

Abstract
Purpose:
Poor prognosis in triple-negative breast cancer (TNBC) is due to an aggressive phenotype and lack of biomarker-driven targeted therapies. Overexpression of cyclin E and phosphorylated-CDK2 are correlated with poor survival in TNBC patients, and the absence of CDK2 desensitizes cells to inhibition of Wee1 kinase, a key cell cycle regulator. We hypothesize that cyclin E expression can predict response to therapies, which include the Wee1 kinase inhibitor, AZD1775.
Experimental Design:
Mono and combination therapies with AZD1775 were evaluated in TNBC cell lines and multiple patient derived xenograft (PDX) models with different cyclin E expression profiles. The mechanism(s) of cyclin E-mediated replicative stress were investigated following cyclin E induction or CRISPR/Cas9 knockout by a number of assays in multiple cells lines.
Results:
Cyclin E overexpression (1) is enriched in TNBCs with high recurrence rates, (2) sensitizes TNBC cell lines and PDX models to AZD1775, (3) leads to CDK2-dependent activation of DNA replication stress pathways and (4) increases Wee1 kinase activity. Moreover, treatment of cells with either CDK2 inhibitors or carboplatin leads to transient transcriptional induction of cyclin E (in cyclin E-low tumors) and result in DNA replicative stress. Such drug mediated cyclin E induction in TNBC cells and PDX models sensitizes them to AZD1775 in a sequential treatment combination strategy.
Conclusions:
Cyclin E is a potential biomarker of response (1) for AZD1775 as monotherapy in cyclin E high TNBC tumors and (2) for sequential combination therapy with CDK2 inhibitor or carboplatin followed by AZD1775 in cyclin E low TNBC tumors.
Translational relevance:
TNBC is a subtype of invasive breast cancer with an aggressive phenotype that has decreased survival compared to other types of breast cancers due, in part, to the lack of biomarker-driven targeted therapies. Here we show that TNBCs can be separated into cyclin E high or low tumors and those with high cyclin E have a significantly worse prognosis. We show that cyclin E high tumors are very sensitive to Wee1 kinase inhibition by AZD1775 as monotherapy. To this end, we report on the relationship between cyclin E levels and the sensitivity to Wee1 kinase inhibition providing the mechanistic evidence in support of a new clinical trial (NCT03253679). We also identified cyclin E as a potential predictor of response for the sequential combination therapy with a CDK2 inhibitor or carboplatin followed by AZD1775 in cyclin E low tumors, providing the scientific rationale for future biomarker-driven clinical trials in TNBC.
Keywords: cyclin E, CDK2, triple-negative breast cancer, Wee1 kinase, AZD1775, MK-1775, dinaciclib, DNA replication stress, patient-derived xenografts
Introduction:
Triple-negative breast cancer (TNBC) is a subtype of invasive breast cancer that lacks estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 (HER2) expression (1). TNBC accounts for up to 17% of breast cancers and represents an aggressive phenotype with higher histologic grade, larger tumor size and more advanced stage at diagnosis, and more likely to affect younger women and African-American women. Patients with TNBC have decreased overall survival compared with patients with other breast cancer subtypes. This decreased survival is due in part to the lack of targeted therapies for TNBCs. While about 40–50% of TNBC patients will achieve complete pathological response (pCR) to standard therapy, there are no biomarkers to select those from the patients who need other targeted approaches. As a result, TNBC patients have a higher likelihood of developing recurrence within the first 5 years of follow-up mainly due to lack of specific targets (2,3).
Over 60% of TNBCs harbor TP53 mutations (4–6) and the cell cycle of these tumors is deregulated at the G1/S and G2/M checkpoints. This deregulation often leads to defects in DNA repair pathways, whose proper function is required to respond to DNA replicative stress (7,8). Exploiting the machinery that regulates cell cycle checkpoints and DNA repair for cancer therapy has been the focus of numerous investigations; however, translation to patient care has not been very successful. A key challenge has been the lack of consistent and validated predictive biomarkers that can readily identify tumors likely to respond to inhibitors of the DNA repair pathways, specifically those that target DNA replicative stress. One such inhibitor, AZD1775, targets Wee1 kinase, a key regulator of the G2/M checkpoint that negatively regulates entry into mitosis through inactivation of CDK1, arresting cells in G2/M and allowing for DNA repair (9). Wee1 kinase inhibition abrogates the G2/M arrest leading to premature mitosis and also deregulates DNA replication at S phase leading to DNA damage. (10–13). Cells with DNA replicative stress are therefore vulnerable to Wee1 kinase inhibition (14). Although AZD1775 has been proposed for treatment of TP53-defective cancers (15,16), studies have shown that the therapeutic efficacy of Wee1 inhibition is independent of TP53 status (11,17,18), suggesting alternative pathways that can deregulate G1-to-S transition (19). In a recent phase I study with AZD1775, none of the patients with a documented TP53 mutation exhibited a response to this agent (20). Given the complex regulation of cell cycle checkpoints, these seemingly contradictory results are not unexpected, indicating alternative pathways that can deregulate the cell cycle and biomarkers are needed to help predict response to Wee1 kinase inhibition.
Cyclin E, the key regulator of the G1 to S transition, activates CDK2 to progress through the S phase and promote DNA replication (21,22). Cyclin E levels are tightly cell cycle regulated, with peak CDK2-associated kinase activity occurring at the G1-to-S boundary in normally proliferating cells and tissues (23). Cyclin E overexpression has been observed in a broad spectrum of human malignancies, including breast cancer, and is associated with poor outcomes (24–31). Overexpression of cyclin E deregulates the cell cycle at both G1/S and G2/M transitions by accelerating S phase entry (32), affecting centrosome amplification (33) and stimulating premature mitosis (34,35). An overactive cyclin E/CDK2 complex also increases DNA origin firing and induces re-replication, causing DNA replication stress in cancer cells (36,37), potentially sensitizing these cells to Wee1 kinase inhibition. Consistently, CDK2 deletion desensitizes cancer cells to inhibition of Wee1 kinase (38). In this study, we hypothesized that overexpression of cyclin E induces DNA replicative stress and stimulates DNA repair responses, thereby sensitizing cells to AZD1775. We also interrogated whether cyclin E expression levels can predict the response to AZD1775 as a single agent or in combination with agents that can transiently increase cyclin E levels in preclinical studies.
Materials and Methods
Cell lines
76NE6 cyclin E–inducible cell lines were generated from the immortalized human mammary epithelial cell line 76NE6 and cultured in Tet-free medium as previously described (39). HEK-293T cells for lentiviral packaging and the TNBC cell lines HCC1806, MDA436, MDA468, and MDA231 were obtained from the American Type Culture Collection and cultured as described previously (30,40). The TNBC cell lines SUM149 and SUM159 were obtained from Asterand Bioscience and cultured in Ham F12 medium with 5% fetal bovine serum, 5 μg/mL insulin, and 1 μg/mL hydrocortisone. All cells were free of mycoplasma contamination. Cell lines were regularly (every 6 months) authenticated by karyotype and short tandem repeat analysis at the MD Anderson Cancer Center Characterized Cell Line Core facility.
Mouse and in vivo studies
All animal studies were approved by the MD Anderson Institutional Animal Care and Use Committee and strictly followed the recommendations in the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health. The generation and preparation of patient derived xenograft (PDX) models were described in the supplementary methods as reported previously (41). A total of 4×106 SUM149 or MDA231 cells were injected into the mammary fat pad to generate xenograft models. The mice were given 50 mg/kg AZD1775 (prepared in 0.5w/v% Methyl Cellulose 400 Solution) orally or 25 mg/kg dinaciclib (prepared in 20% (2-hydroxypropyl)-b-cyclodextrin) or 30 mg/kg carboplatin (prepared in sterile water) by intraperitoneal injection. The length and width of tumor xenografts were measured by caliper twice per week and the volume of tumor was calculated by the formula volume = length × (width)2/2. The specific treatment conditions for each experiment is provided in supplementary methods.
