

Abstract
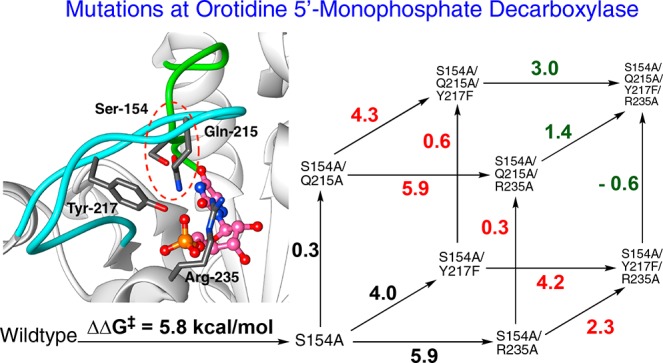
We report the results of a study of the catalytic role of a network of four interacting amino acid side chains at yeast orotidine 5′-monophosphate decarboxylase (ScOMPDC), by the stepwise replacement of all four side chains. The H-bond, which links the −CH2OH side chain of S154 from the pyrimidine umbrella loop of ScOMPDC to the amide side chain of Q215 in the phosphodianion gripper loop, creates a protein cage for the substrate OMP. The role of this interaction in optimizing transition state stabilization from the dianion gripper side chains Q215, Y217, and R235 was probed by determining the kinetic parameter kcat/Km for 16 enzyme variants, which include all combinations of single, double, triple, and quadruple S154A, Q215A, Y217F, and R235A mutations. The effects of consecutive Q215A, Y217F, and R235A mutations on ΔG⧧ for wild-type enzyme-catalyzed decarboxylation sum to 11.6 kcal/mol, but to only 7.6 kcal/mol when starting from S154A mutant. This shows that the S154A mutation results in a (11.6–7.6) = 4.0 kcal/mol decrease in transition state stabilization from interactions with Q215, Y217, and R235. Mutant cycles show that ca. 2 kcal/mol of this 4 kcal/mol effect is from the direct interaction between the S154 and Q215 side chains and that ca. 2 kcal/mol is from a tightening in the stabilizing interactions of the Y217 and R235 side chains. The sum of the effects of individual A154S, A215Q, F217Y and A235R substitutions at the quadruple mutant of ScOMPDC to give the corresponding triple mutants, 5.6 kcal/mol, is much smaller than 16.0 kcal/mol, the sum of the effects of the related four substitutions in wild-type ScOMPDC to give the respective single mutants. The small effect of substitutions at the quadruple mutant is consistent with a large entropic cost to holding the flexible loops of ScOMPDC in the active closed conformation.
Introduction
Orotidine 5′-monophosphate decarboxylase (OMPDC) employs no metal ions or other cofactors, yet results in an enormous 31 kcal/mol stabilization of the transition state for the chemically very difficult decarboxylation of orotidine 5′-monophosphate (OMP) to give uridine 5′-monophosphate (UMP).1−3 This 31 kcal/mol transition state stabilization is the sum of 12, 10, and 9 kcal/mol stabilizing interactions (Scheme 1) from the phosphodianion, ribosyl ring, and pyrimidine ring fragments of the substrate, respectively.4 The enzymatic reaction proceeds by a stepwise mechanism, through a UMP carbanion reaction intermediate (Scheme 1), that is stabilized by ≥16 kcal/mol, relative to UMP, by interactions with the protein catalyst.5,6
Scheme 1. Contribution of Individual Substrate Fragments to the Total Stabilization of the Transition State for OMPDC-Catalyzed Decarboxylation of OMP through the UMP Carbanion Reaction Intermediate.

The X-ray crystal structure for the complex between yeast OMPDC (ScOMPDC) and the tight-binding intermediate analog 6-azauridine 5′-monophosphate (6-azaUMP) shows a network of interactions between the protein and ligand.7,8 We are interested in understanding the mechanism by which interactions of the three-substrate fragments are funneled toward stabilization of the transition state for ScOMPDC-catalyzed decarboxylation of the pyrimidine ring of OMP.1,9
A crucial network of amino acid side chains (Figure 1A,B) extend from the phosphodianion gripper loop shown in blue (P202 to V220) to the pyrimidine umbrella shown in green (A151 to T165) and consists of Q215, Y217, R235, and S154. These side-chains are poorly placed to interact with a hypothetical enzyme-bound transition state at the open enzyme (Figure 1A). Ligand binding drives a large enzyme conformational change to give the closed enzyme (Figure 1B), in which the side chains provide strong transition state stabilization.10−15 This network plays at least two roles in catalysis of the decarboxylation of OMP. (1) The side chains of Q215, Y217, and R235 are distant from the pyrimidine ring and do not interact directly with the site of ScOMPDC-catalyzed decarboxylation,13 but promote decarboxylation by stabilizing the catalytically active closed form of ScOMPDC (EC, Scheme 2) relative to the inactive open form EO.9,16−18 (2) The −CH2OH side chain of S154 bridges the phosphodianion and the pyrimidine binding sites at EC by donating an H-bond to the amide side chain of Q215 and accepting an H-bond from the C-5 pyrimidine −NH. These bridging interactions lock the substrate in a protein cage, by stabilizing the active closed form of ScOMPDC (EC) relative to EO (Figure 1), while holding the side chains from Q215, Y217, and R235 close to the dianion.
Figure 1.

Comparison of the X-ray crystal structures for (A) unliganded ScOMPDC (Protein Databank (PDB) entry 3GDL) and (B) the complex between ScOMPDC and the tight-binding substrate analog 6-azauridine 5′-monophosphate (6-azaUMP) (PDB entry 3GDK). The hypothetical 6-azaUMP ligand has been inserted in panel A to mimic the position of the ligand at panel B. (B) shows the ligand-driven movement of the phosphodianion gripper loop (P202 to 220, shaded blue) toward the pyrimidine umbrella loop (A151 to T165, shaded green) and of R235 toward the phosphodianion. The −CH2OH side chain of S154 bridges these loops and holds Q215 near the dianion by donating an H-bond to the amide side chain of Q215 and accepting an H-bond from the C-5 pyrimidine −NH (dashed red circle).
Scheme 2. Two-Step Mechanism for Substrate Binding to OMPDC.
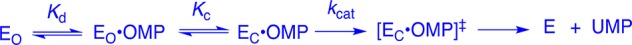
We have shown that the S154A mutation of ScOMPDC results in a large 5.8 kcal/mol destabilization of the transition state for decarboxylation of OMP and provided evidence that S154 plays an important role in orienting Q215 to interact with the substrate phosphodianion.15 We now address the more complex question of whether there is cooperativity between S154 and the dianion gripper side chains Y217 and R235 in stabilizing the decarboxylation transition state. We have probed the functioning of these four interacting side chains by site-directed mutagenesis. Such analyses are straightforward for small networks of two amino acid side chains,19 but studies on networks larger that two side chains are relatively rare. These include an analysis of the role of the three lysine side chains at the KMSKS mobile loop, in catalysis by tyrosyl amino acid synthase,20 and our recent examination of the network of three side chains (Q215, Y217, and R235) that interact with the phosphodianion of substrates bound to OMPDC.11
We are not aware of systematic mutagenesis studies on networks larger than three amino acids. This study on the S154, Q215, Y217, and R235 network (Figure 1) reports data to extend the following earlier work: (a) Studies on the effect of all single, double, and triple Q215A, Y217F, and R235A mutations on the kinetic parameters for OMPDC catalyzed reactions of whole substrates OMP,11,21 and 5-fluoroorotidine 5′-monophosphate (FOMP).10 (b) A restricted study on S154A, Q215A and S154A/Q215A mutants of OMPDC.15 We report here kinetic parameters for the six additional mutant enzymes required to obtain a complete set of kinetic data for all single, double, triple, and quadruple Q215A, Y217F, R235A, and S154A mutations of ScOMPDC. Our analyses of the kinetic parameter kcat/Km for decarboxylation of OMP and FOMP catalyzed by these 16 variants of ScOMPDC (Scheme 3) extends protocol developed in earlier work.10,11,15 Several lines of analysis are explored in describing the role played by the −CH2OH side chain of S154 in the efficient functioning of phosphodianion gripper side chains at the transition state for OMPDC-catalyzed decarboxylation.
