

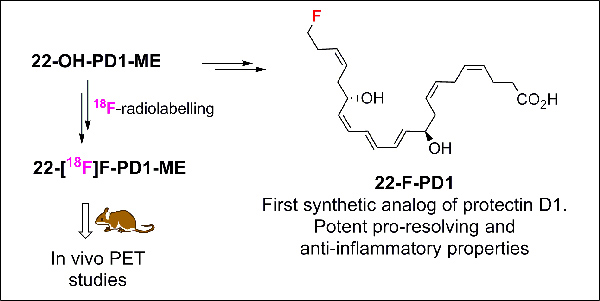
Abstract
Protectin D1 is a specialized pro-resolving mediator with potent pro-resolving and anti-inflammatory effects in vivo in several human disease models. Herein the preparation of the first synthetic analog of protectin D1, named 22-F-PD1, is presented together with data from in vivo investigations. This analog showed potent pro-resolving and anti-inflammatory properties. These results inspired the preparation of the radiotracer 22-[18F]F-PD1-ME that was used in a positron emission tomography proof of concept study. Altogether, the findings presented contribute to new knowledge on the biomolecular properties of protectin D1 analogs. In addition, an improved formal synthesis of the metabolite 22-OH-PD1 is reported.
Graphical Abstract
Introduction
Specialized pro-resolving mediators (SPMs) are enzymatically oxygenated natural products derived from polyunsaturated fatty acids.1 These endogenously formed mediators govern the return to physiology, i.e. catabasis, by acting as potent resolution agonists resulting in pro-resolving and anti-inflammatory effects.2 The SPMs are stereoselectively biosynthesized from arachidonic acid (AA), eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA) and n-3 docosapentaenoc acid (n-3 DPA)3 and participates in the cellular, biochemical and biomolecular mechanisms behind catabasis.4,5 SPMs have potential in immunoresolving pharmacology4 by acting on specific G protein-coupled receptors (GPCRs).6 Protectin D1 (PD1, 1), also named neuroprotectin D1 when isolated from neuron cells,7 has been the subject of numerous biological8 and synthetic studies9 (Figure 1).
Figure 1.

Chemical structures of protectin D1 (1), 22-OH-PD1 (2) and synthetic analogs 3 and 4.
The polyunsaturated di-hydroxylated fatty acid 1 display anti-inflammatory actions in several inflammatory human disease models as a pro-resolving agonist.2,8 PD1 has entered clinical trial development programs against neurodegenerative conditions, such as Alzheimer´s and Parkinson disease.10
PD1 display potent pro-resolution agonist effects11 (EC50 ~ 1 nM, Kd ~31 pmol/mg of cell protein) as well as numerous interesting bioactivities in several human disease models.8 Accordingly, 1 is an interesting lead compound in drug discovery efforts as a biotemplate for the development of new, small molecular anti-inflammatory drugs and immunoresolvents.12 In such endeavors, access to synthetic analogs will provide useful information on structure function and biomolecular relationships.2 These analogs should display potent in vivo pro-resolving and anti-inflammatory agonist effects against GPCRs. As of today, the receptor of 1 remains to be fully elucidated.10,11
Positron emission tomography (PET) is a noninvasive in vivo imaging technique using radiotracers to visualize, characterize and quantify physiological processes at the molecular and receptor level.13 PET is suitable for brain imaging.14 Based on the neuroprotective activities associated with PD1 (1),8 the development of a radiotracer that would mimic the bioactions, bioavailability and physical-chemical characteristic of 1 may provide a better understanding of its neurological functions. PET is also a powerful tool for gaining information on receptor characteristics and interactions.15 Tritium-labeled PD1 showed specific and high-affinity stereoselective binding to retinal pigment epithelial cells as well as high affinity binding with isolated human neutrophils.11 Neither of the SPMs resolvin E1 nor lipoxin A4 competed for this specific binding with human neutrophils rendering support for specific binding of PD1 (1). These findings suggest specific receptors for this SPM in both the immune and in visual systems. Development of a PD1-derived PET radiotracer as well as novel synthetic analogs may aid drug discovery efforts.
