

Abstract
The Federal Pain Research Strategy recommended development of non-opioid analgesics as a top priority in its strategic plan to address the significant public health crisis and individual burden of chronic pain facing >100 million Americans. Motivated by this challenge, a natural product extracts library was screened and identified a plant extract that targets activity of voltage-gated calcium channels. This profile is of interest as a potential treatment for neuropathic pain. The active extract derived from the desert lavender plant native to southwestern United States, when subjected to bioassay-guided fractionation afforded three compounds identified as pentacyclic triterpenoids, betulinic acid (BA), oleanolic acid, and ursolic acid. BA inhibited depolarization-evoked calcium influx in dorsal root ganglion (DRG) neurons predominantly through targeting low-voltage gated (Cav3 or T-type) and CaV2.2 (N-type) calcium channels. Voltage clamp electrophysiology experiments revealed a reduction of Ca2+, but not Na+, currents in sensory neurons following BA exposure. BA inhibited spontaneous excitatory post synaptic currents and depolarization-evoked release of calcitonin gene-related peptide (CGRP) from lumbar spinal cord slices. Notably, BA did not engage human mu, delta, or kappa opioid receptors. Intrathecal administration of BA reversed mechanical allodynia in rat models of chemotherapy-induced peripheral neuropathy (CIPN) and HIV-associated peripheral sensory neuropathy as well as a mouse model of partial sciatic nerve ligation without effects on locomotion. The broad-spectrum biological and medicinal properties reported, including anti-HIV and cancer activities of BA and its derivatives, position this plant-derived small molecule natural product as a potential non-opioid therapy for management of chronic pain.
Keywords: Desert lavender extract, Betulinic acid, T-type calcium channels, Cav3.2/3.3, non-opioid, HIV-associated sensory neuropathy, paclitaxel-induced peripheral neuropathy
Summary
Betulinic acid, derived from the desert lavender, targets voltage-activated T- and N-type calcium channels to curb neuropathic pain.
1. Introduction
Chronic pain affects about 20–30% of the population worldwide [33]. In 2014, pain ranked 3rd in global therapy areas accounting for US prescription sales of $59.7 billion [47]. Opioids are the cornerstone of pain management for many types of pain [20; 31; 85]. However, opioids are only partially effective in some pain types and patient populations, including neuropathic pain, and are often associated with serious side effects - including tolerance, addiction, and death that limit their clinical utility [3]. In February 2018, the International Association for the Study of Pain stated “Opioids are indispensable for the treatment of severe short-lived pain” and “Currently, no other oral medication offers immediate and effective relief of severe pain”. This highlights an urgent need for developing novel therapeutic strategies for the treatment of chronic pain.
Clinically used medications either originate from the repurposing of drugs initially designed for depression and epilepsy (gabapentin [17], amitriptyline [2]) or are natural products like morphine and ziconotide [82]. In this context, it is noteworthy that the first opioid was not synthetic but derived from plants. We thus investigated if using plant extracts could uncover novel natural compounds beneficial for pain. We screened 88 small molecules extracted from plants for their ability to inhibit voltage gated Ca2+ channels (a downstream target of opioids [8]) in dorsal root ganglia (DRG) sensory neurons. This led us to identify betulinic acid from Hyptis emoryi as a novel natural inhibitor of low-voltage activated Ca2+ channels.
H. emoryi (Lamiaceae), commonly known as desert lavender, is a medium to tall tender perennial shrub found in washes and canyons of the southwestern United States in Arizona, Nevada, Colorado, Mojave, California, and northwestern Mexico in Sonora and Baja California [58]. This plant was reported to act as an anesthetic to the esophagus and stomach and help with irritated stomach linings [53]. Previous studies have identified terpenes, flavonoids, lignans, α-pyran derivatives and fatty acids from Hyptis species [19; 24; 43; 62]. Investigation of the essential oil derived from H. emoryi has led to the identification of 34 volatile components [75] whereas investigation of the aerial parts of the plant has afforded betulinic acid [70].
In this study, we discovered that betulinic acid from H. emoryi is a novel natural inhibitor of N- and T-type voltage gated Ca2+ channels. We further characterized this small molecule as a preferential inhibitor for Cav3.2 and Cav2.2. Betulinic acid had no inhibitory effect on voltage gated Na+ channels and did not show any binding to opioid receptors. We found that betulinic acid treatment resulted in inhibition of miniature excitatory post synaptic currents (mEPSCs) and of depolarization-evoked neurotransmitter release. Finally, we identified the antinociceptive potential of betulinic acid in preclinical rat models of chemotherapy induced peripheral neuropathy and HIV-sensory neuropathy as well as a mouse model of partial sciatic nerve ligation without effects on locomotion. Our results show the unexplored potential of small molecule natural plant extracts for chronic pain.
2. Methods
2.1. Animals.
Pathogen-free, adult male and female Sprague-Dawley rats (225–250g; Envigo) or female C57BL/6NHsd (22g; Envigo) were housed in temperature-controlled (23±3˚C) and light-controlled (12-h light/12-h dark cycle; lights on 07:00–19:00) rooms with standard rodent chow and water available ad libitum. The Institutional Animal Care and Use Committee of the College of Medicine at the University of Arizona approved all experiments. All procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health and the ethical guidelines of the International Association for the Study of Pain. Animals were randomly assigned to treatment or control groups for the behavioral experiments. Animals were initially housed 3 per cage but individually housed after the intrathecal cannulation on a 12-hour light-dark cycle with food and water ad libitum. All behavioral experiments were performed by experimenters who were blinded to the experimental groups and treatments.
2.2. Materials
All chemicals, unless noted were purchased from Sigma (St. Louis, MO).
2.3. Analytical Instrumentation, Extraction, and Isolation of Betulinic Acid.
2.4. Preparation of acutely dissociated dorsal root ganglion neurons.
Dorsal root ganglia from all levels were acutely dissociated using methods as described previously [28]. Rat DRG neurons were isolated from 150 to 174 g Sprague-Dawley rats using previously developed procedures [54]. In brief, removing dorsal skin and muscle were removed and vertebral bone processes parallel to the dissection stage-exposed DRG were cut. Dorsal root ganglia were then collected, trimmed at their roots, and enzymatically digested in 3 mL bicarbonate-free, serum-free, sterile DMEM (Cat# 11965, Thermo Fisher Scientific) solution containing neutral protease (3.125 mg.ml-1, Cat#LS02104; Worthington, Lakewood, NJ) and collagenase type I (5 mg/mL, Cat# LS004194, Worthington, Lakewood, NJ) and incubated for 60 minutes at 37˚C under gentle agitation. Dissociated DRG neurons (~1.5 × 106) were then gently centrifuged to collect cells and washed with DRG media (DMEM containing 1% penicillin/streptomycin sulfate from 10,000 μg/mL stock, 30 ng/mL nerve growth factor, and 10% fetal bovine serum (Hyclone)) before plating onto poly-D-lysine– and laminin-coated glass 12- or 15-mm coverslips. All cultures were used within 48 hours.
2.5. Calcium imaging in acutely dissociated dorsal root ganglion (DRG) neurons.
Dorsal root ganglion neurons were loaded for 30 minutes at 37˚C with 3 µM Fura-2AM (Cat# F1221, Thermo Fisher, stock solution prepared at 1mM in DMSO, 0.02% pluronic acid, Cat#P-3000MP, Life Technologies) to follow changes in intracellular calcium([Ca2+]c) in a standard bath solution containing 139 mM NaCl, 3 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 10 mM Na HEPES, pH 7.4, 5 mM glucose, exactly as previously described [9]. Fluorescence imaging was performed with an inverted microscope, NikonEclipseTi-U (Nikon Instruments Inc., Melville, NY), using objective Nikon Fluor 4X and a Photometrics cooled CCD camera Cool SNAP ES2 (Roper Scientific, Tucson, AZ) controlled by Nis Elements software (version 4.20, Nikon Instruments). The excitation light was delivered by a Lambda-LS system (Sutter Instruments, Novato, CA). The excitation filters (340 ± 5 nm and 380 ± 7 nm) were controlled by a Lambda 10 to 2 optical filter change (Sutter Instruments). Fluorescence was recorded through a 505-nm dichroic mirror at 535 ± 25 nm. To minimize photobleaching and phototoxicity, the images were taken every ~10 seconds during the time-course of the experiment using the minimal exposure time that provided acceptable image quality. The changes in [Ca2+]c were monitored by following a ratio of F340/F380, calculated after subtracting the background from both channels.
2.5.1. Dorsal root ganglia neuron transfection.
Collected cells were re-suspended in Nucleofector transfection reagent containing siRNA at 500 nM and 2 µg of the provided GFP plasmid as detailed previously [21]. Then, cells were subjected to electroporation protocol O-003 in an Amaxa Biosystem (Lonza, Basel, Switzerland) and plated onto poly-D-lysine - and laminin-coated glass 12-mm coverslips. Transfection efficiencies were routinely between 20% and 30% with ~10% cell death. Small diameter neurons were selected to target Aδ- and c- fiber nociceptive neurons. For rat DRG culture small cells were considered to be ~ < 30 µm as determined by an eyepiece micrometer within the objective lens. Successfully transfected cells were identified by GFP fluorescence. The siRNA sequences used were: CAGCCAUCUUCGUGGUGGAGAUGAU (for Cacna1h/Cav3.2; (Cat# RSS350286, Thermofisher)); CAGCAUCCUUGGGAUGCAUAUCUUU (for Cacna1i/Cav3.3; Cat# RSS367566); and siRNA Negative Control, Med GC was used as a scrambled siRNA control (Cat# 12935300).
2.5.2. Constellation pharmacology.
These experiments were performed as described previously [54; 77], but with the following modifications. Dorsal root ganglia neurons were loaded at 37˚C with 3 µM Fura-2AM for 30 minutes in Tyrode’s solution (at ~310 mOsm) containing 119 mM NaCl, 2.5 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 25 mM HEPES, pH 7.4, and 30 mM glucose. After a 1-minute baseline measurement, Ca2+ influx was stimulated by the addition of the following receptor agonists: 400 nM menthol, 50 µM histamine, 10 µM adenosine triphosphate (ATP), 200 µM allyl isothiocyanate (AITC), 1 mM acetylcholine (ACh), and 100 nM capsaicin diluted in Tyrode’s solution. At the end of the constellation pharmacology protocol, cell viability was assessed by depolarization-induced Ca2+ influx using an excitatory KCl solution comprised of 32 mM NaCl, 90 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 25 mM HEPES, pH 7.4, and 30 mM glucose. After the 1-minute baseline measurement, each trigger was applied for 15 seconds in the order indicated above in 6-minute intervals. After each trigger, bath solution was continuously perfused over the cells to wash off excess of the trigger. This process was automated using the ValveBank II perfusion system that controlled the perfusion of the standard bath solution and triggers (Automate Scientific). For the BA condition, DRGs were incubated overnight with a 20 μM concentration of the compound. Fluorescence imaging was performed under the same conditions noted above for calcium imaging. A cell was defined as a “responder” if its fluorescence ratio of 340 nm/380 nm was greater than 10% of the baseline value calculated using the average fluorescence in the 30 seconds preceding application of the trigger.
2.6. Semi quantitative real time polymerase chain reaction.
Semi quantitative real time polymerase chain reaction was performed as described previously [10; 55]. After transfection with the indicated siRNA, RNA was extracted from cultured DRGs using Tri reagent (Cat# TR118, MRC, Cincinnati, OH) according to the manufacturer’s protocol. Briefly, tri reagent was added to the DRGs. The homogenates were centrifuged at 5000 x g for 5 min at 4°C to eliminate insoluble materials and chloroform was added to the supernatants at 20 % (v/v). After centrifugation at 12000xg, 15 min, 4°C, the upper, RNA containing, aqueous fraction was harvested in a new tube and the total RNA precipitated by adding isopropanol (50 %, v/v). The precipitates were centrifuged at 20000xg, 10 min, 4°C then the RNA pellet was washed twice with 75% ethanol. Finally purified RNA was resuspended in ultra-pure water and stored at −20°C until analysis. To generate cDNA, 2 µg of purified RNA was retrotranscribed using Maxima Reverse Transcriptase (Cat# EP0743, Thermo Fisher) according to manufacturer’s instructions. qRT-PCR analysis was performed using 5X HOT FIREPol Evagreen (Cat# 08–24-00020, Solis Biodyne, Estonia) on a CFX connect (Biorad, Hercules, CA) according to manufacturer’s instructions. Validated primer sequences used were from [12]: Cav3.2 (Forward: GAGTGTGCCTTGCCCCCTG, Reverse GGTGGCCTATCCCTCCTG) and Cav3.3 (Forward: CCATCAGCGTAGCCACAGCA, Reverse GCTGAGGAGCCCAAGCCT). L27 ribosomal RNA expression (Forward: ATCGCCAAGCGATCCAAGAT, Reverse ACAGAGTACCTTGTGGGCA) was measured as a housekeeping gene to normalize target mRNA expression between animal samples. The mRNA level for each gene (Cav3.x) relative to L27 mRNA (internal control) was calculated using the ΔΔCt method [48].