Scheme 3. Wild-Type ScOMPDC and All Possible Single, Double, Triple and Quadruple Q215A, Y217F, R235A, and S154A Mutants Enzymes.

Asterisks show the new mutant enzymes prepared for this work.
Experimental Section
Materials
OMP(12,22) and FOMP(12,23) were prepared enzymatically by literature procedures. 3-(N-Morpholino)propanesulfonic acid (MOPS) and imidazole were purchased from Sigma (St. Louis, MO). Sodium hydroxide (1.0 N), hydrochloric acid (1.0 N), sodium chloride, tris(hydroxymethyl)-aminomethane (basic form), and Amicon centrifugal filter units with a 10K molecular weight cutoff (MWCO) were purchased from Fisher (Hampton, NH). Nickel chloride hexahydrate was a generous gift from Prof. Andrew Murkin (University at Buffalo, Buffalo, NY). Chelating Sepharose Fast-Flow and Q-Sepharose were purchased from GE Healthcare (Marlborough, MA). Water was purified using a Milli-Q Academic purification system from EMD Millipore (Burlington, MA). All other commercial chemicals were reagent-grade or better and were used without further purification.
Preparation of Wild-Type and Mutant Yeast Orotidine 5′-Monophosphate Decarboxylases
The plasmid pScODC-15b containing the gene encoding wild-type orotidine 5′-monophosphate decarboxylase from Saccharomyces cerevisiae (ScOMPDC) with an N-terminal His6-tag was available from earlier studies.15,24 The protein sequence differs from the published sequence for wild-type ScOMPDC by the following mutations: S2H, C155S, A160S, and N267D.15,24 The kinetic parameters for this preparation of wild-type ScOMPDC are in agreement with those reported by Porter and Short.25 The C155S mutation has been shown to result in a more stable protein.26
Site-directed mutagenesis on pScODC-15b was carried out using the QuikChange II kit from Stratagene (San Diego, CA). The procedures for the preparation of the S154A,15 Q215A,15 R235A,14 Y217F,13 S154A/Q215A,15 Q215A/Y217F,13 Q215/R235A,13 Y217F/R235A,13 and Q215A/Y217F/R235A13 mutants were described in earlier work. The following primers, with the altered codons underlined, were used to prepare the new mutant enzymes from previously prepared mutants of ScOMPDC.
5′-GACGCATTGGGTCAACAGTTTAGAACCGTGGATGATGTGG-3′ was used to introduce the Y217F mutation into the plasmid for the S154A mutant enzyme to yield the plasmid for the S154A/Y217F mutant.
5′-GGATCTGACATTATTATTGTTGGAGCAGGACTATTTGCAAAGGG-3′ was used to introduce the R235A mutation into the plasmid for the S154A/Q215A mutant enzyme to yield the plasmid for the S154A/Q215A/R235A mutant.
5′-GGCCTTTTGATGTTAGCAGAATTGGCATCTAAGGGCTCCC-3′ was used to introduce the S154A mutation into the plasmids for the R235A, Q215A/Y217F, Y217F/R235A, and Q215A/Y217F/R235A mutant enzymes to yield the plasmids for the S154A/R235A, S154A/Q215A/Y217F, S154A/Y217F/R235A, and S154A/Q215A/Y217F/R235A mutant enzymes, respectively.
The mutants of ScOMPDC were overexpressed, after transformation of E. coli BL21 (DE3) with the appropriate plasmid. The proteins were purified as described previously,11,15 except that passage over the Chelating Sepharose Fast-Flow column was repeated a second time to ensure complete removal of host E. coliOMPDC. The fractions from the second Sepharose Fast-Flow column were treated with thrombin to remove the N-terminal His6-tag, and this tag was separated from the protein by purification over a column of Q-sepharose. Wild-type OMPDC from E. coli (EcOMPDC) was prepared and purified according to published procedures.27
Preparation of Solutions
The solution pH was determined at 25 °C using an Orion Model 720A pH meter equipped with a Radiometer pHC4006–9 combination electrode that was standardized at pH 4.00, 7.00, and 10.00 at 25 °C. Stock solutions of OMP (20 mM) and FOMP (20 mM) were prepared by dissolving the solid nucleotide in water and adding 1 N HCl or 1 N NaOH to adjust the solution to the desired pH. These solutions were stored at −20 °C. The concentration of OMP in 0.1 M HCl was determined from the absorbance at 267 nm using a value of ε = 9430 M–1 cm–1.28 The concentration of FOMP in 0.1 M HCl was determined from the absorbance at 271 nm using a value of ε = 10 200 M–1 cm–1.5
Solutions of MOPS buffers (50% free base) were prepared by addition of measured amounts of 1 N NaOH and solid NaCl to give the desired ratio of acidic and basic buffer forms, and solution ionic strength. Stock solutions (20–40 mg/mL) of mutant ScOMPDC were dialyzed exhaustively against 10 mM MOPS at pH 7.0 (50% free base), 4 °C and I = 0.105 (NaCl). The solutions were further concentrated using Amicon filter units, when necessary. The concentration of ScOMPDC in these solutions was determined from the absorbance at 280 nm for wild type OMPDC, and values of ε = 29 900 or 28 400 M–1 cm–1 for Y217F mutant enzymes, that were calculated using the ProtParam tool available on the ExPASy server.29,30
Kinetic Parameters for Decarboxylation of OMP
The decarboxylation of OMP was monitored spectrophotometrically by following the decrease in absorbance at 279, 290, 295, or 300 nm [Δε, λ: – 2400 M–1 cm–1, 279 nm; – 1620 M–1 cm–1, 290 nm; – 840 M–1 cm–1, 295 nm; – 344 M–1 cm–1, 300 nm]. The wavelength was chosen to give an initial absorbance of ≤2.0. These reactions were monitored at the following wavelengths: [OMP] ≤ 0.08 mM, 279 nm; [OMP] = 0.1–0.3 mM, 290 nm; [OMP] = 0.3–0.8 mM, 295 nm; [OMP] ≥ 1 mM, 300 nm. Assays for mutant ScOMPDC-catalyzed decarboxylation of OMP were at 25 °C, pH 7.1 (10 mM MOPS) and I = 0.105 (NaCl). Reactions (1.0 mL) were initiated by addition of 10–60 μL of a stock solution of mutant ScOMPDC to give the following concentrations of mutant enzyme: 0.025 mM, S154A/Y217F mutant; 0.2 mM, S154A/R235A mutant; 0.1 mM, S154A/Q215A/Y217F mutant; and 0.2 mM, S154A/Q215A/R235A mutant. The initial velocity v (M s–1) for mutant ScOMPDC-catalyzed decarboxylation of OMP (0.03–3.0 mM) was determined by monitoring the decrease in absorbance at the chosen wavelength for the decarboxylation of 5–10% of total OMP. Values of kcat/Km (M–1 s–1) were obtained from the nonlinear least-squares fits of four or more values of v/[E] (s–1) to the appropriate kinetic equation.
Kinetic Parameters for Decarboxylation of FOMP
Assays for wild-type EcOMPDC-catalyzed decarboxylation of FOMP were conducted at 25 °C and pH 7.1 (10 mM MOPS) and at I = 0.105 (NaCl). Reactions (1.0 mL) were initiated by the addition of 2–4 μL of a stock solution of wild-type EcOMPDC to give a final enzyme concentration of 5–10 nM. The initial velocity v (M s–1) for wild-type EcOMPDC-catalyzed decarboxylation of FOMP (10–200 μM) was determined by monitoring the decrease in absorbance at 282 nm [Δε = −2400 M–1 cm–1] for the decarboxylation of 5–10% total FOMP. The value of the kinetic parameter kcat/Km (M–1 s–1) was obtained as the slope of the linear plot of values of v/[E] (s–1) against [FOMP].
