

Here, Moquin et al. show that fusion of the promiscuous biotin ligase BirAR118G with RAD18 leads to localized protein biotinylation at DNA damage sites and identify ZPET/ZNF280C as a potential DNA damage response protein. Their findings show that ZPET is an HR repressor and also suggest that localized protein biotinylation at DNA damage sites is a useful strategy to identify DDR proteins.
Keywords: DNA damage response, DNA end resection, homologous recombination
Abstract
Numerous DNA repair and signaling proteins function at DNA damage sites to protect the genome. Here, we show that fusion of the promiscuous biotin ligase BirAR118G with RAD18 leads to localized protein biotinylation at DNA damage sites, allowing identification of ZPET (zinc finger protein proximal to RAD eighteen)/ZNF280C as a potential DNA damage response (DDR) protein. ZPET binds ssDNA and localizes to DNA double-strand breaks (DSBs) and stalled replication forks. In vitro, ZPET inhibits MRE11 binding to ssDNA. In cells, ZPET delays MRE11 binding to chromatin after DSB formation and slows DNA end resection through binding ssDNA. ZPET hinders resection independently of 53BP1 and HELB. Cells lacking ZPET displayed enhanced homologous recombination (HR), accelerated replication forks under stress, and increased resistance to DSBs and PARP inhibition. These results not only reveal ZPET as an HR repressor but also suggest that localized protein biotinylation at DNA damage sites is a useful strategy to identify DDR proteins.
The DNA damage response (DDR) is a complex cellular process crucial for genomic stability. In response to different types of DNA damage, a large number of proteins involved in DNA repair, DNA damage signaling, and other damage-regulated processes are recruited to sites of DNA lesions (Ciccia and Elledge 2010). These proteins are assembled into stable or transient complexes, allowing them to execute their functions. The enrichment of distinct groups of DDR proteins at sites of different DNA lesions is important for not only their biochemical activities but also their functional specificities. Taking advantage of the interactions among DDR proteins, affinity-based biochemical approaches have been successfully used in numerous studies to extend the DDR protein network (Wang et al. 2000; Cortez et al. 2001; Meetei et al. 2003; Xia et al. 2006; Marechal et al. 2014; Tkac et al. 2016). However, the affinity-based approaches are less effective for capturing proteins that weakly or transiently interact with known DDR proteins. Furthermore, some DDR proteins may not interact with any known DDR proteins at all, precluding them from identification by the affinity-based approaches. The limitations of the affinity-based approaches raise the possibility that many uncharacterized DDR proteins might have escaped previous proteomic screens and that an alternative approach to identify DDR proteins may significantly extend the DDR protein network.
BirA is an Escherichia coli protein biotin ligase that biotinylates a specific substrate (Chapman-Smith and Cronan 1999). A BirA acceptor peptide (BAP) has been identified from the substrate and fused to other proteins, enabling BirA to biotinylate these fusion proteins when they are present in close proximity (Duffy et al. 1998). The BirA R118G (BirAR118G) mutant lacks the substrate specificity of wild-type BirA (Choi-Rhee et al. 2004), giving it the ability to biotinylate proteins promiscuously in a proximity-dependent manner. When fused to a bait protein, BirAR118G biotinylates the proteins in close proximity to the bait, allowing biochemical isolation and identification of these proteins (Roux et al. 2012). Fusion proteins containing BirAR118G have been successfully used to study proteins in various cellular compartments, such as cell–cell junctions, nuclear envelope, chromatin, centrosomes, telomeres, and DNA replication forks (Roux et al. 2012; Firat-Karalar et al. 2014; Lambert et al. 2015; Dong et al. 2016; Dubois et al. 2016; Garcia-Exposito et al. 2016). The enrichment of DDR proteins at sites of DNA damage presents an attractive opportunity for using BirAR118G to identify uncharacterized DDR proteins based on their proximity to known DDR proteins, overcoming the limitations of affinity-based approaches. Notably, APEX2, another biotin ligase, was used recently to capture DDR proteins in the absence of exogenous DNA damage (Gupta et al. 2018). In this study, we specifically tested the feasibility of using BirAR118G to identify DDR proteins at DNA damage sites.
As a proof of principle, we fused BirAR118G to the ubiquitin ligase RAD18 (Ting et al. 2010). RAD18 was chosen for this study because it has a unique ability to localize to different DNA lesions, including DNA double-strand breaks (DSBs) and lesions that stall replication forks, which provides a potential strategy to identify DDR proteins that function in distinct contexts. Furthermore, a fusion of wild-type BirA and RAD18 has been used to modify BAP-tagged histones (Shoaib et al. 2013). In response to UV-induced DNA damage, RAD18 monoubiquitinates PCNA, enabling translesion DNA synthesis (TLS) at or behind replication forks (Kannouche et al. 2004; Watanabe et al. 2004). In response to DNA interstrand cross-links (ICLs), RAD18 promotes ubiquitination of the FANCD2–FANCI complex and its accumulation at DNA damage sites (Williams et al. 2011). Furthermore, in response to DSBs, RAD18 is recruited to DNA damage sites in an RNF8-dependent manner, allowing RAD18 to promote homologous recombination (HR) through its interaction with RAD51C (Huang et al. 2009). RAD18 was also shown to ubiquitinate 53BP1 at DSBs, promoting its retention at DSBs in G1 and repair through nonhomologous end joining (NHEJ) (Watanabe et al. 2009). The ability of RAD18 to function in different repair pathways makes BirAR118G-RAD18 a potentially versatile bait protein in multiple DDR contexts.
In this study, we focused on using BirAR118G-RAD18 to identify DDR proteins at DSBs. We successfully detected a number of known DDR proteins that function at DSBs, including MDC1, RNF8, RAP80, and others. In addition, we identified a previously uncharacterized protein, ZNF280C, as a potential player in the DDR. We show that ZNF280C, which we renamed ZPET (zinc finger protein proximal to RAD eighteen), localizes to sites of DSBs and stalled replication forks. ZPET binds ssDNA directly and inhibits MRE11 binding to ssDNA in cell extracts. In response to DSBs, ZPET delays the recruitment of MRE11 and CtIP to chromatin and slows DNA end resection in a ssDNA binding-dependent manner. The function of ZPET in antagonizing resection is independent of 53BP1 and HELB. Loss of ZPET increases HR efficiency, accelerates replication forks under stress, and renders cells resistant to DSBs and PARP inhibition. Together, these results suggest that ZPET functions in a ssDNA-triggered feedback loop to restrict DNA end resection and HR, demonstrating that BirAR118G is a powerful tool to identify DDR proteins at sites of DNA damage.
Results
BirAR118G-RAD18 promotes localized protein biotinylation at DNA damage sites
To identify proteins that localize to sites of DNA damage, we fused BirAR118G with the N terminus of RAD18. U2OS derivative cell lines that stably express BirAR118G-RAD18 were generated. RAD18 is known to localize to sites of DSBs and stalled DNA replication forks (Watanabe et al. 2004; Huang et al. 2009). In cells microirradiated with a UV laser, BirAR118G-RAD18 was efficiently recruited to laser stripes (Fig. 1A). Importantly, when BirAR118G-RAD18-expressing cells were supplemented with exogenous biotin, robust biotin signals were detected in laser stripes (Fig. 1A), suggesting that BirAR118G-RAD18 induces localized protein biotinylation. Due to intrinsic replication stress, RAD18 forms spontaneous foci in a fraction of proliferating cells (Inagaki et al. 2009). In BirAR118G-RAD18-expressing cells, spontaneous BirAR118G-RAD18 foci colocalized with biotin foci in the presence of exogenous biotin (Fig. 1B), suggesting that BirAR118G-RAD18 biotinylates proteins at stalled or collapsed replication forks. After exposure to ionizing radiation (IR), foci containing both BirAR118G-RAD18 and biotin were drastically increased (Fig. 1B), indicating localized protein biotinylation at DSBs. Consistent with the idea that BirAR118G-RAD18 biotinylates proteins at sites of DNA damage, biotin signals colocalized with γH2AX and RPA32, two DNA damage markers, in laser stripes (Fig. 1C). In marked contrast to BirAR118G-RAD18, a fusion protein of BirAR118G and GFP (BirAR118G-GFP) did not colocalize with γH2AX and RPA32 and was unable to induce biotin signals in laser stripes (Fig. 1D; Supplemental Fig. S1). Together, these results suggest that BirAR118G-RAD18 provides an efficient and specific means to induce protein biotinylation at sites of DNA damage.
Figure 1.

