

Summary
Human phosphoglucomutase 1 (PGM1) plays a central role in cellular glucose homeostasis, catalyzing the conversion of glucose 1-phosphate and glucose 6-phosphate. Recently, missense variants of this enzyme were identified as causing an inborn error of metabolism, PGM1 deficiency, with features of a glycogen storage disease and a congenital disorder of glycosylation. Previous studies of selected PGM1 variants have revealed various mechanisms for enzyme dysfunction, including regions of structural disorder and side chain rearrangements within the active site. Here, we examine variants within a substrate-binding loop in domain 4 (D4) of PGM1 that cause extreme impairment of activity. Biochemical, structural, and computational studies demonstrate multiple detrimental impacts resulting from these variants, including loss of conserved ligand binding interactions and reduced mobility of the D4 loop, due to perturbation of its conformational ensemble. These potentially synergistic effects make this conserved ligand-binding loop a hotspot for disease-related variants in PGM1 and related enzymes.
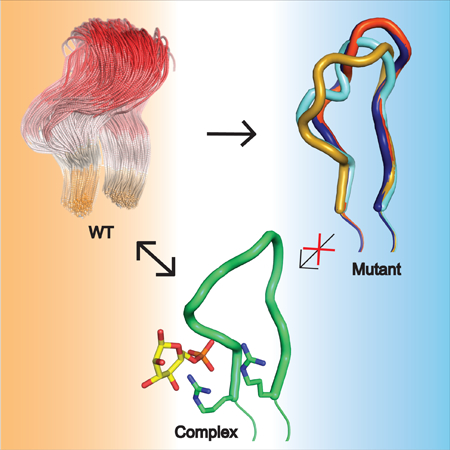
Graphical Abstract
Disease-related variants phosphoglucomutase 1 (PGM1) cause an inherited metabolic deficiency in humans. Stiers, et al., show that that mutations in a key active site region of the enzyme produce multiple detrimental impacts, including reduced flexibility of a loop required for substrate binding.
Introduction
The effects of disease-related variants on the structure and function of human proteins are often not fully understood (Stefl et al., 2013). Because of the large number of such variants, comprehensive experimental investigation is prohibitive, leading to the widespread use of computational algorithms for predicting impact (Gnad et al., 2013; Thusberg et al., 2011). Even in systems where experimental studies are initiated, many variants prove intractable due to problems with protein folding, stability, or cellular trafficking (Tokuriki and Tawfik, 2009; Yue et al., 2005). In a few cases, however, biochemical and atomic resolution structural studies of disease-related variants have proven feasible, shedding light on the molecular basis of disease. One such example is a key metabolic enzyme in humans known as phosphoglucomutase 1 (PGM1), which is responsible for maintaining cellular glucose homeostasis. Approximately 20 missense variants of this enzyme are associated with inherited PGM1 deficiency, an autosomal recessive disease with characteristics of both a glycogen storage disorder (GSD XIV, OMIM 614921) and a congenital disorder of glycosylation of types I and II (Kucukcongar et al., 2015; Loewenthal et al., 2015; Ondruskova et al., 2014; Pérez et al., 2013; Stojkovic et al., 2009; Tegtmeyer et al., 2014; Timal et al., 2012; Wong et al., 2015). Recent X-ray crystallographic studies of several PGM1 missense variants revealed distinct mechanisms for enzyme dysfunction, providing insights well beyond and sometimes differing from their predicted effects (Stiers et al., 2016, 2017a). This work highlights the importance of direct structural characterization for understanding disease-related variants and the value of human PGM1 as a model system for such studies.
The identification of PGM1 deficiency as an inherited disease also provides opportunities for understanding this critical enzyme and its role in human metabolism. PGM1 is a cytosolic enzyme found in most tissues (Muenks et al., 2017). Its catalytic reaction entails two consecutive phosphoryl transfers and proceeds via a bisphosphorylated intermediate (Figure 1A). The first phosphoryl transfer takes place from a conserved phosphoserine residue (Ser117) to substrate, creating glucose 1,6-bisphosphate. The intermediate must then reorient in the active site, allowing the second phosphoryl transfer to occur from the intermediate to the protein, creating product and regenerating the active (phosphorylated) version of the enzyme. PGM1 has 562 residues, comprising four domains of approximately equal size (Figure 1B). Its active site is found within the large central cleft and has four conserved loops with critical functional roles: i) the phosphoserine loop that participates in phosphoryl transfer; ii) a loop that binds a Mg2+ ion needed for activity; iii) a sugar-binding loop; and iv) the phosphate-binding loop that interacts with the phosphate group of the substrates (see (Beamer, 2015) for a detailed review).
Figure 1. Overview of the mechanism and structure of human PGM1.

(A) A schematic of the catalytic reaction, showing the reversible conversion of glucose 1-phosphate to glucose 6-phosphate. Glucose 1,6-bisphosphate undergoes a 180° reorientation in between the two phosphoryl transfer steps of the reaction (gray line indicates axis of rotation). (B) The crystal structure of WT human PGM1. Ser117, Arg503 and Arg515 are highlighted as sticks; bound metal ion is shown as black sphere. The missing residues in the D4 loop are shown with dashed line. (C) A close-up of the active site of PGM1. The bound sulfate ion in WT enzyme that acts as a structural mimic for the phosphate group of the substrate is shown with spheres; bound glucose 6-phosphate (in yellow, this report) is superimposed. The Mg2+ ion near the site of phosphoryl transfer is shown for reference. Missing residues (507–509) in the D4 loop are shown with dashed line.
In the present study we focus on PGM1 variants affecting residues within loop (iv) in the C-terminal domain (domain 4 or D4) of the enzyme: the 13-residue D4 loop. This region is composed of two β-strands and a connecting loop, and includes a number of conserved residues with proposed roles in binding the phosphate group of substrate, a key determinant of ligand recognition for the entire α-D-phosphohexomutase enzyme superfamily (Stiers et al., 2017b). In the 1.85 Å crystal structure of wild-type (WT) PGM1, three residues within this loop (507–509) were missing in the electron density maps, consistent with high mobility (Stiers et al., 2016). This suggested a potential functional role during catalysis, whereby the mobile flap would close upon substrate binding and open to release product. Movement of the flap could also be necessary to permit the 180° reorientation of the intermediate in the midst of the catalytic cycle (Figure 1A).
The functional relevance of the mobile flap and the impacts of missense variants affecting conserved ligand-binding residues in the D4 loop are further investigated herein, using biochemical, structural and computational approaches. Variants affecting this region, identified either from patients with PGM1 deficiency or from exome sequencing databases, are shown to have multiple detrimental effects on the enzyme, culminating in a profound decrease in catalytic activity. These studies provide insights into key functional regions of PGM1 and further expand its utility for probing the molecular effects of disease-related mutations.