2.7. Whole-cell patch recordings of Ca2+ currents in acutely dissociated DRG neurons.
Recordings were obtained from acutely dissociated DRG neurons as described previously [32; 56]. To isolate calcium currents, Na+ and K+ currents were blocked with 500 nM tetrodotoxin (TTX; Alomone Laboratories) and 30 mM tetraethylammonium chloride (TEA-Cl; Sigma). To isolate T-type specific calcium currents, the following compounds were used: SNX482 (200 nM, R-type voltage-gated Ca2+ channel blocker), ω-conotoxin GVIA (500 nM, N-type voltage-gated Ca2+ channel blocker), ω-agatoxin (200 nM, P/Q-type voltage-gated Ca2+ channel blocker), and Nifedipine (10 µM, L-type voltage-gated Ca2+ channel blocker). Extracellular recording solution (at ~310 mOsm) consisted of the following (in mM): 110 N-methyl-D-glucamine (NMDG), 10 BaCl2, 30 TEA-Cl, 10 HEPES, 10 glucose, pH at 7.4, 0.001 TTX, 0.01 nifedipine. The intracellular recording solution (at ~310 mOsm) consisted of the following (in mM): 150 CsCl2, 10 HEPES, 5 Mg-ATP, 5 BAPTA, pH at 7.4. Activation of ICa was measured by using a holding voltage of −90 mV with voltage steps 200 milliseconds in duration applied at 5-second intervals in + 10 mV increments from −70 to +60 mV. Current density was calculated as peak ICa/cell capacitance. Steady-state inactivation of ICa was determined by applying an 800-millisecond conditioning prepulse (−100 to −20mV in +10mV increments) after which the voltage was stepped to −20 mV for 200 milliseconds; a 15-second interval separated each acquisition. Whole-cell voltage clamp recordings were performed at room temperature (RT) using an EPC 10 Amplifier-HEKA as previously described [22]. The internal solution for voltage clamp sodium current recordings contained (in mM): 140 CsF, 1.1 CsEGTA, 10 NaCl, and 15 HEPES (pH 7.3, 290–310 mOsm/L) and external solution contained (in mM): 140 NaCl, 3 KCl, 30 tetraethylammonium chloride, 1 CaCl2, 0.5 CdCl2, 1 MgCl2, 10 D-glucose, and 10 HEPES (pH 7.3, 310–315 mosM/L).
DRG neurons were subjected to current-density (I-V) and fast-inactivation voltage protocols as previously described [21; 28]. In the I-V protocol, cells were held at a −80 mV holding potential before depolarization by 20-millisecond voltage steps from −70 to +60 mV in 10-mV increments. This allowed for collection of current density data to analyze activation of sodium channels as a function of current vs voltage and also peak current density, which was typically observed near ~0 to 10 mV and normalized to cell capacitance (pF). In the fast-inactivation protocol, cells were held at a −80 mV holding potential prior to hyperpolarizing and repolarizing pulses for 500 milliseconds between −120 to −10 mV in 5 mV increments. This step conditioned various percentages of channels into fast-inactivated states so that a 0-mV test pulse for 20 milliseconds could reveal relative fast inactivation normalized to maximum current. Fire-polished recording pipettes, 2 to 5 MΩ resistance were used for all recordings. Whole-cell recordings were obtained with a HEKA EPC-10 USB (HEKA Instruments Inc., Bellmore, NY); data were acquired with a Patchmaster (HEKA) and analyzed with a Fitmaster (HEKA). Capacitive artifacts were fully compensated, and series resistance was compensated by ~70%. Recordings made from cells with greater than a 5mV shift in series resistance compensation error were excluded from analysis. All experiments were performed at room temperature (~23°C).
The Boltzmann relation was used to determine the voltage dependence for activation of ICa and INa wherein the conductance-voltage curve was fit by the equation G/Gmax = 1/ [1 + exp (V0.5 − Vm)/k], where G is the conductance G=I/(Vm−ECa or ENa), Gmax is the maximal conductance obtained from the Boltzmann fit under control conditions, V0.5 is the voltage for half-maximal activation, Vm is the membrane potential, and k is a slope factor. ECa is the reversal potential for ICa; ENa is the reversal potential for INa and was determined for each individual neuron. The values of ICa and INa around the reversal potential were fit with a linear regression line to establish the voltage at which the current was zero. The Boltzmann parameters were determined for each individual neuron and then used to calculate the mean ± S.E.M.
2.8. Cell culture and transient transfection.
Human embryonic kidney (HEK) tsA-201 cells were cultured and transfected using the calcium phosphate method. Cells were plated on glass coverslips, transfected with 3 ug of either of the channel cDNAs. GFP (0.5 μg) was added to the mix as a transfection reporter. All the experiments were performed 72 hrs after cell transfection.
2.9. Voltage clamp recordings from transfected tsA-201 cells.
Whole cell patch clamp recordings were using an Axopatch 200B amplifier linked to a computer with pCLAMP 9.2 software. The cell capacitance and series resistance were compensated. Recording were obtained using the following external solution (in mM): 5 CaCl2, 132.5 CsCl, 1 MgCl2, 10 HEPES and 10 Glucose, pH 7.4. The internal pipette solution contained (in mM): 130 CsCl, 2.5 MgCl2, 10 HEPES, 5 EGTA, 3 ATP, 0.5 GTP, pH 7.4. Currents were evoked by applying 100 ms test pulses from a holding potential of −100 mV to −30. The effects of BA (20 µM) were assessed by normalizing the current amplitude in the presence of the drug to that observed during vehicle administration (0.1 % DMSO).
2.10. Preparation of spinal cord slices
As described previously [95], young rats (postnatal 10–14 days) were deeply anesthetized with diethyl ether and the lumbar region of the spinal column was quickly removed, euthanizing the animal. For spinal nerve blocking, 0.3 mL of 2% lidocaine was injected to both sides of L4 to 5 lumbar vertebrae. Laminectomy was performed from mid-thoracic to low-lumbar levels, and the spinal cord was quickly removed into cold modified artificial cerebrospinal fluid (ACSF) oxygenated with 95% O2 and 5% CO2. The ACSF contained (in millimolar): 80 NaCl, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 3.5 MgCl2, 25 NaHCO3, 75 sucrose, 1.3 ascorbate, 3.0 sodium pyruvate, with pH at 7.4 and osmolarity at 310 mOsm. Transverse 350-μm thick slices were obtained by a vibratome (VT1200S; Leica, Nussloch, Germany). Slices were then incubated for at least 1 hour at room temperature in an oxygenated recording solution containing (in millimolar): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 25 D-glucose, 1.3 ascorbate, 3.0 sodium pyruvate, with pH at 7.4 and osmolarity at 320 mOsm. The slices were then positioned in a recording chamber and continuously perfused with oxygenated recording solution at a rate of 3 to 4 mL/min before electrophysiological recordings at room temperature.
2.10.1. Electrophysiological recording in spinal cord slices by whole-cell patch clamp
Substantia gelatinosa neurons in the superficial layers of the spinal dorsal horn were visualized and identified in the slices by means of infrared differential interference contrast video microscopy on an upright microscope (FN1; Nikon, Tokyo, Japan) equipped with a 3× 40/0.80 water-immersion objective and a charge-coupled device camera. Patch pipettes with resistance at 6 to 10 MΩ were made from borosilicate glass (Sutter Instruments, Novato, CA) on a 4-steps micropipette puller (P-90; Sutter Instruments, Novato, CA). The pipette solution contained the following (in millimolar): 120 potassium gluconate, 20 KCl, 2 MgCl 2, 2 Na 2 -ATP, 0.5 Na-GTP, 20 HEPES, 0.5 EGTA, with pH at 7.28 and osmolarity at 310 mOsm. The membrane potential was held at −60 mV using PATCHMASTER software in combination with a patch clamp amplifier (EPC10; HEKA Elektronik, Lambrecht, Germany).
The whole-cell configuration was obtained in voltage-clamp mode. To record spontaneous excitatory postsynaptic currents (sEPSCs), bicuculline methiodide (10 μM) and strychnine (1 μM) were added to the recording solution to block γ-aminobutyric acid-activated and glycine-activated currents, respectively. Hyperpolarizing step pulses (5 mV in intensity, 50 milliseconds in duration) were periodically delivered to monitor the access resistance (15–25 MΩ), and recordings were discontinued if the access resistance changed by more than 20%. For each neuron, sEPSCs were recorded for a total duration of 2 minutes. Currents were filtered at 3 kHz and digitized at 5 kHz. Data were further analyzed by the Mini-Analysis (Synatosoft Inc, NJ) and Clampfit 10.7 Program. In all experiments measurements were made from one neuron per slice. The amplitude and frequency of sEPSCs were compared between neurons from animals in control and BA groups.
2.11. Calcitonin gene–related peptide release from lumbar slices.
Rats were deeply anesthetized with 5% isofluorane and then decapitated. Two vertebral incisions (cervical and lumbar) were made in order to expose the spinal cord. Pressure was applied to a saline-filled syringe inserted into the lumbar vertebral foramen, and the spinal cord was extracted. Only the lumbar region of the spinal cord was used for the CGRP release assay. Baseline treatments (#1 and #2) involved bathing the spinal cord in Tyrode’s solution. The excitatory solution consisting of 90 mM KCl was paired with the treatment for fraction #4. These fractions (10 minutes, 400 μL each) were collected for measurement of CGRP release. Samples were immediately flash frozen and stored in a −20 °C freezer. BA (20 μM) or vehicle (0.9% saline) was added to the pretreatment and co-treatment fractions (#3 and 4). The concentration of CGRP released into the buffer was measured by enzyme-linked immunosorbant assay (Cat# 589001, Cayman Chemical, Ann Arbor, MI).
2.12. Implantation of intrathecal catheter.
For intrathecal (i.t.) drug administration, rats were chronically implanted with catheters as described by Yaksh and Rudy [88]. Rats were anesthetized with halothane and placed in a stereotactic head holder. The occipital muscles were separated from their occipital insertion and retracted caudally to expose the cisternal membrane at the base of the skull. Polyethylene tubing was passed caudally from the cisterna magna to the level of the lumbar enlargement. Animals were allowed to recover and were examined for evidence of neurologic injury. Animals with evidence of neuromuscular deficits were excluded.
2.13. Testing of allodynia.
The assessment of tactile allodynia (i.e., a decreased threshold to paw withdrawal after probing with normally innocuous mechanical stimuli) consisted of testing the withdrawal threshold of the paw in response to probing with a series of calibrated fine (von Frey) filaments. Each filament was applied perpendicularly to the plantar surface of the paw of rodents held in suspended wire mesh cages. Withdrawal threshold was determined by sequentially increasing and decreasing the stimulus strength (the “up and down” method), and data were analyzed with the nonparametric method of Dixon, as described by Chaplan et al [14] and expressed as the mean withdrawal threshold
2.14. Radioligand Binding Cell Lines and Cell Culture.
The mu opioid receptor (MOR)-CHO cell line was purchased from PerkinElmer (#ES-542-C). The delta-OR (DOR) cell line was created and characterized in our lab, as reported in [73]. The kappa-OR (KOR) cell line was also created in our lab; an N-terminal 3X-hemagglutinin tagged human KOR expression clone from Genecopoeia was electroporated into parental CHO cells and selected with 500 μg/mL G418. The resulting selected population was enriched for receptor expression by live cell labeling with anti-HA-Alexa488 antibody and separating the top 2% of the population by flow cytometry. This high expressing population was characterized by immunocytochemistry and Western blot to establish receptor expression and expected signal transduction activation. All 3 cell lines were characterized by saturation radioligand binding with 3H-diprenorphine, and the measured KD used in competition binding experiments to calculate the KI (MOR = 5.23 nM; DOR = 0.93 nM; KOR = 1.81 nM; all the mean of N ≥ 3 independent experiments). All cells were cultured in 50:50 DMEM/F12 media with 10% heat-inactivated FBS and 1X penicillin/streptomycin supplement (all Gibco/ThermoFisher brand) in a 37°C humidified incubator with 5% CO2 atmosphere; propagation cultures were further maintained in 500 μg/mL G418. Cultures were propagated for no more than 20 passages before discarding. Cell pellets for experiments were prepared by growth in 15 cm plates, harvest with 5 mM EDTA in dPBS (no calcium or magnesium), and the storage of cell pellets at −80°C prior to use.
