

Abstract
Background:
Inflammation is associated with cardiac remodeling and heart failure, but how it is initiated in response to non-ischemic interventions in the absence of cell death is not known. We tested the hypothesis that activation of CaMKIIδ in cardiomyocytes in response to pressure overload elicits inflammatory responses leading to adverse remodeling.
Methods:
Mice in which CaMKIIδ was selectively deleted from cardiomyocytes (CMs) (Cardiac specific knockout; CKO) and floxed control (CTL) mice were subjected to transverse aortic constriction (TAC). The effects of CM-specific CaMKIIδ deletion on inflammatory gene expression, inflammasome activation, macrophage accumulation and fibrosis were assessed by qPCR and histochemistry and ventricular remodeling by echocardiography.
Results:
TAC induced increases in cardiac mRNA levels for pro-inflammatory chemokines and cytokines within 3 days and these responses were significantly blunted when cardiomyocyte CaMKIIδ was deleted. Apoptotic and necrotic cell death were absent at this time. CMs isolated from TAC hearts mirrored these robust increases in gene expression which were markedly attenuated in CKO. Priming and activation of the NLRP3 inflammasome, assessed by measuring IL-1β and NLRP3 mRNA levels, caspase-1 activity and IL-18 cleavage, were increased at day 3 post-TAC in CTL hearts and in CMs isolated from these hearts. These responses were dependent on CaMKIIδ and associated with activation of NFkB and ROS. Accumulation of macrophages observed at day 7–14 post-TAC was diminished in CKO and by blocking MCP-1 signaling, deletion of CM MCP-1 or inhibition of inflammasome activation. Fibrosis was also attenuated by these interventions and in the CKO heart. Ventricular dilation and contractile dysfunction observed at day 42 post-TAC were diminished in the CKO. Inhibition of CaMKII, NFkB, inflammasome or MCP-1 signaling in the first one or two weeks after TAC decreased remodeling but inhibition of CaMKII after two weeks did not.
Conclusions:
Activation of CaMKIIδ in response to pressure overload triggers inflammatory gene expression and activation of the NLRP3 inflammasome in CMs. These responses provide signals for macrophage recruitment, fibrosis and myocardial dysfunction in the heart. Our work suggests the importance of targeting early inflammatory responses induced by cardiomyocyte CaMKIIδ signaling to prevent progression to heart failure.
Keywords: Cardiac remodeling, Inflammation, NLRP3 inflammasome, CaMKIIδ
Introduction
Heart failure is associated with adverse ventricular remodeling and remains a leading cause of death worldwide. Inflammation is an accepted driver of adverse remodeling after acute myocardial infarction1, 2. Cardiac inflammation is triggered, in response to ischemic stress, by release of alarmins or damage associated molecular patterns (DAMPs) from dying cardiomyocytes which induce expression of genes for pro-inflammatory chemokines and cytokines, resulting in immune cell recruitment and a cascade of inflammatory events2–5. The NLRP3 inflammasome, a multiprotein signaling complex, is also assembled and activated in response to DAMPs to catalyze generation of the active forms of the potent pro-inflammatory cytokines IL-1β and IL-18 and is thus another critical component of cardiac inflammation6, 7.
There is growing evidence that cardiac inflammation can also occur under non-ischemic conditions. An early study demonstrated that inflammatory genes and inflammatory cell infiltration occurred in hearts of mice following transverse aortic constriction (TAC)8, 9, and other work shows striking increases in accumulation of both macrophages and T-cells in the heart within days to weeks of TAC which have been linked to fibrosis and adverse ventricular remodeling10–14. The question of how pressure overload triggers inflammatory gene expression and immune cell recruitment in the absence of ischemia induced cell death and accompanying release of factors from dying cells has not, however, been addressed. Recent papers have also linked activation of the NLRP3 inflammasome to ischemic stress and subsequent adverse remodeling15–17. Whether the inflammasome is assembled and activated in cardiomyocytes is controversial, and whether or how this might be triggered in the absence of cell damage signals is unclear.
The multifunctional Ca2+/calmodulin regulated kinase δ, CaMKIIδ is rapidly activated in response to pressure overload18. However our studies and those of others using CaMKIIδ knockout (KO) mice revealed that CaMKIIδ is not required for development of pathological hypertrophy, but instead for the progression from hypertrophy to heart failure19–21. We also reported that cardiomyocyte (CM) CaMKIIδ overexpression induces heart failure and that the resulting phenotypic changes are not rescued by normalizing sarcoplasmic reticulum calcium handling, inhibiting the mitochondrial permeability transition pore or blocking β-adrenergic receptors22–24. These findings suggest that there is an as yet undefined pathway by which CaMKIIδ activation mediates progression to heart failure.
Work from several groups established that CaMKIIδ contributes to inflammatory gene expression in response to ischemic injury and that this occurs through activation of NFkB25–27. This lead us to hypothesize that inflammation could be initiated in response to non-ischemic stress, in the absence of significant cardiomyocyte cell death and DAMP release, by activation of CaMKIIδ in cardiomyocytes. In the present study we used conditional CM-specific CaMKIIδ KO (CKO) mice to demonstrate that cardiomyocytes generate inflammatory chemokines and cytokines and are the initial site of NLRP3 inflammasome activation. We further identify a causal role for CaMKIIδ mediated activation of the NLRP3 inflammasome and inflammatory responses in macrophage recruitment, cardiac fibrosis and development of heart failure induced by pressure overload.
Methods
The data, analytic methods, and study materials will be made available to other researchers as requested, for purposes of reproducing the results, replicating the procedures or for collaborative studies. Detailed Materials and Methods are presented in the Online Data Supplement.
Statistical analysis
All results are presented as mean ± SEM. Comparisons of two groups were accomplished using Mann-Whitney U test or unpaired Student’s t test where appropriate. Experiments with more than two groups were compared by one-way or two-way ANOVA followed by Tukey’s or Bonferroni’s multiple comparisons test. All statistics were calculated using GraphPad Prism 7 (GraphPad Software Inc., San Diego, California, USA). P-values less than 0.05 were considered statistically significant.
Study approval
All animal protocols were approved by the Institutional Animal Care and Use Committee of University of California San Diego. All investigations using human heart were approved by Stritch School of Medicine, Loyola University and the Veterans Administration San Diego.
Results
TAC induces inflammatory genes through cardiomyocyte CaMKIIδ
To determine if inflammation is an early response to pressure overload-induced CaMKIIδ activation we examined the time course of inflammatory gene expression in response to transverse aortic constriction (TAC) in CaMKIIδ fl/fl (control: CTL) and cardiomyocyte-specific CaMKIIδ KO (CKO) mice. Cre expression in cardiomyocytes (CM) results in nearly complete loss of CM CaMKIIδ (Supplemental Figure 1A, 1B). There were no baseline differences in ventricular structure or function between CaMKIIδfl/fl (control: CTL) mice and CKO26. Heart rate and pressure gradients after TAC were also equivalent in the two lines (Supplemental Table 1). Hearts were isolated at various times following TAC, and mRNA prepared for qPCR analysis of a range of proinflammatory genes (Figure 1). The mRNA levels for MCP-1, MIP1α, CXCL-1 and IL-6 were significantly elevated by day 1.5 and peaked at day 3, with 5 – 35 fold increases in CTL vs. sham. All of these responses were decreased by ~40% in CKO hearts. Other genes evaluated including CXCL2, IL-1β, IL-18 and TNF-α increased somewhat more slowly and less transiently but also showed significantly attenuated responses in hearts of CKO mice.
Figure 1. Pressure overload induces inflammatory genes through cardiomyocyte CaMKIIδ.