BirAR118G-RAD18 promotes localized protein biotinylation at DNA damage sites and identifies ZPET as a potential DDR protein. (A) U2OS cells stably expressing BirAR118G-RAD18 were damaged with a UV laser, recovered for 4 h in the presence or absence of exogenous biotin, and subjected to immunofluorescence to analyze colocalization of BirAR118G-RAD18 and biotin at sites of DNA damage. (B) As indicated, cells were either left untreated, incubated with biotin alone, or treated with 10 Gy of IR and recovered for 4 h in the presence of biotin. Cells were stained as in A. (C) Cells were stained for biotin, γH2AX, and RPA32 as indicated. (D) U2OS lines stably expressing BirAR118G-GFP cells were analyzed as in C. (E) Cells were irradiated (10 Gy), allowed to recover for 1 h, and then incubated for an additional 4 h in the presence or absence of biotin. Cell lysates were subjected to a pull-down with streptavidin-conjugated beads. Samples were TMT-labeled followed by analysis using multiplexed mass spectrometry. A volcano plot was generated from the mass spectrometry results (biological duplicate). A Z-score for each P-value was determined. A cutoff of ≥2 was established and is indicated by the enlarged data points in the volcano plot (Lapek et al. 2017). (F,G) Cells were incubated with biotin, treated or untreated with 10 Gy of IR, and recovered for 4 h (F) or 6 h (G). Cells were fractionated into a crude chromatin fraction and subjected to pull-downs with streptavidin-conjugated beads. The indicated proteins in input extracts and pull-downs were analyzed by Western blot.
Identification of ZPET/ZNF280C as a potential DDR protein
We next sought to use BirAR118G-RAD18 to identify proteins that are present in proximity to RAD18 after DNA damage. BirAR118G-RAD18-expressing cells were briefly cultured in the presence of exogenous biotin, exposed to 10 Gy of IR or left untreated, and lysed to generate whole-cell extracts. Biotinylated proteins in cell extracts were captured with streptavidin-coated beads under denaturing conditions and subjected to multiplexed quantitative mass spectrometry analysis (Ting et al. 2011; Lapek et al. 2017). Our analysis identified a number of proteins that were increasingly biotinylated after IR (Fig. 1E; Supplemental Table 1). Among these proteins were several known DDR proteins, including MDC1, RNF8, RNF169, RAP80, BRCA1, 53BP1, and Polη (Fig. 1E; Supplemental Table 1). All of these DDR proteins, including Polη, are known to function at DSBs (Scully et al. 1996; Wang et al. 2002; Goldberg et al. 2003; Lou et al. 2003; Stewart et al. 2003; Huen et al. 2007; Kim et al. 2007; Kolas et al. 2007; Sobhian et al. 2007; Wang et al. 2007; McIlwraith and West 2008; Chen et al. 2012; Buisson et al. 2014). Importantly, BirAR118G-GFP, which was unable to localize to sites of DNA damage, did not lead to preferential biotinylation of DDR proteins after IR (Supplemental Table 2), confirming the specificity of our approach. To validate the mass spectrometry results, we used streptavidin-coated beads to capture biotinylated proteins from cells treated or untreated with IR and analyzed the proteins by Western blot. Indeed, an increased amount of MDC1 was captured after IR (Fig. 1F), confirming that MDC1 was biotinylated in an IR-stimulated manner. Interestingly, we also found that BirAR118G-RAD18 was one of the major biotinylated proteins, although its biotinylation was not induced by IR as the aforementioned DDR proteins (Supplemental Table 1). These results validate the use of BirAR118G-RAD18 to enrich DDR proteins that function at DSBs.
In our mass spectrometry data sets, we noticed that a previously uncharacterized protein, ZNF280C, was biotinylated in an IR-stimulated manner similarly to RAP80 and RNF169 (Fig. 1E). In addition, ZNF280C was biotinylated more efficiently by BirAR118G-RAD18 than by BirAR118G-GFP after IR (Supplemental Table 1). We renamed ZNF280C protein ZPET to reflect its proximity to RAD18. Using an anti-ZPET antibody, we confirmed that ZPET was increasingly biotinylated by BirAR118G-RAD18 after IR (Fig. 1G).
ZPET is recruited to DSBs and stalled replication forks
To investigate whether ZPET functions in the DDR, we next analyzed the localization of ZPET after DNA damage. In cells microirradiated with a UV laser, both endogenous RAD18 and HA-tagged ZPET were detected in laser stripes (Fig. 2A). An IR-induced colocalization of HA-ZPET and γH2AX was also detected by proximity ligation assay (PLA) using both HA and γH2AX antibodies but not either antibody alone (Fig. 2B; Supplemental Fig. S2A). In cells where DSBs are conditionally induced by the restriction enzyme AsiSI (Aymard et al. 2014), an increase of GFP-ZPET levels at DSBs was detected by chromatin immunoprecipitation (ChIP) 60–120 min after DSB induction (Fig. 2C). GFP-ZPET was preferentially enriched at DSBs compared with distal loci (Supplemental Fig. S2B). Furthermore, in a cell line that carries an array of tetracycline response elements (TREs) inserted with 96 I-SceI sites (Wei et al. 2015; Teng et al. 2018), GFP-ZPET formed a single focus in the nucleus when I-SceI was expressed (Fig. 2D). This focus of GFP-ZPET was positive for both γH2AX and TA-Cherry, a marker of the array, showing that GFP-ZPET colocalized with I-SceI-generated DSBs. Together, these results demonstrate that ZPET is recruited to DSBs.
Figure 2.
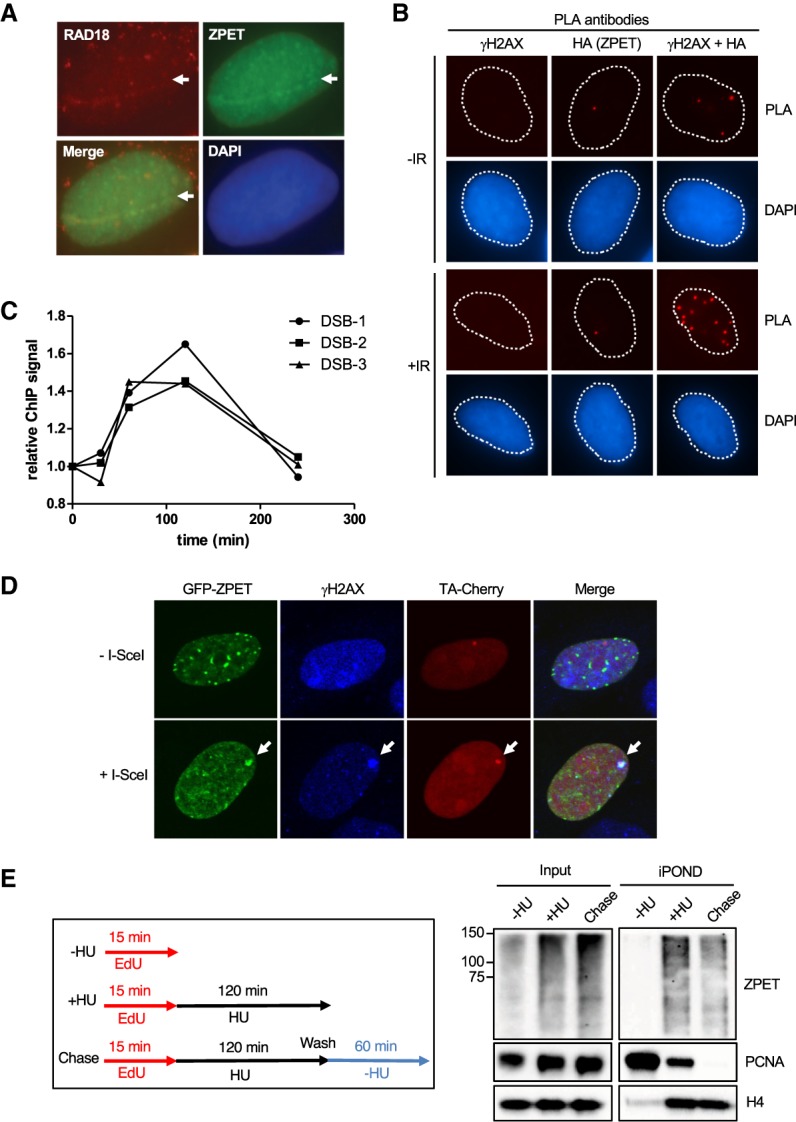
ZPET localizes to sites of DSBs and stalled replication forks. (A) U2OS cells stably expressing doxycycline (Dox)-inducible HA-ZPET were incubated with BrdU and Dox for 24 h. The cells were then microirradiated with a UV laser and allowed to recover for 1 h. Cells were fixed and stained for HA-ZPET and RAD18. (B) The same cells as in A were either left untreated or irradiated (10 Gy) and allowed to recover for 1 h. The cells were then analyzed by PLA using the indicated antibodies. (C) U2OS cells expressing ER-AsiSI were transfected with a linearized plasmid encoding GFP-ZPET, and GFP+ cells were enriched by FACS. These stable cells were treated with 4OHT for 0.5–4 h to induce DSBs. ChIP of GFP-ZPET was carried out with anti-GFP antibody. The relative levels of GFP-ZPET at three AsiSI sites were analyzed by quantitative PCR using primers recognizing adjacent sequences. (D) U2OS cells carrying a TRE/I-SceI array were transfected with plasmids expressing I-SceI, TA-Cherry, and GFP-tagged ZPET. The array and GFP-ZPET were visualized by TA-Cherry and GFP, respectively. (E) HEK293T cells were labeled with EdU for 15 min followed by mock treatment (−HU) or 2 h of hydroxyurea (HU) treatment (+HU). For the chase sample, HU-treated cells were washed and treated with thymidine for 60 min. iPOND (isolation of proteins on nascent DNA) was conducted, and ZPET recruitment to nascent DNA was determined by Western blot.