Recently, we reported the total synthesis and in vivo biological evaluations of the PD1 ω-oxidation metabolite 22-OH-PD1 (2), see Figure 1.16 These studies revealed that 2 displayed in vivo pro-resolution effects in the nanomolar range towards efferocytosis and phagocytosis, hallmark bioactions associated with SPMs. These results warranted our interest in preparing the synthetic analogs depicted in Figure 1.
The C-22 natural occurring compound 22-OH-PD1 (2), produced in nanogram quantities in vivo,3b,16 exhibits several sensitive structural features. In particular the E,E,Z-triene system, embedded by two chiral allylic alcohols at C-10 and C-17, is a challenge to prepare and handle.9e,17 PD1 (1) exhibit its bioactions in a stereodefined fashion where the geometrical and stereoisomers of PD1 (1) display significantly lower anti-inflammatory and pro-resolution effects.3c,8,18
Results and discussion
Our synthetic investigations commenced by developing a modified and high yielding synthesis of the methyl ester 4 of 22-OH-PD1 (2). Commercially available primary alkyl bromide 5 was first reacted with triphenylphosphine in refluxing acetonitrile affording the Wittig-salt 6. Then, aldehyde 719 was added to a cold solution of the preformed ylide of 6. The resulting bis-protected diol was then treated with TBAF in THF yielding the alkyne 9 in 60% yield over the three steps (Scheme 1). Alkyne 9 was subsequently reacted with known 10,16 followed by deprotection (TBAF, THF) and Boland reduction to afford 11. Analysis of UV, 1H NMR, 13C NMR and MS data allowed the assignment of the E,E,Z-triene in methyl ester 11 (Scheme 1). The chemical yield of 11 was 13% over six steps, compared to 4% over 11 steps from our earlier synthesis.16 The physical and spectral data of 11 were in accordance with literature.16 Overall, these efforts also constitute a new formal synthesis of 2, since saponification of 11 has been reported to yield 2.16
Scheme 1.

Formal synthesis of 22-OH-PD1 (2) and preparation of analogs 3, 4 and [18F]4. a) PPh3, MeCN, Δ, 83%; b) NaHMDS, HMPA, THF, 7, 78%, c) TBAF, THF, 92%; d) (i) Pd(PPh3)4, CuI, 10, Et2NH, 69%; (ii) TBAF, THF, 86%; (iii) Zn(Cu/Ag), MeOH, H2O, 40%; e) TsCl, pyridine, 82%; f) TBA[18F]F, MeCN, HPLC purification; g) KF, Kryptofix222, MeCN, 85%; h) LiOH (1.0 M), THF, H2O, −78 °C.
Towards the development of a mild synthetic protocol for obtaining 3 and 4, several conditions, leaving groups and fluorinating reagents were attempted (Electronic Supplementary Information). The best conditions that retained all sensitive functional groups in preparing 3 and 4 from 11 were tosylation (TsCl, pyridine) that yielded with excellent chemoselectivity the product 12 in 82% isolated yield. The tosylate 12 was treated with KF and Kryptofix222 in MeCN that afforded 22-F-PD1-ME (4) in 83% yield after chromatographic purification. The spectral data of 12 and 4 (UV, 1H NMR, 13C NMR and MS) were in accordance with the assigned structures (Electronic Supplementary Information). The chemical purity of 4 was > 96% (HPLC-analysis). Hydrolysis of the methyl ester in 4 into 3 was performed just prior to in vivo experiments as earlier reported.20 Protectins, including 3, are sensitive to air, light and heat.9e
With multi-milligram amounts of 4 and 11 available, in vivo studies were conducted. During the acute phase of inflammation, circulating polymorphonuclear cells (PMNs) extravasate into the inflamed tissue.21 Inhibition of PMN infiltration as well as a decrease of pro-inflammatory mediators are key processes in the resolution of inflammation and are defining actions of SPMs.2 Hence, mice were administered vehicle, PD1 (1) (100 ng/mouse) and 22-F-PD1 (3) (100 ng/mouse) via intraperitoneal injection (i. p.) 10 min after mice were infected with E. coli (1 × 105 c. f. u. delivered i. p.). After a 12 hour post-infection period, exudates were collected and analyzed. The results are presented in Figure 2. The administration of 3 resulted in a significant reduction of neutrophil recruitment comparable to those displayed by PD1 (1) (Figure 2A).