2.15. Competition Radioligand Binding.
Competition radioligand binding experiments were performed as reported in [73]. Membrane preparations of MOR-, DOR-, or KOR-CHO cells were combined with a fixed concentration of 3H-diprenorphine (MOR = 5.33 nM; DOR = 1.43 nM; KOR = 1.95 nM) and a concentration curve of competitor ligand. These reactions were miniaturized to a 200 μL volume in 96 well plates. The reaction proceeded at room temperature for 60 minutes. The reactions were terminated by rapid filtration through 96 well format GF/B filter plates (PerkinElmer) with cold water, washed, dried, and Microscint PS (PerkinElmer) added. The plates were read in a MicroBeta2 96 well format 6 detector scintillation counter (PerkinElmer). The data was normalized to the specific binding caused by 3H-diprenorphine alone (100%) or non-specific binding determined by a 10 μM concentration of known competitor ligand (0%; MOR = naloxone; DOR = SNC80; KOR = naloxone). KI values were calculated using the IC50 of each competitor ligand and the previously established KD of 3H-diprenorphine in each cell line (GraphPad Prism 7.0).
2.16. HIV-induced sensory neuropathy (HIV SN).
Mechanical allodynia is produced by intrathecal administration of the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein, gp120 [52]. Seven days after implantation of an intrathecal catheter, baseline behavioral measurements were obtained and then rats were randomly assigned to two groups. On days 10, 12 and 14, rats were injected i.t. with 300 ng of gp120 (Cat#4961, HIV-1 BaL gp120 recombinant protein, NIH-AIDS Reagent program) in a final volume of 20 µl in 0.9% saline and 0.1% BSA.
2.17. Paclitaxel-induced peripheral neuropathy.
Rats were given paclitaxel (Cat# P-925–1, Goldbio, Olivette, MO) based on the protocol described by Polomano et al. [63]. In brief, pharmaceutical-grade paclitaxel (Taxol) was re-suspended at a concentration of 2 mg/ml in 30% 1:1 Cremophor EL: ethanol, 70 % Saline and given to the rats at 2 mg/kg intraperitoneally (i.p.) every other day for a total of 4 injections (days 0, 2, 4, and 6), resulting in a final cumulative dose of 8 mg/kg. No abnormal spontaneous behavioral changes in the rats were noted during or after the treatment. Animals developed mechanical hyperalgesia within 10 days after the first paclitaxel injection.
2.18. Partial sciatic nerve ligation.
Partial sciatic nerve ligation was performed as described previously [50]. Briefly, mice were anesthetized under isoflurane. A cut was made through the skin to expose the right sciatic nerve. A partial ligation was made by tying one-third to one-half of the dorsal sciatic nerve. The right sciatic nerve of sham-operated mice was exposed, but ligation was not performed. Mice were monitored, adequately rehydrated, and kept in a controlled temperature (37 °C) until fully recovered from anesthesia.
2.19. Assessment of open field behavior.
Rats were randomly assigned to receiving the compound or its vehicle. Ninety to 180 minutes after intrathecal injection of the compound or its vehicle, rats were placed in a round open-field (diameter: 110 cm, walls: 35cm) made of wood and painted black, under bright illumination [64]. Animals were allowed to behave freely in the apparatus for 10 minutes and their behavior was video-recorded. Total distance traveled, maximum speed and time spent in the center of the apparatus (defined by a round 75cm zone in the center of the apparatus) were analyzed by an automated activity monitoring software (Any-Maze, Stoelting, U.S.A.). All behavioral experiments were performed by experimenters who were blinded to experimental groups and treatments.
2.20. Statistical Analyses.
All values represent the mean ± S.E.M, unless noted otherwise. All data was first tested for a Gaussian distribution using a Shapiro-Wilk test (Graphpad Prism 7 Software). The statistical significance of differences between means was determined by either Student’s t test, parametric analysis of variance (ANOVA) followed by post hoc comparisons (Tukey) using GraphPad Prism 7 Software. All behavioral data were analyzed by non-parametric two-way ANOVA (post hoc: Student-Neuman–Kuels) in FlashCalc (Dr. Michael H. Ossipov, University of Arizona, Tucson, AZ, USA). Differences were considered to be significant if p≤ 0.05. All data were plotted in GraphPad Prism 7. No outlier data was removed.
3. Results
3.1. Hyptis emoryi extracts affect depolarization-evoked Ca2+ influx in rat sensory neurons.
To discover novel natural compounds with a therapeutic potential for pain relief, we screened a library of natural product extracts for small molecules capable of inhibiting the voltage gated Ca2+ channels. Of those screened, a chloroform-methanol (1:1) extract derived from desert lavender (Hyptis emoryi) (Figure 1A) was found to have promising potential to decrease depolarization-evoked Ca2+ influx in rat DRG sensory neurons. Bioactivity-guided fractionation of this extract by solvent-solvent partitioning with chloroform and 50% aqueous methanol yielded 2 fractions, F1 and F2 (Figure 1B). We next tested these by Ca2+ imaging (Figure 1C) to further isolate the fraction retaining the biological activity. We sequentially used 2 different concentrations of KCl to evoke Ca2+ influx and analyze independently the activity of low-voltage-activated Ca2+ channels (Cav3.x, trigger 40 mM KCl) or high-voltage activated Ca2+ channels (Cav1.x and Cav2.x, trigger 90 mM KCl) [84] (Figure 1D). Extract and fractions were tested at concentrations of 440 µg.ml−1 for H. emoryi extract and 88.4 µg.ml−1 for F1 and F2. Fraction F1 specifically inhibited 40 mM KCl-evoked Ca2+ influx in rat dorsal root ganglia (DRG) neurons (55.4 ± 2.1% inhibition compared to 0.1% DMSO control, n=188–482 neurons analyzed) while fraction F2 did not show any inhibitory potential for either KCl concentrations (Figure 1E). These results show the potential presence of a natural compound inhibitor of voltage-gated Ca2+ channels (possibly T-type) in fraction F1 derived from Hyptis emoryi extract.
Figure 1. Fractionation of Hyptis emoryi (H. emoryi) extract and the effect of the extract and its fractions on depolarization-evoked Ca2+ influx in DRG sensory neurons.

(A) Photograph of the plant, H. emoryi used in this study. (B) Fractionation of H. emoryi yielding fractions F1 and F2. (C) Differential interference contrast (DIC) and pseudocolored fluorescent images visualized for Fura2-AM in Dorsal Root Ganglia (DRG) neurons. Neurons were sequentially stimulated with 40 mM and 90 mM KCl for a period of 15 seconds following an initial 1-minute baseline measurement, and response was measured for 3 minutes after each challenge. Scale bar is 20 µm. Fluorescence scale indicates Fura-2AM ratio-metric fluorescence value (F340/F380). High values correspond to high intracellular [Ca2+] indicated in red. (D) Traces of response average of >300 neurons treated with H. emoryi extract (440 µg.ml−1) or its fractions F1and F2 (88.4 µg.ml−1). Arrows indicate the beginning of a 15-second stimulation period with 40 or 90 mM KCl as stated. (E) Peak calcium responses of sensory neurons incubated overnight with H. emoryi extract, derived fractions F1 and F2, and 0.1% DMSO (vehicle) in response to 40 and 90 mM KCl (n= 271–518 neurons). Responses were normalized to that of the vehicle and show average response ± S.E.M. Asterisks indicate statistical significance compared with cells treated with the vehicle (p<0.05, one-way ANOVA with Dunnett’s post-hoc test).
3.2. Selection of betulinic acid for inhibition of depolarization-evoked calcium influx in rat DRGs.
Further separation of the active fraction F1 by high-pressure liquid chromatography (HPLC) yielded three pure compounds, identified as betulinic acid (BA), oleanolic acid (OA), and ursolic acid (UA) (Figure 2 and Supplementary Methods). We tested the 3 compounds BA, UA, and OA using the same Ca2+ imaging protocol as described above to identify the small molecule responsible for voltage-gated Ca2+ channel inhibition in DRG sensory neurons (Figure 2A-B). Rat sensory neurons treated with 20 µM of BA, UA, or OA (Figure 2C) (equivalent to 8.84 µg.ml−1 for BA). BA inhibited 40 mM KCl evoked Ca2+ influx (89.8 ± 3.5% inhibition compared to 0.1% DMSO control, n=188) (Figure 2D) and 90 mM KCl evoked Ca2+ influx (40.7 ± 5.0 % inhibition compared to 0.1% DMSO control) (Figure 2E). Oleanolic acid and ursolic acid did not significantly inhibit Ca2+ influx for either of the KCl challenges (Figure 2D-E). Ursolic acid-treated neurons showed an increase (198.7 ± 9.1% when triggered with 40mM KCl and 174.3 ± 4.1% when triggered with 90 mM KCl compared to 0.1% DMSO control, n=129) of K+-evoked Ca2+ influx. The opposing effects of UA and the inhibitory effects of BA on low- (preferential) and high-voltage activated Ca2+ channels supported use of BA, as opposed to the extract, for the studies below.
Figure 2. BA inhibits depolarization-evoked Ca2 influx in DRG sensory neurons.

(A) Differential interference contrast (DIC) and pseudocolored fluorescent images visualized for Fura2-AM before and after stimulations with 40 mM and 90 mM KCl. A stimulation period of 15 seconds each with 40 and 90 mM KCl followed an initial 1-minute baseline measurement; response was subsequently measured for 3 minutes after each stimulation. Scale bar is 20 µm. Fluorescent scale shows relative intracellular calcium [Ca2+] in each Dorsal Root Ganglia (DRG) neuron (F340/F380). (B) Traces of response average of compounds BA, UA, OA and vehicle (0.1% DMSO). Arrows indicate the initiation of a 15 second stimulation period of the DRG sensory neurons with 40 or 90 mM KCl as indicated. (C) Structures and molecular formulas of compounds BA, UA, and OA. (D-E) Bar graphs show normalized peak calcium response average ± S.E.M. of DRG sensory neurons incubated overnight with a 20 µM concentration of compounds BA, UA, OA and vehicle in response to 40 (D) and 90mM KCl (E). Responses were normalized to that of the DMSO. Asterisks indicate statistical significance compared with cells treated with the vehicle (p<0.05, one-way ANOVA with Dunnett’s post-hoc test).
3.3. Betulinic acid inhibits Ca2+ currents in Rat Dorsal Root Ganglia.
We then investigated the effects of betulinic acid on total Ca2+ currents in rat DRG sensory neurons. Neurons were treated overnight with a 20 μM concentration of betulinic acid or control (0.1% DMSO) as indicated and Ca2+ currents were subsequently recorded (Figure 3). When compared with the control (0.1% DMSO), BA-treated neurons exhibited an 51.4% inhibition of total Ca2+ current (Figure 3C). No change in biophysical properties of Ca2+ currents was observed in BA-treated sensory neurons (Figure 3D). This inhibition in Ca2+ currents, consistent with the above shown inhibition of KCl-evoked Ca2+ influx, confirmed the function of betulinic acid as an inhibitor of voltage-gated calcium channels.
Figure 3. BA diminishes Ca2+ currents in DRG sensory neurons.