Mutant ScOMPDC-catalyzed decarboxylation of FOMP was monitored spectrophotometrically over 5–10 half-reaction times and working at a wavelength (282 or 300 nm) where the total initial absorbance for FOMP and mutant ScOMPDC is ≤2.0. Time courses for mutant ScOMPDC-catalyzed decarboxylation of FOMP were obtained at 25 °C and pH 7.1 (10 mM MOPS) and at I = 0.105 (NaCl). Reactions (1.0 mL) containing 60–150 μM FOMP were initiated by addition of 3–40 μL of a stock solution of the mutant ScOMPDC to give the following enzyme concentrations: 3 μM, S154A/Y217F mutant; 20 μM, S154A/R235A mutant; 10 μM, S154A/Q215A/Y217F mutant; 5–10 μM, S154A/Q215A/R235A mutant; 0.2 mM, S154A/Y217F/R235A mutant; and 0.1 mM, S154A/Q215A/Y217F/R235A mutant. The rate constants kobs (s–1) for the pseudo-first-order decay of FOMP to FUMP were obtained by fitting the plot of absorbance A against time to the equation for an exponential decay. Values of the second-order rate constant kcat/Km (M–1 s–1) for ScOMPDC-catalyzed decarboxylation of FOMP were then calculated from the relationship kcat/Km= kobs/[E].
Results
Figure S1 shows the linear plot of v/[E] against [FOMP] for EcOMPDC-catalyzed decarboxylation of FOMP, with the slope of kcat/Km= 2.7 × 106 M–1 s–1. The observation of a linear plot through [FOMP] = 0.2 mM shows that Km ≫ 0.2 mM. By comparison, values of kcat = 14 s–1 and kcat/Km= 6.4 × 105 M–1 s–1 were reported for EcOMPDC-catalyzed decarboxylation of OMP.27 We conclude that the C-5 fluorine of FOMP results in a 4-fold increase in kcat/Km for EcOMPDC-catalyzed decarboxylation.
Figure 2A shows plots of v/[E] against [OMP] for decarboxylation catalyzed by the S154A/Q215A/Y217F (green symbols) and S154A/R235A (purple symbols) mutants of ScOMPDC, and Figure 2B show similar plots for the S154A/Y217F (maroon symbols) and S154A/Q215A/R235A (blue symbols) mutants. The solid lines in Figure 2 show the fits of these experimental data to the full Michaelis–Menten equation (Figure 2A) or the linear form of this equation, where Km ≫ [OMP] (Figure 2B). The kinetic parameters from Figure 2A require that <10% of total OMP is tied up as the Michaelis complex (Table S1). The concentration of free OMP is therefore essentially equal to total [OMP] (Figure 2A).
Figure 2.
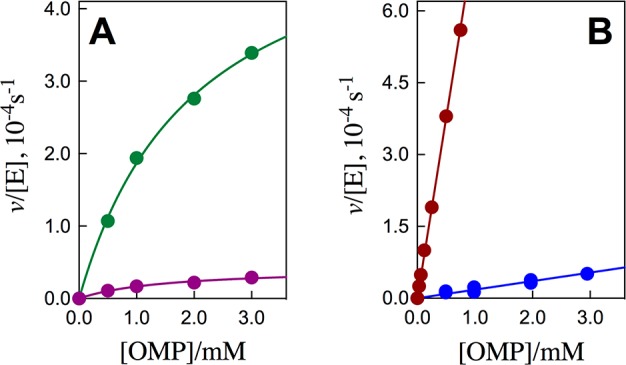
Dependence of v/[E] (s–1) for decarboxylation of OMP catalyzed by mutant forms of ScOMPDC on the concentration of OMP for reactions at 25 °C, pH 7.1 (10 mM MOPS) and at I = 0.105 (NaCl). (A) Green symbols, S154A/Q215A/Y217F mutant; purple symbols, S154A/R235A mutant. (B) Maroon symbols, S154A/Y217F mutant; blue symbols, S154A/Q215A/R235A mutant.
Figure 3 shows time courses for the change in absorbance at 300 nm for decarboxylation of FOMP catalyzed by S154A/Y217F/R235A (Figure 3A) and S154A/Q215A/Y217F/R235A mutants of ScOMPDC (Figure 3B). Figure S2A–D shows time-courses for the change in absorbance at 282 nm for decarboxylation of FOMP catalyzed by the S154A/Y217F (Figure S2A), S154A/R235A (Figure S2B), S154A/Q215/Y217F (Figure S2C), and the S154A/Q215A/R235A (Figure S2D) mutants of ScOMPDC. The fits of the experimental data for each Figure to the exponential decay for the pseudo-first-order enzyme-catalyzed reaction of FOMP gives the observed first order rate constant kobs (s–1) for ScOMPDC-catalyzed decarboxylation. The fits to a simple exponential decay show that these mutant ScOMPDC-catalyzed decarboxylation reactions are first order in [FOMP] (0.06–0.15 mM ≪ Km). Each of the experiments from Figures 3 and S2 were repeated two or more times, and the values of kobs/[E] for each mutant ScOMPDC-catalyzed decarboxylation were found to be reproducible to better than ±10%.
Figure 3.
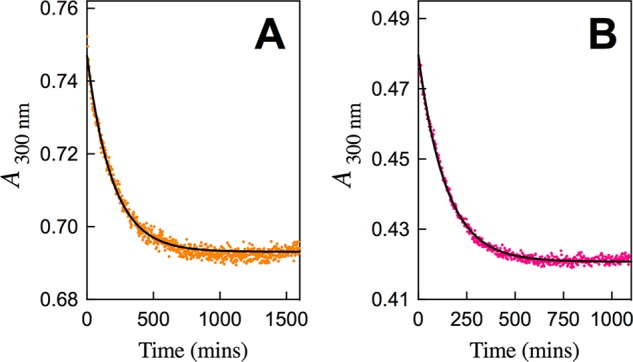
Full time-course for the decarboxylation of FOMP catalyzed by mutants of ScOMPDC for reactions at 25 °C, pH 7.1 (10 mM MOPS) and at I = 0.105 (NaCl). (A) Decarboxylation of 0.10 mM FOMP catalyzed by 0.20 mM of the S154A/Y217F/R235A mutant of ScOMPDC. (B) Decarboxylation of 0.10 mM FOMP catalyzed by 0.10 mM of the S154A/Q215A/Y217F/R235A mutant of ScOMPDC.
The new values of kcat/Km for decarboxylation of OMP catalyzed
by
mutant forms of ScOMPDC, which were obtained from
the fits of data shown in Figure 2 to the appropriate kinetic equation, are shown in
bold type in Table 1. The fits from Figure 2A also gave values of Km= 2.0 ±
0.2 mM for the S154A/R235A mutant and 1.5 ± 0.05 mM for the S154A/Q215A/Y217F
mutant. Table 1 reports
values of kcat/Km determined in earlier work for decarboxylation of OMP catalyzed by wild-type ScOMPDC and mutants of ScOMPDC.11,15 The new values of kcat/Km for decarboxylation
of FOMP catalyzed by mutant forms of ScOMPDC, obtained from the fits of data shown in Figures 3 and S1, are shown
in bold type Table 1. Table 1 reports
values of kcat/Km determined in earlier work for decarboxylation of FOMP catalyzed by wild-type ScOMPDC and mutants of ScOMPDC, to give data for all possible single, double, triple,
and quadruple S154A, Q215A, Y217F, and R235A mutations.10Table 1 also reports rate constant ratios  for wild-type and mutant ScOMPDC-catalyzed decarboxylation
of FOMP and OMP.
for wild-type and mutant ScOMPDC-catalyzed decarboxylation
of FOMP and OMP.