Figure 2.

22-F-PD1 decreases PMN recruitment in vivo. A) Total PMN counts in peritoneal lavages. B) Quantification of macrophage phagocytosis of E. coli in the peritoneum. C) Quantification of E. coli colony forming units (c. f. u.) present in the peritoneal cavity. Results shown as mean ± SEM, n = 4 mice per group. Statistical significance was defined * p ≤ 0.05, ** p ≤ 0.01 using one-way ANOVA with a Tukey’s post-test.
Moreover, experiments using 100 ng of 3 revealed an increase in macrophage phagocytosis of fluorescence labeled yeast cell wall particles (Figure 2 B). Similar effects were also observed for PD1 (1). Quantification of E. coli colony forming units present in the peritoneal cavity was also performed, showing that compound 3 was able to reduce the number of colonies as efficient as PD1 (1) (Figure 2C).
In addition, 22-F-PD1 (3) potently, at concentrations in the pico- to nanomolar range, increased macrophage efferocytosis of apoptotic human neutrophils (Figure 3). The trend for a bell-shaped dose-response curve was obtained with isolated human cells. Overall, these results verified that 3 exhibited both potent anti-inflammatory and pro-resolving actions similar to PD1. Macrophage phagocytosis and efferocytosis are both key pro-resolving biological actions of interest in drug discovery and development.2,8,12
Figure 3.

22-F-PD1 (3) enhances human macrophage efferocytosis of apoptotic PMN. Human macrophages were incubated with either vehicle control (PBS+/+, EtOH 0.01%), PD1 (1) or 22-F-PD1 (3) for 15 min at 37 °C, followed by fluorescent-labeled apoptotic PMN at a 5:1 ratio of PMN:macrophage for 60 min. Results showed as ± SEM, n = 6 independent experiments. Results were compared to vehicle control using a paired t-test, statistical significance defined as * p ≤ 0.05, ** p ≤ 0.01.
Pleased with the synthetic and biological results outlined above, we next attempted the synthesis of the radiotracer 22-[18F]F-PD1. Unfortunately, all attempts to prepare 22-[18F]F-PD1 (3) failed, owing to the extreme chemical instability of the precursor 3 and the the general incompatibility of carboxylic acid residues with standard 18F-fluorination conditions.13 Hence, the ester [18F]4 was investigated as a PET tracer since methyl esters of PUFAs, including SPMs, are rapidly hydrolyzed in vivo to their free fatty acids.1,22 Gratifyingly, treatment of the tosylate 12 in acetonitrile with TBA[18F]F, the latter obtained after azeotropic drying of [18F]fluoride released with aqueous tetra-butylammonium bicarbonate from a Chromabond PS-HCO3 cartridge,23 afforded 22-[18F]F-PD1-ME (4) (Scheme 1). The radiochemical purity of the final product was determined by radio-HPLC analysis. The chemical identity was verified by co-injection of collected radioactive material (specific activity > 50 GBq/μmol) and non-radioactive 4 (See Figure S21 in the Electronic Supplementary Information). Then, as a proof-of-concept, 4–5 MBq of [18F]4 was administrated to mice via a tail vein injection. For dynamic PET scans, mice were prepositioned in the microPET scanner under isoflurane anesthesia, injected with tracers, and imaged continuously from time 0 to 120 min. The results are depicted in Figure 4.