(A) Representative traces of Ca2+ currents evoked from 200 millisecond prepulses between −70 mV and +60 mV, illustrated from DRG sensory neurons treated with 0.1% DMSO or 20 µM of BA. (B) Summary of the normalized (pA/pF) calcium current density and (C) bar graph showing peak Ca2+ current density at +10 mV (mean ± s.e.m.) from DRG sensory neurons treated with 0.1% DMSO (vehicle) or 20 µM of BA as indicated (n = 10 cells per condition). Asterisks indicate statistical significance compared with cells treated with 0.1% DMSO (*p<0.05, unpaired 2-tailed t test). (D) Boltzmann fits for normalized conductance G/Gmax-voltage relations for voltage dependent inactivation and activation of sensory neurons treated with vehicle or a 20 µM concentration of BA as indicated. (E) Representative traces of T-type Ca2+ currents, evoked from 200 millisecond prepulses between −60 mV and +60 mV, from DRG sensory neurons treated with 0.1% DMSO (black) or 20 µM BA (red). T-type currents were pharmacologically isolated using blockers of all other channels (see Methods). (F) Summary of the normalized (pA/pF) T-type calcium current density versus voltage and peak Ca2+ current density at +10 mV (mean ± s.e.m.) (G) from DRG neurons treated with 0.1% DMSO (vehicle, n=9) or 20 µM BA (n=6). Asterisks indicate statistical significance compared with cells treated with 0.1% DMSO (*p<0.05, unpaired 2-tailed t test).
To specifically examine if voltage-gated calcium (i.e. T-type) channels were affected by BA, we pharmacologically isolated T-type currents using blockers of all other channels (SNX482 (200 nM, R-type voltage-gated Ca2+ channel blocker), ω-conotoxin GVIA (500 nM, N-type voltage-gated Ca2+ channel blocker), ω-agatoxin (200 nM, P/Q-type voltage-gated Ca2+ channel blocker), and Nifedipine (10 µM, L-type voltage-gated Ca2+ channel blocker) and then applied BA. Whole-cell Ba2+ currents were elicited from a holding potential of −90 mV to depolarizing test potentials (ranging from–60 mV to +50 mV in 5 mV increments) for 200 ms. Figure 3E shows representative voltage-dependent inward currents (IBa) from DRG neurons with fast kinetic properties (i.e. T-type) similar to those reported previously [80]. Compared with control (0.1% DMSO), BA-treated neurons exhibited an ~50% inhibition of T-type Ca2+ currents (Figure 3F, G). Thus, the data show inhibition of both high- and low-voltage activated calcium channels by BA.
3.4. Betulinic acid inhibits Cav3.2 and Cav3.3 T-type Ca2+channels
The above experiments identify the natural small molecule betulinic acid as an inhibitor of voltage-gated Ca2+ channels. As earlier results (Figure 2) showed predominant block of low-voltage activated channels evoked by 40 mM KCl depolarization, we initially focused our efforts on T-type channels. In DRG sensory neurons, the α subunits of Cav3.2 and Cav3.3 represent the majority of T-type Ca2+ channels [91]. To test if betulinic acid could preferentially target a specific T-type Ca2+ channel subunit we used a knockdown strategy (using short interfering RNA (siRNA)) to eliminate either Cav3.2 or Cav3.3 in DRG sensory neurons (Figure 4). DRG neurons were electroporated with the indicated siRNA (or a scrambled negative control) in combination with a GFP expressing plasmid (to identify transfected cells) (Figure 4A). The cells were cultured for 24 hours before adding 20µM of betulinic acid overnight (or 0.1% DMSO as control) and then tested using 40 mM KCl as a trigger (Figure 4A). In scramble siRNA-transfected cells, betulinic acid strongly inhibited 40 mM KCl evoked Ca2+ influx (Figure 4B). Knocking down Cav3.2 or Cav3.3 decreased 40mM KCl evoked Ca2+ influx. This indicates that both T-type Ca2+ channel subunits are important for low voltage activated Ca2+ influx in DRG neurons. After the knockdown of either Cav3.2 or Cav3.3, as evidence by decreased mRNA expression levels (Figure 4C), betulinic acid failed to inhibit further the 40 mM KCl evoked Ca2+ influx.
Figure 4. Downregulation of Cav3.2 and Cav3.3 channels blocks BA-mediated inhibition of depolarization-evoked Ca2 influx through T-type Ca2+ Channels.

Dorsal root ganglion neurons were transfected during plating with a GFP construct and a scramble siRNA or with siRNAs against Cav3.2 or Cav3.3. A representative experiment (bright field with GFP fluorescence (left panels), and pseudocolored fluorescent images visualized for Fura2-AM before (middle panels) and after stimulations with 40 mM KCl (right panels). In this experiment, vehicle (DMSO)-treated neurons with GFP responds to KCl, whereas BA-treated neuron with GFP fluorescence demonstrates significantly decreased response. (B) Bar graph shows peak calcium response averages ± S.E.M. of DRG sensory neurons treated as indicated. Responses were normalized to that of DMSO (vehicle) in the siRNA scramble condition. Statistical significance compared to vehicle-treated cells is indicated via asterisks (p<0.05, 2-way ANOVA with Sidak’s post hoc test) (n = 8–22 cells per condition). (C) Bar graph showing quantitative RT–PCR analysis of Cav3.2 and Cav3.3 transcripts in DRGs treated with scramble, Cav3.2 or Cav3.3 siRNAs. (n=3). mean ± s.e.m., *p<0.05 Mann-Whitney test. These experiments were done in a blinded fashion.
To complement these data, we explored the inhibitory effect of betulinic acid on tsA-201 cells individually transfected with either Cav3.1, Cav3.2 or Cav3.3 (Figure 5). BA (20 µM) was applied to the transfected cells and the indicated Cav3.x currents were recorded (Figure 5A). We found that BA had no inhibitory effect on Cav3.1 (Figure 5B) while this natural compound inhibited Cav3.2 (Figure 5C) and Cav3.3 (Figure 5D) currents. The comparison of the percentage of Cav3.x current inhibition elicited by BA treatment revealed a stronger inhibitory effect on Cav3.2 than on Cav3.3 (Figure 5E). Maximal inhibition of Cav3.2 was observed with 20 µM BA, although no further inhibition was observed with a doubling of the compound; we could not test higher concentrations because 0.2% DMSO by itself was inhibitory (Table 1). For Cav3.3, a maximal ~31% inhibition was observed with either the 20 or 40 µM concentrations BA. Consistent with the above data in DRGs, 20 µM BA also inhibited Cav2.2 currents by 69.8±2.5% (n=4) in tsA-201 cells (Figure 5F, G). Thus, we conclude that betulinic acid preferentially inhibits Cav3.2 (T-type) and Cav2.2 (N-type) Ca2+ channel subunits.
Figure 5. BA inhibits T- and N-type currents in heterologous cells.

(A) Representative traces before (black) and after (red) application of 20 µM BA. Currents were evoked from −100 to −30 mV for 100ms. Top hCav3.1, middle hCav3.2 and bottom hCav3.3. Representative example of time course of BA-mediated inhibition of hCav3.1 (B), hCav3.2 (C), and hCav3.3 (D). (E) Effect of BA on current amplitude of transiently expressed hCav3.1, hCav3.2 and hCav3.3 channels. (F) Representative traces before (black) and after (red) application of 20 µM BA. Currents were evoked from −100 to +10 mV for 250 ms in cells (n=4) expressing rCav2.2. (G) Representative example of time course of BA-mediated inhibition of rCav2.2. Data are presented as mean ± s.e.m. Number of cells are shown in parentheses.
Table 1:
: Table summarizing percent inhibition of Cav3.x currents by the indicated concentrations of BA.
| 4µM | 10µM | 20µM | 40µM | |
|---|---|---|---|---|
| hCaV3.1 | n.t.1 | n.t. | 1.7 ± 2.4 (4) | 5.8 ± 1.4 (4) |
| hCaV3.2 | 10.9 ± 4.9 (6) | 49.3 ± 8.7 (6) | 66.1 ± 4.6 (6) | 42.4 ± 6.8 (5) |
| hCaV3.3 | 10.9 ± 3.6 (4) | 16.5 ± 2.2 (4) | 31.4 ± 1.8 (8) | 30.8 ± 3.0 (5) |
n.t. = not tested
Values were obtained after DMSO effect was subtracted. Numbers in parentheses represent numbers of cells recorded from.
3.5. Betulinic acid’s effect on the functional landscape of DRG sensory neurons
The results presented thus far collectively demonstrate the potential for inhibition of Ca2+ influx by betulinic acid through control of Cav3.2 and Cav3.3 T-type Ca2+ channels. These results do not uncover cell-specific neuronal classes that may be targeted by betulinic acid. We used the previously described constellation pharmacology protocol [76; 77] to explore cell-specific functionality as a result of key signaling proteins that define precise cell types. The constellation pharmacology assay poses 6 successive stimulations, each 6 minutes apart, to compare Ca2+ influx due to activity of Ca2+-associated membrane proteins: Ca2+ permeable ligand-gated ion channels, metabotropic receptors and voltage-gated Ca2+ channels. Following these 6 challenges, KCl-evoked response due to membrane depolarization is used to assess viability of neurons; neurons not responsive to KCl are excluded from analysis.
The heterogeneity of neuronal responsivity is demonstrated via example traces of sensory neurons treated with control (0.1% DMSO) and with a 20 µM concentration of betulinic acid (Figure 6A-B) as indicated. Notably, inhibition of depolarization-evoked Ca2+ influx was again inhibited as a result of treatment with betulinic acid; this is consistent with our previous data (Figures 2 and 3). Sensory neurons were incubated overnight with the specified treatment, control (n=761) or betulinic acid (n=835), and subsequently imaged with constellation pharmacology protocol. Data was collected from 3 independent experiments, and individual neuronal responses to each constellation trigger were analyzed. Neurons with responses under 10% of baseline fluorescence were excluded from our analyses.
Figure 6. Functional ‘fingerprinting’ of DRG neuronal subclasses following treatment with BA.

Representative traces of sensory neurons treated with 0.1% DMSO (vehicle). (A) or BA (20µM) (B) responding to constellation pharmacology triggers (menthol (400 nM), histamine (50 µM), ATP (10 µM), AITC (200 µM), acetylcholine (1 mM), capsaicin (100 nM) and KCl (90 mM)) during Ca2+ imaging. Each trace represents an individual neuron; a typical experimental trial records the responses of >200 neurons concurrently. The x-axis represents time in seconds, the y-axis shows the relative intracellular calcium [Ca2+] in each DRG neurons (i.e. the F340/F380 ratio). (C) Percentage of DRG sensory neurons that responded to indicated number of triggers. “0” indicates those neurons that only responded to none other than KCl stimulus. (D) Percentage of sensory neurons responding to major classes of constellation triggers. (E) Number of overall functional DRG sensory neuronal classes as a result of treatment with vehicle or BA (20 µM). Average peak response (F) and area under the curve (G) is shown for calcium response in sensory neurons post-indicated treatment, after stimulation by major classes of constellation triggers. Area under the curve was calculated with Graphpad Prism software using the trapezoid rule. (H) Average peak KCl-evoked response of sensory neurons post-indicated treatment. Statistical significance compared with cells treated with 0.1% DMSO are indicated by asterisks (*p<0.05; Student’s t-test). Abbreviations for constellation triggers are as follows: ACh = acetylcholine; AITC = allyl isothiocyanate; ATP = adenosine triphosphate; Hist = histamine; Ment = menthol; Cap = capsaicin; KCl = potassium chloride. Data was acquired from a total of 3 independent experiments with an overall n of 761 (from 4 coverslips) for vehicle (0.1% DMSO) and 835 (from 3 coverslips) for BA (20 µM).
We first inquired how treatment with betulinic acid would change overall functionality of sensory neurons. To test this, we assessed whether the response of neurons to the number of stimulatory challenges, independently of which specific agonists triggered response, was altered in response to BA treatment (Figure 6C). Compared to control-treated cells, more cells treated with betulinic acid responded to 2 stimulatory challenges and less cells responded to only 1 challenge. Next, we asked if betulinic acid affected the sensitivity of DRGs to the different constellation triggers, by analyzing the percent of cells responding to a specific receptor agonist, independently of any other constellation triggers these neurons may have responded to (Figure 6D). Furthermore, more functional cell subclasses were present in the population of neurons treated with betulinic acid (39 different functional classes) in comparison to the control population (30 different functional classes) (Figure 6E). This analysis showed that treatment with betulinic acid increases sensitivity to ATP stimulation. Overall, betulinic acid increased functional competence of cells.