Table 1. Kinetic Parameters for ScOMPDC-Catalyzed Decarboxylation of OMP and FOMPa.
| OMPb |
FOMPc |
|||||
|---|---|---|---|---|---|---|
| ScOMPDC | kcat/Km (M–1 s–1) | ΔΔG⧧ (kcal/mol)d | kcat/Km (M–1 s–1) | ΔΔG⧧ (kcal/mol)d |
|
|
| wild-type | 1.1 × 107 | 1.2 × 107 | 1.1 | |||
| Q215A | 2.6 × 105 | 2.2 | 2.0 × 106 | 1.1 | 7.7 | |
| Y217F | 1.8 × 105 | 2.4 | 1.1 × 106 | 1.4 | 6.1 | |
| R235A | 910 | 5.6 | 1.6 × 105 | 2.6 | 180 | |
| S154A | 630 | 5.8 | 2.9 × 105 | 2.2 | 460 | |
| S154A/Q215A | 380 | 6.1 | 7.7 × 104 | 3.0 | 200 | |
| S154A/Y217F | 0.75 ± 0.01 | 9.8 | 520 ± 10 | 5.9 | 700 | |
| S154A/R235A | 0.027 ± 0.004 | 11.7 | 20 ± 2 | 7.9 | 740 | |
| Q215A/Y217F | 3.4 × 103 | 4.8 | 5.3 × 104 | 3.2 | 16 | |
| Q215A/R235A | 14 | 8.0 | 7200 | 4.4 | 510 | |
| Y217F/R235A | 4.1 | 8.8 | 820 | 5.7 | 200 | |
| S154A/Q215A/Y217F | 0.28 ± 0.02 | 10.4 | 250 ± 10 | 6.4 | 890 | |
| S154A/Q215A/R235A | 0.018 ± 0.001 | 12.0 | 10 ± 1 | 8.3 | 560 | |
| S154A/Y217F/R235A | 0.0006 ± 0.0002f | 14.0 | 0.44 ± 0.02 | 10.1 | ||
| Q215A/Y217F/R235A | 0.037 | 11.6 | 28 | 7.7 | 760 | |
| S154A/Q215A/Y217F/R235A | 0.0016 ± 0.0005f | 13.4 | 1.1 ± 0.1 | 9.6 | ||
Conditions: pH 7.1 (10 mM MOPS), 25 °C and I = 0.105 (NaCl).
The new values for this manuscript are in bold type. The other rate constants are from ref (11) or (15). The quoted uncertainty is the standard error from the least-squares fit of the kinetic data to the appropriate kinetic equation.
The new values for this manuscript are in bold type. The other rate constants are from ref (10).The quoted uncertainty in the original values is the average of two or more determinations of kcat/Km.
Calculated from the ratio of the values of kcat/Km for wild-type and mutant ScOMPDC-catalyzed decarboxylation of OMP or FOMP.
The ratio of the values of kcat/Km for OMPDC-catalyzed decarboxylation of OMP and FOMP.
No detectable activity
toward decarboxylation
of OMP: kcat/Km < 0.006 M–1 s–1. The rate constants were estimated from kcat/Km for mutant enzyme-catalyzed decarboxylation
of FOMP (see text). The range of values is calculated
from the estimated uncertainty of ±50% in 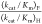 for OMPDC-catalyzed decarboxylation
of OMP and FOMP.
for OMPDC-catalyzed decarboxylation
of OMP and FOMP.
There was no detectable decarboxylation of 1.0 mM OMP ([ΔA300 ≤ 0.03)
catalyzed
by 0.2 mM of the S154A/Y217F/R235A or S154A/Q215A/Y217F/R235A mutants
over a 20 h reaction time. This shows that kcat/Km < 0.006 M–1 s–1 for the mutant enzyme-catalyzed decarboxylation
reactions. Large limiting ratios 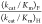 are observed for reactions catalyzed by
severely impaired mutants of ScOMPDC, where the relative
reaction barriers are determined by chemical reactivity of enzyme-bound OMP and FOMP.10 The
ratio of
are observed for reactions catalyzed by
severely impaired mutants of ScOMPDC, where the relative
reaction barriers are determined by chemical reactivity of enzyme-bound OMP and FOMP.10 The
ratio of 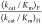 = 700 ± 200 is the average of the
ratios from Table 1 for severely impaired mutants where (kcat/Km)H ≤ 1.0 M–1 s–1 (S154A/Y217F, S154A/R235A, S154A/Q215A/Y217F,
S154A/Q215A/R235A, and Q215A/Y217F/R235A). The values of (kcat/Km)H for decarboxylation catalyzed by the S154A/Y217F/R235A and S154A/Q215A/Y217F/R235A
mutants, shown in underlined type in Table 1, were estimated from the value (kcat/Km)F for decarboxylation of FOMP and the rate constant ratio
= 700 ± 200 is the average of the
ratios from Table 1 for severely impaired mutants where (kcat/Km)H ≤ 1.0 M–1 s–1 (S154A/Y217F, S154A/R235A, S154A/Q215A/Y217F,
S154A/Q215A/R235A, and Q215A/Y217F/R235A). The values of (kcat/Km)H for decarboxylation catalyzed by the S154A/Y217F/R235A and S154A/Q215A/Y217F/R235A
mutants, shown in underlined type in Table 1, were estimated from the value (kcat/Km)F for decarboxylation of FOMP and the rate constant ratio 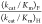 = 700 ± 200.
= 700 ± 200.
Discussion
All of the newly reported
mutants of ScOMPDC were
expressed in E. Coli that contained host EcOMPDC. These mutants were prepared with an N-terminal
His6-tag, and with two passes over a chelating column to
ensure complete removal of EcOMPDC host enzyme. The
value of 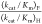 > 67 (Table 1)
for the most severely impaired S154A/Y217F/R235A
mutant, which shows no activity toward decarboxylation of OMP, is much larger than the value for EcOMPDC,
> 67 (Table 1)
for the most severely impaired S154A/Y217F/R235A
mutant, which shows no activity toward decarboxylation of OMP, is much larger than the value for EcOMPDC, 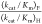 = 4. This shows that there is no significant
(<10%) decarboxylation of FOMP by wild-type EcOMPDC from the host organism. If, for example, 10% of
the observed activity of this preparation toward decarboxylation of FOMP were due to wild-type host OMPDC, then a
detectable value of
= 4. This shows that there is no significant
(<10%) decarboxylation of FOMP by wild-type EcOMPDC from the host organism. If, for example, 10% of
the observed activity of this preparation toward decarboxylation of FOMP were due to wild-type host OMPDC, then a
detectable value of 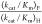 = (1.00)/[0.10][0.25] = 40 would
have been
observed.
= (1.00)/[0.10][0.25] = 40 would
have been
observed.
The <2-fold variation in the 5-F substituent effect
on decarboxylation
reactions catalyzed by severely impaired mutants of ScOMPDC [(kcat/Km)OMP ≤ 1.0 M–1 s–1, 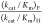 = 560–890) shows that the value
of (kcat/Km)F for these mutant enzymes establishes (kcat/Km)H to within
a factor of 2. The rate constants (kcat/Km)H for decarboxylation
reactions catalyzed by the S154A/Y217F/R235A and S154A/Q215A/Y217F/R235A
mutants, that were too slow to be detected [(kcat/Km)H < 0.006
M–1 s–1], were estimated from
the value of (kcat/Km)F (Table 1) and
= 560–890) shows that the value
of (kcat/Km)F for these mutant enzymes establishes (kcat/Km)H to within
a factor of 2. The rate constants (kcat/Km)H for decarboxylation
reactions catalyzed by the S154A/Y217F/R235A and S154A/Q215A/Y217F/R235A
mutants, that were too slow to be detected [(kcat/Km)H < 0.006
M–1 s–1], were estimated from
the value of (kcat/Km)F (Table 1) and 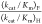 = 700.
= 700.
This study of the effect of mutations of S154, Q215, Y217, and R235 will provide a clear description of the role of this network in catalysis, provided the effects are restricted to the elimination of interactions between the transition state and the excised side chain(s). The following observations provide strong evidence that there only minimal effects of these mutations on [(kcat/Km)H that arise from protein structural changes that perturb protein-transition state interactions. (1) Single Q215A, Y217F, and R235A mutations result in negligible (≤0.5 kcal/mol) changes in the activation barrier to OMPDC-catalyzed decarboxylation of the phosphodianion truncated substrate 1-(β-erythrofuranosyl)orotic acid (EO, Scheme 4).13 This provides strong evidence that the single mutations have no effect on the structural integrity and catalytic activity at the site of decarboxylation of the pyrimidine ring, and are restricted to the loss of protein dianion interactions at the dianion activation site. (2) The sum of the effects of the four single S154A, Q215A, Y217F, and R235A mutations on the activation barrier ΔG⧧ for decarboxylation catalyzed by wild-type OMPDC is 16.0 kcal/mol (Table 1). By comparison, there is only a 13.4 kcal/mol difference between the activation barriers ΔG⧧ for wild type and the S154A/Q215A/Y217F/R235A quadruple mutant enzyme-catalyzed decarboxylation of OMP (Table 1) This shows that there can be no large additional increase in the activation barrier of OMPDC-catalyzed decarboxylation associated with the simultaneous mutations of the S154A, Q215A, Y217F, and R235A, compared with the mutation of single side chains. (3) Q215A mutations result in a ca. 2 kcal/mol destabilization of the transition state for decarboxylation of OMP when S154 is present and in a smaller ca. 0.3 kcal/mol destabilization when S154 is absent, as discussed in detail below. The exception is the Q215A mutation of the S154A/Y217F/R235A variant, which results in a ca. 0.5 kcal/mol stabilization or the transition state for ScOMPDC-catalyzed decarboxylation of FOMP. This is consistent with a shift in the position of the Q215 side chain at the S154A/Y217F/R235A triple mutant, which alters the interaction of the protein with the decarboxylation transition state. Aside from this minor effect, we are able to rationalize the other effects of S154A, Q215A, Y217F, and R235A mutations by a consideration of the loss in transition state stabilization from interaction with the deleted side chains.