In parallel with the experiments using BirAR118G-RAD18, we used iPOND (isolation of proteins on nascent DNA) and mass spectrometry to characterize the proteins recruited to stalled DNA replication forks (Sirbu et al. 2011). ZPET was identified as one of the proteins recruited to stalled forks in hydroxyurea (HU), a replication inhibitor (Supplemental Fig. S2C). To confirm the mass spectrometry data, we analyzed the iPOND samples by Western blot using ZPET and PCNA antibodies (Fig. 2E). As expected, PCNA was readily detected at unperturbed replication forks and reduced after HU treatment (Sirbu et al. 2011). Due to the cross-linking step of iPOND and the large size of ZPET, the ZPET in iPOND samples was detected as smears in Western blots. In contrast to PCNA, ZPET was detected only after HU treatment, suggesting that ZPET is recruited to stalled forks. The levels of ZPET captured by iPOND were reduced but not eliminated after thymidine chase, suggesting that some ZPET may remain associated with chromatin after replication (see the Discussion). The topoisomerase I inhibitor camptothecin (CPT), which induces replication-associated DSBs, also triggered ZPET binding to replication forks (Supplemental Fig. S2D). Thus, ZPET is recruited not only to DSBs but also to stalled and collapsed replication forks.
ZPET binds ssDNA in vitro and in cells
The localization of ZPET to DSBs and stalled replication forks suggests that ZPET may associate with a DNA structure commonly induced in these contexts. ssDNA is generated at DSBs by DNA end resection and also increasingly exposed at replication forks stalled by HU. To test whether ZPET binds ssDNA, we used biotinylated ssDNA to pull down proteins from cell extracts. ssDNA captured HA-ZPET along with endogenous RPA and ATR (Fig. 3A). In contrast, dsDNA did not bind HA-ZPET (Supplemental Fig. S3A). Furthermore, in electrophoresis mobility shift assay (EMSA), the HA-ZPET isolated with the HA tag bound to ssDNA but not dsDNA (Fig. 3B; Supplemental Fig. S3B). Together, these results suggest that ZPET is a ssDNA-binding protein.
Figure 3.

ZPET localizes to stalled forks by binding ssDNA. (A) HA-ZPET expression was induced by Dox in U2OS cells as indicated. Biotinylated ssDNA was used to capture proteins from cell extracts. The HA-ZPET, ATR, and RPA32 captured by ssDNA and in input extracts were detected by Western blot. (B, top panel) Increasing concentrations of HA-ZPET (35, 70, 140 nM) were incubated with 20 nM ssDNA (63 nucleotides) or 20 nM dsDNA (55 base pairs), and analyzed by EMSA. (Bottom panel) The complex formation of ZPET and DNA was quantified. The error bar represents the SD from two independent experiments. (C) HA-tagged wild-type ZPET (ZPETWT), ZPETΔZF2–4, and ZPETΔZF3-5 were transiently expressed in HEK293T cells and analyzed by biotin-ssDNA pull-down. (Top panel) A schema of the domains of ZPET and the ZPET mutants is shown. (Bottom panel) The levels of ZPET variants captured by ssDNA and in input extracts were analyzed by Western blot. (D) HA-tagged ZPETWT, ZPETΔZF2-4, and ZPETΔZF3-5 were transiently expressed in HEK293T cells and analyzed by iPOND after EdU labeling and HU treatment. The levels of HA-ZPET in iPOND samples and input extracts were analyzed by Western blot.
ZPET contains a domain of unknown function (DUF) at the N terminus and a cluster of five zinc fingers in the central region (Fig. 3C). The DUF of ZPET was not necessary or sufficient for ssDNA binding (Supplemental Fig. S3C). However, two ZPET mutants lacking three of the zinc fingers (ZPETΔZF2-4 and ZPETΔZF3-5) were defective for ssDNA binding compared with wild-type ZPET (ZPETWT) (Fig. 3C), suggesting that ZPET binds ssDNA through its zinc fingers. To test whether the ssDNA binding of ZPET is important for its localization, we compared ZPETWT, ZPETΔZF2-4, and ZPETΔZF3-5 using iPOND and immunostaining. In HU-treated cells, ZPETΔZF2-4 and ZPETΔZF3-5 were detected less efficiently than ZPETWT by iPOND (Fig. 3D). However, ZPETΔZF2-4 and ZPETΔZF3-5 were still detected at the TRE/I-SceI array when I-SceI was expressed (Supplemental Fig. S3D). Thus, the ssDNA binding of ZPET is important for its localization to stalled replication forks but not for its accumulation at DSBs. Although ZPET may bind ssDNA at resected DNA ends, its accumulation around DSBs likely occurs through a ssDNA-independent mechanism.
ZPET depletion accelerates resection-driven RPA phosphorylation
To assess the function of ZPET in the DDR, we knocked down ZPET with three independent siRNAs (Fig. 4A; Supplemental Fig. S4A). Knockdown of ZPET did not significantly alter the cell cycle (Supplemental Fig. S4B). ZPET knockdown cells and control cells were treated with 1 µM CPT to induce replication-associated DSBs. After 45 min of CPT treatment, γH2AX was similarly induced in control cells and ZPET knockdown cells (Fig. 4A), suggesting that the same levels of DSBs were generated in these cells. However, the phosphorylation of RPA32 at Ser4/8 was significantly enhanced in ZPET knockdown cells compared with control cells (Fig. 4A). Consistently, immunofluorescence analysis revealed that ZPET knockdown cells displayed an increase in phosphorylated RPA32 (p-RPA32 Ser33) foci after IR compared with control cells (Fig. 4B). RPA32 is progressively phosphorylated by ATR and DNA-PK during DNA end resection (Shiotani et al. 2013). The increase of RPA32 phosphorylation in ZPET knockdown cells after CPT or IR treatment suggests that ZPET may modulate DNA end resection.
Figure 4.

ZPET depletion accelerates resection-driven RPA phosphorylation. (A) U2OS cells were transfected with control and three independent ZPET siRNAs as indicated. Cells were treated with 1 µM CPT for 45 min, and cell lysates were analyzed by Western blot using the indicated antibodies. (B) U2OS cells were transfected as in A, untreated or treated with 10 Gy of IR, and analyzed by immunofluorescence using the p-RPA32 (S33) antibody. (C) Wild-type or ZPET knockout U2OS cells were treated with 1 µM CPT for the indicated durations. Cell lysates were analyzed by Western blot using the indicated antibodies. (D,E) Cells were analyzed as in A except where indicated and were pretreated with mirin (D) or transfected with CtIP siRNA (E) prior to CPT treatment.
To confirm the results from ZPET knockdown cells, we generated several independent ZPET knockout cell lines using CRISPR/Cas9. Consistent with ZPET knockdown cells, ZPET knockout cells also displayed an increase in RPA32 phosphorylation after 45–60 min of CPT treatment (Fig. 4C). Resection of CPT-induced DSBs is dependent on the MRE11 nuclease and CtIP, which associates with the MRE11–RAD50–NBS1 (MRN) complex (Sartori et al. 2007). Notably, while CPT-induced RPA32 phosphorylation was increased in ZPET knockdown cells, the p-RPA32 was nearly abolished by mirin, an inhibitor of the MRE11 nuclease (Fig. 4D). Furthermore, the p-RPA32 in ZPET knockdown cells was also significantly reduced by CtIP knockdown (Fig. 4E). These results suggest that the heightened RPA32 phosphorylation in ZPET-depleted cells is driven by MRE11- and CtIP-mediated DNA end resection. Thus, in the absence of ZPET, the resection of DNA ends by MRN-CtIP may be enhanced.