In a more specific inquiry, we also explored how betulinic acid altered the extent of Ca2+ influx following specific stimulation by each constellation trigger. Thus, we analyzed peak Ca2+ responses (Figure 6F) and area under the curve (AUC) of these responses in sensory neurons as a result of the treatment. Notably, while betulinic acid did not significantly alter peak Ca2+ responses due to stimulation by any of the constellation triggers, the AUC of Ca2+ response due to stimulation by Acetylcholine (ACh) and (ATP) was increased, and AUC of Ca2+ response due to stimulation by allyl isothiocyanate (AITC) was decreased in BA-treated neurons (Figure 6G). Stimulation by histamine, menthol, or capsaicin, did not alter the AUC of Ca2+ response in sensory neurons after treatment with betulinic acid (Figure 6G).
We then asked if betulinic acid mediated inhibition of depolarization-evoked Ca2+ response would be altered in a functional class-specific manner. To test this, we assessed average peak Ca2+ response due to KCl challenge in sensory neurons that specifically responded to a particular constellation trigger, independent of any other constellation triggers they might have responded to. Treatment with betulinic acid significantly decreased KCl evoked Ca2+ response in sensory neurons that responded to an ATP stimulus; However, DRGs that responded to ACh, AITC, Histamine, menthol, and capsaicin did not have altered KCl-evoked response due to betulinic acid treatment (Figure 6H). These results assist in uncovering the mechanism of Ca2+ inhibition by betulinic acid and warrant further exploration into the potential for betulinic acid as an anti-nociceptive agent.
3.6. Betulinic acid does not bind to the orthosteric site of the opioid receptors.
To confirm that betulinic acid does not produce potential anti-nociception through off-target binding to the opioid receptors, we performed competition radioligand binding at all 3 opioid receptors in vitro. We competed betulinic acid and a positive control compound (naloxone for MOR and DOR, U50,488 for KOR) vs. a fixed concentration of 3H-diprenorphine in Chinese Hamster Ovary (CHO) cells expressing the human μ (MOR), δ (DOR), or κ (KOR) opioid receptor. We found that betulinic acid did not bind to any opioid receptor up to a 10 μM concentration (Figure 7A-C). In contrast, the positive control compounds bound to all 3 targets with expected affinity (Figure 7A-C). These results strongly suggest that betulinic acid is not engaging the opioid receptors, and that potential anti-nociception would thus not be as a result of opioid receptor association.
Figure 7. BA does not bind to the opioid receptors.

Competition radioligand binding was performed in CHO cells expressing the human MOR, DOR, or KOR (see Methods for details). BA or a positive control compound was competed against 3H-diprenorphine in all 3 cell lines. Curves reported as the mean ± SEM of the mean value from each individual experiment in n = 3 independent experiments. The KI also reported as the mean ± SEM of the individual value from each of n = 3 independent experiments. BA did not produce competition binding up to 10 μM in any cell line. (A) MOR: Naloxone KI = 33.9 ± 1.7 nM. (B) DOR: Naloxone KI = 55.7 ± 6.7 nM. (C) KOR: U50,488 KI = 22.4 ± 4.1 nM.
3.7. Sodium currents are resistant to betulinic acid.
To further explore the mechanism of betulinic acid, we investigated its possible effect on voltage gated Na+ channel currents in rat sensory neurons. Sensory neurons were treated overnight with a 20μM concentration of betulinic acid or control (0.1% DMSO) as indicated, and subsequently recorded (Figure 8A-B). In comparison to the control, sensory neurons exhibited no change in both total sodium current density and TTX-sensitive current density (Figure 8C) after betulinic acid treatment. Biophysical properties of Na+ currents in sensory neurons treated with betulinic acid remained unaltered compared with 0.1% DMSO control (Figure 8D).
Figure 8. Treatment with BA does not affect Na+ currents in DRG sensory neurons.

(A) Representative traces of Na + currents evoked from 200 millisecond prepulses between −70 mV and +60 mV with voltage protocol, illustrated from DRG sensory neurons treated with 0.1% DMSO or 20 µM BA. (B) Normalized current (pA/pF) versus voltage relationship of the sodium current density. (C) Scatter graph showing total peak Na+ current density at −10 mV (left bars) and TTX-sensitive Na+ current (right bars) (mean ± s.e.m.) from DRG sensory neurons treated with 0.1% DMSO (vehicle) or 20 µM BA (n = 8 cells per condition). No statistical significance was detected compared with cells treated with 0.1% DMSO (p>0.05, Mann-Whitney test). (D) Boltzmann fits for normalized conductance G/Gmax-voltage relations for voltage dependent fast-inactivation and activation of sensory neurons treated with vehicle or a 20 µM BA.
3.8. Betulinic acid inhibits spontaneous electric post-synaptic currents via presynaptic suppression.
Since T- and N-type calcium channels contribute to action potential firing and neurotransmitter release [13], we performed electrophysiological recordings in substantia gelatinosa neurons in the superficial layers (within laminae I-II) of the spinal dorsal horn (Figure 9A) to measure whether BA could inhibit spontaneous excitatory post-synaptic currents (sEPSC). There was no significant decrease in spontaneous EPSC amplitude (post-synaptic effect) (Figure 9C) or input resistance (Figure 9E) of neurons treated with 0.1% DMSO or BA (20 µM). However, BA treatment decreased sEPSC frequency (DMSO, 2.18 ± 0.24 Hz; BA, 1.26 ± 0.25 Hz, P<0.05) (Figure 9D), suggesting presynaptic suppression of neurotransmitter release by BA.
Figure 9. Betulinic acid decreases spontaneous excitatory post-synaptic current in substantia gelatinosa neurons.

(A) Anatomy and location of neurons with rat slices used for electrophysiology patch-clamp recordings. Arrow indicates the cell being patched (shadow of the pipette is also seen atop the cell). (B) Representative traces recorded from control (0.1% DMSO) and BA (20 µM)-treated groups. (C) Spontaneous EPSC amplitude as a result of treatment with DMSO or BA. (D) Spontaneous EPSC frequency as a result of treatment with DMSO or BA. (E) Input resistance of neurons recorded from both groups. Asterisks indicate significance compared with cells (n=13–14 per condition) treated with 0.1% DMSO (*p<0.05, unpaired 2-tailed t test).
3.9. Betulinic acid inhibits potassium-evoked neuropeptide release in spinal cord ex-vivo.
Presynaptic release of the neurotransmitter CGRP from sensory neurons is a known mediator of pro-nociceptive neuronal signaling. Therefore, we investigated whether or not betulinic acid inhibited depolarization-evoked CGRP-release from rat spinal cord. To test this, we used an ex vivo method for evoked CGRP release from the lumbar region of rat spinal cord. Enzyme-linked immunosorbent assay (ELISA) was used to measure CGRP content; samples were collected every 10 min. Basal CGRP levels were 1.85 ± 0.32 pg/ml/mg of tissue (Figure 10, fractions #1 & 2). Vehicle (0.1% DMSO) or a 20 μM concentration of betulinic acid was added (Figure 10, fraction #3) 10 min prior to stimulation with 90 mM KCl (Figure 10, fraction #4). Treatment with betulinic acid did not elicit any CGRP release from the spinal cords (Figure 10, fraction #3). Under depolarization, treatment with betulinic acid resulted in a ~40% decrease (evoked CGRP level for vehicle was 53.89 ±9.9 and for BA 32.10 ± 6.97 pg/ml/mg of tissue) of the depolarization-evoked increase in CGRP release seen in vehicle-treated tissue (Figure 10, fraction #4). These results show that inhibiting T-type voltage gated Ca2+ channels with BA results in decreased depolarization-evoked CGRP release.
Figure 10. CGRP release from spinal cord is inhibited by BA.

KCl (90 mM) depolarization-evoked CGRP release was measured from spinal cord tissue isolated from naïve adult rats as a result of pre- and co-incubation with 0.1% DMSO or 20 µM BA as indicated. Bar graph shows immunoreactive CGRP levels observed in bath solution normalized to the weight of each spinal cord tissue. Statistical significance is indicated by asterisks for fraction 4 (*p<0.05; 2-way ANOVA with Sidak’s post hoc test) in comparison with control treatment.
3.10. Chemotherapy-induced peripheral neuropathy is alleviated by treatment with Betulinic acid.
The above results identify BA as a novel compound inhibiting T-type voltage gated Ca2+ channels with a preference for Cav3.2 and Cav3.3. These channels are known to participate in neuropathic pain signal transmission [34; 37; 69]. In a model of chemotherapy-induced peripheral neuropathy, increased Cav3.2 expression underlies the painful behavior induced by paclitaxel administration [46]. We next tested if Cav3.2 inhibition by BA could be beneficial to reverse nociceptive behaviors in a rat model of paclitaxel induced peripheral neuropathy. Rats received 4 injections of paclitaxel (2mg/kg) and developed mechanical allodynia as measured by decreased paw withdrawal threshold (Figure 11A). We injected betulinic acid (2 µg/5 μL, i.th.) and observed a significant reversal of mechanical allodynia at 1 hour after injection (Figure 11A). This effect lasted for 2 additional hours (Figure 11A) before returning back to pre-drug baseline. Area under the curve shows a significant antinociceptive effect of BA compared to vehicle (saline) injected animals (Figure 11B). These results show that betulinic acid has the potential for pain relief in paclitaxel induced peripheral neuropathy.
Figure 11. BA reduces paclitaxel-induced and gp120-induced mechanical allodynia.

(A) Paw withdrawal threshold of adult male rats (n=6) was measured 15 days after 4 intraperitoneal injections of paclitaxel. Rats were treated intrathecally (i.th.) with saline (vehicle) or BA (2 µg/5 μL) as indicated. Asterisks indicate statistical significance compared with tissue treated with saline (*p<0.05; 2-way ANOVA with a Student-Neuman–Kuels post hoc test). (B) Area under the curve was derived again as indicated before using Graphpad Prism. Statistical significance is indicated by asterisks (*p<0.05, Mann-Whitney test) in comparison to vehicle-treated rats. (C) Paw withdrawal threshold of adult male rats (n=6) was measured 15 days after 3 intrathecal injections of glycoprotein-120. Rats were treated with saline (vehicle) or BA (20μM) as indicated. Asterisks indicate statistical significance compared with tissue treated with saline (*p<0.05; 2-way ANOVA with a Student-Neuman–Kuels post hoc test). (D) Area under the curve was derived as indicated before using Graphpad Prism. Statistical significance is indicated by asterisks (*p<0.05, Mann-Whitney test) in comparison to vehicle-treated rats. Chemotherapy induced peripheral neuropathy (CIPN); HIV-induced sensory neuropathy (HIV-SN).
3.11. Betulinic acid reverses HIV-induced sensory neuropathy behavior in rats.
Sensory neuropathy is a frequent complication of Human Immunodeficiency Virus (HIV) infection [51]. Because voltage-gated Ca2+ channels have previously been found to contribute to neuropathic pain [34; 37; 61; 69], we investigated the potential of betulinic acid to provide to anti-nociception in HIV-induced sensory neuropathy. To test this, we submitted the rats to 3 intrathecal injections of the HIV-1 envelope glycoprotein (gp120) resulting in the development of mechanical allodynia [52; 56; 90]. Intrathecal injection of BA (2 µg/5 μL, i.th.) reversed mechanical allodynia at 2 and 3 hours (Figure 11C). This reversal of mechanical allodynia is supported by a significant increase of the area under the curve between BA treated compared to vehicle (saline) treated animals (Figure 11D). We conclude that BA has an anti-nociceptive potential for HIV-induced sensory neuropathy.
3.12. Betulinic Acid reverses pSNL-induced sensory neuropathy behavior in mice.
To explore the species and gender related effects of betulinic acid in known animal pain models, we intrathecally administered betulinic acid 7 days post pSNL or sham surgeries in female C57BL/6NHsd mice. pSNL surgery evokes neuropathic pain in animals [50] and has been reported to involve plasticity in Cav2.2-mediated calcium currents [89]. Animals receiving a pSNL or sham surgery were administered intrathecally (via a lumbar puncture) 0.1% DMSO or betulinic acid (2 µg/5 μL). Betulinic acid had no effect on mechanical allodynia in sham mice, but significantly reversed mechanical allodynia at 2 and 3 hours post pSNL surgery (Figure 12A). Area under the curve analysis also revealed a reversal in mechanical allodynia in pSNL mice treated with BA (Figure 12B). Thus, we conclude that BA acts to suppress pSNL-induced neuropathic pain.