Figure 4.

In vivo PET-experiments with [18F]4 and mouse brain. Times specified A-D is averaged activity reconstructed from the acquired list mode data.
As seen from Figure 4A, a rapid and robust extraction into brain with [18F]4 was observed five minutes into the dynamic PET experiment (Figure 4A) followed by rapid wash-out reaching equilibrium after 35 min (Figure 4B). A slowly increasing signal, albeit low, from bone structures indicate some release of [18F]fluoride (Figure 4C). In these present conditions, direct evidence of specific binding in brain or other organs was not observed, even when PD1 (1) was used in blocking experiments (See Figure S22 in Electronic Supplementary Information). Hence, further investigations are deemed necessary to clarify the in vivo stability and specific binding of [18F]4 and its free acid.
Conclusions
In conclusion, the potent pro-resolving agonist PD1 (1) and the ω−22 oxidation metabolite product of 1, i.e. 22-OH-PD1 (2), were used as biotemplates for the preparation of the synthetic analogs. An in vivo proof of concept study with [18F]4 a rapid uptake in the central nervous system (CNS) enabled PET-visualization in mice, while the synthetic analog 22-F-PD1 (3) displayed key pro-resolving bioactions similar to PD1 (1). Tritium labeled C15-C16 protectin D1 provided the first evidence for specific high affinity receptors in retinal epithelial pigment cells and human neutrophils.11 The novel PET-tracer 22-[18F]F-PD1-ME (4) did not display any specific binding in naive mice. However, the synthetic and biological results presented provide new knowledge on the biomolecular mechanisms and structure function of analogs of SPMs. Development of new immunoresolvents, including those derived from SPMs, are of current interest in drug discovery.8,12 Recent evidence has been provided for the crosstalk between the CNS and leukocytes in regulating SPM biosynthesis,24 where the novel analog 22-F-PD1 (3) will be of interest. Herein we have also presented a greatly improved formal total synthesis of 22-OH-PD1 (2) with respect to yield and number of steps. This SPM will be used for future structure function relationship studies that will be reported in due time.
Experimental
Methyl (4Z,7Z,10R,11E,13E,15Z,17S,19Z)-10,17-dihydroxy-22-(tosyloxy)docosa-4,7,11,13,15,19-hexaenoate (12)
The alcohol 11 was prepared according to literature.16 To a solution of alcohol 11 (6 mg, 0.015 mmol, 1.0 equiv) in pyridine (0.2 mL), TsCl (3.5 mg, 0.018 mmol, 1.2 equiv.) was added and the reaction was stirred for 16 h. The mixture was quenched with saturated aq. NH4Cl (1.5 mL) and EtOAc (1.5 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (2 × 2 mL). The combined organic layers were dried (Na2SO4) and then the bulk of the solvent was carefully removed in vacuo. The crude product was purified by column chromatography on silica (hexanes/EtOAc/MeOH 58:40:2) to afford the title compound as a colorless oil. Yield: 7 mg (82%). The chemical purity (>94%) was determined by HPLC analysis (Eclipse XDB-C18, MeOH/H2O, 7:3, 1.0 mL/min): tr = 12.25 min; TLC (hexanes/EtOAc/MeOH 58:40:2, CAM stain): Rf = 0.53; [α] = - 17 (c = 0.11, MeOH); 1H NMR (300 MHz, MeOH) δ 7.78 (d, J = 8.3 Hz, 2H), 7.44 (d, J = 8.6 Hz, 2H), 6.59 – 6.46 (m, 1H), 6.36 – 6.18 (m, 2H), 6.06 (t, J = 10.9 Hz, 1H), 5.75 (dd, J = 14.4, 6.7 Hz, 1H), 5.56 – 5.27 (m, 7H), 4.61 – 4.51 (m, 1H), 4.14 (q, J = 6.5 Hz, 1H), 4.02 (t, J = 6.6 Hz, 2H), 3.65 (s, 3H), 2.87 – 2.73 (m, 2H), 2.46 (s, 3H), 2.41 – 2.11 (m, 10H). 13C NMR (75 MHz, MeOH) δ 175.3, 146.4, 138.0, 135.1, 134.7, 134.6, 131.5, 131.1 (2C), 131.0, 130.7, 130.3, 129.7, 129.0 (2C), 129.0, 128.9, 126.7, 126.5, 73.0, 71.2, 68.3, 52.1, 36.5, 36.3, 34.8, 28.3, 26.7, 23.8, 21.6. HRMS (TOF ES+): Exact mass calculated for C30H40NaO7S [M+Na]+: 567,2387, found 567.2392.
