

Abstract
Bacterial nucleotidyl cyclase toxins are potent virulence factors that upon entry into eukaryotic cells are stimulated by endogenous cofactors to catalyze the production of large amounts of 3′5′-cyclic nucleoside monophosphates. The activity of the effector ExoY from Pseudomonas aeruginosa is stimulated by the filamentous form of actin (F-actin). Utilizing yeast phenotype analysis, site-directed mutagenesis, functional biochemical assays, and confocal microscopy, we demonstrate that the last nine amino acids of the C terminus of ExoY are crucial for the interaction with F-actin and, consequently, for ExoY's enzymatic activity in vitro and toxicity in a yeast model. We observed that isolated C-terminal sequences of P. aeruginosa ExoY that had been fused to a carrier protein bind to F-actin and that synthetic peptides corresponding to the extreme ExoY C terminus inhibit ExoY enzymatic activity in vitro and compete with the full-length enzyme for F-actin binding. Interestingly, we noted that various P. aeruginosa isolates of the PA14 family, including highly virulent strains, harbor ExoY variants with a mutation altering the C terminus of this effector. We found that these naturally occurring ExoY variants display drastically reduced enzymatic activity and toxicity. Our findings shed light on the molecular basis of the ExoY–F-actin interaction, revealing that the extreme C terminus of ExoY is critical for binding to F-actin in target cells and that some P. aeruginosa isolates carry C-terminally mutated, low-activity ExoY variants.
Keywords: actin, bacterial toxin, protein-protein interaction, cyclic nucleotide, Pseudomonas aeruginosa (P. aeruginosa), confocal microscopy, protein cross-linking, type III secretion system (T3SS), ExoY
Introduction
Pseudomonas aeruginosa is an opportunistic human pathogen causing severe infections of both healthy and immunocompromised individuals (for review see Refs. 1 and 2). To manipulate eukaryotic host cells, P. aeruginosa injects effector proteins by a type 3 secretion system (T3SS)3 to the target cells (for review see Refs. 3 and 4). Upon delivery, the effector proteins interact with specific eukaryotic cofactors and become potent enzymes that hijack cellular processes (5–7). ExoY, one of the most prevalent T3SS effectors of P. aeruginosa (8, 9), generates cGMP and cAMP upon interaction with its specific cofactor, F-actin (10). The binding of ExoY to F-actin stabilizes actin filaments and alters their turnover (10). In cell culture experiments, ExoY causes the formation of gaps between endothelial cells, thus increasing permeability of the endothelial monolayer in lungs (11, 12) (yet these effects were not observed by other groups (13, 14)). Very recently, two groups independently demonstrated that ExoY activity also leads to the inhibition of the host immune responses by suppressing the activation of TAK1 and decreasing the production of IL-1β (15, 16). In vivo in animal models, ExoY was shown to cause severe lung damage in rats (17) and mice (18).
ExoY was identified by Yahr et al. (19) as a 378-amino acid-long protein that shares sequence similarity with well-characterized calmodulin-activated adenylyl cyclases from Bordetella pertussis (CyaA) and Bacillus anthracis (edema factor). These toxins share highly conserved regions responsible for catalysis but are different in regions predicted to bind the activator (19, 20). Structural models of the CyaA–calmodulin and edema factor–calmodulin complexes, solved by X-ray diffraction and supplemented by numerous biophysical studies and mutagenesis analysis, give insight into the activator recognition and the rearrangements of the active center upon calmodulin binding (21–24). A partial crystal structure of ExoY was recently published by Khanppnavar and Datta (25), but unfortunately the protein was extensively trimmed by proteases before crystallization, and many important regions were lost and not seen in the structure. In this study we characterize an ExoY region located at the C terminus of the enzyme, which is required for the interaction with F-actin and therefore enzymatic activity in vitro.
Results
C-terminal deletions abolish enzymatic activity of ExoY in vitro and affect toxicity in a yeast model
Overexpression of ExoY from P. aeruginosa PAO1 strain impairs yeast growth (10, 26). To define the ExoY polypeptide regions, which are necessary for toxicity in Saccharomyces cerevisiae, we performed an ExoY deletion analysis. We cloned different ExoY variants under a galactose-dependent promoter (PGal1,10), transformed the ExoY-bearing plasmids in yeast, and assessed cell growth under conditions of high ExoY expression (galactose-containing medium), intermediate expression (a mixture of galactose and glucose), or no ExoY expression (glucose-containing medium).
Yeast expressing WT ExoY or the variants deleted of the first 10 or 20 N-terminal residues failed to grow (Fig. 1A) on galactose-containing (inducing) media. In contrast, a yeast strain expressing an ExoY variant lacking the 30 N-terminal amino acids displayed a moderate growth on galactose-containing media indicating loss of toxicity of this variant. Hence, whereas the N-terminal 20 amino acids are clearly dispensable for ExoY toxicity in yeast (actually these residues may correspond to a T3SS secretion signal (27)), deletion of 10 additional amino acids at the N terminus likely impairs the catalytic core of ExoY. Indeed, according to a secondary structure prediction performed by PSIPRED server (28) (data not shown) and the recently published ExoY crystal structure (25), the first α-helix of the catalytic core is predicted to start at Asp-25 of ExoY, whereas the extreme N terminus is predicted to be intrinsically disordered.
Figure 1.

Toxicity of ExoY variants in yeast S. cerevisiae. A and D, the viability of yeast strains possessing an empty vector, complete ExoY, ExoY bearing N-terminal (ΔN10, ΔN20, ΔN30) or C-terminal deletions (ΔC3, ΔC5, ΔC7, ΔC9), ExoY variant from PA14 strain, and 19 ExoY mutants bearing single or double mutations (Fig. 1D) was assessed by a drop-test technique, by applying a normalized yeast culture in several dilutions on the plate containing 2% glucose (no ExoY expression), a mixture of 2% galactose and 0.02% of glucose (intermediate expression level), or 2% galactose (high ExoY expression level). D is composed of two agar plate photos, corresponding to one experiment. B and E, the toxin expression levels in yeast grown on 2% galactose were determined by Western blotting (WB). Following SDS-PAGE with yeast lysates and blotting to PVDF, the membrane was cut into two parts. The upper part was incubated with anti-Myc antibodies (9B11), and the lower part was incubated with anti-RPS9 serum for loading control. The figure is composed of two gels corresponding to one experiment. C, sequence alignment of the C terminus of ExoY variants from P. aeruginosa PAO1 and PA14.
ExoY toxicity in yeast decreased progressively with increasing length of the deletion from the C terminus. Whereas the five C-terminal amino acids, seemed to be dispensable showing no effect on toxicity, yeast cells expressing an ExoY variant with deletion of the last seven residues (ΔC7) grew moderately on galactose, whereas a strain expressing a variant lacking the last nine amino acids (ΔC9) showed unimpaired growth on galactose-containing media. This indicates that the ΔC7 and ΔC9 deletions severely decreased or abolished the ExoY toxicity in yeast. Interestingly, these ExoY variants with C-terminal deletions appeared to be much more highly expressed than the WT form in vivo (Fig. 1B). Likely, yeast cells could tolerate a much higher expression of these truncated toxin variants because of the drastic drop of their toxicity. To confirm these observations, we purified the corresponding recombinant ExoY variants and measured their guanylyl cyclase activity in vitro in the presence of F-actin. As shown in Table 1, deletion of the five C-terminal amino acids resulted in >99% loss of enzymatic activity, and deletion of the last nine amino acids fully abolished the enzymatic activity of the protein. It is noteworthy that ExoY ΔC5 remained highly toxic in vivo despite its strongly reduced enzymatic activity (yet partly balanced by a higher expression level). These observations highlight the high sensitivity of yeast cells to ExoY toxicity.