Results
Biochemical studies of variants in the D4 loop
Eight mutants affecting the D4 loop of PGM1 were studied (Table 1). Two of these were identified from patients affected by PGM1 deficiency: R503Q and R515L (Wong et al., 2015). Four others (G508R, G511R, and R515Q/W) were identified as likely detrimental to enzyme function, from a survey of human PGM1 variants found in various genome databases (Methods and (Muenks et al., 2017)). Two additional mutants, R503A and R515A, were engineered to further examine the effects of the amino acid side chain at these residue positions. Mutant proteins were expressed recombinantly in E. coli and purified as described in Methods. Results of the biochemical characterization are summarized in Table 1. Soluble protein was obtained for all of the variants, most at levels similar to that of WT enzyme. Potential aggregation was assessed with dynamic light scattering. Six of the eight variants showed hydrodynamic radii (Rh) consistent with the expected molecular weight of the protein. However, G508R and G511R had larger Rh and correspondingly increased estimated molecular weights in solution. Due to apparent aggregation, neither of these variants was studied further.
Table 1.
Summary of biochemical data on PGM1 loop mutants
| Name | Source mutation | Sol. protein (%) |
MW (kD) |
Polydispersity (%) |
Rh (nm) |
T0.5 (°) |
kcat (1/s) |
Km (μM) |
kcat / Km (μM/s) |
Phosphorylation before/after G16P (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| WTa | - | 90 | 67 | 20 | 3.7 | 46.3 | 143+/−2 | 80 +/−4 | 1.8+/−0.1 | 35 / 90 |
| R503A | in-house | 90 | 63 | 21 | 3.8 | n.d. | n.c. | n.c. | n.c. | 40 / 85 |
| R503Qb | patient | 90 | 72 | 0 | 3.8 | 48.7 | n.c. | n.c. | n.c. | 25 / 92 |
| G508R | ExAC | 80 | 10 2 |
18 | 4.3 | - | - | - | - | 34 / 79 |
| G511R | ExAC, Cosmic |
80 | 22 2 |
0 | 6.0 | - | - | - | - | 13 / 82 |
| R515A | in-house | 90 | 51 | 19 | 3.3 | n.d. | n.c. | n.c. | n.c. | 31 / 87 |
| R515Lb | patient | 75 | 68 | 15 | 3.7 | 46.8 | n.c. | n.c. | n.c. | 33 / 49 |
| R515Q | Cosmic | 90 | 53 | 19 | 3.3 | 47.6 | n.c. | n.c. | n.c. | 34 / 82 |
| R515W | ExAC, Cosmic | 70 | 66 | 16 | 3.6 | 44.9 | n.c. | n.c. | n.c. | 17 / 36 |
Abbreviations: n.c. – not characterizable; n.d. – not done; (−) unable to assess due to aggregation;
ExAC: Exome Aggregation Consortium (Lek et al., 2016); Cosmic: Catalog of Somatic Mutations in Cancer (Forbes et al., 2017).
Data for WT PGM1 previous published (Lee et al., 2014) except for T (this study).
0.5 Kinetic data previously published (Wong et al., 2015).
Phosphorylation assessed via electrospray ionization mass spectrometry as described in Methods.
The six remaining variants were characterized for thermal stability, enzyme activity, and the competence of their catalytic serine for phosphorylation by the known activator and reaction intermediate glucose 1,6-bisphosphate (Table 1) (Lee et al., 2014). As assessed by thermal shift assays, none of the variants showed significant changes in stability relative to WT enzyme: T0.5 values range from 44.9 to 48.7° C, generally within 1–2° C of WT enzyme (T0.5 = 46.3° C). In contrast, all six variants had profoundly impaired enzyme activity. While low phosphoglucomutase activity could be observed over extended time (hours) at very high enzyme concentration (100 times WT), it was too low to permit characterization of the steady state kinetic parameters. For comparison, other missense variants with kcat as low as 0.1% of the WT enzyme are still quantifiable (Lee et al., 2014). Despite their impaired activity, all of the variants were nevertheless competent for phosphorylation of the catalytic serine (P-Ser117) as assessed by electrospray ionization mass spectrometry (Figure S1), although for several this was less efficient than for WT enzyme (Table 1) under the conditions tested. Thus, it appears that impaired catalysis in these variants is not attributable to lack of phosphorylation of Ser117, which is required but not sufficient for enzyme activity.
Arg503 and Arg515 variants cause unexpected ordering of the D4 loop
The crystal structures of four variants of human PGM1, with substitutions at either residue 503 or 515, were determined (Methods). The proteins crystallized under similar conditions as WT enzyme and with isomorphous crystal lattices (Table 2). Two identical copies of the polypeptide chain are found in the asymmetric unit: chain A is more highly ordered in all PGM1 crystals, and is therefore used as the reference in following descriptions. Resolution of the diffraction data for the different variants ranged from 1.75 to 2.6 Å.
Table 2.
Crystallographic data collection and refinement statistics
| Protein | R503Q | R515L | R515Q | R515W | G6P complex |
|---|---|---|---|---|---|
| Space Group | P41212 | P41212 | P41212 | P41212 | P41212 |
| Unit cell | 172.1 172.1 | 171.3 171.3 | 174.0 174.0 | 172.3 172.3 | 171.9 171.9 |
| parameters (Å) | 99.8 | 99.4 | 99.5 | 99.4 | 99.4 |
| Resolution (Å) | 60.95 (1.95) | 60.68 (2.20) | 61.5 (2.60) | 60.93 (1.75) | 60.82 (2.30) |
| Observations | 1557274 (74492) | 893507 (42745) | 660415 (62010) | 2043537 (59107) | 878402 (37536) |
| Unique | 108994 (5314) | 75286 (4379) | 47453 (4566) | 150294 (6946) | 64976 (3790) |
| reflections | |||||
| Rmerge (I) | 0.14 (1.902) | 0.279 (2.141) | 0.185 (1.846) | 0.089 (1.58) | 0.160 (1.774) |
| Rpim (I) | 0.038 (0.526) | 0.084 (0.72) | 0.052 (0.518) | 0.025 (0.561) | 0.045 (0.573) |
| Mean I/σ(I) | 15.1 (1.5) | 9.8 (1.2) | 15.2 (1.4) | 20.1 (1.1) | 14.8 (1.3) |
| CC1/2 | 0.999 (0.709) | 0.997 (0.693) | 0.998 (0.796) | 0.999 (0.595) | 0.998 (0.656) |
| Completeness (%) | 100.0 (99.9) | 100.0 (99.9) | 100.0 (100.0) | 99.7 (93.6) | 97.8 (82.6) |
| Multiplicity | 14.3 (14) | 11.9 (9.8) | 13.9 (13.6) | 13.6 (8.5) | 13.5 (9.9) |
| No. of protein residues | 1126 | 1128 | 1124 | 1125 | 1128 |
| No. of atoms | 9725 | 9316 | 8281 | 9898 | 9259 |
| No. of sulfate ions | 18 | 6 | 13 | 5 | 0 |
| No. of water molecules | 949 | 635 | 91 | 1180 | 571 |
| Rcryst | 0.1693 | 0.1897 | 0.2386 | 0.1652 | 0.1674 |
| Rfree | 0.2129 | 0.2507 | 0.2954 | 0.1909 | 0.2263 |
| RMSD bond lengths (Å) | 0.007 | 0.007 | 0.010 | 0.006 | 0.007 |
| RMSD bond angles (°) | 0.812 | 0.853 | 1.384 | 0.783 | 0.894 |
| Ramachandran | |||||
| Plot | |||||
| Favored (%) | 98.03 | 96.44 | 96.78 | 98.21 | 96.80 |
| Outliers (%) | 0.18 | 0.18 | 0.63 | 0.09 | 0.18 |
| MOLPROBITY | 98 | 98 | 98 | 99 | 99 |
| score (%-tile) | |||||
| Average B (Å2) | 41.82 | 52.17 | 71.27 | 32.07 | 41.11 |
| Protein | 40.57 | 52.03 | 70.94 | 30.31 | 40.89 |
| Water | 45.53 | 50.04 | 52.38 | 42.13 | 42.48 |
| Sulfate ions | 112.54/18 | 108.43/6 | 129.77/13 | 67.15/5 | - |
| (<B>/#) | |||||
| Glucose 6-P | - | - | - | - | 79.21/2 |
| (<B>/#) | |||||
| Coordinate | 0.197 | 0.295 | 0.454 | 0.211 | 0.270 |
| Error (Å) | |||||
| PDB code | 5VG7 | 5VEC | 5VIN | 5VBI | 6BJ0 |
Values for the outer resolution shell of data are given in parentheses.
