

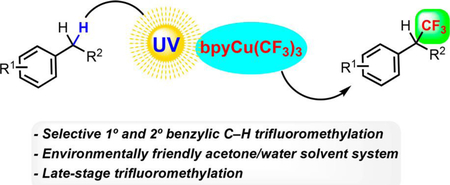
Abstract
The installation of trifluoromethyl groups has become an essential step across a number of industries such as agrochemicals, drug discovery, and materials. Consequently, the rapid introduction of this critical functional group in a predictable fashion would benefit current practitioners in those fields. This communication describes a mild trifluoromethylation of benzylic C−H bonds with high selectivity for the least hindered hydrogen atom. The reaction provides monotrifluoromethylation and proceeds in an environmentally friendly acetone/water solvent system. The method can be used to install benzylic trifluoromethyl groups on highly functionalized drug molecules.
Graphical Abstract
Facile installation of the trifluoromethyl group remains a critical goal for organic synthesis. The unusual and often desirable properties of the trifluoromethyl group (e.g., high electronegativity and Teflon-like stability) have propelled this moiety into the structures of a striking number of drugs, drug-candidates, agrochemicals, and materials.1 Ever since the McLoughlin−Thrower reaction,2 chemists have searched for easily implemented couplings to install the trifluoromethyl group.3 Excitingly, a number of recent reports describe novel trifluoromethylating reagents that offer the potential for new opportunities to trifluoromethylate organic molecules. The wide availability of the Ruppert−Prakash,4 Togni,5 Umemoto,6 Langlois,7 Grushin,8 Chen,9 Shibata,10 Baran11 and other reagents12 have provided chemists with numerous useful trifluoromethyl synthons with unique properties. Evaluating such reagents under traditional reaction conditions should enable rapid access to new chemical matter.
For aromatic trifluoromethylation, methods have primarily focused on metal-mediated couplings related to the McLoughlin−Thrower2,13 or radical addition to electron-deficient aromatic system to supplant individual Csp2−H bonds (Figure 1c).11,14 The introduction of aliphatic trifluoromethyl groups has lagged by comparison.3b,e, 15 For example, while fluoroform is reasonably acidic (pKa = 28),16 the simple SN2 reaction to install aliphatic trifluoromethyl groups remains troublesome due to the rapid decomposition of the CF3 anion to fluoride and difluorocarbene.17 Recent work has demonstrated that the use of radicals generated from aliphatic carboxylic acids,18 esters,19 or halides20 offers a more convenient path to aliphatic trifluoromethylation compared to metal-mediated coupling.15b A more direct approach would be to target Csp3−H bonds for trifluoromethylation without the use of preinstalled functional groups,21 a variant of which was published by Liu and co-workers during the preparation of this work.22
Figure 1.

The switch from aromatic bromination (a) to benzylic bromination (b) requires relatively minor changes to the reaction conditions. The modern switch from aromatic trifluoromethylation (c) to benzylic trifluoromethylation (d) has yet to be realized.
Classic halogenation chemistry uses subtle changes in reaction conditions to select for aromatic or benzylic functionalization (Figure 1a,b). While the halogenation of alkenes and aromatics was known in the 19th century (Figure 1a), it was the Wohl−Ziegler reaction that revolutionized our thinking on how subtle changes in reagents and conditions can have a drastic effect on the outcome of halogenation chemistry (Figure 1b).23 Mysterious at the time, the divergent selectivity stemmed from the unknown, but critical, mechanistic switch from two-electron to radical chemistry. Based on this classic work, we were curious whether some modern trifluormethylating reagents might be capable of quenching benzylic radicals generated directly from C−H bonds (Figure 1d).
