

Summary
Recent evidence indicates that specific RNAs promote the formation of ribonucleoprotein condensates by acting as scaffolds for RNA-binding proteins (RBPs). We systematically investigated RNA-RBP interaction networks to understand ribonucleoprotein assembly. We found that highly contacted RNAs are structured, have long UTRs, and contain nucleotide repeat expansions. Among the RNAs with such properties, we identified the FMR1 3′ UTR that harbors CGG expansions implicated in fragile X-associated tremor/ataxia syndrome (FXTAS). We studied FMR1 binding partners in silico and in vitro and prioritized the splicing regulator TRA2A for further characterization. In a FXTAS cellular model, we validated the TRA2A-FMR1 interaction and investigated implications of its sequestration at both transcriptomic and post-transcriptomic levels. We found that TRA2A co-aggregates with FMR1 in a FXTAS mouse model and in post-mortem human samples. Our integrative study identifies key components of ribonucleoprotein aggregates, providing links to neurodegenerative disease and allowing the discovery of therapeutic targets.
Keywords: phase separation, scaffolding RNA, CGG repeat expansion, FMR1 premutation, fragile X-associated tremor/ataxia syndrome, FXTAS, RNA aggregates, RNA binding proteins, RBP, TRA2A splicing regulator, neurodegeneration
Graphical Abstract
Highlights
-
•
Highly contacted granule RNAs are structured and contain long, repetitive UTRs
-
•
Mutations related with FXTAS increase FMR1 scaffolding propensity
-
•
TRA2A co-aggregates with FMR1 in FXTAS mouse model and in post-mortem human samples
-
•
TRA2 sequestration has both transcriptomic and post-transcriptomic implications
Cid-Samper et al. analyze protein-RNA networks and identify properties of RNA scaffolds within biological condensates. They find that CGG repeats in the 3′ UTR of FMR1 attract several proteins, including the splicing factor TRA2A that co-aggregates in fragile X-associated tremor/ataxia syndrome (FXTAS).
Introduction
Proteins and RNAs coalesce in large phase-separated condensates that are implicated in several cellular processes (Jiang et al., 2015, Woodruff et al., 2017).
Among the most studied condensates are ribonucleoprotein (RNP) granules that assemble in liquid-like cellular compartments composed of RNA-binding proteins (RBPs) (Hyman et al., 2014, Maharana et al., 2018) that are in dynamic exchange with the surrounding environment (Bolognesi et al., 2016). RNP granules, such as processing bodies and stress granules (SGs), are evolutionarily conserved from yeast to human (Brangwynne et al., 2009, Jain et al., 2016, Riback et al., 2017) and contain constitutive protein components, such as G3BP1 (yeast: Nxt3), TIA1 (Pub1), and TIAR (Ngr1) (Buchan et al., 2008). Several granule-forming RBPs are prone to form amyloid aggregates upon amino acid mutations (Hyman et al., 2014, Kato et al., 2012) that induce a transition from a liquid droplet to a solid phase (Qamar et al., 2018). This observation has led to the proposal that a liquid-to-solid phase transition is a mechanism of cellular toxicity (Patel et al., 2015) in diseases, such as amyotrophic lateral sclerosis (ALS) (Murakami et al., 2015) and myotonic dystrophy (Pettersson et al., 2015).
All the components of molecular complexes need to be physically close to each other to perform their functions. One way to achieve this, while keeping selectivity in a crowded cell, is to use platform or scaffold molecules that piece together components of a complex or a pathway. Indeed, RBPs are known to act as scaffolding elements promoting RNP assembly through protein-protein interactions (PPIs) (Banani et al., 2017); yet, protein-RNA interactions (PRIs) also play a role in the formation of condensates. Recent work based on G3BP1 pull-down indicates that 10% of the human transcripts can assemble into SGs (Khong et al., 2017). If distinct RNA species are present in the condensates, a fraction of them could be involved in mediating RBP recruitment. In this regard, we previously observed that different RNAs act as scaffolds for RNP complexes (Ribeiro et al., 2018), which indicates that specific transcripts might promote the formation of RNP condensates.
Combining PPI and PRI networks revealed by enhanced cross-linking and immunoprecipitation (eCLIP) (Van Nostrand et al., 2016) and mass spectrometric analysis of SGs (Jain et al., 2016), we identified a class of transcripts that bind to a large number of proteins and, therefore, qualify as potential scaffolding elements. In agreement with recent literature reports, we found that UTRs have a particularly strong potential to bind proteins in RNP granules, especially when they contain specific homo-nucleotide repeats (Saha and Hyman, 2017). In support of this observation, several diseases, including myotonic dystrophy (MD) and a number of ataxias (spinocerebellar ataxia [SCA]), have been reported to be linked to expanded trinucleotide repeats that trigger the formation of intranuclear condensates in which proteins are sequestered and functionally impaired. Specifically, expanded RNA repeats lead to RNA-mediated condensate formation in DM1 (Mooers et al., 2005), SCA8 (Mutsuddi et al., 2004), and SCA10 (White et al., 2010).
By understanding the characteristics of RNAs involved in RNP assembly, we aim to unveil the molecular details of specific human diseases. Indeed, the appearance of RNP condensates, often called inclusions or foci, is not only linked to ALS, Huntington’s disease, and MD but also other diseases, such as fragile X-associated tremor/ataxia syndrome (FXTAS) (Tassone et al., 2004, Sellier et al., 2017). The onset and development of FXTAS is currently explained by two main mechanisms (Botta-Orfila et al., 2016): (1) RNA-mediated recruitment of proteins attracted by CGG trinucleotide repeats in the 5′ UTR of fragile X mental retardation protein (FMR1) RNA and (2) aggregation of repeat-associated non-AUG (RAN) polyglycine peptides translated from the FMR1 5′ UTR (FMRpolyG) (Todd et al., 2013). Previous work indicates that FMR1 inclusions contain specific proteins, such as HNRNP A2/B1, MBNL1, LMNA, and INA (Iwahashi et al., 2006). Also, FMRpolyG peptides (Sellier et al., 2017) have been found in the inclusions, together with CUGBP1, KHDRBS1, and DGCR8 that are involved in splicing regulation and mRNA transport regulation of microRNA regulation (Sellier et al., 2010, Sellier et al., 2013). Although KHDRBS1 does not bind physically (Sellier et al., 2010), its protein partner DGCR8 interacts with CGG repeats (Sellier et al., 2013), indicating that sequestration is a process led by a pool of proteins that progressively attract other networks.
Notably, CGG repeats contained in the FMR1 5′ UTR are of different lengths (the most common allele in Europe being of 30 repeats). At over 200 repeats, methylation and silencing of the FMR1 gene block FMRP protein expression (Todd et al., 2013). The premutation range (55–200 CGG repeats) is instead accompanied by appearance of foci that are the typical hallmark of FXTAS (Todd et al., 2013). These foci are highly dynamic and behave as RNP condensates that phase separate in the nucleus forming inclusions (Tassone et al., 2004). Although long lived, they rapidly dissolve upon tautomycin treatment, which indicates liquid-like behavior (Strack et al., 2013).
The lability of FMR1 inclusions, which impedes their biochemical characterization (Mitchell et al., 2013, Marchese et al., 2016), complicates the identification of RBPs involved in FXTAS. As shown in previous studies of RNP networks (Cirillo et al., 2017, Marchese et al., 2017), computational methods can be exploited to identify key partners of RNA molecules. New contributions from other research areas are needed, especially because FXTAS pathological substrate is still under debate and there is still insufficient knowledge of targets for therapeutic intervention (Todd et al., 2013, Sellier et al., 2017). Here, we propose an integrative approach to identify new markers based on the properties of PRI networks and characteristics of scaffolding RNAs.
Results
In this work, we exploited a high-throughput computational approach to investigate the physico-chemical properties of scaffolding RNAs (Figure 1A). We focused on the experimental characterization of FMR1 that we predict to bind a large number of RBPs. Among the FMR1 partners that we identified, we selected the splicing regulator TRA2A and studied the biological consequences of its recruitment in RNP condensates. We used murine and post-mortem human tissues to assess TRA2A involvement in FXTAS.
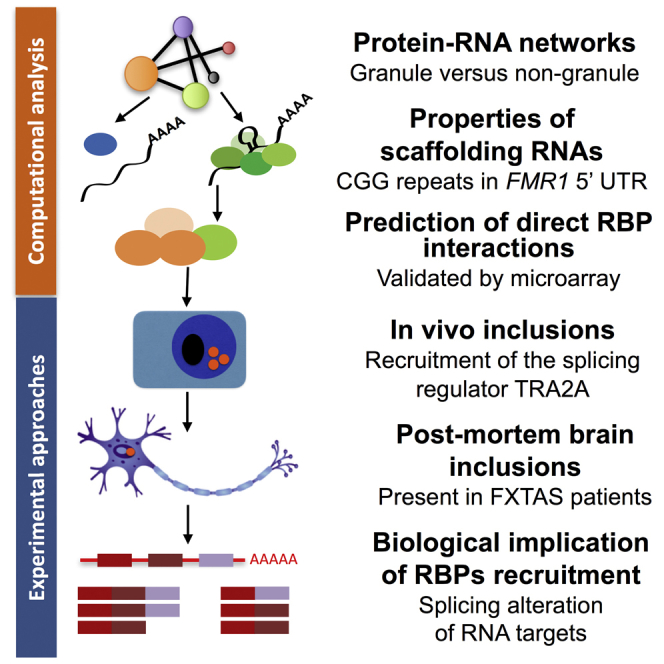
Figure 1.

RNA as a Key Element in RNP Condensates
(A) We explored the differences between granule and non-granule RBPs by using an interaction network approach. We first studied the physico-chemical properties of scaffolding RNAs and prioritized FMR1 5′ UTR for experimental characterization. We retrieved known RBPs and identified FMR1 partners involved in FXTAS, including splicing factors. We evaluated the biological consequences of RBP recruitment in cellular context and the presence of ribonucleoprotein (RNP) complexes in FXTAS brain inclusions.
(B) Statistical differences between granule and non-granule elements (proteins or RNA) in protein-protein interaction (PPI) and protein-RNA interaction (PRI) networks. Only when analyzing RNA interactions, granule and non-granule networks show different topologies (Figures S1C and S1D).
(C) Independently of RNA length, granule RBPs contact more transcripts that non-granule RBPs (“total” indicates all RBPs regardless of their granule or non-granule definition; ∗ indicates p value < 0.01; Kolmogorov-Smirnov [KS] test).
(D) Transcripts interact more frequently with granule than non-granule RBPs. The fractions of granule and non-granule RBP contacts, monitored at different lengths, show consistent enrichments (p value < 0.01; KS test). Highly contacted transcripts are enriched in small nuclear RNAs (snRNAs) and small nucleolar RNAs (snoRNAs) (p value < 2.2e−16, Wilcoxon rank-sum test). Already described scaffolding RNAs such as NEAT1 are also identified.
Protein-Protein Networks Do Not Discriminate Granule and Non-Granule RBPs
We first studied if RBPs phase-separating in RNP condensates interact with specific sets of proteins and RNAs. To discriminate proteins that are in RNP condensates (granule RBPs) from other RBPs (non-granule RBPs), we relied on recent proteomics data on human and yeast SGs (STAR Methods; Table S1A) as well as computational methods. The PRI datasets were identified through eCLIP (human) (Van Nostrand et al., 2016) and microarray (yeast) (Mittal et al., 2011) studies (Table S1B).
We analyzed if granule and non-granule RBPs show different interaction network properties. To this aim, we used available PPI datasets (STAR Methods) (Huttlin et al., 2015, Mittal et al., 2011). We based the topological analysis on three centrality measures describing the importance of a node (protein) within the network. For each protein, we computed the degree (number of protein interactions), betweenness (number of paths between protein pairs), and closeness centrality (how close one protein is to other proteins in the network). We found that granule and non-granule RBP networks display very similar topology both in yeast and human datasets (Figure 1B; Figures S1A and S1B).