Ramachandran plots generated with Molprobity via the PDB validation server.
Maximum likelihood-based coordinate error estimate reported by PHENIX.
A superposition of the polypeptide backbones of the four mutants with that of WT enzyme shows that the structures are very similar overall (Figure 2A). For example, the overall root-mean-square-deviation (RMSD) between R503Q and WT enzyme is 0.51 Å for 540 Cα pairs. In each structure, the substituted residue (i.e., Gln, Trp, Leu) is observed in the electron density maps and makes no direct interactions with other residues in the protein. Nevertheless, all of the structures show one clear difference compared to WT enzyme: ordering of the mobile flap (residues 507–509) within the D4 loop (Figure 2B–E). In the two high-resolution structures of R503Q and R515W, all residues of the loop are clearly defined (Figure 2D–E). In the lower resolution structures, partial ordering is seen (Figure S2A), which is nonetheless significant relative to the WT structure in 2Fo-Fc maps (see Figure S2B for omit maps). The conformers of the D4 loops vary somewhat in the different structures (Figure 2B), but include additional inter-residue interactions and regions of ordered secondary structure. For instance, in the R503Q and R515W variants, residues 509–510 form a β-turn connecting two β-strands, with residues 505–507 forming a bulge within this region (Figure 2D,E).
Figure 2. Crystal structures of missense variants of Arg503 and Arg515.

(A) Backbone superposition of WT PGM1 (gray) with R503Q (red), R515L (gold), R515W (purple), R515Q (cyan), and the enzyme-substrate complex with G6P (green). (B) A close-up view of the D4 loop of each structure in (A). The break in the WT chain is indicated with spheres and highlighted by arrows. 2Fo-Fc electron density maps calculated from the final models and contoured at 1.0 σ for the D4 loop in (C) WT PGM1, and the (D) R503Q and (E) R515Q variants. For omit maps, see Figure S2B.
A structural explanation for the reduced mobility of the flap in these missense variants is not immediately obvious. In all cases, the site of the mutation (residue 503 or 515) is outside of the flap, and none of the introduced side chains in the variants interact with residues in the flap. The ordering of the flap also occurs regardless of the type of substitution (e.g., Trp vs. Leu). Indeed, even alanine mutants at these positions result in highly impaired enzyme activity, arguing against steric effects from the introduced side chains (Table 1). Taken together, these observations raised the following possibility: perhaps the conformer(s) observed in the variants exist within the WT conformational ensemble of this region, but become more energetically favorable in the missense variants. A corollary to this hypothesis would be that the active site environment of the WT enzyme is poised to encourage many possible conformers of the D4 loop, presumably for functional reasons. Given the known association of the D4 loop with ligand binding, we pursued the crystal structure of a PGM1-substrate complex.
The D4 loop changes conformation to bind ligand
The crystal structure of PGM1 in complex with glucose 6-phosphate (G6P) was determined at 2.3 Å resolution (Table 2). (Since the enzyme reaction is reversible, it is equally valid to consider G6P as either substrate or product.) G6P was soaked into crystals of WT enzyme grown in a low salt condition (Methods). Clear evidence for ligand binding was observed in initial electron density maps; an omit map calculated from the final refined structure is shown in Figure S2B. Differences between the polypeptide backbones of apo- and ligand-bound PGM1 are minor, with some small shifts in residues of domains 1 and 4 (Figure 3A). G6P is bound deep within the active site, with approximately 50% of its surface area inaccessible to solvent (Figure 3B). Notably, the flap region of the D4 loop is mostly ordered in the ligand complex, although its conformation is distinctly different from that seen in the missense variants (Figures 2A and S2B). Significant changes in ϕ/Ψ angles occur, especially for Gly506, Gly508, and Ala510, producing an overall closure of the loop. The D4 loop in the enzyme-G6P complex has no regular secondary structure, but is rather wrapped around the phosphate group of ligand (Figure 3C). Unlike the R503Q and R515W complexes, no inter-residue contacts are observed between residues in the loop, highlighting the significant conformational change that would be needed to convert between the ligand-bound and ordered conformers of the variants.
Figure 3. Crystal structure of the PGM1 complex with glucose 6-phosphate.

(A) A backbone superposition of the apo-enzyme (gray) and enzyme-ligand complex (green). G6P is shown in a space-filling model. (B) Space-filling representation of the PGM1-G6P complex, looking down into the active site. View is a 90° rotation relative to (A). Residues in the mobile flap are in magenta. (C) A close-up view of the PGM1-G6P interactions involving residues in the D4 loop (dashed orange lines). An additional interaction with the backbone amide of Gly506 is not shown. Ser117 and the bound metal ion (black sphere) are shown for reference. For an omit map of the ligand, see Figure S2B.