Figure 12. BA reduces pSNL-induced mechanical allodynia.

(A) Paw withdrawal threshold of adult female mice (n=3–4) was measured 7 days after pSNL or sham surgery as indicated. Mice were intrathecally (via lumbar puncture) treated with saline (vehicle) or BA (2 µg/5 μL, i.th.) as indicated. Asterisks indicate statistical significance compared with mice treated with saline (*p<0.05; 2-way ANOVA with a Student-Neuman–Kuels post hoc test). (B) Area under the curve was derived again as indicated before using Graphpad Prism. Statistical significance is indicated by asterisks (*p<0.05, Mann-Whitney test) in comparison to vehicle-treated rats.
3.13. Systemic administration of betulinic acid is ineffective in reducing paclitaxel-induced mechanical allodynia.
To further assess betulinic acid’s efficacy in the model of chemotherapy-induced peripheral neuropathy, we tested betulinic acid via intraperitoneal injection at two doses (5mg/kg; 20mg/kg). However, we found that betulinic acid failed to significantly reverse paclitaxel-induced mechanical allodynia at both of these doses (Figure 13). These experiments were largely limited by the solubility of the compound as betulinic acid is fairly insoluble. At doses higher than 20mg/kg, betulinic acid precipitated out of solution, and thus we propose that systemic administration of betulinic acid is ineffective in this paclitaxel model due to a dose-limiting issue. As betulinic acid could be a lead compound, upon which effective antinociceptive drugs may be modeled, the insolubility of betulinic acid must be taken into account when developing future pharmacotherapies based on this natural compound.
Figure 13. Systemic BA administration does not reverse paclitaxel-induced mechanical allodynia.

Paw withdrawal threshold of adult male rats (n=6) was measured 15 days after 4 intraperitoneal injections of paclitaxel. Rats were treated intraperitoneally (i.p.) with saline (vehicle) or two doses of BA (5 (A, B) or 20 mg/kg (C, D)). Time course and AUC are shown. No statistical significance was seen with BA (p>0.05; 2-way ANOVA with a Student-Neuman–Kuels post hoc test).
3.13. Betulinic acid has no effects on sedation or locomotion in the open field test.
To assess if betulinic acid had any effects on sedation or locomotion at therapeutic doses, we injected rats intrathecally with 0.1% DMSO or BA (2 µg/5 μL, i.th.) and monitored their activity in the open field test. Here, we chose rats treated with paclitaxel to assess if BA had any beneficial effect on locomotion. There was no difference in the times after i.th. injection that the rats entered the open field apparatus: 131.5 ± 15 min for 0.1% DMSO and 134.5 ± 12min for BA. Importantly, BA treatment did not modify total distance traveled, time in center zone or the rats’ maximum speed suggesting that this compound does not affect exploratory behavior, anxiety and locomotor activity, respectively, in animal previously treated with paclitaxel (Figure 14).
Figure 14. BA does not affect anxiety, locomotion, or exploratory behaviors of paclitaxel-treated rats.
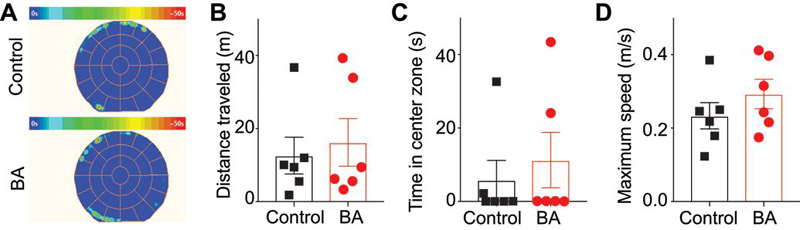
(A) Average heat maps of animals’ position for animals (n=6) treated with 0.1% DMSO (top) or BA (2 µg/5 μL) (bottom). Scatter plots of total distance traveled as a result of treatment with DMSO or BA (p=0.9372 Mann-Whitney test) (B), time in center zone as a result of treatment with DMSO or BA (p=0.8485, Mann-Whitney test) (C), and maximum speed of animals as a result of treatment with DMSO or BA (p=0.4848, Mann-Whitney test) (D). There were no statistical differences between the two conditions for any of the parameters tested.
4. Discussion
The results described here uncovered a new role for the natural product betulinic acid in antinociception in experimental models of neuropathic pain. We elucidated signaling mechanisms responsible for these effects of betulinic acid providing evidence for inhibition of low voltage-activated (Cav3, T-type) channels and release of calcitonin gene related peptide (CGRP). Importantly, we also showed that betulinic acid does not inhibit sodium channels nor does it engage opioid receptors. The present findings provide compelling evidence that blockade of Cav3.2, Cav3.3, and Cav2.2 by betulinic acid is sufficient to reverse mechanical allodynia induced by two neuropathic pain states without any motoric effects and further substantiate the development of betulinic acid as a possible naturally-derived therapeutic for chronic pain.
T-type Ca2+ channels are low-voltage-activated calcium channels (Cav3) with a half activation voltage of ~ –45 mV [79]. They conduct relatively small and transient currents that support their involvement in neuronal excitability. Mammals express three different T-type calcium channel α1 subunits – Cav3.1, Cav3.2 and Cav3.3 – with overlapping pharmacological properties, but somewhat distinct biophysical properties and distributions [42; 45]. The predominant Cav3 channel isoform involved in pain signaling is Cav3.2 [4; 6], with expression in specific subpopulations of primary afferent neurons [27]. T-type Ca2+ channel activity is increased in afferent pain fibers in various chronic pain states, such as traumatic nerve injury, diabetic neuropathy, and chemotherapy-induced neuropathies [92]. Given the roles of these channels in regulating afferent fiber excitability and synaptic function in the spinal dorsal horn and their dynamic regulation during pain states, it is likely that blocking or depleting Cav3.2 channels in these tissues should mediate analgesic effects [7]. Silencing of Cav3.2 calcium channels (but not Cav3.1 or Cav3.3 channels) protects from mechanical hypersensitivity in nerve-injured mice [6]. Intrathecal administration of antisense oligonucleotide of Cav3.2 and Cav3.3, but not Cav3.1, significantly relieved tactile allodynia and thermal hyperalgesia in rats with chronic compression of the dorsal root ganglion [83]. Importantly, systemic or intrathecal delivery of T-type calcium channel blockers such as ethosuximide [15; 29; 72], mibefradil, TTA-A2, and TTA-P2 [16; 78] mediates analgesia in rodents. Therefore, T-type Ca2+ channels represent high profile drug targets for pain therapies including, but not limited to, chemotherapy-induced pain [39; 72]. Despite good efficacy in rodent models of acute, inflammatory and chronic pain, T-type Ca2+ channel blockers that have advanced to human pain clinical trials remain limited. Two novel T-type channel blockers (ABT-639 [78] and MK-8998 [23]) have failed in human clinical trials, and it is unclear if another compound (Z944)[44; 81] will be advanced into human Phase II trials and, if so, whether it would show efficacy. Betulinic acid, by targeting both Cav3.2 and Cav3.3 channels, may offer an advantage over targeting either channel alone. That BA also targets N-type Ca2+ channels, which are well-established to play a role in nociceptive signaling [93], increases the therapeutic promise of this natural compound. While betulinic acid does not inhibit sodium channels in sensory neurons, the constellation pharmacology results point to a possible effect on ATP- and acetylcholine-responding dorsal root ganglion neurons, suggesting that other cell populations contribute to the anti-nociceptive effect of this natural compound. Betulinic acid blocks N-type calcium channels inferred from our electrophysiological recordings in sensory neurons (Figure 3), the modest inhibition of calcium influx evoked by 90 mM KCl (Figure 2), which presumably recruits high-voltage activated calcium channels, including N-type [84], and the inhibition of N-type calcium channel current in tsA-201 cells (Figure 5). The inhibition of sEPSC frequency, but not amplitude, further supports a presynaptic mechanism for betulinic acid.
Multiple lines of evidence suggest that betulinic acid has broad-spectrum actions and is thus important in diverse biological pathways. For example, betulinic acid has been reported to possess anti-cancer, anti-viral, anti-HIV, anti-inflammatory, anti-leukemic, anti-diabetic, anti-hyperlipidemic, anti-bacterial, and anti-mycobacterial activities [5; 25; 26; 38; 41; 71; 86; 87; 94]. Betulinic acid has also been shown to be effective in reducing effects of metabolic syndrome and preventative in diabetic nephropathy [1; 65]. It was reportedly protective against ischemic complications as well as a potential therapy for Alzheimer’s disease, atherosclerosis and other vascular diseases [35; 36; 59]. Betulinic acid promotes osteogenesis and inhibits adipogenesis [11]. Our study is the first to demonstrate an antinociceptive activity of betulinic acid in neuropathic pain. It is tempting to speculate that the antinociceptive actions of betulinic acid in reversing mechanical allodynia in gp120-induced model of HIV-induced peripheral neuropathy may, in part, be due to its action as an HIV-1 entry inhibitor [86]. Thus, by intervening with both HIV-1’s viral activity and associated neuropathy, betulinic acid might serve as a disease-modifying therapy to provide more than just symptomatic relief.
While betulinic acid is broadly effective, its poor solubility in aqueous environments, limited half-life [67; 68], and complex purification protocols [74] have precluded its development as a therapeutic. To work around these limitations of betulinic acid, studies have focused on enhancing the half-life and solubility of the compound by using delivery systems such as nanoparticles, nanoemulsions, liposomes, etc. [66]. Yet others are structurally modifying betulinic acid to identify derivatives that have anti-HIV/cancer activity with improved pharmacokinetic profiles and high safety index [18].
The high rate of chronic pain in the US has contributed in part to the current opioid epidemic. Results from a recent randomized clinical trial do not support initiation of opioid therapy for moderate to severe chronic back pain or hip or knee osteoarthritis pain [40]. Furthermore, likely due to the issues of addiction associated with opioids, misuse of opioid prescriptions affects a large population of the country; upwards of 12 million people in the United States reported prescription misuse in 2015 [30]. While the control of opioid prescription is likely improving, the demand for pain relief necessitates other effective measures for pain relief to assist in the amelioration of adverse effects of opioids, and to drive down the necessity for opioid prescription to begin with. Here, natural products such as betulinic acid could be an answer. Mechanistically, betulinic acid offers a departure from opioid mechanisms in that first, it lacks perceivable binding to opioid receptors, and second, it targets T-type Ca2+ channels. While N-type calcium channels have been shown to associate with opioid receptors, there are no reports on specifically linking T-type Ca2+ channels to opioid signaling pathways [49]. Between 1981 and 2014, 21% (n=326) of all drugs approved by the FDA (n=1562) were natural products and an additional 20% (n=320) were derivatives from natural products [60]. Thus, plant-based small molecule natural products such as betulinic acid and its soluble derivatives, may have the potential as non-opioid therapeutics for management of chronic pain.
Supplementary Material
Acknowledgments
Acknowledgements – This work was supported by National Institutes of Health awards (R01NS098772 from the National Institute of Neurological Disorders and Stroke and R01DA042852 from the National Institute on Drug Abuse to R.K.); and a Neurofibromatosis New Investigator Award from the Department of Defense Congressionally Directed Military Medical Research and Development Program (NF1000099); and a Children’s Tumor Foundation NF1 Synodos award (2015–04-009A) to R.K. A.M. was supported by a Young Investigator’s Award from the Children’s Tumor Foundation (2015–01-011). G.W.Z. is a Canada Research Chair and supported by a Foundation grant from the Canadian Institutes for Health Research, M.A.G. is supported by a Fellowship from Alberta Innovates and Canadian Institutes for Health Research. We thank the College of Agriculture and Life Sciences (University of Arizona) for financial support (to A.A.L.G.). The picture of the desert lavender was taken by Gabriel Jude Khanna.
Footnotes
Conflict of interest – There is no conflict of interest for any of the authors.