High-throughput survival assay (HTSA)
Cells were treated and their survival examined in 96-well plates over an 11-day period, a method that allows analysis of cytotoxicity of one or more drugs in a wide range of adherent cell lines and provides results that are highly consistent with classic clonogenic assays as described previously (40,42). At the end of the 11-day assay, the plates were subjected to an MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide; RPI Corp.) assay as described previously (40,42). MTT was solubilized, and the absorbance of each well was read at 590 nm using an Epoch microplate spectrophotometer (BioTek). The combination index (CI) for each combination treatment was calculated using the CalcuSyn program (Biosoft). CI < 0.9 indicates the synergy of a combination treatment; 0.9<CI<1.1 indicates additivity and CI>1.1 indicates antagonism (40,42). The agents subjected to this assay are AZD1775 (obtained from the Institute of Applied Cancer Science, MD Anderson Cancer Center), dinaciclib (Merck & Co., Inc.), meriolin5 (ManRos Therapeutics), SNS032 (Selleck Chemicals), roscovitine (ManRos Therapeutics), palbociclib (Pfizer), MLN8237 (Selleck Chemicals), carboplatin (Sigma-Aldrich), cisplatin (Sigma-Aldrich), paclitaxel (Sigma-Aldrich), epirubicin and doxorubicin (obtained from the pharmacy at The University of Texas MD Anderson Cancer Center). Additional details regarding the individual treatment conditions and doses of each drug used per cell line are provided in supplementary methods.
CRISPR/Cas9 KO
The sgRNA targeting human cyclin E and CDK2 were cloned into pX330GFP, which was transfected into MDA231 cells or MDA231 cyclin E–inducible cells using PEI (polyethylenimine). Green fluorescent protein–positive cells were sorted into 96-well plates at one cell per well. Successful cyclin E knockout (KO) or CDK2 KO in clones was confirming using Western blot.
The cyclin E sgRNA sequence was 5´GGACGGCGGCGCGGAGTTCT3´. The CDK2 sgRNA sequences were 5´CACCGCATGGGTGTAAGTACGAACA3´ and 5´CACCGGATTTCGGCCAAGGCAGCCC3´.
Statistical analysis
For patient cohort studies, the relationship between cyclin E and p-CDK2 scores on immunohistochemistry (IHC) was evaluated by using Stata SE statistical software version 12.0 (StataCorp LP). All tests were two-tailed, and statistical significance was set at p<0.05. End points were recurrence-free survival and overall survival and were calculated from the time of surgery to breast cancer recurrence. Breast cancer cases from the TCGA and METABRIC database were analyzed using the cbioportal website (www.cbioportal.org).
Each cell culture experiment had at least three technical and three biological repeats. Continuous outcomes were summarized with means and standard error. Comparisons between two groups were analyzed by two-sided t-test using MS Excel or Prism 6. Comparisons among more than 2 groups were analyzed by one-way ANOVA using Prism 6. Tumor growth in the mouse models was compared by two-sided t-test with MS Excel. Kaplan-Meier survival curves in the mouse studies were generated by Prism 6 software (GraphPad), and the significance was determined by using a log-rank (Mantel-Cox) test. All graphs were generated either in MS Excel or Prism 6.
Results
High cyclin E levels predict poor prognosis in TNBC.
To determine whether overexpression of cyclin E is enriched in TNBC and its relationship with prognosis, we compared the percentages of patient samples with cyclin E alteration (CCNE1 gene amplification and/or RNA upregulation) among all breast cancer subtypes in The Cancer Genome Atlas (TCGA) and METABRIC databases (fig. 1A, fig. S1A–B) (43,44). We found a higher prevalence of cyclin E alteration in TNBC patients (52% in TCGA and 40% in METABRIC) than in patients with the estrogen receptor–positive (ER+) subtype (3% in TCGA and 2.3% in METABRIC). Cyclin E DNA/RNA alterations were also correlated with worse outcomes in the TCGA TNBC subset (fig. 1B). Next we examined the expression of cyclin E protein levels using immunohistochemical staining in 248 TNBC samples in tissue microarrays from the National Cancer Institute and the United Kingdom (fig. 1C, fig. S1C, table S1) (30). Of these patients, 77% had high expression of cyclin E and significantly shorter time to recurrence than those with low cyclin E expression (fig. 1D). Together, these findings suggest that TNBCs with elevated cyclin E levels have a higher recurrence rate than those with low cyclin E levels.
Figure 1. Cyclin E overexpression sensitizes TNBC to Wee1 inhibition in vitro and in vivo.
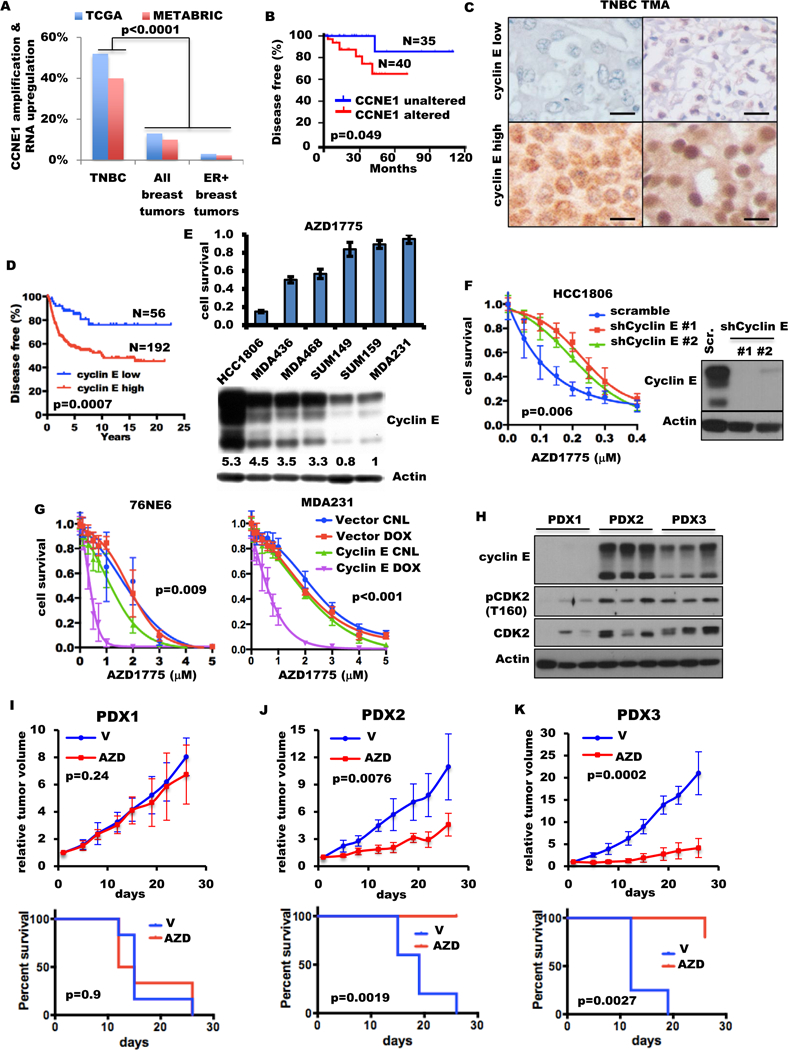
(A) Percentage of breast tumors with CCNE1 amplification and/or RNA upregulation in TNBC, all tumors and ER positive (ER+) tumors from TCGA and METABRIC databases. Fisher exact test was used to compare two groups. (B) Kaplan-Meier survival plot of cyclin E alterations (mRNA and DNA) of the TNBC patient cohort from the TCGA database. (C) Cyclin E immunohistochemical analysis of TNBC tumor tissues from two representative patient samples with low (top) or high (bottom) cyclin E. (Scale: 600 μm). (D) Kaplan-Meier survival plot of cyclin E immunohistochemical results (high/low) from 248 TNBC samples from two different patient cohorts. (E) Six TNBC cell lines were immunoblotted (bottom) with the indicated antibodies and subjected to high-throughput survival assay (HTSA) (top) with 0.4 μM AZD1775 for 48 hours. (F) HCC1806-shRNA cyclin E cells were subjected to HTSA (left) with increasing concentrations of AZD1775 for 48 hours as shown in fig. S2C and were immunoblotted (right) with the indicated antibodies. (G) Cyclin E expression in the inducible 76NE6 cells and MDA231 cells treated with (DOX) or without (control, or CNL) 10 ng/mL doxycycline for 10 days. After cyclin E was induced for 24 hours, cells were treated with AZD1775 for 48 hours and subjected to MTT on day 11, as shown in fig. S2E. (H) Immunoblot of three different tumors from TNBC PDX models with the indicated antibodies. (I-K) Mice harboring (I) PDX1, (J) PDX2, or (K) PDX3 models were treated with either vehicle (V) or 50 mg/kg AZD1775 (AZD) for 4 cycles as shown in fig. S4A, presented as (top) relative tumor volume and (bottom) ethical endpoint survival (n=4–6). One-way analysis of variance (ANOVA) was used for multiple-group comparison. Log-rank test was used in survival experiments.
Cyclin E overexpression sensitizes TNBC to Wee1 kinase inhibition in vitro and in vivo.