Protein-RNA Networks Robustly Discriminate Granule and Non-Granule RBPs
In both yeast and human, we found that PRIs significantly increase the centrality measures of the granule network (Figure 1B; Figures S1C and S1D). Importantly, human and yeast granule RBPs interact with more transcripts than other RBPs (Figure 1C; Figure S2; Tables S1C–S1F; yeast p value = 0.02, human p value = 0.003, Wilcoxon rank-sum test; STAR Methods). Such a difference holds even when looking independently at either coding or non-coding RNAs (Figure S2; coding p value = 0.003, non-coding p value = 0.01, Wilcoxon rank-sum test) and upon normalization by transcript length (yeast p value = 0.02; human p value = 0.002, Wilcoxon rank-sum test).
Granule RBPs Share RNA Networks
In both yeast and human proteomes, we found that granule-forming RBPs share a larger number of transcripts (Figure S3A; Tables S1G–S1J; yeast p value < 2.2e−16, K562 p value < 2.2e−16, KS test). Independent of their length, RNAs contacting granule RBPs preferentially interact with other granule RBPs (Figure 1D, p value < 2.2e−16, Wilcoxon rank-sum test). In agreement with this finding, RNAs interacting exclusively with granule RBPs have a higher number of protein contacts than RNAs associating only with non-granule RBPs (Figure S3B, p value = 0.04, Wilcoxon rank-sum test). This observation is consistent with a picture in which RNAs share a high number of RBP interactions to promote recruitment into RNP granules. Using a high confidence threshold to select RBP partners (number of reads normalized by expression levels in the third quartile of the statistical distribution) (Armaos et al., 2017), we found that our list of RNAs overlaps with a recently published atlas of transcripts enriched in SGs (area under the receiver operating characteristic [ROC] curve [AUC] of 0.89; sensibility of 81.3% and specificity of 85.2%; Figure S3C; Table S1K) (Khong et al., 2017).
Non-Coding RNAs Are Contacted by Granule RBPs
Among the most contacted RNAs, we found an enrichment of small nuclear and nucleolar RNAs that are known to be associated with paraspeckles and Cajal bodies formation (Figure 1D; Table S2A; p value < 2.2e−16, Wilcoxon rank-sum test). We also identified a few highly contacted long non-coding RNAs, such as NEAT1, that interacts with all the proteins present in our dataset (Figure 1D). In agreement with this finding, NEAT1 has been described as an architectural RNA (West et al., 2016) implicated in scaffolding RBPs (Maharana et al., 2018) for paraspeckle nucleation. We hypothesize that other highly contacted long non-coding RNAs may have similar functions within cytoplasmic RNP granules. For instance, NORAD, a recently described long non-coding RNA involved in genomic stability, interacts with the large majority of proteins in our dataset (Lee et al., 2016). NORAD has repetitive sequence regions, is activated upon stress, has ability to recruit proteins (Tichon et al., 2016), and aggregates in SGs (Khong et al., 2017).
Characteristic Features of Candidate Scaffolding RNAs
We next studied which properties support the scaffolding activity of RNAs within granules. In this analysis, we define as granule transcripts those contacted by a larger number of granule-forming RBPs than non-granule forming RBPs (vice versa for non-granule transcripts; STAR Methods; Table S2B and S2C), we found that RNAs enriched in granule RBP contacts are more expressed (Figure 2A; p value = 5e−11, Kolmogorov-Smirnov, KS test), structured in UTRs (Figures 2B, and 2C; parallel analysis of RNA structure [PARS] data; p values = 0.005 and 0.05; we note that the signal is enriched at the 3′ UTRs with p value < 0.001; the 5′ UTRs is associated with a p value of 0.02; KS test; STAR Methods), and have longer UTRs (Figure 2D; 5′UTR is shown; p value = 0.005, KS test; 3′ UTR is reported in Figure S3D). This result, also valid in yeast (Figures S3E and S3F; Tables S2B and S2C), is consistent with previous observations that length (Zhang et al., 2015), structure (Reineke et al., 2015), and abundance (Jain and Vale, 2017) contribute to RNA assembly into RNP granules (Khong et al., 2017).
Figure 2.

Properties of Scaffolding RNAs
(A–D) Properties of RNAs contacted by granule-proteins. Granule transcripts are more abundant (A, p value = 4.65e-11, KS test), and structured (B, p value = 0.005; C, p value = 0.04; KS test) with longer UTRs (D, p value 5′UTR = 0.005, KS test) than non-granule RNAs.
(E) Occurrence of CCG, UGC, CGC, GGU, and CGG repeats discriminate the 5′ UTRs of granule and non-granule transcripts (the area under the ROC curve, AUC, is used to separate the two groups).
(F) Increasing the length of CGG repeats results in stronger secondary structural content (the CROSS algorithm [Delli Ponti et al., 2017] is used to measure the amount of double-stranded RNA).
Triplets prone to assemble into hairpin-like structures (Krzyzosiak et al., 2012), including CCG, UGC, CGC, GGU, and CGG, discriminate granule and non-granule transcripts in the 5′ UTRs (AUCs, >0.60; Figure 2E). In agreement with these findings, predictions of RNA structure performed with the Computational Recognition of Secondary Structure (CROSS) algorithm (Delli Ponti et al., 2017) indicate that the structural content (presence of double-stranded regions) is enriched in granule-associated transcripts (Figure S4A) and increase proportionally to CGG repeat length (Figure 2F), which is in line with UV-monitored structure melting experiments (Krzyzosiak et al., 2012).
In Silico Predictions Indicate a Large Number of Partners for FMR1 Scaffolding RNA
To further investigate the scaffolding ability of homo-nucleotide expansions, we selected the FMR1 transcript that contains CGG repetitions. Using catRAPID omics (STAR Methods) (Agostini et al., 2013), we computed interactions between the 5′ FMR1 UTR (containing 79 CGG repeats) and a library of nucleic-acid binding proteins (3,340 DNA-binding, RNA-binding, and structurally disordered proteins) (Livi et al., 2015). Previously identified CGG-binding proteins (Sellier et al., 2010), such as HNRNP A1, A2/B1, A3, C, D, and M, and SRSF 1, 4, 5, 6, 7, and 10, as well as MBNL1 and KHDRBS3, were predicted to interact strongly (discriminative power, >0.90) and specifically (interaction strength, >0.90; Figure 3A; Table S3; empirical p values < 0.01). Similar binding propensities were also found for a set of 92 RBPs reported to assemble in SGs (Jain et al., 2016) (Table S3). In addition, our calculations identify a group of 37 RBPs that are predicted to form granules by the catGRANULE algorithm (Bolognesi et al., 2016) (STAR Methods; Figures S4B and S4C). Among the RBPs prone to associate with FMR1, we found a class of splicing factors, including TRA2A (interaction score, 0.99; specificity, 1.00; Table S3).
Figure 3.

Protein Interactions of CGG Repeats
(A) Using catRAPID omics (Agostini et al., 2013), we computed protein interactions with the first FMR1 exon (containing 79 CGG repeats). Previously identified partners, such as HNRNP A1, A2/B1, A3, C, D, and M; SRSF 1, 4, 5, 6, 7, and 10; as well as MML1 and KHDRBS3 show strong binding propensities and specificities (blue dots) (Sellier et al., 2010). A previously unknown interactor, TRA2A (red dot), shows comparable binding propensities.
(B) We validated RBP interactions with FMR1 exon (“pre”containing 79 CGG repeats) through protein arrays (Cirillo et al., 2017, Marchese et al., 2017). We obtained high reproducibility between replicas (Pearson’s correlations > 0.75 in log scale) and identified strong-affinity interactions (signal to background ratio > 2.5; red dots). The same procedure was applied to the FMR1 exon containing 21 CGG repeats (Table S4).
(C) We measured catRAPID omics (Agostini et al., 2013) performances on protein array data selecting an equal number of strong- (highest signal to background ratios) and poor-affinity (lowest signal to background ratios) candidates.
(D) Out of 27 candidates binding to both 79 and 21 CGG repeats (signal to background ratio > 2.5), 15 are highly prone to form granules (blue bars) (Bolognesi et al., 2016), and the splicing regulator TRA2A (red bar) shows the highest propensity. The black bars indicate non-specific partners interacting also with SNCA 3′ UTR (Cirillo et al., 2017, Marchese et al., 2017) or showing poor RNA-binding propensity (Livi et al., 2015).
High-Throughput Validation of CGG Partners and Identification of TRA2A Interaction
We used protein arrays (Cirillo et al., 2017, Marchese et al., 2017) to perform a large in vitro screening of RBP interactions with the first FMR1 exon (STAR Methods). We probed both expanded (79 CGG) and normal (21 CGG) range repeats on independent replicas, obtaining highly reproducible results (Pearson’s correlations, >0.75 in log scale; Figure 3B; Table S4). We used the 3′ UTR of a similar length transcript, SNCA (575 nucleotides), to control for the specificity of RBP interactions (Marchese et al., 2017).
Using fluorescence intensities (signal to background ratio) to measure binding affinities, we found that previously identified partners SRSF 1, 5, and 6 rank in the top 1% of all interactions (out of 8,900 proteins), followed by KHDRBS3 (2%) and MBNL1 (5%). We observed strong intensities (signal to background ratio, >1.5 corresponding to top 1% of all interactions) for 85 RBPs interacting with expanded repeats (60 RBPs for normal-range repeats) and using more stringent cutoff (signal to background ratio, >2.5 for top 1% of all interactions) we identified 27 previously unreported interactions (binding to both expanded and normal range repeats).
The list of 85 RBPs showed enrichment in Gene Ontology (GO) terms related to splicing activity (FDR, <10−7), as reported by cleverGO (Klus et al., 2015) as well as GeneMANIA server (https://genemania.org/) and includes SRSF 1, 5, 6, and 10; PCBP 1 and 2; HNRNP A0 and F; NOVA1;PPIG; and TRA2A. catRAPID omics predictions are in agreement with protein array experiments: from low- to high-affinity interactions, catRAPID performances increase, reaching AUCs of 0.80 (Figure 3C), indicating a strong predictive power. Notably, although KHDRBS1 (not present in the protein array) is predicted to have poor binding propensity to CGG repeats, two of its RBP partners, CIRBP and PTBP2, rank in the top 1% of all fluorescence intensities, as predicted by catRAPID (Cirillo et al., 2013), and DGCR8, which interacts with KHDRBS1 through DROSHA (Sellier et al., 2013), is found to interact (top 7% of all fluorescence intensities).
Out of 27 high-confidence candidates, 24 were predicted by catGRANULE (Bolognesi et al., 2016) to form granules, and among them, the splicing regulator TRA2A showed the highest score (granule propensity, 2.15; Figure 3D; Figure S4D; Table S3). In agreement with our predictions, eCLIP experiments indicate that the FMR1 transcript ranks in the top 25% of strongest interactions with TRA2A (Van Nostrand et al., 2016).
TRA2A Recruitment in FMR1 Inclusions Is Driven by CGG Hairpins In Vivo
As splicing defects have been reported to occur in FXTAS disease (Botta-Orfila et al., 2016, Sellier et al., 2010), we decided to further investigate the recruitment of the splicing regulator TRA2A. B lymphocytes are often used for initial investigations because of their easy accessibility from blood samples from patients. Expansions of CGG from 55 to 200 CGG repeats result in mRNA levels in B lymphocytes that can exceed by 2–10 fold (Tassone et al., 2007). Therefore, B lymphocytes are considered a good model to recapitulate some of the events occurring due to the permutation (i.e., higher expression levels of FMR1), and to explore new biomarkers. We measured RNA and protein levels of TRA2A in B lymphocytes of a normal individual (41 CGG repeats; Coriell repository number NA20244A) and a FXTAS premutation carrier (90 CGG repeats; Coriell repository number GM06906B). RNA and protein levels of TRA2A were found significantly increased 2.9 and 1.4 times in the FXTAS premutation carrier compared with a normal individual, which indicates that the TRA2A is significantly altered in disease (Figure S5).