22-F-PD1-ME (4)
To a solution of tosylate 12 (2.5 mg, 0.005 mmol, 1.0 equiv) in MeCN (0.1 mL), was added Kryptofix222 (K222) (3.8 mg, 0.01 mmol, 2 equiv.) and KF (0.6 mg, 0.01 mmol, 2.0 equiv) and the reaction mixture was stirred for 1h. The solution was filtered through a pad of Celite and the resulting filtrate was concentrated in vacuo. The crude product was purified by column chromatography on silica (hexanes/EtOAc/MeOH 54.5:45:0.5) to afford the title compound as a colorless oil. Yield: 1.5 mg, 83%. The chemical purity (>96%) was determined by HPLC analysis (Eclipse XDB-C18, MeOH/H2O, 7:3, 1.0 mL/min): tr = 9.27 min; TLC (hexanes/EtOAc/MeOH 59.5:40:0.5, CAM stain): Rf = 0.23; [α] = - 22 (c = 0.12, MeOH); 1H NMR (600 MHz, MeOD) δ 6.55 (dd, J = 13.9, 11.4 Hz, 1H), 6.34 – 6.23 (m, 2H), 6.10 (t, J = 11.2 Hz, 1H), 5.78 (dd, J = 14.6, 6.5 Hz, 1H), 5.60 – 5.34 (m, 7H), 4.64 – 4.57 (m, 1H), 4.46 (t, J = 6.4 Hz, 1H), 4.38 (t, J = 6.4 Hz, 1H), 4.19 – 4.12 (m, 1H), 3.67 (s, 3H), 2.88 – 2.77 (m, 2H), 2.56 – 2.22 (m, 10H). 13C NMR (151 MHz, MeOD) δ 175.3, 138.0, 135.0, 134.7, 131.4, 130.9, 130.6, 130.3, 129.1, 128.9, 128.9, 127.3 (d, 3JCF = 6.5 Hz), 126.5, 84.1 (d, 1JCF = 166.9 Hz), 73.0, 68.4, 52.1, 36.6, 36.3, 34.8, 30.0 (d, 2JCF = 20.5 Hz), 26.7, 23.8. HRMS (TOF ES+): Exact mass calculated for C23H33FNaO4 [M+Na]+: 415.2255, found 415.2260.
(4Z,7Z,10R,11E,13E,15Z,17S,19Z)-22-Fluoro-10,17-dihydroxydocosa-4,7,11,13,15,19-hexaenoic acid (3)
A stock solution of ester 4 (10 μg) in EtOH was dried under a gentle stream of nitrogen and then dissolved in THF (500 μL) and cooled to −78 °C using a dry ice/isopropanol cooling bath. Then aqueous 1.0 M LiOH (100 μL) was slowly added via a Hamilton syringe at −78 °C. Additional H2O (100 μL) was added and the vial was covered with aluminium foil and left stirring for 10 h. From the solution above the precipitate, the lithium salt was gently removed by using a syringe. Next, the THF-solution was dried under a gentle stream of nitrogen before quantification using UV (EtOH) λmax 262 (log ε 3.51), 271 (log ε 4.12), 282 (log ε 2.64) nm. The chemical purity was >98% (HPLC-analysis). After quantification and removal of THF under a gentle stream of nitrogen, 3 was suspended in 50 μL PBS+/+ pH = 7.45. The PBS solution was kept on solid dry ice in a closed container prior to use in incubation experiments.