Overexpression of cyclin E has been shown to promote genomic instability by causing DNA replication stress and deregulation of the G1-to-S transition in various in vitro and in vivo model systems (33,34,45–47). Wee1 kinase, which prevents premature mitotic entry by inhibiting CDK1 and CDK2, may be essential for preventing massive DNA damage and cell death when cyclin E is overexpressed (48–50). To test whether overexpression of cyclin E sensitizes TNBC cells to Wee1 kinase inhibition, we evaluated the sensitivity of six TNBC cell lines to the clinically available Wee1 inhibitor AZD1775 (51). Expression of cyclin E was highly predictive (r=0.90) of response to AZD1775 in all six TNBC cell lines (fig. 1E, fig. S2A). Among other key cell cycle modulators examined, only p-CDK1 (Y15), which is a known downstream effector of Wee1 kinase, and p-CDK2 (T160) were moderately predictive of AZD1775 sensitivity in only two (HCC1806 and MDA231) of six cell lines (fig. S2B). Knockdown of cyclin E in HCC1806 and MDA231 desensitized them to AZD1775 (fig. 1F, fig. S2C–D, table S2), while induction of cyclin E in 76NE6 and MDA231 cells significantly increased sensitivity to AZD1775 (fig. 1G fig. S2E–F, table S2). Induction of tumor-specific cytoplasmic low-molecular-weight cyclin E (39,52) in 76NE6 cells also increased sensitivity to AZD1775 (fig. S2G).
To examine the duration of cyclin E overexpression required to sensitize cells to Wee1 kinase inhibition, we evaluated the response of MDA231 and 76NE6 cyclin E–inducible cells to AZD1775 by inducing the expression of cyclin E from 24 hours to 10 days (fig. S3A). Induction of cyclin E for 48 hours was sufficient to maximally sensitize cells to Wee1 kinase inhibition (fig. S3B–D, table S2). These results suggest that as long as cyclin E can be transiently induced during treatment with AZD1775, the cells can be sensitized to Wee1 kinase inhibition.
Next, we examined the sensitivity to AZD1775 in vivo in one cyclin E–low and two cyclin E–high TNBC PDX models which were selected from 25 TNBC PDXs on the basis of the cyclin E levels (41) (fig. S4). PDX1 has a normal CCNE1 copy number and low cyclin E expression, while PDX2 and PDX3 exhibit CCNE1 amplification and cyclin E overexpression (fig. 1H table S3). The histology of these PDX models were similar to the original patient tumors (fig. S5). CDK2 and p-CDK2 are co-overexpressed with cyclin E in PDX2 and PDX3 (fig. 1H). Mice representing each PDX model were treated with either vehicle or 50 mg/kg AZD1775 for 4 cycles and euthanized at the end of treatment or when the maximum allowable tumor burden was reached (schema, fig. S6A). Consistent with our in vitro results, AZD1775 treatment significantly decreased tumor burden and improved survival of the cyclin E–overexpressing PDX2 and PDX3 models but not the PDX1 model (fig. 1I–K fig. S6B). Collectively, these results suggest that cyclin E overexpression in cell lines and PDX models predicts response to Wee1 kinase inhibition.
Cyclin E–overexpressing cells rely on Wee1 kinase to suppress DNA replicative stress and stimulate DNA damage repair.
Since both cyclin E and Wee1 kinase are involved in maintaining normal DNA replication fork progression (12,36,47,53), we next examined whether the progression of DNA replication in TNBC cells with high cyclin E is altered upon treatment with AZD1775 using DNA fiber assays. MDA231 cyclin E–inducible cells were pulse labeled with iododeoxyuridine for 60 minutes followed by chlorodeoxyuridine labeling for 30 minutes, and the speed of DNA replication was quantitated. Compared with control cells (0.59 kb/minute), DNA replicative fork progression was slower in cells with either cyclin E overexpression (0.43 kb/minute) or Wee1 kinase inhibition (0.29 kb/minute) (fig. 2A–B fig. S7A–B). Moreover, the replication forks in cyclin E induced cells treated with AZD1775 not only progressed at only 25% the speed of those in control cells (fig. 2A–B) but also showed severe stress (i.e., 75% of these replication forks progressed slower than 0.15 kb/minute) (fig. S7B).
Figure 2. Wee1 kinase protects the cyclin E-overexpressed cells from DNA replication stress and DNA damage.
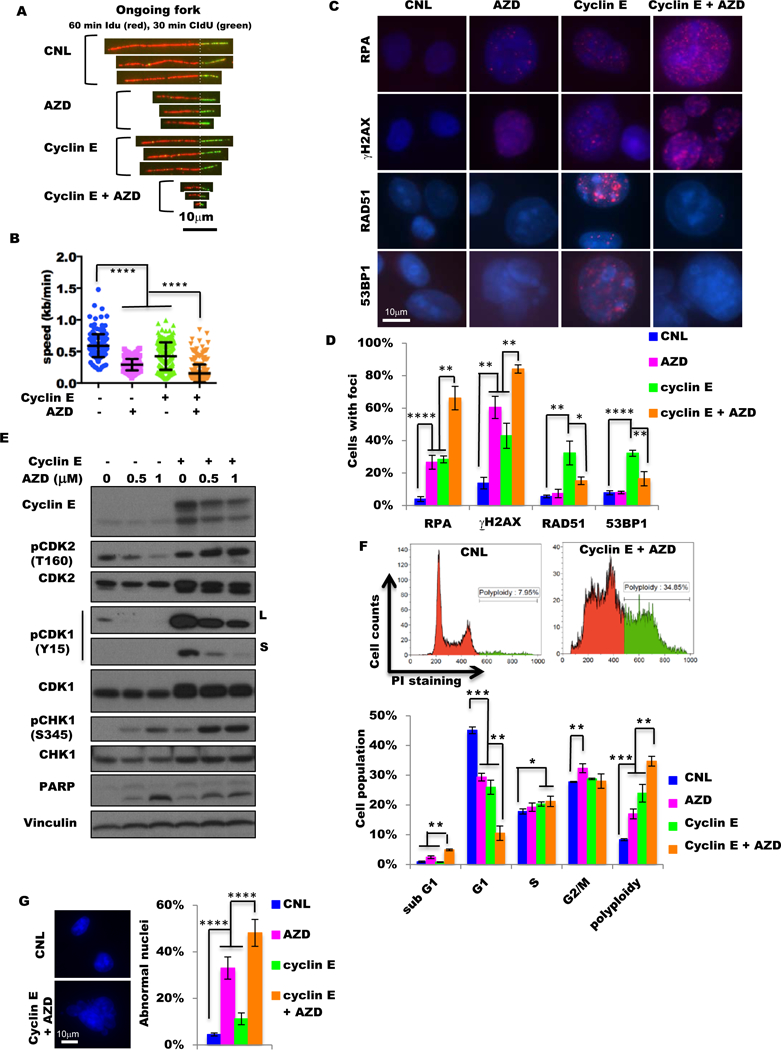
(A) Representative ongoing replication forks of DNA tracts pulse-labeled with iododeoxyuridine (IdU) and chlorodeoxyuridine (CIdU) from control cells (CNL), cells treated with 48 hours of 1 μM AZD1775 (AZD), cells with 48-hour induction of cyclin E by 10 ng/mL doxycycline (cyclin E), and cells with both cyclin E induction and AZD1775 treatment for 48 hours (cyclin E + AZD) in MDA231 cyclin E-inducible cells. Scale bar, 10 μm. (B) Analysis of replication fork progression of the ongoing replication forks in the samples as shown in (A). More than 190 DNA fibers were analyzed for each sample. (C) Representative images of RPA, γH2AX, RAD51, and 53BP1 foci in control cells (CNL), cells treated with 48 hours of 1 μM AZD1775 (AZD), cells with 48-hour induction of cyclin E by 10 ng/mL doxycycline (cyclin E), and cells with both cyclin E induction and AZD1775 treatment for 48 hours (cyclin E + AZD) in MDA231 cyclin E-inducible cells. Scale bar, 10 μm. (D) Quantification of foci-positive cell (>5 foci per cell) population in the samples as in (C), n=3. (E) Immunoblot of indicated antibodies of MDA231 cyclin E–inducible cells with (+) or without (−) cyclin E induction by 10 ng/mL doxycycline and with different concentrations (0, 0.5, and 1 μM) of AZD1775 for 48 hours. L, long exposure; S, short exposure. Vinculin is the loading control. (F) The representative histogram images (upper) and the cell-cycle analysis with flow cytometry (lower) in control cells (CNL), cells treated with 48 hours of 1 μM AZD1775 (AZD), cells with 48-hour induction of cyclin E by 10 ng/mL doxycycline (cyclin E), cells with both cyclin E induction and AZD1775 treatment for 48 hours (cyclin E + AZD) in MDA231 cyclin E-inducible cells. Green in histogram image indicates polyploidy. (G) Representative images (left) of normal (CNL) and abnormal (cyclin E + AZD) nuclei and quantification of the frequency of abnormal nuclei in control cells (CNL), cells treated with 48 hours of 1 μM AZD1775 (AZD), cells with 48-hour induction of cyclin E by 10 ng/mL doxycycline (cyclin E), and cells with both cyclin E induction and AZD1775 treatment for 48 hours (cyclin E + AZD) in MDA231 cyclin E-inducible cells. Scale bar, 10 μm. A two-tailed unpaired t-test was used to compare two groups. Error bars represent standard error of the mean. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
We also quantitated replication protein A (RPA) foci, an indicator of replicative stress (54,55), in the MDA231 cyclin E–inducible cells. Consistent with the results of the DNA fiber assay, the RPA foci–positive cells were significantly more frequent (66%) in the cyclin E–induced cells treated with AZD1775 than in the control cells (4%), the untreated cyclin E–induced cells (28%) or the AZD1775–treated uninduced (27%) cells (fig. 2C–D fig. S7C). Furthermore, the levels of key regulators of DNA replicative stress: ATR, p-CHK1(S345), Wee1, and p-CDK1(Y15), also increased upon cyclin E induction and decreased upon cyclin E downregulation in 76NE6 and MDA231 cyclin E inducible and HCC1806 cyclin E knockdown cell lines (fig. 2E and fig. S7D). Collectively, these results suggest that replication fork progression and replicative regulation pathways in cells with high cyclin E is protected by Wee1 kinase activity.