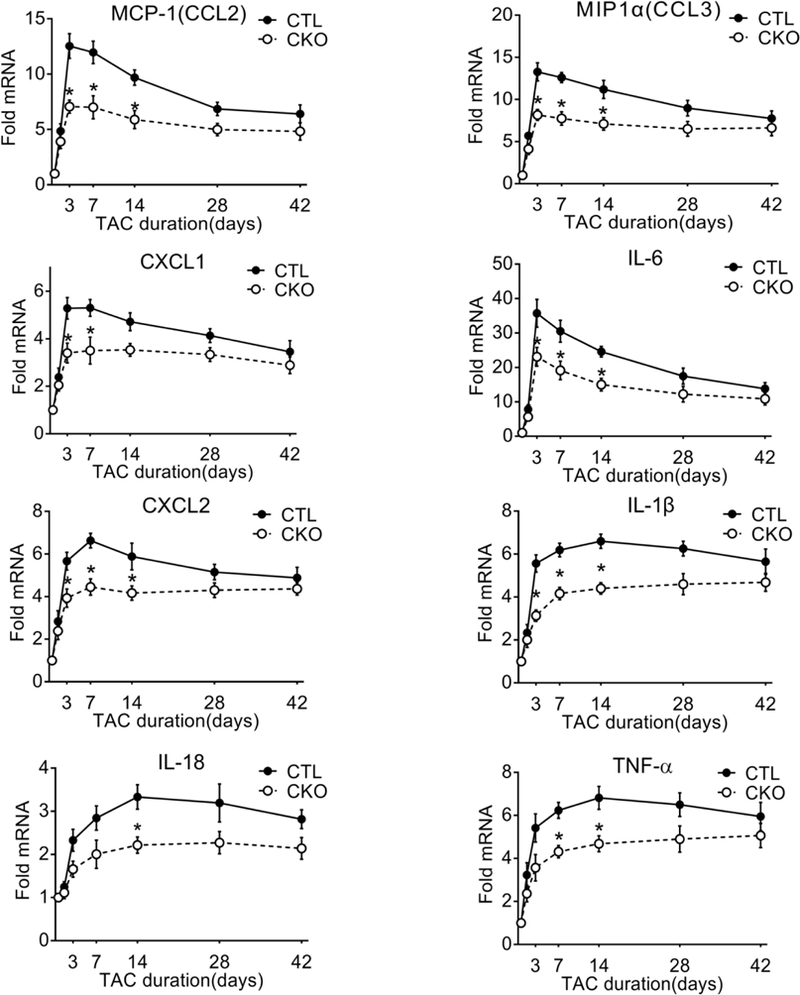
Monocyte chemoattractant protein 1 (MCP-1/CCL2), macrophage inflammatory protein 1α (MIP1α/CCL3), C-X-C motif ligand 1 (CXCL1), C-X-C motif ligand 2 (CXCL2), interleukin 6 (IL-6), interleukin 1β (IL-1β), interleukin 18 (IL-18) and tumor necrosis factor α (TNFα) mRNA in whole ventricular lysates as measured by qPCR, normalized for the internal control GAPDH and expressed as fold increase over sham. N=6–8 per group (Male=3–4, Female=3–4). Data are presented as mean ± SEM. *P<0.05 vs CTL (fl/fl) TAC by two-way ANOVA followed by Bonferroni’s multiple comparisons test.
TAC activates CaMKIIδ and NFkB signaling required for early regulation of inflammatory gene expression
CaMKII activation was assessed using a 32P enzymatic activity assay, to extend our previous work using autophosphorylation as a readout18. The fraction of autonomously active (CaM independent) CaMKIIδ activity was demonstrated to be increased 1.6 fold (Supplemental Figure 2). We next determined whether NFkB, a master regulator of inflammation, was activated through CaMKIIδ as an early response to TAC. NFkB activation, assessed by immunoblotting for increases in the p65 NFkB subunit in nuclear fractions, was increased in CTL hearts at day 1.5 and day 3 post-TAC and this response was significantly attenuated in CKO hearts (Figure 2A). To assess the involvement of NFkB activation in the induction of inflammatory genes, mice were injected with BMS-345541 (BMS), an inhibitor of IkB kinase, an upstream regulator of NFkB. This led to nearly complete inhibition of TAC-induced increases in inflammatory gene mRNA (Figure 2B).
Figure 2. Cardiomyocyte CaMKIIδ mediates pressure overload-induced NFkB activation which is required for inflammatory gene expression.
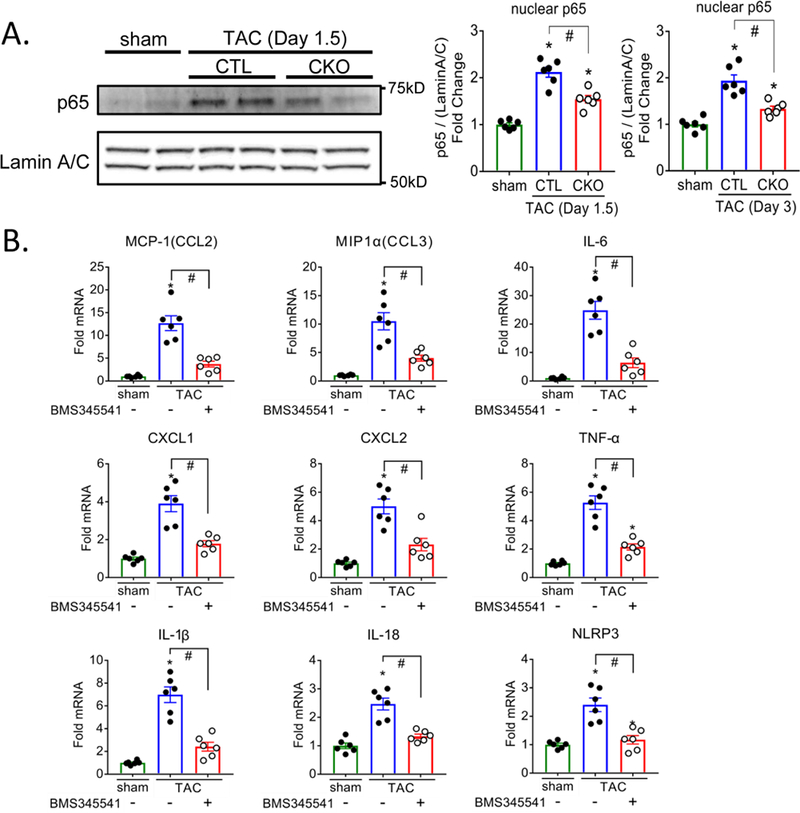
(A) Nuclear fractions were prepared from ventricles and immunoblotted for p65 NFKB subunit. Lamin A/C was used as a nuclear loading control. Representative immunoblot image and quantification of pooled data. N=6 per group (Male=3, Female=3) (B) Mice were injected intraperitoneally every 12 hours with saline (-) or 45 mg/kg BMS345541(+) starting 1hr prior to TAC surgery and for the 3 days prior to termination of the experiment. Inflammatory gene expression in whole ventricular lysates as measured by qPCR, normalized for the internal control GAPDH and expressed as fold increase over sham. N=6 per group (Male=3, Female=3). Data are presented as mean ± SEM. *P<0.05 vs sham, #P<0.05 vs CTL TAC by one-way ANOVA followed by Tukey’s multiple comparisons test.
CaMKIIδ-dependent inflammatory gene expression is initiated in cardiomyocytes versus non-cardiomyocytes
To demonstrate that CMs are the initial site of inflammatory gene expression in the heart, we isolated both adult mouse ventricular CMs and non-cardiomyocytes from CTL and CKO mice at day 3 post-TAC. The isolation procedures, detailed in materials and methods, yielded CM preparations with minimal contamination with immune cells, endothelial cells or fibroblasts, as assessed by Western blotting for CD45, CD31(PECAM-1) and PDGFRα respectively (Supplemental Figure 1C). Inflammatory gene mRNA levels in the isolated CMs were increased to an extent similar in magnitude to that observed in the whole heart lysate, and attenuated by 70–80 % in CKO vs CTL cells (Figure 3). In contrast, significant increases in MCP-1, MIP1α and IL-6 gene expression were not seen in the non-cardiomyocyte cells separated from the CMs at day 3 post-TAC (Supplemental Figure 3). These data provide evidence that TAC regulates inflammatory gene expression within the CMs, and through activation of CaMKIIδ, at the onset of pressure overload stress.
Figure 3. Robust CaMKIIδ dependent inflammatory gene expression is evident in ventricular myocytes isolated from hearts at day 3 after TAC.
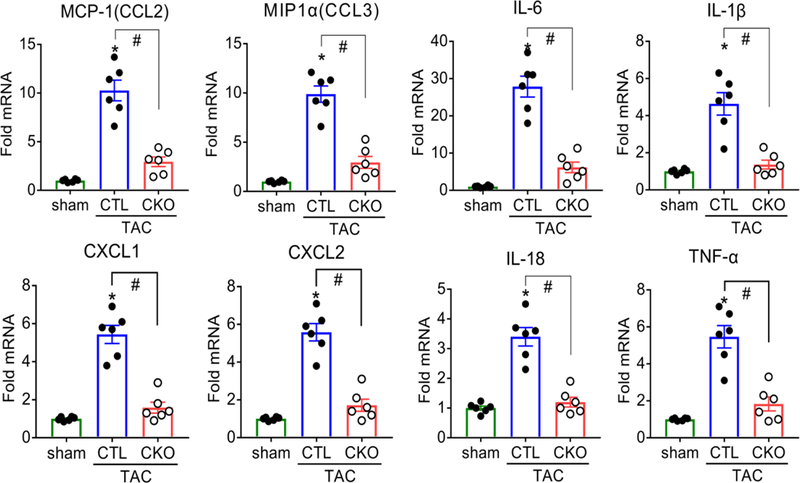
Monocyte chemoattractant protein 1 (MCP-1/CCL2), macrophage inflammatory protein 1α (MIP1α/CCL3), C-X-C motif ligand 1 (CXCL1), C-X-C motif ligand 2 (CXCL2), interleukin 6 (IL-6), interleukin 1β (IL-1β), interleukin 18 (IL-18) and tumor necrosis factor α (TNFα) mRNA in adult mice ventricular cardiomyocytes (CMs) isolated at day 3 of TAC as measured by qPCR, normalized for the internal control GAPDH and expressed as fold increase over sham. *P<0.05 vs sham, #P<0.05 vs CTL TAC. N=6 per group (Male=3, Female=3). Data are presented as mean ± SEM. *P<0.05 vs sham, #P<0.05 vs CTL TAC by one-way ANOVA followed by Tukey’s multiple comparisons test.