Microbial-initiated peritonitis
FVB male mice (6–8 weeks old, Charles River Laboratories, Wilmington, MA) were fed ad libitum Laboratory Rodent Diet 20–5058 (Lab Diet, Purina Mills, Great Summit, MO). E. coli (serotype O6:K2:H1) was cultured in Luria-Bertani (LB) broth, collected at mid-log phase (OD600 nm ≈ 0.5; 5 × 108 CFU/mL). Mice were administered vehicle (saline containing 0.01% EtOH), 22-F-PD1 (3) (100 ng/mouse) or PD1 (1) (100 ng/mouse) via intraperitoneal injection (i. p.). 10 min after mice were infected with E. coli (1 × 105 c. f. u. delivered i. p.) peritoneal lavage was performed up to 12 h post infection. Bacterial clearance was measured by serially diluting exudates onto LB agar plates and cultured overnight at 37 oC. PMN were enumerated using light microscopy and flow cytometry analysis. Cells were surface-stained with antibodies against CD11b (clone M1/70; eBioscience, San Diego, CA), F4/80 (clone BM8; eBioscience, San Diego, CA) and Ly6G (clone RB6–8C; eBioscience, San Diego, CA). Cells were also intracellular stained with anti-E coli antibody (clone GTX408556; GeneTex, Irvine, CA). Fluorescence minus one (FMO) controls were included during staining procedures. Cell staining was evaluated using a FACSCanto II flow cytometer (BD Biosciences, San Jose, CA) and results were analyzed using FlowJo software (Tree Star, Ashland, OR). All mouse experiments were approved by the Standing Committee on Animals of Harvard Medical School (Protocol 02570) and complied with institutional and US National Institutes of Health guidelines.
Macrophage efferocytosis
Monocytes were isolated from leukopacks obtained from Children’s Hospital Blood Bank (Boston, MA) using Ficoll-Histopaque 1077–1 (Sigma Aldrich, St. Louis, MO). Macrophage differentiation was induced over 7 days of culture with GM-CSF (10 ng/mL, R&D Systems, Minneapolis, MN) in RMPI 1640 (Life Technologies, Carlsbad, CA) supplemented with 10% FBS (Invitrogen, Grand Island, NY), 2 mM L-glutamine (Lonza, Basel, Switzerland) and Penicillin-Streptomycin (Lonza, Basel, Switzerland). Before each experiment (24 h), macrophages were adhered on a 96-well plate (5 × 104 cells/well) in culture medium deprived of GM-CSF. Macrophages were incubated with vehicle (PBS with 0.01% ethanol by volume) alone, PD1 (1) or 22-F-PD1 (3) (15 min at 37 °C). Next, fluorescently labeled apoptotic PMN (Bisbenzimide H 33342, Sigma Aldrich, St. Louis, MO; 10 ng/mL) were added to macrophages (1:3 macrophage:PMN; 1 h at 37 °C). Plates were gently washed, extracellular fluorescence quenched using trypan blue, and fluorescence was measured on a SpectraMax M3 plate reader (Molecular Devices Inc, Sunnyvale, CA).
