

SUMMARY
Prevailing dogma holds that ribosomes are uniform in composition and function. Here, we show that nutrient limitation-induced stress in E. coli changes the relative expression of rDNA operons to alter the rRNA composition within the actively translating ribosome pool. The most upregulated operon encodes the unique 16S rRNA, rrsH, distinguished by conserved sequence variation within the small ribosomal subunit. rrsH-bearing ribosomes affect the expression of functionally coherent gene sets and alter the levels of the RpoS sigma factor, the master regulator of the general stress response. These impacts are associated with phenotypic changes in antibiotic sensitivity, biofilm formation, and cell motility and are regulated by stress response proteins, RelA and RelE, as well as the metabolic enzyme and virulence-associated protein, AdhE. These findings establish that endogenously encoded, naturally occurring rRNA sequence variation can modulate ribosome function, central aspects of gene expression regulation, and cellular physiology.
Graphical Abstract
In Brief
Most organisms encode multiple, distinct copies of rRNA genes, rendering the composition of the ribosome pool intrinsically heterogeneous. Here, Kurylo et al. show that nutrient limitation in E. coli upregulates the expression of ribosomes bearing conserved sequence variation in 16S rRNA that can regulate gene expression and phenotype.
INTRODUCTION
The ribosome is a two-subunit, multi-megadalton RNA protein complex that translates mRNA into protein through temporally coordinated transient interactions with cellular factors and tRNA. Although ribosomes are widely considered homogeneous assemblies that only passively contribute to gene expression, emerging evidence suggests that the composition of the actively translating ribosome may contribute to mRNA-specific changes in gene expression (Dinman, 2016; Genuth and Barna, 2018; Sauert et al., 2015).
In bacteria, stress-induced cleavage of the anti-Shine-Dal-garno sequence from rRNA and antibiotic-induced shedding of ribosomal proteins from assembled ribosomes have been suggested to enhance the translation of leaderless mRNAs (Sauert et al., 2015). In eukaryotes, imbalances in the stoichiometry of specific ribosomal proteins within the assembled ribosome have been linked to alterations in gene expression (Ferretti et al., 2017; Shi et al., 2017). Varied levels of post-transcriptional rRNA modification have also been associated with changes in ligand binding and translational fidelity (Jack et al., 2011). The potential link between translation efficiency and ribosome concentration has, however, been raised as a confounding factor in the proposed mechanisms of gene-specific translational control (Culviner and Laub, 2018; Lodish, 1974; Mills and Green, 2017). Questions as to how changes in ribosome composition could be efficiently regulated have also been voiced (Briggs and Dinman, 2017; Leslie, 2017).
Although the roles of rRNA in diverse aspects of ribosome function are firmly established (Noller, 2005), the potential contributions of endogenously encoded rRNA sequence variation to gene expression regulation has received relatively little attention. The majority of organisms natively encode multiple highly homologous yet distinct genes for the rRNA components of the ribosome (Prokopowich et al., 2003; Sun et al., 2013). For instance, the E. coli K-12 MG1655 (K12) genome encodes seven rDNA operons (rrnA-E, G, and H), each of which contains genes for the core 16S, 23S, and 5S rRNA elements of the two-subunit ribosome (Figures 1A–1C; Blattner et al., 1997). The rRNA gene products encoded by these operons are distinguished by sequence variation at 23 positions in the 16S rRNA, 35 positions in the 23S rRNA, and 4 positions in the 5S rRNA (Figures 1C, 1D, and S1A–S1E). As each of these operons is constitutively expressed (Condon et al., 1992), the translating ribosome pool in E. coli—as in most organisms—is intrinsically heterogeneous.
Figure 1. E. coli rDNA Operons Are Located Non-contiguously on the Chromosome and Encode Unique rRNAs.

(A) E. coli rDNA operon organization. The prerRNA transcript is expressed from two tandem promoters (P1 and P2), encodes a 16S rRNA, one or more tRNA genes, a 23S rRNA, and a 5S rRNA and is terminated at two tandem terminators (t1 and t2).
(B) Map of the E. coli chromosome (Blattner et al., 1997) indicating the location of all seven rDNA operons. The origin of replication is indicated by ‘‘Ori.’’
(C) 70S ribosome structure with a tRNA
(red) in the peptidyl (P) site. Large subunit (50S) ribosomal proteins (blue), small subunit (30S) ribosomal proteins (tan), and rRNA (gray) are shown, and rRNA variant positions are rendered as spheres and colored by element.
(D) 30S ribosomal subunit (solvent-side view) with major structural regions labeled, where rRNA variant positions are rendered as spheres and colored by element. PDB: 3R8T (Dunkle et al., 2011).
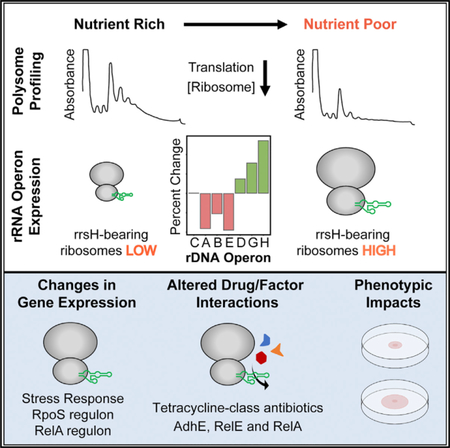
(E and F) Polysome profiles ofwild-type E. coli grown in (E) complex or (F) minimal media.
(G) The percent change of rDNA operons that are up-(green) or downregulated (red) in minimal media relative to complex media (p < 0.01). rDNA operons are listed on the x axis in order of their proximity to the origin of replication (Figure 1B).
rDNA copy number is typically associated with cellular growth and proliferation rates (Condon et al., 1995; Gyorfy et al., 2015). However, E. coli rDNA operons possess functionally distinct promoters and are located non-contiguously within the genome (Figure 1B), suggesting that they may be differentially regulated (Condon et al., 1992; Hillebrand et al., 2005; Hirvonen et al., 2001). Context-dependent expression of specific rDNA operons has been documented in bacteria, parasites, zebrafish, and mammals (Condon et al., 1992; Gunderson et al., 1987; Kim et al., 2007; Locati et al., 2017; Lགྷpez-López et al., 2007; Parks et al., 2018). The physiological significance of these changes and the impacts of the encoded rRNA sequence variation are, however, not presently known.
Here, we show that the E. coli rDNA operon, rrnH, becomes more highly expressed on a relative basis in response to nutrient limitation. The rrnH operon encodes the 16S rRNA gene, rrsH, which is conserved in enterobacteria and is distinguished by ten sequence variants in the small ribosomal subunit head domain. We find that rrsH-bearing ribosomes causally impact transcriptional and translational expression of coherent gene sets, including stress response genes regulated by the RpoS sigma factor, the master regulator of the general stress response (Battesti et al., 2011; Fredriksson et al., 2007). Functional analyses reveal that the expression of rrsH-bearing ribosomes alters RpoS protein levels and mediates pheno-typic changes in antibiotic sensitivity, bio-film formation, and cell motility. These effects are shown to involve the stress-related protein factors, RelA and RelE, as well as the metabolic enzyme and viru-lence-associated protein, AdhE, each of which interacts with the ribosome near the region of sequence variation within the small subunit head domain (Brown et al., 2016; Neubauer et al., 2009; Shasmal et al., 2016). These findings establish that an endogenously encoded rDNA operon in bacteria with conserved sequence variation exhibits context-dependent expression and that ribosomes bearing this variant rRNA have the capacity to alter stress response gene expression and physiology.
RESULTS
Relative rDNA Operon Expression Levels Can Be Quantified via RNA-Seq
Accurate quantification of the relative intragenomic expression of rDNA alleles using high-throughput sequencing methods is challenged by read mapping uncertainty that arises from the high sequence homology of rRNA genes (Treangen and Salz-berg, 2011). Conventional protocols for RNA sequencing (RNA-seq) library preparation also include steps designed to deplete rRNA. Here, we employed RNA-seq without rRNA depletion, together with an expectation-maximization algorithm that ac-counts for read mapping uncertainty (Li et al., 2010), to infer the relative expression of rRNA variants in E. coli. We validated this approach by sequencing mixtures of ribosome populations of known proportions isolated from E. coli exclusively expressing distinct 16S rRNAs from a multi-copy plasmid (Asai et al., 1999; Brosius et al., 1981) to correctly recall the mixing proportions with high accuracy (Figure S1F; STAR Methods). The approach also reliably tracked the expression of heat-shock-induced, plasmid-borne, MS2-tagged ribosomes (Youngman and Green, 2005) in wild-type E. coli cells and determined the extent of their purification from a mixed ribosome pool (Figure S1G). These controls demonstrate that RNA-seq coupled with expectation maximization can accurately infer rRNA expression levels even among genes that are >99% homologous.
The rRNA Composition of the Ribosome Pool Is Modulated during Nutrient Limitation
With a robust method for quantifying the relative expression levels of rRNA variants, we sought to determine whether nutrient limitation-induced stress alters the distribution of rDNA allele expression. In E. coli, growth in nutrient-limiting minimal media is a well-established, natural stress (Hengge-Aronis, 1993) that induces broad, adaptive changes in gene expression relative to growth in complex media via the general stress response (Tao et al., 1999). To examine rDNA operon expression, we grew E. coli in complex or minimal media and performed RNA-seq on total RNA, representing the entire expressed transcrip-tome, and on polysomal RNA, representing RNAs actively engaged in protein synthesis (STAR Methods). Consistent with a stress-induced reduction in global translation, the ribosome and polysome levels decreased for cells grown in minimal media (Figures 1E and 1F). As anticipated, we also observed widespread differences in gene expression, where 1,314 and 1,427 genes were found to be differentially expressed in the total and polysomal RNA pool, respectively (Figure S1H; STAR Methods).
As expected, the genes observed to be differentially expressed significantly overlapped with those previously associated with nutrient limitation-induced stress, respecting direction of change (p < 1e-6; STAR Methods; Tao et al., 1999). Confirming that nutrient limitation activates canonical aspects of the general stress response, promoters targeted by RpoS (Gama-Castro et al., 2016) were enriched among upregulated transcriptional units (TUs) (Cho et al., 2009; p = 4.5e-5). Notably, RpoS is one of only two genes commonly upregulated among studies of global gene expression during acute and chronic nutrient stress (Figure S1I).
In both minimal and complex media, all seven rDNA operons were expressed, and their relative expression levels were >99.6% correlated between total and polysomal fractions (Figures S1J and S1K). Thus, all expressed rRNAs are assembled into mature, translation-competent ribosomes with equivalent efficiency. Strikingly, six of the seven E. coli rDNA operons were differentially expressed between minimal and complex media (Figures 1G and S1L). These data unexpectedly reveal that modulation of the rRNA composition of the actively translating ribosome pool is a feature of nutrient limitation-induced stress.
The rDNA operons located proximal to the origin of replication, oriC (rrnA, rrnB, and rrnE), with the exception of rrnC, were relatively downregulated in minimal media, and those located distally (rrnD, rrnG, and rrnH), particularly the rrnH operon located farthest from the origin of replication, were relatively up-regulated (Figures 1B and 1G). Consequently, ribosomes assembled from the rRNA components of the rrnH operon become relatively enriched in the actively translating ribosome pool under nutrient limitation-induced stress.
The rrsH Variant Allele Is Evolutionarily Conserved and Uniquely Positioned in the Genome
In E. coli MG1655, the rrnD, rrnG, and rrnH operons are distin-guished by variants in distinct structural domains of the 16S rRNA component of the small subunit. rrsD and rrsG 16S rRNAs possess sequence variants within the ‘‘foot’’ domain comprising helices 6–11 (h6–h11). The rrsH 16S rRNA is distinguished by ten variant nucleotides within the ‘‘head’’ domain, nine of which cluster within the helix 33 (h33) ‘‘beak’’ region (Figures 1C, 1D, and S1A–S1C). The presence of identical h33 variants in the highly divergent E. coli MRE600 strain (Kurylo et al., 2016) prompted us to examine further the conservation of these variants.