Table 1.
Guanylate cyclase activity of ExoY and different variants
| Activity |
||
|---|---|---|
| ExoY variant | μmol of cGMP/min/mga | % from the WT |
| WT | 74.4 ± 16.0 | 100.0 ± 21.5 |
| ΔC5 | 0.4 ± 0.2 | 0.5 ± 0.3 |
| ΔC9 | <0.05 | <0.07 |
| F367A | 2.2 ± 0.6 | 3.0 ± 0.8 |
| L371G | 8.2 ± 4.2 | 11.0 ± 5.6 |
| PA14 | 0.9 ± 0.6 | 1.2 ± 0.8 |
| PA14–PAO1 | 75.6 ± 8.7 | 101.6 ± 11.7 |
a Enzymatic activities of WT PAO1 ExoY (WT), C-terminal deletion mutants (ΔC5 and ΔC9), two variants with single mutations (F367A or L371G), a PA14 ExoY variant, and a PAO1–PA14 chimera were measured as described under “Material and Methods” in the presence of 1 μm F-actin and 2 mm GTP. Mean values and standard deviations were calculated as a result of three independent measurements.
We then employed site-directed mutagenesis to identify within the C-terminal region the residues that might be crucial for ExoY activation by F-actin. Nineteen ExoY variants with single or double substitutions in the region spanning residues Ser-366 to Val-378 were cloned and expressed in S. cerevisiae. ExoY variants possessing single mutations in F367A or L371G remained toxic in the conditions of high ExoY expression but grew better than the WT ExoY-expressing yeast strain on the intermediate expression conditions (Fig. 1, D and E), suggesting that these mutations decreased ExoY toxicity. In agreement with the in vivo data, the variants with single mutations F367A or L371G showed decreased activity in vitro compared with the WT (Table 1). As noted above for ExoY ΔC5, comparison of the in vitro measured guanylyl cyclase activities and in vivo toxicity assays points to high sensitivity of yeast toward ExoY toxicity, because toxin variants with low enzymatic activity drastically decreased yeast viability. Altogether, these results clearly indicate that the C terminus of ExoY is crucial for the enzymatic activity and toxicity in yeast.
The PA14 variant of ExoY has diminished enzymatic activity and is less toxic when expressed in yeast
Interestingly, the ExoY protein expressed in the P. aeruginosa reference strain UCBPP PA14 (here called PA14) contains several amino acid substitutions as compared with that of PAO1, including an altered and extended C terminus (Fig. 1C) that results from a frameshift mutation close to the 3′ end of the gene. To study the biological properties of this naturally occurring variant, we expressed the corresponding protein in yeast. When expressed at intermediate levels in yeast, we observed diminished toxicity of this ExoY variant compared with the WT ExoY of P. aeruginosa PAO1 (Fig. 1A). However, in vitro the guanylyl cyclase activity of the purified PA14 ExoY variant amounted to only 1% of that of the WT toxin (Table 1). The PA14 ExoY variant has several additional amino acid substitutions (D110N, R141Q, S227N, and V320L) in addition to the changes affecting the C terminus. To exclude the possibility that these amino acid substitutions contribute to the largely diminished activity of the PA14 ExoY variant, we created a PA14–PAO1 chimera, possessing the C terminus of the WT PAO1 toxin (PA14 ExoY residues Met-1 to Phe-373, then five amino acids from PAO1 ExoY, Phe-374–Val-378). The C-terminal substitution restored full activity of the PA14 variant, indicating that the modified C terminus was the only reason for reduced activity of this toxin variant (Table 1). Interestingly, a blast search in the database of P. aeruginosa genomes (https://www.patricbrc.org/; Ref. 45)4 revealed 118 strains harboring the same frameshift mutation at the C terminus of ExoY (Table S1). Our results thus indicate that a large number of P. aeruginosa strains from environmental or clinical sources may express an ExoY protein with strongly reduced enzymatic activity and consequently a much reduced toxicity in yeast.
C-terminal deletions abolish ExoY binding to actin in vitro
To test whether the alteration of the C terminus of ExoY could affect the interaction with F-actin, we performed cosedimentation experiments (Fig. 2, A and B) with ExoY variants and preformed F-actin. The single mutation L371G largely impaired the interaction of ExoY with F-actin (estimated Kd > 10 μm), whereas the C-terminal deletions of five or nine residues (ΔC5 and ΔC9) completely abolished binding to F-actin. The F367A and PA14 ExoY variants appeared to be less stable in solution and were partly found in the pellet fraction already in the absence of actin thus precluding accurate F-actin–binding analysis. More noticeably, the PA14–PAO1 chimera showed similar binding to actin as WT PAO1 ExoY (Kd = 1.9 ± 0.4 and 1.1 ± 0.1 μm, respectively). Additional F-actin–binding assays were also performed in experimental conditions favoring the solubility of the ExoY proteins (i.e. high NaCl concentrations instead of low KCl; Fig. 2, D and E). These data confirmed the impaired interaction of the PA14, F367A, and L371G variants with F-actin (all exhibited Kd values of >10 μm), and similar binding of PA14–PAO1 chimera and WT ExoY to F-actin (Kd = 1.2 ± 0.2 and 1.1 ± 0.3 μm, respectively).
Figure 2.

F-actin–binding properties of ExoY variants. A, SDS-PAGE analysis of supernatant (sup) and pellet (pel) fractions after ultracentrifugation of 2 μg (3 μm) of ExoY variants in the absence or presence of actin. The upper band corresponds to ExoY, and the lower band corresponds to actin. B, the fractions of ExoY variants that cosedimented with F-actin were quantified by densitometry using ImageJ software and plotted against actin concentrations. Error bars correspond to standard deviations of three independent experiments. C, pulldown experiment with F-actin and ExoY variants performed as follows: 12 μg of ExoY variants and/or 12 μg of F-actin were added to nickel–nitrilotriacetic acid–agarose beads and incubated for 1 h at 4 °C. The samples were washed three times with a buffer containing 20 mm imidazole and eluted by addition of hot Laemmli buffer. Corresponding amounts of bound (El) and unbound (flow through, FT) fractions were analyzed by SDS-PAGE, stained with Coomassie, or blotted and incubated with anti-actin antibodies (WB, anti-actin C4 Abcam). The upper band corresponds to ExoY, and the lower band corresponds to actin. The figure is composed of three parts of two gels corresponding to the same experiment. D, SDS-PAGE analysis of supernatant and pellet fractions after ultracentrifugation of 2 μg (3 μm) of ExoY variants in the absence or presence of actin in high ionic strength conditions. The upper band corresponds to ExoY, and the lower band corresponds to actin. In this set of experiments, ExoY proteins (3 μm) were incubated with actin (indicated concentrations) polymerized to a steady state in the presence of 150 mm Tris, pH 9.0, 360 mm NaCl, 20 mm MgCl2, 1 mm DTT, and 2.5 mm ATP. These experimental conditions were chosen to maintain ExoY variants in a soluble form and prevent nonspecific aggregation/sedimentation. E, the fractions of ExoY variants that cosedimented with F-actin were quantified by densitometry using ImageJ software and plotted against actin concentrations. Error bars correspond to standard deviations of three independent experiments.
Finally, we carried out independent binding studies using pulldown assays that fully corroborate the results from cosedimentation experiments (Fig. 2C). Therefore, we conclude that the C-terminal mutations or deletions impair or completely abolish F-actin binding and, in turn, result in decreased enzymatic activity and diminished toxicity in our yeast model.