ZPET delays MRN-CtIP recruitment and slows DNA end resection
The increase of RPA32 phosphorylation in ZPET-depleted cells prompted us to test whether ZPET is an inhibitor of DNA end resection. To analyze the effects of ZPET protein on resection, we generated stable cell lines that inducibly express HA-ZPET. Cells were treated with doxycycline (Dox) to induce HA-ZPET expression and exposed to CPT for different lengths of time (Fig. 5A). Opposite to that in ZPET-depleted cells, CPT-induced RPA32 phosphorylation was reduced in cells expressing HA-ZPET compared with control cells. Notably, the reduction of RPA32 phosphorylation in HA-ZPET-expressing cells was most evident 30–60 min after CPT treatment (Fig. 5A). At 120 min after CPT treatment, the levels of p-RPA32 were similar in HA-ZPET-expressing cells and control cells (Fig. 5A), suggesting that ZPET slows but not does not block resection. Consistent with the Western results, induction of HA-ZPET reduced the immunofluorescence of p-RPA32 (Ser33) after CPT treatment (Fig. 5B). To rule out the possibility that HA-ZPET exerts dominant-negative effects on endogenous ZPET, we used a ZPET knockout cell line to generate secondary cell lines that inducibly express HA-ZPET (Fig. 5C). Even in the absence of endogenous ZPET, HA-ZPET reduced RPA32 phosphorylation 30–60 min after CPT treatment. These results further support the notion that ZPET slows DNA end resection.
Figure 5.
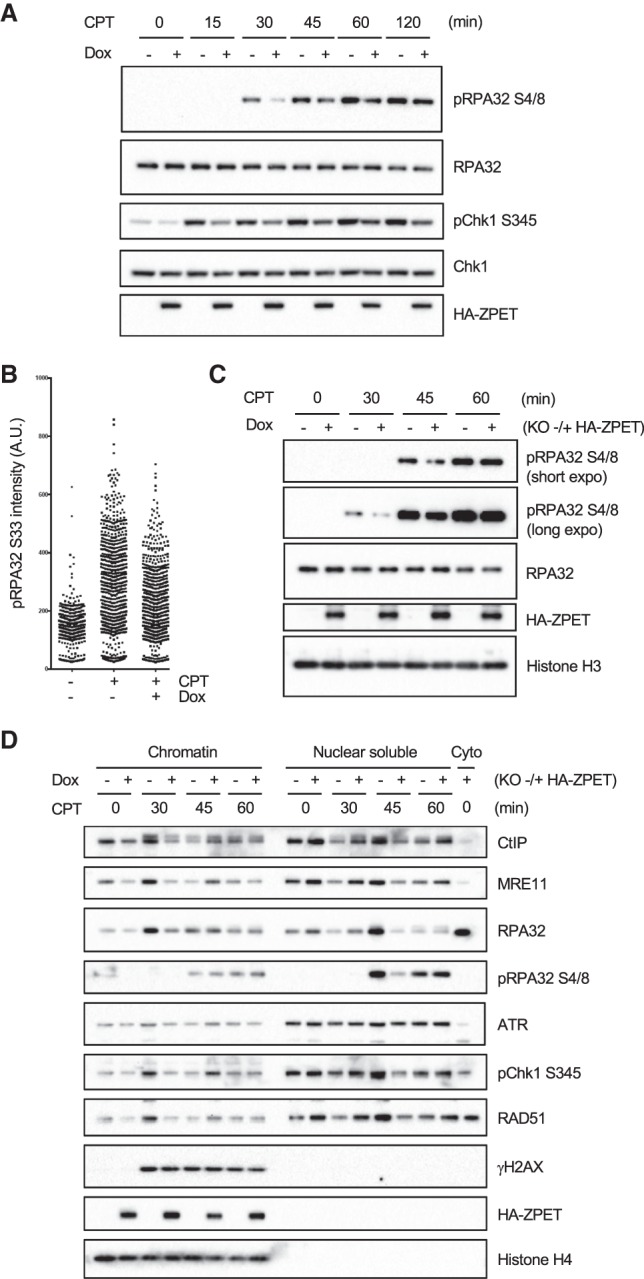
ZPET delays the binding of MRE11 and CtIP to chromatin and slows resection. (A) HA-ZPET expression was induced by Dox in U2OS cells as indicated. Cells were treated with CPT for the indicated durations. Cell lysates were analyzed by Western blot using the indicated antibodies. (B) Cells were plated on coverslips, treated with Dox and CPT as indicated, and analyzed by immunofluorescence using the p-RPA32 (S33) antibody. (C) ZPET knockout cells reconstituted with Dox-inducible HA-ZPET were cultured in the presence or absence of Dox. The cells were then treated with 1 µM CPT for the indicated durations, and whole-cell extracts were analyzed by Western blot using the indicated antibodies. (D) Cells were treated as in C and fractionated into chromatin, nuclear-soluble, and cytoplasmic fractions. The levels of the indicated proteins in these fractions were analyzed by Western blot.
To understand how ZPET slows DNA end resection, we analyzed the chromatin binding of a number of DDR proteins in ZPET knockout cells with or without HA-ZPET (Fig. 5D). In ZPET knockout cells without HA-ZPET, the levels of both MRE11 and CtIP on chromatin were increased 30 min after CPT treatment, indicating the recruitment of MRN-CtIP to DSBs. Concomitant with the recruitment of MRN-CtIP, RPA32 levels also increased on chromatin, indicating the initiation of resection. Levels of ATR, phosphorylated Chk1 (p-Chk1), and RAD51 on chromatin were also elevated 30 min after CPT treatment, suggesting that ssDNA triggered ATR activation and HR. Interestingly, p-RPA32 appeared in soluble nuclear fractions 45 min after CPT treatment, suggesting that phosphorylation of RPA promotes its turnover on chromatin. Importantly, the recruitment of MRN-CtIP to chromatin was significantly reduced by HA-ZPET in ZPET knockout cells 30 min after CPT treatment (Fig. 5D). At 45 min after CPT treatment, a delayed recruitment of MRE11 and CtIP was observed in ZPET knockout cells expressing HA-ZPET, although this recruitment was less efficient than that in ZPET knockout cells without HA-ZPET at 30 min. Consistent with the delay in MRN-CtIP recruitment, the binding of RPA32, ATR, p-Chk1, and RAD51 to chromatin were all delayed in ZPET knockout cells expressing HA-ZPET. Furthermore, the release of p-RPA32 from chromatin was also delayed by HA-ZPET in ZPET knockout cells. Thus, ZPET delays and dampens the recruitment of MRN-CtIP to chromatin during DNA end resection.
ZPET inhibits the association of MRE11 with ssDNA
ZPET may delay the recruitment of MRN-CtIP to DSBs by inhibiting its binding to DNA or chromatin. Since ZPET binds ssDNA, we next tested whether the ssDNA binding of ZPET is important for its role in slowing resection. At 45 min after CPT treatment, the levels of p-RPA32 were lower in cells expressing ZPETWT than in cells expressing ZPETΔZF2-4 or ZPETΔZF3-5 (Fig. 6A), suggesting that ZPET needs to bind ssDNA to inhibit resection.
Figure 6.

ZPET inhibits resection by binding ssDNA and functions independently of 53BP1 and HELB. (A) HA-tagged ZPETWT, ZPETΔZF2-4, and ZPETΔZF3-5 were transiently expressed in HEK293T cells. Cells were treated with 1 µM CPT for 45 min or left untreated, and the levels of the indicated proteins were analyzed by Western blot. (B) HA-ZPET expression was induced by Dox in U2OS cells as indicated. Biotinylated ssDNA was used to capture proteins from cell extracts containing both chromatin-bound and soluble fractions. The levels of HA-ZPET and MRE11 on ssDNA and in input extracts were analyzed by Western blot. (C) Wild-type or 53BP1 knockout U2OS cells were transfected with control or ZPET siRNA as indicated. Cells were treated with 1 µM CPT for 45 min or left untreated. Cell lysates were analyzed by Western blot using the indicated antibodies. (D) Wild-type and ZPET knockout lines were transfected with either control or HELB siRNA as indicated and analyzed by Western blot using the indicated antibodies.
During DNA end resection, MRN-CtIP first generates DNA nicks near DNA ends and then converts them into ssDNA gaps and 3′ ssDNA overhangs (Garcia et al. 2011; Cannavo and Cejka 2014; Anand et al. 2016). The association of MRN with ssDNA may be important for the conversion of ssDNA gaps into ssDNA overhangs. To test whether ZPET affects the ssDNA binding of MRN, we generated extracts from HA-ZPET-expressing cells and control cells and used biotinylated ssDNA to capture MRE11 from these extracts. Lower levels of MRE11 were captured by ssDNA from extracts of HA-ZPET-expressing cells (Fig. 6B), suggesting that ZPET inhibits the binding of MRE11 to ssDNA. This result provides a possible mechanism that reduces the association of MRN-CtIP with resection intermediates and hinders its function in resection.