A broader phylogenetic analysis of the family Enterobacteri-aceae, which includes E. coli and other enteric bacteria, revealed that 20.8% (1,773/8,511) of sequenced genomes encode at least one 16S rRNA with an h33 sequence identical to that of rrsH (Figure 2A). This includes Salmonella enterica, which diverged from E. coli over 120 million years ago (Ochman and Wilson, 1987). In contrast, only 3.6% (309/8,511) encode a 16S rRNA with a foot domain sequence identical to that of rrsG, the second most highly upregulated 16S rRNA. We also found that rDNA operons encoding the rrsH h33 sequence are more distally located from the origin of replication relative to other rDNA operons than expected by chance (p < 1e-4; STAR Methods). Given the inverse correlation between rRNA gene conversion and genomic distance (Hashimoto et al., 2003), we speculate that conservation of the rrnH operon sequence variation may relate to its tendency to occupy a relatively distal position within the bacterial genome.
Figure 2. rrsH-Bearing Ribosomes Regulate Gene Expression.

(A) Conservation of rrsH h33 (16S positions 997–1,044) and rrsG h9–10 (16S positions 180–230) sequence variation in multiple genera within the family Enterobacteriaceae.
(B) RNA-seq analysis of Δ7prrn or K12 strains expressing either the rrsH or rrsB allele from pKK3535-HBB or pKK3535-BBB plasmids, respectively. Polysome fractions from each strain were isolated under minimal growth conditions in biological triplicate.
(C) Overlap of differentially expressed genes between Δ7prrn-HBB/BBB with (p)ppGpp- and RpoS-regulated genes (Dong and Schellhorn, 2009; Durfee et al., 2008).
(D and E) Comparison of RpoS protein levels between (D) Δ7prrn-BBB and Δ7prrn-HBB and between (E) K12-BBB+ and K12-HBB+ measured by western blot and normalized to total protein (n = 5). Error bars indicate SEM, and * indicates p < 0.05. Representative blot (left) and a loading control visualized by the Colloidal Blue Staining Kit (right) are shown.
rrsH-Bearing Ribosomes Regulate the Transcriptional Expression of Stress Response Genes
In light of the relative increase in rrnH expression observed to accompany nutrient limitation-induced stress and the evolutionary conservation of the rrsH h33 sequence, we hypothesized that rrsH-bearing ribosomes may contribute to the modulation of gene expression regulation. To test this hypothesis, we engineered two Δ7prrn E. coli strains that lack all endogenous rDNA operons and express plasmid-borne copies of either the rrnB operon or the rrnB operon containing the ten rrsH variants within the small-subunit head domain (STAR Methods). We refer to these plasmids as pKK3535-BBB and pKK3535-HBB, where the gene of origin for the 16S, 23S, and 5S rRNAs is listed in 5’ to 3’ orientation. The corresponding strains are referred to as Δ7prrn-BBB and Δ7prrn-HBB, respectively. This plasmid-based expression system bypasses complexities associated with chromosome position-dependent impacts on rDNA expression and controls for tRNA gene copy number (Figures 1A and 1B). As described below, this system also enabled the purification of pure ribosome populations for in vitro investigations and pro-vides a consistent means of examining the impacts of rRNA sequence variation across distinct strains and genetic contexts. The Δ7prrn-BBB and Δ7prrn-HBB strains were confirmed by deep sequencing to be isogenic, except for the intended 16S rRNA variants, and exhibited indistinguishable doubling times in minimal media (STAR Methods). We therefore used the Δ7prrn-BBB and Δ7prrn-HBB strains as a controlled setting in which to investigate the impacts of endogenously encoded rRNA sequence variation on gene expression.
To examine whether sequence variants in the ribosome’s head domain regulate transcriptional and translational aspects of gene expression, we performed RNA-seq on total and polysomal RNA, respectively, harvested from Δ7prrn-BBB and Δ7prrn-HBB strains, which were both grown in minimal media. Strikingly, this analysis revealed that 864 genes (19% of the annotated transcriptome) were differentially expressed at the level of transcription, with 440 genes exhibiting similar differences in the actively translating polysome pool (Figures 2B and S2A). The altered expression of a subset of these genes was verified by real-time qPCR (Figure S2B). These findings reveal that evolutionarily conserved, endogenously encoded sequence variation within the small subunit head domain of the ribosome has the capacity to substantially alter gene expression at the levels of both tran-scription and translation.
Notably, the genes that were differentially expressed in Δ7prrn-HBB relative to Δ7prrn-BBB strains were overrepre-sented for those regulated during acute and chronic nutrient stress (p < 1e0–6; STAR Methods; Durfee et al., 2008; Tao et al., 1999; Traxler et al., 2008). They also significantly overlapped with those differentially expressed between wild-type cells grown in minimal versus complex media, respecting direction of change (p < 1e-6 for both polysomal and total RNA; Figure S2C; STAR Methods). Hence, two bacterial strains, whose only sequence distinctions are ten naturally encoded variant nu-cleotides within the small subunit head domain of the ribosome, exhibit substantial differences in the expression of gene sets associated with nutrient limitation-induced stress.
The observed differences in stress response-related gene expression led us to hypothesize that rrsH-bearing ribosomes contribute to transcriptional aspects of the general stress response. RpoS and the (p)ppGpp alarmone mediate important transcriptional impacts on the stress response whose own regulation occurs, at least in part, during protein synthesis (Battesti et al., 2011; Hauryliuk et al., 2015). In line with RpoS and (p) ppGpp contributions to the observed distinctions in gene expression, genes differentially expressed in the Δ7prrn-HBB strain significantly overlapped with RpoS- and (p)ppGpp-dependent gene sets identified in genetic knockout studies, respecting direction of change (p < 1e 6 for all tests; Figures 2C and 2D; STAR Methods; Dong and Schellhorn, 2009; Traxler et al., 2008). TUs more highly expressed in Δ7prrn-HBB relative to Δ7prrn-BBB were also enriched for those whose promoters are regulated by RpoS (p = 2.9e-12; Gama-Castro et al., 2016). As anticipated from these analyses, RpoS protein levels were found to be higher in the Δ7prrn-HBB strain (Figure 2D; STAR Methods), while significant changes in RpoS mRNA levels were not observed in either the total or polysomal RNA pools (Ta-ble S4). We therefore conclude that the ten-nucleotide variants within the head domain of rrsH-bearing ribosomes contribute to RpoS-mediated changes in the stress response gene expression program during nutrient limitation and that RpoS protein levels are altered by post-transcriptional control mechanisms.
rrsH-Bearing Ribosome Dosage Enhances the Stress Response in Wild-Type Cells
To examine whether the dosage of rrsH-bearing ribosomes af-fects gene expression in wild-type cells that possess all seven endogenous rDNA operons, we transformed pKK3535-BBB or pKK3535-HBB plasmids into E. coli K12 cells (K12-BBB+ and K12-HBB+ strains, respectively), grew them identically in mini-mal media, and performed RNA-seq (STAR Methods). In these strains, rrsB and rrsH rRNAs comprise ∼70% of the rRNA composition of the respective ribosome pool, providing a 3.5-fold increase over the ∼20% present in wild-type cells grown un-der the same conditions (Figure S1H).
Consistent with the hypothesis that rrsH-bearing ribosomes causally alter gene expression, we identified 114 and 183 genes as differentially expressed at the level of total and polysomal RNA, respectively (Figures 2B and S2D). As for the Δ7prrn-HBB and Δ7prrn-BBB strains, the differentially expressed genes significantly overlapped with RelA-driven gene expression programs during acute nutrient stresses, respecting direction of change (p < 1e-6; STAR Methods; Durfee et al., 2008; Traxler et al., 2008). They also significantly overlapped with the genes differentially expressed in wild-type cells grown in complex or minimal media and between Δ7prrn-BBB and Δ7prrn-HBB strains grown in minimal media (p < 1e-6 for all tests; Figure S2C; STAR Methods). Moreover, upregulated TUs were again enriched for those with promoters regulated by RpoS (p = 1.2e-9; Gama-Castro et al., 2016). Western blot analyses further confirmed that the K12-HBB+ strain exhibited higher levels of the RpoS protein (Figure 2E). Notably, the number of differentially expressed genes and the magnitude of difference in RpoS protein levels correlated between the Δ7prrn and K12 genetic backgrounds. These findings substantiate the conclusion that rrsH-bearing ribosomes affect gene expression and suggest that the observed enrichment of rrsH-bearing ribosomes during nutrient limitation contributes to the general stress response gene expression program (Figure 1G).
The rrsH Variants Alter the Ribosome’s Sensitivity to Tetracycline-Class Antibiotics
To examine the impact of rrsH sequence variants in the ribosome on cellular phenotype, we quantified the growth profiles of Δ7prrn-HBB and Δ7prrn-BBB strains under hundreds of distinct growth conditions through a comparative BIOLOG Phenotype Microarray (Bochner et al., 2001). We found that the two strains exhibited growth differences in eight conditions associated with stress (Figures 3A and S3A). Strikingly, four of these conditions reflected differential sensitivities to tetracycline-class antibiotics. Tetracyclines block tRNA accommodation into the A site of the ribosome during the elongation step of translation by binding to helix 34 (h34) of the small subunit head domain proximal to the h33 variants (Jenner et al., 2013). These findings suggest that the rrsH-encoded sequence variants within the small sub-unit head domain may allosterically alter drug binding to h34 and/or aspects of the elongation mechanism targeted by the drug’s interaction with the ribosome. Alternatively, resistance to tetracycline-class antibiotics could reflect secondary effects, such as altered cellular pH, ionic strength, or rates of drug efflux.
Figure 3. rrsH-Bearing Ribosomes Are Comparatively Resistant to Tetracycline.

(A) BIOLOG analysis comparing the growth rate of Δ7prrn-HBB (cyan) to that of Δ7prrn-BBB (red) in the presence of tetracycline. Shading indicates SD of three biological replicates.
(B) Schematic of tRNA selection where FRET is monitored between aminoacyl- and peptidyl-tRNA on individual ribosomes. Ternary complex containing an LD650-labeled Phe-tRNAPhe molecule is delivered to surface-immobilized ribosomes programmed with a cognate mRNA codon in the A site and enters the ribosome through distinct intermediate states on pathway to a fully accommodated state where peptide-bond formation occurs.
(C) Representative smFRET trace showing the process of aminoacyl-tRNA entry into the ribosome. Prior to ternary complex binding to the ribosome, no FRET is observed (zero FRET; left). Aminoacyl-tRNA entry into the ribosome occurs through two intermediate states: a low-FRET, codon-recognition (CR) state and an intermediate-FRET, GTPase-activated (GA) state. Transitions between the CR and GA state and the GA to AC (accommodated) state are highlighted. Productive formation of the GA state facilitates entry into the AC state.
(D and E) Post-synchronized, ensemble smFRET histograms of tRNA selection on rrsB-(D) and rrsH-bearing (E) ribosomes in the presence of tetracycline. Plots show the sum of 12 repeats for each experiment and are comprised of >4,000 individual FRET trajectories.
(F) Log odds of tRNA accommodation in the presence of tetracycline (p = 1.2e 20). Error bars indicate SEM and ***p < 0.01 (STAR Methods).