Radiochemistry and Radiosynthesis of 22-[18F]F-PD1-ME (4)
Isotope production and radiochemistry were performed at Norwegian Medical Cyclotron Center (Gaustad, Oslo) [18F]Fluoride was produced on GE PETtrace 6 cyclotron via the 18O(p,n)18F reaction and delivered as [18F]fluoride in 18O-enriched-water. Radiosynthesis and azeotropic drying were performed on a Scansys radiochemistry module (Værløse, Denmark). Radio-HPLC analysis was performed using an Agilent 1200 coupled in series with a Gabi star NaI-radiodetctor (Raytest, Straubenhardt, Germany). [18F]Fluoride (10–20 GBq) was separated from 18O-enriched-water using a Chromafix PS-HCO3 (45 mg) and subsequently released into a 3 mL glass V-vial using 600 μL solution of water/MeCN (1:1) containing 30 μL 0.75 M aqueous tetra-butylamminium bicarbonate (TBAHCO3). The solution was dried with two cycles of azeotropic drying with MeCN (600 μL) under a flow of He at 105 °C. The dried [18F]FTBA residue was redissolved in anhydrous MeCN (300 μL) containing 1 mg of tosylate precursor 12. The sealed vial was heated at 65 °C for 30 minutes. The reaction mixture was quenched by addition of water (1.7 mL) and loaded onto a semi-prep column (Phenomenex Onyx Monolithic C18, 100 × 100 mm), flow 4.0 mL/min, using 45% (v/v) ethanol in water as mobile phase. Isolated non-decay corrected radiochemical yields ranged between 8 to 20% (n = 4). For in vivo studies, the collected fraction typically 2 mL, was diluted with 10 mL isotonic saline and passed through a 0.22 μm filter into sealed sterile vial. The total time for synthesis including purification was 70 min. The radiochemical purity and product identity was determined by radio-HPLC analysis (Figure S21 in the Electronic Supporting Information). Analysis was performed using an isocratic eluent (MeOH/H2O, 70:30) with an ACE C18-AR column (3 μm, 50 × 4.6 mm) at a flow rate 1.0 mL/min.
Supplementary Material
Acknowledgements
We are thankful for skilful expertise and technical assistance from Hong Qu, Institute of Basic Medical Sciences, Faculty of Medicine, University of Oslo. The Norwegian Research Council is gratefully acknowledged for generous funding to T.V.H (FRIPRO-FRINATEK 230470). C.N.S. is supported by the National Institutes of Health GM Grant PO1GM095467.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: [Additional experimental procedures and characterization data, 1H-, 13C-NMR, HRMS and UV/VIS spectra as well as chromatograms of HPLC analyses.]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Serhan CN, Petasis NA, Chem. Rev, 2011, 111, 5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Serhan CN, Nature, 2014, 510, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tabas I, Glass CK Science, 2013, 339, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Serhan CN, Hamberg M, Samuelsson B. B, Proc. Natl. Acad. Sci. U. S. A, 1984, 81, 5335. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert J K. Exp. Med, 2000, 192, 1197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac RL, J. Exp. Med, 2002, 196, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN, J. Biol. Chem 2003, 278, 14677. [DOI] [PubMed] [Google Scholar]; (e) Ariel A, Pin-Lan L, Wang W, Tang WX, Fredman G, Hong S, Gotlinger KH, Serhan CN, J. Biol. Chem, 2005, 280, 43079. [DOI] [PubMed] [Google Scholar]; (f) Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS, Porter TF, Oh SF, Spite M, J. Exp. Med 2009, 206, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Dalli J, Colas RA, Serhan CN, Sci. Rep, 2013, 3, 1940. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Dalli J, Zhu M, Vlasenko NA, Deng B, Haeggstrӧm JZ, Petasis NA, Serhan CN, FASEB J., 2013, 27, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Aursnes M, Tungen JE, Colas R, Vlasakov I, Dalli J, Serhan CN, Hansen J TV. Nat. Prod, 2015, 78, 2924. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Primdahl KG, Tungen JE, Walker ME, Colas RA, Dalli J, Hansen J,TV, Vik A, Org. Biomol. Chem 2017, 15, 8606. [DOI] [PubMed] [Google Scholar]; (k) Vik A, Dalli J, Hansen Bioorg TV. Med. Chem. Lett, 2017, 27, 2259. [DOI] [PubMed] [Google Scholar]