Within the active site, G6P binds with its O1 hydroxyl near Ser117 (the catalytic phosphoserine), as would be required for phosphoryl transfer, and with its phosphate group proximal to the D4 loop (Figure 3C). As expected (Beamer, 2015), multiple residues in the D4 loop, including one from the flap, are involved in direct interactions with G6P. These include both Arg503 and Arg515, which make bidentate interactions with the phosphate, as well as additional phosphate contacts made by Ser505, Gly506, and Thr507. Interactions are also made with the hydroxyl groups of the sugar by Arg293, Glu376, and Ser378, although these vary somewhat in the two copies of the polypeptide chain. Some of the enzyme-ligand interactions in the PGM1-G6P complex are similar to those in two unpublished structures of rabbit PGM (97% sequence identity to human enzyme) bound to ligands, but analysis of the electron density maps of these structures indicates multiple problems, including incorrectly modeled D4 loops (Figure S3), so detailed comparisons are not possible.
The structure of the PGM1-G6P complex highlights the crucial role of the D4 loop in ligand binding, and also helps explain the detrimental impact of the 503 and 515 missense variants. First, the essential roles of Arg503 and Arg515 in contacting the phosphate group of the substrate are clear: these two residues are part of an intricate network of interactions between the protein and bound ligand (Figure 3C), and it seems certain that substitution of either residue would disrupt binding. Support for this is evident in the changed position (or loss) of a bound sulfate ion, a mimic for the phosphate group of the substrate, seen in the crystal structures of missense variants as compared to WT enzyme (Figure 2C–E and S2A). Second, the conformer of the D4 loop present in the G6P complex is incompatible with the conformer(s) of D4 observed in the mutant structures (Figure S2B), which would present an additional impediment to ligand binding. Finally, the ordering of the D4 loop observed in the mutant structures, relative to its mobility in WT enzyme, suggests that an unfavorable conformational transition would be necessary to achieve the productive ligand-binding conformer of the loop in the variant proteins. Overall, observations from the PGM1-G6P complex support the functional importance of flap mobility, and suggest that the conformers observed in the missense variants would be unfavorable for ligand binding.
Evaluation of loop mobility by molecular dynamics
To further examine the intrinsic mobility of the D4 loop, we conducted 10 ns molecular dynamics (MD) simulations of both WT PGM1 and the R503Q variant (Methods). The proteins remained stable throughout the course of the simulation, with RMSD values of the structures generally between 1–2 Å (Figure S4A,B). An analysis of the Cα root mean square fluctuations (RMSF), relative to the energy-minimized starting structures, shows that the D4 loop (residues 505–511) is the most highly mobile region of the protein, even in the R503Q variant (Figures 4 and S4C–D). (A previous study of PGM1 mechanism by MD showed a similar result for WT enzyme (Bras et al., 2018)). We further examined the range of conformers of the D4 loop sampled during the MD simulation, and find that the conformers observed in the crystal structures of the missense variants and PGM1-G6P complex are encompassed within this range (Figure 4B).
Figure 4. Analyses of D4 loop flexibility based on the MD simulations of human PGM1.

(A) RMSF values for WT enzyme mapped onto its structure. Increasing tube radius and a change from blue to red indicate higher RMSF values. (B) A sampling of the D4 loop conformers from the MD trajectory (one structure shown per 10 ps cycle) of WT PGM1. Sampled conformers (white) are shown only for the D4 loop; other regions of the polypeptide backbone are from the crystal structures of the missense variants and PGM1-G6P complex. Colors are as in Figure 2. View is 180° rotation relative to (A). Residence density analysis of the MD trajectories for the D4 loop (residues 505–513) of WT PGM1 (C) and the R503Q variant (D). Maps were calculated as described in Methods. Note the discontinuity of the density for WT D4 loop (blue) versus the continuous density for the R503Q loop (orange).
The RMSF analyses suggest generally similar behavior of WT PGM1 and the R503Q variant during the MD simulations, in contrast to the obvious differences seen in the electron density maps of this region. To resolve this apparent paradox, we utilized calculations of the residence density (Michaud-Agrawal et al., 2011), a time-averaged distribution of atoms within a particular volumetric region over the course of the simulation (Methods). These calculations reveal distinct differences between the D4 loop in these two proteins (Figure 4C,D). In the case of WT enzyme, the loop has discontinuous density, showing that these residues sample a wide range of conformers during the simulations, consistent with the lack of electron density for this region in the WT structure. On the other hand, the D4 loop of the R503Q variant has well-defined residence density that generally matches the conformer observed in its crystal structure. The density calculations show that, despite the overall high RMSF of the D4 loop in the R503Q variant (Figure S2B), this region tends to occupy fewer conformational states for relatively longer times, as compared to WT enzyme. Thus, the single amino change of the missense variant perturbs the conformational ensemble of the D4 loop.
Discussion
The D4 loop is one of four highly conserved regions of the active site of phosphoglucomutases. Its sequence conservation is evident across a wide range of organisms, including bacteria, plants, and animals. In particular, the residues corresponding to Arg503 and Arg515, as well as the subsequent -Thr-Gly-Ser-, are nearly invariant across evolutionary space (Figure 5). This is not surprising, based on the critical role of these residues in ligand binding, as confirmed in the first PGM1-substrate complex presented here. The importance of this region in substrate recognition was previously recognized in related enzymes in the α-D-phosphohexomutase superfamily, where the corresponding loop was shown to interact with the phosphate group of the ligand regardless of whether the 1- or 6-phosphosugar is bound (Nishitani et al., 2006; Regni et al., 2004). These critical protein-phosphate contacts allow enzymes in this superfamily to accommodate both 1- and 6-phosphosugars in the same binding site, which is key to their reversible reaction (Figure 1).
Figure 5. Sequences of the D4 loop of phosphoglucomutase in diverse eukaryotic organisms.

Top: a multiple sequence alignment (spanning residues 503 to 515 of human PGM) highlighting identical residues with red background. Bottom: A consensus Web Logo (Crooks et al., 2004) of the D4 loop. R503 and R515 are highlighted by yellow star; variants relevant to this study are indicated with arrows at bottom. Those with confirmed roles in PGM1 deficiency are marked with an asterisk.
The profound functional impairment of the Arg503 and Arg515 variants is consistent with the critical roles of these two residues in phosphate recognition. Nevertheless, the PGM1 variants at residues 503 and 515 appear to have no direct structural impact on the enzyme, beyond the site of substitution. Unexpectedly, however, we find that mutations at these residue positions result in the ordering of the nearby, mobile flap of D4. This indirect effect, which occurs regardless of the site or nature of the substitution, suggests an additional contributing factor to enzyme dysfunction in these variants: changes to the functional dynamics of this key ligand-binding loop. This proposal is supported by the MD simulations, which show that the R503Q mutation shifts the conformeric ensemble of the D4 loop, producing a favored, major conformer that is incompatible with ligand-binding. Ordering of this loop might also impede access of the substrate to the active site, the release of product, and reorientation of the intermediate (Figure 3C).