Fusion proteins containing the very C terminus of ExoY bind to F-actin
We designed and produced a set of eight fusion proteins in which various segments of the ExoY polypeptide chains (excluding the conserved catalytic core regions, as predicted by a Phyre 2 homology model (29), highly similar to the recently published ExoY structure; see Fig. 3A and Fig. S1) were fused to the maltose-binding protein (MBP), via a glycine-serine linker (Fig. 3, A and B). The MBP-fusion proteins were tested for their ability to bind to F-actin in cosedimentation assays (Fig. 3C). SDS-PAGE analysis of supernatant and pellet fractions demonstrated that two proteins containing either the 39 or 19 last amino acids of ExoY (MBP–ExoY340–378 or MBP–ExoY360–378 respectively) interact with F-actin. The cosedimentation of these MBP fusions varied in a dose-dependent manner with the concentration of F-actin (Fig. 3D).
Figure 3.

F-actin–binding properties of the set of the fusion proteins with ExoY-derived peptides. A, Phyre 2 (29) predicted structural model of ExoY. Parts colored brown were incorporated in the MBP-fusion proteins. B, schematic representation of the set of fusion proteins with ExoY-derived peptides used for experiments described for C. C, SDS-PAGE analysis of cosedimentation experiments of F-actin with the MBP-fusion proteins (2 μg, 3 μm). The upper band corresponds to ExoY, and the lower band corresponds to actin. sup, supernatant; pel, pellet. D, the fractions of the fusion proteins 2 μg (3 μm) that cosedimented with F-actin were quantified by densitometry using ImageJ software and were plotted against actin concentrations. Error bars correspond to standard deviations of three independent experiments. E, SDS-PAGE analysis of cosedimentation experiments with 3 μm of MBP–ExoY346–378 and increasing concentrations of F-actin polymerized at steady state in the presence of a 2.5-fold excess of phalloidin over actin (see “Materials and methods”). F, the increasing amounts of MBP–ExoY346–378 bound to filaments were estimated by quantifying by densitometry the amounts of unbound protein in the supernatant (upper panel in E), which could be reliably determined because there are few unpolymerized actins in these fractions. The bound MBP–ExoY fractions were then deduced and plotted as a function of phalloidin-stabilized F-actin concentrations. The data were fitted to a single binding isotherm to derive the equilibrium dissociation constant (Kd). Error bars correspond to standard errors of four independent experiments.
To estimate the binding affinity of the C-terminal extremity of ExoY to F-actin, we used another MBP–ExoY fusion, MBP–ExoY346–378, and performed cosedimentation assays with phalloidin-stabilized F-actin (Fig. 3E). The presence of phalloidin decreased the concentration of nonpolymerized actin at steady-state to nearly zero and thus allowed us to reliably quantify the unbound MBP–ExoY346–378 fusion in the supernatant fractions (instead of the bound fractions in the above experiments; in this latter case, the bound MBP–ExoY fusions could not be accurately determined by SDS-PAGE because they migrated too close to actin). From these experiments, performed under nearly physiological ionic conditions, a dissociation constant (Kd) of 10 ± 1 μm was estimated for the interaction of MBP–ExoY346–378 with phalloidin-stabilized F-actin (Fig. 3F). Under similar conditions WT ExoY interacted with F-actin with a Kd of ∼1.0 μm (10).
To confirm the direct interaction between the C-terminal extremity of ExoY and F-actin, we performed cross-linking experiments with a photoactivatable cross-linker benzophenone-4-maleimide (BPM). This reagent is a heterobifunctional chemical compound consisting of a maleimide group that can form a covalent bond with cysteines and a UV-activatable benzophenone group that can react with various side chains of proteins. Because the fusion protein MBP–ExoY360–378 does not possess a native cysteine, we designed and produced three MBP-fusion proteins in which we introduced a single cysteine either by substituting the residues Leu-371 (MBP-fusionL371C) or Phe-373 (MBP-fusionF373C) with a cysteine or by adding an extra cysteine at the very C terminus, Cys-379 (MBP-fusionC379). The L371C and F373C substitutions decreased binding of the MBP fusion to F-actin (Fig. 4A), and therefore, the corresponding proteins were not suitable for the following cross-linking experiments. On the other hand, the added cysteine at the C terminus, Cys-379 (MBP-fusionC379), did not interfere with binding to F-actin (Fig. 4A). The MBP-fusionC379 protein was first incubated with BPM in the dark; then the reagent was removed; and F-actin was added. After a short UV exposure to photoactivate the benzophenone group, the mixture was ultracentrifuged, and supernatant and pellet fractions were analyzed by SDS-PAGE. As shown in Fig. 4B, a band of 110 kDa, which was observed in these conditions, (Fig. 4B) likely corresponds to a photocross-linked product between actin (42 kDa) and the MBP-fusion protein (45 kDa, migrates as a 55-kDa protein), because it was not detected in the control sample not exposed to UV. The presence of both MBP-fusion protein and actin in the 110-kDa band was confirmed by Western blotting with either HisProbe (to detect the MBP-fusion) or actin-specific antibodies (Fig. 4B). This experiment thus demonstrates a direct interaction between the C terminus of ExoY and F-actin.
Figure 4.

Photocross-linking of MBP-fused ExoY-derived peptides to F-actin. A, SDS-PAGE analysis of cosedimentation experiments with 10 μm F-actin and 10 μm of MBP-fusion proteins. The fractions of MBP-fusion protein cosedimented with F-actin were quantified by densitometry using ImageJ software and are marked above the corresponding bands. B, SDS-PAGE and Western blotting analysis (WB) of cosedimentation experiments of F-actin and cysteine-containing MBP-fusion proteins at different steps of BPM-cross-linking. HisProbe-HRP and anti-actin antibodies (C4, Abcam) were used to detect fusion proteins and actin, respectively. The bands marked with asterisks correspond to the MBP-fusion–F-actin cross-link. sup, supernatant; pel, pellet.
Peptides from the C terminus of ExoY, inhibit activity of the toxin in vitro
Two peptides of 32 or 20 amino acids (pep1 and pep2), derived from the C terminus of ExoY and corresponding to residues Lys-347–Val-378 and Trp-359–Val-378, respectively (Fig. 5A), were synthesized, and their effects on ExoY activity and/or ExoY–F-actin interaction were tested. A third peptide (pep3), corresponding to a distinct ExoY sequence (residues Pro-169–Pro-182 are part of the fusion protein MBP–ExoY155–194 that did not bind F-actin in cosedimentation experiments; Fig. 3C), was used as a control. Pep1 and pep2, when added to the reaction mixture, significantly inhibited ExoY's guanylyl cyclase activity in a dose-dependent manner (Fig. 5B). The control peptide pep3 had a limited effect on the guanylyl cyclase activity only at the highest concentration tested.
Figure 5.