TAC does not induce apoptotic or necrotic cell death at times up to 7 days
CM cell death leads to release of DAMPs which stimulate inflammation2, 4, thus we sought to rule out the possibility that TAC induces inflammation through such a mechanism. We examined hearts after 3 and 7 days of TAC and observed no increase in Evans blue dye uptake, TUNEL staining, caspase-3 cleavage, or caspase-3 activation (Supplemental Figure 4). Only at later times of TAC were caspase-3 cleavage and caspase-3 activity increased (Supplemental Figure 4B, 4C). We also examined the IRF3-dependent gene program shown to be activated in response to cytosolic dsDNA released from CMs following myocardial infarction28. Increases in these genes were seen after myocardial infarction (MI) surgery but not at either day 3 or 7 after TAC (Supplemental Figure 5).
Deletion of CaMKIIδ in cardiomyocytes attenuates TAC-induced accumulation of CD68+ macrophages
To determine if macrophage recruitment was elicited by TAC through activation of CaMKIIδ in cardiomyocytes, we immunostained cardiac tissue sections with the macrophage marker CD68. CD68+ macrophage accumulation was markedly increased at day 7 and 14 post-TAC and significantly attenuated in CKO mice (Figure 4A, 4B). A similar response was observed using F4/80 staining (Supplemental Figure 6A, 6B). Treatment with the CaMKII inhibitor KN-93 for the first week of TAC also reduced TAC-induced CD68 positive cell accumulation at day 14 (Figure 4C) confirming that the role of CaMKIIδ in TAC-induced macrophage accumulation was not due to secondary effects of CaMKIIδ gene deletion. There was also upregulation of mRNA for IL-10 and TGF-β, cytokines considered to be products of macrophages29, 30 which were attenuated in the CKO vs CTL (Figure 4D).
Figure 4. Deletion of CaMKIIδ in cardiomyocytes attenuates pressure overload-induced accumulation of CD68+ macrophages and their products in the heart.
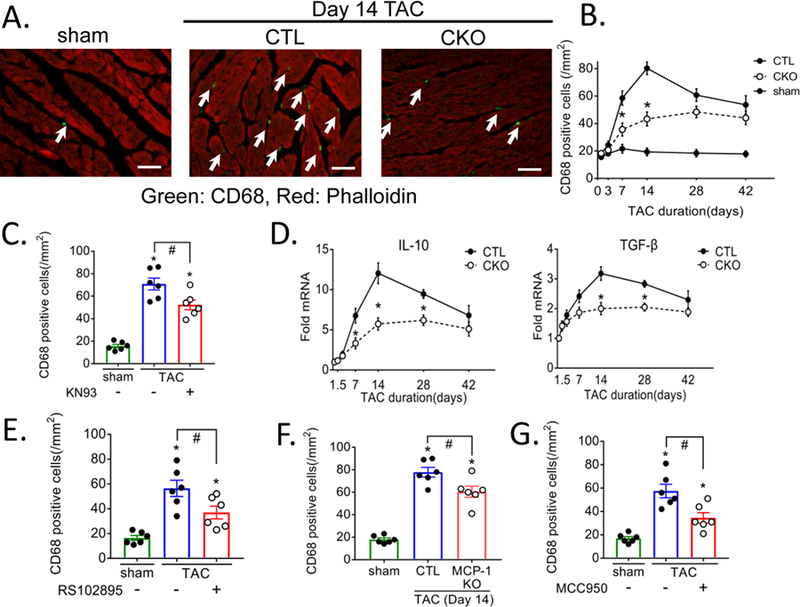
(A) Representative image of cardiac section stained with CD68+ antibody (Green: marked by arrows) for macrophage and phalloidin (Red) for cardiomyocyte, obtained at day 14 of TAC, scale bar= 50 μm. (B) Quantification of CD68+ cells in cardiac sections from sham, CTL and CKO mice subjected to 3, 7, 14, 28 and 42 days TAC. Data were quantified from 4–6 images per sample. N=6 per group (Male=3, Female=3). Data are presented as mean ± SEM. *P<0.05 vs CTL (fl/fl) TAC by two-way ANOVA followed by Tukey’s multiple comparisons test. (C) Mice injected with saline (-) or 5 mg/kg of the CaMKII inhibitor KN93 (+) starting 1hr prior to and daily up to 7 days TAC were harvested at day 14 and sections were stained for CD68, N=6 per group (Male=3, Female=3). Data is presented as mean ± SEM. *P<0.05 vs sham, #P<0.05 vs CTL TAC by one-way ANOVA followed by Tukey’s multiple comparisons test. (D) TGF-β and IL-10 mRNA in whole ventricular lysates as measured by qPCR, normalized for the internal control GAPDH and expressed as fold increase over sham. N=6 per group (Male=3, Female=3). Data are presented as mean ± SEM. *P<0.05 vs CTL (fl/fl) TAC by two-way ANOVA followed by Bonferroni’s multiple comparisons test. (E-G) CD68 positive cells quantitated at day 14 of TAC. (E) Mice were injected intraperitoneally with saline (-) or 10 mg/kg RS102895 (+) 1hr prior to TAC and every 6 hours for 3 days to block the MCP-1 receptor. (F) Cardiac specific MCP-1 KO mice were subjected to TAC. (G) Mice were injected intraperitoneally with saline (-) or 10 mg/kg MCC950 (+) 1hr prior to TAC and twice daily for 7 days to inhibit the inflammasome. N=6 per group (Male=3, Female=3). Data are presented as mean ± SEM. *P<0.05 vs sham, #P<0.05 vs CTL TAC by one-way ANOVA followed by Tukey’s multiple comparisons test.
The NLRP3 inflammasome is activated in response to TAC through cardiomyocyte CaMKIIδ
The NLRP3 inflammasome is a critical mediator of inflammatory responses but whether it is activated in CMs remains to be established. We demonstrated above (Figure 2B and Figure 3) that mRNA levels for IL-1β and IL-18, proteins processed by the NLRP3 inflammasome, were increased by through CaMKIIδ and NFkB signaling. NLRP3, a main component of the NLRP3 inflammasome was also increased at the mRNA level in whole heart homogenates at days 1.5 and 3 post-TAC, and attenuated in CKO (Figure 5A) or when NFkB was inhibited using BMS345541 (Figure 2B). NLRP3 protein was also increased at day 3 post-TAC and significantly lower in CKO mice (Supplemental Figure 7).
Figure 5. Inflammasome is activated in response to pressure overload through cardiomyocyte CaMKIIδ.
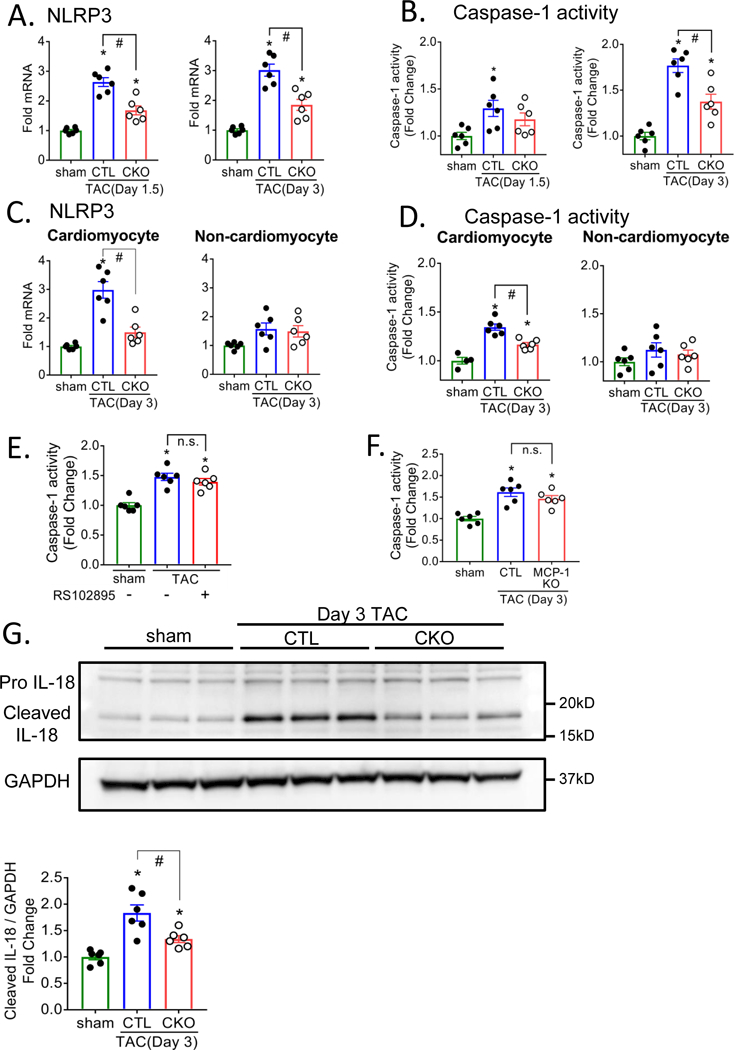
NOD-like pyrin domain-containing protein 3 (NLRP3) mRNA in whole heart homogenate (A) or isolated CMs (C) as measured by qPCR, normalized for the internal control GAPDH and expressed as fold increase over sham. Caspase-1 activity in whole heart homogenate (B, E, F) or isolated CMs (D) was assessed using a fluorometric caspase-1 activity assay. (G) Pro IL-18 and cleaved IL-18 was determined by immunoblotting in isolated CMs. (E) CTL mice were injected intraperitoneally with saline (-) or 45 mg/kg BMS345541 (+) 1hr prior to TAC surgery and twice daily for 3 days. (F) Cardiac specific MCP-1 KO mice were subjected to TAC. N=6 per group (Male=3, Female=3). Data are presented as mean ± SEM. *P<0.05 vs sham, #P<0.05 vs CTL TAC by one-way ANOVA followed by Tukey’s multiple comparisons test.