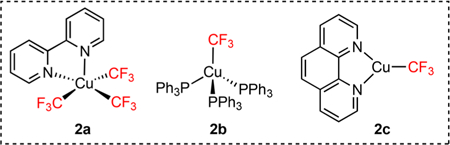
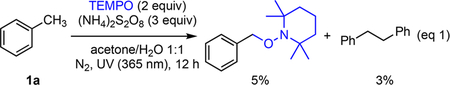
To start our investigations, we evaluated a variety of mild peroxides for their ability to generate benzylic radicals from toluene (1a). We found ammonium persulfate forms the TEMPO inclusion product and bibenzyl in modest yield (eq 1). Based on a number of recent reports on copper-catalyzed benzylic C−H functionalization,24 we reasoned that one of the newly introduced CuCF3 complexes 2a−2c might work in synergy with benzylic radical formation.8,12 To test this hypothesis, ammonium persulfate and Grushin’s reagent (2a) were heated to 50 °C in the presence of toluene (Table 1, entry 3). Interestingly, 6% of the desired product 3a formed in the reaction. Since UV light facilitates radical C−H halogenation,23a we exposed the reaction to a 365 nm LED bulb (Table 1, entry 4), which dramatically increased the yield to 58%. A variety of strong acids proved beneficial, with trifluoroacetic acid being the most convenient and high yielding (Table 1, entry 5). The presence of acid may facilitate the homolysis of Grushin’s reagent (2a) through the protonation of the bipy ligand. No product formed under a variety of reaction conditions with copper complex 2b (Table 1, entry 7), but complex 2c did provide detectable 4% yield (Table 1, entry 8). Based on the observation that aromatic trifluoromethylation was a competing pathway (Figure 1a), we reasoned that the CF3 radical byproduct from Grushin’s reagent (2a) was consuming 1a. Consequently, we sought a mild radical quenching agent that would be degenerate with benzyl radical. After evaluating a series of silanes (see Supporting Information), we found both triisopropylsilane and t-butyldimethylsilane dramatically increased the yield (Table 1, entries 1, 5, 9−10) while suppressing aromatic trifluoromethylation (Figure 1a). Consequently, the direct trifluoromethylation of toluene (1a) could be conducted in near quantitative yield based on 2a with equimolar ammonium persulfate/triisopropylsilane and 8 equiv of TFA25 in acetone/water solvent system (Table 1, entry 1).
Table 1.
Optimization of Pertinent Reaction Parametersa
| Entry | conditions | Yield of 3a (%)b |
|---|---|---|
| 1 | standard conditions | 99% |
| 2 | without (NH4)2S208 | 0% |
| 3 | 50°C intead of UV | 6% |
| 4 | without TFA andiPr3SiH | 58% |
| 5 | withoutiPr3SiH | 78% |
| 6 | without TFA | 62% |
| 7 | 2b instead of 2a | 0% |
| 8 | 2c instead of 2a | 4% |
| 9 | Et3SiH instead ofiPr3SiH | 60% |
| 10 | tBuMe2SiH instead of’iPr3SiH | 81% |
 | ||
Unless otherwise noted, all the reactions were run with 1a (0.6 mmol) and 2a (0.3 mmol) in 3.0 mL of solvent for 18 h.
Yields were determined by19F NMR spectroscopy with 1-chloro-4-fluorobenzene as the internal standard.
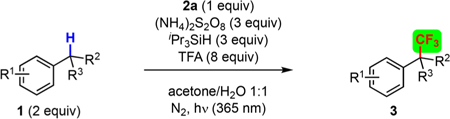
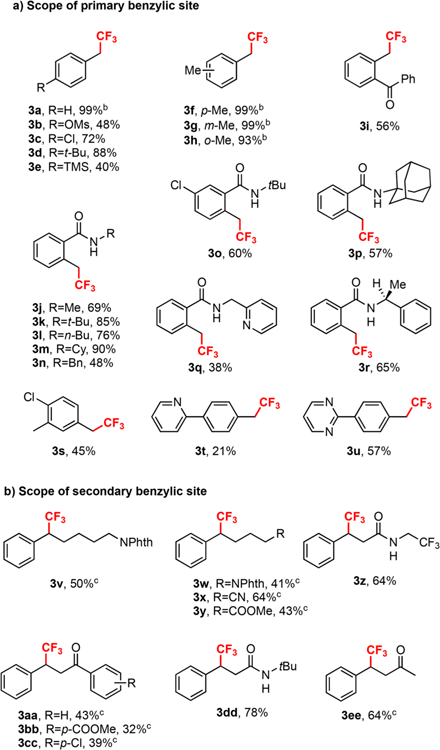
After establishing the optimal reaction conditions for the trifluoromethylation of toluene, we next evaluated a range of benzylic C−H bonds (Table 2). Interestingly, the reaction did not track the reactivity expected of benzylic C−H radical abstraction.23a That is to say, the reaction showed a clear dependence on the steric environment of the benzylic C−H bond, whereby primary benzylic C−H bonds reacted faster and in higher yield than secondary benzylic C−H bonds (Table 2a vs 2b). Moreover, tertiary benzylic or tertiary unactivated C−H bonds showed no trifluoromethylation under our standard reaction conditions. This surprising result allows practitioners to reliably and selectively target specific benzylic C−H bonds in more complicated systems. Moreover, the reaction offers a monoselective trifluoromethylation, with excess Grushin’s reagent (2a) providing only minimal ditrifluoromethylation. Even in cases with multiple, identical benzylic C−H sites, the monotrifluoromethylation could be achieved in high yield (Table 2a, 3f−3h). The reaction tolerated a range of functional groups, including halides (3c, 3o, 3s, and 3cc), pseudohalides (3b), ketones (3i, 3aa−3cc, and 3ee), esters (3y and 3bb), amides (3j−3r, 3z, and 3dd), aliphatic nitriles (3x), phthalimides (3v and 3w), pyridines (3q and 3t), pyrimidines (3u) and silanes (3e). Moreover, substrates 1 could be used as the limiting reagent with only 10−15% loss in yield (1.5 equiv 2a), if the substrate is of particular value. Interestingly, there were occasions where seemingly valid substrates (simple primary or secondary benzylic C−H bonds with similar structural features) failed to undergo trifluoromethylation (see SI). Consequently, a greater mechanistic understanding was needed to reliably target requisite C−H bonds.