Inhibition of ExoY activity and actin binding by peptides derived from the C terminus of ExoY. A, sequences of three peptides derived from the C terminus of ExoY used in the study. B, the ExoY guanylyl cyclase activity was measured as described under “Material and methods” in the presence of the indicated concentrations of the different peptides and 1 μm F-actin. ExoY was present at the concentration of 2.2 nm, and 100% of activity corresponds to 35 μmol of cGMP/min/mg. Error bars correspond to standard deviations of three independent experiments. C and D, SDS-PAGE analysis of cosedimentation experiments of 1.5 μm full-length ExoY and 1.5 μm F-actin in the presence of peptides. Fractions of ExoY cosedimented with F-actin in the presence of the peptides were quantified by densitometry using ImageJ software and are presented in D. Error bars correspond to standard deviations of three independent experiments. The difference between the no-peptide (no pep) value and other values was analyzed by the two-tail Student's t test. *, p < 0.05; **, p < 0.01. E and F, SDS-PAGE analysis of cosedimentation experiments with 8 μm fusion protein MBP–ExoY360–378 and 8 μm F-actin in the presence of peptides. The fractions of MBP-fusion360–378 cosedimented with F-actin in the presence of peptides were quantified by densitometry using ImageJ software and are presented in F. Error bars correspond to standard deviations of four independent experiments. The difference between the no-peptide value and other values was analyzed by the two-tail Student's t test. **, p < 0.01; ***, p < 0.001. sup, supernatant; pel, pellet; ns, not significant.
The addition of peptides to the mixture of ExoY and F-actin slightly decreased the amount of ExoY that could bind to F-actin in cosedimentation experiments (Fig. 5, C and D). The effect of peptides was more evident using the MBP–ExoY360–378 fusion protein, demonstrating a direct competition with the MBP-fusion protein for actin binding (Fig. 5, E and F). In both cosedimentation experiments, the control peptide pep3 did not affect the interaction of F-actin with ExoY or MBP fusion.
Actin mutation D25N abolishes binding of F-actin to ExoY
We have previously identified and characterized a yeast strain bearing a mutation D25N in actin that rendered yeast resistant to ExoY. In addition, yeast lysate prepared from this strain did not activate ExoY in vitro, suggesting defects in the ExoY–actin interaction (10). Asp-25 of actin is a solvent-exposed residue distant from actin–actin contacts in F-actin. Its mutation disturbs the interaction of F-actin–binding proteins with filaments without preventing actin polymerization (30). Here we purified WT and D25N actin from S. cerevisiae to directly assess the ExoY–F-actin interaction using cosedimentation assays. As expected, ExoY was found mostly in the pellet when WT yeast actin was used (Fig. 6, A and B). On the contrary, ExoY did not cosediment with D25N–F-actin, indicating that the mutation D25N in actin abolished ExoY–F-actin interaction. Then we performed the same cosedimentation experiments with yeast F-actin using a MBP–ExoY360–378 fusion, instead of the full-size toxin. In agreement with the above results, we observed cosedimentation of the fusion protein with WT yeast actin, whereas the D25N mutation fully abolished binding of the MBP-fusion protein to F-actin (Fig. 6, C and D). These experiments indicate the critical importance of Asp-25 of actin for the interaction with ExoY and suggest that the extreme C terminus of ExoY interacts with F-actin in close proximity to actin Asp-25.
Figure 6.

ExoY and MBP-fusion binding to WT or D25N actin. A, SDS-PAGE analysis of cosedimentation experiments of 2 μg (3 μm) of full-length ExoY and yeast F-actin. B, the fractions of ExoY that cosedimented with F-actin were quantified by densitometry using ImageJ software and were plotted against actin concentrations. Error bars correspond to standard deviations of three independent experiments. C, SDS-PAGE analysis of cosedimentation experiments of 2 μg (3 μm) of fusion protein MBP–ExoY360–378 and yeast F-actin. D, the fractions of the fusion protein MBP–ExoY360–378 that cosedimented with F-actin were quantified by densitometry using ImageJ software and were plotted against actin concentrations. Error bars correspond to standard deviations of two independent experiments. sup, supernatant; pel, pellet.
Actin D25N mutation and the C-terminal deletion of ExoY impair GFP–ExoY–actin colocalization in yeast
Previously we observed the colocalization of GFP–ExoYK81M with F-actin–rich structures in transfected mammalian cells by confocal microscopy (10). To confirm the key role of actin Asp-25 and the C-terminal extremity of ExoY in ExoY–F-actin interactions, we examined the colocalization of these proteins in yeast cells by confocal microscopy. For these experiments, we used the catalytically impaired K81M ExoY variant to circumvent the high toxicity of the WT toxin. Plasmids encoding GFP, GFP–ExoYK81M, or a GFP–ExoYK81MΔC9 (GFP fused to ExoYK81M lacking the nine C-terminal residues) were transformed either in the S. cerevisiae SC489 strain that expresses WT actin or in the strain SC691 that expresses the D25N actin mutant. The cells were fixed, and F-actin was stained with Alexa Fluor 647 phalloidin, a high-affinity F-actin fluorescent probe (Fig. 7A). We observed clustering and peripheral location of GFP–ExoYK81M in the presence of WT actin, whereas GFP or GFP–ExoY fusions expressed in the presence of WT or D25N actin, respectively, were uniformly distributed in the cells (Fig. 7, A and B). F-actin distribution was not modified in any of these strains, although yeast cells expressing GFP–ExoYK81M and WT actin were found to be slightly larger than the other three strains (Fig. 7B). In the strain expressing GFP–ExoYK81M and WT actin, the phalloidin fluorescence intensity was slightly higher (n = 20 cells; p < 0.001; two-tail Student's t test) than in others (Fig. 7C). This is consistent with our previous observations that ExoY recruitment along the side of F-actin increases the overall content of F-actin in transfected mammalian cells by stabilizing filaments and disturbing the balance of their assembly and disassembly by eukaryotic actin-binding proteins (10). Calculated Mander's coefficient, a correlation between the fluorescence signals in two channels, of 0.75 ± 0.13, confirmed colocalization of ExoYK81M with F-actin (Fig. 7D). However, in the other three yeast strains, the low value of the coefficient indicated a lack of colocalization of either ExoYK81M with D25N F-actin or ExoYK81M ΔC9 with WT F-actin.
Figure 7.

Colocalization of ExoY with F-actin in yeast cells. A, confocal microscopy images of phalloidin-stained yeast (670-nm fluorescent phalloidin), expressing either GFP (as control), a fusion of GFP to ExoYK81M, or a fusion of GFP to ExoYK81M, lacking its nine C-terminal amino acids (GFP–ExoYK81M ΔC9). The images are representatives of two independent experiments. B, distribution of actin (red dashed lines) and GFP (green solid lines) in yeast cells. The data shown represent mean values from four individual yeast cells. C, F-actin content in yeast cells. The values correspond to the mean intensity per pixel in the phalloidin channel for 20 individual cells. Error bars correspond to the standard deviation of 20 analyzed yeast cells from two independent experiments (n = 20 cells; ***, p < 0.001; two-tail Student's t test). D, colocalization of GFP or GFP–ExoY fusions with actin in yeast cells, calculated from 10 cells analyzed in two independent experiments (***, p < 0.001; two-tail Student's t test).
Discussion
Previously we demonstrated that F-actin is the eukaryotic cofactor necessary for activation of ExoY after delivery into target cells (10). Here we further characterized the molecular basis of the ExoY–actin interaction and show that the extreme C terminus of ExoY is crucial for binding to F-actin. Deletion of the last nine amino acids of ExoY (ΔC9) completely abolished its toxicity in a yeast model and abrogated ExoY binding to F-actin in vitro, leading to a complete loss of its enzymatic activity. However, the ΔC9 deletion did not affect the apparent stability of the protein because the ΔC9 variant accumulated to high levels in yeast, was well-produced in Escherichia coli, and remained stable in a soluble form after purification. Notably, the C-terminal region is absent from the recently published partial structure of ExoY (25), which was partially proteolysed before crystallization. It is likely that in the absence of the F-actin activator, the C-terminal region is too flexible to allow crystallization. A flexible C-terminal polypeptide may indeed facilitate the molecular recognition of F-actin according to a fly-casting mechanism (31).