Yet, nuclear inclusions do not form in B lymphocytes and we used the COS-7 cellular model to study FMR1 inclusions (Sellier et al., 2010). We observed that transfection of a plasmid containing CGG expansions (triplet repeated 60 times) induces significant increases in RNA and protein levels of TRA2A after 48 hours (Figure S5) (Sellier et al., 2010). By means of RNA fluorescence in situ hybridization (FISH) coupled to immunofluorescence (STAR Methods), we found that CGG expansions and endogenous TRA2A significantly co-localize in nuclear inclusions (45 out of 50 screened cells showed unambiguous match). By contrast, TRA2A shows a diffuse nuclear pattern in cells that do not overexpress CGG repeats (Figure 4A).
Figure 4.

Endogenous TRA2A Is Recruited in Nuclear RNA Inclusions upon CGG Overexpression
This specific recruitment is validated by experiments with TRA2A overexpression and TRA2A knockdown.
(A) COS-7 cells were transfected with either CGG (60×) or the empty vector as control. After 24 hr of transfection, cells were immunostained with primary antiTRA2A antibody and secondary 488 and hybridized with a Cy3-GGC (8×) probe for RNA FISH. The graph represents the 488 Cy3 intensity co-localization in the section from the white line.
(B) After 24 hr of transfection, cells were immunostained with antiTRA2A antibody and hybridized with a Cy3-GGC (8×) probe for RNA FISH; relative TRA2A protein levels in COS-7 cells were treated as in (B) (p < 0.028, unpaired t test).
(C) COS-7 cells were transfected with empty vector or CGG (60×) and GFP-TRA2A. After 48h, cells were hybridized with Cy3-GGC (8×) probe for RNA FISH. The graph represents the GFP/Cy3 intensities co-localization in the section from the white line.
Upon knockdown of TRA2A using small interfering RNA (siRNA) (STAR Methods), we observed that the nuclear aggregates still form (Figures 4B), whereas overexpression of TRA2A attached to GFP (GFP-TRA2A) results in strong recruitment in CGG inclusions (Figure 4C; the control GFP plasmid and GFP-TRA2A in the absence of CGG repeats does not give a granular pattern).
To further characterize the recruitment of TRA2A in CGG repeats, we treated COS-7 cells with two different chemicals. By incubating COS-7 cells with 9-hydroxy-5,11-dimethyl-2-(2-(piperidin-1-yl)ethyl)-6H-pyrido[4,3-b]carbazol-2-ium (also named 1a) that binds to CGG repeats preventing interactions with RBPs (Disney et al., 2012), TRA2A recruitment was blocked (Figure 5A). Using TmPyP4 to specifically unfold CGG repeats (Morris et al., 2012), we found that the aggregates are disrupted and TRA2A remains diffuse (Figure 5B).
Figure 5.

Disrupting CGG Hairpins and Dissolving RNA Inclusions Impair TRA2A Sequestration
(A) COS-7 cells were co-transfected with empty vector or CGG (60×), and after 24 hr of transfection, cells were treated with 1a to block protein binding.
(B) COS-7 cells were treated similarly as in (A) but with the TmPyP4 molecule instead of 1a to disrupt CGG structure. In both cases, cells were immunostained with primary anti TRA2A antibody and hybridized with the Cy3-GGC (8×) probe for RNA FISH.
Our experiments show that the aggregation of TRA2A is caused by CGG repeats and tightly depends on the hairpin structure.
TRA2A Recruitment in RNA Inclusions Is Independent of its Partner TRA2B
Using RNA FISH coupled to immunofluorescence, we found that TRA2B, which interacts with TRA2A (Huttlin et al., 2015) and binds to CGG repeats (Sellier et al., 2010), aggregates in COS-7 cells transfected with CGG repeats (×60; Figure 6A). Notably, endogenous TRA2B is recruited by CGG inclusions upon TRA2A knock down (Figure 6B, upper row; Figure 6C; the result is also observed when TRA2B is overexpressed; Figures S6A and 6B). Similarly, endogenous TRA2A co-localizes with CGG repeats upon TRA2B knock down (Figure 6B, lower row; see also Figures S6C–S6E). Thus, upon knock down of one of the two proteins, the other one is still recruited by the overexpressed CGG repeats (Figure 6B; Figures S6F and S6G). By contrast, in absence of CGG overexpression, neither TRA2A nor TRA2B localize within the inclusions (Figures S6A–S6E).
Figure 6.

Endogenous TRA2B Is Recruited in CGG Inclusions but TRA2A Recruitment Is Independent from TRA2B
(A) COS-7 cells were transfected with CGG (60×). After 24 hr of transfection, cells were immunostained with antiTRA2B antibody and hybridized with a Cy3-GGC (8×) probe for RNA FISH.
(B) COS-7 cells were transfected with CGG (60×) and siTRA2A or siTRA2B. After 24 hr of transfection, cells were immunostained with antiTRA2 antibodies and hybridized with a Cy3-GGC probe for RNA FISH.
(C) TRA2A protein levels in COS-7 cells were treated as described in (B).
Alterations in RNA Splicing by FMR1 Inclusions Correlate with Alterations in RNA Splicing by TRA2A Knockdown
When assessing TRA2A levels in response to CGG overexpression in COS-7 cells, we found that around 20%–25% of TRA2A is recruited in condensates that are positive for CGG FISH signal (STAR Methods), which is fully compatible with SOD1 accumulation in SGs (Mateju et al., 2017). To study the functional implications of TRA2A recruitment in FMR1 inclusions, we analyzed changes in RNA splicing (STAR Methods).
Splicing alterations due to TRA2A sequestration were investigated through microarrays and RNA sequencing (RNA-seq) experiments (both in triplicate experiments) to identify events (1) occurring upon CGG aggregate formation (CGG+ TRA2A+; 74 instances) and (2) altered when TRA2A is knocked down (CGG+ TRA2A-, 82 instances). With respect to events occurring in the absence of CGG aggregates (CGG− TRA2A+, i.e., physiological conditions), 59 exons are spliced in CGG+ TRA2A+ and not in CGG+ TRA2A− (CGG+ TRA2A+∖CGG+ TRA2A−) and thus depend on TRA2A sequestration (39 skipped and 20 included exons; q-value, <0.10; Figure 7A; Table S5). Notably, 67 events occur exclusively in CGG+ TRA2A− and can be ascribed to perturbations in the splicing factor network (Tan and Fraser, 2017), while 15 (i.e., 3+12) occur in both CGG+ TRA2A+ and CGG+ TRA2A− (Figure 7B) and are, therefore, TRA2A independent (Figure 7B).
Figure 7.

TRA2A Recruitment in FMR1 Inclusions Affects Splicing
(A) With respect to events occurring in the absence of CGG aggregates (CGG− TRA2A+), 59 exons are spliced in CGG+ TRA2A+ and not in CGG+ TRA2A− (i.e., CGG+ TRA2A+∖CGG+ TRA2A−; red points). We investigated cases caused by TRA2A depletion in absence of CGG aggregates (CGG− TRA2A−) and identified 11 events that are present in CGG+ TRA2A+ and not in CGG+ TRA2A− (i.e.: CGG− TRA2A− ∩ CGG+ TRA2A+∖CGG+ TRA2A−; orange points; Table S5; STAR Methods).
(B) Muscle proteins, including PIP5K1A, TPM1, and autophagy-related SQSTM1, are subjected to exon skipping upon TRA2A recruitments in RNP condensates (Table S5; Figure 7B). Events occurring when TRA2A is depleted are linked to neuro-pathogenesis (GADD45B and CCT7) and skeletal development (KIAA1217 and TM4SF19).
(C) TRA2A immunohistochemistry in wild-type (WT) and premutated mouse model (counterstaining was done with hematoxylin; the arrow points to the inclusion).
(D) TRA2A immunohistochemistry in human hippocampus from control and FXTAS (counterstained with hematoxylin; the arrow points to the inclusion).
(E) Double immunofluorescence of TRA2A as well as FMRpolyG peptides in human FXTAS (STAR Methods).
To better understand TRA2A-dependent effects, we studied events caused by TRA2A depletion in the absence of CGG aggregates (CGG− TRA2A−; Figure 7A): 11 out of 59 CGG+ TRA2A+ cases are present in CGG− TRA2A− but not in CGG+ TRA2A− (CGG− TRA2A− ∩ CGG+ TRA2A+∖CGG+ TRA2A−), which is statistically highly significant. Indeed, by shuffling the splicing events reported in one of the two experiments (i.e., randomizing the association between exons and q-values), we found that the intersection CGG− TRA2A− ∩ CGG+ TRA2A+∖CGG+ TRA2A−, never contains 11 events (we found 1 event in 288 out 10,000 randomizations and 2 events in 3 out 10,000 randomizations, but never > 3 events; p value < 10−4; Figure 7B). Notably, 17 cases occur in CGG+ TRA2A− and CGG− TRA2A− but not in CGG+ TRA2A+, which is expected because splicing factors work together on common targets (Tan and Fraser, 2017).
Using the cleverGO algorithm (Klus et al., 2015), we found that the largest GO cluster of affected genes includes RBPs (18 genes; Table S5; “RNA-binding”; fold enrichment of 24; p value < 10−8; calculated with Bonferroni correction; examples: HNRNPL, CIRBP, and DDX24) and, more specifically, spliceosome components (“mRNA splicing via spliceosome”; fold enrichment of 5; p value < 10−3, examples: HNRNP A2/B1, and SRSF 10) or genes related to alternative splicing activity (“regulation of splicing”; fold enrichment of 6; p value < 10−3, examples: RBM5 and THOC1).
Intriguingly, genes associated with mental retardation, such as UBE2A (Budny et al., 2010), ACTB (Procaccio et al., 2006), and ACTG1 (Rivière et al., 2012), have splicing patterns affected by TRA2A sequestration. Similarly, muscle-related proteins, including PIP5K1A (Chen et al., 2018) and TPM1 (Erdmann et al., 2003), and genes linked to intellectual disabilities, such as DOCK3 (de Silva et al., 2003), and craniofacial development, such as WWP2 (Zou et al., 2011), are subjected to exon skipping upon TRA2A recruitments in RNP condensates (Table S5; Figure 7B). Out of 59 splicing events occurring in CGG+ TRA2A+ and CGG+ TRA2A− conditions, 23 (including ACTG1, TMP1, and WWP2) involve transcripts that physically bind to FMRP protein, as also detected in CLIP experiments (available from http://starbase.sysu.edu.cn/), which unveils an important link (significance: p value < 10−4; Fisher’s exact test) to fragile X syndrome (Maurin et al., 2014).
In the 11 CGG+ TRA2A+ cases present in CGG− TRA2A− but not in CGG+ TRA2A− (Figure 7B) there is GADD45B linked to synaptic plasticity (Ma et al., 2009), as well as g KIAA1217 (Semba et al., 2006) and TM4SF19 (de la Rica et al., 2013) associated with skeletal development pathways. We also found the molecular chaperone CCT that is known to restrict neuro-pathogenic protein aggregation via autophagy (Pavel et al., 2016).
TRA2A Is Present in Murine and Human FXTAS Brain Inclusions
We tested if TRA2A co-aggregates with FMR1 inclusions in two mouse models with repeats containing more than 90 CGG: (1) the 5′ UTR was expressed under the control of doxycycline (Hukema et al., 2015) and (2) the CGG repeat has been replaced by the human expanded repeat (Willemsen et al., 2003). Immunohistochemistry experiments with sections of paraffin-embedded neurons and astrocytes indicated that the TRA2A protein is present in the inclusions (Figure 7C; STAR Methods).