- 4.Serhan CN, Annu. Rev. Immunol, 2007, 25, 101. [DOI] [PubMed] [Google Scholar]
- 5.Serhan CN, Chiang N,N, Van Dyke TE, Nat. Rev. Immunol 2008, 8, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serhan CN, Chiang N, Curr. Opin. Pharmacol 2013, 13, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukherjee PK, Marcheselli VL, Serhan CN, Bazan NG, Proc. Natl. Acad. Sci. U.S.A 2004, 101, 8491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serhan CN, FASEB J, 2017, 31, 1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Ogawa N, Kobayashi Y, Tetrahedron Lett. 2011, 52, 3001 [Google Scholar]; (b) Petasis NA, Yang R, Winkler JW, Zhu M, Uddin J, Bazan JNG, Serhan CN, Tetrahedron Lett. 2012, 53, 1695. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Aursnes M, Tungen JE, Vik A, Dalli J, Hansen TV, T. V., Org. Biomol. Chemistry 2014, 12, 432. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rodriguez AR, Spur BW Tetrahedron Lett. 2014, 55, 6011 [Google Scholar]; (e) Hansen TV, Dalli J, Serhan CN, Prostaglandins Other Lipid Mediat. 2017, 133, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duvall MG, Levy BD. Eur J Pharmacol, 2016, 785, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marcheselli VL, Mukherjee PK, Makto A, Hong S, Antony R, Sheets K, Winkler JW, Bazan NG, Serhan CN, Prostaglandins Leukot. Essent. Fatty Acids 2010, 82, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fullerton JN, Gilroy DW, Nat. Rev. Drug Discov, 2016, 15, 551. [DOI] [PubMed] [Google Scholar]
- 13.Long N, T- Wong W (Ed.) The Chemistry of Molecular Imaging, 2015, Hoboken, New Jersey, Wiley, ISBN-978–1-118–09327. [Google Scholar]
- 14.Nasrallah I, Dubroff J Semin. Nuc. Med 2013, 43, 449. [DOI] [PubMed] [Google Scholar]
- 15.Krishnan HS, Ma L, Vasdev N, Liang Chem SH. Eur. J, 2014, 20,14575 and references cited therein. [Google Scholar]
- 16.Tungen JE, Aursnes M, Vik A, Ramon S, Colas RA, Dalli J, Serhan CN, Hansen TV, J. Nat. Prod 2014, 77, 2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nolsøe JM, Aursnes M, E Tungen J, Hansen TV, J. Org. Chem, 2015, 80, 5377. [DOI] [PubMed] [Google Scholar]
- 18.Serhan CN, Gotlinger K, Hong S, Lu Y, Siegelman J, Baer T, Yang R, Colgan SP, Petasis NA, Immunol J. 2006, 176, 1848. [DOI] [PubMed] [Google Scholar]
- 19.Tungen JE, Aursnes M, Dalli J, Arnardottir CN, Serhan H,CN, Hansen TV, Chem. Eur. J, 2014, 20,14575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Tungen JE, Aursnes M, Hansen TV, Tetrahedron Lett, 2015, 56, 1843 [Google Scholar]; (b) Aursnes M, Tungen JE, Vik A, Colas RA, Cheng C-YC, Dalli J, Serhan CN, Hansen TV, J. Nat. Prod 2014, 77, 910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janeway CA, Travers P, Walport M, Shlomick MJ 2005, Immunobiology: The immune system in health and disease (6th ed.). New York: Garland Science Publishing; pp. 76–84. [Google Scholar]
- 22.Saghir M, Werner J, Laposata M, Am. J. Physiol 1997, 273, G184. [DOI] [PubMed] [Google Scholar]
- 23.Brichard L, Aigbirhio FI, Eur. J. Org. Chem 2014, 28, 6145. [Google Scholar]
- 24.Dalli J, Serhan CN, Curr. Opin. Immunol 2017, 50, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.