Such potentially compounding effects may help explain the prevalence of missense variants in the D4 loop that are associated with PGM1 deficiency (Muenks et al., 2017). In addition to those identified in patients (R503Q and R515L), we show here that other variants found in the human population (G506R, G511R, R515W, R515Q) also deleteriously impact PGM1 function in vitro. While these variants have not yet been identified in patients with PGM1 deficiency, our results suggest they would likely cause disease in homozygous individuals. Although not yet biochemically or structurally characterized, disease-associated variants in the D4 loop have also been identified in a related human enzyme, PGM3, consistent with the conserved functional role of this region (Muenks et al., 2017; Zhang et al., 2014). Thus this region may be a hotspot for disease-associated mutations in the entire enzyme superfamily.
The importance of missense variants in inherited human disease is well appreciated, but a molecular understanding of the effects of the mutation is often lacking (Stefl et al., 2013). Despite their functional impairment, the D4 variants described herein lack the obvious structural impacts seen in several other PGM1 missense variants, including several that cause direct and profound structural changes within the active site (Stiers et al., 2016). Instead, the effects of the D4 variants appear to be explained by the loss of key ligand interactions, with a potentially compounding, indirect effect on loop dynamics. Dynamic changes due to mutations, which act at both short and long-range, have been previously observed, in both computational and experimental studies (Nicchia et al., 2011; Sekhar et al., 2016; Verma et al., 2012; Wang et al., 2013). The possible contribution of a dynamic defect in PGM1 deficiency may facilitate alternative therapeutic options, such as the use of small molecules to modulate the conformational ensemble of the protein (Encarnación et al., 2016; Gupta et al., 2016; Street et al., 2010).
STAR Methods
Contact for Reagent and Resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lesa J Beamer (beamerl@missouri.edu).
Experimental Model and Subject Details
We used E.coli BL21(DE3) cells for recombinant expression of His-tagged PGM1 for biochemical and biophysical experiments. The cells were cultured using standard practices in LB media.
Methods Details
Mutagenesis, protein expression and purification.
Variants R503Q and R515L were identified in patients with PGM1 deficiency and previously shown to lack quantifiable activity (Wong et al., 2015). The G508R, G511R, R515Q, and R515W mutants were identified as potentially deleterious to PGM1 function from a computational analysis of known human variants of PGM1 (Muenks et al., 2017) found in the ExAc (Lek et al., 2016) or Cosmic sequence databases (Forbes et al., 2017). Each of these variants is rare in the human population. R503A and R515A were engineered for this study. All PGM1 mutants were constructed using the QuikChange kit (Agilent) and verified by automated DNA sequencing.
Missense variants were expressed recombinantly in E. coli and purified to homogeneity via an N-terminal His6 affinity tag, as previously described (Lee et al., 2014). The purified proteins were dialyzed into a solution of 50 mM MOPS, pH 7.4, with 1 mM MgCl2, and concentrated to ~10 mg/mL or higher. If not used immediately, samples were flash-frozen in liquid nitrogen and stored at −80 °C.
Biochemical studies
Dynamic light scattering—
Protein samples at 1 mg/mL in 50 mM MOPS, pH 7.4, and 1 mM MgCl2 were prepared and centrifuged prior to data collection. Data were collected on a Protein Solutions DynaPro 99 instrument at a wavelength of 8363 Å for 200 s (10 s each for 20 acquisitions) at 25 °C. Polydispersity of samples ranged from 0 to 21%.
Kinetic characterization—
Phosphoglucomutase activities for the missense mutants were assessed by coupling the formation of glucose 6-phosphate from glucose 1-phosphate to NADH formation via glucose 6-phosphate dehydrogenase (G6PDH). Leuconostoc mesenteroides G6PDH, α-D-glucose 1-phosphate, and glucose 1,6-bisphosphate were obtained from Sigma. Reactions were conducted at 25 °C in 50 mM MOPS, pH 7.4, with 1 mM dithiothreitol, 1.5 mM MgSO4, and 0.9 mM NAD+. The activator glucose 1,6-bisphosphate was present at 1.0 μM and the substrate (α-D-glucose 1-phosphate) at 200 μM. Assays of variants were done in parallel with a control (WT enzyme at 4 – 7.8 nM) and monitored for at least one hour. Enzyme concentrations for the variants at 1×, 10×, and 100× of the WT control were assessed, all of which failed to produce quantifiable activity.
Assessment of phosphorylation by mass spectrometry—
The phosphorylation state of the active site phosphoserine (Ser117) was analyzed before and after treatment with glucose 1,6-bisphosphate using electrospray ionization mass spectrometry (Figure S1). Protein samples at concentrations between 75 and 150 μM in 50 mM MOPS, pH 7.4, 1 mM MgCl2 were incubated with a 6-fold molar excess of glucose 1,6-bisphosphate for 16 h at 4 °C. For mass spectrometric analyses, protein samples at 1 pmol/μl in 1% formic acid were analyzed by NanoLC Nanospray QTOF (Agilent 6520) in positive ion mode with a Zorbax C8 trap column. Data were examined using the Qual software provided with the instrument. The mass error between samples is 0.11 Da (2.1 ppm) and quantification error is 2%. Percent phosphorylation was calculated by normalizing the sum of the dephosphorylated and phosphorylated peak heights to 1.0.
Thermal Shift Assays—
Dephosphorylated WT PGM1 and missense variants were diluted to 0.5 mg/mL (~8 μM) in 50 mM MOPS, pH 7.4 supplemented with 1 mM MgCl2. Samples were incubated with dye from the Applied Biosystems® Protein Thermal Shift kit per manufacturer’s recommendation for 1 hour at 4 °C. A QuantStudio 3 Real-Time PCR System (ThermoFisher Scientific) was used to ramp from 4 °C to 95 °C in 0.3 °C increments with 10 second holds between ramping steps. Fluorescence values were normalized as in (Andreotti et al., 2015) and T0.5 calculated as the midpoint of the normalized fluorescence response. Samples were run in duplicate.
Crystallization
Proteins destined for crystallization trials were purified as above, with the additional step of cleavage of the histidine tag, as in (Stiers et al., 2016). Successful cleavage was confirmed by electrospray ionization mass spectrometry. Initial crystallization screens were set up at 19 °C with Crystal Screen kits 1 and 2 (Hampton Research) and Wizard screen kits 1 and 2 (Emerald BioSystems Inc.) at a protein concentration of 9–16 mg/mL using the hanging drop vapor diffusion method at 20° C. Drops containing 2 μl protein solution and 2 μl crystallization buffer were sealed over a 0.5 mL reservoir. Crystals of R503Q used for data collection were grown in 1.8 M ammonium sulfate and 0.1 M TRIS HCl, pH 7.5. Final R515L and R515Q crystals were grown in 0.01 M cobalt (II) chloride hexahydrate, 0.1 M MES monohydrate pH 6.5, and 1.8–2.1 M ammonium sulfate. R515W crystals were grown in 0.1 M HEPES 7.5, 0.1 M sodium chloride, and 1.5–1.7 M ammonium sulfate. Crystals were cryoprotected using a solution of well buffer supplemented with 30% glycerol (v/v), mounted on Hampton loops, and flash cooled in liquid nitrogen. Crystals grew in approximately one week in space group P41212 with ~60% solvent (VM = 3.04 Å3/Da). The asymmetric unit contains two copies of the polypeptide chain.