To differentiate between these two possibilities, we compared the rates of tRNA accommodation into ribosomes isolated from the Δ7prrn-HBB and Δ7prrn-BBB strains using in vitro single-molecule fluorescence resonance energy transfer (smFRET) imaging techniques (Geggier et al., 2010; Juette et al., 2016). Consistent with our BIOLOG data (Figure 3A) and corroborating the model that the rrsH sequence variation physically alters the ribosome, we found that the entry of aminoacyl-tRNA (aa-tRNA) into rrsH-bearing ribosomes (Figures 3B and 3C) was comparatively resistant to tetracycline (p = 1.2e-20; Figures 3D–3F; STAR Methods). In line with its established mode of ac-tion (Blanchard et al., 2004a; Jenner et al., 2013), tetracycline was specifically less efficient at impeding the transition between the codon recognition state (low-FRET) and productive forma-tion of the guanosine triphosphatase (GTPase)-activated state (intermediate-FRET) on rrsH-bearing ribosomes. Consequently, aa-tRNA accommodated more rapidly into the peptidyl transferase center (high-FRET; Figure 3F). By contrast, significant differences with respect to aa-tRNA selection were not detected in the absence of drug (p = 0.1; Figure S3B; STAR Methods). We conclude from these findings that the rrsH variants either alter the kinetics of tetracycline binding to the ribosome in a manner that reduces its probability of inhibition or that they alter the selection process in a manner that allows aa-tRNA to bypass the steric block that normally prevents tRNA entry into the drug-bound ribosome. Hence, the relative resistance of the Δ7prrn-HBB strain to tetracycline arises, at least in part, from physical distinctions in the ribosome itself, which stem from the endoge-nously encoded rRNA sequence variation.
Ribosomes Bearing the rrsH 16S rRNA Differentially Associate with Extra-ribosomal Factors
The gene expression differences resulting from rrsH-bearing ribosome expression may arise through specific contacts between the variant nucleotides and ribosome-associated factors or indirectly through structural alterations in ribosome composition or dynamics. To examine whether the rrsH variants induce alterations in rRNA secondary or tertiary structure in the ribosome, we performed a dimethyl sulfate (DMS) modification assay, DMS-MaP-seq (Zubradt et al., 2017), on small (30S) ribosomal subunits isolated from Δ7prrn-HBB or Δ7prrn-BBB strains. This chemical probing analysis revealed no differences in the DMS reactivity of adenosine or cytosine nucleotides (Table S3; STAR Methods). Thus, according to DMS-MaPseq, rrsB-and rrsH-bearing ribosomes are compositionally and structurally indistinguishable.
To examine whether the ribosomes in Δ7prrn-HBB and Δ7prrn-BBB strains exhibit distinct protein compositions, we performed a comparative mass spectrometry analysis on high-salt washed rrsB- and rrsH-bearing ribosomes (STAR Methods). This analysis did not identify significant differences in the core ribosomal protein compositions (Table S1). We did, however, observe that ribosomes isolated from Δ7prrn-HBB contained a non-ribosomal protein, aldehyde-alcohol dehydrogenase (AdhE), that was not found in those isolated from Δ7prrn-BBB (Figure S4A; Table S2). No difference in AdhE expression was detected in whole-cell lysate from these strains (Figure S4B). AdhE is a multifunctional metabolic enzyme essential for growth under anaerobic conditions that has also been implicated as an ATP-dependent RNA helicase that binds the small subunit head domain through contacts with h33 and ribosomal proteins S3, S10, and S14 (Shasmal et al., 2016). We thus infer that the rrsH sequence variants may enhance the ribosome’s interaction with AdhE. These observations suggest that the differences in gene expression exhibited by rrsH-bearing ribosomes may manifest, at least in part, via distinctions in the ribosome’s association with cellular factors that are either directly influenced by contacts with the ten-nucleotide variants in the small subunit head domain or through compositional or structural distinctions in the ribosome that cannot be discerned using the methods applied.
AdhE, RelE, and RelA Are Functionally Linked to rrsH-Bearing Ribosomes
Like AdhE, the stress-related proteins RelE and RelA bind the ribosome in close proximity to the rrsH variants in the small sub-unit head domain. RelA binds at the interface between large and small subunits and contacts intersubunit bridge B1a, which links the large subunit with the small subunit head domain (Brown et al., 2016). RelE binds the ribosome at a site that overlaps with both aa-tRNA and tetracycline (Brodersen et al., 2000; Neubauer et al., 2009). To examine the hypothesis that the ribosome’s interactions with AdhE, RelE, and RelA are affected by rRNA sequence elements within the small subunit head domain, we performed genetic interaction studies using ΔadhE, ΔrelE, and ΔrelA knockout strains from the Keio collection (Yamamoto et al., 2009) transformed with either the pKK3535-BBB or pKK3535-HBB plasmid (STAR Methods). As the strains compared are isogenic aside from the ten variant nucleotides within the plasmid-expressed rrsH and rrsB alleles, these investigations specifically query the impact of sequence variation in the ribosome on gene expression in each genetic background. The identification of rRNA variant-dependent differences in gene expression in these contexts would thereby establish the existence of functional linkages (Kelley and Ideker, 2005) between the ten-nucleotide-sequence variants within the small subunit head domain of the ribosome and proteins AdhE, RelE, and RelA.
To provide context for these genetic interaction studies, we compared the gene expression profiles and RpoS levels be-tween BW25113 (Keio parent) strains expressing pKK3535-HBB or -BBB plasmids. Differentially expressed genes (n = 17) included those associated with biofilm formation and oxidative stress response originating from the CP4–44 cryptic prophage element (Figure 4A). Consistent with the observed correlation between gene expression impacts and RpoS protein levels, significantly different levels of the RpoS protein were not observed (Figure S4C; STAR Methods). We attribute these dis-tinctions from K12 and Δ7prrn strains to the unique genetic and phenotypic features of the Keio parent strain with regard to growth and stress response activation (Grenier et al., 2014; Tamar et al., 2016; Wood et al., 2006; Yu et al., 2013; Figure S4D). Consistent with the notion that RpoS protein levels are a deter-mining feature of the gene expression changes associated with rrsH-bearing ribosome expression, no changes in gene expres-sion were evidenced when analogous experiments were performed in Keio strains lacking the rpoS gene (data not shown).
Figure 4. Functional Linkages between the Ribosome Head Domain Protein Factors RelA, RelE, and AdhE Regulate Gene Expression and Phenotype.

(A) RNA-seq analysis of Keio parent strain (BW25113) or isogenic strains lacking relA, relE, or adhE genes. The compared strains are identical except for the ten nucleotide variants encoded by the pKK3535-BBB and pKK3535-HBB rDNA plasmids. All experiments were performed on isolated polysome fractions in biological triplicate.
(B)Gene ontology enrichment analysis of differentially expressed genes from (A).
(C) Quantification of RpoS protein levels measured by western blot and normalized to total protein (n = 5). Representative blot and loading control of total protein visualized by the Colloidal Blue Staining Kit are shown.
(D–I) The impacts of ribosome sequence variation on (D–G) cell motility and (H and I) biofilm formation. Genetic backgrounds are listed on the x axis. Error bars indicate SEM. *** indicates p < 0.01.
Consistent with functional interactions of AdhE, RelE, and RelA proteins with the small subunit head domain, we observed rRNA sequence-dependent differences in gene expression in the ΔadhE, ΔrelE, and ΔrelA genetic backgrounds (Figure 4A). These differences were most extensive in the ΔadhE and ΔrelE back-grounds, where we observed 1,651 and 1,338 differentially expressed genes, respectively. Between ΔadhE strains, rrsH-upregulated genes were overrepresented for those related to motility (locomotion, p = 9.1e 10; chemotaxis, p = 4.7e-5), and downregulated genes were overrepresented for those related to the stress response (amino acid biosynthesis, p = 1.2e-15; polysaccharide metabolism, p = 9.0e-10; Figure 4B). As expected from these findings, RpoS levels were lower in the ΔadhE strain expressing rrsH-bearing ribosomes (p < 0.05; Figure 4C). As no significant differences were observed in polysome-associated RpoS mRNA levels (Table S4), we attribute the observed changes in gene expression to mechanisms that modulate RpoS protein levels post-transcriptionally. Between ΔrelE strains, rrsH-upregulated genes were overrepre-sented for transport-related processes and RNA metabolism (active ion transmembrane transporter activity, p = 7.3e 7; RNA processing, p = 4.9e 6), and downregulated genes were overrepresented for cellular respiration (p = 1.7e-11) and heterocycle biosynthetic processes (p = 7.7e-7; Figure 4B).
Between ΔrelA strains, fewer genes were differentially expressed than observed between ΔadhE or ΔrelE strains (Figure 4A), suggesting a weaker functional linkage between RelA and rRNA sequence in the Keio parent background. Nonetheless, rrsH-upregulated genes were overrepresented for those related to the stress response (nitrogen utilization, p = 6.6e-8; branched-chain amino acid biosynthesis, p = 5.4e 8), and upregulated TUs were overrepresented for those with promoters targeted by RpoS (p = 8.5e-8; Gama-Castro et al., 2016). rrsH-downregulated genes were enriched for those related to motility (chemotaxis, p = 3.9e-11; locomotion, p = 3.6e-10; Figure 4B). These gene sets also significantly overlapped with those differentially expressed during the wild-type response to nutrient limitation (Figure S4E) and with gene expression changes observed between wild-type and ∆rpoS strains grown under nutrient limitation-induced stress, respecting direction of change (p < 1e-6 for both tests; Figure S4F; STAR Methods; Dong and Schellhorn, 2009; Tao et al., 1999). Consistent with RelA-independent path-ways of RpoS and stress response regulation (Battesti et al., 2011), western blot analyses confirmed higher RpoS protein levels in the ΔrelA strain expressing rrsH-bearing ribosomes (p < 0.05; Figure 4C). As no significant differences were observed in polysome-associated RpoS mRNA levels (Table S4), we again attribute these effects to post-transcriptional mechanisms.
In light of structural data showing that AdhE, RelE, and RelA associate with the small subunit head domain of the ribosome, we propose that AdhE, RelE, and RelA functionally interact with translating ribosomes in an rRNA-sequence-dependent manner to differentially affect the steady-state levels of the RpoS protein and stress response gene expression.
rRNA Sequence Variation in the Ribosome Alters Biofilm Formation and Cell Motility
In bacteria, reduced cell motility and increased biofilm formation are fundamental aspects of the general stress response (Dong and Schellhorn, 2009). We therefore set out to examine the impacts on locomotion inferred by our gene expression studies using semi-solid agar cell motility assays. Consistent with prediction, cells expressing the rrsH-bearing ribosomes exhibited a 27% decrease in motility in the ΔrelA background (p = 4.9e-5), a 58% increase in the ΔadhE background (p = 1.5e-4), and no effects in Keio parent or ΔrelE backgrounds (Figures 4D–4G).
Keio parent and ΔadhE cells expressing the rrsH-bearing ribosomes also exhibited gene expression differences that significantly overlapped with those associated with biofilm formation (p < 0.002 for both tests; Schembri et al., 2003). The direction of change observed for these genes suggested that cells expressing rrsH-bearing ribosomes should exhibit an enhancement of biofilm formation in Keio parent background and a deficiency in biofilm formation in the ΔadhE background. Consistent with prediction, the Keio parent strain expressing rrsH-bearing ribosomes exhibited an 11% enhancement in biofilm formation (p = 7.9e 3), and in the ΔadhE background, we observed a 78% reduction in biofilm formation (p = 2.4e-8; Figures 4H and 4I). Although analogous experiments were pursued in the ΔrelA and ΔrelE backgrounds, no biofilm formation could be measured in these genetic contexts (STAR Methods). These functional analyses collectively demonstrate that the expression of rrsH-bearing ribosomes gives rise to context-dependent changes in phenotype directly related to adaptive responses to stress and cell survival.
DISCUSSION
The data presented demonstrate that the relative expression levels of distinct, endogenously encoded rDNA operons in E. coli modulate in response to nutrient limitation-induced stress, thereby altering the balance of intrinsic heterogeneities that are constitutively present in the rRNA component of the ribosome pool. The relative upregulation of rrnH operon expression accompanying nutrient limitation-induced stress may reflect replication-associated gene dosage effects (Couturier and Ro-cha, 2006), nucleoid restructuring, and promoter-specific differences in transcriptional activation (Condon et al., 1992; Jin et al., 2012). These impacts may include comparatively strong interactions with the factor for inversion stimulation (FIS) transcriptional activator and relatively weak interactions with the histone-like nucleoid structuring protein (H-NS) transcriptional repressor, the latter of which is upregulated under nutrient-limiting conditions (Hillebrand et al., 2005; Hirvonen et al., 2001). Importantly, the observed increase in rrnH operon expression represents an average of the cell population. The stochastic nature of gene expression, as well as drug resistance and persistence in bacteria (Raj and van Oudenaarden, 2008; Shan et al., 2017), suggests that rDNA operon expression may also exhibit substantial cell-to-cell and time-dependent variations that relate to the specific cellular stress encountered.