Activation of inflammasomes leads to cleavage of pro-caspase-1 to its catalytically active form thus increases in caspase-1 activity are indicative of inflammasome activation. We observed significant increases in caspase-1 activity in whole heart homogenates at day 1.5 and 3 post-TAC (Figure 5B) and this response was decreased in hearts of CKO mice. A proposed mechanism for activation of the inflammasome is through as yet undefined effects of reactive oxygen species (ROS)31. Accordingly we tested the hypothesis that CaMKIIδ regulates inflammasome activation following pressure overload through ROS generation. ROS levels assessed by DCFDA fluorescence staining of CTL and CKO mouse ventricle sections indicated that 3 day TAC increases cardiac ROS and that this occurs through CaMKIIδ (Supplemental Figure 8A). We also assessed and quantitated mitochondrial ROS generation by isolating mitochondria from hearts following TAC and measuring mitochondrial protein carbonylation as an indicator of mitochondrial ROS production (Supplemental Figure 8B). Mitochondrial protein carbonylation was increased by TAC in CTL but not in CKO. To test the role of mitochondrial ROS in cardiac inflammasome activation mice were injected with the mitochondrial targeted ROS scavenger MitoTEMPO. TAC induced ROS accumulation was inhibited (Supplemental Figures 8A and 8B) concomitant with significant inhibition of TAC induced caspase-1 activation (Supplemental Figure 8C).
To assess the extent to which the observed inflammasome activation occurs in CMs we separated CMs and non-cardiomyocytes from hearts harvested at day 3 post-TAC. NLRP3 mRNA and caspase-1 activity were both increased in CMs but not in the non-cardiomyocyte fraction expected to contain fibroblasts, immune cells and endothelial cells (Figure 5C, 5D). Changes in this fraction did occur later as shown for day 7 post-TAC (Supplemental Figure 9). Interleukin 18 (IL-18) cleavage to its active product by caspase-1 is an additional indicator of inflammasome activation. Western blotting of CMs isolated from CTL mouse heart revealed that a significant fraction of IL-18 protein was present in its cleaved form at 3 day post-TAC (Figure 5G) and this response diminished in CMs from CKO mice.
To directly visualize activation of caspase-1 following TAC we used a fluorescence indicator of caspase-1 activity (FAM-FLICA™ caspase-1) to image caspase activation in tissue sections (Figure 6A) and quantitated this in isolated CMs (Figure 6B, 6C). Caspase-1 activation was significantly increased by TAC in CTL mouse CMs and virtually abolished in those from CKO (Figure 6B, 6C). The specificity of the FLICA fluorescence signal for caspase-1 was demonstrated by loss of this response in mice treated with MCC950, an inhibitor of NLRP3 inflammasome activation. Finally expressing constitutively active CaMKIIδ in neonatal rat ventricular myocytes at an MOI which does not induce cell death increased FLICA fluorescence in an NLRP3 dependent manner (Figure 6D and 6E).
Figure 6. Visualization of inflammasome activation in cardiomyocytes in response to pressure overload.
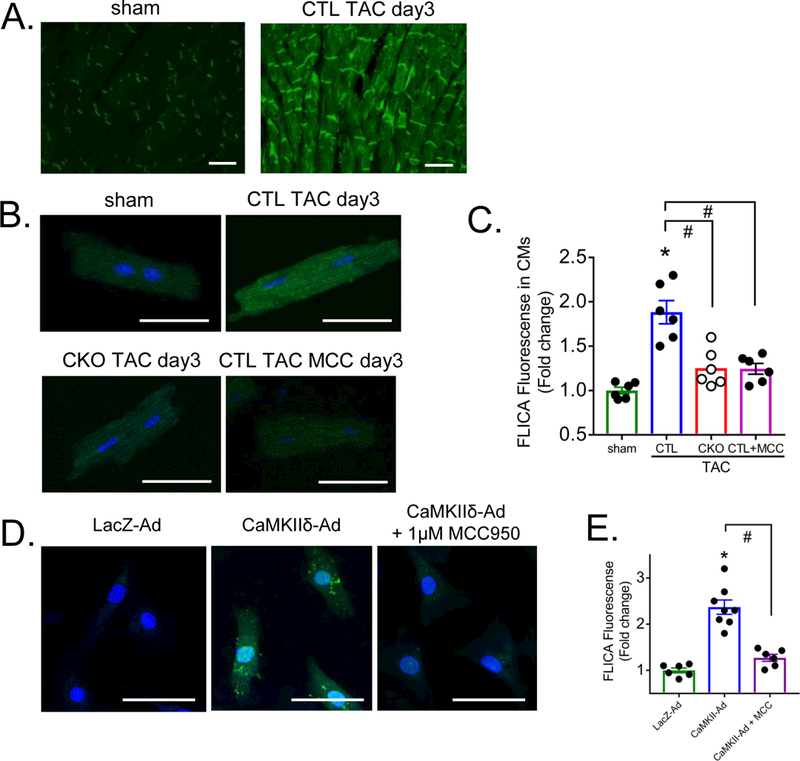
Representative image in cardiac section (A) and CMs (B) at day 3 of TAC. FAM-FLICA™ Caspase-1 assay kit was used to visualize caspase-1 activity. Scale bar = 50 μm. Mice were injected intraperitoneally with saline (-) or 10 mg/kg MCC950 (+) 1hr prior to TAC and twice daily for 3 days. (C) Quantification of FAM-FLICA Caspase-1 fluorescent signal in CMs. Data were quantified from 4–8 images per sample. N=6 per group (Male=3, Female=3). Data are presented as mean ± SEM. *P<0.05 vs sham, #P<0.05 vs CTL TAC by one-way ANOVA followed by Tukey’s multiple comparisons test. (D) Representative image and (E) quantification of fluorescence in NRVMs infected with AdLacZ or the constitutively active δC isoform of CaMKII (AdCaMKIIδ) at a multiplicity of infection of 50. NRVMs were starved for 5 hours prior to a 3-hour infection then washed with serum-free medium and cultured overnight. MCC950 (1 μM) was applied at the time of virus infection and medium exchange. FAM-FLICA Caspase-1 assay kit was used to visualize caspase-1 activity, scale bar= 50 μm. N=6–8 per group. Data are presented as mean ± SEM. *P<0.05 vs LacZ-Ad, #P<0.05 vs CaMKIIδ-Ad by one-way ANOVA followed by Tukey’s multiple comparisons test.
Chronic models of heart failure
To extend the findings obtained using the relatively acute TAC model, we also examined cardiac inflammatory responses in a chronic mouse model of heart failure achieved by 4 weeks of daily isoproterenol (ISO) injection. We have demonstrated that development of fibrosis and contractile dysfunction in this model depend on CaMKIIδ32. The possibility that inflammatory responses also play a role in CaMKIIδ signaling to heart failure in the ISO model is suggested by our finding that the inflammatory genes MCP-1 and IL-6, as well as the inflammasome component NLRP3 and cleaved IL-18, are significantly upregulated in a CaMKIIδ dependent manner (Supplemental Figure 10). As further validation that similar inflammatory responses occur in a clinically relevant chronic heart failure model we compared human non-ischemic cardiomyopathy and normal hearts (Supplemental Figure 11). We detected increased MCP-1, IL-6, IL-10 and IL-1β mRNA, as well as increased NLRP-3 mRNA and cleaved IL-18 in association with upregulated CaMKIIδ in the heart failure samples.