For preparation of a PGM1-substrate complex, a novel, low salt crystallization condition was identified for WT PGM1. Crystallization conditions were screened at 19°C using Intelli-Plate 96–3 well sitting drop plates (0.2 μL protein + 0.2 μL well buffer) at a protein concentration of 12 mg/mL. Crystals of WT PGM1 were obtained in condition C12 from PEG Ion Screen HT (Hampton Research), which contains 0.2 M sodium tartrate dibasic dihydrate and 20% PEG 3,350 at pH 7.3. Despite the different crystallization conditions, these crystals were isomorphous with those grown previously from ammonium sulfate (Stiers et al., 2016) (Table S1). For ligand soaks, single crystals were extracted from drops, resuspended in well buffer, and the concentration of PEG 3,350 increased to 30%. Step-wise additions of 10 mM glucose-6-phosphate were made to the crystals over a period of five minutes to a final concentration of ~7 mM; crystals were flash-cooled directly into liquid nitrogen for storage without additional cryoprotection.
X-ray diffraction data collection and refinement
Diffraction data were collected at a wavelength of 1.00003 Å from single crystals on beamline 4.2.2 of the Advanced Light Source using a Taurus-1 CMOS detector in shutterless mode. The data were processed using XDS (Kabsch, 2010) and AIMLESS (Evans and Murshudov, 2013) via CCP4i (Potterton et al., 2003). Data processing statistics are listed in Table S1. Values of CC1/2 > 0.30 (Karplus and Diederichs, 2012) and Rpim (Weiss, 2001) were used to determine the high resolution cutoff due to the large number of images (1800–3600 per data set) and high redundancy obtained with the shutterless data collection.
Crystallographic refinement calculations were initiated using coordinates of WT PGM1 (PDB code: 5EPC). Refinement was performed with PHENIX (Adams et al., 2010); progress was monitored by following R free with 5% of each data set was set aside for cross validation. The B-factor model consisted of an isotropic B-factor for each atom; TLS refinement was used as automated in PHENIX. COOT (Emsley and Cowtan, 2004) was used for model building. The Rfree data sets for the mutants were constrained to match those of the WT data. Structures were validated using MolProbity (Chen et al., 2010) and refinement statistics are listed in Table 2. In the R503Q and R515W structures, Ser117 was modeled as a mixture of serine and phosphoserine, based on the electron density maps. Structural figures were prepared with PYMOL (DeLano, 2002). Coordinates and structure factor amplitudes have been deposited in the PDB under the accession number listed in Table 2.
Molecular dynamics simulations and trajectory analyses
The structure of WT PGM1 (PDB ID 5EPC, chain A) and the R503Q mutant (PDB ID 5VG7, chain A) were prepared for MD simulations. Missing side chains were modeled using Swiss-PDBViewer (Guex and Peitsch, 1997). Initial coordinates for the missing residues in the flap of WT enzyme were taken from the structure of the R503Q mutant. Topologies were generated using the Amber (amber99sbff) force-field (Hornak et al., 2006) with GROMACS 2016.3 (Abraham et al., 2015; Berendsen et al., 1995; Hess et al., 2008; Pall et al., 2015; Pronk et al., 2013; Van Der Spoel et al., 2005). The system was solvated with the TIP3P water model and neutralized by addition of counter ions in a dodecahedral box (Miyamoto and Kollman, 1992). The neutralized system was energy minimized by the steepest descent algorithm with a tolerance of force (Fmax) less than 1000 kJ mol−1 nm−1, and both converged to an energy below −1.0e+06 kJ mol−1. The energy-minimized structures were subjected to consecutive 100 ps NVT and NPT equilibration steps. Each system remained stable at 300 K using a modified Berendsen thermostat, and pressure was maintained using the Parrinello-Rahman coupling method (Bussi et al., 2007). Production MD simulations were continued from the final frame of the NPT equilibration for 10 ns. Equilibration and production MD used the LINCS constraint algorithm (Hornak et al., 2006) with long-range electrostatics calculated using the Particle Mesh Ewald method (Essmann et al., 1995). Trajectories were analyzed with the GROMACS energy, rms, and rmsf utilities (Abraham et al., 2015; Pronk et al., 2013). RMSF values were mapped to the B-factor field of the initial frame from production MD and visualized in PyMOL (DeLano, 2002).
Trajectory density analysis — Raw trajectories were corrected for periodic boundary conditions, rotation, and translation with the trjconv utility of GROMACS (Abraham et al., 2015). The analysis.density submodule of the MDAnalysis python package (Michaud-Agrawal et al., 2011) was used to generate densities from the trajectories using the default grid spacing of 1.0 Å. Density maps were visualized against the initial production MD frame in PyMOL (DeLano, 2002) using the volume representation with carve set to 1.6 Å (default).