Importantly, RAN translation has been shown to occur in FMR1 5′ UTR, resulting in the production of FMRpolyG and FMRpolyA peptides (Todd et al., 2013). The main RAN translation product, FMRpolyG, co-localizes with ubiquitin in intranuclear inclusions (Sellier et al., 2017).
In agreement with our murine model, we found positive staining for TRA2A in nuclear inclusions from two FXTAS post-mortem human brain donors (Figure 7D), and remarkably, we observed co-localization with FMRpolyG (Figure 7E). We observe that FMRpolyG reaches its highest abundance in hippocampus co-aggregating in 20% total nuclei (Glineburg et al., 2018). In the same tissue, TRA2A co-localizes with inclusions in 2%–3% of total nuclei, thus indicating strong sequestration (Figure S7). Interestingly, TRA2A-positive cells aggregate in groups that are in close proximity, which provides precious information on the biochemical behavior of aggregates as well as the spreading nature of the disease across brain districts (Figure S7).
Thus, TRA2A sequestration by CGG repeats is not only observed in cell lines but also in FXTAS animal models and human post-mortem brain samples.
Discussion
Previous evidence indicates that proteins are the main cohesive elements within RNP granules (Banani et al., 2017). Yet, specific RNAs can act as structural scaffolds to assemble proteins in RNP condensates, as recently reported in the literature (Langdon et al., 2018), and RNA-RNA interactions play an important role in the formation of RNP assemblies (Van Treeck and Parker, 2018). Our analysis of PRI networks reveals that scaffolding RNAs have a large number of RBP contacts, increased length, and high structural content. In agreement with our computational analysis, two works published at the time of writing indicate that UTR length and structural content (Khong et al., 2017, Maharana et al., 2018) are important properties of RNAs aggregating in RNP condensates. Moreover, nucleotide repeats (Jain and Vale, 2017), changes in RNA levels (Tartaglia and Vendruscolo, 2009), and RNA binding abilities (Zhang et al., 2015) are known factors modulating phase transitions in the cell.
Our PRI networks were retrieved from eCLIP experiments (Van Nostrand et al., 2016) that have been performed in conditions different from those promoting the formation of physiological SGs. Similarly, PPI networks were taken from the BioPlex database that includes highly curated, multi-source experiments (Huttlin et al., 2015). Yet, our underlying hypothesis is that PPI and PRI are governed by physico-chemical forces that are in place regardless of the environmental conditions, and we assume that the ability of proteins and RNAs to assemble is impaired when the molecules are poorly expressed or chemically modified. Indeed, to control for the contribution of RNA abundance, we used it to normalize the number of CLIP reads in our calculations. Supporting our assumptions, the RNA list retrieved from the network analysis shows a very significant overlap with a recently published atlas of transcripts enriched in SGs (Khong et al., 2017). We note that our analysis would be more accurate if the protein and RNA interaction networks were known for the different biological condensates.
Combining computational approaches with large-scale in vitro experiments, we unveiled the scaffolding ability of FMR1 5′ UTR, recovering previously known partners relevant in FXTAS, such as SRSF1, 5, and 6; KHDRBS3; and MBNL1 (Sellier et al., 2010), and identifying additional interactions involved in alternative splicing, such as PCBP 1 and 2, HNRNP A0 and F, NOVA1, PPIG, and TRA2A. At the time of writing, TRA2A has been reported to be a component of ALS granules (Markmiller et al., 2018). Yet, TRA2A does not appear in TAU inclusions (Maziuk et al., 2018), which indicates that its sequestration occurs only in specific neurodegenerative diseases.
To prove the implication of TRA2A sequestration in FXTAS pathogenesis and overcome the technical limitation of our cellular model in which the non-AUG codon downstream the 5′ UTR of FMR1 is lacking, we tested TRA2A colocalization with FMRpolyG in patients’ brain samples. Our experiments showed that the two proteins colocalize, providing additional information on TRA2A involvement in FXTAS disease. This result indicates that CGG could interact with both TRA2A and FMRpolyG and supports our previous work indicating that interactions between proteins and cognate RNAs are frequent in aggregation-prone genes (Cirillo et al., 2013).
Through splicing microarrays and RNA-seq analysis, we found that TRA2A sequestration induces changes in the splicing of genes associated with mental retardation, including ACTB (Procaccio et al., 2006) and ACTG1 (Rivière et al., 2012), intellectual disabilities, such as DOCK3 (de Silva et al., 2003), and craniofacial development, such as WWP2 (Zou et al., 2011), which are relevant in the context of fragile X syndrome (Maurin et al., 2014). Thus, the identification of TRA2A opens the avenue for new therapeutic intervention to correct the splicing defects of deregulated transcripts or to restore the functional role by clustered regularly interspaced short palindromic repeat (CRISPR)-Cas technology.
In the future, it will be very important to analyze genome-wide data in different bio-specimens from patients to see the expression of differently spliced variants of TRA2A targets. Nevertheless, because FXTAS is a rare disease with low penetrance, the number of samples from patients is very limited. Therefore, more work should be done in this direction to promote biomarker discovery in patients and, ultimately, promote personalized treatment. Yet, our theoretical framework is also applicable to other diseases in which RNAs promote the formation of phase-separated condensates that could be used by the pathologist to identify the proteins that are specifically sequestered.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-TRA2A | Abcam | ab72625; RRID:AB_1524507 |
| Anti-TRA2B | Abcam | ab31353; RRID:AB_778565 |
| Anti-rabbit 647 | Abcam | ab150115; RRID:AB_2687948 |
| Anti-rabbit 488 | Abcam | ab11008; RRID:AB_297665 |
| Anti-ubiquitin | Dako | Z0458; RRID:AB_2315524 |
| Anti-rabbit Fab 488 | Molecular probes | A11070; RRID:AB_142134 |
| Anti-mouse cy3 | Jackson | 715-165-150; RRID:AB_2340813 |
| Anti-tubulin | Abcam | ab7291; RRID:AB_2241126 |
| Anti-mouse | Abcam | ab97046; RRID:AB_10680920 |
| FMRpolyG antibodies 8FM | Dr. Renate Hukema PMID: 26060190 | N/A |
| Anti-rabbit Protein G | Merk | 18-161; RRID:AB_2756347 |
| Anti TMPyP4 tosylate | Abcam | ab120793; RRID:AB_2756346 |
| Biological Samples | ||
| Post-mortem Human Tissue | Netherlands Brain Bank, Netherlands Institute for Neuroscience, Amsterdam | https://www.brainbank.nl/ (project nr. 1084) |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine | Invitrogen | 13778150 |
| DAB susbtrate | Dako | K3468 |
| Brightvision poly-HRP-linker | Immunologic | DPVO-HRP 55 |
| Label IT Cy5 | Mirus | MIR 3725 |
| DNEM – Dulbecco’s Modified Eagle Medium | Sigma Aldrich | D9785 |
| FBS – Fetal Bovine Serum | Sigma Aldrich | F9665 |
| MEM - Minimun Essential Medium Eagle | Sigma Aldrich | M2279 |
| Trypan blue solution | Sigma Aldrich | T8154 |
| Sudan black B | Abcam | Ab146284 |
| TBS – Tris Buffered Saline | Sigma Aldrich | T5912 |
| Tween 20 | Sigma Aldrich | P1379 |
| PBS – Phosphate buffered saline | Sigma Aldrich | 79378 |
| 1a molecule | Dr. Mathew D. Disney, Scripps Research Institute, CA 92037, USA | N/A |
| BSA – Bovine Serum Albumins | Sigma Aldrich | A2058 |
| Triton X-100 | Sigma Aldrich | 11332481001 |
| DAPI histology mounting médium | Sigma Aldrich | F6057 |
| Critical Commercial Assays | ||
| RNA extraction kit | QIAGEN | 74106 |
| SuperScript III First Strand Synthesis SuperMix for qRT-PCR | Invitrogen | 11752250 |
| SyBr Green MasterMix | Invitrogen | 4367659 |
| MiniElute PCR Purification kit | QIAGEN | 28004 |
| MEGAScript T7, High yield transcription kit | Invitrogen | AM1334 |
| Cy5 label IT uArray Labeling kit | Mirus | MIR3700 |
| Agencourt RNAclean XP magnetic beads | Beckman Coulter | A63987 |
| TURBODNase 2U/uL | Invitrogen | AM2238 |
| Human Protein microarrays v5.2 | Life Technologies | PAH0525101 |
| Stellaris RNA FISH | Biosearch Technologies | N/A |
| Bradford Protein Assay | BioRad | 5000205 |
| Luminata Starter kit | Millipore | WBLUM0100 |
| Affimetrix Human Clariom D Array | Thermo Fisher | 902922 |
| TruSeq total RNA-rRNA depletion | Illumina | 20020596 |
| NuPAGE 4-12% Bis-Tris Protein Gel | Thermo Fisher | NP0321BOX |
| Deposited Data | ||
| Splicing arrays | This paper | GEO:GSE108007 |
| RNA-Seq | This paper | GEO:GSE121304 |
| Experimental Models: Cell Lines | ||
| Human lymphocytes (CGG(41X)) | Coriell repository | NA20244A |
| Human lymphocytes (CGG(90X)) | Coriell repository | GM06906B |
| COS-7 | ATCC | CRL-1651 |
| Experimental Models: Organisms/Strains | ||
| Mouse C57BL/6 |
Dr. Renate Hukema, PMID: 26060190 | N/A |
| PMID:12700164 | ||
| Oligonucleotides | ||
| siTRA2A | Ambion | AM16704 |
| siTRA2B | Ambion | S12749 |
| Recombinant DNA | ||
| FMR1 5′-UTR and control pCAGIG vector | Dr. Eulàlia Martí | N/A |
| PMID: 24418349 | ||
| GFP-TRA2A, GFP-TRA2B, GFP vectors |
Dr. Eulàlia Martí | N/A |
| PMID: 24418349 | ||
| Software and Algorithms | ||
| GenePix Pro 6.1 | Molecular Devices | http://mdc.custhelp.com/app/answers/detail/a_id/18691/∼/genepix%C2%AE-pro-6-microarray-acquisition-%26-analysis-software-download-page |
| Robust Multi-array Average (RMA) | PMID: 12925520 | http://rmaexpress.bmbolstad.com/ |
| LIMMA | PMID: 24485249 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Transcriptome Analysis Console Software | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/life-science/microarray-analysis/microarray-analysis-instruments-software-services/microarray-analysis-software/affymetrix-transcriptome-analysis-console-software.html |
| STAR_2.5.2 | PMID: 23104886 | https://github.com/alexdobin/STAR |
| EventPointer v1.0.0 | PMID: 27315794 | https://bioconductor.org/packages/release/bioc/html/EventPointer.html |
| DEXSeq v.24.2 package (R 3.4.0) | PMID: 22722343 | https://bioconductor.org/packages/release/bioc/html/DEXSeq.html |
| ImageJ | PMID: 22930834 | https://imagej.nih.gov/ij/download.html |
| igraph | http://igraph.org/ | http://igraph.org/r/ |
| R Studio 1.1.456 | Rstudio | https://www.rstudio.com/ |
| R version 3.5.1 | R | https://www.r-project.org/ |
| CROSS | PMID: 27899588 | http://service.tartaglialab.com/new_submission/cross |
| catRAPID omics | PMID: 23975767 | http://s.tartaglialab.com/page/catrapid_omics_group |
| catGRANULE | PMID: 27320918 | http://service.tartaglialab.com/new_submission/catGRANULE |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gian Gaetano Tartaglia (gian.tartaglia@crg.eu).
Experimental Model and Subject Details
Human lymphocytes
Human lymphocytes cells from Coriell repository (CGG(41X); Coriell repository number NA20244A and CGG(90X) Coriell repository number GM06906B) were grown in suspension in DMEM 10% fetal bovine serum (FBS) 1% Penicillin/Streptomycin, 2mM Glutamine at 37°C with a 5% CO2 atmosphere. Cell counting was performed with Neubauer chamber.