References
- [1].Ahangarpour A, Oroojan AA, Khorsandi L, Shabani R, Mojaddami S. Preventive effects of betulinic acid on streptozotocinnicotinamide induced diabetic nephropathy in male mouse. Journal of nephropathology 2016;5(4):128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Barsa JA, Sauders JC. Amitriptyline (Elavil), a new antidepressant. Am J Psychiatry 1961;117:739–740. [DOI] [PubMed] [Google Scholar]
- [3].Benyamin R, Trescot AM, Datta S, Buenaventura R, Adlaka R, Sehgal N, Glaser SE, Vallejo R. Opioid complications and side effects. Pain physician 2008;11(2 Suppl):S105–120. [PubMed] [Google Scholar]
- [4].Bernal Sierra YA, Haseleu J, Kozlenkov A, Begay V, Lewin GR. Genetic Tracing of Cav3.2 T-Type Calcium Channel Expression in the Peripheral Nervous System. Frontiers in molecular neuroscience 2017;10:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bildziukevich U, Rarova L, Saman D, Wimmer Z. Picolyl amides of betulinic acid as antitumor agents causing tumor cell apoptosis. European journal of medicinal chemistry 2018;145:41–50. [DOI] [PubMed] [Google Scholar]
- [6].Bourinet E, Alloui A, Monteil A, Barrere C, Couette B, Poirot O, Pages A, McRory J, Snutch TP, Eschalier A, Nargeot J. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J 2005;24(2):315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bourinet E, Francois A, Laffray S. T-type calcium channels in neuropathic pain. Pain 2016;157 Suppl 1:S15–22. [DOI] [PubMed] [Google Scholar]
- [8].Bourinet E, Soong TW, Stea A, Snutch TP. Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proceedings of the National Academy of Sciences of the United States of America 1996;93(4):1486–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, Liu N, Xiong W, Ripsch MS, Wang Y, Fehrenbacher JC, Fitz SD, Khanna M, Park CK, Schmutzler BS, Cheon BM, Due MR, Brustovetsky T, Ashpole NM, Hudmon A, Meroueh SO, Hingtgen CM, Brustovetsky N, Ji RR, Hurley JH, Jin X, Shekhar A, Xu XM, Oxford GS, Vasko MR, White FA, Khanna R. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca(2)(+) channel complex. Nature medicine 2011;17(7):822–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. The Journal of biological chemistry 2009;284(45):31375–31390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brusotti G, Montanari R, Capelli D, Cattaneo G, Laghezza A, Tortorella P, Loiodice F, Peiretti F, Bonardo B, Paiardini A, Calleri E, Pochetti G. Betulinic acid is a PPARgamma antagonist that improves glucose uptake, promotes osteogenesis and inhibits adipogenesis. Scientific reports 2017;7(1):5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Caceres AI, Gonzalez-Obeso E, Gonzalez C, Rocher A. RT-PCR and pharmacological analysis of L-and T-type calcium channels in rat carotid body. Advances in experimental medicine and biology 2009;648:105–112. [DOI] [PubMed] [Google Scholar]
- [13].Catterall WA. Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release. Cell calcium 1998;24(5–6):307–323. [DOI] [PubMed] [Google Scholar]
- [14].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994;53(1):55–63. [DOI] [PubMed] [Google Scholar]
- [15].Chen YL, Tsaur ML, Wang SW, Wang TY, Hung YC, Lin CS, Chang YF, Wang YC, Shiue SJ, Cheng JK. Chronic intrathecal infusion of mibefradil, ethosuximide and nickel attenuates nerve ligation-induced pain in rats. British journal of anaesthesia 2015;115(1):105–111. [DOI] [PubMed] [Google Scholar]
- [16].Choe W, Messinger RB, Leach E, Eckle VS, Obradovic A, Salajegheh R, Jevtovic-Todorovic V, Todorovic SM. TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. MolPharmacol 2011;80(5):900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Crawford P, Ghadiali E, Lane R, Blumhardt L, Chadwick D. Gabapentin as an antiepileptic drug in man. J Neurol Neurosurg Psychiatry 1987;50(6):682–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Csuk R Betulinic acid and its derivatives: a patent review (2008–2013). Expert opinion on therapeutic patents 2014;24(8):913–923. [DOI] [PubMed] [Google Scholar]
- [19].Deng Y, Balunas MJ, Kim JA, Lantvit DD, Chin YW, Chai H, Sugiarso S, Kardono LB, Fong HH, Pezzuto JM, Swanson SM, de Blanco EJ, Kinghorn AD. Bioactive 5,6-dihydro-alpha-pyrone derivatives from Hyptis brevipes. Journal of natural products 2009;72(6):1165–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dowell D, Haegerich TM, Chou R. CDC Guideline for Prescribing Opioids for Chronic Pain--United States, 2016. JAMA : the journal of the American Medical Association 2016;315(15):1624–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dustrude ET, Moutal A, Yang X, Wang Y, Khanna M, Khanna R. Hierarchical CRMP2 posttranslational modifications control NaV1.7 function. Proceedings of the National Academy of Sciences of the United States of America 2016;113(52):E8443–E8452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dustrude ET, Wilson SM, Ju W, Xiao Y, Khanna R. CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. The Journal of biological chemistry 2013;288(34):24316–24331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Egan MF, Zhao X, Smith A, Troyer MD, Uebele VN, Pidkorytov V, Cox K, Murphy M, Snavely D, Lines C, Michelson D. Randomized controlled study of the T-type calcium channel antagonist MK-8998 for the treatment of acute psychosis in patients with schizophrenia. Hum Psychopharmacol 2013;28(2):124–133. [DOI] [PubMed] [Google Scholar]
- [24].Facey PC, Porter RB, Reese PB, Williams LA. Biological activity and chemical composition of the essential oil from Jamaican Hyptis verticillata Jacq. Journal of agricultural and food chemistry 2005;53(12):4774–4777. [DOI] [PubMed] [Google Scholar]
- [25].Fadipe VO, Mongalo NI, Opoku AR, Dikhoba PM, Makhafola TJ. Isolation of anti-mycobacterial compounds from Curtisia dentata (Burm.f.) C.A.Sm (Curtisiaceae). BMC complementary and alternative medicine 2017;17(1):306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fontanay S, Grare M, Mayer J, Finance C, Duval RE. Ursolic, oleanolic and betulinic acids: antibacterial spectra and selectivity indexes. Journal of ethnopharmacology 2008;120(2):272–276. [DOI] [PubMed] [Google Scholar]
- [27].Francois A, Schuetter N, Laffray S, Sanguesa J, Pizzoccaro A, Dubel S, Mantilleri A, Nargeot J, Noel J, Wood JN, Moqrich A, Pongs O, Bourinet E. The Low-Threshold Calcium Channel Cav3.2 Determines Low-Threshold Mechanoreceptor Function. Cell Rep 2015. [DOI] [PubMed] [Google Scholar]
- [28].Francois-Moutal L, Wang Y, Moutal A, Cottier KE, Melemedjian OK, Yang X, Wang Y, Ju W, Largent-Milnes TM, Khanna M, Vanderah TW, Khanna R. A membrane-delimited N-myristoylated CRMP2 peptide aptamer inhibits CaV2.2 trafficking and reverses inflammatory and postoperative pain behaviors. Pain 2015;156(7):1247–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hamidi GA, Ramezani MH, Arani MN, Talaei SA, Mesdaghinia A, Banafshe HR. Ethosuximide reduces allodynia and hyperalgesia and potentiates morphine effects in the chronic constriction injury model of neuropathic pain. European journal of pharmacology 2012;674(2–3):260–264. [DOI] [PubMed] [Google Scholar]
- [30].Han B, Compton WM, Blanco C, Crane E, Lee J, Jones CM. Prescription Opioid Use, Misuse, and Use Disorders in U.S. Adults: 2015 National Survey on Drug Use and Health. Annals of internal medicine 2017;167(5):293–301. [DOI] [PubMed] [Google Scholar]
- [31].Hassett AL, Aquino JK, Ilgen MA. The risk of suicide mortality in chronic pain patients. Current pain and headache reports 2014;18(8):436. [DOI] [PubMed] [Google Scholar]
- [32].Ibrahim MM, Patwardhan A, Gilbraith KB, Moutal A, Yang X, Chew LA, Largent-Milnes T, Malan TP, Vanderah TW, Porreca F, Khanna R. Long-lasting antinociceptive effects of green light in acute and chronic pain in rats. Pain 2017;158(2):347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Institute of Medicine Report from the Committee on Advancing Pain Research C, Education. Relieving Pain in America, A Blueprint for Transforming Prevention, Care, Education and Research: The National Academies Press, 2011. [PubMed] [Google Scholar]
- [34].Jacus MO, Uebele VN, Renger JJ, Todorovic SM. Presynaptic Cav3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 2012;32(27):9374–9382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jiao S, Zhu H, He P, Teng J. Betulinic acid protects against cerebral ischemia/reperfusion injury by activating the PI3K/Akt signaling pathway. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2016;84:1533–1537. [DOI] [PubMed] [Google Scholar]
- [36].Jin SW, Choi CY, Hwang YP, Kim HG, Kim SJ, Chung YC, Lee KJ, Jeong TC, Jeong HG. Betulinic Acid Increases eNOS Phosphorylation and NO Synthesis via the Calcium-Signaling Pathway. Journal of agricultural and food chemistry 2016;64(4):785–791. [DOI] [PubMed] [Google Scholar]
- [37].Kang XJ, Chi YN, Chen W, Liu FY, Cui S, Liao FF, Cai J, Wan Y. Increased expression of CaV3.2 T-type calcium channels in damaged DRG neurons contributes to neuropathic pain in rats with spared nerve injury. Mol Pain 2018;14:1744806918765808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kim J, Lee YS, Kim CS, Kim JS. Betulinic acid has an inhibitory effect on pancreatic lipase and induces adipocyte lipolysis. Phytotherapy research : PTR 2012;26(7):1103–1106. [DOI] [PubMed] [Google Scholar]
- [39].Kopecky BJ, Liang R, Bao J. T-type calcium channel blockers as neuroprotective agents. Pflugers Arch 2014;466(4):757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Krebs EE, Gravely A, Nugent S, Jensen AC, DeRonne B, Goldsmith ES, Kroenke K, Bair MJ, Noorbaloochi S. Effect of Opioid vs Nonopioid Medications on Pain-Related Function in Patients With Chronic Back Pain or Hip or Knee Osteoarthritis Pain: The SPACE Randomized Clinical Trial. JAMA : the journal of the American Medical Association 2018;319(9):872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kumar D, Mallick S, Vedasiromoni JR, Pal BC. Anti-leukemic activity of Dillenia indica L. fruit extract and quantification of betulinic acid by HPLC. Phytomedicine : international journal of phytotherapy and phytopharmacology 2010;17(6):431–435. [DOI] [PubMed] [Google Scholar]
- [42].Lambert RC, Bessaih T, Leresche N. Modulation of neuronal T-type calcium channels. CNSNeurolDisordDrug Targets 2006;5(6):611–627. [DOI] [PubMed] [Google Scholar]
- [43].Lamblin F, Hano C, Fliniaux O, Mesnard F, Fliniaux MA, Laine E. [Interest of lignans in prevention and treatment of cancers]. Medecine sciences : M/S 2008;24(5):511–519. [DOI] [PubMed] [Google Scholar]
- [44].Lee M Z944: a first in class T-type calcium channel modulator for the treatment of pain. Journal of the peripheral nervous system : JPNS 2014;19 Suppl 2:S11–12. [DOI] [PubMed] [Google Scholar]
- [45].Leresche N, Lambert RC. T-type calcium channels in synaptic plasticity. Channels (Austin) 2017;11(2):121–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li Y, Tatsui CE, Rhines LD, North RY, Harrison DS, Cassidy RM, Johansson CA, Kosturakis AK, Edwards DD, Zhang H, Dougherty PM. Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. Pain 2017;158(3):417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lindsley CW. 2014 global prescription medication statistics: strong growth and CNS well represented. ACS chemical neuroscience 2015;6(4):505–506. [DOI] [PubMed] [Google Scholar]