The broad conservation of the rrsH h33 sequence, combined with the positive relationship evidenced between rrsH-bearing ribosome expression, RpoS protein levels, and the expression of RpoS-regulated genes, supports a model wherein rrnH operon expression contributes to bacterial fitness during natural cycles of nutrient-rich and nutrient-poor growth. These findings do not stipulate that the rrnH operon is necessary for the general stress response or RpoS expression, as both occur in cell strains in which the rrnH operon is absent. Additional experiments will be necessary to delineate the contribution of rrsH-bearing ribosomes to stress responses in natural settings.
The molecular basis of the observed inversion of this positive correlation between rrsH-bearing ribosome and RpoS protein levels in the absence of the metabolic enzyme AdhE is not presently clear. We note, however, that the virulence of the human pathogen E. coli 0157:H7 EDL933, in which 6 of 7 rDNA operons bear the exact rrsH h33 sequence, is attenuated by deletion of the AdhE protein (Beckham et al., 2014). Our finding that the small subunit beak domain of 16S rRNA affects cell motility and biofilm formation is particularly intriguing in this regard as this region of rRNA, often referred to as hypervariable region V6, is a distinguishing feature of pathogenic bacteria (Chakravorty et al., 2007). Examination of ribosome-associated factor interactions with the V6 variant nucleotides present in the ribo-somes of pathogens may therefore lead to deeper mechanistic understanding of antibiotic resistant and persister populations.
Our finding that the rRNA composition of the assembled ribo-some can regulate stress response gene expression is consistent with established links between transcription and translation in bacteria. The ribosome serves as a focal point of the nutrient limitation-induced stress response via RelA, which binds to translating ribosomes together with deacylated tRNA to stimulate the production of the (p)ppGpp alarmone (Jin et al., 2012; Figure 5). The resulting accumulation of (p)ppGpp alters gene expression by redirecting RNA polymerase (RNAP) away from genes involved in energy-intensive processes, including ribo-some biogenesis and cell motility, to those that promote survival (Battesti et al., 2011; Mauri and Klumpp, 2014; österberg et al., 2011). This effect is mediated by the direct binding of (p)ppGpp to RNAP, which releases it from rDNA operons to enable its association with the cellular pool of sigma factors (österberg et al., 2011). Our finding that the ribosome can contribute to modulating the cellular concentration of the RpoS sigma factor likely affects the competition between sigma factors for binding to the RNAP core particle. The ribosome also serves as the plat-form for post-transcriptional control of gene expression through its interactions with RelE and other normally repressed endonucleases that compete with tRNA and factor binding at the A site of the ribosome to degrade mRNAs during protein synthesis (Christensen et al., 2001; Durfee et al., 2008; Schureck et al., 2016; Figure 5). The ribosome also influences the speed and processivity of co-transcriptional translation through direct inter-actions with RNAP (McGary and Nudler, 2013; Figure 5). Interestingly, this interaction includes contacts between RNAP and the C1120T variant of rrsH-bearing ribosomes (Kohler et al., 2017; Figure S1A). It is presently unclear whether, and in what manner, AdhE interactions with the small subunit head domain impact RNAP-ribosome interactions and whether RNAP itself differentially interacts with rrsH-bearing ribosomes (Figure 5).
Figure 5. Protein Synthesis Reactions Are Mediated by Transient, Competing Factor Interactions with the Dynamic Small Subunit Head Domain of the Ribosome.
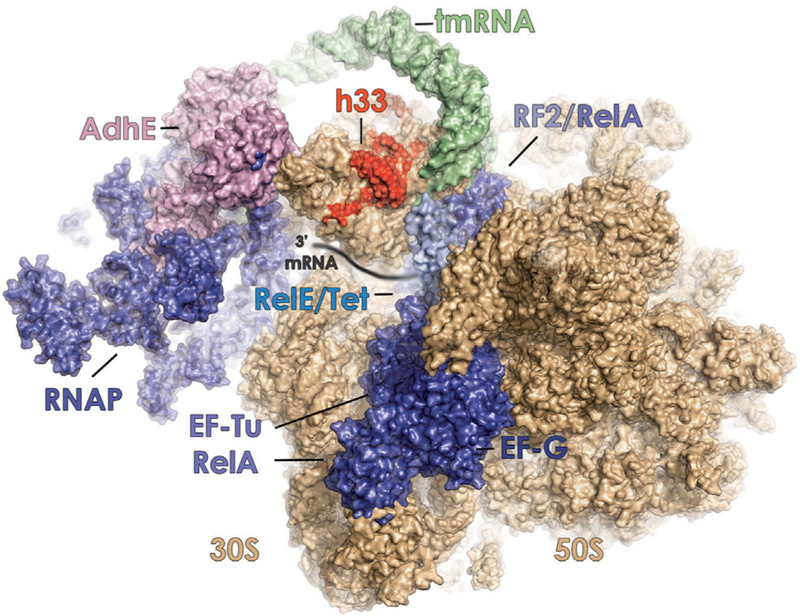
Schematic representation showing known interactions of elongation factor-G (EF-G) (PDB: 4V90), elongation factor-Tu (EF-Tu) (PDB: 4V68), RelA (PDB: 5KPV), release factor 2 (RF2) (PDB: 5DEF), RelE (PDB: 4V7J), RNA polymerase (RNAP) (PDB: 5MY1), tetracycline (Tet) (PDB: 4V9A, blue), and tmRNA (PDB: 4V6T, green), with the leading edge of the bacterial ribosome (30S and 50S subunits, tan). The 30 end of the mRNA substrate is schematically shown as it enters the aminoacyl (A) site. The proposed interaction of the metabolic enzyme alcohol dehydrogenase (AdhE) (pink) with the small subunit head domain is also shown (Shasmal et al., 2016). The positions of variant nucleo-tides within helix 33 (h33) of rrsH-bearing ribosomes are depicted in red.
Given the findings presented and extant structural data, we speculate that the rrsH sequence variants alter kinetic features of the competing binding interactions of AdhE, RelE, RelA, and other translation factors at the leading edge of the ribosome to influence both initiation and elongation phases of protein synthesis (Figure 5). This model is supported by the presented evidence that the A site of the rrsH-bearing ribosome differentially interacts with tetracycline. It is also consistent with the proposed mechanism by which gene-specific changes arise from altered interactions between the ribosome, mRNA, and translation initiation factors (Mills and Green, 2017). It is not yet clear whether the impacts of rrsH-bearing ribosomes affect global translation or specific subsets of cellular mRNAs.
RpoS regulation can occur at transcriptional, translational, proteolytic, and functional levels (Battesti et al., 2011). In this light, it is notable that the observed capacity of rrsH-bearing ribosomes to modulate RpoS protein levels largely occurs in the absence of significant changes in RpoS mRNA (Table S4). This finding suggests rrsH-bearing ribosome-dependent mechanisms of post-transcriptional control. Such impacts may include altered production rates of the RpoS protein from a given transcript and/or altered post-translational degradation of the RpoS protein (Battesti et al., 2011). In this context, we note that the production of the RpoS protein is enhanced by the presence of rare leucine codons distributed throughout its open reading frame (Kolmsee and Hengge, 2011) and that the genes upregulated by rrsH-bearing ribosomes are enriched for rare codons (Figure S5). RpoS protein levels are also strongly influenced by the actions of transfer-mRNA (tmRNA), which interacts with the small subunit head domain (Ramrath et al., 2012; Ranquet and Gottesman, 2007; Figure 5) to mediate the rescue of ribosomes stalled at rare codons or on truncated mRNAs (Ranquet and Gottesman, 2007). These observations collectively suggest that rrsH-bearing ribosomes may influence the fidelity of translation in a manner that either directly alters RpoS protein production from a given transcript or indirectly alters RpoS protein degradation by affecting its proteolytic degradation. These latter impacts can arise from global changes in translation fidelity and oxidative protein damage, which are critical features of bacterial physiology during nutrient limitation-induced stress (Fredriksson et al., 2007).
The observation that the rRNA component of the ribosome can impact gene expression is consistent with central tenets of the ‘‘specialized ribosome’’ hypothesis (Dinman, 2016; Genuth and Barna, 2018). An important distinction from previous studies, however, is that the heterogeneities we document are encoded in the RNA template for ribosome biogenesis. In this context, we note that variation in rDNA operon copy number and rRNA sequence can provide a regulable basis for introducing compositional and functional changes in the ribosome that contribute to central aspects of gene expression. Future investigations will be needed to examine the full extent of rRNA-sequence-dependent impacts on gene expression in bacteria at different stages of the growth cycle, in response to different environmental cues, and in the presence of distinct ribosome-interacting factors. Investigations into the extent and role of rRNA sequence variation on central physiological programs in higher eukaryotic organisms are also warranted, as mammalian rDNA operons exhibit intra-individual sequence variation and tissue-specific expression patterns (Parks et al., 2018).
STAR*METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-AdhE | Agrisera | Cat# AS10748 RRID# AB_10751783 |
| Mouse anti-RpoS | Biolegend | Cat # 663703 RRID# AB_1134163 |
| Bacterial and Virus Strains | ||
| E. coli K-12 MG1655 | ATCC | #700926 |
| E. coli MC338 | (O’Connor, 2009) | N/A |
| E. coli BW25113 | (Yamamoto et al., 2009) | N/A |
| E. coli JW2755–3 | (Yamamoto et al., 2009) | N/A |
| E. coli JW1555–2 | (Yamamoto et al., 2009) | N/A |
| E. coli JW1228–1 | (Yamamoto et al., 2009) | N/A |
| E. coli JW5437–1 | (Yamamoto et al., 2009) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tetracycline hydrochloride | Millipore Sigma | 58346 |
| Ampicillin sodium salt | VWR | 0339 |
| Kanamycin sulfate | VWR | 0408 |
| Chloramphenicol | Sigma | C0378 |
| Spectinomycin sulfate | Millipore | 158993 |
| Kirromycin | Sigma | 50935–71-2 |
| Fusidic acid | Sigma | F0881 |
| Crystal Violet | Sigma | 548–62-9 |
| Critical Commercial Assays | ||
| QIAzol Lysis Reagent | QIAGEN | 79306 |
| RNeasy Mini Kit | QIAGEN | 74104 |
| TruSeq Stranded Total RNA Sample Preparation Kit | Illumina | 20020597 |
| QIAamp DNA Mini Kit | QIAGEN | 53104 |
| Sigma | GERPN2235 | |
| SuperScript III First-Strand Synthesis System | Invitrogen | 18080051 |
| PowerUp SYBR Green Master Mix | Thermo Fisher | A25741 |
| Deposited Data | ||
| DMS-MaPseq Data | This Study | GSE105112 |
| RNA-Sequencing Data | This Study | PRJNA487814 |
| Experimental Models: Organisms/Strains | ||
| E. coli Δ7prrn-BBB | This study | N/A |
| E. coli Δ7prrn-HBB | This study | N/A |
| E. coli K12-BBB | This study | N/A |
| E. coli K12-HBB | This study | N/A |
| E. coli BW25113-BBB | This study | N/A |
| E. coli BW25113-HBB | This study | N/A |
| E. coli ΔrelA-BBB | This study | N/A |
| E. coli ΔrelA-HBB | This study | N/A |
| E. coli ΔrelE-BBB | This study | N/A |
| E. coli ΔrelE-HBB | This study | N/A |
| E. coli ΔadhE-BBB | This study | N/A |
| E. coli ΔadhE-HBB | This study | N/A |
| E. coli DrpoS-BBB | This study | N/A N/A |
| E. coli DrpoS-HBB | This study | N/A |
| Oligonucleotides | ||
| rrsH_1655_F: 5’–ACAGCCGGTTCGGTTGAAGAG–3’ | This study | N/A |
| rrsH_1655_R: 5’– CCTGAGCTACAAGCCTGTAGAGG – 3’ | This study | N/A |
| aceE_F: 5’-ATGACGTGGATCCGATCGAA-3’ | This study | N/A |
| ace_R: 5’-GAATACGGCGTTCCAGTTCC-3’ | This study | N/A |
| gabD_F: 5’-TTCTTCCAGCCGACCATTCT-3’ | This study | N/A |
| gabD_R: 5’-GGCTTAAATCACGGGCGTAG-3’ | This study | N/A |
| hcaT_F: 5’ – GCTGCTCGGCTTTCTCATCC – 3’ | (Peng et al., 2014) | N/A |
| hcaT_R: 5’ – CCAACCACGCTGACCAACC – 3’ | (Peng et al., 2014) | N/A |
| Recombinant DNA | ||
| pKK3535 | (Brosius et al., 1981) | N/A |
| pKK3535-HBB | This study | N/A |
| Software and Algorithms | ||
| GEM | (Marco-Sola et al., 2012) | N/A |
| RSEM | (Li and Dewey, 2011) | N/A |
| voom | (Law et al., 2014) | N/A |
| limma | (Ritchie et al., 2015) | N/A |
| OPM | (Vehkala et al., 2015) | N/A |
| MaxQuant | (Cox et al., 2011) | N/A |
| SKM | (Qin, 2007) | N/A |
| SPARTAN | (Juette et al., 2016) | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Dr. Scott C. Blanchard (scb2005@med.cornell.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
E. coli K-12 MG1655 cells were purchased from ATCC (#700926). E. coli Δ7prrn strain MC338 was a gift from Dr. Michael O’Connor (UMKC). E. coli strains from the Keio Collection were obtained from the Coli Genetic Stock Center at Yale University. Standard cell culture conditions were LB Broth, Miller, under aerated conditions at 225 rpm. All cell strains were stored at 80°C in 25% glycerol.