Quantification and Statistical Analysis
Thermal shift analysis -
Relative fluorescence units were normalized to fraction unfolded using the minimum and maximum fluorescence reading for the well as in (Andreotti et al., 2015) with the formula , where Funfolded is the relative fractional portion unfolded at a temperature point, RFUi is the fluorescence reading at a temperature point, RFUmin is the minimum reading for that sample, and RFUmax is the maximum reading for that sample. The point at which the protein is half unfolded (Funfolded = 0.50), designated T0.5, is used for comparison of samples.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Glucose-6-phosphate Dehydrogenase from Leuconostoc mesenteroides | Sigma-Aldrich | CA#: G8404–2KU |
| α-D-Glucose-1,6-bisphosphate | Sigma-Aldrich | CA#: G6893–25MG |
| DL-Dithiothreitol | Sigma-Aldrich | CA#: D9779–1G |
| α-D-Glucose-1 -phosphate | Sigma-Aldrich | CA#: G7000–5G |
| Magnesium sulfate | Sigma-Aldrich | CA#: M7506–500G |
| NAD+ | Sigma-Aldrich | CA#: N0632–5G |
| Critical Commercial Assays | ||
| Applied Biosystems Protein Thermal Shift Dye Kit | ThermoFisher Scientific | CA#: 4461146 |
| Crystal Screen 1 & 2 | Hampton Research | CA#: HR2–110 & HR2–112 |
| Wizard Classic Crystallization Screen 1 & 2 | Emerald BioSystems Inc. | CA#: 1009530 & 1009531 |
| QuikChange II Site-Directed Mutagenesis Kit | Agilent | CA#: 200523 |
| Deposited Data | ||
| R503Q variant structure | This paper | PDB: 5VG7 |
| R515L variant structure | This paper | PDB: 5VEC |
| R515Q variant structure | This paper | PDB: 5VIN |
| R515W variant structure | This paper | PDB: 5VBI |
| PGM1-G6P complex structure | This paper | PDB: 6BJ0 |
| Wild-type PGM1 | (Stiers et al., 2016) | PDB: 5EPC |
| Experimental Models: Organisms/Strains | ||
| Escherichia coli: BL21DE3 | New England Biosciences | CA#: C2527H |
| Recombinant DNA | ||
| Plasmid: Wild-type human PGM1 | (Lee et al., 2014) | N/A |
| Plasmid: PGM1-R503Q missense variant | This paper | N/A |
| Plasmid: PGM1-R515L missense variant | This paper | N/A |
| Plasmid: PGM1-R515Q missense variant | This paper | N/A |
| Plasmid: PGM1-R515W missense variant | This paper | N/A |
| Software and Algorithms | ||
| GROMACS molecular dynamics software | (Abraham et al., 2015) | http://dx.doi.Org/10.1016/j.softx.2015.06.001 |
| PyMOL | (DeLano, 2002) | http://www.pymol.org |
| MDAnalysis python package | (Michaud-Agrawal et al., 2011) | https://doi.org/10.1002/jcc.21787 |
| R | (R Core Team, 2014) | https://cran.r-project.org/ |
| Bio3D R-package | (Grant et al., 2006) | https://doi.org/10.1093/bioinformatics/btl461 |
| Other | ||
Highlights.
Crystal structures of PGM1 missense variants in an active site loop are presented
Mutations cause direct effects on ligand binding and indirect effects on loop mobility
The conformational ensemble of the wild-type enzyme is altered in the variants
The loop is a hotspot for disease-related mutations in PGM1
ACKNOWLEDGMENTS.
We thank Abigail Graham and Zachary Addison for assistance with protein purification and crystallization, Jay Nix of the Advanced Light Source beamline 4.2.2 for assistance with data collection and processing, Ritcha Mehra-Chaudhary of the University of Missouri Molecular Interactions Core for assistance with protein purification, and Brian Mooney of the University of Missouri Charles W. Gehrke Proteomics Center for mass spectrometry. KMS was supported by National Institutes of Health training grant T32 GM008396–26 and a predoctoral fellowship 17PRE33400210 from the American Heart Association. This work was supported by grants to LJB from the University of Missouri Research Board and the National Science Foundation (MCB-0918389). Molecular dynamics studies were performed on the high performance computing infrastructure provided by Research Computing Support Services and in part by the National Science Foundation under grant number CNS-1429294 at the University of Missouri, Columbia MO. Part of this work was performed at the Advanced Light Source. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract DE-AC02–05CH11231.
Abbreviations not defined in text:
- MOPS
3-morpholinopropane-1-sulfonic acid
- MES
2-(N-morpholino)ethanesulfonic acid
- TRIS
2-Amino-2-hydroxymethyl-propane-1,3-diol
- PDB
Protein Data Bank
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service toour customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare that they have no conflict of interest.
Data and Software Availability
Accession Numbers
The atomic coordinates and structure factors for the structures described in this study have been deposited to the RCSB PDB (www.rcsb.org) with the PDB ID codes: 5VG7, 5VEC, 5VIN, 5VBI, and 6BJ0.
Additional Resources
N/A
Supplemental Information
REFERENCES
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindah E (2015). Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. [Google Scholar]
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreotti G, Monticelli M, and Cubellis MV (2015). Looking for protein stabilizing drugs with thermal shift assay. Drug Test. Anal 7, 831–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beamer LJ (2015). Mutations in hereditary phosphoglucomutase 1 deficiency map to key regions of enzyme structure and function. J. Inherit. Metab. Dis 38, 243–256. [DOI] [PubMed] [Google Scholar]
- Berendsen HJC, van der Spoel D, and van Drunen R (1995). GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun 91, 43–56. [Google Scholar]
- Bras N, Fernandes P, Ramos M, and Schwartz S (2018). Mechanistic insights on human phosphoglucomutase revealed by transition path sampling and molecular dynamics calculations. Chemistry 24, 1978–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G, Donadio D, and Parrinello M (2007). Canonical sampling through velocity rescaling. J. Chem. Phys 126, 014101. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D, Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: A sequence logo generator. Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL (2002). The PyMOL Molecular Graphics System. Schrödinger LLC Wwwpymolorg Version 1, http://www.pymol.org. [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Encarnación MC, Palomino-Morales RJ, Fuchs JE, Esperanza PG, Noel MT, Salido E, Timson DJ, and Pey AL (2016). Conformational dynamics is key to understanding loss-of-function of NQO1 cancer-associated polymorphisms and its correction by pharmacological ligands. Sci. Rep 6, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, and Pedersen LG (1995). A smooth particle mesh Ewald method. J. Chem. Phys 103, 8577–8593. [Google Scholar]
- Evans PR, and Murshudov GN (2013). How good are my data and what is the resolution? Acta Crystallogr. Sect. D, Biol. Crystallogr 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al. (2017). COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res 45, D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnad F, Baucom A, Mukhyala K, Manning G, and Zhang Z (2013). Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genomics 14 Suppl 3, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, and Caves LSD (2006). Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 221, 2695–2696. [DOI] [PubMed] [Google Scholar]