Signaling to macrophage recruitment
MCP-1, which was upregulated by TAC through CaMKIIδ in CMs (Figures 1, 3) is known to recruit monocytes to tissues where they differentiate to macrophages. Monocyte recruitment by MCP-1 occurs through its interaction with its receptor C-C chemokine receptor type 2 (CCR2). To determine whether MCP-1 plays a major role in regulation of macrophage accumulation following TAC we treated CTL mice with RS102895, a highly selective small molecule antagonist of CCR2. This treatment decreased TAC-induced CD68 positive cell accumulation assessed at day 14 by 50 % (Figure 4E). To directly determine whether the MCP-1 involved in this response was derived from CMs we generated cardiomyocyte specific MCP-1 KO (MCP1-KO), as shown in Supplemental Figure 1D, by crossing αMHC-Cre to B6.Cg-Ccl2tm1.1Pame/J mice. CD68 positive cell accumulation in response to TAC was also significantly attenuated in these mice (Figure 4F). Inflammasome activation also contributes to macrophage recruitment as we found that treatment with MCC950 leads to ~60 % inhibition of the TAC-induced accumulation of CD68 positive macrophages (Figure 4G). Thus inflammasome-generated cytokines, as well as MCP-1, contribute to TAC and CaMKIIδ induced macrophage recruitment. These data support our hypothesis that pro-inflammatory gene expression and inflammasome activation in cardiomyocytes are the initiating CaMKIIδ mediated responses in the pathway leading to macrophage recruitment. Notably blocking macrophage recruitment with MCP-1 or inflammasome inhibitors did not decrease inflammatory gene expression assessed at day 3 post-TAC (Supplemental Figure 12A, 12B, 12C) nor did inhibition of MCP-1 decrease caspase-1 activation (Figure 5E, 5F).
Deletion of CaMKIIδ in cardiomyocytes attenuates TAC-induced fibrosis
Pressure overload leads to robust induction of myocardial fibrosis, as demonstrated by Masson’s trichrome staining (Figure 7A). Fibrosis was diminished by 50% in hearts from CKO vs. CTL mice (Figure 7A). The fibrotic gene markers collagen type 1a1 (Col1a1), collagen type 3a1 (Col3a1), and periostin were also upregulated by TAC in CTL hearts and these increases, evident at day 7, 14 and 28, were significantly attenuated in CKO hearts (Figure 7B). Inhibition of cardiomyocyte MCP-1 signaling assessed in cardiac MCP-1 KO, or reduction of inflammasome activation (by administering MCC950) also attenuated TAC-induced fibrosis (Figure 7C, 7D), paralleling the effects of these inhibitors on macrophage accumulation. These results support a critical role for cardiomyocyte CaMKIIδ signaling in development of fibrosis in the heart.
Figure 7. Deletion of CaMKIIδ in cardiomyocytes attenuates pressure overload-induced fibrosis.
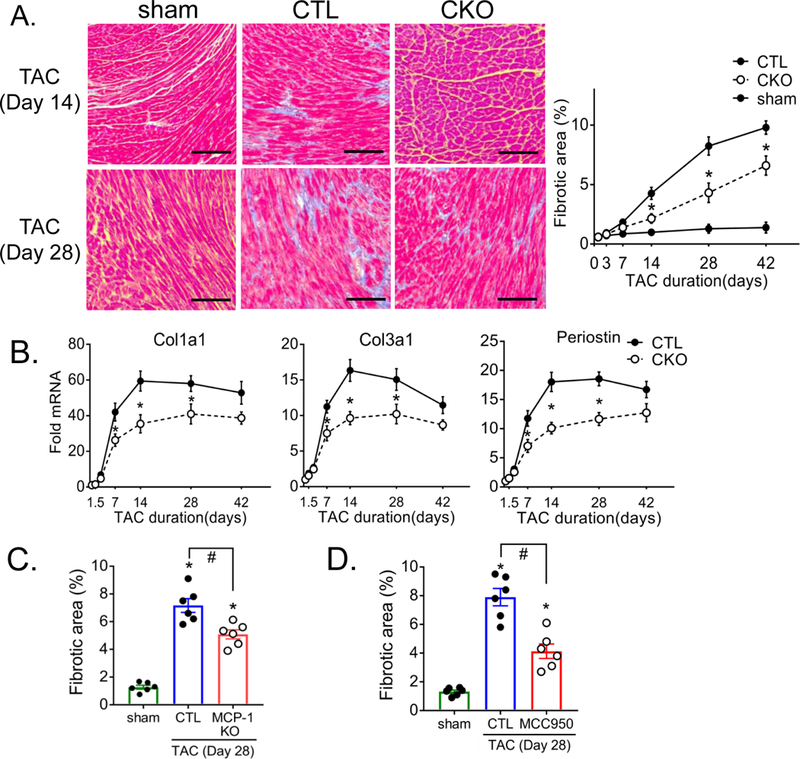
(A) Representative image of cardiac sections stained with Masson’s trichrome at 2 or 4 weeks following TAC. Connective tissue is stained blue. Scale bar= 100 μm. Quantification of % area showing fibrosis was calculated using ImageJ software. Data were quantified from 6–8 images per sample. N=6 per group (Male=3, Female=3). Data are presented as mean ± SEM. *P<0.05 vs CTL (fl/fl) TAC by two-way ANOVA followed by Tukey’s multiple comparisons test. (B) Fibrotic gene expression, Collagen Type I Alpha 1 Chain (Col1a1), Collagen Type III Alpha 1 Chain (Col3a1) and Periostin in whole ventricular lysates as measured by qPCR, normalized for the internal control GAPDH and expressed as fold increase over sham. N=6 per group (Male=3, Female=3). Data are presented as mean ± SEM. *P<0.05 vs CTL (fl/fl) TAC by two-way ANOVA followed by Bonferroni’s multiple comparisons test. (C, D) Cardiac sections quantitated for % area showing fibrosis at day 28 of TAC. (C) Cardiac specific MCP-1 KO mice were subjected to TAC. (D) Mice were injected with saline or 10mg/kg MCC950 1hr prior to TAC and twice daily for 7 days. N=6 per group (Male=3, Female=3). Data are presented as mean ± SEM. *P<0.05 vs sham, #P<0.05 vs CTL TAC by one-way ANOVA followed by Tukey’s multiple comparisons test.
Early CaMKIIδ activation in cardiomyocytes initiates TAC-induced cardiac remodeling
We implicated CaMKIIδ in the transition of pressure overload induced hypertrophy to ventricular dilation and dysfunction in the global CaMKIIδ KO mouse19. Here we demonstrate that ventricular dilation and decreases in ejection fraction at day 42 of TAC are also significantly diminished when CaMKIIδ is selectively deleted from cardiomyocytes (Figure 8A). To implicate early activation of CaMKIIδ signaling pathways in development of TAC-induced fibrosis and subsequent ventricular remodeling and dysfunction, we inhibited downstream mediators of the effects of CaMKIIδ: NFkB, NLRP3 and MCP-1. Pharmacological inhibition of NFkB with BMS345541 or NLRP-3 with MCC950 1hr prior to and for 7 days post TAC attenuated development of cardiac dilation and contractile dysfunction at 42 days post-TAC (Supplemental Figure 13A, 13B) as did specific deletion of MCP-1 from CMs (Supplemental Figure 13C).
Figure 8. CaMKIIδ activation at early times after TAC is required for subsequent cardiac remodeling.