ZPET inhibits resection independently of 53BP1 and HELB
In addition to ZPET, 53BP1 and HELB are also important inhibitors of resection. 53BP1 restricts DSB resection by recruiting RIF1 and PTIP (Bunting et al. 2010; Callen et al. 2013; Chapman et al. 2013; Di Virgilio et al. 2013; Escribano-Diaz et al. 2013; Zimmermann et al. 2013; Wang et al. 2014). The HELB helicase is recruited to DSBs by RPA, where it restricts resection by inhibiting EXO1 and DNA2 (Tkac et al. 2016). To understand whether ZPET functions with 53BP1 or HELB to inhibit resection, we depleted ZPET, 53BP1, and HELB individually or in combination and analyzed the effects on CPT-induced p-RPA32. In contrast to ZPET knockdown, loss of 53BP1 did not increase p-RPA32 after 45 min of CPT treatment (Fig. 6C, lanes 5–7), suggesting that ZPET and 53BP1 affect resection differently. In addition, p-RPA32 was similarly increased by ZPET knockdown in control cells and 53BP1 knockout cells (Fig. 6C, cf. lanes 5 and 7 and lanes 6 and 8), lending further support to the notion that ZPET inhibits resection independently of 53BP1. In contrast to 53BP1 depletion but similar to ZPET knockdown, knockdown of HELB increased p-RPA32 after 45 min of CPT treatment (Fig. 6D, lanes 5–7). Notably, codepletion of HELB and ZPET further increased p-RPA32 compared with depletion of HELB or ZPET alone (Fig. 6D, cf. lanes 6,7 and 8), suggesting that the roles of HELB and ZPET in resection regulation are not epistatic. Thus, ZPET functions independently of 53BP1 and HELB to restrict resection.
ZPET loss enhances HR and fork recovery
To determine whether the impact of ZPET on DNA end resection is functionally significant, we used the mClover assay to measure HR efficiency in ZPET knockout cells. In the mClover assay, a DSB is generated in the endogenous LAMINA gene by CRISPR/Cas9, and the coding sequence of mClover is inserted into the DSB via HR (Pinder et al. 2015). The HR-mediated insertion of mClover results in a fluorescent mClover-Lamin A fusion protein, which is used to score HR efficiency. Using the mClover assay, we found that HR efficiency was increased in ZPET knockout cell lines compared with a control line (Fig. 7A). Furthermore, expression of HA-ZPET in ZPET knockout cells reduced HR efficiency (Fig. 7A), confirming that the effects of ZPET knockout on HR are specific. Together, these results suggest that ZPET not only inhibits resection but also reduces HR efficiency.
Figure 7.

ZPET represses HR, slows replication forks under stress, and increases cellular sensitivity to CPT and olaparib. (A) Wild-type cells, ZPET knockout cells, and ZPET knockout cells reconstituted with HA-ZPET were analyzed for HR efficiency using the mClover assay. The left panel is a schema of the mClover assay. In the right panel, each data point represents an independent sample. Red and blue data points were generated using two subclones of reconstituted ZPET knockout cells (>900 cells were analyzed for each sample). (B) ZPET knockout cells reconstituted with Dox-inducible HA-ZPET were sequentially labeled with CIdU and IdU as indicated. Cells were treated with 250 µM HU or 2.5 µM CPT during IdU labeling. (Right panel) The length of replication tracts before HU/CPT treatments (CIdU-labeled) was analyzed. (Left and middle panels) The effects of HU/CPT on replication tracts (IdU/CIdU ratios) were analyzed. The length of 200 replication tracts were measured. n = 200. (Black bars) Mean length or mean ratio. Significance was determined by Mann-Whitney test. (****) P < 0.0001; (ns) not significant. (C) Wild-type and ZPET knockout U2OS lines were treated with increasing doses of CPT. Cell viability was analyzed in 7 d. Error bars represent SD. n = 3 technical triplicates. (D) ZPET knockout and reconstituted cells were treated with increasing doses of CPT. Cell viability was analyzed in 7 d. (E) Wild-type and ZPET knockout cells were treated with increasing concentrations of olaparib. Cell viability was analyzed in 7 d. (F) Wild-type and ZPET knockout U2OS cells were transfected with control or BRCA1 siRNA as indicated and treated with increasing doses of olaparib. Cell viability was analyzed in 5 d.
Since ZPET is recruited to stalled and collapsed replication forks, we asked whether ZPET is functionally important for fork dynamics. We analyzed replication fork progress in ZPET knockout cells with or without HA-ZPET using DNA fiber assay. Before cells were exposed to HU or CPT, HA-ZPET did not significantly alter fork speed (Fig. 7B, right panel). However, after cells were treated with HU or CPT, HA-ZPET reduced fork progression (Fig. 7B, left and middle panels). These results suggest that ZPET suppresses the processing of stalled/collapsed replication forks, thereby slowing fork recovery and progression.
ZPET loss promotes cellular resistance to DSBs and PARP inhibition
The impact of ZPET on HR and fork progression in CPT prompted us to test whether ZPET affects CPT sensitivity. We treated ZPET knockout and control cells with increasing concentrations of CPT and analyzed cell survival in 7 d (Fig. 7C). Multiple independent ZPET knockout cell lines displayed increased survival after CPT treatment compared with the control line, showing that the lack of ZPET promotes cell survival in response to replication-associated DSBs. Furthermore, expression of HA-ZPET in ZPET knockout cells reduced cell survival after CPT treatment (Fig. 7D), confirming that the effects of ZPET knockout on CPT sensitivity are specific. Similar cell survival results were also obtained when ZPET knockout and control cells were briefly treated with high concentrations of CPT (Supplemental Fig. S5A,B). Thus, consistent with the role of ZPET in restricting HR and fork recovery, cells lacking ZPET are more resistant to CPT-induced DSBs.
Cells defective in HR are susceptible to PARP inhibition. To assess whether ZPET influences the sensitivity of cells to PARP inhibition, we analyzed the survival of ZPET knockout and control cells in the presence of olaparib, a PARP inhibitor (PARPi) (Fig. 7E). Consistent with the increased HR in ZPET knockout cells, ZPET knockout cells were more resistant to olaparib than control cells in the 7-d cell survival assay. Furthermore, expression of HA-ZPET in ZPET knockout cells reduced cell survival in olaparib (Supplemental Fig. S5C), confirming that the effects of ZPET knockout on olaparib sensitivity are specific. The increase of olaparib resistance in ZPET knockout cells raises the question of whether loss of ZPET causes PARPi resistance in BRCA1-deficient cells. To test this possibility, we used siRNA to knock down BRCA1 in ZPET knockout and control cells (Fig. 7F). Because BRCA1 depletion reduces cell survival in long-term assays, we analyzed olaparib sensitivity in 5 d instead of 7 d. As expected, knockdown of BRCA1 drastically increased olaparib sensitivity (Fig. 7F; Yazinski et al. 2017). Unlike in the 7-d assay, ZPET knockout cells did not display an increase in olaparib resistance in the 5-d assay, probably owing to the smaller number of cell divisions in olaparib. However, compared with control cells, ZPET knockout cells were significantly more resistant to olaparib after BRCA1 knockdown in the 5-d assay. Together, these results suggest that loss of ZPET may contribute to the PARPi resistance of BRCA1-deficient cells by enhancing HR.
Discussion
The use of localized protein biotinylation to identify DDR proteins
Identification of DDR proteins is one of the major tasks for the research of DDR. Numerous DDR proteins have been identified by coimmunoprecipitations and tandem affinity purifications of known DDR proteins (Wang et al. 2000; Cortez et al. 2001; Meetei et al. 2003; Xia et al. 2006; Marechal et al. 2014; Tkac et al. 2016). These biochemical approaches rely on stable protein–protein interactions and may not be effective for identifying proteins that transiently or weakly interact with bait proteins. In addition, some DDR proteins may not interact with any known DDR proteins at all, making them impossible to capture with the available baits. Many known DDR proteins localize to sites of DNA damage to execute their functions in DNA repair and DNA damage signaling (Bekker-Jensen et al. 2006). This common spatial feature of DDR proteins provides another opportunity to identify unknown proteins involved in the DDR. The promiscuous BirAR118G mutant offers a unique strategy to biotinylate proteins in proximity to BirAR118G fusions. In this study, we used BirAR118G-RAD18 as a proof of principle to show that localized protein biotinylation at DNA damage sites can be used to identify previously unknown DDR proteins. This strategy may capture proteins that transiently or weakly interact with any given bait and even proteins at DNA damage sites that do not interact with the bait at all, overcoming the limitations of affinity-based approaches. Recently, the biotin ligase APEX2 has also been used to capture complexes of DDR proteins (Gupta et al. 2018). Together, these studies have opened a new avenue for the identification of uncharacterized DDR proteins.