COS-7 cells
COS-7 cell lines were cultured in DMEM 10% FBS, 0,1% non-essential aminoacids, pyruvate and glutamine, at 37°C with a 5% CO2 atmosphere. Cells were counted with a Neubauer chamber.
Mouse model
We employed two established FXTAS mouse models: 1) the knock-in mouse in which the murine CGG repeat has been replaced by a human expanded repeat of 100-150 CGGs (Willemsen et al., 2003) and 2) an inducible model in which the 5′UTR containing 90 CGG repeats was expressed under the control of doxycycline (Hukema et al., 2015). Both male and female mice were used, since both genders form the characteristic inclusions. All experiments were conducted with the permission of the local institute of animal welfare (IVD) and the study was complying with ethical permission CCD license AVD101002015290. The knock-in mice were aged about 70 weeks. These mice were previously shown to contain ubiquitin positive intra-nuclear inclusions. For the doxycycline inducible mice, the dox treatment was of 12-28 week, which was sufficient to allow formation of inclusions.
Human post-mortem samples
We collected samples from one male (died at age 73) and one female (84 years old), both in the premutation range (100 CGG repeats). The brain samples were obtained from the Netherlands Brain Bank (NBB; project nr. 1084), Netherlands Institute for Neuroscience, Amsterdam (open access https://www.brainbank.nl/). All material has been collected from donors for or from whom a written informed consent for a brain autopsy and the use of the material and clinical information for research purposes had been obtained by the NBB.
Method Details
RNA IVT and Protein arrays
FMR1 5′-UTR expanded and control pCAGIG vectors, with 79 and 21 repeats, respectively, were generated by Dr. Marti’s group. The UTRs were subcloned in PBSK plasmid containing promoters suitable for in vitro transcription. The plasmid was digested in final volume reaction of 30ul with restriction enzymes and the digestion was ensured by loading 1ul in a 1% agarose gel. The reaction was purified with the MinElute PCR Purification Kit following manufacturer’s instructions. In vitro transcription was performed with the T7 Megascript T7, High Yield Transcription Kit, Invitrogen, Thermo Scientific according to standard procedure with the addition of 1% DMSO and 1% ribolock, overnight at 37°C. The synthetized RNA was treated with TURBODNase 2U/ul (Invitrogen) at 37°C for 15min. The RNA was purified with magnetic beads (Agencourt RNA Clean XP) eluting in 30ul of nuclease-free water.The integrity and specificity of the RNA was checked by means of RNA denaturing agarose gel and Bioanalyzer quality control.
The CGGxRNA was fluorescently labeled with Cy5 Label IT uArray Labeling Kit (Mirus) with slight modifications from standard protocol. Briefly, 5ug of RNA were mixed with 1:5 Label IT Cy5 reagent and incubated in a final volume of 25ul at 37°C for 70min. The reaction was stopped by adding 2.5ul of 10X Stop solution. Again the labeled RNA was purified with magnetic beads (Agencourt RNA Clean XP).
The RNA concentration and labeling density were measured with Nanodrop 1000 spectrophotometer (Thermo Scientific) and calculated as follows.
Only reactions with an RNA labeling density of 1 Cy5 dye per 700-900 nt were used.
Base:dye = (Abase∗ε dye)/(Adye∗ε base)
Abase = A260-(Adye∗CF260)
Constants:
ε dye = 250000
CF260 = 0.05
ε base = 8250
Labeled RNA integrity was verified with the Agilent 2100 Bioanalyzer.
50 pmoles of labeled RNA were hybridized in the protein arrays Human Protein Microarrays v5,2, Life Technologies.
The arrays were dried and immediately scanned at 635nm in Microarray Scanner G2505B (Agilent). GenePix Pro 6.1 software (Molecular Devices) was used to determine the signal at 635nm of each spotted protein location and therefore quantify the RNA-protein interaction. Specifically, the local background intensity (B635) was subtracted from the intensity (F635) at each of the duplicate spots for a given protein, to quantify. Data was filtered based on signal to background ratio for each of the duplicate feature to be greater than 2.5 fold and Z-Score ≥ 3 from the global mean signal from all the spotted proteins. Finally, the intersection of technical replicates was considered as the final value for quantification.
IF-RNA FISH in COS-7 cells
COS-7 cells were grown on 13mm coverslips until a 70% confluence. Cells were transfected with lipofectamine 2000 (Invitrogen, #13778150) according to manufacturer’s instructions and stained after 24 hours.
SiRNA treatments were performed with lipofectamine 2000 (Invitrogen, 11668019) using different siRNA sequences (siTRA2A, Ambion AM16704, siTRA2B, Ambion S12749, compared to water). Overexpression of proteins was achieved by transfection of GFP-TRA2A, GFP-TRA2B, compared with GFP-vector, all plasmids kindly given by Dr. Nicolas Charlet-Berguerand. For further treatments, cells were incubated with 25nM TMP4yP (Abcam ab120793) or 15uM of 1a molecule (this latter kindly given by Matthew D. Disney).
Prior to immunostaining, cells were fixed with 4% paraformaldehyde for 10 minutes and washed three times with PBS. Permeabilization was done with Triton X-100 0.1% for 5 minutes. Cells were washed 3 times with PBS and then blocked with BSA 1% solution for 20 minutes and washed again with PBS. Primary antibodies were used in a 1:50 dilution (antiTRA2A, Abcam, ab72625, antiTRA2B, Abcam, ab31353). Secondary antibodies (anti Rabbit 647, ab150115, anti-rabbit 488, ab11008) were used in a 1:200 dilution after three washes with PBS solution. RNA FISH assay was done after the immunostaining according to manufacturer’s protocol (Stellaris, Biosearch Technologies). The RNA FISH probe ((GGC)8X-Cy3, Sigma) was used at 125nM final concentration and cells were then incubated at 37°C overnight, as reported in Sellier et al., 2010. Finally, cells were mounted directly in Fluoroshield with DAPI histology mounting medium (Sigma, #F6057). All coverslips were examined using a fluorescence microscope (Leica) coupled to a DMI600 camera. Intensity graphs were generated with ImageJ software to assess levels of colocalization of signals from different fluorescence channels.
q-PCR
Human lymphocytes were washed and pelleted by centrifugation at 800rpm for 2 min. COS-7 cells were tripsinized and pelleted by centrifugation at 1200rpm for 2 minutes. RNA extraction from the different cultured cells was done according to manufacturer’s instructions (QIAGEN, #50974136). cDNA was generated by RT-PCR using SuperScript III First-Strand Synthesis SuperMix for qRT-PCR (Invitrogen, # 11752250) to quantify mRNAs. q-PCR was performed using Sybr Green master mix (Invitrogen, # 4367659) and analyzed by AB7900HT (Leica). In all experiments, GAPDH was used as internal control in all experiments.
Western Blot
Total proteins from human lymphocytes and COS-7 cells (one day post-transfection) were extracted. The level of protein was measured by the Bio-Rad Protein Assay according to manufacturer’s instructions. All lysates were resolved in a 4%–12% gel (NuPAGE, Invitrogen) according to the molecular size of the proteins and then transferred to a nitrocellulose membrane 0.2 μm. The membranes were blocked with 5% non-fat dry milk in TBS-Tween 1% then washed with PBS and incubated with anti TRA2A (1:1000, Abcam ab72625), anti TRA2B (1:500, Abcam ab31353) or anti Tubulin (1:5000, Abcam ab7291) overnight at 4°C. After primary antibodies treatment, membranes were washed three times with TBS-Tween 1% and then incubated with the secondary peroxidase antibody 1h with an anti-mouse (Abcam ab97046) or an anti-rabbit antibody (Protein G, Merk #18-161). Visualization of the signal was achieved by Luminata Starter kit (Millipore, WBLUM0100) according to manufacturer’s recommended instructions, and with Amersham Imager 600.
Immunohistochemistry and immunofluorescence from murine and human brain tissue
Tissues were fixed overnight in 4% paraformaldehyde and embedded in paraffin according to standard protocols. Sections (6μm) were deparaffinized followed by antigen retrieval using microwave treatment in 0.01M sodium citrate. Endogenous peroxidase activity was blocked and immunostaining was performed overnight at 4°C using TR2A (Abcam, ab72625) and 8FM 1:10 antibodies (Buijsen et al., 2014). In order to better visualize inclusions an extra antigen retrieval step was added, using proteinase K. Antigen-antibody complexes were visualized by incubation with DAB substrate (Dako, K3468) after incubation with Brightvision poly-HRP-linker (Immunologic, DPVO-HRP 55). Slides were counterstained with hematoxylin and mounted with Entellan.
For (double) immunofluorescence, slides were blocked for auto-fluorescence with Sudan Black in 70% ethanol. Primary antibodies include TR2A (Abcam, ab72625), 8FM 1:10 (Buijsen et al., 2014) and ubiquitin (Dako, Z0458). Secondary antibodies were antirabbit Fab 488 (Molecular Probes, A11070) and antimouse cy3 (Jackson, 715-165-150). Nuclei were visualized with Hoechst (Figure S7).
Quantification of TRA2A levels in presence of CGG aggregates
We quantified the amount of soluble protein in presence of CGG aggregates by microscopy in COS-7 cells, and found that around 20%–25% of TRA2A protein is present in the inclusions. Specifically, we assessed the intensity of the signal from endogenous TRA2A in cells from four different experiments upon overexpression of CGG (ImageJ), we measured the signal from TRA2A antibody for each cell from by selecting i) the Intensity Density (ID) from the Area from all the aggregates colocalizing with CGG signal; ii) the total ID for each cell; iii) the substraction of the total ID minus the sum of all the aggregates, then meaning the signal background; and finally iv) the fraction of protein that is in the aggregates in respect to the total of protein diffused.
Levels of functional protein are altered, in a magnitude that ranges from 20%–25%, which is fully compatible with previous reports in literature (Colomer et al., 1996, Grousl et al., 2018).
Quantification and Statistical Analysis
Data acquisition and composition
Granule-forming proteins were extracted from a previous publication (Jain et al., 2016) that reports the most exhaustive list of components for cytoplasmic RNP granules to date, comprising 205 yeast and 411 human granule-forming proteins (Figure S1A; Tables S1A–S1F).
Human PPIs were taken from the BioPlex database (Huttlin et al., 2015) that includes highly curated data produced by high-throughput affinity-purification mass spectrometry. BioPlex contains 56,554 interactions among 510,882 different proteins (Figure 1A; Figure S1C). Human PRIs were identified through eCLIP experiments (Van Nostrand et al., 2016). The dataset contains 1,103,800 interactions of 78 proteins in the K562 cell line (Figure 1B; Table S1B; Figure S1C). We processed the eCLIP data normalizing the number of reads by gene expression (Armaos et al., 2017). We considered interactions having values of number of reads by expression level higher than the first, second and third quartile of the distribution of the whole dataset (Figure S2; Tables S1C–S1E). After data normalization and filtering, the dataset includes 22,961 transcripts interacting with at least one protein (20,724 coding and 2,237 non-coding). We extracted the expression levels for K562 transcripts from the ENCODE project (Armaos et al., 2017).
For yeast analysis we used the dataset reported in a previous article (Mittal et al., 2011) that includes both protein-protein and PRIs. The protein-protein network is based on the integration of two mass spectrometry studies that comprise a total of 5,303 proteins and 401,821 interactions (Mittal et al., 2011) (Figure 1A). PRIs were extracted from the integration of four different studies on immunoprecipitation of RBPs followed by microarray analysis of the bound transcripts (Table S1F). The data includes a total of 24,932 interactions from 69 RPBs to 6,159 transcripts.