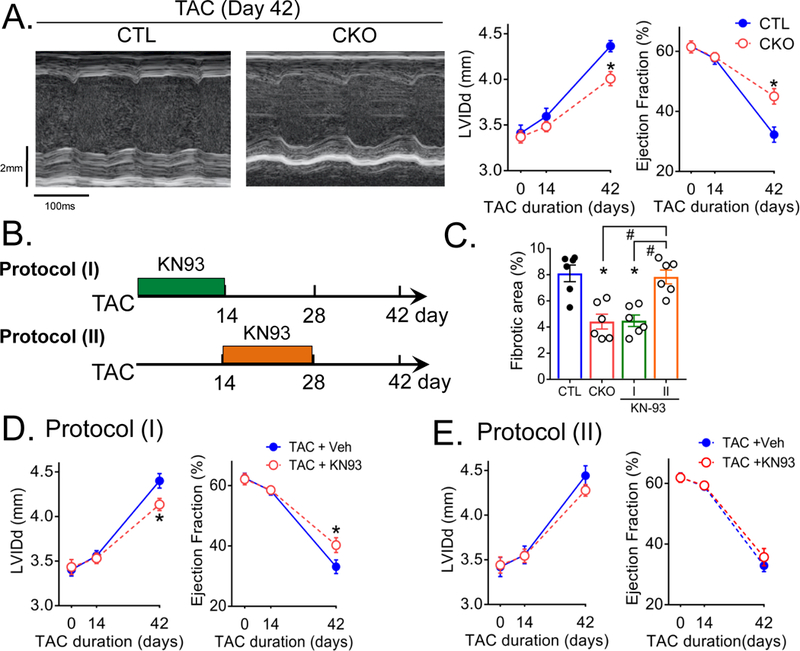
(A) Left: Representative echocardiographs from CTL and CKO at 6 weeks of TAC. Scale bars, vertical 2.0 mm and horizontal 100 msec. Right: Echocardiographic analysis of ventricular dilation and contractile function at various times following TAC. Cardiac dilation was evaluated by left ventricular end-diastolic internal dimension (LVIDd) and contractile function by Ejection Fraction. Heart rate values ranged from 495 to 540 bpm and were not significantly different between CTL TAC and CKO TAC. N=6 per group (Male=3, Female=3). Data are presented as mean ± SEM. *P<0.05 vs CTL (fl/fl) TAC by two-way ANOVA followed by Bonferroni’s multiple comparisons test. (B) Mice were injected intraperitoneally with 5 mg/kg of KN93 starting 1hr prior to TAC and daily for 14 days (Protocol I), or injected with 5 mg/kg of KN93 starting at 14 days and daily up to 28 days (Protocol II). (C) Quantification of % fibrotic area at day 28 of TAC. Data are presented as mean ± SEM. *P<0.05 vs CTL, #P<0.05 vs protocol (II) by one-way ANOVA followed by Tukey’s multiple comparisons test. (D, E) Echocardiographic analysis of ventricular dilation and contractile function at various times following TAC. Heart rate values ranged from 490 to 530 bpm and were not significantly different between TAC + Veh and TAC + KN93. Data are presented as mean ± SEM. *P<0.05 vs TAC + Veh by two-way ANOVA followed by Bonferroni’s multiple comparisons test.
To provide further evidence that early signaling actions of CaMKIIδ in CMs trigger the ensuing maladaptive inflammation and remodeling, we compared the effects of blocking CaMKIIδ signaling at various times. Mice treated with the CaMKII inhibitor KN-93 beginning 1 hour prior to and daily up to day 14 post-TAC (Figure 8B, Protocol I) showed significantly inhibited fibrosis assessed at day 28, i.e. 2 weeks after KN93 treatment was discontinued (Figure 8C). Ventricular dilation was also diminished as was contractile dysfunction (Figure 8D). Remarkably when the two week treatment with KN-93 was delayed (Protocol II) until 14 days after initiation of TAC (a time at which macrophage accumulation is maximal) neither fibrosis nor cardiac dysfunction were attenuated (Figure 8C, 8E).
Discussion
A small but compelling body of evidence has accumulated in recent years implicating inflammation in cardiac remodeling in response to pressure-overload10, 13, 14, 33–36. The mechanism(s) by which maladaptive inflammatory responses to pressure overload are initiated however, remain unknown. The studies presented here lead to several important new concepts relevant to this question. As summarized in the schematic in Supplemental Figure 14, our data show that 1) signals generated in CMs, as opposed to the non-cardiomyocyte cell population in the heart, are the initiators of inflammatory gene expression in response to pressure overload; 2) activation of CaMKIIδ in CMs triggers NFkB mediated pro-inflammatory gene expression; 3) activation of CM CaMKIIδ primes and activates the inflammasome through NFkB and ROS signaling; 4) chemokines (e.g. MCP-1) and products of inflammasome activation produced in CMs contribute to macrophage infiltration in the heart; 5) activation of CM CaMKIIδ signaling pathways contributes to development of fibrosis and adverse remodeling in response to pressure overload; 6) fibrosis, ventricular dilation and dysfunction can be attenuated by blocking CaMKIIδ inflammatory signaling within the first two weeks of pressure overload but not later, after the onset of inflammatory cell accumulation.
Evidence that the TAC induced inflammatory pathway is unique from that triggered by cell death
In response to ischemia, as occurs in myocardial infarction, dying cardiomyocytes release DAMPs or alarmins that trigger robust inflammatory cascades2, 4, 5, 37, 38. We present several lines of evidence which argue that CM death does not occur during the first days to week following TAC, when activation of NFkB, inflammatory gene expression and the NLRP3 inflammasome are evident. Cardiomyocyte necrosis and apoptosis, assessed by Evans blue dye uptake and TUNEL staining, as well as measurement of caspase-3 activity were not changed during the first week, although cell death does occur at later times as previously reported19. Work from the Otsu laboratory, using mice in which DNase was deleted, provided evidence that TAC can induce release of mitochondrial DNA which, if not eliminated by autophagy-mediated degradation, can lead to inflammation and heart failure39. This did not occur in wild type mice, nonetheless we considered the possibility of dsDNA serving as a DAMP following TAC. Induction of IRF-3 dependent gene products, recently shown to result from signaling through release of dsDNA in the mouse heart following MI28, were examined and found to not be induced at day 3 or 7 following TAC. Thus while CaMKII activation has been associated with CM cell death40, the attenuated inflammation that we see in the CKO is unlikely to reflect diminished CaMKIIδ signaling to apoptotic or necrotic responses.
Evidence that cardiomyocytes are the initiating site of inflammatory gene expression
There is considerable evidence that “cardiokines” released from the heart can serve autocrine and paracrine functions38, 41. While CMs could serve as sites of cardiokine generation most studies have considered cardiac fibroblasts, endothelial cells or resident immune cells as those that contribute to production of inflammatory mediators. Our studies considered the question of whether the CM is the predominant site at which proinflammatory chemokines and cytokines are generated at early times following TAC. Remarkably CMs isolated from hearts at day 3 post-TAC showed TAC induced increases in cytokine and chemokine mRNA expression levels equivalent to those seen in the whole heart homogenate, and dependent on CaMKIIδ. In contrast increase in mRNA for these mediators was not observed in the non-cardiomyocyte fraction (e.g., fibroblast, endothelial cells and immune cells) isolated from the heart subjected to 3 day TAC. Thus while a portion of the increase in inflammatory gene expression seen in the whole heart homogenate at day 3 post-TAC and at later times occurs independent of cardiomyocyte CaMKIIδ signaling, the cardiomyocyte, through CaMKIIδ, serves as a significant generator of pro-inflammatory mediators.
Studies using cardiomyocyte specific MCP-1 KO mice provide evidence for the role of cardiomyocytes in subsequent inflammation and cardiac remodeling. Specifically we demonstrate that TAC-induced macrophage recruitment and fibrosis are diminished, and heart failure development is attenuated when cardiomyocyte MCP-1 is deleted. While loss of these TAC-mediated responses is not complete, these findings support the conclusion that an early increase in MCP-1 gene expression in cardiomyocytes plays a role in initiating inflammatory responses and subsequent cardiac remodeling.
Site and mechanism of activation of the NLRP3 inflammasome
There are established mechanisms by which NLRP3 inflammasome signaling in the heart would be triggered in response to ischemic cell death2, 4, 42, when dying cells release substances that activate NFKB (e.g. dsDNA HMGB-1), and mediators of inflammasome activation (e.g. ATP, ROS) are generated. How this might occur in the absence of cell death is not clear29, but recent papers demonstrate inflammasome activation in the heart following pressure overload and acute ISO treatment35, 43. These reports suggest that the inflammasome is activated in CMs, specifically demonstrating increased CM-associated ASC (apoptosis-associated speck like protein containing a caspase recruitment domain), NLRP3 and cleaved IL-18. Here we use visualization and enzymatic activity of caspase-1, as well as isolated cardiomyocytes, to provide stronger evidence that CMs are the site at which the inflammasome is initially primed and activated in response to pressure overload and further demonstrate a mechanistic role for CaMKIIδ in this response.
Using an enriched preparation of cardiomyocytes isolated from hearts following TAC, we show that NLRP-3 induction and caspase-1 activation, assessed by a caspase-1 enzymatic activity and by visualization of activated caspase-1 in CMs, are evident in the CM compartment at day 3. We also demonstrate increases in cleaved IL-18 in the CM compartment at this time. Notably neither caspase-1 activity nor NLRP-3 mRNA are significantly elevated in the non-cardiomyocyte fraction at day 3 post-TAC. Thus neither resident nor recruited macrophages appear to contribute to inflammasome activation at this early time nor do cardiac fibroblasts appear to be involved. These data provide strong support for the concept that the cardiomyocyte is the site at which inflammasome activation is initiated to recruit immune cells and propagate inflammatory signaling in non-cardiac cells.
Mechanistically, we demonstrate that the induction of IL-1β, IL-18 and NLRP-3, as well as the activation of caspase-1 and cleavage of IL-18 apparent in the heart within 3 days of TAC, are attenuated when CaMKIIδ is deleted from cardiomyocytes. In addition, our studies using BMS345541 show that CaMKIIδ activation in myocytes primes the inflammasome through activation of NFkB. We also elucidate the mechanism by which the second step, inflammasome activation, occurs. TAC increases mitochondrial ROS, an event that we show to be diminished in the CKO, i.e. to be dependent on CaMKIIδ signaling in cardiomyocytes. The mechanism by which CaMKIIδ increases ROS levels has not been clearly established but earlier work from our group and studies from the Anderson lab demonstrate effects of CaMKII signaling on mitochondria that can affect Ca2+ uptake and thus mitochondrial ROS generation21, 44. Our experiments using Mito-TEMPO show that scavenging mitochondrial ROS attenuates TAC induced caspase-1 activation, implicating ROS in this process. Our conclusion that CaMKIIδ mediates inflammasome activation, in addition to the extensive evidence that this occurs in cardiomyocytes, substantially advance understanding of the sites and pathways by which cardiac inflammation is initiated in response to non-ischemic stress.