Table 2.
Substrate Scope for Trifluoromethylationa
|
All reactions were run on 0.3 mmol scale with 1a (0.6 mmol) and 2a(0.3 mmol) in 3.0 mL of solvent for 18 h unless otherwise noted. Isolated yield.
Yield determined by19F NMR spectroscopy with 1-bromo-4-fluorobenzene as the internal standard.
2.0 equiv of tBuMe2SiH instead of 3.0 equiv of iPr3SiH, and 4.0 equiv of (NH4)2S2O8 were used. Cy = cyclohexyl, Phth = phthalimido.
To understand the observed C−H selectivity, we probed the underlying mechanism for trifluoromethylation of primary, secondary, and tertiary benzylic C−H bonds (Figure 2). Experimentally, we observed that tertiary benzylic substrates react to produce benzylic hydroxylation and styrene (see SI), providing evidence for the formation of a tertiary benzylic radical that is slow to recombine with Cu(II). To better understand this observation, we calculated the relative energies for the reaction coordinate of primary (1p), secondary (1s), and tertiary (1t) radicals. While the recombination with bipyCu(CF3)2 (4) is thermodynamically favorable for primary (1p) and secondary (1s) by −4.2 and −4.0 kcal/mol, respectively, the lowest energy cumenyl-copper(III) species (5t) was +5.6 kcal/mol less stable than 1t. This thermodynamically uphill radical Cu−C recombination is surprising, but reflects the unfavorable steric hindrance in 5t. Moreover, the rate-determining reductive elimination is definitively higher for tertiary 5t-TS but not so unfavorable as to subvert the overall reaction at 23 °C. Consequently, the energetically uphill recombination of the tertiary radicals with Cu(II) (4) allows unfavorable side reactions (e.g., oxidation/elimination to form styrene) before productive reductive elimination can occur.
Figure 2.

Computed energy profile of C−H selectivity. Bipyridineligated Cu illustrated here possessed the highest energy barriers relative to water- or unligated Cu.
While we constructed this reaction through analogy to the Wohl−Ziegler reaction, a detailed picture for our current mechanistic understanding arose through key mechanistic experiments (Figure 3). The UV light serves multiple roles, facilitating persulfate cleavage and homolysis of 2a to form the active Cu(II) species (4) and CF3 radical (Figure 3).20 While both sulfate radical anion and silyl radical could form the benzylic radical (1-rad), H-atom abstraction with sulfate is −12.9 kcal/mol more favorable thermodynamically than with silyl radical (see SI). We were intrigued by the production of significant quantities of fluoroform in the reaction. Examination of deuterated toluene (d8−1a), silane, and water revealed CDCF3 forms from the reaction of CF3 radical with silane (see SI). These experiments explain the underlying mechanism for the ability of silane to suppress aromatic trifluoromethylation (Figure 1c), through the quenching of CF3 radical (see SI). Subsequent steps remain favorable whether the Cu(II) species is unligated, complexed with bpy, or the aqua complex 5 (see SI). Since the Cu(II) forms in acidic water, aqua complex 5 represents the most relevant intermediate along the reaction coordinate. Since 1-rad recombination with the Cu(CF3)2 (4) is only ~4 kcal/mol, we ruled out the outersphere mechanism (i.e., where 1-rad abstractsa CF3 directly), which would requirea >61 kcal/mol transition state (see SI). Finally, the reductive elimination continues through a low 10.5−21.8 kcal/mol barrier (depending on ligation state, see SI) to form desired product 3 and unreactive 6 (cf. 2c in Table 1).