We considered non-granule forming those RBPs present in the protein-RNA dataset and not described as granule-forming in a previous article (Jain et al., 2016) (Figure S1; Tables S1A–S1E). There are 22,571 transcripts interacting with at least one granule-forming protein, 287 transcripts interacting only with granule-forming proteins and 390 with only non-granule forming proteins.
In the case of the granule and non-granule protein-protein networks comparison, we included RBP lists provided by (Brannan et al., 2016, Gerstberger et al., 2014) for yeast and human. These datasets comprise a total of 690 yeast RBPs and 1,795 human RBPs.
Network analysis
Protein-protein and protein-RNA networks consisted of a set of nodes (protein or RNAs) that are connected through edges (interactions). All network analyses were performed in the R environment (http://www.r-project.org) using the igraph package (http://igraph.org/)(Csárdi and Nepusz, 2006). We employed build-in functions to compute degree, betweenness and closeness measures of centrality. Networks were considered directionless and unweighted. Degree centrality is defined as the number of edges a node has. The other centrality measures were based on the shortest path length between nodes in the network (i.e., minimum number of edges between two certain nodes). In this sense, betweenness is defined as the number of shortest paths in the network that go through a certain node. Closeness centrality is the inverse of the average of the shortest path between a certain node and all the other nodes in the network. We compared the distribution of centrality values for granule and non-granule RBPs in the same global protein-protein or protein-RNA network (Figure 1A; Figures S1C and S1D).
We used the Jaccard index (Tables S1G–S1J) as a measure of the overlap of RNA targets between pairs of proteins. The Jaccard index of a specific couple of proteins a and b (Ja,b) was computed as:
were A is the set of RNA targets of the first protein of the pair, B is the set of RNA targets of the second protein of the pair, |A B| is the size of the intersection of A and B (i.e., number of RNA targets shared by the two proteins) and |A U B| is the size of the union of A and B (i.e., the total number of RNA targets of A and B minus the number of shared RNA targets).
RNA properties analysis
To study features of granule and non-granule transcripts, we compared RNAs with the same number of total protein contacts (Tables S2A–S2C). We used a cut-off of 10 RBP contacts in the human analysis and 5 contacts in the yeast case. The total number of contacts was chosen to have granule and non-granule sets of comparable size (Tables S2B and S2C), yet we note that the results are independent of a particular cut-off. Granule transcripts are contacted by a larger number of granule-forming RBPs than non-granule forming RBPs (viceversa for non-granule transcripts).
The UTR analysis is based on Ensembl annotation (Tables S2B and S2C).
RNA secondary structure
To profile the secondary structure of granule and non-granule transcripts (Figures 2B and 2C; Figure S3E; Tables S2B and S2C), we used PARS data (Kertesz et al., 2010, Wan et al., 2014). PARS distinguishes double- and single-stranded regions using the catalytic activity of two enzymes, RNase V1 (able to cut double-stranded nucleotides) and S1 (able to cut single-stranded nucleotides). Nucleotides with a PARS score higher than 0 indicate double-stranded conformation, while values lower than 0 are considered single-stranded (Kertesz et al., 2010, Wan et al., 2014). Undetermined nucleotides with a PARS score of 0 were discarded from our analysis. For each transcript, we counted the number of nucleotides with PARS score above zero divided by total length of the sequence.
We also predicted the secondary structure of granule and non-granule transcripts using CROSS (PARS human model; Figure 2F; Figures S4A). Input sequences were the same employed for the granule RNA properties analysis. For each sequence, structural content is defined as the percentages of nucleotides with a CROSS score bigger than 0.5 (double-stranded prone).
Statistical analysis
To assess whether granule RBPs exhibit different trends compared to non-granule RBPs, we used the Wilcoxon test (also called Mann-Whitney U test). Wilcoxon test is a non-parametric test used to compare the mean of two distributions without any given assumption about them. To compare properties of highly versus lowly contacted RNAs and difference in target overlap between granule and non-granule pairs, we used the Kolmogorov-Smirnov test (KS test). KS test is also a non-parametric test used to compare the distance between two cumulative distribution functions (CDFs).
Data and Software Availability
catRAPID omics analysis
catRAPID omics was used to compute the interaction propensity (Figures 3A and 3C) of CGG repeats with proteins (Agostini et al., 2013). catRAPID omics ranks predictions based on interactions score as well as presence of motifs and RBDs (Bellucci et al., 2011). For RNA sequences > 1000 nt, the uniform fragmentation procedure is applied to determine the binding regions of a protein. The software is available at http://s.tartaglialab.com/page/catrapid_omics_group.
catGRANULE analysis
Structural disorder, nucleic acid binding propensity and amino acid patterns such as arginine-glycine and phenylalanine-glycine are key features of proteins coalescing in granules (Bolognesi et al., 2016). These features were combined in a computational approach, catGRANULE, that we employed to identify RBPs assembling into granules (scores > 0 indicate granule propensity; Figure 2D) (Bolognesi et al., 2016). The software is available at http://service.tartaglialab.com/new_submission/catGRANULE.
Splicing Arrays experiments and analysis
COS-7 cells were grown in P10 plates and cultured in different conditions and in three biological replicates each: control, (CGG)60X 185ng, siTRA2A 50nM, (CGG)60X 185ng+siTRA2A 13.6ng, GFP-TRA2A 200ng, (CGG)60X 185ng + GFP-TRA2A 200ng.
Total RNA extraction was performed with QIAGEN RNeasy Mini Kit including DNase treatment according to manufacturer’s instructions. RNA amount was quantified and controlled with Nanodrop and Bioanalyzer. 100ng of total RNA from each sample were labeled according to the Affymetrix GeneChip® Whole Transcript Plus protocol, and hybridized to Affymetrix Human Clariom D array using a Affymetrix GeneChip Hybridization Oven 645, in Servei de Microarrays (IMIM-Barcelona). GeneChip was scanned using Affymetrix GeneChip Scanner 3000 7G. The data were analyzed using the RMA algorithm and then LIMMA was applied to calculate significant differential expression between samples. Splicing arrays were analyzed with the Transcriptome Analysis Console Software (Thermo Fisher Scientific), setting the following thresholds and methods: Gene-Level Fold Change < −2 or > 2, Gene-Level P value < 0,05, Splicing Index < −2 or > 2, Exon-Level P value < 0,05, Anova Method: ebayes, Probeset (Gene/Exon) considered expressed if ≥ 50% samples have DABG values below DABG Threshold (DABG < 0,05), event Pointer P value < 0,1 and event score > 0,2. Data are deposited in GEO repository with accession number GEO: GSE108007.
RNA-seq experiments and analysis
An aliquot of the same RNA extracted from COS-7 cells for splicing arrays (previous section) was used for RNASEQ, in three biological replicates each: control, (CGG)60X 185ng, siTRA2A 50nM, (CGG)60X 185ng+siTRA2A 13.6ng, GFP-TRA2A 200ng, (CGG)60X 185ng + GFP-TRA2A 200ng. 500ng of total RNA were used for library preparation with TruSeq total RNA-rRNA depletion (Illumina). The sequencing was performed with HiSeq3000, paired-end, 2X125, 3samples/lane.
Nucleotide alignments were performed using STAR_2.5.2 with reference genome and annotations taken from Gencode Release 27 (GRCh38.p10) (http://www.gencodegenes.org/releases/current.html. Pre-processing of bam files was made with python scripts (“dexseq_prepare_annotation.py” and “dexseq_count.py”) provided in the DEXSeq v 1.24.2 R package. Statistical analysis of alternative splicing events was done with R 3.4.0 using two methods: EventPointer v1.0.0 and DEXSeq v 1.24.2. Data are deposited in GEO repository with accesion number GEO: GSE121304.
Acknowledgments
The authors are deeply in debt to the brain donors and their relatives. We acknowledge specimen donations from Dr. Nicolas Charlet-Berguerand (pcDNA3 CGG 60X), Dr. David Elliott and Dr. Caroline Dalgliesh (plasmid GFP TRA2A), Dr. Eulàlia Martí (plasmids CGG 21X and 79X), and Dr. Matthew D. Disney (molecule 1a). We thank Martin Vabulas for the “schemino” of Figure 1A and all members of Tartaglia’s lab, especially Dr. Elias Bechara, for the interpretation of splicing experiments, Laura Padovani and Andrea Vandelli for the computational analyses. We are grateful to Lara Nonell Mazelón and Magdalena Arnal Segura for RNA-seq and splicing arrays analysis, and Dr. Fatima Gebauer, Dr. Natàlia Sánchez de Groot, Dr. Davide Cirillo, and Dr. Domenica Marchese for stimulating discussions. The research leading to these results has been supported by the European Research Council (RIBOMYLOME_309545), Spanish Ministry of Economy and Competitiveness (BFU2014-55054-P and BFU2017-86970-P), and “Fundació La Marató de TV3” (PI043296). We acknowledge support of the Spanish Ministry of Economy and Competitiveness, ‘Centro de Excelencia Severo Ochoa 2013-2017’. We acknowledge the support of the CERCA Programme, Generalitat de Catalunya and Spanish Ministry for Science and Competitiveness (MINECO) to the EMBL partnership.
Author Contributions
G.G.T. and T.B.-O. conceived the study together with the help of B.B.; F.C.-S. performed the calculations; T.B.-O. supervised M.G.-B. and performed all the experiments as well as analyzed samples from R.K.H., L.-A.W.S., and E.G.; T.B.-O., N.L.-G., G.G.T., and B.L. analyzed the data; T.B.-O., F.C.-S., B.B., and G.G.T. wrote the manuscript.
Declaration of Interests
The authors declare no conflict of interest.
Published: December 18, 2018
Footnotes
Supplemental Information includes seven figures and five tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.11.076.
Contributor Information
Teresa Botta-Orfila, Email: teresa.botta@idibaps.org.
Gian Gaetano Tartaglia, Email: gian@tartaglialab.com.
Supplemental Information
A) List of human and yeast granule proteins. B, C, D, E) Granule and non-granule RBPs, RNA partners of human RBPs identified at different cut-offs of the reads/expression distribution (first, second and third quartile are indicated with Q1, Q2 and Q3). Names starting by NM indicate coding transcripts and names starting by NR indicate non-coding transcripts. F) RNA partners of yeast RBPs. G, H, I) Overlap between interactomes of human RBPs calculated using the Jaccard index (first, second and third quartile are indicated with Q1, Q2 and Q3) and J) Overlap between yeast RBP interactomes. K) Intersection between Q3 RNA interactome and granule transcripts reported in (Khong et al., 2017).
A) RBP contacts of human RNAs. Names starting by NM indicate coding transcripts and names starting by NR indicate non-coding transcripts. B) Number of total, granule and non-granule contacts, structural content, length and UTR size of human transcripts; C) Number of total, granule and non-granule contacts, structural content, length of yeast transcripts.
catRAPID scores (discriminative power DP or interaction score, interaction strength IS or specificity) (Agostini et al., 2013), name of the gene, catGRANULE score (Bolognesi et al., 2016), granule ability (predicted / validated) and empirical p value indicating the ability of proteins to interact with CGG repeats (calculated on 3340 DNA-binding, RNA-binding and structurally disordered proteins).
We employed protein arrays to perform a large in vitro screening of RBP interactions with the first FMR1 exon (Cirillo et al., 2017; Marchese et al., 2017). We probed both expanded (“PRE”; 79 CGG) and normal (“WT”; 21 CGG) on three independent arrays, obtaining highly reproducible results.
A) CGG overexpression vs CTL; B) CGG over-expression and TRA2A knockdown vs control. Fold Changes, significance and (sub)-exon names are reported. Microarray: Transcriptome Analysis Console (TAC) 4.0 software (ThermoFisher) was used to identify splicing events. RNA-seq: Statistical analysis of alternative splicing events was done using EventPointer v1.0.0 and DEXSeq v 1.24.2 (GO analysis: http://www.tartaglialab.com/GO_analyser/render_GO_universal/2105/64ce4f8d1d/, http://www.tartaglialab.com/GO_analyser/render_GO_universal/2108/eef220536a/).