In this study we demonstrated that ExoY can bind to F-actin in the presence of a high ionic strength, which suggests that the binding is mediated in part by hydrophobic interaction. This is fully consistent with our observation that two hydrophobic amino acids, Phe-367 and Leu-371 of ExoY, are implicated in the interaction with F-actin.
We further demonstrated that the last 20–30 C-terminal residues of ExoY can directly interact with F-actin. However, the lower affinity of an MBP-fusion protein, containing the last 32 amino acids, compared with the full-length ExoY, suggests that other region(s) also contribute to the toxin's interaction with its activator. A similar pattern of enzyme–activator interactions involving several spatially separated interacting regions has been reported for the calmodulin-activated adenylyl cyclase toxins from B. pertussis: peptides corresponding to a H-helix peptide or loop-helix-loop motif of CyaA (residues 234–254 and 341–364) independently contribute to high-affinity calmodulin binding (21, 23).
Based on our present results, we can propose a schematic model for ExoY–F-actin interaction (Fig. 8) and subsequent activation of the toxin nucleotidyl cyclase activity. In the absence of activator, in P. aeruginosa, ExoY is inactive likely because key catalytic loops are not properly folded to permit catalysis as has been reported for the related calmodulin-activated adenylate cyclases (32). Once being delivered by the T3SS into the target eukaryotic cell, ExoY may first bind to actin filament via its C-terminal extremity, which may work as a flexible bait to capture F-actin. Our present data further suggest that the C terminus may contact actin in the proximity of actin Asp-25, a key residue for ExoY–F-actin interaction (Fig. 8, step 1). In a second step the core ExoY domain may then interact with an adjacent actin molecule on the filament and contribute to the stabilization of the complex. This additional interaction would trigger a conformational change resulting in the reshaping of the catalytic site into enzymatically active conformation (Fig. 8, step 2). This simple model would thus explain why ExoY is activated only in the presence of F-actin.
Figure 8.
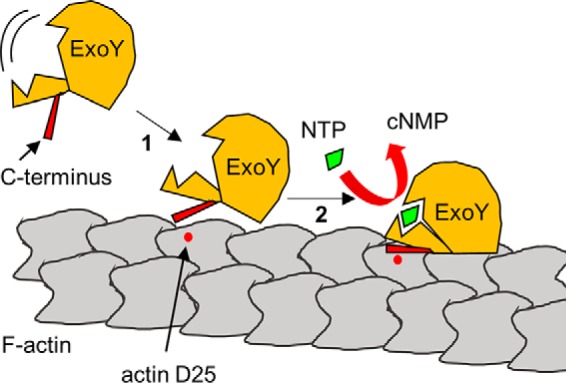
Model for ExoY–F-actin interaction. ExoY is inactive in the absence of activator, F-actin, possibly because of a highly flexible catalytic center. Activation of the toxin by F-actin may occur in two steps. First, the C terminus of ExoY interacts with F-actin in proximity to the Asp-25 residue of an actin molecule. Second, a second interaction of the ExoY catalytic core with an adjacent actin molecule on the filament may trigger conformational changes of ExoY, resulting in the reorganization of its catalytic site and stimulation of enzymatic activity.
Several effectors from different Gram-negative bacteria have also been shown to interact with F-actin. For example, Salmonella enterica SipC and Chlamydia trachomatis Tarp effectors are known to nucleate actin and bundle F-actin to promote bacterial invasion (33, 34). Although high-resolution SipC-F-actin or Tarp-F-actin structures are not yet available, biochemical studies have identified short amino acid sequences in these toxins that are involved in their binding to F-actin (34, 35). However, no obvious conserved motifs can be found between the actin-binding regions of these effectors and the C terminus of ExoY.
Another Salmonella effector, SipA, binds and stabilizes actin filaments hijacking target cell cytoskeleton to promote pathogen internalization. Low-resolution electronic microscopy studies revealed that a single SipA polypeptide connects adjacent actin protomers inside the interstrand cleft of actin filaments (36). Similar to SipA, Vibrio parahemeolyticus VopV, an effector that is required for bacteria-induced enterotoxicity, binds to the interstrand cleft of actin filaments (37). Unlike SipA or VopV, the C terminus of ExoY interacts with actin in close proximity to Asp-25 of actin. Therefore, these effectors unlikely share the same interaction sites with ExoY on F-actin.
In silico analysis of ExoY-bearing strains present in the Patric database revealed a frequently occurring frameshift mutation in the exoY gene, which alters the extreme C terminus of the toxin. This mutation occurs in a region containing five consequent thymidines, which might be prone to deletions during DNA replication. These variants are found in particular in various P. aeruginosa isolates belonging to the PA14 family. We show here that because of this mutation of the very C terminus, the PA14 ExoY variant has impaired toxicity in the yeast model and drastically decreased guanylyl cyclase activity in vitro. Virulent P. aeruginosa strains bearing T3SS are divided into two major groups, termed PAO1 and PA14-like, based on the presence of the respective effectors: ExoS, a multifunctional toxin, which modulates cytoskeleton organization, trafficking, etc., or ExoU, a phospholipase. In our analysis, all 118 strains with this particular ExoY allele were ExoU-positive. The toxicity of these strains is largely determined by the activity of ExoU, causing a rapid host cell death by cell lysis (38, 39). One could speculate that strains that bear this powerful phospholipase do not need a fully functional ExoY anymore. Alternatively, the ExoY activity in these strains might be adjusted to maintain an appropriate interplay between the different effectors. Our present results thus raise intriguing questions about the potential advantages for ExoU-producing P. aeruginosa strains of harboring a disabled ExoY effector.
The precise role of ExoY in P. aeruginosa infection still remains largely unclear. ExoY has been proposed to alter cell integrity leading to loss of endothelial cell barrier integrity (11, 17), although more recent results suggest that it may also contribute to dampen the host innate immune responses (15, 16). Inactivation of these ExoY functions may be beneficial at certain stages or forms of the bacterial infection. This should be addressed in future studies.
Materials and methods
Bacteria and yeast strains, plasmids, and growth conditions
Strains, plasmids and primers are listed in Table S2. Plasmid constructions are described in the supporting Materials and methods. E. coli strains were grown in lysogeny broth medium. Ampicillin (100 μg/ml) or kanamycin (50 μg/ml) was added for plasmid maintenance in E. coli. S. cerevisiae strains were grown in rich yeast extract-peptone-dextrose or in minimal medium containing yeast nitrogene base without amino acids (Difco) containing galactose or glucose, and supplemented if required with uracil, histidine, leucine, tryptophane, or adenine. Glucose and galactose were present at 2% unless specified. For medium solidification, agar was added at 1.5%. S. cerevisiae strains were transformed using the lithium-acetate method (40). Yeast viability upon ExoY expression was analyzed by drop test: 5-fold serial dilutions of cell suspensions prepared from overnight agar cultures normalized by A600 measurements were spotted onto agar plates containing the required supplements. The plates were incubated for 2–3 days at 30 °C.
Analysis of ExoY expression levels in yeast was performed as follows: yeast cells were incubated in a liquid galactose-containing medium for 16h at 30 °C. Cells corresponding to 1 ml of A600 1.0 were washed with 0.1 m NaOH, resuspended in 50 μl of 1× Laemmli sample buffer, and boiled for 5 min at 95 °C. 10 μl of the extracts were separated by SDS-PAGE, followed by Western blotting analysis and incubation with anti-Myc (9B11, Abcam) or anti-RPS9 serum (polyclonal rabbit antibodies were a generous gift of Prof. S. Rospert).