The products of the NLRP3 inflammasome, IL-1β and IL-18, are potent pro-inflammatory cytokines which have been implicated in macrophage recruitment, amplifying and sustaining cardiac inflammation43. These cytokines, as well as IL-10, have been shown to contribute to systolic and diastolic dysfunction through indirect effects on development of myocardial fibrosis and cardiomyocyte apoptosis, as well as via direct effects on cardiac contractile function30, 45. Thus NLRP3 inflammasome activation could function not only as an initiator of cardiac inflammation but also as a generator of cytokines that play a local role in causing cardiomyocyte dysfunction. In this regard, it is interesting to note that we also observe increases in IL-1β, active cleaved IL-18, and IL-10 in hearts from patients with non-ischemic cardiomyopathy, concomitant with elevated levels of CaMKII expression.
Temporal aspects of inflammatory responses and their therapeutic implications
To support our conclusion that activation of CaMKIIδ in cardiomyocytes triggers events that initiate ensuing inflammatory responses and cardiac remodeling, we asked whether blocking CaMKIIδ signaling at early times in the course of pressure overload affected the subsequent development of heart failure. Remarkably when mice received daily injections of inhibitors of either NFkB or the NLRP3 inflammasome during the first week following TAC, contractile dysfunction and dilation monitored 6 weeks later were significantly attenuated. Additionally treatment with the CaMKII inhibitor, KN93, during the first two weeks of TAC blocked the fibrotic response seen at 6 weeks after TAC to an extent equivalent to that seen in the CKO (i.e. when CaMKIIδ activation was abolished throughout the entire time period). In contrast, delayed treatment with KN93 beginning two weeks after the onset of pressure overload, had no significant effect on development of fibrosis, dilation or contractile dysfunction. Consistent with these findings a study published by the Prabhu laboratory demonstrated that when macrophage function was inhibited by inducible macrophage deletion (in macrophage Fas-induced apoptosis transgenic mice) at two weeks post-TAC, subsequent cardiac fibrosis and remodeling were unaffected46. Whereas earlier treatment appeared to be effective47. These data are consistent with and support the notion that early inhibition of inflammatory signals is most effective at preventing heart failure development following TAC. This could also explain why attempts to treat established heart failure by blocking inflammation have been largely unsuccessful48.
We conclude that CaMKIIδ activation in cardiomyocytes is responsible for initiating cardiac inflammation through cytokine generation and NLRP3 inflammasome activation. While we have focused on MCP-1 actions and generation of IL-18, other products generated through NFkB and inflammasome signaling could also contribute to cardiac dysfunction. These include IL-6 which has been reported to mediate pressure-overload induced cardiac remodeling in the mouse heart33, 36, as well as IL-1β, the target of blockade by canakinumab which was shown to diminish adverse cardiac events in patients with coronary artery disease 49 and anakinra, an inhibitor of the IL-1 receptor, which improved cardiac performance in patients with decompensated systolic HF 50.
Study Limitation
The observed effects of genetic deletion did not completely normalize the phenotype, thus other signaling pathways not delineated here must also occur in response to pressure overload. How CMs signal to other non-CM and which specific immune cells are involved has not been determined. We also acknowledged the possibility other isoforms of CaMKII may be upregulated in the human failing heart and contribute to disease.
Supplementary Material
Clinical Perspective
What is new?
We establish a mechanism by which cardiac inflammation is initiated in response to hemodynamic stress, in the absence of significant cardiomyocyte cell death.
This is the first study to reveal a central and critical role for cardiomyocytes, and signaling through CaMKIIδ, in initiating inflammatory responses in the heart.
The NLRP3 inflammasome is activated in cardiomyocytes and contributes to macrophage recruitment, fibrosis and ventricular dysfunction in response to pressure overload.
What are the clinical implications?
Results of recent clinical trials implicate interleukins, generated through the NLRP3 inflammasome, in adverse cardiac effects of associated with coronary artery disease and heart failure.
Our studies reveal sites and mechanisms of pro-inflammatory gene and inflammasome activation within cardiomyocytes which could serve as targets for early intervention or disease prevention.
Acknowledgments
We thank Jeffrey Smith for laboratory management, Nicole Purcell for training on TAC surgery, Melissa Barlow for animal husbandry, Valerie Tan for MI surgery, Richard Daneman for use of his cryostat and the UC San Diego Cancer Center Histology Core for paraffin embedding and sectioning.
Sources of Funding
This work was supported by National Institutes of Health (NIH) grants R37HL028143, R56HL097037 and TRDRP grant to J.H. Brown, the American Heart Association grant 15GRNTZ297009 to S. Miyamoto, and NIH and Veterans Administration grants HL127806 and BX003260 to RS. Ross. T. Suetomi was supported by postdoctoral fellowships from the American Heart Association 17POST33680017 and the Uehara Memorial Foundation (Japan). A. Willeford was supported by NIH Research Training Grants T32 GM007752, T32DK007541 and the UC San Diego Skaggs School of Pharmacy and Pharmaceutical Sciences. CS. Brand was supported by the NIH fellowship T32HL007444 and F32HL140851. Y. Cho was supported by the American Heart Association 14SDG17790005.
Footnotes
Disclosures None.
References
- 1.Westman PC, Lipinski MJ, Luger D, Waksman R, Bonow RO, Wu E and Epstein SE. Inflammation as a Driver of Adverse Left Ventricular Remodeling After Acute Myocardial Infarction. J Am Coll Cardiol. 2016;67:2050–2060. [DOI] [PubMed] [Google Scholar]
- 2.Epelman S, Liu PP and Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. 2015;15:117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaefer L Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem. 2014;289:35237–35245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dick SA and Epelman S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circ Res. 2016;119:159–176. [DOI] [PubMed] [Google Scholar]
- 5.Rider P, Voronov E, Dinarello CA, Apte RN and Cohen I. Alarmins: Feel the Stress. J Immunol. 2017;198:1395–1402. [DOI] [PubMed] [Google Scholar]
- 6.Toldo S and Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018;15:203–214. [DOI] [PubMed] [Google Scholar]
- 7.Guo H, Callaway JB and Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia Y, Lee K, Li N, Corbett D, Mendoza L and Frangogiannis NG. Characterization of the inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem Cell Biol. 2009;131:471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song X, Kusakari Y, Xiao CY, Kinsella SD, Rosenberg MA, Scherrer-Crosbie M, Hara K, Rosenzweig A and Matsui T. mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am J Physiol Cell Physiol. 2010;299:C1256–C1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salvador AM, Nevers T, Velazquez F, Aronovitz M, Wang B, Abadia Molina A, Jaffe IZ, Karas RH, Blanton RM and Alcaide P. Intercellular Adhesion Molecule 1 Regulates Left Ventricular Leukocyte Infiltration, Cardiac Remodeling, and Function in Pressure Overload-Induced Heart Failure. J Am Heart Assoc. 2016;5:e003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damilano F, Franco I, Perrino C, Schaefer K, Azzolino O, Carnevale D, Cifelli G, Carullo P, Ragona R, Ghigo A, Perino A, Lembo G and Hirsch E. Distinct effects of leukocyte and cardiac phosphoinositide 3-kinase gamma activity in pressure overload-induced cardiac failure. Circulation. 2011;123:391–399. [DOI] [PubMed] [Google Scholar]
- 12.Weisheit C, Zhang Y, Faron A, Kopke O, Weisheit G, Steinstrasser A, Frede S, Meyer R, Boehm O, Hoeft A, Kurts C and Baumgarten G. Ly6C(low) and not Ly6C(high) macrophages accumulate first in the heart in a model of murine pressure-overload. PLoS One. 2014;9:e112710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velazquez F, Aronovitz M, Kapur NK, Karas RH, Blanton RM and Alcaide P. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circ Heart Fail. 2015;8:776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, Delage C, Calise D, Dutaur M, Parini A and Pizzinat N. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. 2014;129:2111–2124. [DOI] [PubMed] [Google Scholar]
- 15.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S and Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. [DOI] [PubMed] [Google Scholar]