ZPET mediates a ssDNA-triggered feedback loop to restrict resection
In this study, we identified ZPET as a repressor of DNA end resection. Interestingly, ZPET is a ssDNA-binding protein, and its function in slowing resection is dependent on its ssDNA binding. Together, these findings suggest that ZPET mediates a ssDNA-triggered feedback loop to restrict resection. Although ZPET and HELB function independently to restrict resection, their roles are remarkably similar. Whereas ZPET binds ssDNA, HELB is recruited by RPA (Tkac et al. 2016). Both proteins are required for preventing hyperresection during the early phase of DSB response. The existence of these feedback loops suggests that the proper control of resection is critical for the DDR.
Why do cells need a ZPET-mediated feedback loop to restrict resection? The restriction of resection by ZPET may control the choice between different DSB repair pathways. Alternatively, ZPET may inhibit HR during a specific window of the cell cycle or in specific regions of the genome. Furthermore, ZPET may prevent excessive resection, which may lead to aberrant or inaccurate repair. We cannot exclude the possibility that ZPET has other functions and that its impacts on DNA end resection are merely side effects. Understanding whether ZPET has a physiological role in protecting the genome in specific contexts is an important task for future investigations. In pathological and therapeutic contexts, such as in BRCA1-deficient cancer cells and in response to PARPi and CPT treatments, ZPET may interfere with the HR pathway, and loss of ZPET may allow cells to tolerate genomic instability and develop drug resistance.
It is worth noting that the ZPET may be regulated by multiple mechanisms during the DDR. In previous proteomics studies, ZNF280C was identified as a potential substrate of ATM/ATR kinases (Matsuoka et al. 2007), a protein ubiquitinated in a DNA damage-induced manner (Mandemaker et al. 2017), and a protein deSUMOylated after DNA damage (Hendriks et al. 2015). These results suggest that ZPET may be a node of regulation during the DDR.
ZPET inhibits resection as a ssDNA-binding protein
The ability of ZPET to bind ssDNA is important for its function to slow resection. During resection, MRN-CtIP first generates DNA nicks near DSB ends and then acts as a 3′-to-5′ exonuclease to convert DNA nicks into 3′ ssDNA overhangs (Garcia et al. 2011; Cannavo and Cejka 2014; Anand et al. 2016). The association of MRN-CtIP with resection intermediates, such as ssDNA gaps, may be important for its function in resection. In cell extracts, ZPET inhibits the binding of MRE11 to ssDNA. In cells, ZPET delays and attenuates the recruitment of MRE11 and CtIP to chromatin. These results provide a possible mechanism to explain the inhibitory effects of ZPET on resection. We noted that many of the DDR proteins that were biotinylated by BirAR118G-RAD18 after IR are known to associate with the chromatin flanking DSBs, suggesting that ZPET may be present in this chromatin environment. Consistent with the possibility that ZPET spreads on chromatin, induction of AsiSI-mediated DSB formation led to small increases of ZPET at loci distal to AsiSI sites (Supplemental Fig. S2B), and ZPET remained bound to replicated DNA in iPOND after thymidine chase (Fig. 2E). The binding of ZPET to chromatin may also explain the localization of ZPET ssDNA-binding mutants to DSBs (Supplemental Fig. S3D). MDC1, the top hit of BirAR118G-RAD18, mediates the recruitment of MRN to chromatin (Chapman and Jackson 2008; Wu et al. 2008), raising the possibility that ZPET may act on chromatin to hinder MRN recruitment. In a previous proteomic study, ZPET was shown to bind major satellites in a SUV39H-dependent manner, suggesting that it may have an affinity for H3K9me3-marked chromatin (Saksouk et al. 2014). Although the induction of H3K9me3 by DSBs was observed only by some studies but not others (Ayrapetov et al. 2014; Clouaire et al. 2018), the H3K9me3 reader protein HP1α is recruited to DNA damage sites (Baldeyron et al. 2011).
In addition to DSBs, ZPET is also recruited to stalled and collapsed replication forks. In the absence of ZPET, replication forks progress faster in HU and CPT, suggesting that ZPET slows a rate-limiting event during the recovery of stalled/collapsed forks. It is tempting to speculate that ZPET inhibits the resection of reversed replication forks and/or fork-derived one-ended DSBs. While loss of ZPET allows efficient fork recovery under stress, the function of ZPET at stalled/collapsed forks may be important for suppressing mutagenic processes, such as break-induced replication (BIR) and single-strand annealing (SSA), in specific regions of the genome.
Although ZPET and 53BP1 function independently to restrict resection, they share a surprising similarity. Recently, several studies suggested that the 53BP–RIF1–REV7 pathway inhibits resection through the Shieldin complex (Dev et al. 2018; Ghezraoui et al. 2018; Gupta et al. 2018; Mirman et al. 2018; Noordermeer et al. 2018). SHLD2, a component of Shieldin, is a ssDNA-binding protein like ZPET (Noordermeer et al. 2018). Notably, the ssDNA binding of SHLD2 is critical for Shieldin function, suggesting that Shieldin, like ZPET, also inhibits resection by binding ssDNA. Furthermore, another ssDNA-binding complex, CST (CTC1–STN1–TEN1), also contributes to the inhibition of resection (Mirman et al. 2018). Together, these studies on Shieldin, CST, and ZPET highlight the importance of ssDNA-binding proteins in the regulation of resection.
Resection kinetics and HR efficiency
Interestingly, while the effects of ZPET on resection are largely restricted to the early phase of this process, ZPET affects HR efficiency significantly. This observation raises several non-mutually exclusive possibilities. First, the rapid formation of long ssDNA overhangs may be important for high HR efficiency. RPA may be recruited more efficiently when long ssDNA is rapidly exposed, favoring HR over other resection-dependent repair pathways, such as alternative NHEJ (Alt-NHEJ), SSA, and BIR. Second, the swift activation of the ATR–Chk1 pathway may be important for efficient HR. The ATR–Chk1 pathway is known to elicit a PP2A-mediated feedback loop to restrict Chk1 activity (Leung-Pineda et al. 2006). Fast resection may allow ATR to promote HR more efficiently before the PP2A-mediated feedback loop is fully active. Finally, the HR pathway may be more efficient during the early phase of DSB response because the functionality of certain HR proteins may decline over time. For example, RAD51 is ubiquitinated by the E3 ligase RFWD3 after DSB formation (Inano et al. 2017), but RAD51 ubiquitination interferes with the BRCA2–RAD51 interaction (Luo et al. 2016). In light of this study, it is tempting to speculate that the kinetics of DNA end resection during the early phase of DSB response is an important determinant of HR efficiency, presenting an enticing model for future investigations.
Materials and methods
Cell lines
U2OS cells lines stably expressing BirAR118G-RAD18 or BirAR118G-GFP were generated using linearized plasmids and selected with 1 µg/mL puromycin. Clones expressing similar levels of BirAR118G-RAD18 or BirAR118G-GFP were chosen for comparison.
Antibodies
The following antibodies were used in this study: histone H4 (Abcam, ab31830), ZNF280C (Abcam, ab83287), PCNA (Santa Cruz Biotechnology, Sc-56), p-Chk1 (S345) (Cell Signaling, 2348), GAPDH (Millipore), histone H3 (Abcam), RPA34 (Thermo Scientific, MA1-26418), MDC1 (Bethyl Laboratories, A300-051A), ATR (Bethyl Laboratories, A300-138A), CtIP (Abcam, ab155988), MRE11 (Genetex), RPA32 (S4/8) (Bethyl Laboratories, A300-245), phospho-histone H2A.X (Ser139) (20E3), HA (Covance, HA.11), and GFP (Invitrogen, A11122).
Biotinylation assay
Cells were either left untreated or treated with 10 Gy of IR and allowed to recover for 1 h. Cells were incubated in 0.5–1 µg/mL biotin for 4 h and then harvested. Where indicated, crude chromatin extracts were generated by first pre-extracting cells with 0.5% Triton X-100 PBS for 5 min. Pre-extracted cells or intact cells were lysed in 1% SDS, 20 mM Tris (pH 8.0) supplemented with 10 mM DTT, and protease inhibitor cocktail and a phosphatase inhibitor cocktail for 10 min. Cell lysates were sonicated three times at 60% power using a cup sonicator following a cycle of 30 sec on and 30 sec off. Lysates were boiled for 10 min and vortexed every 3 min. The lysates were then diluted in NETN lysis buffer (20 mM Tris at pH 8.0, 150 mM NaCl, 0.5% NP-40, 10% glycerol), precleared with protein A-conjugated magnetic beads, and then subjected to biotin pull-down with streptavidin-conjugated magnetic beads (M-280). Pull-downs were washed twice with 1% SDS RIPA, twice with 1% SDS RIPA high salt, and once with 1% SDS RIPA. Pull-downs were then boiled in sample buffer and subjected to mass spectrometry analysis or Western blot.