Protein purifications
Recombinant ExoY variants containing a C-terminal FLAG–His6 tag were purified under denaturing conditions as described before and were kept in buffer, containing 500 mm NaCl, 20 mm Tris, pH 9.0, 10% glycerol, 1 mm DTT (10). Maltose-binding protein fusions with complete ExoY or ExoY fragments were purified from BL21 DE3 E. coli transformed with corresponding plasmids. The cells were grown at 37 °C for 3 h, and then IPTG was added to a final concentration of 1 mm, followed by 2 h of expression at 37 °C. The cells were harvested by centrifugation; resuspended in buffer A containing 300 mm NaCl, 20 mm Tris, pH 8.0; and lysed by sonication. The soluble fraction was applied on nickel-iminodiacetic acid resin and eluted by buffer A, supplemented with 250 mm imidazole. The eluates were dialyzed against buffer B, containing 150 mm NaCl, 20 mm Tris, pH 8.0, 10% glycerol and kept at −20 °C.
Yeast actin was purified according to the DNase I affinity method (41, 42) and was kept on ice in G buffer (5 mm Tris, pH 7.8, 0.1 mm CaCl2, 0.2 mm ATP, 1 mm DTT). To ensure complete actin functionality, after purification the protein was recycled by polymerization/depolymerization (10, 43). Rabbit skeletal muscle α-actin was purified according to the method of Spudich and Watt as described previously (10, 43).
Measurement of ExoY activity in vitro
ExoY-catalyzed synthesis of cGMP was measured as described previously (10). Reactions were carried out for 10 min at 30 °C in the presence of 1 μm of F-actin, 50 ng (22.7 nm) of ExoY, and 2 mm GTP spiked with 0.1 μCi of [α-33P]GTP.
Pulldowns and cosedimentation assays
Pulldown experiments with ExoY variants were performed as described previously (10). F-actin cosedimentation assays with complete ExoY proteins or with MBP fusions were performed using rabbit skeletal muscle α-actin or yeast actin. For Figs. 2, 3 (C and D), 4, 5, and 6, actin was polymerized to steady state by adding Tris, pH 8.0, KCl, MgCl2, DTT, and ATP to a final concentration of 150, 150, 20, 1, and 2.5 mm, respectively. To study F-actin–binding properties of the full-length ExoY (Fig. 2, A and B), 5 μl of F-actin was mixed with 10 μl of ExoY at 0.2 mg/ml (in 20 mm Tris, pH 9.0, 500 mm NaCl). To study F-actin–binding properties of the fusion proteins (Figs. 3, C and D, and 4A), 13 μl of F-actin was mixed with 2 μl of fusion protein at 1.0 mg/ml (in 20 mm Tris, pH 8.0, 150 mm NaCl). For the competition assays (Fig. 5), 5.5 μl of preformed F-actin was mixed with 7.5 μl of peptide (in 20 mm Tris, pH 8.0, 150 mm NaCl; synthesized by Genosphere with >95% purity) and 2 μl of 1 mg/ml of ExoY (in 20 mm Tris, pH 9.0, 500 mm NaCl) or 2 μl of the 1 mg/ml fusion protein (in 20 mm Tris, pH 8.0, 150 mm NaCl). For the cosedimentation experiments with yeast actin (Fig. 6), 13 μl of preformed F-actin was mixed with 2 μl of 1 mg/ml of ExoY (in 20 mm Tris, pH 9.0, 500 mm NaCl) or 2 μl of the 1 mg/ml fusion protein (in 20 mm Tris, pH 8.0, 150 mm NaCl). For cosedimentation assays with complete ExoY proteins in the conditions of high ionic strength (Fig. 2, D and E), actin was polymerized to a steady state by adding Tris, pH 9.0, NaCl, MgCl2, DTT, and ATP to a final concentration of 150, 360, 20, 1, and 2.5 mm, respectively. High ionic strength and pH 9 were chosen to maintain ExoY in a soluble form. Then 4 μl of 0.5 mg/ml ExoY variant (in 20 mm Tris, pH 9.0, 500 mm NaCl) was added to 11 μl of F-actin. The mixtures of preformed F-actin, MBP-fusion proteins, full-length ExoY variants, or peptides were incubated for 5 min at room temperature and were centrifuged at 100,000 × g for 60 min at 4 °C. Supernatants and resuspended pellet fractions were analyzed by SDS-PAGE on 4–12% precast gradient gels. Because of similar protein sizes of ExoY and actin (44 and 42 kDa, respectively), quantification of ExoY in the pellet fraction was derived from the amount of unbound ExoY measured by plotting intensity values across lanes, followed by calculation of the area under the curve using ImageJ software.
The measurement of the binding affinity of MBP–ExoY360–378 to phalloidin-stabilized F-actin by cosedimentation assays (Fig. 3, E and F) was performed from F-actin polymerized at steady state overnight in 100 mm KCl, 10 mm Tris, pH 7.8, 4 mm ATP, 6 mm MgCl2, 5 mm tris-(2-carboxyethyl) phosphine hydrochloride in the presence of a 2.5-fold excess of phalloidin over actin. Phalloidin did not modify the binding affinity of ExoY for F-actin (10). MBP–ExoY348–378 was incubated with phalloidin-stabilized F-actin at steady state for 45 min at room temperature and ultracentrifuged at 200,000 × g for 40 min at 4 °C. Aliquots of supernatant and resuspended pellet fractions were loaded on gradient (4–15%) SDS gel. The bound fractions were derived from the gradual depletion of the unbound protein in the supernatants (Fig. 3E), quantified by densitometry using ImageJ, and fitted as described previously (10).
BPM cross-linking
The cysteine-containing MBP-fusion protein was reduced by incubation with 2 mm DTT at room temperature for 15 min. The sample buffer was exchanged to 300 mm NaCl and 20 mm Hepes, pH 7.4, on a desalting column. To perform the first step of cross-linking, BPM was added at a ratio of one MBP-fusion protein to two BPM molecules and kept in the dark on the ice overnight. The following day, excess of BPM molecules was inactivated by the addition of 2 mm DTT, followed by buffer exchange to 300 mm NaCl and 20 mm Hepes, pH 7.4, on a desalting column. The sample was mixed with steady state-polymerized F-actin at a 1 to 1 ratio and was radiated with UV light of 365 nm for 30 min to perform the cross-linking.
Confocal microscopy of yeast cells and image processing
Yeast cells bearing plasmids encoding GFP–ExoY fusions or GFP alone were grown overnight on a liquid minimal medium supplemented with 2% glucose at 30 °C. The following day, the cells were washed twice with liquid minimal medium without sugars, diluted to an A600 of 0.03 in minimal medium, supplemented with 2% galactose and 0.02% glucose, and grown for 16–18 h until the A600 reached 0.2–0.4. The cells were harvested by centrifugation at 8,000 rpm, resuspended in PBS buffer, fixed by incubating with 4% formaldehyde for 20 min at room temperature, and then washed ones with PBS buffer. Alexa Fluor 647 phalloidin (Molecular Probes, Invitrogen) staining was performed by incubation of yeast cells with 0.66 μm fluorescent phalloidin buffered with PBS and 0.5% of Triton X-100 at room temperature in the dark for 30 min, followed by two washes with PBS. We chose the phalloidin conjugated to a far red 670-nm fluorophore to ensure a signal separation from GFP. The cells were applied on concanavalin A–coated slides, mounted with FluoromontG, covered with round 1.5-thickness cover glass, and immediately processed. Image acquisition was performed in Leica TCS SP5 confocal laser scanning microscope, with 63× oil-immersion objective (NA 1.4). Excitation was performed using argon (488-nm line) and HeNe lasers (633-nm line), and emission was recorded with hybrid (HyD) detectors. Leica Application Suite Advance fluorescence software was used to operate the microscope.