- [48].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25(4):402–408. [DOI] [PubMed] [Google Scholar]
- [49].Mahmoud S, Farrag M, Ruiz-Velasco V. Ggamma7 proteins contribute to coupling of nociceptin/orphanin FQ peptide (NOP) opioid receptors and voltage-gated Ca(2+) channels in rat stellate ganglion neurons. Neuroscience letters 2016;627:77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Malmberg AB, Basbaum AI. Partial sciatic nerve injury in the mouse as a model of neuropathic pain: behavioral and neuroanatomical correlates. Pain 1998;76(1–2):215–222. [DOI] [PubMed] [Google Scholar]
- [51].Manji H Neuropathy in HIV infection. Current opinion in neurology 2000;13(5):589–592. [DOI] [PubMed] [Google Scholar]
- [52].Milligan ED, O’Connor KA, Nguyen KT, Armstrong CB, Twining C, Gaykema RP, Holguin A, Martin D, Maier SF, Watkins LR. Intrathecal HIV-1 envelope glycoprotein gp120 induces enhanced pain states mediated by spinal cord proinflammatory cytokines. The Journal of neuroscience : the official journal of the Society for Neuroscience 2001;21(8):2808–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Moore M Medicinal plants of the desert and canyon west, 1989. [Google Scholar]
- [54].Moutal A, Chew LA, Yang X, Wang Y, Yeon SK, Telemi E, Meroueh S, Park KD, Shrinivasan R, Gilbraith KB, Qu C, Xie JY, Patwardhan A, Vanderah TW, Khanna M, Porreca F, Khanna R. (S)-lacosamide inhibition of CRMP2 phosphorylation reduces postoperative and neuropathic pain behaviors through distinct classes of sensory neurons identified by constellation pharmacology. Pain 2016;157(7):1448–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Moutal A, Honnorat J, Massoma P, Desormeaux P, Bertrand C, Malleval C, Watrin C, Chounlamountri N, Mayeur ME, Besancon R, Naudet N, Magadoux L, Khanna R, Ducray F, Meyronet D, Thomasset N. CRMP5 Controls Glioblastoma Cell Proliferation and Survival through Notch-Dependent Signaling. Cancer Res 2015;75(17):3519–3528. [DOI] [PubMed] [Google Scholar]
- [56].Moutal A, Li W, Wang Y, Ju W, Luo S, Cai S, Francois-Moutal L, Perez-Miller S, Hu J, Dustrude ET, Vanderah TW, Gokhale V, Khanna M, Khanna R. Homology-guided mutational analysis reveals the functional requirements for antinociceptive specificity of collapsin response mediator protein 2-derived peptides. British journal of pharmacology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Moutal A, Wang Y, Yang X, Ji Y, Luo S, Dorame A, Bellampalli SS, Chew LA, Cai S, Dustrude ET, Keener JE, Marty MT, Vanderah TW, Khanna R. Dissecting the role of the CRMP2-neurofibromin complex on pain behaviors. Pain 2017;158(11):2203–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Munz PA. A California flora. Berkeley,: Published for the Rancho Santa Ana Botanic Garden by the University of California Press, 1959. [Google Scholar]
- [59].Navabi SP, Sarkaki A, Mansouri E, Badavi M, Ghadiri A, Farbood Y. The effects of betulinic acid on neurobehavioral activity, electrophysiology and histological changes in an animal model of the Alzheimer’s disease. Behavioural brain research 2018;337:99–106. [DOI] [PubMed] [Google Scholar]
- [60].Newman DJ, Cragg GM. Natural Products as Sources of New Drugs from 1981 to 2014. Journal of natural products 2016;79(3):629–661. [DOI] [PubMed] [Google Scholar]
- [61].Newshan G HIV neuropathy treated with gabapentin. AIDS 1998;12(2):219–221. [PubMed] [Google Scholar]
- [62].Novelo M, Cruz JG, Hernandez L, Pereda-Miranda R, Chai H, Mar W, Pezzuto JM. Cytotoxic constituents from Hyptis verticillata. Journal of natural products 1993;56(10):1728–1736. [DOI] [PubMed] [Google Scholar]
- [63].Polomano RC, Mannes AJ, Clark US, Bennett GJ. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain 2001;94(3):293–304. [DOI] [PubMed] [Google Scholar]
- [64].Prut L, Belzung C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur J Pharmacol 2003;463(1–3):3–33. [DOI] [PubMed] [Google Scholar]
- [65].Rios JL, Manez S. New Pharmacological Opportunities for Betulinic Acid. Planta medica 2018;84(1):8–19. [DOI] [PubMed] [Google Scholar]
- [66].Saneja A, Arora D, Kumar R, Dubey RD, Panda AK, Gupta PN. Therapeutic applications of betulinic acid nanoformulations. Annals of the New York Academy of Sciences 2018. [DOI] [PubMed] [Google Scholar]
- [67].Saneja A, Kumar R, Singh A, Dhar Dubey R, Mintoo MJ, Singh G, Mondhe DM, Panda AK, Gupta PN. Development and evaluation of long-circulating nanoparticles loaded with betulinic acid for improved anti-tumor efficacy. International journal of pharmaceutics 2017;531(1):153–166. [DOI] [PubMed] [Google Scholar]
- [68].Saneja A, Sharma L, Dubey RD, Mintoo MJ, Singh A, Kumar A, Sangwan PL, Tasaduq SA, Singh G, Mondhe DM, Gupta PN. Synthesis, characterization and augmented anticancer potential of PEG-betulinic acid conjugate. Materials science & engineering C, Materials for biological applications 2017;73:616–626. [DOI] [PubMed] [Google Scholar]
- [69].Sekiguchi F, Kawara Y, Tsubota M, Kawakami E, Ozaki T, Kawaishi Y, Tomita S, Kanaoka D, Yoshida S, Ohkubo T, Kawabata A. Therapeutic potential of RQ-00311651, a novel T-type Ca2+ channel blocker, in distinct rodent models for neuropathic and visceral pain. Pain 2016;157(8):1655–1665. [DOI] [PubMed] [Google Scholar]
- [70].Sheth K, Jolad S, Wiedhopf R, Cole JR. Tumor-inhibitory agent from Hyptis emoryi (Labiatae). J Pharm Sci 1972;61(11):1819. [DOI] [PubMed] [Google Scholar]
- [71].Silva FS, Oliveira PJ, Duarte MF. Oleanolic, Ursolic, and Betulinic Acids as Food Supplements or Pharmaceutical Agents for Type 2 Diabetes: Promise or Illusion? Journal of agricultural and food chemistry 2016;64(15):2991–3008. [DOI] [PubMed] [Google Scholar]
- [72].Snutch TP, Zamponi GW. Recent advances in the development of T-type calcium channel blockers for pain intervention. British journal of pharmacology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Stefanucci A, Lei W, Hruby VJ, Macedonio G, Luisi G, Carradori S, Streicher JM, Mollica A. Fluorescent-labeled bioconjugates of the opioid peptides biphalin and DPDPE incorporating fluorescein-maleimide linkers. Future Med Chem 2017;9(9):859–869. [DOI] [PubMed] [Google Scholar]
- [74].Sun ZL, He JM, Lan JE, Mu Q. High-Speed Counter-Current Chromatography with an Online Storage Technique for the Preparative Isolation and Purification of Dihydroflavonoids from Sophora alopecuroides L. Phytochemical analysis : PCA 2017;28(6):496–504. [DOI] [PubMed] [Google Scholar]
- [75].Tanowitz BD, Junak SA, Smith DM. Terpenoids of Hyptis emoryi. J Nat Prod 1984;47(4):739–740. [DOI] [PubMed] [Google Scholar]
- [76].Teichert RW, Memon T, Aman JW, Olivera BM. Using constellation pharmacology to define comprehensively a somatosensory neuronal subclass. Proceedings of the National Academy of Sciences of the United States of America 2014;111(6):2319–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Teichert RW, Schmidt EW, Olivera BM. Constellation pharmacology: a new paradigm for drug discovery. Annu Rev Pharmacol Toxicol 2015;55:573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Teleb M, Zhang FX, Huang J, Gadotti VM, Farghaly AM, AboulWafa OM, Zamponi GW, Fahmy H. Synthesis and biological evaluation of novel N3-substituted dihydropyrimidine derivatives as T-type calcium channel blockers and their efficacy as analgesics in mouse models of inflammatory pain. Bioorganic & medicinal chemistry 2017;25(6):1926–1938. [DOI] [PubMed] [Google Scholar]
- [79].Todorovic SM, Jevtovic-Todorovic V. T-type voltage-gated calcium channels as targets for the development of novel pain therapies. British journal of pharmacology 2011;163(3):484–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Todorovic SM, Jevtovic-Todorovic V. T-type voltage-gated calcium channels as targets for the development of novel pain therapies. BrJPharmacol 2011;163(3):484–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Tringham E, Powell KL, Cain SM, Kuplast K, Mezeyova J, Weerapura M, Eduljee C, Jiang X, Smith P, Morrison JL, Jones NC, Braine E, Rind G, Fee-Maki M, Parker D, Pajouhesh H, Parmar M, O’Brien TJ, Snutch TP. T-type calcium channel blockers that attenuate thalamic burst firing and suppress absence seizures. Science translational medicine 2012;4(121):121ra119. [DOI] [PubMed] [Google Scholar]
- [82].Wallace MS. Ziconotide: a new nonopioid intrathecal analgesic for the treatment of chronic pain. Expert review of neurotherapeutics 2006;6(10):1423–1428. [DOI] [PubMed] [Google Scholar]
- [83].Wen XJ, Li ZJ, Chen ZX, Fang ZY, Yang CX, Li H, Zeng YM. Intrathecal administration of Cav3.2 and Cav3.3 antisense oligonucleotide reverses tactile allodynia and thermal hyperalgesia in rats following chronic compression of dorsal root of ganglion. Acta PharmacolSin 2006;27(12):1547–1552. [DOI] [PubMed] [Google Scholar]
- [84].Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW. Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell 2012;149(5):1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Woolf CJ, Hashmi M. Use and abuse of opioid analgesics: potential methods to prevent and deter non-medical consumption of prescription opioids. CurrOpinInvestigDrugs 2004;5(1):61–66. [PubMed] [Google Scholar]
- [86].Worachartcheewan A, Toropova AP, Toropov AA, Siriwong S, Prapojanasomboon J, Prachayasittikul V, Nanatasenamat C. Quantitative structure-activity relationship study of betulinic acid derivatives against HIV using SMILES-based descriptors. Curr Comput Aided Drug Des 2018. [DOI] [PubMed] [Google Scholar]
- [87].Xiao S, Tian Z, Wang Y, Si L, Zhang L, Zhou D. Recent progress in the antiviral activity and mechanism study of pentacyclic triterpenoids and their derivatives. Medicinal research reviews 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiology & behavior 1976;17(6):1031–1036. [DOI] [PubMed] [Google Scholar]
- [89].Yang L, Stephens GJ. Effects of neuropathy on high-voltage-activated Ca(2+) current in sensory neurones. Cell calcium 2009;46(4):248–256. [DOI] [PubMed] [Google Scholar]
- [90].Yuan SB, Shi Y, Chen J, Zhou X, Li G, Gelman BB, Lisinicchia JG, Carlton SM, Ferguson MR, Tan A, Sarna SK, Tang SJ. Gp120 in the pathogenesis of human immunodeficiency virus-associated pain. Annals of neurology 2014;75(6):837–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Yue J, Liu L, Liu Z, Shu B, Zhang Y. Upregulation of T-type Ca2+ channels in primary sensory neurons in spinal nerve injury. Spine (Phila Pa 1976) 2013;38(6):463–470. [DOI] [PubMed] [Google Scholar]
- [92].Zamponi GW. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nature reviews Drug discovery 2016;15(1):19–34. [DOI] [PubMed] [Google Scholar]
- [93].Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacological reviews 2015;67(4):821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Zeng AQ, Yu Y, Yao YQ, Yang FF, Liao M, Song LJ, Li YL, Yu Y, Li YJ, Deng YL, Yang SP, Zeng CJ, Liu P, Xie YM, Yang JL, Zhang YW, Ye TH, Wei YQ. Betulinic acid impairs metastasis and reduces immunosuppressive cells in breast cancer models. Oncotarget 2018;9(3):3794–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Zhang SH, Yu J, Lou GD, Tang YY, Wang RR, Hou WW, Chen Z. Widespread pain sensitization after partial infraorbital nerve transection in MRL/MPJ mice. Pain 2016;157(3):740–749. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.