References
- Agostini F., Zanzoni A., Klus P., Marchese D., Cirillo D., Tartaglia G.G. catRAPID omics: a web server for large-scale prediction of protein-RNA interactions. Bioinformatics. 2013;29:2928–2930. doi: 10.1093/bioinformatics/btt495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armaos A., Cirillo D., Gaetano Tartaglia G. omiXcore: a web server for prediction of protein interactions with large RNA. Bioinformatics. 2017;33:3104–3106. doi: 10.1093/bioinformatics/btx361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani S.F., Lee H.O., Hyman A.A., Rosen M.K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017;18:285–298. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci M., Agostini F., Masin M., Tartaglia G.G. Predicting protein associations with long noncoding RNAs. Nat. Methods. 2011;8:444–445. doi: 10.1038/nmeth.1611. [DOI] [PubMed] [Google Scholar]
- Bolognesi B., Lorenzo Gotor N., Dhar R., Cirillo D., Baldrighi M., Tartaglia G.G., Lehner B. A concentration-dependent liquid phase separation can cause toxicity upon increased protein expression. Cell Rep. 2016;16:222–231. doi: 10.1016/j.celrep.2016.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botta-Orfila T., Tartaglia G.G., Michalon A. Molecular pathophysiology of fragile x-associated tremor/ataxia syndrome and perspectives for drug development. Cerebellum. 2016;15:599–610. doi: 10.1007/s12311-016-0800-2. [DOI] [PubMed] [Google Scholar]
- Brangwynne C.P., Eckmann C.R., Courson D.S., Rybarska A., Hoege C., Gharakhani J., Jülicher F., Hyman A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–1732. doi: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- Brannan K.W., Jin W., Huelga S.C., Banks C.A.S., Gilmore J.M., Florens L., Washburn M.P., Van Nostrand E.L., Pratt G.A., Schwinn M.K. SONAR discovers RNA-binding proteins from analysis of large-scale protein-protein interactomes. Mol. Cell. 2016;64:282–293. doi: 10.1016/j.molcel.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan J.R., Muhlrad D., Parker R. P bodies promote stress granule assembly in Saccharomyces cerevisiae. J. Cell Biol. 2008;183:441–455. doi: 10.1083/jcb.200807043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budny B., Badura-Stronka M., Materna-Kiryluk A., Tzschach A., Raynaud M., Latos-Bielenska A., Ropers H.H. Novel missense mutations in the ubiquitination-related gene UBE2A cause a recognizable X-linked mental retardation syndrome. Clin. Genet. 2010;77:541–551. doi: 10.1111/j.1399-0004.2010.01429.x. [DOI] [PubMed] [Google Scholar]
- Buijsen R.A., Sellier C., Severijnen L.-A.W., Oulad-Abdelghani M., Verhagen R.F., Berman R.F., Charlet-Berguerand N., Willemsen R., Hukema R.K. FMRpolyG-positive inclusions in CNS and non-CNS organs of a fragile X premutation carrier with fragile X-associated tremor/ataxia syndrome. Acta Neuropathol. Commun. 2014;2:162. doi: 10.1186/s40478-014-0162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Wan J., Yu B., Diao Y., Zhang W. PIP5K1α promotes myogenic differentiation via AKT activation and calcium release. Stem Cell Res. Ther. 2018;9:33. doi: 10.1186/s13287-018-0770-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo D., Agostini F., Klus P., Marchese D., Rodriguez S., Bolognesi B., Tartaglia G.G. Neurodegenerative diseases: quantitative predictions of protein-RNA interactions. RNA. 2013;19:129–140. doi: 10.1261/rna.034777.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo D., Blanco M., Armaos A., Buness A., Avner P., Guttman M., Cerase A., Tartaglia G.G. Quantitative predictions of protein interactions with long noncoding RNAs. Nat. Methods. 2017;14:5–6. doi: 10.1038/nmeth.4100. [DOI] [PubMed] [Google Scholar]
- Colomer V., Kicska G.A., Rindler M.J. Secretory granule content proteins and the luminal domains of granule membrane proteins aggregate in vitro at mildly acidic pH. J. Biol. Chem. 1996;271:48–55. doi: 10.1074/jbc.271.1.48. [DOI] [PubMed] [Google Scholar]
- de la Rica L., Rodríguez-Ubreva J., García M., Islam A.B., Urquiza J.M., Hernando H., Christensen J., Helin K., Gómez-Vaquero C., Ballestar E. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 2013;14:R99. doi: 10.1186/gb-2013-14-9-r99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva M.G., Elliott K., Dahl H.-H., Fitzpatrick E., Wilcox S., Delatycki M., Williamson R., Efron D., Lynch M., Forrest S. Disruption of a novel member of a sodium/hydrogen exchanger family and DOCK3 is associated with an attention deficit hyperactivity disorder-like phenotype. J. Med. Genet. 2003;40:733–740. doi: 10.1136/jmg.40.10.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delli Ponti R., Marti S., Armaos A., Tartaglia G.G. A high-throughput approach to profile RNA structure. Nucleic Acids Res. 2017;45 doi: 10.1093/nar/gkw1094. e35–e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disney M.D., Liu B., Yang W.-Y., Sellier C., Tran T., Charlet-Berguerand N., Childs-Disney J.L. A small molecule that targets r(CGG)(exp) and improves defects in fragile X-associated tremor ataxia syndrome. ACS Chem. Biol. 2012;7:1711–1718. doi: 10.1021/cb300135h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann J., Daehmlow S., Wischke S., Senyuva M., Werner U., Raible J., Tanis N., Dyachenko S., Hummel M., Hetzer R., Regitz-Zagrosek V. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin. Genet. 2003;64:339–349. doi: 10.1034/j.1399-0004.2003.00151.x. [DOI] [PubMed] [Google Scholar]
- Gerstberger S., Hafner M., Tuschl T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014;15:829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glineburg M.R., Todd P.K., Charlet-Berguerand N., Sellier C. Repeat-associated non-AUG (RAN) translation and other molecular mechanisms in fragile x tremor ataxia syndrome. Brain Res. 2018;1693:43–54. doi: 10.1016/j.brainres.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grousl T., Ungelenk S., Miller S., Ho C.-T., Khokhrina M., Mayer M.P., Bukau B., Mogk A. A prion-like domain in Hsp42 drives chaperone-facilitated aggregation of misfolded proteins. J. Cell Biol. 2018;217:1269–1285. doi: 10.1083/jcb.201708116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukema R.K., Buijsen R.A.M., Schonewille M., Raske C., Severijnen L.-A.W.F.M., Nieuwenhuizen-Bakker I., Verhagen R.F.M., van Dessel L., Maas A., Charlet-Berguerand N. Reversibility of neuropathology and motor deficits in an inducible mouse model for FXTAS. Hum. Mol. Genet. 2015;24:4948–4957. doi: 10.1093/hmg/ddv216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin E.L., Ting L., Bruckner R.J., Gebreab F., Gygi M.P., Szpyt J., Tam S., Zarraga G., Colby G., Baltier K. The BioPlex network: a systematic exploration of the human interactome. Cell. 2015;162:425–440. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman A.A., Weber C.A., Jülicher F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014;30:39–58. doi: 10.1146/annurev-cellbio-100913-013325. [DOI] [PubMed] [Google Scholar]
- Iwahashi C.K., Yasui D.H., An H.-J., Greco C.M., Tassone F., Nannen K., Babineau B., Lebrilla C.B., Hagerman R.J., Hagerman P.J. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129:256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- Jain A., Vale R.D. RNA phase transitions in repeat expansion disorders. Nature. 2017;546:243–247. doi: 10.1038/nature22386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S., Wheeler J.R., Walters R.W., Agrawal A., Barsic A., Parker R. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell. 2016;164:487–498. doi: 10.1016/j.cell.2015.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Wang S., Huang Y., He X., Cui H., Zhu X., Zheng Y. Phase transition of spindle-associated protein regulate spindle apparatus assembly. Cell. 2015;163:108–122. doi: 10.1016/j.cell.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M., Han T.W., Xie S., Shi K., Du X., Wu L.C., Mirzaei H., Goldsmith E.J., Longgood J., Pei J. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M., Wan Y., Mazor E., Rinn J.L., Nutter R.C., Chang H.Y., Segal E. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong A., Matheny T., Jain S., Mitchell S.F., Wheeler J.R., Parker R. The stress granule transcriptome reveals principles of mrna accumulation in stress granules. Mol. Cell. 2017;68:808–820.e5. doi: 10.1016/j.molcel.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klus P., Ponti R.D., Livi C.M., Tartaglia G.G. Protein aggregation, structural disorder and RNA-binding ability: a new approach for physico-chemical and gene ontology classification of multiple datasets. BMC Genomics. 2015;16:1071. doi: 10.1186/s12864-015-2280-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyzosiak W.J., Sobczak K., Wojciechowska M., Fiszer A., Mykowska A., Kozlowski P. Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res. 2012;40:11–26. doi: 10.1093/nar/gkr729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langdon E.M., Qiu Y., Ghanbari Niaki A., McLaughlin G.A., Weidmann C.A., Gerbich T.M., Smith J.A., Crutchley J.M., Termini C.M., Weeks K.M. mRNA structure determines specificity of a polyQ-driven phase separation. Science. 2018;360:922–927. doi: 10.1126/science.aar7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Kopp F., Chang T.-C., Sataluri A., Chen B., Sivakumar S., Yu H., Xie Y., Mendell J.T. Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO proteins. Cell. 2016;164:69–80. doi: 10.1016/j.cell.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livi C.M., Klus P., Delli Ponti R., Tartaglia G.G. catRAPID signature: identification of ribonucleoproteins and RNA-binding regions. Bioinformatics. 2015;32:773–775. doi: 10.1093/bioinformatics/btv629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D.K., Jang M.-H., Guo J.U., Kitabatake Y., Chang M.-L., Pow-Anpongkul N., Flavell R.A., Lu B., Ming G.-L., Song H. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–1077. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharana S., Wang J., Papadopoulos D.K., Richter D., Pozniakovsky A., Poser I., Bickle M., Rizk S., Guillén-Boixet J., Franzmann T.M. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science. 2018;360:918–921. doi: 10.1126/science.aar7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese D., de Groot N.S., Lorenzo Gotor N., Livi C.M., Tartaglia G.G. Advances in the characterization of RNA-binding proteins. Wiley Interdiscip. Rev. RNA. 2016;7:793–810. doi: 10.1002/wrna.1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese D., Botta-Orfila T., Cirillo D., Rodriguez J.A., Livi C.M., Fernández-Santiago R., Ezquerra M., Martí M.J., Bechara E., Tartaglia G.G., Catalan MSA Registry (CMSAR) Discovering the 3′ UTR-mediated regulation of alpha-synuclein. Nucleic Acids Res. 2017;45:12888–12903. doi: 10.1093/nar/gkx1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S., Soltanieh S., Server K.L., Mak R., Jin W., Fang M.Y., Luo E.-C., Krach F., Yang D., Sen A. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell. 2018;172:590–604.e13. doi: 10.1016/j.cell.2017.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateju D., Franzmann T.M., Patel A., Kopach A., Boczek E.E., Maharana S., Lee H.O., Carra S., Hyman A.A., Alberti S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017;36:1669–1687. doi: 10.15252/embj.201695957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurin T., Zongaro S., Bardoni B. Fragile X syndrome: from molecular pathology to therapy. Neurosci. Biobehav. Rev. 2014;46:242–255. doi: 10.1016/j.neubiorev.2014.01.006. [DOI] [PubMed] [Google Scholar]