The cyclin E–induced DNA replication stress may also increase DNA damage and activate DNA repair pathways (36,47,48). Consistent with this hypothesis, immunofluorescence staining of γH2AX foci (marker of DNA double-strand breaks) and RAD51 or 53BP1 foci (markers of DNA repair) revealed that cyclin E induction significantly increased frequencies of foci-positive cells for all three markers compared with uninduced MDA231 cells (fig. 2C–D fig. S7C). Treatment with AZD1775 increased γH2AX-positive cells, but not RAD51 or 53BP1-posivitve cells. Specifically, the majority (84%) of cyclin E–induced MDA231 plus AZD1775-treated cells are γH2AX positive. However, only 15% and 16% of these cells were RAD51 and 53BP1 positive, respectively (fig. 2C–D fig. S7C). These results suggest that the majority of the cyclin E–induced, AZD1775-treated MDA231 cells harbor unrepairable DNA damage.
Treatment of cyclin E–induced MDA231 and 76NE6 cells with AZD1775 resulted in significant accumulation of sub-G1 (fig. 2F fig. S8A) and cleaved PARP (fig. 2E) compared with control cells. Furthermore, in these cyclin E–induced AZD1775-treated MDA231 and 76NE6 cells, the percentage of cells in G1 phase dropped to 10.5%−13.5% while the polyploid population increased up to 4-fold compared with controls (fig. 2F fig. S8A). Measurement of multinucleation and micronucleation (indicators of mitotic catastrophe) revealed that treatment of high cyclin E MDA231 cells with AZD1775 resulted in accumulation of cells with abnormal nuclei in 48% of these cells but only 4.5% of uninduced and/or untreated controls (fig. 2G fig. S8B). Collectively, these results suggest that cyclin E overexpression induces DNA replicative stress and increases DNA double-strand breaks, which can be repaired via Wee1 kinase activation and the ATR-CHK1 pathway. However, when Wee1 kinase was inhibited by AZD1775 in the cyclin E–high cells, DNA repair pathways were unable to repair DNA damage sufficiently, leading to accumulation of abnormal nuclei and cell death.
CDK2 is required for cyclin E mediated sensitization to Wee1 kinase inhibition in TNBC cells.
Kinase activities of CDK2 and cyclin E in both 76NE6 cyclin E–inducible cells and HCC1806 cyclin E–downregulated cells (fig. S9A) were positively associated with levels of p-CDK2 (T-160) (fig. S7D), suggesting that the kinase associated function of cyclin E is CDK2 dependent in these cells. Moreover, in patient samples (from our two TNBC patient cohorts fig. 1C–D), 85% (164/192) of all cyclin E–overexpressing TNBC tumors also overexpressed p-CDK2 (p=0.0001; table S4) and that co-overexpression of cyclin E and p-CDK2 (but not p-CDK2 alone) was significantly (p=0.0004) associated with recurrence (fig. 3A fig. S9B). Based on these findings we next interrogated if CDK2 is essential in sensitizing cyclin E–overexpressing cells to Wee1 kinase inhibition.
Figure 3. Cyclin E overexpression depends on CDK2 to sensitize cells to Wee1 kinase inhibition.
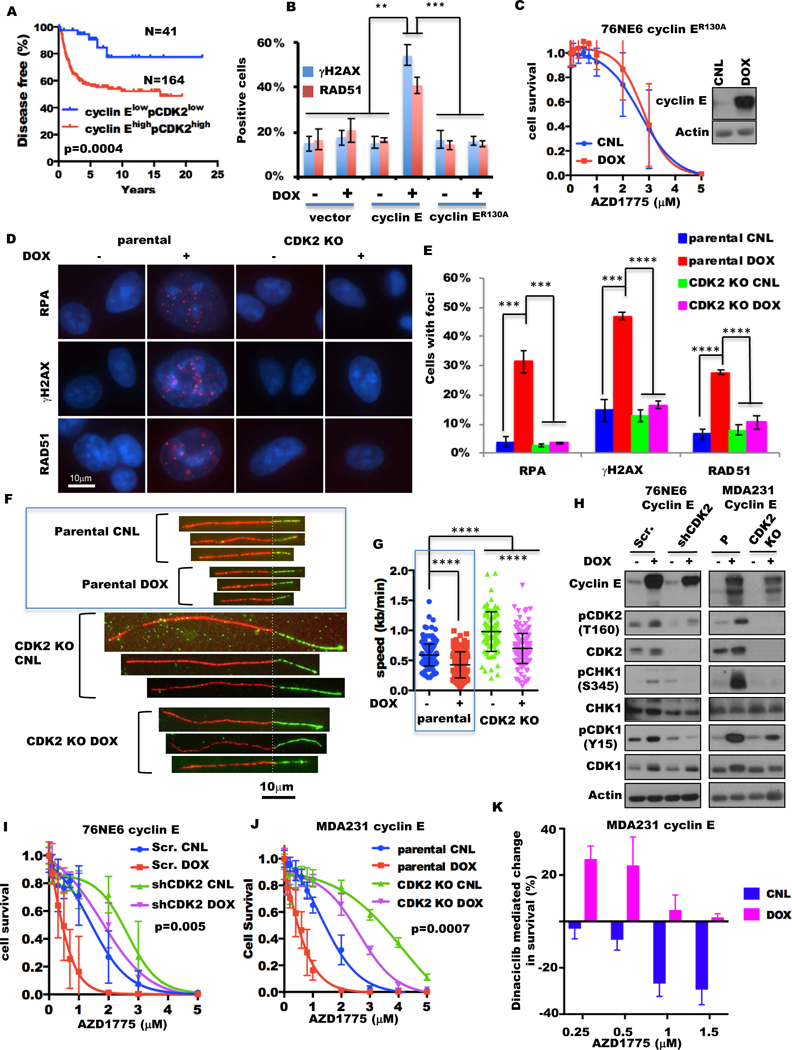
(A) Kaplan-Meier survival plots of cyclin EhighpCDK2high and cyclin ElowpCDK2low tumors from the TNBC patient cohorts from fig. 1D. (B) Percentages of γH2AX- and RAD51-positive cells of 76NE6 cyclin E-inducible cells cultured with (+) or without (−)10 ng/mL doxycycline (DOX) for 24 hours by IHC staining. (C) 76NE6 cyclin ER130A inducible cells cultured with (DOX) or without (CNL) 10 ng/mL doxycycline for 24 hours were treated with AZD1775 for 48 hours and subjected to HTSA as in fig. S2C. Inset shows immunoblot of cyclin E expression. (D) Representative images of RPA, γH2AX, RAD51, and 53BP1 foci in MDA231 cyclin E-inducible cells as in (A). Scale bar, 10 μm. (F) Quantification of foci-positive cell (>5 foci per cell) population in the samples as in (D), n=3. (F) Representative ongoing replication forks of DNA tracts pulse-labeled with IdU and CIdU from the parental cells and CDK2 knockout (KO) cells (via CRISPR/CAS9) of MDA231 with or without cyclin E induction for 48 hours by 10 ng/mL doxycycline (DOX). Scale bar, 10 μm. The data in the blue box are presented in fig 2A. (G) Analysis of replication fork progression of the ongoing replication forks in the samples as in (F). More than 89 DNA fibers were analyzed for each sample. The data in the blue box were presented in fig 2B. (H) 76NE6 cyclin E–inducible cells with scrambled shRNA (Scr.) or cyclin E shRNA knockdown (shCyclin E) (left) with (+) or without (−) 10 ng/mL doxycycline (DOX) for 24 hours (left) and MDA231 cyclin E-inducible cells as in (F) (right) were subjected to immunoblot with the indicated antibodies. Actin is the loading control. (I) 76NE6 cyclin E–inducible cells with CDK2 or scramble shRNA were subjected to HTSA as in fig. S2E. (J) Parental and CDK2 knockout cells of MDA231 cyclin E–inducible cells were subjected to HTSA as in fig. S10F. (K) MDA231 cyclin E–inducible cells were concomitantly treated with 6 nM dinaciclib and different concentrations of AZD1775 with or without 10 ng/mL doxycycline for 48 hours as in fig. S10H. The bar graph shows the changes in cell survival between the cells without and with 6nM dinaciclib at absence (CNL) or presence (DOX) of 10 ng/mL doxycycline at the different AZD1775 doses.