Scheme 4. Stepwise OMPDC-Catalyzed Decarboxylation of Phosphodianion Truncated Substrate EO.

Mutant Boxes
We begin by examining two-dimensional mutant cycles (Figure 4) constructed from data in Table 1. These cycles show the effects of mutations of S154, which links the pyrimidine umbrella and phosphodianion gripper loops (Figure 1), on the stabilizing interactions between individual phosphodianion gripper side chains Q215, Y217, and R235 and the decarboxylation transition state (Figure 5).
Figure 4.

Mutant cycles that show the effect of S154A mutations on: (A) stabilization of the transition state for wild-type ScOMPDC-catalyzed decarboxylation of OMP by interactions with Q215; (B) stabilization of the transition state for wild-type ScOMPDC-catalyzed decarboxylation of OMP by interactions with Y217F; (C) stabilization of the transition state for Y217F ScOMPDC-catalyzed decarboxylation of OMP by interactions with R235; (D) stabilization of the transition state for wild-type ScOMPDC-catalyzed decarboxylation of OMP by interactions with R235A; (E) stabilization of the transition state for R235A ScOMPDC-catalyzed decarboxylation of OMP by interactions with Y217F.
Figure 5.
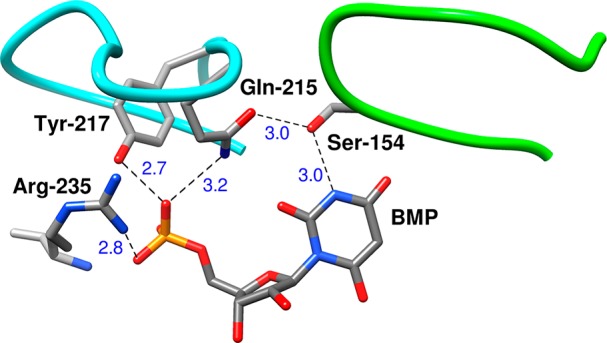
Representation of the X-ray crystal structure of a complex between OMPDC and the intermediate analog 6-hydroxyuridine 5′-monophosphate (BMP, PDB entry 1DQX). Phosphodianion gripper loop is shaded blue, and the pyrimidine umbrella loop is shaded green. These loops are clamped over the substrate by the following network of hydrogen bonding interactions: (a) H-bonds between the Q215, Y217, and R235 side chains and the ligand phosphodianion; (b) H-bonds that link the −CH2OH side chain of S154 to the amide side chain of Q215 and to the pyrimidine nitrogen. All distances are in Ångstroms.
Q215 (Figure 4A)
The Q215A mutation results in a 2.2 kcal/mol destabilization of the transition state for wild-type ScOMPDC-catalyzed decarboxylation of OMP, but the same mutation of the S154A mutant enzyme results in a much smaller 0.3 kcal/mol transition state destabilization.15 This provides strong evidence that a hydrogen bond between the −CH2OH group of S154 and the amide side-chain of Q215 (Figure 1) positions the Q215 side to interact with the phosphodianion of OMP.
Y217 (Figure 4B,C)
The Y217F mutation results in a 2.4 kcal/mol destabilization of the transition state for wild-type OMPDC-catalyzed decarboxylation of OMP, but the same mutation of S154A mutant ScOMPDC results in a larger 4.0 kcal/mol destabilization (Figure 4B). We conclude that the H-bond between the CH2OH group of S154 and the amide side-chain of Q215 at wild-type ScOMPDC fixes the position of Y217 and R235 at the phosphodianion. This clamp is largely maintained at the Y217F mutant, where the effect is limited to the loss of the interactions of the single side chain. The clamp is released by the S154A mutation, so now the second Y217F mutation results in both the loss of the interactions to Y217 and in a weakening of the interactions to R235. The R235A mutation of Y217F ScOMPDC results in a 6.4 kcal/mol transition state destabilization, but the same mutation of the S154A/Y217F mutant results in a smaller 4.2 kcal/mol transition state destabilization (Figure 4C). This provides direct evidence that the double S154A/Y217F mutation leads to a weakening of the transition state stabilization by the remaining R235 side chain.
Interaction of R235 (Figure 4D,E)
The R235A mutation results in a 5.6 kcal/mol destabilization of the transition state for wild-type ScOMPDC-catalyzed decarboxylation of OMP, but the same mutation of the S154A mutant enzyme results in a larger 5.9 kcal/mol transition state destabilization (Figure 4D), consistent with conclusion that the R235A mutation of S154A ScOMPDC results in the loss of the interaction of the mutated R235 side chain, and a small weakening of the interaction with Y217F. The Y217F mutation of R235A ScOMPDC results in a 3.2 kcal/mol kcal/mol transition state destabilization, but the same mutation of the S154A/R235A mutant results in only an estimated 2.3 kcal/mol destabilization (Figure 4E). This suggests that the double S154A/R235A mutation leads to a weakening of the transition state stabilization by the remaining Y217 side chain.
In conclusion, S154 plays both a specific and a general role in promoting interactions between OMPDC and dianion gripper side chains. The −CH2OH side chain plays the specific role of holding the amide side chain of Q215 in a position to interact with the substrate dianion. The side chain acts globally to optimize interactions with Y217 and R235 by locking the substrate in the protein cage created by the interactions between the pyrimidine umbrella and phosphodianion gripper side chains (Figures 1 and 5).
Reconstruction of Wild-Type OMPDC
Table 2 shows the effects of single S154A Q215A, Y217F, and R235A mutations on (ΔG⧧)OMP for decarboxylation of OMP determined for mutations at wild-type ScOMPDC and various single, double, and triple mutants of ScOMPDC. Table 2 defines a total of 32 different effects of single mutations of S154A Q215A, Y217F, and R235A on the activity of wild-type and variant ScOMPDC. We focus here on the recovery in enzymatic activity observed upon substitution of wild-type amino acid side chains at the quadruple S154A/Q215A/Y217F/R235A mutant.
Table 2. Effect of Single Substitutions of Amino Acid Residues X on the Activation Barrier (ΔG⧧)OMP for Decarboxylation of OMP Catalyzed by Wild-Type ScOMPDC and by Previously Mutated ScOMPDCa.
| (ΔΔG⧧X)OMP (kcal/mol)b |
|||||
|---|---|---|---|---|---|
| enzyme | X = Q215A | X = Y217F | X = R235A | X = S154A | Σ(ΔΔG⧧X)OMPc |
| wild typed | 2.2 | 2.4 | 5.6 | 5.8 | 16.0 |
| triple mutante | –0.6 | 1.4 | 3.0 | 1.8 | 5.6 |
| single mutantf | 2.4 (Y217F) | 2.6 (Q215A) | 5.8 (Q215A) | 3.9 (Q215A) | |
| 2.4 (R235A) | 3.2 (R235A) | 6.4 (Y217F) | 7.4 (Y217F) | ||
| 0.3 (S154A) | 4.0 (S154A) | 5.9 (S154A) | 6.1 (R235A) | ||
| double mutantg | 2.8 (Y217F/R235A) | 3.6 (Q215A/R235A) | 6.8 (Q215A/Y217F) | 5.6 (Q215A/Y217F) | |
| 0.6 (S154A/Y217F) | 4.3 (S154A/Q215A) | 5.9 (S154A/Q215A) | 4.0 (Q215A/R235A) | ||
| 0.3 (S154A/R235A) | 2.3 (S154A/R235A) | 4.2 (S154A/Y217F) | 5.2 (Y217F/R235A) | ||
For OMPDC-catalyzed decarboxylation at 25 °C, pH 7.1 (10 mM MOPS), and ionic strength of 0.105 (NaCl). The respective substitutions that cause the largest and smallest changes in (ΔG⧧)OMP are shown in underlined and bold text, respectively.