- Maziuk B.F., Apicco D.J., Cruz A.L., Jiang L., Ash P.E.A., da Rocha E.L., Zhang C., Yu W.H., Leszyk J., Abisambra J.F. RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol. Commun. 2018;6:71. doi: 10.1186/s40478-018-0574-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell S.F., Jain S., She M., Parker R. Global analysis of yeast mRNPs. Nat. Struct. Mol. Biol. 2013;20:127–133. doi: 10.1038/nsmb.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal N., Scherrer T., Gerber A.P., Janga S.C. Interplay between posttranscriptional and posttranslational interactions of RNA-binding proteins. J. Mol. Biol. 2011;409:466–479. doi: 10.1016/j.jmb.2011.03.064. [DOI] [PubMed] [Google Scholar]
- Mooers B.H.M., Logue J.S., Berglund J.A. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc. Natl. Acad. Sci. USA. 2005;102:16626–16631. doi: 10.1073/pnas.0505873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M.J., Wingate K.L., Silwal J., Leeper T.C., Basu S. The porphyrin TmPyP4 unfolds the extremely stable G-quadruplex in MT3-MMP mRNA and alleviates its repressive effect to enhance translation in eukaryotic cells. Nucleic Acids Res. 2012;40:4137–4145. doi: 10.1093/nar/gkr1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T., Qamar S., Lin J.Q., Schierle G.S.K., Rees E., Miyashita A., Costa A.R., Dodd R.B., Chan F.T.S., Michel C.H. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron. 2015;88:678–690. doi: 10.1016/j.neuron.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutsuddi M., Marshall C.M., Benzow K.A., Koob M.D., Rebay I. The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with staufen in Drosophila. Curr. Biol. 2004;14:302–308. doi: 10.1016/j.cub.2004.01.034. [DOI] [PubMed] [Google Scholar]
- Csárdi G., Nepusz T. The igraph software package for complex network research. InterJournal Complex Syst. 2006:1695. [Google Scholar]
- Patel A., Lee H.O., Jawerth L., Maharana S., Jahnel M., Hein M.Y., Stoynov S., Mahamid J., Saha S., Franzmann T.M. A liquid-to-solid phase transition of the als protein fus accelerated by disease mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- Pavel M., Imarisio S., Menzies F.M., Jimenez-Sanchez M., Siddiqi F.H., Wu X., Renna M., O’Kane C.J., Crowther D.C., Rubinsztein D.C. CCT complex restricts neuropathogenic protein aggregation via autophagy. Nat. Commun. 2016;7:13821. doi: 10.1038/ncomms13821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson O.J., Aagaard L., Jensen T.G., Damgaard C.K. Molecular mechanisms in DM1—a focus on foci. Nucleic Acids Res. 2015;43:2433–2441. doi: 10.1093/nar/gkv029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procaccio V., Salazar G., Ono S., Styers M.L., Gearing M., Davila A., Jimenez R., Juncos J., Gutekunst C.-A., Meroni G. A mutation of beta -actin that alters depolymerization dynamics is associated with autosomal dominant developmental malformations, deafness, and dystonia. Am. J. Hum. Genet. 2006;78:947–960. doi: 10.1086/504271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamar S., Wang G., Randle S.J., Ruggeri F.S., Varela J.A., Lin J.Q., Phillips E.C., Miyashita A., Williams D., Ströhl F. FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-π interactions. Cell. 2018;173:720–734.e15. doi: 10.1016/j.cell.2018.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke L.C., Kedersha N., Langereis M.A., van Kuppeveld F.J.M., Lloyd R.E. Stress granules regulate double-stranded RNA-dependent protein kinase activation through a complex containing G3BP1 and Caprin1. MBio. 2015;6:e02486. doi: 10.1128/mBio.02486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riback J.A., Katanski C.D., Kear-Scott J.L., Pilipenko E.V., Rojek A.E., Sosnick T.R., Drummond D.A. Stress-triggered phase separation is an adaptive, evolutionarily tuned response. Cell. 2017;168:1028–1040.e19. doi: 10.1016/j.cell.2017.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro D.M., Zanzoni A., Cipriano A., Delli Ponti R., Spinelli L., Ballarino M., Bozzoni I., Tartaglia G.G., Brun C. Protein complex scaffolding predicted as a prevalent function of long non-coding RNAs. Nucleic Acids Res. 2018;46:917–928. doi: 10.1093/nar/gkx1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivière J.-B., van Bon B.W.M., Hoischen A., Kholmanskikh S.S., O’Roak B.J., Gilissen C., Gijsen S., Sullivan C.T., Christian S.L., Abdul-Rahman O.A. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat. Genet. 2012;44:440–444. doi: 10.1038/ng.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S., Hyman A.A. RNA gets in phase. J. Cell Biol. 2017;216:2235–2237. doi: 10.1083/jcb.201706034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C., Rau F., Liu Y., Tassone F., Hukema R.K., Gattoni R., Schneider A., Richard S., Willemsen R., Elliott D.J. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010;29:1248–1261. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C., Freyermuth F., Tabet R., Tran T., He F., Ruffenach F., Alunni V., Moine H., Thibault C., Page A. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2013;3:869–880. doi: 10.1016/j.celrep.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C., Buijsen R.A.M., He F., Natla S., Jung L., Tropel P., Gaucherot A., Jacobs H., Meziane H., Vincent A. Translation of expanded CGG repeats into fmrpolyg is pathogenic and may contribute to fragile x tremor ataxia syndrome. Neuron. 2017;93:331–347. doi: 10.1016/j.neuron.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba K., Araki K., Li Z., Matsumoto K., Suzuki M., Nakagata N., Takagi K., Takeya M., Yoshinobu K., Araki M. A novel murine gene, Sickle tail, linked to the Danforth’s short tail locus, is required for normal development of the intervertebral disc. Genetics. 2006;172:445–456. doi: 10.1534/genetics.105.048934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack R.L., Disney M.D., Jaffrey S.R. A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat-containing RNA. Nat. Methods. 2013;10:1219–1224. doi: 10.1038/nmeth.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J.H., Fraser A.G. The combinatorial control of alternative splicing in C. elegans. PLoS Genet. 2017;13:e1007033. doi: 10.1371/journal.pgen.1007033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia G.G., Vendruscolo M. Correlation between mRNA expression levels and protein aggregation propensities in subcellular localisations. Mol. Biosyst. 2009;5:1873–1876. doi: 10.1039/b913099n. [DOI] [PubMed] [Google Scholar]
- Tassone F., Iwahashi C., Hagerman P.J. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS) RNA Biol. 2004;1:103–105. doi: 10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]
- Tassone F., Beilina A., Carosi C., Albertosi S., Bagni C., Li L., Glover K., Bentley D., Hagerman P.J. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA. 2007;13:555–562. doi: 10.1261/rna.280807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichon A., Gil N., Lubelsky Y., Solomon T.H., Lemze D., Itzkovitz S., Stern-Ginossar N., Ulitsky I. A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nat. Commun. 2016;7:12209. doi: 10.1038/ncomms12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd P.K., Oh S.Y., Krans A., He F., Sellier C., Frazer M., Renoux A.J., Chen K.C., Scaglione K.M., Basrur V. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron. 2013;78:440–455. doi: 10.1016/j.neuron.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand E.L., Pratt G.A., Shishkin A.A., Gelboin-Burkhart C., Fang M.Y., Sundararaman B., Blue S.M., Nguyen T.B., Surka C., Elkins K. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP) Nat. Methods. 2016;13:508–514. doi: 10.1038/nmeth.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Treeck B., Parker R. Emerging roles for intermolecular RNA-RNA interactions in RNP assemblies. Cell. 2018;174:791–802. doi: 10.1016/j.cell.2018.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y., Qu K., Zhang Q.C., Flynn R.A., Manor O., Ouyang Z., Zhang J., Spitale R.C., Snyder M.P., Segal E., Chang H.Y. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014;505:706–709. doi: 10.1038/nature12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West J.A., Mito M., Kurosaka S., Takumi T., Tanegashima C., Chujo T., Yanaka K., Kingston R.E., Hirose T., Bond C. Structural, super-resolution microscopy analysis of paraspeckle nuclear body organization. J. Cell Biol. 2016;214:817–830. doi: 10.1083/jcb.201601071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White M.C., Gao R., Xu W., Mandal S.M., Lim J.G., Hazra T.K., Wakamiya M., Edwards S.F., Raskin S., Teive H.A.G. Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet. 2010;6:e1000984. doi: 10.1371/journal.pgen.1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen R., Hoogeveen-Westerveld M., Reis S., Holstege J., Severijnen L.-A.W.F.M., Nieuwenhuizen I.M., Schrier M., van Unen L., Tassone F., Hoogeveen A.T. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum. Mol. Genet. 2003;12:949–959. doi: 10.1093/hmg/ddg114. [DOI] [PubMed] [Google Scholar]
- Woodruff J.B., Ferreira Gomes B., Widlund P.O., Mahamid J., Honigmann A., Hyman A.A. The centrosome is a selective condensate that nucleates microtubules by concentrating tubulin. Cell. 2017;169:1066–1077.e10. doi: 10.1016/j.cell.2017.05.028. [DOI] [PubMed] [Google Scholar]
- Zhang H., Elbaum-Garfinkle S., Langdon E.M., Taylor N., Occhipinti P., Bridges A.A., Brangwynne C.P., Gladfelter A.S. RNA controls PolyQ protein phase transitions. Mol. Cell. 2015;60:220–230. doi: 10.1016/j.molcel.2015.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W., Chen X., Shim J.-H., Huang Z., Brady N., Hu D., Drapp R., Sigrist K., Glimcher L.H., Jones D. The E3 ubiquitin ligase Wwp2 regulates craniofacial development through mono-ubiquitylation of Goosecoid. Nat. Cell Biol. 2011;13:59–65. doi: 10.1038/ncb2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A) List of human and yeast granule proteins. B, C, D, E) Granule and non-granule RBPs, RNA partners of human RBPs identified at different cut-offs of the reads/expression distribution (first, second and third quartile are indicated with Q1, Q2 and Q3). Names starting by NM indicate coding transcripts and names starting by NR indicate non-coding transcripts. F) RNA partners of yeast RBPs. G, H, I) Overlap between interactomes of human RBPs calculated using the Jaccard index (first, second and third quartile are indicated with Q1, Q2 and Q3) and J) Overlap between yeast RBP interactomes. K) Intersection between Q3 RNA interactome and granule transcripts reported in (Khong et al., 2017).
A) RBP contacts of human RNAs. Names starting by NM indicate coding transcripts and names starting by NR indicate non-coding transcripts. B) Number of total, granule and non-granule contacts, structural content, length and UTR size of human transcripts; C) Number of total, granule and non-granule contacts, structural content, length of yeast transcripts.
catRAPID scores (discriminative power DP or interaction score, interaction strength IS or specificity) (Agostini et al., 2013), name of the gene, catGRANULE score (Bolognesi et al., 2016), granule ability (predicted / validated) and empirical p value indicating the ability of proteins to interact with CGG repeats (calculated on 3340 DNA-binding, RNA-binding and structurally disordered proteins).
We employed protein arrays to perform a large in vitro screening of RBP interactions with the first FMR1 exon (Cirillo et al., 2017; Marchese et al., 2017). We probed both expanded (“PRE”; 79 CGG) and normal (“WT”; 21 CGG) on three independent arrays, obtaining highly reproducible results.
A) CGG overexpression vs CTL; B) CGG over-expression and TRA2A knockdown vs control. Fold Changes, significance and (sub)-exon names are reported. Microarray: Transcriptome Analysis Console (TAC) 4.0 software (ThermoFisher) was used to identify splicing events. RNA-seq: Statistical analysis of alternative splicing events was done using EventPointer v1.0.0 and DEXSeq v 1.24.2 (GO analysis: http://www.tartaglialab.com/GO_analyser/render_GO_universal/2105/64ce4f8d1d/, http://www.tartaglialab.com/GO_analyser/render_GO_universal/2108/eef220536a/).