To this end we compared the DNA-damage and response abilities of the mutated cyclin ER130A, which prevents its interaction with CDK2 (39), to wild-type cyclin E in 76NE6 inducible cells. While the induction of wild-type cyclin E resulted in significant (p<0.0003) accumulation of γH2AX and RAD51 foci-positive cells (fig. 3B) and sensitized cells to AZD1775 (fig. 1G), the induction of the mutant cyclin ER130A did not sensitize cells to AZD1775 nor increase the γH2AX and RAD51 foci-positive cells compared with the vector alone and the uninduced control cells (fig. 3B–C fig. S9C). Furthermore, knockdown of CDK2 in HCC1806 cells significantly (p=0.0001) reduced their sensitivity to AZD1775 (fig. S9D) and decreased the levels of DNA replication stress regulators such as p-CHK1, ATR, and Wee1 (fig. S9E).
Next we explored whether CDK2 depletion could rescue cyclin E–induced DNA replicative stress and AZD1775 sensitivity. CDK2 was knocked out (KO via CRISPR/CAS9) in MDA231 cyclin E–inducible cells, and the indicators of DNA replicative stress (RPA foci), DNA damage (γH2AX foci), and DNA repair (RAD51 foci) were measured. The percentages of cells with foci for all three indicators approached that of parental cells with basal levels of CDK2 and cyclin E expression (fig. 3D–E fig. S10A). The progression of DNA replication forks in CDK2 KO cells was faster than that in the parental cells under both cyclin E–uninduced (0.98 kb/minute in CDK2 KO vs 0.59 kb/minute in parental) and cyclin E–induced (0.69 kb/minute in CDK2 KO vs 0.46 kb/minute in parental) conditions (fig. 3F–G fig. S10B–C). Although the replicative forks of CDK2 KO cells with cyclin E overexpression (mean 0.69 kb/minute) progressed slower than those of the uninduced CDK2 KO cells, these forks still progressed faster than those of the parental cyclin E–uninduced cells (mean 0.56 kb/minute). These results suggest that cyclin E- induced replicative stress, DNA damage and DNA repair are all dependent on a functional CDK2.
Similarly, the KO or knockdown (shRNA) of CDK2 inhibited the cyclin E–mediated increase of DNA replication stress regulators (such as p-CHK1 (S345), p-CDK1 (Y15), ATR, and Wee1 kinase) in both MDA231 and 76NE6 cyclin E–inducible cells (fig. 3H fig. S10D–E). To examine whether the ablation or knockdown of CDK2 can also rescue cells from cyclin E–induced sensitivity to AZD1775, 76NE6 CDK2 shRNA and MDA231 CDK2 KO cells were treated with increasing concentrations of AZD1775 in the presence (induced) or absence (uninduced) of cyclin E. The results revealed that KO or knockdown of CDK2 (in the presence or absence of cyclin E) made the cells more resistant to AZD1775 compared with their respective controls (fig. 3I–J, fig. S10F–G). To explore the effect of pharmacologic inhibition of CDK2 on sensitivity to AZD1775 induced by cyclin E overexpression, MDA231 cyclin E–inducible cells were treated with dinaciclib (a CDK1/2/5/9 inhibitor) and AZD1775 (fig. S10H). The addition of dinaciclib increased the cell survival of the cyclin E–induced (but not the control) cells to AZD1775 across all concentrations we examined (fig. 3K, fig. S10I). Collectively these results (fig 3, fig S9, and fig S10) underscore the requirement of CDK2 for cyclin E–induced sensitivity to Wee1 kinase inhibition.
Sequential combination treatment of dinaciclib followed by AZD1775 in TNBC with low cyclin E levels is synergistic in vitro and in vivo.
Since in the uninduced (cyclin E–low) MDA231 cells, the combination treatment of dinaciclib and AZD1775 results in more cell killing than the AZD1775 treatment alone (fig. 3K fig. S10I), we next hypothesized that the combination of these two agents is likely to be synergistic in cyclin E–low TNBC cells. To test this hypothesis, we compared the efficacy of the dinaciclib and AZD1775 combination in MDA231 cyclin E–inducible cells, using a high-throughput survival assay that allows comprehensive evaluation of two or more drugs given either in sequence or concomitantly (schematically depicted in fig. S10H and S11A) (40,42).
The results revealed that only when cyclin E–low (uninduced) cells were treated sequentially with dinaciclib followed by AZD1775 was the combination therapy synergistic, with a CI of 0.8. On the other hand, the concomitant treatment with both agents, in the presence or absence of cyclin E, resulted in an antagonistic response (CI 1.14–1.5). Furthermore, the sequential combination of dinaciclib followed by AZD1775 was only additive (CI of 1.0) in cyclin E–induced cells (fig. 4A table S5). To validate these findings, we treated the panel of six TNBC cell lines (from fig. 1E) either with sequential treatment of dinaciclib followed by AZD1775 (D→AZD) or with concomitant treatment of both agents (D+AZD) (fig. S11B). The only condition resulting in a synergistic interaction of the two agents, with CIs of 0.3–0.8, was the sequential administration of the drugs (D→AZD) in the cyclin E–low TNBC cell lines (SUM149, SUM159, and MDA231). All other conditions—sequential or concomitant in cyclin E–high TNBC cells (HCC1806, MDA436, MDA468) or concomitant in cyclin E–low TNBC cells—resulted in either antagonistic or additive interactions, with CIs of 1.0–1.7 (fig. 4B table S5), which were also confirmed by clonogenic assays (fig. 4C). The D→AZD treatment resulted in apoptosis in the cyclin E–low TNBC cells, as shown by (i) higher percentages of sub-G1, (ii) an increased annexin V–positive population, and (iii) accumulation of cleaved PARP in the combination-treated cells compared with the single agent–treated cells (fig. 4D fig. S11C, table S6). Cell cycle analyses revealed that the changes in G1, S, or G2/M phases between the high and low cyclin E cells did not correlate with the efficacy of the D→AZD treatment (fig. S11D).
Figure 4. Sequential combination treatment with CDK2 inhibitor followed by AZD1775 in cyclin E–low TNBC is synergistic in vitro and in vivo.