Figure 3.

Mechanistic details.
The primary kinetic isotope effect (KIE) of kH/kD = 2.8 observed for the intermolecular, same-flask competition of toluene (1a) and toluene-d8 (d8-1a) suggests that the C−H bond-cleavage step or a prior step is rate determining (see SI).26 In combination with the kH/kD = 4.2 for the intramolecular KIE of mono-CD3 p-xylene (d3−1f), the homolysis of persulfate at 23 °C under 365 nm light offers the most likely rate-determining step for the overall reaction. While similar to radical halogenation,27 the facility with which the benzylic radical is quenched with trifluoromethylcopper reagent 4 is remarkable.
The clear advantage of this reaction is the ability to predictably incorporate the trifluoromethyl group at benzylic sites of elaborated molecules. To test this methodology, we subjected several biologically relevant small molecules trifluoromethylation (Scheme 1). Trifluoromethylation of protected 4-methylphenylglycine, meclizine, or celecoxib provided novel, undisclosed CF3 analogs in all cases. Clearly, the method should find immediate use for the facile trifluoromethylation of important molecules.
Scheme 1. Late-Stage C−H Trifluoromethylation of Bioactive Molecules.

aThe reactions were run on 0.3 mmol scale with substrate (0.6 mmol) and 2a (0.3 mmol) in 3.0 mL of solvent for 18 h unless otherwise noted. Isolated yield.b10 equiv of TFA was used.
In summary, we developed a mild method for the trifluoromethylation of unhindered benzylic C−H bonds. The method tolerates a wide range of functional groups and basic heterocycles and can be used in the late-stage trifluoromethylation of bioactive molecules. Moreover, the reaction proceeds in an environmentally friendly 1:1 acetone/water mixture. Detailed mechanistic analysis offers a framework to understand selectivity of Grushin’s reagent 2a toward trifluoromethylation of primary and secondary benzylic hydrogens.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge funds from Indiana University in partial support of this work. We also gratefully acknowledge the NSF CAREER Award (CHE-1254783), NIH (GM121668) and the NSF MRI CHE-1726633. Eli Lilly & Co. and Amgen supported this work through the Lilly Grantee Award and the Amgen Young Investigator Award.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b08547.
Experimental details and spectroscopic data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Muller K; Faeh C; Diederich F Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]; (b) Purser S; Moore PR; Swallow S; Gouverneur V Chem. Soc. Rev 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]; (c) Kirk KL Org. Process Res. Dev 2008, 12, 305–321. [Google Scholar]; (d) Wang J; Sánchez-Roselló M; Aceña JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Chem. Rev 2014, 114, 2432–2506. [DOI] [PubMed] [Google Scholar]
- (2).(a) Mcloughlin VCR; Thrower J Tetrahedron 1969, 25, 5921–5940. [Google Scholar]; (b) Kobayashi Y; Kumadaki I Tetrahedron Lett. 1969, 10, 4095. [Google Scholar]