Calculated from the ratio of values of kcat/Km for the precursor and mutant enzymes.
Sum of the effects of the S154A, Q215A, Y217F, and R235A substitutions on (ΔΔGX⧧)OMP.
Effect of the first mutation X on the activation barrier for wild-type OMPDC-catalyzed decarboxylation of OMP.
Effect of the final mutation X on the activation barrier for the decarboxylation reaction catalyzed by the corresponding triple mutant of OMPDC.
Effect of the mutation X on the activation barrier for the decarboxylation reaction catalyzed by the OMPDC mutant given in parentheses.
Effect of the mutation X on the activation barrier for the decarboxylation reaction catalyzed by the OMPDC double mutant given in parentheses.
The substitution of single wild-type amino acids at the quadruple mutant to give triple mutants has only small effects on the stability of the decarboxylation transition state. These are shown as the bold entries in Table 2. For example, the 3.0 and 1.8 kcal/mol effects of substitution of S154 and R235 are much smaller than the 5.6 and 5.8 kcal/mol effects of these substitutions at the S154A and R235A mutant enzymes. Consequently, the sum of the effects of single S154A, Q215A, Y217F, and R235A substitutions at triple mutants of OMPDC, 5.6 kcal/mol (Table 2) is only 35% of the sum of the effects of these deletions at wild-type ScOMPDC (16.0 kcal/mol). The small restorative effects of single substitutions at the S154A/Q215A/Y217F/R235A quadruple mutant are consistent with a large entropic cost to holding OMPDC in the active closed conformation. We propose that this represents the cost of fixing the positions of the pyrimidine umbrella and phosphodianion gripper loops (Figure 1) relative to the transition state.27 In every case, a large fraction of the total binding energy of the single side chain at the triple mutants is used to pay this entropic cost, so only a small fraction remains for expression as transition state stabilization.
By contrast, the largest effects of A215Q, F217Y, and A235R substitutions on (ΔG⧧)OMP (underlined text in Table 2) are observed when these side chains are incorporated into triple mutants of OMPDC to give the corresponding double mutant (bottom row, Table 2). For example, the A235R substitution at the Q215A/Y217F/R235A triple mutant provides for a 6.8 kcal/mol decrease in (ΔG⧧)OMP compared with the smaller 5.6 kcal/mol effect of the A235R substitution at the R235A single mutant; and, the F217Y substitution at the S154A/Q215A/Y217F triple mutant provides a 4.3 kcal/mol decrease in (ΔG⧧)OMP compared with the smaller 2.4 kcal/mol effect of the F217Y substitution at the F217Y single mutant (Table 2).
The 6.8 kcal/mol effect of the A235R substitution at Q215A/Y217F/R235A ScOMPDC on (ΔG⧧)OMP represents the stabilizing interaction of the R235 side chain, plus an improvement in the functioning of S154 already present at the triple mutant. This cooperativity between the expression of side chain interactions reflects the roughly constant entropic cost for immobilizing the phosphodianion gripper and pyrimidine gripper loops at the active form of ScOMPDC. This full price is paid by single amino acids added to the quadruple mutant, but it is split when two amino acids are added to the quadruple mutant, with the result that a larger fraction of the total binding energy of each side chain is expressed as stabilization of the decarboxylation transition state at the double mutant. An exception to this trend is the large 7.4 kcal/mol effect of the A154S substitution at the S154A/Y217F mutant shown in Figure 4B. This reflects the key role played by the clamping −CH2OH side chain of S154 in optimizing protein dianion interactions, and the large effect of the S154A mutation on the interaction of the remaining R235 side chains with the substrate dianion, as discussed above.
Smaller effects of the Q215, Y217, and R235 substitutions on (ΔG⧧)OMP are observed when these side chains are inserted at double mutants of ScOMPDC to give the single mutant (third row, Table 2), compared with their insertions at triple mutants to give the double mutants (bottom row, Table 2). The effects of these substitutions are then only slightly reduced when they result in wild-type ScOMPDC. We propose that the interactions between any two of four side chains results in a large fractional reduction of entropy associated with loop motion and that the further entropic penalty paid upon substitution of the third side chain is small and upon substitution of the fourth side chain is negligible. Such effects are well-documented for the formation of intermolecular supramolecular complexes that are stabilized linear arrays of hydrogen bonds. The supramolecular complex stabilized by a single hydrogen bond is relatively weak, due to the large entropic penalty to the reaction between monomers to form the dimer. A larger fraction of the total stabilization from subsequent H-bonds is expressed at complexes stabilized by more than one hydrogen bond, because of the relatively small effect of each new H-bond on the total entropy of the supramolecular complex.31
Cubic Mutant Cycles
The data from Table 2 were used to construct the cubes shown in Figure 6. These show 24 of the 32 different effects of single substitutions from Table 2. All 32 effects may be shown on a pseudo-four-dimensional hypercube, obtained by placing the cube from Figure 6B at the center of a larger cube for Figure 6A, and drawing eight lines to connect the corners of the two cubes.32 The last eight connections are not shown on Figure 6A,B, but several are given and discussed in our analysis of Figure 4. This hypercube is composed of eight distinct cubic cells, two of which are shown in Figure 6A,B.
Figure 6.

Triple mutant cubes that show the effects of single amino acid mutations on (ΔG⧧)OMP (kcal/mol)for reactions catalyzed by wild-type ScOMPDC (black values), by single mutants of ScOMPDC (red values), and by double mutants of ScOMPDC (green values). (A) Effect of these mutations on wild-type ScOMPDC. (B) Effect of mutations on ScOMPDC, previously mutated at position S154. The values of (ΔΔGX⧧)OMP were calculated from the ratio of the kinetic parameters kcat/Km for precursor and mutated ScOMPDC-catalyzed decarboxylation of OMP (Table 1) and are reported in Table 2.
Figure 6A is the previously reported cube for the effects of Q215A, Y217F, and R235A mutations on (ΔG⧧)OMP for wild-type ScOMPDC-catalyzed decarboxylation of OMP.11 The effect of these same mutations on (ΔG⧧)OMP for ScOMPDC previously mutated at S154 is illustrated by Figure 6B. We concluded earlier that the effect of mutations of residue X = Q215, Y217, or R235 on (ΔΔGX⧧)OMP is approximately the same when the mutation is carried out at wild-type OMPDC or singly mutated or doubly mutated forms of OMPDC.11 This shows that when S154 is present at ScOMPDC, the loss of transition state stabilization from replacement of one or two side chains does not result in large changes in the stabilization by the remaining side chain(s).
By contrast, the effect of single mutations of X = Q215, Y217, or R235 on (ΔΔGX⧧)OMP is dependent on the context of the mutation, when the S154A mutant is the parent enzyme (Figure 6B), so that the presence of S154 is required for the regular behavior observed for Figure 6A. Figure 6B may be used to illustrate points made above in the discussion of Table 2. For example, the smallest changes in (ΔΔGX)OMP (shown in green) are observed for single mutations of triple mutants to form the quadruple mutant; while, significantly larger changes in (ΔΔGX⧧)OMP (shown in red) are observed for the same mutations at double mutants to form the corresponding triple mutant.