Raw data were analyzed and processed in ImageJ. For colocalization analysis, multicolor beads were used to correct for chromatic shift. Raw images were corrected and filtered using median filter with a pixel radius of 2, followed by background subtraction with a rolling ball radius of 10 pixels. Next, images were analyzed for colocalization with JACoP plugin (44). Mander's colocalization coefficients of 10 cells are reported. This value is a correlation between the fluorescence signal in two channels (44). Fluorescence distribution on green (GFP or GFP–ExoY) and red (actin) channel was assessed by plotting intensity profile of a straight cross-section of a yeast cell. To assess the yeast actin content, the fluorescence intensity in the phalloidin channel was measured using ImageJ software in randomly chosen individual cells.
Author contributions
A. B., D. L., and U. M. conceptualization; A. B., I. S., L. R., D. L., and U. M. formal analysis; A. B., I. S., L. R., D. L., and U. M. validation; A. B., I. S., L. R., B.B., and U. M. investigation; A. B. and I. S. visualization; A. B., I. S., L. R., B.B., D. L., and U. M. methodology; A. B. and D. L. writing-original draft; A. B., I. S., L. R., D. L., and U. M. writing-review and editing; L. R. resources; B. B. data curation; D. L. and U. M. supervision; L. R., D. L., and U. M. funding acquisition; D. L. and U. M. project administration.
Supplementary Material
Acknowledgments
A. B. thanks Dr. Cosmin Saveanu, Dr. Ina Attree, and Dr. Yury Belyi for fruitful discussions. We thank Dorothée Raoux-Barbot for technical support, Dr. Violaine David for critical reading, Dr. Alexandre Chenal for help in peptide design, and Prof. Sabine Rospert for the anti-RPS9 serum. We acknowledge the Imagopole France-BioImaging infrastructure, which is supported by French National Research Agency Grant ANR 10-INSB-04-01 (Investments for the Future), for advice and access to the Leica TCS SP5 system.
This work was supported by the Institut Pasteur under Grant PTR 43-16, by French Infrastructure for Integrated Structural Biology (FRISBI) Grant ANR-10-INBS-05, and by CNRS. This work was also supported in part by a stipend from the Pasteur-Paris University International Ph.D. program (to A. B. and I. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1 and S2, Fig. S1, and supporting text and references.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- T3SS
- type 3 secretion system
- MBP
- maltose-binding protein
- BPM
- benzophenone-4-maleimide.
References
- 1. Gellatly S. L., and Hancock R. E. (2013) Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog Dis. 67, 159–173 10.1111/2049-632X.12033 [DOI] [PubMed] [Google Scholar]
- 2. Klockgether J., and Tümmler B. (2017) Recent advances in understanding Pseudomonas aeruginosa as a pathogen. F1000Res. 6, 1261 10.12688/f1000research.10506.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Galle M., Carpentier I., and Beyaert R. (2012) Structure and function of the type III secretion system of Pseudomonas aeruginosa. Curr. Protein Pept. Sci. 13, 831–842 10.2174/138920312804871210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berube B. J., Rangel S. M., and Hauser A. R. (2016) Pseudomonas aeruginosa: breaking down barriers. Curr. Genet. 62, 109–113 10.1007/s00294-015-0522-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fu H., Coburn J., and Collier R. J. (1993) The eukaryotic host factor that activates exoenzyme S of Pseudomonas aeruginosa is a member of the 14-3-3 protein family. Proc. Natl. Acad. Sci. U.S.A. 90, 2320–2324 10.1073/pnas.90.6.2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yasmin L., Veesenmeyer J. L., Diaz M. H., Francis M. S., Ottmann C., Palmer R. H., Hauser A. R., and Hallberg B. (2010) Electrostatic interactions play a minor role in the binding of ExoS to 14-3-3 proteins. Biochem. J. 427, 217–224 10.1042/BJ20100043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tessmer M. H., Anderson D. M., Buchaklian A., Frank D. W., and Feix J. B. (2017) Cooperative substrate–cofactor interactions and membrane localization of the bacterial phospholipase A2 (PLA2) enzyme, ExoU. J. Biol. Chem. 292, 3411–3419 10.1074/jbc.M116.760074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feltman H., Schulert G., Khan S., Jain M., Peterson L., and Hauser A. R. (2001) Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa. Microbiology 147, 2659–2669 10.1099/00221287-147-10-2659 [DOI] [PubMed] [Google Scholar]
- 9. Haghi F., Zeighami H., Monazami A., Toutouchi F., Nazaralian S., and Naderi G. (2018) Diversity of virulence genes in multidrug resistant Pseudomonas aeruginosa isolated from burn wound infections. Microb. Pathog. 115, 251–256 10.1016/j.micpath.2017.12.052 [DOI] [PubMed] [Google Scholar]
- 10. Belyy A., Raoux-Barbot D., Saveanu C., Namane A., Ogryzko V., Worpenberg L., David V., Henriot V., Fellous S., Merrifield C., Assayag E., Ladant D., Renault L., and Mechold U. (2016) Actin activates Pseudomonas aeruginosa ExoY nucleotidyl cyclase toxin and ExoY-like effector domains from MARTX toxins. Nat. Commun. 7, 13582 10.1038/ncomms13582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sayner S. L., Frank D. W., King J., Chen H., VandeWaa J., and Stevens T. (2004) Paradoxical cAMP-induced lung endothelial hyperpermeability revealed by Pseudomonas aeruginosa ExoY. Circ. Res. 95, 196–203 10.1161/01.RES.0000134922.25721.d9 [DOI] [PubMed] [Google Scholar]
- 12. Balczon R., Prasain N., Ochoa C., Prater J., Zhu B., Alexeyev M., Sayner S., Frank D. W., and Stevens T. (2013) Pseudomonas aeruginosa exotoxin Y-mediated Tau hyperphosphorylation impairs microtubule assembly in pulmonary microvascular endothelial cells. PLoS One 8, e74343 10.1371/journal.pone.0074343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ganter M. T., Roux J., Su G., Lynch S. V., Deutschman C. S., Weiss Y. G., Christiaans S. C., Myazawa B., Kipnis E., Wiener-Kronish J. P., Howard M., and Pittet J.-F. (2009) Role of small GTPases and αvβ5 integrin in Pseudomonas aeruginosa-induced increase in lung endothelial permeability. Am. J. Respir. Cell Mol. Biol. 40, 108–118 10.1165/rcmb.2007-0454OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huber P., Bouillot S., Elsen S., and Attrée I. (2014) Sequential inactivation of Rho GTPases and Lim kinase by Pseudomonas aeruginosa toxins ExoS and ExoT leads to endothelial monolayer breakdown. Cell Mol. Life Sci. 71, 1927–1941 10.1007/s00018-013-1451-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jeon J., Kim Y.-J., Shin H., and Ha U.-H. (2017) T3SS effector ExoY reduces inflammasome-related responses by suppressing bacterial motility and delaying activation of NF-kappaB and caspase-1. FEBS J. 284, 3392–3403 10.1111/febs.14199 [DOI] [PubMed] [Google Scholar]