- (3).(a) Furuya T; Kamlet AS; Ritter T Nature 2011, 473, 470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Merino E; Nevado C Chem. Soc. Rev 2014, 43, 6598–6608. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Besset T; Schneider C; Cahard D Angew. Chem., Int. Ed 2012, 51, 5048–5050. [DOI] [PubMed] [Google Scholar]; (d) Barata-Vallejo S; Lantaño B; Postigo A Chem. - Eur.J 2014, 20, 16806–16829. [DOI] [PubMed] [Google Scholar]; (e) Alonso C; Martínez de Marigorta E; Rubiales G; Palacios F Chem. Rev 2015, 115, 1847–1935. [DOI] [PubMed] [Google Scholar]; (f) Xu X-H; Matsuzaki K; Shibata N Chem. Rev 2015, 115, 731–764. [DOI] [PubMed] [Google Scholar]; (g) Egami H;Sodeoka M Angew. Chem., Int. Ed 2014, 53, 8294–8308. [DOI] [PubMed] [Google Scholar]; (h) Chu L; Qing F-L Acc. Chem. Res 2014, 47, 1513–1522. [DOI] [PubMed] [Google Scholar]; (i) Tomashenko OA; Grushin VV Chem. Rev 2011, 111, 4475–4521. [DOI] [PubMed] [Google Scholar]; (j) Shibata N; Mizuta S; Kawai H Tetrahedron: Asymmetry 2008, 19, 2633–2644. [Google Scholar]; (k) Ma J-A; Cahard D Chem. Rev 2008, 108, PR1–PR43. [DOI] [PubMed] [Google Scholar]; (l) Ma J-A; Cahard D Chem. Rev 2004, 104, 6119–6146. [DOI] [PubMed] [Google Scholar]; (m) Kumadaki I; Ando A; Sato K; Tarui A; Omote M Synthesis 2010, 2010, 1865–1882. [Google Scholar]
- (4).(a) Rubiales G; Alonso C; de Marigorta EM; Palacios F Arkivoc 2014, 362–405. [Google Scholar]; (b) Liu X; Xu C; Wang M; Liu Q Chem. Rev 2015, 115, 683–730. [DOI] [PubMed] [Google Scholar]; (c) Ruppert I; Schlich K; Volbach W Tetrahedron Lett. 1984, 25, 2195–2198. [Google Scholar]; (d) Prakash GKS; Krishnamurti R; Olah GA J. Am. Chem. Soc 1989, 111, 393–395. [Google Scholar]
- (5).(a) Charpentier J; Fruh N; Togni A Chem. Rev 2015, 115, 650–682. [DOI] [PubMed] [Google Scholar]; (b) Eisenberger P; Gischig S; Togni A Chem. -Eur. J 2006, 12, 2579–2586. [DOI] [PubMed] [Google Scholar]
- (6).(a) Umemoto T; Ishihara S J. Am. Chem. Soc 1993, 115, 2156–2164. [Google Scholar]; (b) Umemoto T Chem. Rev 1996, 96, 1757–1778. [DOI] [PubMed] [Google Scholar]
- (7).(a) Zhang C Adv. Synth. Catal 2014, 356, 2895–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tordeux M; Langlois B; Wakselman CJ Org. Chem 1989, 54, 2452–2453. [Google Scholar]; (c) Langlois BR; Laurent E; Roidot N TetrahedronLett. 1991, 32, 7525–7528. [Google Scholar]
- (8).(a) Romine AM; Nebra N; Konovalov AI; Martin E; Benet-Buchholz J; Grushin VV Angew. Chem., Int. Ed 2015, 54, 2745–2749. [DOI] [PubMed] [Google Scholar]; (b) Tomashenko OA; Escudero-Adan EC; Belmonte MM; Grushin VV Angew. Chem., Int. Ed 2011, 50, 7655–7659. [DOI] [PubMed] [Google Scholar]
- (9).Chen Q-Y; Wu S-WJ Chem. Soc., Chem. Commun 1989, 705. [Google Scholar]
- (10).Noritake S; Shibata N; Nakamura S; Toru T; Shiro M Eur. J. Org. Chem 2008, 2008, 3465–3468. [Google Scholar]
- (11).Fujiwara Y; Dixon JA; O’Hara F; Funder ED; Dixon DD; Rodriguez RA; Baxter RD; Herle B; Sach N; Collins MR; Ishihara Y; Baran PS Nature 2012, 492, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Morimoto H; Tsubogo T; Litvinas ND; Hartwig JF Angew. Chem., Int. Ed 2011, 50, 3793–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Cho EJ; Senecal TD; Kinzel T; Zhang Y; Watson DA; Buchwald SL Science 2010, 328, 1679–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Roy S; Gregg BT; Gribble GW; Le V-D; Roy S Tetrahedron 2011, 67, 2161–2195. [Google Scholar]