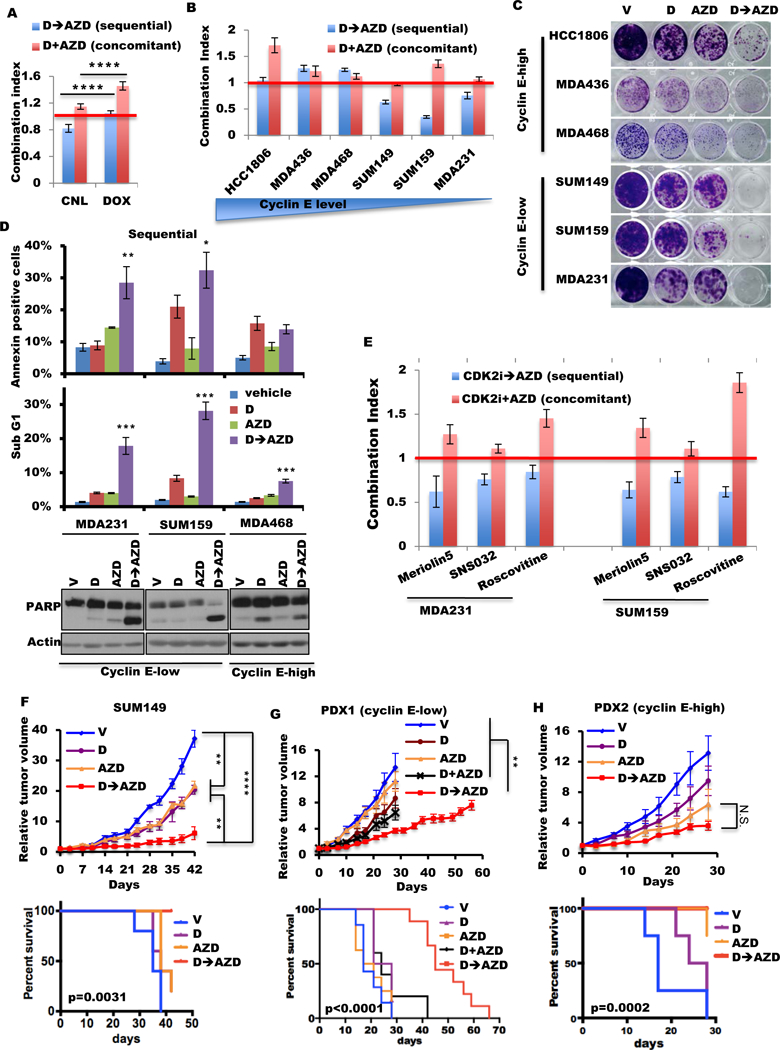
(A) Combination index (CI) of sequential (D→AZD) and concomitant (D+AZD) combination of dinaciclib and AZD1775 in MDA231 cyclin E–inducible cells with (DOX) or without (CNL) 10 ng/mL doxycycline. CI=1.0 indicates an additive effect (red line). (B) Mean CI values of sequential (D→AZD) and concomitant (D+AZD) treatment using dinaciclib and AZD1775 in TNBC cells by HTSA. CI=1.0 indicates an additive effect (red line). The concentrations of inhibitors of this assay are listed in table S5. (C) The clonogenic assay of TNBC cells with dinaciclib (D) and/or AZD1775 (AZD), as single agents or in combination sequentially (D→AZD). (D) Cells were treated with vehicle, dinaciclib (24 hours, D), AZD1775 (48 hours, AZD), or sequential dinaciclib (24 hours) followed by AZD1775 (48 hours) (D→AZD) at concentrations listed in table S6. Cells subjected annexin V assay (top), quantification of the sub-G1 population (middle), or immunoblot with PARP antibody (bottom). Differences between combination treatment and other groups were analyzed statistically for significance. (E) Mean CI values of sequential (CDK2i→AZD) and concomitant (CDK2i+AZD) treatment using CDK2 inhibitor (meriolin5, SNS032, or roscovitine) and AZD1775 in MDA231 and SUM159 by HTSA as in (B). CI=1.0 indicates an additive effect (red line). (F, G, H) Tumor volumes (top) and mouse survival (bottom) of mice harboring xenograft tumors derived from (F) SUM149 cells or patient tumors, (G) PDX1, or (H) PDX2 were treated with 25 mg/kg dinaciclib once per day for 2 days and 50 mg/kg AZD1775 twice per day for 2 days of each 7-day cycle. (Schematic in fig. S12A.) V, mouse treated by vehicle only; D, mouse treated by dinaciclib and vehicle of AZD1775; AZD, mouse treated by AZD1775 and vehicle of dinaciclib; D+AZD, mouse concomitantly treated by dinaciclib and AZD1775; D→AZD, mouse sequentially treated by dinaciclib and AZD1775; N.S., not significant. SUM149 xenografts were treated for 6 cycles; PDX1 and PDX2 were treated for 4 cycles, with the exception of the D+AZD arm and the D→AZD arm in PDX1, which were treated for 6 cycles and 10 cycles respectively. A two-tailed unpaired t-test was use to compare two groups (n=4–20). Error bars represent standard error of the mean. The log-rank (Mantel-Cox) test was used in survival experiments. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
To confirm the synergism of CDK2 inhibition followed by Wee1 kinase inhibition in the cyclin E–low TNBC cells, we evaluated the efficacy of the sequential combination of other CDK2 inhibitors (meriolin5, SNS032, and roscovitine) with AZD1775. Results revealed that only the sequential combination of these CDK2 inhibitors followed by AZD1775, but not the concomitant treatment combination, caused synergistic interaction in the cyclin E–low TNBC cells (fig. 4E table S7).
To evaluate the efficacy of D→AZD treatment in vivo, we administered these drugs sequentially in orthotopic SUM149 xenograft and TNBC PDX models with either low or high cyclin E expression. For each model, we treated the mice first with 25 mg/kg dinaciclib once per day for 2 days, followed by 50 mg/kg AZD1775 twice per day for 2 days, followed by 3 days off; this 7-day cycle was repeated for 4–10 cycles (fig. S12A). Consistent with the in vitro results, D→AZD sequential treatment prolonged the mouse survival in PDX1 model (low cyclin E) synergistically (p=0.0006) and in SUM149 xenograft model with a trend toward synergism (p=0.097) (fig. 4F–G table S8). In both models, the D→AZD sequential treatment also significantly decreased tumor growth compared with those treated with either of the single agents (fig. 4F–G fig. S12B–C, table S9). In the cyclin E–amplified PDX2 model, which was sensitive to AZD1775 alone (fig. 1J), the D→AZD combination showed no significant improvements compared with AZD1775 alone (fig. 4H fig. S12B–C, Tables S8, S9).
We next interrogated whether the concomitant treatment (D+AZD) of the cyclin E–low PDX1 model could also result in a survival benefit compared with single-drug treatment. To this end, mice were treated concomitantly with 25 mg/kg dinaciclib once per day and 50 mg/kg AZD1775 twice per day on days 1 and 2 of each cycle, followed by 5 days off (fig. S12A). This 7-day treatment cycle was repeated for up to 6 cycles. The D+AZD arm did not show a significant difference in tumor growth compared with the single-drug arm and showed much less effectiveness compared with the D→AZD arm (fig. 4G fig. S12B–C). Furthermore, the D+AZD treatment arm had no additional survival benefit compared with no treatment or single-drug treatment arm (fig. 4G). Lastly, the D→AZD treatment was well tolerated in mice, as indicated by the lack of weight loss in any of the treatment arms during the treatment (fig. S12D) as well as normal whole-blood counts measured at the end of 6–10 cycles (fig. S13). Collectively these results suggest that the sequential combination treatment using dinaciclib followed by AZD1775 is much more effective than the concomitant combination in vivo in the cyclin E low PDX model.
Transient CDK2 inhibition–induced cyclin E sensitizes cyclin E–low TNBC cells to Wee1 kinase inhibition.
To examine the mechanism of the sequential treatment with dinaciclib followed by AZD1775, we interrogated whether dinaciclib could induce the transient upregulation of cyclin E levels in the TNBC cells since cyclin E induction sensitized these cells to Wee1 kinase inhibitor (fig. 1G, fig S3B–D). Therefore, we compared the cyclin E levels among all six TNBC cell lines with or without 24-hour dinaciclib treatment and after the treatment withdrawal. Cyclin E protein levels in TNBC cells that were synergistic to the sequential D→AZD treatment were increased and remained high, even after dinaciclib was withdrawn for 24 hours (fig. 5A). On the other hand, in TNBC cells that did not have synergistic response to the sequential D→AZD treatment, cyclin E levels did not increase.
Figure 5. Transactivation of cyclin E by dinaciclib sensitizes TNBC cells to AZD1775.