- 16.Sandanger O, Ranheim T, Vinge LE, Bliksoen M, Alfsnes K, Finsen AV, Dahl CP, Askevold ET, Florholmen G, Christensen G, Fitzgerald KA, Lien E, Valen G, Espevik T, Aukrust P and Yndestad A. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2013;99:164–174. [DOI] [PubMed] [Google Scholar]
- 17.Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF and Abbate A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A. 2011;108:19725–19730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J Jr., Bers DM and Brown JH. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–919. [DOI] [PubMed] [Google Scholar]
- 19.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D and Brown JH. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreusser MM, Lehmann LH, Keranov S, Hoting MO, Oehl U, Kohlhaas M, Reil JC, Neumann K, Schneider MD, Hill JA, Dobrev D, Maack C, Maier LS, Grone HJ, Katus HA, Olson EN and Backs J. Cardiac CaM Kinase II genes delta and gamma contribute to adverse remodeling but redundantly inhibit calcineurin-induced myocardial hypertrophy. Circulation. 2014;130:1262–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westenbrink BD, Ling H, Divakaruni AS, Gray CB, Zambon AC, Dalton ND, Peterson KL, Gu Y, Matkovich SJ, Murphy AN, Miyamoto S, Dorn GW 2nd and Heller Brown J. Mitochondrial reprogramming induced by CaMKIIdelta mediates hypertrophy decompensation. Circ Res. 2015;116:e28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang T, Guo T, Mishra S, Dalton ND, Kranias EG, Peterson KL, Bers DM and Brown JH. Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiac dysfunction in CaMKIIdelta(C) transgenic mice. Circ Res. 2010;106:354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, Karch J, Gabel S, Farber J, Force T, Brown JH, Murphy E and Molkentin JD. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest. 2010;120:3680–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dewenter M, Neef S, Vettel C, Lammle S, Beushausen C, Zelarayan LC, Katz S, von der Lieth A, Meyer-Roxlau S, Weber S, Wieland T, Sossalla S, Backs J, Brown JH, Maier LS and El-Armouche A. Calcium/Calmodulin-Dependent Protein Kinase II Activity Persists During Chronic beta-Adrenoceptor Blockade in Experimental and Human Heart Failure. Circ Heart Fail. 2017;10:e003840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh MV, Kapoun A, Higgins L, Kutschke W, Thurman JM, Zhang R, Singh M, Yang J, Guan X, Lowe JS, Weiss RM, Zimmermann K, Yull FE, Blackwell TS, Mohler PJ and Anderson ME. Ca2+/calmodulin-dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. J Clin Invest. 2009;119:986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ling H, Gray CB, Zambon AC, Grimm M, Gu Y, Dalton N, Purcell NH, Peterson K and Brown JH. Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ Res. 2013;112:935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weinreuter M, Kreusser MM, Beckendorf J, Schreiter FC, Leuschner F, Lehmann LH, Hofmann KP, Rostosky JS, Diemert N, Xu C, Volz HC, Jungmann A, Nickel A, Sticht C, Gretz N, Maack C, Schneider MD, Grone HJ, Muller OJ, Katus HA and Backs J. CaM Kinase II mediates maladaptive post-infarct remodeling and pro-inflammatory chemoattractant signaling but not acute myocardial ischemia/reperfusion injury. EMBO Mol Med. 2014;6:1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP Jr., Kohler RH, Arlauckas SP, Iwamoto Y, Savol A, Sadreyev RI, Kelly M, Fitzgibbons TP, Fitzgerald KA, Mitchison T, Libby P, Nahrendorf M and Weissleder R. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. 2017;23:1481–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hulsmans M, Sam F and Nahrendorf M. Monocyte and macrophage contributions to cardiac remodeling. J Mol Cell Cardiol. 2016;93:149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hulsmans M, Sager HB, Roh JD, Valero-Munoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, Osborne MT, Hung J, Vinegoni C, Naxerova K, Sosnovik DE, Zile MR, Bradshaw AD, Liao R, Tawakol A, Weissleder R, Rosenzweig A, Swirski FK, Sam F and Nahrendorf M. Cardiac macrophages promote diastolic dysfunction. J Exp Med. 2018;215:423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tschopp J and Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. [DOI] [PubMed] [Google Scholar]
- 32.Grimm M, Ling H, Willeford A, Pereira L, Gray CB, Erickson JR, Sarma S, Respress JL, Wehrens XH, Bers DM and Brown JH. CaMKIIdelta mediates beta-adrenergic effects on RyR2 phosphorylation and SR Ca(2+) leak and the pathophysiological response to chronic beta-adrenergic stimulation. J Mol Cell Cardiol. 2015;85:282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lai NC, Gao MH, Tang E, Tang R, Guo T, Dalton ND, Deng A and Tang T. Pressure overload-induced cardiac remodeling and dysfunction in the absence of interleukin 6 in mice. Lab Invest. 2012;92:1518–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ehrentraut H, Felix Ehrentraut S, Boehm O, El Aissati S, Foltz F, Goelz L, Goertz D, Kebir S, Weisheit C, Wolf M, Meyer R and Baumgarten G. Tlr4 Deficiency Protects against Cardiac Pressure Overload Induced Hyperinflammation. PLoS One. 2015;10:e0142921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li R, Lu K, Wang Y, Chen M, Zhang F, Shen H, Yao D, Gong K and Zhang Z. Triptolide attenuates pressure overload-induced myocardial remodeling in mice via the inhibition of NLRP3 inflammasome expression. Biochem Biophys Res Commun. 2017;485:69–75. [DOI] [PubMed] [Google Scholar]
- 36.Zhao L, Cheng G, Jin R, Afzal MR, Samanta A, Xuan YT, Girgis M, Elias HK, Zhu Y, Davani A, Yang Y, Chen X, Ye S, Wang OL, Chen L, Hauptman J, Vincent RJ and Dawn B. Deletion of Interleukin-6 Attenuates Pressure Overload-Induced Left Ventricular Hypertrophy and Dysfunction. Circ Res. 2016;118:1918–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christia P and Frangogiannis NG. Targeting inflammatory pathways in myocardial infarction. European journal of clinical investigation. 2013;43:986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghigo A, Franco I, Morello F and Hirsch E. Myocyte signalling in leucocyte recruitment to the heart. Cardiovasc Res. 2014;102:270–280. [DOI] [PubMed] [Google Scholar]
- 39.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I and Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng N and Anderson ME. CaMKII is a nodal signal for multiple programmed cell death pathways in heart. J Mol Cell Cardiol. 2017;103:102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimano M, Ouchi N and Walsh K. Cardiokines: recent progress in elucidating the cardiac secretome. Circulation. 2012;126:e327–e32. [DOI] [PubMed] [Google Scholar]
- 42.Toldo S, Mezzaroma E, Mauro AG, Salloum F, Van Tassell BW and Abbate A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid Redox Signal. 2015;22:1146–1161. [DOI] [PubMed] [Google Scholar]
- 43.Xiao H, Li H, Wang JJ, Zhang JS, Shen J, An XB, Zhang CC, Wu JM, Song Y, Wang XY, Yu HY, Deng XN, Li ZJ, Xu M, Lu ZZ, Du J, Gao W, Zhang AH, Feng Y and Zhang YY. IL-18 cleavage triggers cardiac inflammation and fibrosis upon beta-adrenergic insult. Eur Heart J. 2018;39:60–69. [DOI] [PubMed] [Google Scholar]
- 44.Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS and Anderson ME. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491:269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toldo S, Mezzaroma E, O’Brien L, Marchetti C, Seropian IM, Voelkel NF, Van Tassell BW, Dinarello CA and Abbate A. Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. Am J Physiol Heart Circ Physiol. 2014;306:H1025–H1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patel B, Ismahil MA, Hamid T, Bansal SS and Prabhu SD. Mononuclear Phagocytes Are Dispensable for Cardiac Remodeling in Established Pressure-Overload Heart Failure. PLoS One. 2017;12:e0170781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patel BD, Bansal S, Ismahil A, Hamid T, Prabhu SD. Abstract 19521: Macrophages Infiltrating into the Heart Early After Pressure Overload Are Indispensable for the Transition to Chronic Heart Failure. AHA Scientific Session 2016 Abstract. Circulation. 2016;134:A19521 [Google Scholar]
- 48.Heymans S, Hirsch E, Anker SD, Aukrust P, Balligand JL, Cohen-Tervaert JW, Drexler H, Filippatos G, Felix SB, Gullestad L, Hilfiker-Kleiner D, Janssens S, Latini R, Neubauer G, Paulus WJ, Pieske B, Ponikowski P, Schroen B, Schultheiss HP, Tschope C, Van Bilsen M, Zannad F, McMurray J and Shah AM. Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2009;11:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ and Group CT. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 50.Van Tassell BW, Canada J, Carbone S, Trankle C, Buckley L, Oddi Erdle C, Abouzaki NA, Dixon D, Kadariya D, Christopher S, Schatz A, Regan J, Viscusi M, Del Buono M, Melchior R, Mankad P, Lu J, Sculthorpe R, Biondi-Zoccai G, Lesnefsky E, Arena R and Abbate A. Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results From REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ Heart Fail. 2017;10: pii: e004373. doi: 10.1161/CIRCHEARTFAILURE.117.004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.