- (14).(a) Studer A Angew. Chem., Int. Ed 2012, 51, 8950–8958. [DOI] [PubMed] [Google Scholar]; (b) Li L; Mu X; Liu W; Wang Y; Mi Z; Li CJ J. Am. Chem. Soc 2016, 138, 5809–5812. [DOI] [PubMed] [Google Scholar]; (c) Liu P; Liu W; Li CJ J. Am. Chem. Soc 2017, 139, 14315–14321. [DOI] [PubMed] [Google Scholar]
- (15).(a) Koike T; Akita M Acc. Chem. Res 2016, 49, 1937–1945. [DOI] [PubMed] [Google Scholar]; (b) Xu J; Liu X; Fu Y Tetrahedron Lett. 2014, 55, 585–594. [Google Scholar]; (c) Li L; Chen Q-Y; Guo YJ Fluorine Chem. 2014, 167, 79–83. [Google Scholar]
- (16).Symons EA; Clermont MJ J. Am. Chem. Soc 1981, 103, 3127–3130. [Google Scholar]
- (17).Prakash GK; Jog PV; Batamack PT; Olah GA Science 2012, 338, 1324–1327. [DOI] [PubMed] [Google Scholar]
- (18).(a) Kautzky JA; Wang T; Evans RW; MacMillan DWC J. Am. Chem. Soc 2018, 140, 6522. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tan X; Liu Z; Shen H; Zhang P; Zhang Z; Li C J.Am. Chem. Soc 2017, 139, 12430–12433. [DOI] [PubMed] [Google Scholar]
- (19).Ambler BR; Zhu L; Altman RA J. Org. Chem 2015, 80, 8449–8457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Shen H; Liu Z; Zhang P; Tan X; Zhang Z; Li CJ Am. Chem. Soc 2017, 139, 9843–9846. [DOI] [PubMed] [Google Scholar]
- (21).(a) Ide T; Masuda S; Kawato Y; Egami H; Hamashima Y Org. Lett 2017, 19, 4452–4455. [DOI] [PubMed] [Google Scholar]; (b) Egami H; Ide T; Kawato Y; Hamashima Y Chem. Commun 2015, 51, 16675–16678. [DOI] [PubMed] [Google Scholar]
- (22).Paeth M; Carson W; Luo JH; Tierney D; Cao Z; Cheng MJ; Liu W Chem. - Eur. J 2018, 24, 11559–11563. [DOI] [PubMed] [Google Scholar]
- (23).(a) Djerassi C Chem. Rev 1948, 43, 271–317. [DOI] [PubMed] [Google Scholar]; (b) Schmid H; Karrer P Helv. Chim. Acta 1946, 29, 573–581. [Google Scholar]; (c) Horner L; Winkelmann EH Angew. Chem 1959, 71, 349–365. [Google Scholar]
- (24).(a) Zhang H-J; Su F; Wen T-B J. Org. Chem 2015, 80, 11322–11329. [DOI] [PubMed] [Google Scholar]; (b) Howard E-L; Guzzardi N; Tsanova VG; Stika A; Patel B Eur. J. Org. Chem 2018, 2018, 794–797. [Google Scholar]; (c) Yu H; Li Z; Bolm C Org. Lett 2018, 20, 2076–2079. [DOI] [PubMed] [Google Scholar]; (d) Samzadeh-Kermani A New J. Chem 2018, 42, 4766–4772. [Google Scholar]; (e) Yamamoto C; Takamatsu K; Hirano K; Miura MJ Org. Chem 2016, 81, 7675–7684. [DOI] [PubMed] [Google Scholar]; (f) Vasilopoulos A; Zultanski SL; Stahl SS J. Am. Chem. Soc 2017, 139, 7705–7708. [DOI] [PubMed] [Google Scholar]
- (25).(a) Li Z; Zhang C; Zhu L; Liu C; Li C Org. Chem. Front 2014, 1, 100–104. [Google Scholar]; (b) Li Z; Wang Z; Zhu L; Tan X; Li CJ Am. Chem. Soc 2014, 136, 16439–16443. [DOI] [PubMed] [Google Scholar]
- (26).(a) Westheimer FH Chem. Rev 1961, 61, 265–273. [Google Scholar]; (b) Simmons EM; Hartwig JF Angew. Chem., Int. Ed 2012, 51, 3066–3072. [DOI] [PubMed] [Google Scholar]
- (27).Wiberg KB; Slaugh LH J. Am. Chem. Soc 1958, 80, 3033–3039. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.