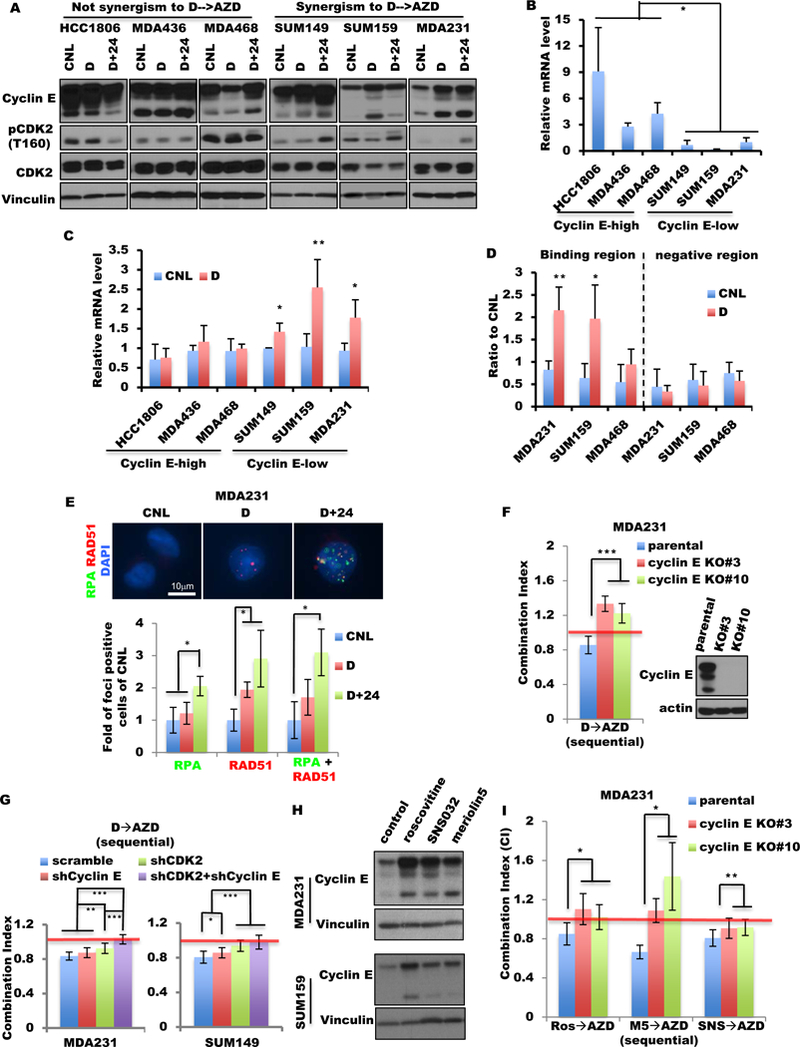
(A) TNBC cells were treated with dinaciclib (D) or vehicle (CNL) for 24 hours or with dinaciclib (24 hours) followed by 24 hours in drug-free medium (D+24) at IC50 concentrations (table S6) and subjected to Western blot with the indicated antibodies. (B) Relative cyclin E mRNA levels of TNBC cell lines were quantified by qRT-PCR using the level in MDA231 cells as a baseline (equivalent to 1). (C) Six TNBC cell lines were treated with dinaciclib (D) or vehicle (CNL) for 24 hours at IC50 concentrations (table S6) and subjected to qRT-PCR of cyclin E. (D) Chromatin immunoprecipitation–qPCR of the enhancer domain (binding region) and downstream negative region of CCNE1 by H3K27AC antibody in cell lines treated with dinaciclib as in (C). (E) Representative images (top) and quantification of foci-positive cell (>5 foci per cell, bottom) population of RPA and RAD51 foci in MDA231 cells treated as in (A). RPA+RAD51 indicates cells with both RPA and RAD51 foci. Scale bar, 10 μm. n=3. (F) Parental cells and two cyclin E CRISPR/CAS9 knockout (KO) clones (#3 and #10) of MDA231 were subjected to sequential combination treatment (D→AZD) as in fig. 5B. Mean CI values are shown. CI=1.0 indicates an additive effect (red line). Inserted panels indicate the protein level of cyclin E of parental and KO cells by immunoblot. (G) The CI of MDA231 (left) and SUM149 (right) cells with single and/or double knockdown of CDK2 and/or cyclin E with the D→AZD treatment as in fig. 5B. (H) MDA231 and SUM159 cells were treated with roscovitine, SNS032, or meriolin5 for 24 hours and subjected to immunoblot with indicated antibodies. The concentrations of each drug used for each cell line are listed in table S6. Vinculin is a loading control. (I) Parental cells and two cyclin E CRISPR/CAS9 KO clones (#3 and #10) of MDA231 were subjected to sequential combination treatment as in fig. 5B. Mean CI values are shown. CI=1.0 indicates an additive effect (red line). Ros→AZD, the sequential combination of roscovitine followed by AZD1175; M5→AZD, the sequential combination of meriolin5 followed by AZD1775; SNS→AZD, the sequential combination of SNS032 followed by AZD1775. A two-tailed unpaired t-test was use to compare two groups. Error bars represent standard error of the mean. *, p<0.05; **, p<0.01; ***, p<0.001.
The synergistic TNBC cells (i.e. cyclin E low cells) had significantly less cyclin E mRNA (p<0.04) than the cyclin E high TNBC cells (fig. 5B). Furthermore, the cyclin E mRNA levels increased upon dinaciclib treatment in the cyclin E–low cells, but not in the cyclin E–high cells (fig. 5C). The binding of H3K27ac (which distinguishes active from inactive/poised enhancers (56)) on the cyclin E enhancer increased significantly in cyclin E–low cells (MDA231 and SUM159) following treatment with dinaciclib, but not in cyclin E–high cells (MDA468) (fig. 5D fig. S14A), indicating increased transcriptional activity on the cyclin E gene in the cyclin E–low cells upon dinaciclib treatment. These results suggest that dinaciclib treatment could induce cyclin E expression transcriptionally in the cyclin E–low TNBC cells.
The increased cyclin E levels, following dinaciclib mediated transient induction, were associated with increased CDK2 kinase activity and p-CDK2 (T160) levels only upon removal of dinaciclib for 24 hours (D+24 lanes of fig. 5A fig. S14B). Moreover, the dinaciclib-induced cyclin E also significantly increased the percentage of cells with RPA foci and cells with both RPA and RAD51 foci at 24 hours after dinaciclib treatment (fig. 5E fig. S14C). These results suggest that while dinaciclib is necessary to induce cyclin E levels, the drug has to be removed for cyclin E to be functional.
KO of cyclin E (CRISPR/CAS9) in MDA231 cells resulted in antagonistic interaction of the sequential D→AZD treatment (fig. 5F), suggesting the requirement of cyclin E for the observed synergism. To examine the contribution of CDK2 to the sequential treatment, cyclin E and/or CDK2 were downregulated (shRNA) in the cyclin E–low TNBC cells (fig. S14D). Downregulation of CDK2 increased the CI values of the D→AZD treatment, and the double knockdown with cyclin E changed the interaction of the sequential combination from synergistic to additive (CI=1) (fig. 5G). Moreover, other CDK2 kinase inhibitors, including roscovitine, SNS032, and meriolin5, increased cyclin E protein levels in MDA231 and SUM159 cells (fig. 5H). Lastly, the sequential combination of roscovitine and meriolin5 with AZD1775 became antagonistic in the cyclin E KO MDA231 cells (fig. 5I). These results suggest that only when the CDK2 inhibitor is removed does the transient induction of cyclin E by CDK2 kinase inhibition lead to a functional cyclin E/CDK2 complex with increased DNA stress, sensitizing TNBC cells to Wee1 kinase inhibition.
Sequential combination treatment of carboplatin followed by Wee1 kinase inhibitor in TNBC is synergistic in vitro and in vivo.
We next examined the pharmacologic generalizability of transient cyclin E upregulation in mediating sensitivity to AZD1775 by conducting a small-scale screening of clinically available targeted and systemic therapeutic agents in two of the cyclin E–low cell lines, MDA231 and SUM159. These agents included a CDK4/6 inhibitor (palbociclib), an aurora kinase A inhibitor (MLN8237), and several chemotherapeutic agents, including carboplatin, cisplatin, paclitaxel, epirubicin, and doxorubicin. The synergistic efficacies of sequential treatment using these agents followed by AZD1775 were significantly correlated with the induction status of cyclin E (p=0.0014) (fig. 6A–B table S6–7 and table S10), suggesting that cyclin E expression could be a potential pharmacodynamic marker for predicting response to these sequential combinations of drugs with AZD1775.
Figure 6. Sequential combination treatment with carboplatin followed by AZD1775 is synergistic in vitro and in vivo.