METHOD DETAILS
Bacterial growth conditions and RNA isolation from total and polysomal fractions
Overnight cultures or E. coli K-12 MG1655 (ATCC #700926) were grown in LB Broth, Miller (EMD Millipore), to saturation and diluted to OD600 = 0.048 in M9 minimal media (M9) (Sigma) or OD600 = 0.012 in Terrific broth (Novagen). M9 salts were supplemented with 0.2% glucose, 1 mM MgSO4, 0.1 mM CaCl2. Cells were grown at 37 C with 225 rpm shaking in baffled flasks. At OD600nm = 0.6–0.65 (M9) or OD600nm = 1.4–1.5 (TB). Cells were harvested using rapid vacuum filtration onto 0.2 mm polyethersulfone membranes (Pall) and flash frozen. Cell pellets were combined with 650 μl of frozen lysis buffer pellets (1X Tris-polymix pH 7.5 (Juette et al., 2016; Wasserman et al., 2016), 200 μM fusidic acid, 10 μM kirromycin, 0.4% Triton X-100, 0.1% NP-40, and 15 ng DNase) and cryolysed using a Mixer Mill MM 400 (Retsch). Crushate was then thawed on a room temperature water bath. A portion was taken for total RNA purification using QIAzol (QIAGEN) and the RNeasy Mini Kit (QIAGEN), while the remainder was used for polysome isolation using sucrose density gradient ultracentrifugation. Sucrose gradients (10%–47% w/v), made using a Gradient Master (Biocomp), contained 1X Tris-polymix (pH 7.5) and 200 μM fusidic acid. Prior to mixing, the 10% sucrose component was supplemented with 1 μM kirromycin. Samples were spun on sucrose gradients in a SW41 rotor (Beckman Coulter) for 2.5 hours at 35,000 rpm and 4°C. Sucrose gradients were analyzed using a Gradient Fractionator (Brandel) and polysome fractions were collected and pelleted for 4 hr at 78,000 rpm and 4°C in a TLA 100.3 rotor (Beckman Coulter). Pelleted polysomes were resuspended in subunit dissociation e2 Cell Reports 25, 236–248.e1–e6, October 2, 2018 buffer (1X PBS pH 7.4, 0.5 M KCl, 2 mM puromycin) and RNA was extracted using QIAzol (QIAGEN) and purified using the RNeasy Mini kit (QIAGEN).
RNA-Sequencing (RNA-Seq) and data analysis
Libraries were prepared using the TruSeq Stranded Total RNA Prep Kit (Illumina) and sequenced on an Illumina HiSeq 2500 at the Genomics Resources Core Facility at Weill Cornell Medicine. Raw Illumina RNA-seq reads were filtered with the fastq_illumina_filter (http://cancan.cshl.edu/labmembers/gordon/fastq_illumina_filter/), and the first base of each read was trimmed with the fastx_trimmer of the fastx toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html). Reads passing these filters were mapped the annotated rRNA transcripts with the GEM aligner (Marco-Sola et al., 2012) with parameters ‘‘-m 0.04 -e 1–max-big-indel-length 0.’’ Reads that did not align to the rRNA transcripts were then mapped to the E. coli transcriptome with GEM, using parameters ‘‘-m 0.04 -e 0.04–max-big-in-del-length 0.’’ We defined the E. coli transcriptome as the transcriptional units (TUs) as annotated by RegulonDB, wherein for each operon defined by RegulonDB, we included only the longest associated transcriptional unit (Gama-Castro et al., 2016). For non-rRNA genes, transcript abundance was calculated with RSEM (Li and Dewey, 2011; Li et al., 2010) Gene expression was variance-stabilized with voom (Law et al., 2014), and differential expression between pairs of conditions was calculated with limma (Ritchie et al., 2015). For a given pair of conditions, we declared genes on TUs with FDR %0.05 to be differentially expressed. To calculate rDNA operon expression, reads that were mapped to the rRNA transcripts were re-aligned with GEM (Marco-Sola et al., 2012), but with high error rate to encourage multimapping of reads to all homologous positions among the rRNAs (parameters: ‘‘-m 4 -e 4–max-big-indel-length 0 –d all’’). rRNA abundance was calculated via RSEM. Note that the RSEM algorithm accounts for multimapping reads in a principled and rigorous fashion to infer abundance (Li et al., 2010). Differential expression of rRNA between pairs of conditions was calculated with limma (Ritchie et al., 2015).
Generation of mutant E. coli strains
The 16S rRNA portion of rrnH operon (rrsH) was PCR amplified from E. coli K12 MG1655 genomic DNA using the following primers: rrsH_1655_F: (5’–ACAGCCGGTTCGGTTGAAGAG–3’) and rrsH_1655_R: (5’– CCTGAGCTACAAGCCTGTAGAGG – 3’). The single PCR product was cloned into the pCR™ –TOPO TA Vector using the TOPO TA Cloning Kit (Thermo-Fisher) and verified using Sanger sequencing. The rrsH beak domain was excised from the resulting plasmid, pCR-TOPO-rrnH, using ApaI (NEB) and RsrII (NEB) and sub-cloned into pKK3535 (‘‘pKK3535-BBB’’) (Brosius et al., 1981) linearized with the same enzymes. The resulting plasmid, pKK3535-HBB, retained all large subunit rRNAs and regulatory features (promoters, spacers, tRNAs, terminators) intrinsic to rrnB. The sequences of pKK3535-BBB and pKK3535-HBB were determined, in their entirety, by the MGH CCIB DNA Core. The pKK3535-BBB and pKK3535-HBB plasmids were transformed into MC388, a strain lacking endogenous rDNA operons (Asai et al., 1999) and containing the pCSacB7 plasmid as described previously (Sun et al., 2011). The resulting strains, Δ7prrn-BBB and Δ7prrn-HBB, express rRNA from either the wild-type rrnB (pKK3535-BBB) or rrnB containing the h33 sequence of rrsH (pKK3535-HBB) and have indistinguishable doubling times in minimal media (∼70 min.). Additional genetic background strains expressing plasmids pKK3535-BBB and pKK3535-HBB were: K-12 MG1655 (Blattner et al., 1997; Kurylo et al., 2016), which exhibited indistinguishable doubling times (∼65 min.), BW25113, JW2755–3 (ΔrelA), JW1555–2 (ΔrelE), JW5437–1 (DrpoS), and JW1228–1 (ΔadhE) (Yamamoto et al., 2009).
Bacterial strain growth experiments
Overnight cultures of all strains were grown in LB broth. When the cells from rrsB and rrsH-expressing strains reached equivalent points in early stationary phase (OD600 = 1.5), cells were diluted in pre-warmed M9 minimal media containing the appropriate selection markers to a final cell density of OD600 = 0.03 and grown at 37°C with 225 rpm shaking in baffled flasks. Antibiotics were added at the following concentrations when required: 50 μg/mL ampicillin, 50 μg/mL kanamycin, 40 μg/mL spectinomycin. At OD600nm = 0.5–0.6, cells were harvested. To harvest cells, µΔ7prrn strains were pretreated with chloramphenicol at 125 mg/mL for 5 minutes with shaking. Then, cells were pelleted for 4 min. at 4,200 rpm (+4°C) in an Allegra X-15R centrifuge (Beckman) and flash frozen on liquid nitrogen. All other strains were harvested using rapid vacuum filtration (as described above). Total and polyso-mal RNA were prepared as described above and sequenced using RNA-Seq (as described above).
Conservation of rRNA variants
Tree-of-life-wide multiple alignments of large and small subunit rRNAs were downloaded as ARB files from the ARB-Silva database (Quast et al., 2013). The rrsH h33 ‘‘beak’’ region was considered conserved in organism X if X contained at least one rRNA transcript R for which positions 997–1044 of rrsH all aligned and matched exactly to nucleotides in R. Conservation of h9-h10 rrsG ‘‘foot’’ region was determined analogously, using positions 180–230 of rrsG. To test if rDNA operons containing the rrsH h33 beak sequence were more distally located from the original of replication than expected by chance, a permutation test was performed on the 188 organ-isms with assembled genomes in GenBank and whose 16S rRNA sequences were represented in the ARB database and which con-tained at least one rDNA operon with an rrsH-matching h33 sequence. The origin of replication for these genomes was determined using the oriloc function from R package seqinr.
DMS-MapSeq
DMS treatment was performed in quadruplicate in a final volume of 25 μl, containing 30 mM K-HEPES (pH 7.5), 7 mM MgOAc2, 100 mM KCl, 2mM DTT, 1.0 μM of 30S ribosomal subunits isolated from Δ7prrn-HBB and Δ7prrn-BBB strains. 1 μL of 2.5 M dimethyl sulfate (sigma D186309, use within 6 months) diluted in ethanol (to 100 mM final concentration) was added, reactions mixed by pipetting, and incubated at 37°C for 4 minutes. Reactions were quenched by addition of 475 μL of (30% 2-mercaptoethanol, 0.3 M sodium acetate, pH 5.5), isopropanol precipitated, resuspended in 200 μL of 0.3M sodium acetate (pH 5.2) and extracted twice with acid phenol-chlo-roform-isoamyl alcohol (Thermo-Fisher AM9722). Reactions were ethanol precipitated again, and libraries were prepared and sequenced as in (McClary et al., 2017) except that after ligation reactions samples were cleaned up with 5’ deadenylase and recJ (NEB) in lieu of gel purification. Raw reads were processed as before, except that Δ7prrn-HBB and Δ7prrn-BBB were separately aligned to their corresponding sequences. The nucleotide misincorporation rate, a proxy for the extent of DMS modification, was defined as the number of sequence mismatches at an rRNA position, divided by the number of reads overlapping position. All A and C positions, excluding the positions that differ in sequence between strains, with DMS signal 10-fold above the no-DMS background were compared by t test. Positions were considered to have different DMS reactivity if the ratio of DMS signal to no-DMS background was statistically significant after Bonferroni correction and the absolute difference between the log2 ratios was greater than 2. Raw sequencing data and mutation rates have been deposited in the gene expression omnibus with accession number GSE 105112.