Quantitative mass spectrometry and data analysis
Proteins enriched by streptavidin beads were suspended in 3% SDA, 4 M urea, and 50 mM HEPES (pH 8.5). Proteins were reduced and alkylated, purified by precipitation, and digested with Lys-C and trypsin (Edwards and Haas 2016). Peptides were labeled with TMT-10plex reagents as follows (McAlister et al. 2012): sample 26B_1 (126), 26BR_1 (127n), 64BR_1 (127c), 26B_2 (128n), 26BR_2 (128c), and 64BR_2 (129n). Labeled peptides were pooled and fractionated by basic reversed-phase high-performance liquid chromatography (HPLC). Twelve fractionations were analyzed by multiplexed quantitative proteomics performed on an Orbitrap Fusion mass spectrometer (Thermo Scientifc) using a simultaneous precursor selection (SPS)-based MS3 method (Ting et al. 2011; McAlister et al. 2014).
MS2 data were annotated using SEQUEST searching data against the UniProt database of human protein sequences (Eng et al. 1994; Huttlin et al. 2010). A <1% false discovery filter for both peptide and protein assignments was applied through the target decoy database search strategy using linear discriminant analysis and posterior error histogram sorting (Elias and Gygi 2007; Huttlin et al. 2010). Peptides with unambiguous protein annotations were assigned to the protein with the most matching peptides (Huttlin et al. 2010). For quantification, we extracted TMT reporter ion intensities as those of the most intense ions within a 0.03-Th window around the predicted MS3 reporter ion intensities. Only MS3 with an average signal to noise value >40 per reporter ion as well as with an isolation specificity >0.75 were considered for quantification (Ting et al. 2011). TMT intensities were first normalized for each protein based on the median average protein intensity calculated for all proteins. Next, a median of the normalized intensities was calculated from all protein intensities in each TMT channel, and the protein intensities were normalized to the median value of these median intensities.
Supplementary Material
Acknowledgments
We thank members of the Zou, Dyson and Elia laboratories for helpful discussions. D.M.M. is supported by a National Institutes of Health (NIH) F32 post-doctoral fellowship (5F32CA192779). R.B. is supported by an NIH K99 award (1K99CA212154). M.-M.G. is partly supported by a post-doctoral fellowship from Fonds de recherche Santé Québec (FRQS). L.Z. is the James and Patricia Endowed Chair in Cancer Research and a Jim and Ann Orr Massachusetts General Hospital (MGH) Research Scholar. This work is supported by NIH grants GM067388 and CA197779 to L.Z.
Author contributions: L.Z. designed and supervised the project. D.M.M., M.-M.G., J.-M.Z., J.O., T.Y., R.B., S.A.Y., J.T., M.B., and J.-P.G. performed the experiments or data analysis. G.G.P., L.L., and W.H. helped supervise the experiments.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.315978.118.
References
- Anand R, Ranjha L, Cannavo E, Cejka P. 2016. Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol Cell 64: 940–950. 10.1016/j.molcel.2016.10.017 [DOI] [PubMed] [Google Scholar]
- Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP, et al. 2014. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol 21: 366–374. 10.1038/nsmb.2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD. 2014. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci 111: 9169–9174. 10.1073/pnas.1403565111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldeyron C, Soria G, Roche D, Cook AJ, Almouzni G. 2011. HP1α recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J Cell Biol 193: 81–95. 10.1083/jcb.201101030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J. 2006. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol 173: 195–206. 10.1083/jcb.200510130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R, Niraj J, Pauty J, Maity R, Zhao W, Coulombe Y, Sung P, Masson JY. 2014. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase η in recombination-associated DNA synthesis at blocked replication forks. Cell Rep 6: 553–564. 10.1016/j.celrep.2014.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 2010. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141: 243–254. 10.1016/j.cell.2010.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M, Starnes LM, et al. 2013. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 153: 1266–1280. 10.1016/j.cell.2013.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Cejka P. 2014. Sae2 promotes dsDNA endonuclease activity within Mre11–Rad50–Xrs2 to resect DNA breaks. Nature 514: 122–125. 10.1038/nature13771 [DOI] [PubMed] [Google Scholar]
- Chapman JR, Jackson SP. 2008. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep 9: 795–801. 10.1038/embor.2008.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. 2013. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 49: 858–871. 10.1016/j.molcel.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman-Smith A, Cronan JE Jr. 1999. The enzymatic biotinylation of proteins: a post-translational modification of exceptional specificity. Trends Biochem Sci 24: 359–363. 10.1016/S0968-0004(99)01438-3 [DOI] [PubMed] [Google Scholar]
- Chen J, Feng W, Jiang J, Deng Y, Huen MS. 2012. Ring finger protein RNF169 antagonizes the ubiquitin-dependent signaling cascade at sites of DNA damage. J Biol Chem 287: 27715–27722. 10.1074/jbc.M112.373530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi-Rhee E, Schulman H, Cronan JE. 2004. Promiscuous protein biotinylation by Escherichia coli biotin protein ligase. Protein Sci 13: 3043–3050. 10.1110/ps.04911804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204. 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouaire T, Rocher V, Lashgari A, Arnould C, Aguirrebengoa M, Biernacka A, Skrzypczak M, Aymard F, Fongang B, Dojer N, et al. 2018. Comprehensive mapping of histone modifications at DNA double-strand breaks deciphers repair pathway chromatin signatures. Mol Cell 72: 250–262.e6. 10.1016/j.molcel.2018.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ. 2001. ATR and ATRIP: partners in checkpoint signaling. Science 294: 1713–1716. 10.1126/science.1065521 [DOI] [PubMed] [Google Scholar]
- Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, Coates J, Sczaniecka-Clift M, Wei W, Ostermaier M, et al. 2018. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol 20: 954–965. 10.1038/s41556-018-0140-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. 2013. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339: 711–715. 10.1126/science.1230624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong JM, Tay FP, Swa HL, Gunaratne J, Leung T, Burke B, Manser E. 2016. Proximity biotinylation provides insight into the molecular composition of focal adhesions at the nanometer scale. Sci Signal 9: rs4 10.1126/scisignal.aaf3572 [DOI] [PubMed] [Google Scholar]
- Dubois ML, Bastin C, Lévesque D, Boisvert FM. 2016. Comprehensive characterization of minichromosome maintenance complex (MCM) protein interactions using affinity and proximity purifications coupled to mass spectrometry. J Proteome Res 15: 2924–2934. 10.1021/acs.jproteome.5b01081 [DOI] [PubMed] [Google Scholar]
- Duffy S, Tsao KL, Waugh DS. 1998. Site-specific, enzymatic biotinylation of recombinant proteins in Spodoptera frugiperda cells using biotin acceptor peptides. Anal Biochem 262: 122–128. 10.1006/abio.1998.2770 [DOI] [PubMed] [Google Scholar]
- Edwards A, Haas W. 2016. Multiplexed quantitative proteomics for high-throughput comprehensive proteome comparisons of human cell lines. Methods Mol Biol 1394: 1–13. 10.1007/978-1-4939-3341-9_1 [DOI] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. 2007. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4: 207–214. 10.1038/nmeth1019 [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR. 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5: 976–989. 10.1016/1044-0305(94)80016-2 [DOI] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. 2013. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 49: 872–883. 10.1016/j.molcel.2013.01.001 [DOI] [PubMed] [Google Scholar]
- Firat-Karalar EN, Rauniyar N, Yates JR III, Stearns T. 2014. Proximity interactions among centrosome components identify regulators of centriole duplication. Curr Biol 24: 664–670. 10.1016/j.cub.2014.01.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia V, Phelps SE, Gray S, Neale MJ. 2011. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 479: 241–244. 10.1038/nature10515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Exposito L, Bournique E, Bergoglio V, Bose A, Barroso-Gonzalez J, Zhang S, Roncaioli JL, Lee M, Wallace CT, Watkins SC, et al. 2016. Proteomic profiling reveals a specific role for translesion DNA polymerase η in the alternative lengthening of telomeres. Cell Rep 17: 1858–1871. 10.1016/j.celrep.2016.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, Fischer R, Deobagkar-Lele M, Sanchiz-Calvo M, Fueyo-Marcos E, et al. 2018. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 560: 122–127. 10.1038/s41586-018-0362-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J, Jackson SP. 2003. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 421: 952–956. 10.1038/nature01445 [DOI] [PubMed] [Google Scholar]
- Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, Typas D, Lammers M, Mailand N, Nussenzweig A, et al. 2018. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell 173: 972–988.e23. 10.1016/j.cell.2018.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks IA, Treffers LW, Verlaan-de Vries M, Olsen JV, Vertegaal AC. 2015. SUMO-2 orchestrates chromatin modifiers in response to DNA damage. Cell Rep 10: 1778–1791. 10.1016/j.celrep.2015.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Huen MS, Kim H, Leung CC, Glover JN, Yu X, Chen J. 2009. RAD18 transmits DNA damage signalling to elicit homologous recombination repair. Nat Cell Biol 11: 592–603. 10.1038/ncb1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. 2007. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131: 901–914. 10.1016/j.cell.2007.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP. 2010. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143: 1174–1189. 10.1016/j.cell.2010.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki A, van Cappellen WA, van der Laan R, Houtsmuller AB, Hoeijmakers JH, Grootegoed JA, Baarends WM. 2009. Dynamic localization of human RAD18 during the cell cycle and a functional connection with DNA double-strand break repair. DNA Repair (Amst) 8: 190–201. 10.1016/j.dnarep.2008.10.008 [DOI] [PubMed] [Google Scholar]
- Inano S, Sato K, Katsuki Y, Kobayashi W, Tanaka H, Nakajima K, Nakada S, Miyoshi H, Knies K, Takaori-Kondo A, et al. 2017. RFWD3-mediated ubiquitination promotes timely removal of both RPA and RAD51 from DNA damage sites to facilitate homologous recombination. Mol Cell 66: 622–634.e8. 10.1016/j.molcel.2017.04.022 [DOI] [PubMed] [Google Scholar]
- Kannouche PL, Wing J, Lehmann AR. 2004. Interaction of human DNA polymerase η with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 14: 491–500. 10.1016/S1097-2765(04)00259-X [DOI] [PubMed] [Google Scholar]
- Kim H, Chen J, Yu X. 2007. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 316: 1202–1205. 10.1126/science.1139621 [DOI] [PubMed] [Google Scholar]
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al. 2007. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318: 1637–1640. 10.1126/science.1150034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JP, Tucholska M, Go C, Knight JD, Gingras AC. 2015. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J Proteomics 118: 81–94. 10.1016/j.jprot.2014.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapek JD Jr, Greninger P, Morris R, Amzallag A, Pruteanu-Malinici I, Benes CH, Haas W. 2017. Detection of dysregulated protein-association networks by high-throughput proteomics predicts cancer vulnerabilities. Nat Biotechnol 35: 983–989. 10.1038/nbt.3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung-Pineda V, Ryan CE, Piwnica-Worms H. 2006. Phosphorylation of Chk1 by ATR is antagonized by a Chk1-regulated protein phosphatase 2A circuit. Mol Cell Biol 26: 7529–7538. 10.1128/MCB.00447-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Wu X, Chen J. 2003. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature 421: 957–961. 10.1038/nature01447 [DOI] [PubMed] [Google Scholar]
- Luo K, Li L, Li Y, Wu C, Yin Y, Chen Y, Deng M, Nowsheen S, Yuan J, Lou Z. 2016. A phosphorylation-deubiquitination cascade regulates the BRCA2–RAD51 axis in homologous recombination. Genes Dev 30: 2581–2595. 10.1101/gad.289439.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandemaker IK, van Cuijk L, Janssens RC, Lans H, Bezstarosti K, Hoeijmakers JH, Demmers JA, Vermeulen W, Marteijn JA. 2017. DNA damage-induced histone H1 ubiquitylation is mediated by HUWE1 and stimulates the RNF8-RNF168 pathway. Sci Rep 7: 15353 10.1038/s41598-017-15194-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, Li JM, Ji XY, Wu CS, Yazinski SA, Nguyen HD, Liu S, Jimenez AE, Jin J, Zou L. 2014. PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol Cell 53: 235–246. 10.1016/j.molcel.2013.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316: 1160–1166. 10.1126/science.1140321 [DOI] [PubMed] [Google Scholar]
- McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, et al. 2012. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem 84: 7469–7478. 10.1021/ac301572t [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP. 2014. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86: 7150–7158. 10.1021/ac502040v [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwraith MJ, West SC. 2008. DNA repair synthesis facilitates RAD52-mediated second-end capture during DSB repair. Mol Cell 29: 510–516. 10.1016/j.molcel.2007.11.037 [DOI] [PubMed] [Google Scholar]
- Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W. 2003. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol 23: 3417–3426. 10.1128/MCB.23.10.3417-3426.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, de Lange T. 2018. 53BP1–RIF1–shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature 560: 112–116. 10.1038/s41586-018-0324-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Alvarez-Quilon A, Moatti N, Zimmermann M, et al. 2018. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560: 117–121. 10.1038/s41586-018-0340-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinder J, Salsman J, Dellaire G. 2015. Nuclear domain ‘knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res 43: 9379–9392. 10.1093/nar/gkv993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, Burke B. 2012. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196: 801–810. 10.1083/jcb.201112098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saksouk N, Barth TK, Ziegler-Birling C, Olova N, Nowak A, Rey E, Mateos-Langerak J, Urbach S, Reik W, Torres-Padilla ME, et al. 2014. Redundant mechanisms to form silent chromatin at pericentromeric regions rely on BEND3 and DNA methylation. Mol Cell 56: 580–594. 10.1016/j.molcel.2014.10.001 [DOI] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. 2007. Human CtIP promotes DNA end resection. Nature 450: 509–514. 10.1038/nature06337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Ganesan S, Brown M, De Caprio JA, Cannistra SA, Feunteun J, Schnitt S, Livingston DM. 1996. Location of BRCA1 in human breast and ovarian cancer cells. Science 272: 123–126. 10.1126/science.272.5258.123 [DOI] [PubMed] [Google Scholar]
- Shiotani B, Nguyen HD, Hakansson P, Marechal A, Tse A, Tahara H, Zou L. 2013. Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep 3: 1651–1662. 10.1016/j.celrep.2013.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoaib M, Kulyyassov A, Robin C, Winczura K, Tarlykov P, Despas E, Kannouche P, Ramanculov E, Lipinski M, Ogryzko V. 2013. PUB-NChIP—‘in vivo biotinylation’ approach to study chromatin in proximity to a protein of interest. Genome Res 23: 331–340. 10.1101/gr.134874.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. 2011. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25: 1320–1327. 10.1101/gad.2053211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. 2007. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316: 1198–1202. 10.1126/science.1139516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. 2003. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421: 961–966. 10.1038/nature01446 [DOI] [PubMed] [Google Scholar]
- Teng Y, Yadav T, Duan M, Tan J, Xiang Y, Gao B, Xu J, Liang Z, Liu Y, Nakajima S, et al. 2018. ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat Commun 9: 4115 10.1038/s41467-018-06586-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L, Jun H, Junjie C. 2010. RAD18 lives a double life: its implication in DNA double-strand break repair. DNA Repair (Amst) 9: 1241–1248. 10.1016/j.dnarep.2010.09.016 [DOI] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, Haas W. 2011. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods 8: 937–940. 10.1038/nmeth.1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkac J, Xu G, Adhikary H, Young JTF, Gallo D, Escribano-Diaz C, Krietsch J, Orthwein A, Munro M, Sol W, et al. 2016. HELB is a feedback inhibitor of DNA end resection. Mol Cell 61: 405–418. 10.1016/j.molcel.2015.12.013 [DOI] [PubMed] [Google Scholar]
- Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. 2000. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 14: 927–939. [PMC free article] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 2002. 53BP1, a mediator of the DNA damage checkpoint. Science 298: 1435–1438. 10.1126/science.1076182 [DOI] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. 2007. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316: 1194–1198. 10.1126/science.1139476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Aroumougame A, Lobrich M, Li Y, Chen D, Chen J, Gong Z. 2014. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev 28: 2693–2698. 10.1101/gad.252478.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. 2004. Rad18 guides polη to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J 23: 3886–3896. 10.1038/sj.emboj.7600383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Iwabuchi K, Sun J, Tsuji Y, Tani T, Tokunaga K, Date T, Hashimoto M, Yamaizumi M, Tateishi S. 2009. RAD18 promotes DNA double-strand break repair during G1 phase through chromatin retention of 53BP1. Nucleic Acids Res 37: 2176–2193. 10.1093/nar/gkp082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Nakajima S, Böhm S, Bernstein KA, Shen Z, Tsang M, Levine AS, Lan L. 2015. DNA damage during the G0/G1 phase triggers RNA-templated, Cockayne syndrome B-dependent homologous recombination. Proc Natl Acad Sci 112: E3495–E3504. 10.1073/pnas.1507105112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SA, Longerich S, Sung P, Vaziri C, Kupfer GM. 2011. The E3 ubiquitin ligase RAD18 regulates ubiquitylation and chromatin loading of FANCD2 and FANCI. Blood 117: 5078–5087. 10.1182/blood-2010-10-311761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Luo K, Lou Z, Chen J. 2008. MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc Natl Acad Sci 105: 11200–11205. 10.1073/pnas.0802885105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. 2006. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 22: 719–729. 10.1016/j.molcel.2006.05.022 [DOI] [PubMed] [Google Scholar]
- Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A, et al. 2017. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev 31: 318–332. 10.1101/gad.290957.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 2013. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339: 700–704. 10.1126/science.1231573 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.