- Guex N, and Peitsch MC (1997). SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- Gupta AN, Neupane K, Rezajooei N, Cortez LM, Sim VL, and Woodside MT (2016). Pharmacological chaperone reshapes the energy landscape for folding and aggregation of the prion protein. Nat. Commun 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B, Kutzner C, Van Der Spoel D, and Lindahl E (2008). GRGMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, and Simmerling C (2006). Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Genet 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). Software XDS for image rotation, recognition and crystal symmetry assignment. Acta Crystallogr., Sect. D Biol. Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus PA, and Diederichs K (2012). Linking crystallographic model and data quality. Science 336, 1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukcongar A, Tumer L, Suheyl Ezgu F, Seher Ksapkara C, Jaeken J, Matthijs G, Rymen D, Dalgic B, Bideci A, and Hasanoglu A (2015). A case with rare type of congential-disorder of glycosylation: PGM1-CDG. Genet. Couns 26, 87–90. [PubMed] [Google Scholar]
- Lee Y, Stiers KM, Kain BN, and Beamer LJ (2014). Compromised catalysis and potential folding defects in in vitro studies of missense mutants associated with hereditary phosphoglucomutase 1 deficiency. J. Biol. Chem 289, 32010–32019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewenthal N, Haim A, Parvari R, and Hershkovitz E (2015). Phosphoglucomutase-1 deficiency: Intrafamilial clinical variability and common secondary adrenal insufficiency. Am. J. Med. Genet. Part A 167, 3139–3143. [DOI] [PubMed] [Google Scholar]
- Michaud-Agrawal N, Denning EJ, Woolf TB, and Beckstein O (2011). MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem 32, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, and Kollman PA (1992). Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem 13, 952–962. [Google Scholar]
- Muenks AG, Stiers KM, and Beamer LJ (2017). Sequence-structure relationships, expression profiles, and disease-associated mutations in the paralogs of phosphoglucomutase 1. PLoS One 12, e0183563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicchia GP, Ficarella R, Rossi A, Giangreco I, Nicolotti O, Carotti A, Pisani F, Estivill X, Gasparini P, Svelto M, et al. (2011). D184E mutation in aquaporin-4 gene impairs water permeability and links to deafness. Neuroscience 197, 80–88. [DOI] [PubMed] [Google Scholar]
- Nishitani Y, Maruyama D, Nonaka T, Kita A, Fukami TA, Mio T, Yamada-Okabe H, Yamada-Okabe T, and Miki K (2006). Crystal structures of N-acetylglucosamine-phosphate mutase, a member of the a-D-phosphohexomutase superfamily, and its substrate and product complexes. J. Biol. Chem 281, 19740–19747. [DOI] [PubMed] [Google Scholar]
- Ondruskova N, Honzik T, Vondrackova A, Tesarova M, Zeman J, and Hansikova H (2014). Glycogen storage disease-like phenotype with central nervous system involvement in a PGM1-CDG patient. Neuro Endocrinol Lett 35, 137–141. [PubMed] [Google Scholar]
- Pall S, Abraham MJ, Kutzner C, Hess B, and Lindahl E (2015). Tackling exascale software challenges in molecular dynamics simulations with GROMACS. In Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), pp. 3–27. [Google Scholar]
- Pérez B, Medrano C, Ecay MJ, Ruiz-Sala P, Martínez-Pardo M, Ugarte M, and Pérez-Cerdá C (2013). A novel congenital disorder of glycosylation type without central nervous system involvement caused by mutations in the phosphoglucomutase 1 gene. J. Inherit. Metab. Dis 36, 535–542. [DOI] [PubMed] [Google Scholar]
- Potterton E, Briggs P, Turkenburg M, and Dodson E (2003). A graphical user interface to the CCP 4 program suite. Acta Crystallogr. Sect. D Biol. Crystallogr 59, 1131–1137. [DOI] [PubMed] [Google Scholar]
- Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, Van Der Spoel D, et al. (2013). GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2014). R: A Language and Environment for Statistical Computing. R Found. Stat. Comput [Google Scholar]
- Regni C, Naught L, Tipton PAPA, and Beamer LJLJ (2004). Structural Basis of Diverse Substrate Recognition by the Enzyme PMM/PGM from P. aeruginosa. Structure 12, 55–63. [DOI] [PubMed] [Google Scholar]
- Sekhar A, Rumfeldt JAO, Broom HR, Doyle CM, Sobering RE, Meiering EM, and Kay LE (2016). Probing the free energy landscapes of ALS disease mutants of SOD1 by NMR spectroscopy. Proc. Natl. Acad. Sci 113, E6939–E6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, and Berendsen HJC (2005). GROMACS: Fast, flexible, and free. J. Comput. Chem 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- Stefl S, Nishi H, Petukh M, Panchenko AR, and Alexov E (2013). Molecular Mechanisms of Disease-Causing Missense Mutations. J. Mol. Biol 425, 3919–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiers KM, Kain BN, Graham AC, and Beamer LJ (2016). Induced structural disorder as a molecular mechanism for enzyme dysfunction in phosphoglucomutase 1 deficiency. J. Mol. Biol 428, 1493–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiers KM, Graham AC, Kain BN, and Beamer LJ (2017a). Asp263 missense variants perturb the active site of human phosphoglucomutase 1 (PGM1). FEBS J 284, 937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiers KM, Muenks AG, and Beamer LJ (2017b). Biology, mechanism, and structure of enzymes in the α-D-phosphohexomutase superfamily. Adv. Protein Chem. Struct. Biol 109, 265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojkovic T, Vissing J, Petit F, Piraud M, Orngreen MC, Andersen G, Claeys KG, Wary C, Hogrel J-Y, and Laforêt P (2009). Muscle Glycogenosis Due to Phosphoglucomutase 1 Deficiency. N. Engl. J. Med 361, 425–427. [DOI] [PubMed] [Google Scholar]
- Street TO, Krukenberg KA, Rosgen J, Bolen DW, and Agard DA (2010). Osmolyte-induced conformational changes in the Hsp90 molecular chaperone. Protein Sci 19, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegtmeyer LC, Rust S, van Scherpenzeel M, Ng BG, Losfeld M-E, Timal S, Raymond K, He P, Ichikawa M, Veltman J, et al. (2014). Multiple Phenotypes in Phosphoglucomutase 1 Deficiency. N. Engl. J. Med 370, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thusberg J, Olatubosun A, and Vihinen M (2011). Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat 32, 358–368. [DOI] [PubMed] [Google Scholar]
- Timal S, Hoischen A, Lehle L, Adamowicz M, Huijben K, Sykut-Cegielska J, Paprocka J, Jamroz E, van Spronsen FJ, Körner C, et al. (2012). Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum. Mol. Genet 21, 4151–4161. [DOI] [PubMed] [Google Scholar]
- Tokuriki N, and Tawfik DS (2009). Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol 19, 596–604. [DOI] [PubMed] [Google Scholar]
- Verma D, Jacobs DJ, and Livesay DR (2012). Changes in lysozyme flexibility upon mutation are frequent, large and long-ranged. PLoS Comput. Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Wen X, Qin X, Wang Z, Tan Y, Shen Y, and Xi Z (2013). Quantitative structural insight into human variegate porphyria disease. J. Biol. Chem 288, 11731–11740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MS (2001). Global indicators of X-ray data quality. J. Appl. Crystallogr 34, 130–135. [Google Scholar]
- Wong SYW, Beamer LJ, Gadomski T, Honzik T, Mohamed M, Wortmann SB, Brocke Holmefjord KS, Mork M, Bowling F, Sykut-Cegielska J, et al. (2015). Defining the Phenotype and Assessing Severity in Phosphoglucomutase-1 Deficiency. J. Pediatr 175, 130–136. [DOI] [PubMed] [Google Scholar]
- Yue P, Li Z, and Moult J (2005). Loss of Protein Structure Stability as a Major Causative Factor in Monogenic Disease. J. Mol. Biol 353, 459–473. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, Jing H, Kim ES, Biancalana M, Wolfe L. a., et al. (2014). Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J. Allergy Clin. Immunol 133, 1400–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.