Generation of mutant E. coli strains
The 16S rRNA portion of rrnH operon (rrsH) was PCR amplified from E. coli K12 MG1655 genomic DNA using the following primers: rrsH_1655_F: (5’–ACAGCCGGTTCGGTTGAAGAG–3’) and rrsH_1655_R: (5’– CCTGAGCTACAAGCCTGTAGAGG – 3’). The single PCR product was cloned into the pCR™ –TOPO® TA Vector using the TOPO TA Cloning Kit (Thermo-Fisher) and verified using Sanger sequencing. The rrsH beak domain was excised from the resulting plasmid, pCR-TOPO-rrnH, using ApaI (NEB) and RsrII (NEB) and sub-cloned into pKK3535 (‘‘pKK3535-BBB’’) (Brosius et al., 1981) linearized with the same enzymes. The resulting plasmid, pKK3535-HBB, retained all large subunit rRNAs and regulatory features (promoters, spacers, tRNAs, terminators) intrinsic to rrnB. The sequences of pKK3535-BBB and pKK3535-HBB were determined, in their entirety, by the MGH CCIB DNA Core. The pKK3535-BBB and pKK3535-HBB plasmids were transformed into MC388, a strain lacking endogenous rDNA operons (Asai et al., 1999) and containing the pCSacB7 plasmid as described previously (Sun et al., 2011). The resulting strains, Δ7prrn-BBB and Δ7prrn-HBB, express rRNA from either the wild-type rrnB (pKK3535-BBB) or rrnB containing the h33 sequence of rrsH (pKK3535-HBB), and have indistinguishable doubling times in minimal media (∼70 min.). Additional genetic background strains expressing plasmids pKK3535-BBB and pKK3535-HBB were: K-12 MG1655 (Blattner et al., 1997; Kurylo et al., 2016), which exhibited indistinguishable doubling times (∼65 min.), BW25113, JW2755–3 (ΔrelA), JW1555–2 (ΔrelE), JW1228–1 (ΔadhE), and JW5437–1 (DrpoS) (Yamamoto et al., 2009).
Bacterial strain growth experiments
Overnight cultures of all strains were grown in LB broth. When the cells from rrsB and rrsH-expressing strains reached equivalent points in stationary phase, cells were diluted in pre-warmed M9 minimal media (as described above) containing the appropriate selection markers and grown at °37 C with 225 rpm shaking in baffled flasks. Antibiotics were added at the following concentrations when required: 50 μg/mL ampicillin, 50 μg/mL kanamycin, 40 μg/mL spectinomycin. At OD600nm = 0.5–0.6, cells were harvested. To harvest cells, Δ7prrn strains were pre-treated with chloramphenicol at 125 μg/mL for 5 minutes with shaking. Then, cells were pelleted for 4 min. at 4,200 rpm (+4°C) in an Allegra X-15R centrifuge (Beckman) and flash frozen on liquid nitrogen. All other strains were harvested using rapid vacuum filtration (as described above). Total and polysomal RNA were prepared as described above and sequenced using RNA-Seq (as described above).
Genome Sequencing and Analysis
Genomic DNA (gDNA) was harvested from overnight cultures of strains Δ7prrn-HBB and Δ7prrn-BBB using the QIAamp DNA Mini Kit (QIAGEN) according to the manufacturer’s instructions. DNA quality and quantity were determined using an Agilent 2200 TapeStation and Qubit dsDNA BR Assay (Life Technologies), respectively. gDNA was sequenced on a Pacific Biosciences RS II instrument at Weill Cornell Medical College and single-contig chromosome assemblies were obtained at greater than 80X coverage for each strain as previously described (Kurylo et al., 2016). De novo genome assemblies were constructed for Δ7prrn-HBB and Δ7prrn-BBB, separately, using the RS_Resequencing.1 module within Pacific Biosciences SMRT Portal. Long reads were then cross-mapped between strains (Δ7prrn-HBB reads mapped to the Δ7prrn-BBB de novo genome assembly, and vice-versa, called ‘‘remapping’’) and analyzed with custom scripts. A position is considered polymorphic if remapping gives reciprocal variant calls at the same position of the genome. Here, a variant call at position p is a reciprocal if the reads from strain A mapped onto the de novo assembly of strain B call an insertion at position p, and the reads from strain B mapped onto the de novo assembly of strain A call a deletion at position p, or vice-versa. There were no reciprocal variants, and therefore we concluded that strains Δ7prrn-HBB and Δ7prrn-BBB possess no chromosomal differences. The complete sequences of the pKK3535 plasmids used in these strains were also determined using Illumina sequencing at the MGH CCIB DNA Core.
Phenotype Microarray Experiment and Analysis
BIOLOG phenotype microarrays (PMs) enable the simultaneous quantitation of growth rate in a large number of conditions by moni-toring cellular respiration using the irreversible reduction of tetrazolium dye to the purple compound formazan in multi-well plates (Bochner et al., 2001). The growth of strains Δ7prrn-HBB and Δ7prrn-BBB on PM11–20 was compared in three separate experiments using the OPM analysis package (Vehkala et al., 2015).
Proteomics and data analysis
Strains Δ7prrn-BBB and Δ7prrn-HBB were grown to mid-log in M9 minimal media (as described above) and tightly-coupled 70S ri-bosomes were isolated as previously described (Blanchard et al., 2004b; Powers and Noller, 1991). Six samples (three biological replicates from each strain) were denatured, reduced, and alkylated followed by proteolytic digestion with LysC (Wako Chemicals) and trypsin (Promega). Approximately 120 femtomol of each desalted (Rappsilber et al., 2007) sample was analyzed by nano-LC-MS/ MS system (Ultimate 3000 coupled to a QExactive Plus, Thermo Scientific). Peptides were separated using a 12 cm x 75mm C18 col-umn (3 mm particles, Nikkyo Technos Co., Ltd. Japan) at a flow rate of 200 nL/min, with a 5%–40% gradient over 130 minutes (buffer 0.1% formic acid, buffer B: 0.1% formic acid in acetonitrile). Data were quantified and searched against E.coli database (July 2014) using MaxQuant (version 1.5.3.28) (Cox et al., 2011). Oxidation of methionine and protein N-terminal acetylation were allowed as variable modifications, cysteine carbamidomethyl was set as a fixed modification and two missed cleavages were allowed. ‘‘Match between runs’’ option was enabled, and false discovery rates for proteins and peptides were set to 1%. Protein abundances were expressed as LFQ (label free quantitation) values. Proteins were considered to be differentially observed if their fold-change ex-ceeded 1.5 and their FDR was less than 0.05.
Western blotting
Cells were grown in M9 minimal media (as described above) and harvested at mid-log. For 96 kDa protein AdhE: cell lysate was pre-pared using cryolysis, as described above. 15 μg of cell lysate from each strain was run on 4%–20% SDS-PAGE gels (Bio-Rad) and samples used for western blot were transferred to a nitrocellulose blotting membrane (Amersham) using a TE 77 Semi-dry Transfer Unit (Amersham). As a loading control for total protein, a second gel with the same samples was stained with the Colloidal Blue Staining Kit (Thermo) according to manufacturer’s instructions. The nitrocellulose membrane was blocked for three hours at room temperature in 1X tris-buffered saline containing 0.1% Tween-20 (TBST) and 5% non-fat dairy milk. Then, the membrane was incubated overnight at +4°C with anti-AdhE antibody (Agrisera #AS10 748) at a dilution of 1:4000, washed with TBST, incubated with goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology) at 1:10k dilution for 1 hour at room temperature, washed with TBST, and imaged (using a G-box imaging system from Syngene) with ECL Select Western Blotting Detection Reagent (Amersham). Quantification was performed in ImageJ. For 38 kDa protein RpoS: cell pellets were directly resuspended in Laemmli buffer normalized to cell number, heat denatured at 95°C for 5 min., and cell lysate was clarified by microcentrifugation. Western blotting was performed using the same protocol as above, but with an anti-RpoS primary antibody (BioLegend #W0009, clone 1RS1) at a dilution of 1:4000 and a goat antimouse IgG-HRP secondary antibody (Santa Cruz Biotechnology) at a dilution of 1:10k.
qRT-PCR
Primer pairs were first validated for amplification efficiency using a standard curve. cDNA was generated using random hexamer priming and the SuperScript III First-Strand Synthesis Kit (Thermo Fisher). qRT-PCR was performed using SYBR® Green Real-Time PCR Master Mix (Thermo Fisher) on a QuantStudio 3 Real-Time PCR System (Thermo Fisher). Quantification and statistical analysis was performed using the ddCT method (Pfaffl, 2001). Primer pairs for aceE and gabD, respectively, were: aceE_F: 5’-ATGACGTGGATCCGATCGAA-3’ and ace_R: 5’-GAATACGGCGTTCCAGTTCC-3’; gabD_F: 5’-TTCTTCCAGCCGACCATTCT-3’ and gabD_R: 5’-GGCTTAAATCACGGGCGTAG-3’. Genes were normalized to hcaT levels as previously described (Peng et al., 2014).
Single-molecule fluorescence (smFRET) microscopy
Single-molecule FRET experiments of aa-tRNA selection were performed as described previously (Blanchard et al., 2004b; Cocozaki et al., 2016; Geggier et al., 2010; Juette et al., 2016) in the presence of 1 μM tetracycline or, as a control, in the absence of the drug. Briefly, 70S initiation complexes isolated as previously described (Blanchard et al., 2004b) from Δ7prrn-BBB or Δ7prrn-HBB and pro-grammed Cy3 s4U8-labeled P-site fMet-tRNAfMet and 5’-biotinylated mRNA were surface-immobilized within a passivated microfluidic chamber via a biotin-streptavidin bridge. Single-molecule imaging was performed on a custom-built prism-based total internal reflection fluorescence microscope (Juette et al., 2016) using custom LabVIEW software. Movies were recorded at 15 ms time resolution with continuous green (532 nm) laser excitation at 300 W/cm2 intensity. After pre-equilibration in 1X Tris-polymix pH7.5 containing 1 mM tetracycline, pre-formed EF-Tu∙ Phe-tRNAPhe∙GTP ternary complex containing LD650-acp3U47-labeled tRNA (Blanchard et al., 2004b), also pre-equilibrated in 1 mM tetracycline, was delivered to the flow cell at a final concentration of 20 nM by manual injection at the beginning of each movie.
smFRET data analysis
FRET traces were calculated from movies and analyzed using the custom software package SPARTAN (https://scottcblanchardlab. com/software) as described previously (Juette et al., 2016). Traces from 7 movies per condition (Δ7prrn-BBB or Δ7prrn-HBB ribosomes), recorded on 4 different days, were combined and pre-filtered using standard criteria (Juette et al., 2016) (signal-to-noise ratio > 8, background noise < 70 photoelectrons, donor-acceptor correlation coefficient between 1.1 and 0.5, removal of traces with multistep photobleaching). FRET events (> 1 frame) were identified by idealizing to a simple two-state (on/off) model using the SKM algorithm (Qin, 2007), extracted and post-synchronized by aligning to the first appearance of FRET. The resulting event traces were idealized to a previously established 4-state model of aa-tRNA selection (Geggier et al., 2010) using SKM. Based on these idealizations, the probability of forward transitions from the low-FRET/codon recognition state to intermediate-FRET/ GTPase-activated or high-FRET/accommodated states were computed. Differences in state transition probabilities were tested with logistic regression. In the absence of tetracycline, we found s forward-sampling probabilities of 0.627 ± 0.022 and 0.643 ± 0.020 for Δ7prrn-BBB and Δ7prrn-HBB ribosomes, respectively (not significantly different, p = 0.1). In the presence of 1 μM tetracycline, the determined forward-sampling probabilities were 0.407 ± 0.010 for Δ7prrn-BBB and 0.458 ± 0.009 Δ7prrn-HBB (p = 1.15e-20).