- 16. He C., Zhou Y., Liu F., Liu H., Tan H., Jin S., Wu W., and Ge B. (2017) Bacterial nucleotidyl cyclase inhibits the host innate immune response by suppressing TAK1 activation. Infect. Immun. 85, e00239–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stevens T. C., Ochoa C. D., Morrow K. A., Robson M. J., Prasain N., Zhou C., Alvarez D. F., Frank D. W., Balczon R., and Stevens T. (2014) The Pseudomonas aeruginosa exoenzyme Y impairs endothelial cell proliferation and vascular repair following lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 306, L915–L924 10.1152/ajplung.00135.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kloth C., Schirmer B., Munder A., Stelzer T., Rothschuh J., and Seifert R. (2018) The role of Pseudomonas aeruginosa ExoY in an acute mouse lung infection model. Toxins (Basel) 10, E185 10.3390/toxins10050185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yahr T. L., Vallis A. J., Hancock M. K., Barbieri J. T., and Frank D. W. (1998) ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc. Natl. Acad. Sci. U.S.A. 95, 13899–13904 10.1073/pnas.95.23.13899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Belyy A., Mechold U., Renault L., and Ladant D. (2018) ExoY, an actin-activated nucleotidyl cyclase toxin from P. aeruginosa: A minireview. Toxicon 149, 65–71 10.1016/j.toxicon.2017.12.046 [DOI] [PubMed] [Google Scholar]
- 21. Guo Q., Shen Y., Lee Y.-S., Gibbs C. S., Mrksich M., and Tang W.-J. (2005) Structural basis for the interaction of Bordetella pertussis adenylyl cyclase toxin with calmodulin. EMBO J. 24, 3190–3201 10.1038/sj.emboj.7600800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen Y., Zhukovskaya N. L., Guo Q., Florián J., and Tang W.-J. (2005) Calcium-independent calmodulin binding and two-metal-ion catalytic mechanism of anthrax edema factor. EMBO J. 24, 929–941 10.1038/sj.emboj.7600574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. O'Brien D. P., Durand D., Voegele A., Hourdel V., Davi M., Chamot-Rooke J., Vachette P., Brier S., Ladant D., and Chenal A. (2017) Calmodulin fishing with a structurally disordered bait triggers CyaA catalysis. PLoS Biol. 15, e2004486 10.1371/journal.pbio.2004486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Selwa E., Davi M., Chenal A., Sotomayor-Pérez A.-C., Ladant D., and Malliavin T. E. (2014) Allosteric activation of Bordetella pertussis adenylyl cyclase by calmodulin: molecular dynamics and mutagenesis studies. J. Biol. Chem. 289, 21131–21141 10.1074/jbc.M113.530410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khanppnavar B., and Datta S. (2018) Crystal structure and substrate specificity of ExoY, a unique T3SS mediated secreted nucleotidyl cyclase toxin from Pseudomonas aeruginosa. Biochim. Biophys. Acta Gen. Subj. 1862, 2090–2103 10.1016/j.bbagen.2018.05.021 [DOI] [PubMed] [Google Scholar]
- 26. Arnoldo A., Curak J., Kittanakom S., Chevelev I., Lee V. T., Sahebol-Amri M., Koscik B., Ljuma L., Roy P. J., Bedalov A., Giaever G., Nislow C., Merrill R. A., Lory S., and Stagljar I. (2008) Identification of small molecule inhibitors of Pseudomonas aeruginosa exoenzyme S using a yeast phenotypic screen. PLoS Genet. 4, e1000005 10.1371/journal.pgen.1000005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maresso A. W., Frank D. W., and Barbieri J. T. (2006) Pseudomonas aeruginosa toxins. In The Comprehensive Sourcebook of Bacterial Protein Toxins, 3rd Ed., pp. 257–269, Academic Press, London [Google Scholar]
- 28. Buchan D. W., Minneci F., Nugent T. C., Bryson K., and Jones D. T. (2013) Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–W357 10.1093/nar/gkt381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. E. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miller C. J., and Reisler E. (1995) Role of charged amino acid pairs in subdomain-1 of actin in interactions with myosin. Biochemistry 34, 2694–2700 10.1021/bi00008a037 [DOI] [PubMed] [Google Scholar]
- 31. Shoemaker B. A., Portman J. J., and Wolynes P. G. (2000) Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. U.S.A. 97, 8868–8873 10.1073/pnas.160259697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Drum C. L., Yan S.-Z., Bard J., Shen Y.-Q., Lu D., Soelaiman S., Grabarek Z., Bohm A., and Tang W.-J. (2002) Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 415, 396–402 10.1038/415396a [DOI] [PubMed] [Google Scholar]
- 33. Hayward R. D., and Koronakis V. (1999) Direct nucleation and bundling of actin by the SipC protein of invasive Salmonella. EMBO J. 18, 4926–4934 10.1093/emboj/18.18.4926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jiwani S., Alvarado S., Ohr R. J., Romero A., Nguyen B., and Jewett T. J. (2013) Chlamydia trachomatis Tarp harbors distinct G and F actin binding domains that bundle actin filaments. J. Bacteriol. 195, 708–716 10.1128/JB.01768-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Myeni S. K., and Zhou D. (2010) The C terminus of SipC binds and bundles F-actin to promote Salmonella invasion. J. Biol. Chem. 285, 13357–13363 10.1074/jbc.M109.094045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lilic M., Galkin V. E., Orlova A., VanLoock M. S., Egelman E. H., and Stebbins C. E. (2003) Salmonella SipA polymerizes actin by stapling filaments with nonglobular protein arms. Science 301, 1918–1921 10.1126/science.1088433 [DOI] [PubMed] [Google Scholar]
- 37. Nishimura M., Fujii T., Hiyoshi H., Makino F., Inoue H., Motooka D., Kodama T., Ohkubo T., Kobayashi Y., Nakamura S., Namba K., and Iida T. (2015) A repeat unit of Vibrio diarrheal T3S effector subverts cytoskeletal actin homeostasis via binding to interstrand region of actin filaments. Sci. Rep. 5, 10870 10.1038/srep10870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sato H., and Frank D. W. (2014) Intoxication of host cells by the T3SS phospholipase ExoU: PI(4,5)P2-associated, cytoskeletal collapse and late phase membrane blebbing. PLoS One 9, e103127 10.1371/journal.pone.0103127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gendrin C., Contreras-Martel C., Bouillot S., Elsen S., Lemaire D., Skoufias D. A., Huber P., Attree I., and Dessen A. (2012) Structural basis of cytotoxicity mediated by the type III secretion toxin ExoU from Pseudomonas aeruginosa. PLoS Pathog. 8, e1002637 10.1371/journal.ppat.1002637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gietz R. D., and Woods R. A. (2002) Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350, 87–96 10.1016/S0076-6879(02)50957-5 [DOI] [PubMed] [Google Scholar]
- 41. Kron S. J., Drubin D. G., Botstein D., and Spudich J. A. (1992) Yeast actin filaments display ATP-dependent sliding movement over surfaces coated with rabbit muscle myosin. Proc. Natl. Acad. Sci. U.S.A. 89, 4466–4470 10.1073/pnas.89.10.4466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goode B. L. (2002) Purification of yeast actin and actin-associated proteins. Methods Enzymol. 351, 433–441 10.1016/S0076-6879(02)51862-0 [DOI] [PubMed] [Google Scholar]
- 43. Spudich J. A., and Watt S. (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 246, 4866–4871 [PubMed] [Google Scholar]
- 44. Bolte S., and Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 224, 213–232 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- 45. Wattam A. R., Davis J. J., Assaf R., Boisvert S., Brettin T., Bun C., Conrad N., Dietrich E. M., Disz T., Gabbard J. L., Gerdes S., Henry C. S., Kenyon R. W., Machi D., Mao C., et al. (2017) Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 45, D535–D542 10.1093/nar/gkw1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.