009) Multiple conformational changes in a GTPase complex regulate protein targeting, Proc. Natl. Acad. Sci. U. S. A. 106, 1754–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Rashid R, Kai W, and Shan S (2010) Sequential checkpoints govern fidelity during co-translational protein targeting, Science 328, 757–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egea PF, Shan S, Napetschnig J, Savage DF, Walter P, and Stroud RM (2004) Substrate twinning activates the signal recognition particle and its receptor, Nature 427, 215–221. [DOI] [PubMed] [Google Scholar]
- 22.Shan S, Chandrasekar S, and Walter P (2007) Conformational changes in the GTPase modules of SRP and its receptor drive initiation of protein translocation, J. Cell Biol. 178, 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaru-Ampornpan P, Shen K, Lam VQ, Ali M, Doniach S, Jia TZ, and Shan S (2010) ATP-independent reversal of a membrane protein aggregate by a chloroplast SRP subunit, Nature Struct. Molec. Biol. 17, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Falk S, Ravaud S, Koch J, and Sinning I (2010) The C-terminus of the Alb3 membrane insertase recruits cpSRP43 to the thylakoid membrane, J. Biol. Chem. 285, 5954–5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan J, Kight A, Goforth RL, Moore M, Peterson EC, Sakon J, and Henry R (2002) ATP stimulates signal recognition particle (SRP)/FtsY-supported protein integration in chloroplasts, J. Biol. Chem. 277, 32400–32404. [DOI] [PubMed] [Google Scholar]
- 26.Jaru-Ampornpan P, Chandrasekar S, and Shan S (2007) Efficient interaction between two GTPases allows the chloroplast SRP pathway to bypass the requirement for an SRP RNA., Mol. Biol. Cell 18, 2636–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peluso P, Shan S, Nock S, Herschlag D, and Walter P (2001) Role of SRP RNA in the GTPase cycles of Ffh and FtsY, Biochemistry 40, 15224–15233. [DOI] [PubMed] [Google Scholar]
- 28.Yuan J, Henry R, Cline K (1993) Stromal factor plays an essential role in protein integration into thylakoids that cannot be replaced by unfolding or by heat shock protein Hsp70, Proc. Natl. Acad. Sci. 90, 8552–8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shepotinovskaya IV, Freymann DM (2001) Conformational change of the N-domain on formation of the complex between the GTPase domains of Thermus aquaticus Ffh and FtsY, Biochem. Biophys. Acta 1597, 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaru-Ampornpan P, Nguyen TX, and Shan S (2009) A distinct mechanism to achieve efficient SRP-SRP receptor interaction by the chloroplast SRP, Mol. Biol. Cell 20, 3965–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shan S, and Walter P (2005) Co-translational protein targeting by the signal recognition particle, FEBS Lett. 579, 921–926. [DOI] [PubMed] [Google Scholar]
- 32.Chandrasekar S, Chartron S, Ampornpan P, and Shan S (2008) Crystal structure of the chloroplast signal recognition particle (SRP) receptor: domain arrangement modulates SRP-receptor interaction, J. Mol. Biol. 375, 425–436. [DOI] [PubMed] [Google Scholar]
- 33.Nilsson R, van Wikj KL . (2002) Transient interaction of cpSRP54 with elongating nascent chains of the chloroplast-encoded D1 protein; ‘cpSRP54 caught in the act’, FEBS Letters 524, 127–133. [DOI] [PubMed] [Google Scholar]
- 34.Lewis NE MN, Kathir KM, Rajalingam D, Kight AD, Daily A, Kumar TK, Henry RL, Goforth RL. (2010) A dynamic cpSRP43-Albino3 interaction mediates translocase regulation of chloroplast signal recognition particle (cpSRP)-targeting components., J Biol Chem 285, 34220–34230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.