Biofilm formation assays
Biofilm formation assays were performed as previously described (Coffey and Anderson, 2014). Overnight cultures of E. coli were grown in LB broth to saturation at 37° C. In the morning, cells were diluted to OD600 = 0.2 and grown to OD600 = 1.4–1.5 in 10 mL LB broth. 50 mL of M9 minimal media (as described above) was then inoculated with 750 of cells and grown to OD600 = 0.5–0.6. At this point, cells were diluted 1:100 in fresh M9 media and 100 μL was added to each well of a ‘‘U’’ bottom polystyrene 96-well plates (VWR) in multiple replicates. A blank well of uninoculated media was used as the control. Plates were incubated at 37 C for 48 h. to allow for biofilm formation. After 48 h., wells were washed with water and biofilms were stained for 10 min. with 0.1% crystal violet. Wells were washed again with water and dried overnight at room temperature. In the morning, 150 μL 30% acetic acid was added to wells for 10 min. After 10 min., well contents were mixed by pipetting and 125 μL was transferred to a transparent, flat-bottom 96-well plate (VWR). Quantification of biofilm was performed by taking an absorbance reading at 595 nm using an Infinite M1000 Pro (TECAN).
Cell motility assays
Cells were grown in the same manner as for biofilm formation assays (described above). Cells were grown to mid-log in M9 media OD600 = 0.5–0.6 before 5 mL was spotted on the center of M9 minimal media (as described above) semi-solid agar plates (0.25% agar) and incubated at room temperature on a lab bench. Cell motility images were captured using a G:Box (Syngene) and quantification was performed by measuring diameter of cell ring using ImageJ. Cell motility was statistically compared using unpaired t tests.
QUANTIFICATION AND STATISTICAL ANALYSIS
Gene Ontology enrichment p values for gene set analyses were calculated via the Fisher Exact Test using the limma package (Ritchie et al., 2015). P values were differential RpoS protein levels quantified from western blots were obtained with the t test.
p values for differential codon usage were obtained using the known population median test (Parks, 2018).
Permutation tests were performed to test for statistically significant overlaps between two differentially expressed gene sets. In particular, for consider differentially expressed gene set Si consisting of nu;i upregulated genes and nd;i downregulated genes. The aim is to test if the total number of commonly regulated genes between S1 and S2, i.e., upregulated in both S1 and S2 or down-regulated in both S1 and S2, is higher than expected by chance. For n = 106 permutations, we sampled nu;2 genes and nd;2 genes and counted the total number overlapping genes by comparing to those upregulated and downregulated in gene set S1, respectively.
Supplementary Material
Highlights.
Nutrient limitation upregulates the relative expression of the rDNA operon, rrnH
Ribosomes bearing the 16S rRNA, rrsH, can modulate the general stress response
rrsH-bearing ribosomes impact drug resistance, cell motility, and biofilm formation
Naturally occurring rRNA sequence variation encodes altered ribosome function
ACKNOWLEDGMENTS
We would like to thank Drs. Henrik Molina and Milica Tesic Mark for their assistance and expertise performing the mass spectrometry investigations presented. We thank Drs. Catherine L. Squires (Stanford) and Michael O’Connor (UMKC) for helpful early discussions. We thank members of the Blanchard lab and Dr. Kyu Rhee for critical comments and discussion during manuscript preparation and Dr. Lucy Skrabanek from the Weill Cornell Medicine Applied Bioinformatics Core for preliminary data analysis efforts. This work was funded by the NIH (R01-GM079238 to S.C.B.), the Tri-Institutional Stem Cell Initiative funded by the Starr Foundation (to S.C.B. and C.T.V.), the Swedish Research Council (Vetenskapsra˚det) (to C.T.V.), and the Jacques Cohenca Predoctoral Fellowship (to C.M.K.). B.Z. is an HHMI Fellow of the Damon Runyon Cancer Research Foundation (DRG-2250–16).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
Data from RNA-Sequencing experiments has been deposited as SRA: PRJNA487814. Raw sequencing data and mutation rates from DMS-MaPseq experiments have been deposited as GEO: GSE105112.
REFERENCES
- Antoniani D, Rossi E, Rinaldo S, Bocci P, Lolicato M, Paiardini A, Raf-faelli N, Cutruzzolá F, and Landini P (2013). The immunosuppressive drug azathioprine inhibits biosynthesis of the bacterial signal molecule cyclic-di-GMP by interfering with intracellular nucleotide pool availability. Appl. Micro-biol. Biotechnol 97, 7325–7336. [DOI] [PubMed] [Google Scholar]
- Asai T, Condon C, Voulgaris J, Zaporojets D, Shen B, Al-Omar M, Squires C, and Squires CL (1999). Construction and initial characterization of Escherichia coli strains with few or no intact chromosomal rRNA operons. J. Bacteriol 181, 3803–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battesti A, Majdalani N, and Gottesman S (2011). The RpoS-mediated general stress response in Escherichia coli. Annu. Rev. Microbiol 65, 189–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckham KSH, Connolly JPR, Ritchie JM, Wang D, Gawthorne JA, Tahoun A, Gally DL, Burgess K, Burchmore RJ, Smith BO, et al. (2014). The metabolic enzyme AdhE controls the virulence of Escherichia coli O157:H7. Mol. Microbiol 93, 199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard SC, Gonzalez RL, Kim HD, Chu S, and Puglisi JD (2004a). tRNA selection and kinetic proofreading in translation. Nat. Struct. Mol. Biol 11, 1008–1014. [DOI] [PubMed] [Google Scholar]
- Blanchard SC, Kim HD, Gonzalez RL Jr., Puglisi JD, and Chu S (2004b). tRNA dynamics on the ribosome during translation. Proc. Natl. Acad. Sci. USA 101, 12893–12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. (1997). The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462. [DOI] [PubMed] [Google Scholar]
- Bochner BR, Gadzinski P, and Panomitros E (2001). Phenotype microar-rays for high-throughput phenotypic testing and assay of gene function. Genome Res 11, 1246–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JW, and Dinman JD (2017). Subtractional heterogeneity: a crucial step toward defining specialized ribosomes. Mol. Cell 67, 3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodersen DE, Clemons WM Jr., Carter AP, Morgan-Warren RJ, Wimberly BT, and Ramakrishnan V (2000). The structural basis for the ac-tion of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell 103, 1143–1154. [DOI] [PubMed] [Google Scholar]
- Brosius J, Ullrich A, Raker MA, Gray A, Dull TJ, Gutell RR, and Noller HF (1981). Construction and fine mapping of recombinant plasmids contain-ing the rrnB ribosomal RNA operon of E. coli. Plasmid 6, 112–118. [DOI] [PubMed] [Google Scholar]
- Brown A, Fernández IS, Gordiyenko Y, and Ramakrishnan V (2016). Ribosome-dependent activation of stringent control. Nature 534, 277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravorty S, Helb D, Burday M, Connell N, and Alland D (2007). A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 69, 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho B-K, Zengler K, Qiu Y, Park YS, Knight EM, Barrett CL, Gao Y, and Palsson BØ (2009). The transcription unit architecture of the Escherichia coli genome. Nat. Biotechnol 27, 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SK, Mikkelsen M, Pedersen K, and Gerdes K (2001). RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. USA 98, 14328–14333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocozaki AI, Altman RB, Huang J, Buurman ET, Kazmirski SL, Doig P, Prince DB, Blanchard SC, Cate JHD, and Ferguson AD (2016). Resistance mutations generate divergent antibiotic susceptibility profiles against translation inhibitors. Proc. Natl. Acad. Sci. USA 113, 8188–8193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey BM, and Anderson GG (2014). Biofilm formation in the 96-well mi-crotiter plate. Methods Mol. Biol 1149, 631–641. [DOI] [PubMed] [Google Scholar]
- Condon C, Philips J, Fu ZY, Squires C, and Squires CL (1992). Comparison of the expression of the seven ribosomal RNA operons in Escherichia coli. EMBO J 11, 4175–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condon C, Liveris D, Squires C, Schwartz I, and Squires CL (1995). rRNA operon multiplicity in Escherichia coli and the physiological implications of rrn inactivation. J. Bacteriol 177, 4152–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier E, and Rocha EPC (2006). Replication-associated gene dosage effects shape the genomes of fast-growing bacteria but only for transcription and translation genes. Mol. Microbiol 59, 1506–1518. [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, and Mann M (2011). Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res 10, 1794–1805. [DOI] [PubMed] [Google Scholar]
- Culviner PH, and Laub MT (2018). Global analysis of the E. coli toxin MazF reveals widespread cleavage of mRNA and the inhibition of rRNA maturation and ribosome biogenesis. Mol. Cell 70, 868–880.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinman JD (2016). Pathways to specialized ribosomes: the brussels lecture. J. Mol. Biol 428 (10 Pt B), 2186–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong T, and Schellhorn HE (2009). Control of RpoS in global gene expression of Escherichia coli in minimal media. Mol. Genet. Genomics 281, 19–33. [DOI] [PubMed] [Google Scholar]
- Dunkle JA, Wang L, Feldman MB, Pulk A, Chen VB, Kapral GJ, Noeske J, Richardson JS, Blanchard SC, and Cate JHD (2011). Struc-tures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science 332, 981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durfee T, Hansen A-M, Zhi H, Blattner FR, and Jin DJ (2008). Transcription profiling of the stringent response in Escherichia coli. J. Bacteriol 190, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti MB, Ghalei H, Ward EA, Potts EL, and Karbstein K (2017). Rps26 directs mRNA-specific translation by recognition of Kozak sequence elements. Nat. Struct. Mol. Biol 24, 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson A, Ballesteros M, Peterson CN, Persson O, Silhavy TJ, and Nystro¨m T (2007). Decline in ribosomal fidelity contributes to the accumula-tion and stabilization of the master stress response regulator sigmaS upon carbon starvation. Genes Dev 21, 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama-Castro S, Salgado H, Santos-Zavaleta A, Ledezma-Tejeida D, Mu-ñiz-Rascado L, Garcı´a-Sotelo JS, Alquicira-Hernández K, Martı´nez-Flores I, Pannier L, Castro-Mondrago´n JA, et al. (2016). RegulonDB version 9.0: high-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res 44 (D1), D133–D143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geggier P, Dave R, Feldman MB, Terry DS, Altman RB, Munro JB, and Blanchard SC (2010). Conformational sampling of aminoacyl-tRNA during selection on the bacterial ribosome. J. Mol. Biol 399, 576–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genuth NR, and Barna M (2018). The discovery of ribosome heterogeneity and its implications for gene regulation and organismal life. Mol. Cell 71, 364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenier F, Matteau D, Baby V, and Rodrigue S (2014). Complete genome sequence of Escherichia coli BW25113. Genome Announc 2, e01038–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson JH, Sogin ML, Wollett G, Hollingdale M, de la Cruz VF, Waters AP, and McCutchan TF (1987). Structurally distinct, stage-specific ribosomes occur in Plasmodium. Science 238, 933–937. [DOI] [PubMed] [Google Scholar]
- Guo MS, and Gross CA (2014). Stress-induced remodeling of the bacterial proteome. Curr. Biol 24, R424–R434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorfy Z, Draskovits G, Vernyik V, Blattner FF, Gaal T, and Posfai G (2015). Engineered ribosomal RNA operon copy-number variants of E. coli reveal the evolutionary trade-offs shaping rRNA operon number. Nucleic Acids Res 43, 1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto JG, Stevenson BS, and Schmidt TM (2003). Rates and con-sequences of recombination between rRNA operons. J. Bacteriol 185, 966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauryliuk V, Atkinson GC, Murakami KS, Tenson T, and Gerdes K (2015). Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Microbiol 13, 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge-Aronis R (1993). Survival of hunger and stress: the role of rpoS in early stationary phase gene regulation in E. coli. Cell 72, 165–168. [DOI] [PubMed] [Google Scholar]
- Hillebrand A, Wurm R, Menzel A, and Wagner R (2005). The seven E. coli ribosomal RNA operon upstream regulatory regions differ in structure and transcription factor binding efficiencies. Biol. Chem 386, 523–534. [DOI] [PubMed] [Google Scholar]
- Hirvonen CA, Ross W, Wozniak CE, Marasco E, Anthony JR, Aiyar SE, Newburn VH, and Gourse RL (2001). Contributions of UP elements and the transcription factor FIS to expression from the seven rrn P1 promoters in Escherichia coli. J. Bacteriol 183, 6305–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack K, Bellodi C, Landry DM, Niederer RO, Meskauskas A, Musal-gaonkar S, Kopmar N, Krasnykh O, Dean AM, Thompson SR, et al. (2011). rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol. Cell 44, 660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner L, Starosta AL, Terry DS, Mikolajka A, Filonava L, Yusupov M, Blanchard SC, Wilson DN, and Yusupova G (2013). Structural basis for potent inhibitory activity of the antibiotic tigecycline during protein synthesis. Proc. Natl. Acad. Sci. USA 110, 3812–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin DJ, Cagliero C, and Zhou YN (2012). Growth rate regulation in Escherichia coli. FEMS Microbiol. Rev 36, 269–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juette MF, Terry DS, Wasserman MR, Altman RB, Zhou Z, Zhao H, and Blanchard SC (2016). Single-molecule imaging of non-equilibrium mo-lecular ensembles on the millisecond timescale. Nat. Methods 13, 341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley R, and Ideker T (2005). Systematic interpretation of genetic interac-tions using protein networks. Nat. Biotechnol 23, 561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-L, Shin E-K, Kim H-M, Ryou S-M, Kim S, Cha C-J, Bae J, and Lee K (2007). Heterogeneous rRNAs are differentially expressed during the morphological development of Streptomyces coelicolor. FEMS Microbiol. Lett 275, 146–152. [DOI] [PubMed] [Google Scholar]
- Kohler R, Mooney RA, Mills DJ, Landick R, and Cramer P (2017). Archi-tecture of a transcribing-translating expressome. Science 356, 194–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmsee T, and Hengge R (2011). Rare codons play a positive role in the expression of the stationary phase sigma factor RpoS (s(S)) in Escherichia coli. RNA Biol 8, 913–921. [DOI] [PubMed] [Google Scholar]
- Kurylo CM, Alexander N, Dass RA, Parks MM, Altman RA, Vincent CT, Mason CE, and Blanchard SC (2016). Genome sequence and anal-ysis of Escherichia coli MRE600, a colicinogenic, nonmotile strain that lacks RNase I and the type I methyltransferase, EcoKI. Genome Biol. Evol 8, 742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie M (2017). There are millions of protein factories in every cell. Surprise, they’re not all the same. Science http://www.sciencemag.org/news/2017/06/there-are-millions-protein-factories-every-cell-surprise-they-re-not-all-same.