The effects of consecutive Q215A, Y217F, and R235A mutations sum to 11.6 kcal/mol, when starting from wild-type ScOMPDC (Figure 6A) but to only 7.6 kcal/mol when starting from S154A mutant ScOMPDC (Figure 6B). This shows that the S154A mutation results in a total (11.6–7.6) = 4.0 kcal/mol decrease in transition state stabilization from the collective interactions with the phosphodianion gripper side chain Q215, Y217, and R235. One-half (2 kcal/mol) of this decrease is due to the loss of interactions with the side chain of Q215 (Figure 4A),15 and 2 kcal/mol results from a weakening in the interactions with Y217 and R235 (Figure 4B,C). We attribute the remaining 2 kcal/mol of the total 5.8 kcal/mol effect of the S154A mutation to the loss of the stabilizing H-bonding interaction between the S154 oxygen and the pyrimidine −NH. By comparison, the loss of this H-bonding interaction results in a 3 kcal/mol destabilization of the transition state for OMPDC-catalyzed decarboxylation of the phosphodianion truncated substrate EO (Scheme 4).33
Summary and Conclusions
This is the first report of an examination of the catalytic role of a network of four interacting amino acid side chains, by determining the effects of stepwise replacement of all four side chains. The four mutations result in a total (2 × 1010)-fold reduction in the activity of wild-type ScOMPDC, which corresponds to the loss of 45% of the 31 kcal/mol intrinsic substrate binding energy.3,34 The modeling of these effects of site-directed mutagenesis is a significant challenge to existing computational methods,35 similar to that for modeling the effects of mutagenesis studies on the activity of triosephosphate isomerase.36−43
The relative ease of the interpretation of these experimental results reflects the choice of mobile structural elements that lie on the protein surface. A similar study is less likely to provide insight into the function of amino acid networks at the interior of protein catalysts, because of unintended structural effects of these mutations on the function of other active site catalytic side chains. The utilization of amino acid side chains, at mobile structural elements, for stabilization of enzymatic transition states is less efficient than the utilization of rigid, preorganized, side chains,44,45 because of the entropic penalty to freezing the mobile elements at the transition state. However, flexible elements are often required to allow for the entrance and departure of ligands from active site cages.17,46 We find that the entropic penalty to fixing the active site loops of ScOMPDC (Figure 5) is a significant fraction of the interaction energy of a single side chain. This penalty is difficult to detect, and therefore easily underestimated, when it is divided between four amino acid side chains.
The mutant cubes from Figure 6A,B show that the single intraloop H-bond between the side chains of Q215 and S154 (Figure 5) has the effect of increasing the total transition state stabilization from the Q215, Y217, and R235 side chains by ca. 4 kcal/mol. These results provide a telling example of the importance of the precise placement of side chains in obtaining efficient enzymatic catalysis48 and illustrates the magnitude of the problems that must be solved by protein engineers who are working to design proteins with enzyme-like activity.
These results are consistent with the notion that the large catalytic rate acceleration by ScOMPDC is the result of many additive electrostatic interactions between the enzyme and decarboxylation transition state at a preorganized enzyme active site,44,45,49 with two wrinkles: (1) Several amino acid side chains are not preorganized at the unliganded enzyme, but only organize after the binding of substrate, through the utilization of enzyme-dianion interactions to hold ScOMPDC in a reactive caged Michaelis complex.1,4,9,17 (2) The conversion of OMPDC from a flexible open form to a stiff and organized Michaelis complex results in the loss of most or all of the entropy associated with the flexible loops at the unliganded enzyme. The intrinsic substrate binding energy is utilized to pay the price for this reduction in entropy.34
Acknowledgments
We acknowledge the National Institutes of Health Grants GM39754 and GM116921 (to J.P.R.) and Grant GM65155 (to J.A.G.) for generous support of this work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b09609.
Calculation of the concentration of OMP present in the Michaelis complex (E·OMP) for the initial velocity determinations; linear plot of v/[E] against [FOMP] for EcOMPDC-catalyzed decarboxylation of FOMP; plots of absorbance at 282 nm against time for decarboxylation of FOMP catalyzed by the S154A/Y217F, S154A/R235A, S154A/Q215A/Y217F, and the S154A/Q215A/R235A mutants of ScOMPDC (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Richard J. P.; Amyes T. L.; Reyes A. C. Orotidine 5′-Monophosphate Decarboxylase: Probing the Limits of the Possible for Enzyme Catalysis. Acc. Chem. Res. 2018, 51, 960–969. 10.1021/acs.accounts.8b00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B. G.; Wolfenden R. Catalytic proficiency: the unusual case of OMP decarboxylase. Annu. Rev. Biochem. 2002, 71, 847–885. 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- Radzicka A.; Wolfenden R. A proficient enzyme. Science 1995, 267, 90–93. 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. Enzyme Architecture: Erection of Active Orotidine 5′-Monophosphate Decarboxylase by Substrate-Induced Conformational Changes. J. Am. Chem. Soc. 2017, 139, 16048–16051. 10.1021/jacs.7b08897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang W.-Y.; Wood B. M.; Wong F. M.; Wu W.; Gerlt J. A.; Amyes T. L.; Richard J. P. Proton Transfer from C-6 of Uridine 5′-Monophosphate Catalyzed by Orotidine 5′-Monophosphate Decarboxylase: Formation and Stability of a Vinyl Carbanion Intermediate and the Effect of a 5-Fluoro Substituent. J. Am. Chem. Soc. 2012, 134, 14580–14594. 10.1021/ja3058474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. Formation and Stability of a Vinyl Carbanion at the Active Site of Orotidine 5′-Monophosphate Decarboxylase: pKa of the C-6 Proton of Enzyme-Bound UMP. J. Am. Chem. Soc. 2008, 130, 1574–1575. 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B. G.; Hassell A. M.; Wolfenden R.; Milburn M. V.; Short S. A. Anatomy of a proficient enzyme: the structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 2011–2016. 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K. K.; Wood B. M.; Fedorov A. A.; Fedorov E. V.; Imker H. J.; Amyes T. L.; Richard J. P.; Almo S. C.; Gerlt J. A. Mechanism of the Orotidine 5′-Monophosphate Decarboxylase-Catalyzed Reaction: Evidence for Substrate Destabilization. Biochemistry 2009, 48, 5518–5531. 10.1021/bi900623r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Malabanan M. M.; Zhai X.; Reyes A. C.; Richard J. P. Enzyme activation through the utilization of intrinsic dianion binding energy. Protein Eng., Des. Sel. 2017, 30, 159–168. 10.1093/protein/gzw064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Ming S.; Amyes T. L.; Gerlt J. A.; Richard J. P. Rate and Equilibrium Constants for an Enzyme Conformational Change during Catalysis by Orotidine 5′-Monophosphate Decarboxylase. Biochemistry 2015, 54, 4555–4564. 10.1021/acs.biochem.5b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman L. M.; Amyes T. L.; Goryanova B.; Gerlt J. A.; Richard J. P. Enzyme Architecture: Deconstruction of the Enzyme-Activating Phosphodianion Interactions of Orotidine 5′-Monophosphate Decarboxylase. J. Am. Chem. Soc. 2014, 136, 10156–10165. 10.1021/ja505037v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Amyes T. L.; Gerlt J. A.; Richard J. P. Role of a Guanidinium Cation–Phosphodianion Pair in Stabilizing the Vinyl Carbanion Intermediate of Orotidine 5′-Phosphate Decarboxylase-Catalyzed Reactions. Biochemistry 2013, 52, 7500–7511. 10.1021/bi401117y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Ming S. A.; Goldman L. M.; Wood B. M.; Desai B. J.; Gerlt J. A.; Richard J. P. Orotidine 5′-monophosphate decarboxylase: Transition state stabilization from remote protein-phosphodianion interactions. Biochemistry 2012, 51, 4630–4632. 10.1021/bi300585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett S. A.; Amyes T. L.; McKay Wood B.; Gerlt J. A.; Richard J. P. Activation of R235A Mutant Orotidine 5′-Monophosphate Decarboxylase by the Guanidinium Cation: Effective Molarity of the Cationic Side Chain of Arg-235. Biochemistry 2010, 49, 824–826. 