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Ruotti V, Stewart RM, Thomson JA, and Dewey CN (2010). RNA-seq gene expression estimation with read mapping uncertainty. Bioinformatics 26, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locati MD, Pagano JFB, Girard G, Ensink WA, van Olst M, van Leeu-wen S, Nehrdich U, Spaink HP, Rauwerda H, Jonker MJ, et al. (2017). Expression of distinct maternal and somatic 5.8S, 18S, and 28S rRNA types during zebrafish development. RNA 23, 1188–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish HF (1974). Model for the regulation of mRNA translation applied to haemoglobin synthesis. Nature 251, 385–388. [DOI] [PubMed] [Google Scholar]
- López-López A, Benlloch S, Bonfa´ M, Rodríguez-Valera F, and Mira A (2007). Intragenomic 16S rDNA divergence in Haloarcula marismortui is an adaptation to different temperatures. J. Mol. Evol 65, 687–696. [DOI] [PubMed] [Google Scholar]
- Marco-Sola S, Sammeth M, Guigo´ R, and Ribeca P (2012). The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods 9, 1185–1188. [DOI] [PubMed] [Google Scholar]
- Mauri M, and Klumpp S (2014). A model for sigma factor competition in bac-terial cells. PLoS Comput. Biol 10, e1003845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClary B, Zinshteyn B, Meyer M, Jouanneau M, Pellegrino S, Yusu-pova G, Schuller A, Reyes JCP, Lu J, Guo Z, et al. (2017). Inhibition of eukaryotic translation by the antitumor natural product agelastatin A. Cell Chem. Biol 24, 605–613.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGary K, and Nudler E (2013). RNA polymerase and the ribosome: the close relationship. Curr. Opin. Microbiol 16, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EW, and Green R (2017). Ribosomopathies: there’s strength in numbers. Science 358, eaan2755. [DOI] [PubMed] [Google Scholar]
- Neubauer C, Gao Y-G, Andersen KR, Dunham CM, Kelley AC, Hent-schel J, Gerdes K, Ramakrishnan V, and Brodersen DE (2009). The struc-tural basis for mRNA recognition and cleavage by the ribosome-dependent endonuclease RelE. Cell 139, 1084–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noller HF (2005). RNA structure: reading the ribosome. Science 309, 1508–1514. [DOI] [PubMed] [Google Scholar]
- O’Connor M (2009). Helix 69 in 23S rRNA modulates decoding by wild type and suppressor tRNAs. Mol. Genet. Genomics 282, 371–380. [DOI] [PubMed] [Google Scholar]
- Ochman H, and Wilson AC (1987). Evolution in bacteria: evidence for a universal substitution rate in cellular genomes. J. Mol. Evol 26, 74–86. [DOI] [PubMed] [Google Scholar]
- österberg S, del Peso-Santos T, and Shingler V (2011). Regulation of alter-native sigma factor use. Annu. Rev. Microbiol 65, 37–55. [DOI] [PubMed] [Google Scholar]
- Parks MM (2018). An exact test for comparing a fixed quantitative property between gene sets. Bioinformatics 34, 971–977. [DOI] [PubMed] [Google Scholar]
- Parks MM, Kurylo CM, Dass RA, Bojmar L, Lyden D, Vincent CT, and Blanchard SC (2018). Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci. Adv 4, eaao0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S, Stephan R, Hummerjohann J, and Tasara T (2014). Transcrip-tional analysis of different stress response genes in Escherichia coli strains subjected to sodium chloride and lactic acid stress. FEMS Microbiol. Lett 361, 131–137. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers T, and Noller HF (1991). A functional pseudoknot in 16S ribosomal RNA. EMBO J 10, 2203–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokopowich CD, Gregory TR, and Crease TJ (2003). The correlation between rDNA copy number and genome size in eukaryotes. Genome 46, 48–50. [DOI] [PubMed] [Google Scholar]
- Qin F (2007). Principles of single-channel kinetic analysis. Methods Mol. Biol 403, 253–286. [DOI] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glóckner FO (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, and van Oudenaarden A (2008). Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135, 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramrath DJF, Yamamoto H, Rother K, Wittek D, Pech M, Mielke T, Loerke J, Scheerer P, Ivanov P, Teraoka Y, et al. (2012). The complex of tmRNA-SmpB and EF-G on translocating ribosomes. Nature 485, 526–529. [DOI] [PubMed] [Google Scholar]
- Ranquet C, and Gottesman S (2007). Translational regulation of the Escher-ichia coli stress factor RpoS: a role for SsrA and Lon. J. Bacteriol 189, 4872–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purifica-tion, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, and Nei M (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol 4, 406–425. [DOI] [PubMed] [Google Scholar]
- Sauert M, Temmel H, and Moll I (2015). Heterogeneity of the translational machinery: Variations on a common theme. Biochimie 114, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schembri MA, Kjaergaard K, and Klemm P (2003). Global gene expression in Escherichia coli biofilms. Mol. Microbiol 48, 253–267. [DOI] [PubMed] [Google Scholar]
- Schureck MA, Repack A, Miles SJ, Marquez J, and Dunham CM (2016). Mechanism of endonuclease cleavage by the HigB toxin. Nucleic Acids Res 44, 7944–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y, Brown Gandt A., Rowe SE, Deisinger JP, Conlon BP, and Lewis K (2017). ATP-dependent persister formation in Escherichia coli. MBio 8, e02267–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shasmal M, Dey S, Shaikh TR, Bhakta S, and Sengupta J (2016). E. coli metabolic protein aldehyde-alcohol dehydrogenase-E binds to the ribosome: a unique moonlighting action revealed. Sci. Rep 6, 19936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Fujii K, Kovary KM, Genuth NR, Róst HL, Teruel MN, and Barna M (2017). Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol. Cell 67, 71–83.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solheim M, La Rosa SL, Mathisen T, Snipen LG, Nes IF, and Brede DA (2014). Transcriptomic and functional analysis of NaCl-induced stress in Enterococcus faecalis. PLoS ONE 9, e94571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S, Weiser B, and Noller HF (1988). Model for the three-dimensional folding of 16 S ribosomal RNA. J. Mol. Biol 204, 447–481. [DOI] [PubMed] [Google Scholar]
- Sun Q, Vila-Sanjurjo A, and O’Connor M (2011). Mutations in the intersubunit bridge regions of 16S rRNA affect decoding and subunit-subunit interactions on the 70S ribosome. Nucleic Acids Res 39, 3321–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D-L, Jiang X, Wu QL, and Zhou N-Y (2013). Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Appl. Environ. Microbiol 79, 5962–5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamar E, Koler M, and Vaknin A (2016). The role of motility and chemotaxis in the bacterial colonization of protected surfaces. Sci. Rep 6, 19616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, and Kumar S (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol 30, 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H, Bausch C, Richmond C, Blattner FR, and Conway T (1999). Functional genomics: expression analysis of Escherichia coli growing on min-imal and rich media. J. Bacteriol 181, 6425–6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxler MF, Summers SM, Nguyen H-T, Zacharia VM, Hightower GA, Smith JT, and Conway T (2008). The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol. Microbiol 68, 1128–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, and Salzberg SL (2011). Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet 13, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vehkala M, Shubin M, Connor TR, Thomson NR, and Corander J (2015). Novel R pipeline for analyzing Biolog Phenotypic MicroArray data. PLoS ONE 10, e0118392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Pulk A, Wasserman MR, Feldman MB, Altman RB, Cate JHD, and Blanchard SC (2012). Allosteric control of the ribosome by small-molecule antibiotics. Nat. Struct. Mol. Biol 19, 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman MR, Alejo JL, Altman RB, and Blanchard SC (2016). Multi-perspective smFRET reveals rate-determining late intermediates of ribosomal translocation. Nat. Struct. Mol. Biol 23, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood TK, Gonza´lez Barrios AF, Herzberg M, and Lee J (2006). Motility influences biofilm architecture in Escherichia coli. Appl. Microbiol. Biotechnol 72, 361–367. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Nakahigashi K, Nakamichi T, Yoshino M, Takai Y, Touda Y, Furubayashi A, Kinjyo S, Dose H, Hasegawa M, et al. (2009). Update on the Keio collection of Escherichia coli single-gene deletion mutants. Mol. Syst. Biol 5, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman EM, and Green R (2005). Affinity purification of in vivo-assem-bled ribosomes for in vitro biochemical analysis. Methods 36, 305–312. [DOI] [PubMed] [Google Scholar]
- Yu F, Liu X, Tao Y, and Zhu K (2013). High saturated fatty acids proportion in Escherichia coli enhances the activity of ice-nucleation protein from Pantoea ananatis. FEMS Microbiol. Lett 345, 141–146. [DOI] [PubMed] [Google Scholar]
- Zubradt M, Gupta P, Persad S, Lambowitz AM, Weissman JS, and Rouskin S (2017). DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 14, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.