10.1021/bi902174q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett S. A.; Amyes T. L.; Wood B. M.; Gerlt J. A.; Richard J. P. Dissecting the Total Transition State Stabilization Provided by Amino Acid Side Chains at Orotidine 5′-Monophosphate Decarboxylase: A Two-Part Substrate Approach. Biochemistry 2008, 47, 7785–7787. 10.1021/bi800939k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Zhai X.; Morgan K. T.; Reinhardt C. J.; Amyes T. L.; Richard J. P. The Activating Oxydianion Binding Domain for Enzyme-Catalyzed Proton Transfer, Hydride Transfer and Decarboxylation: Specificity and Enzyme Architecture. J. Am. Chem. Soc. 2015, 137, 1372–1382. 10.1021/ja5123842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. Enzyme architecture: on the importance of being in a protein cage. Curr. Opin. Chem. Biol. 2014, 21, 1–10. 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K.; Amyes T. L.; Richard J. P. Hydron Transfer Catalyzed by Triosephosphate Isomerase. Products of the Direct and Phosphite-Activated Isomerization of [1-13C]-Glycolaldehyde in D2O. Biochemistry 2009, 48, 5769–5778. 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Goodey N. M. Catalytic Contributions from Remote Regions of Enzyme Structure. Chem. Rev. 2011, 111, 7595–7624. 10.1021/cr100042n. [DOI] [PubMed] [Google Scholar]
- First E. A.; Fersht A. R. Analysis of the role of the KMSKS loop in the catalytic mechanism of the tyrosyl-tRNA synthetase using multimutant cycles. Biochemistry 1995, 34, 5030–5043. 10.1021/bi00015a014. [DOI] [PubMed] [Google Scholar]
- Miller B. G.; Snider M. J.; Short S. A.; Wolfenden R. Contribution of Enzyme-Phosphoribosyl Contacts to Catalysis by Orotidine 5′-Phosphate Decarboxylase. Biochemistry 2000, 39, 8113–8118. 10.1021/bi000818x. [DOI] [PubMed] [Google Scholar]
- Van Vleet J. L.; Reinhardt L. A.; Miller B. G.; Sievers A.; Cleland W. W. Carbon isotope effect study on orotidine 5′-monophosphate decarboxylase: support for an anionic intermediate. Biochemistry 2008, 47, 798–803. 10.1021/bi701664n. [DOI] [PubMed] [Google Scholar]
- Gross A.; Abril O.; Lewis J. M.; Geresh S.; Whitesides G. M. Practical synthesis of 5-phospho-D-ribosyl α-1-pyrophosphate (PRPP): enzymatic routes from ribose 5-phosphate or ribose. J. Am. Chem. Soc. 1983, 105, 7428–7435. 10.1021/ja00363a037. [DOI] [Google Scholar]
- Wood B. M.; Chan K. K.; Amyes T. L.; Richard J. P.; Gerlt J. A. Mechanism of the Orotidine 5′-Monophosphate Decarboxylase-Catalyzed Reaction: Effect of Solvent Viscosity on Kinetic Constants. Biochemistry 2009, 48, 5510–5517. 10.1021/bi9006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter D. J.T.; Short S. A. Yeast Orotidine-5′-Phosphate Decarboxylase: Steady-State and Pre-Steady-State Analysis of the Kinetic Mechanism of Substrate Decarboxylation. Biochemistry 2000, 39, 11788–11800. 10.1021/bi001199v. [DOI] [PubMed] [Google Scholar]
- Sievers A.; Wolfenden R. The effective molarity of the substrate phosphoryl group in the transition state for yeast OMP decarboxylase. Bioorg. Chem. 2005, 33, 45–52. 10.1016/j.bioorg.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Toth K.; Amyes T. L.; Wood B. M.; Chan K. K.; Gerlt J. A.; Richard J. P. An Examination of the Relationship between Active Site Loop Size and Thermodynamic Activation Parameters for Orotidine 5′-Monophosphate Decarboxylase from Mesophilic and Thermophilic Organisms. Biochemistry 2009, 48, 8006–8013. 10.1021/bi901064k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt J. G. The synthesis of orotidine 5′-phosphate. J. Am. Chem. Soc. 1963, 85, 1118–23. 10.1021/ja00891a018. [DOI] [Google Scholar]
- Gasteiger E.; Gattiker A.; Hoogland C.; Ivanyi I.; Appel R. D.; Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E.; Hoogland C.; Gattiker A.; Duvaud A.; Wilkins M. R.; Appel R. D.; Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. Proteomics Protocols Handbook 2005, 571–607. 10.1385/1-59259-890-0:571. [DOI] [Google Scholar]
- Gong B. Molecular Duplexes with Encoded Sequences and Stabilities. Acc. Chem. Res. 2012, 45, 2077–2087. 10.1021/ar300007k. [DOI] [PubMed] [Google Scholar]
- Guthrie J. P. Hydration of Carbonyl Compounds, an Analysis in Terms of Multidimensional Marcus Theory. J. Am. Chem. Soc. 2000, 122, 5529–5538. 10.1021/ja992992i. [DOI] [Google Scholar]
- Barnett S. A.Studies on the Mechanism of Action of Orotidine 5′-Monophosphate Decarboxylase. Ph.D. Thesis. University at Buffalo, Buffalo, NY, 2009. [Google Scholar]
- Jencks W. P. Binding energy, specificity, and enzymic catalysis: the Circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 1975, 43, 219–410. 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- Vardi-Kilshtain A.; Doron D.; Major D. T. Quantum and Classical Simulations of Orotidine Monophosphate Decarboxylase: Support for a Direct Decarboxylation Mechanism. Biochemistry 2013, 52, 4382–4390. 10.1021/bi400190v. [DOI] [PubMed] [Google Scholar]
- Kulkarni Y. S.; Liao Q.; Byléhn F.; Amyes T. L.; Richard J. P.; Kamerlin S. C. L. Role of Ligand-Driven Conformational Changes in Enzyme Catalysis: Modeling the Reactivity of the Catalytic Cage of Triosephosphate. J. Am. Chem. Soc. 2018, 140, 3854–3857. 10.1021/jacs.8b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni Y. S.; Liao Q.; Petrović D.; Krüger D. M.; Strodel B.; Amyes T. L.; Richard J. P.; Kamerlin S. C. L. Enzyme Architecture: Modeling the Operation of a Hydrophobic Clamp in Catalysis by Triosephosphate Isomerase. J. Am. Chem. Soc. 2017, 139, 10514–10525. 10.1021/jacs.7b05576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Malabanan M. M.; Zhai X.; Kim K. J.; Reinhardt C. J.; Wierenga R. K.; Drake E. J.; Gulick A. M. Structure–Function Studies of Hydrophobic Residues That Clamp a Basic Glutamate Side Chain during Catalysis by Triosephosphate Isomerase. Biochemistry 2016, 55, 3036–3047. 10.1021/acs.biochem.6b00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai X.; Amyes T. L.; Richard J. P. Role of Loop-Clamping Side Chains in Catalysis by Triosephosphate Isomerase. J. Am. Chem. Soc. 2015, 137, 15185–15197. 10.1021/jacs.5b09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai X.; Amyes T. L.; Richard J. P. Enzyme Architecture: Remarkably Similar Transition States for Triosephosphate Isomerase-Catalyzed Reactions of the Whole Substrate and the Substrate in Pieces. J. Am. Chem. Soc. 2014, 136, 4145–4148. 10.1021/ja501103b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Koudelka A. P.; Amyes T. L.; Richard J. P. Mechanism for Activation of Triosephosphate Isomerase by Phosphite Dianion: The Role of a Hydrophobic Clamp. J. Am. Chem. Soc. 2012, 134, 10286–10298. 10.1021/ja303695u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. Mechanism for Activation of Triosephosphate Isomerase by Phosphite Dianion: The Role of a Ligand-Driven Conformational Change. J. Am. Chem. Soc. 2011, 133, 16428–16431. 10.1021/ja208019p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai X.; Reinhardt C. J.; Malabanan M. M.; Amyes T. L.; Richard J. P. Enzyme Architecture: Amino Acid Side-Chains That Function To Optimize the Basicity of the Active Site Glutamate of Triosephosphate Isomerase. J. Am. Chem. Soc. 2018, 140, 8277–8286. 10.1021/jacs.8b04367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Parson W. W. Modeling electrostatic effects in proteins. Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764, 1647–1676. 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Warshel A. Electrostatic Origin of the Catalytic Power of Enzymes and the Role of Preorganized Active Sites. J. Biol. Chem. 1998, 273, 27035–27038. 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- Wolfenden R. Enzyme catalysis. Conflicting requirements of substrate access and transition state affinity. Mol. Cell. Biochem. 1974, 3, 207–11. 10.1007/BF01686645. [DOI] [PubMed] [Google Scholar]
- Blomberg R.; Kries H.; Pinkas D. M.; Mittl P. R.E.; Grütter M. G.; Privett H. K.; Mayo S. L.; Hilvert D. Precision is essential for efficient catalysis in an evolved Kemp eliminase. Nature 2013, 503, 418–421. 10.1038/nature12623. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Florian J.; Strajbl M.; Villa J. Circe effect versus enzyme preorganization: what can be learned from the structure of the most proficient enzyme?. ChemBioChem 2001, 2, 109–111. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.