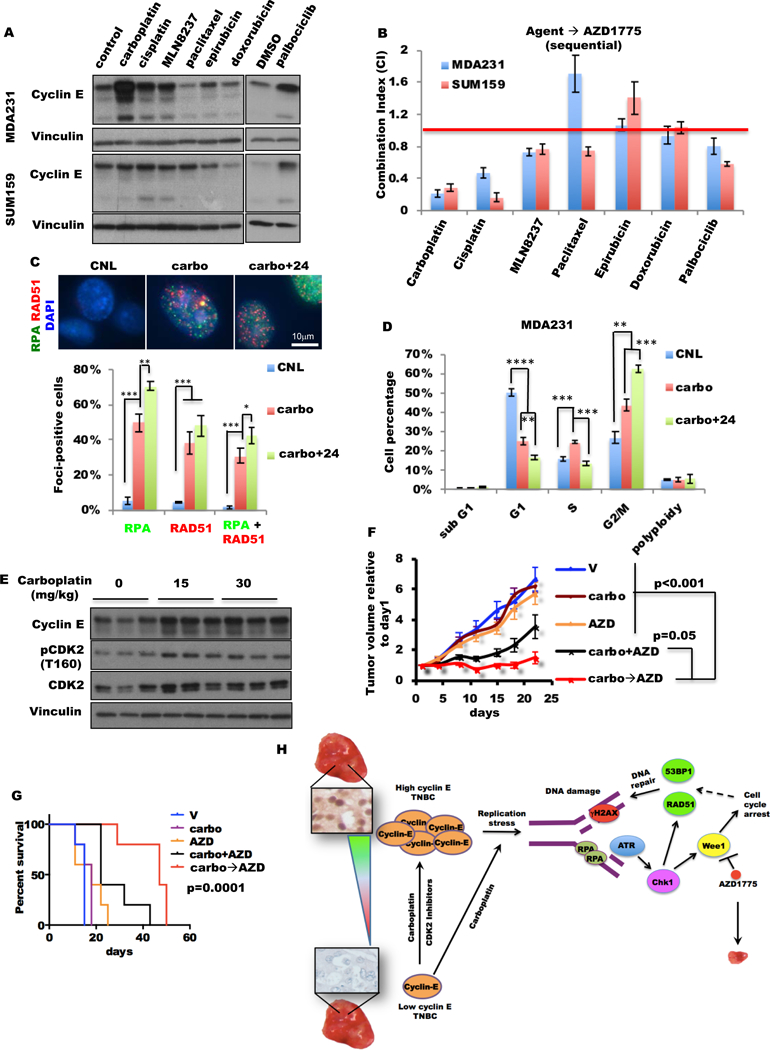
(A) MDA231 and SUM159 cells were treated with palbociclib for 72 hours and other agents as indicated for 24 hours and subjected to immunoblot. Dimethyl sulfoxide (DMSO) was the vehicle control for palbociclib. (The concentrations of each drug used for each cell line are listed in table S6.) (B) The mean CI for each agent followed sequentially by AZD1775 for 48 hours in MDA231 and SUM159 cells was determined by CalcuSyn. Palbociclib was administered for 72 hours, and other agents were administered for 24 hours. The red line represents CI=1. (The concentrations of each drug used for each cell line are listed in table S7.) (C) MDA231 cells were treated with carboplatin (carbo) or vehicle (CNL) for 24 hours, or with carboplatin (24 hours) followed 24 hours in drug-free medium (carbo+24) at the same concentrations as in (A) and subjected to immunofluorescence staining with RPA and RAD51 antibodies. Representative images (top) and quantification of foci-positive cell (>5 foci per cell, bottom) population of RPA and RAD51 foci. RPA+RAD51 indicates cells with both RPA and RAD51 foci. Scale bar, 10 μm. n=3. (D) Cell cycle analysis of MDA231 cells treated as in (C) by FACS, n=3. (E) Mice harboring MDA231T xenograft tumors were treated with 0, 15, or 30 mg/kg carboplatin at 1 dose/week for 3 doses. Tumors were harvested at 24 hours after the last dose of carboplatin. Immunoblot of three different tumors for each dosage with the indicated antibodies. (F, G) Tumor volumes (F) and mouse survival (G) of mice harboring MDA231T xenograft tumors were treated with 30 mg/kg carboplatin once per day for 1 day and 50 mg/kg AZD1775 twice per day for 2 days of each 7-day cycle and repeated for 3–7 cycles. (Schematic in fig. S15D.) V, mouse treated only with vehicle; carbo, mouse treated with carboplatin and vehicle of AZD1775; AZD, mouse treated with AZD1775 and vehicle of dinaciclib; carbo+AZD, mouse concomitantly treated with carboplatin and AZD1775; carbo→AZD, mouse sequentially treated by carboplatin and AZD1775, n=5. (H) Working model. A two-tailed unpaired t-test was use to compare two groups. Error bars represent standard error of the mean. The log-rank (Mantel-Cox) test was used in survival experiments. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
RPA and RAD51 foci staining revealed that 24 hours of carboplatin treatment was sufficient to stimulate DNA replicative stress (RPA foci) in 50% and DNA repair (RAD51 foci) in 38% of the MDA231 cells (fig. 6C). The percentage of cells that were positive for both RPA and RAD51 foci significantly increased upon carboplatin treatment and represented the main population of foci-positive cells (63% of RPA-positive and 82% of RAD51-positive), suggesting that most of cells with DNA replication stress are undergoing DNA repair (fig. 6C fig. S15A–B). These indicators of DNA replicative stress and DNA repair were further increased following the removal of carboplatin for 24 hours (fig. 6C fig. S15A–B). Cell cycle analysis revealed that cells were blocked at both the S phase and the G2/M phase with carboplatin and at the G2/M phase following 24 hours of removal from carboplatin (fig. 6D). Since Wee1 kinase regulates both the DNA replicative checkpoint and the G2/M checkpoint (10,11,14,57), these results suggest that the TNBC cells treated with carboplatin could respond to Wee1 kinase inhibition both before and after the removal of carboplatin. As predicted, the concomitant combination of carboplatin and AZD1775 in cyclin E low TNBC cells were also synergistic in vitro (fig. S15C).
To evaluate the in vivo efficacy of the combination treatment using carboplatin and AZD1775, we first interrogated whether carboplatin treatment could increase cyclin E protein levels in the orthotopic MDA231T (MDA231 cells passaged through mice) xenograft models by treating the mice with various doses (0, 15, or 30 mg/kg) of carboplatin (1 dose/week) for 3 weeks. Consistent with the in vitro studies, cyclin E and p-CDK2 (T160) protein levels increased following carboplatin treatment (fig. 6E). Next we randomized MDA231T tumor–bearing mice into five arms: (1) vehicle (V), (2) carboplatin treatment (carbo), (3) AZD1775 treatment (AZD), (4) concomitant treatment using carboplatin and AZD1775 (carbo+AZD), and (5) sequential treatment using carboplatin followed by AZD1775 (carbo→AZD). The mice were treated with 30 mg/kg carboplatin and 50 mg/kg AZD1775 in a 7-day cycle and repeated for 3–7 cycles (fig. S15D). Consistent with the in vitro studies, the carbo→AZD sequential combination significantly reduced tumor growth compared with the vehicle and single-drug arms and extended the median survival time from 15 days in the vehicle arm to 47 days, which was more effective than the carbo+AZD concomitant combination (fig. 6F–G). Furthermore, the sequential carbo→AZD combination treatment was statistically synergistic using our two study endpoints of survival (p=0.026) and tumor growth rate (p=0.011) (tables S8, S9). However, the concomitant carbo+AZD combination was not synergistic with either study end points (tables S8, S9). All single and combination treatment arms were well tolerated, as indicated by the lack of significant changes in body weight or in complete blood counts of non–tumor-bearing mice at the end of 4 cycles (fig. S15E, S16).
Discussion
Here we report that TNBCs with cyclin E overexpression have a worse prognosis and a higher rate of DNA damage than other TNBCs. We also found that PDX models derived from TNBCs that overexpress cyclin E are very sensitive to AZD1775 owing to CDK2-dependent activation of the DNA replication stress pathway. Moreover, co-expression of cyclin E and p-CDK2 in TNBC tumors is correlated with poor prognosis in >70% of all TNBC cases examined, suggesting AZD1775 could be a promising therapy for these patients. Finally, we found that the sequential treatment of PDX models with low cyclin E levels using agents that induce cyclin E expression followed by AZD1775 causes significant and sustained growth inhibition. Collectively, our results suggest that cyclin E alteration could be used as a potential biomarker to select patients whose tumors are likely to respond to AZD1775 either as a single agent or in combination with agents that can transiently induce cyclin E.
Patients with TNBC have limited treatment options beyond standard chemotherapy which has substantial toxicities. Some therapeutics, including PARP inhibitors given in combination with chemotherapy have shown promise; however, owing to the lack of predictive biomarkers of response, many patients do not experience a response to such therapies. Genomic sequencing for genes involved in DNA repair pathways (in addition to BRCA1/2) across tumor types suggests that many patients with advanced malignancies may be candidates for DNA repair–targeted therapeutics such as AZD1775 (58). We propose that cyclin E expression could be used to identify patients who would either benefit from AZD1775 as monotherapy (in cyclin E high TNBC tumors) or in sequential combination therapies with carboplatin or dinaciclib (in cyclin E low tumors).
Our laboratory has previously reported on the prognostic role of cyclin E in breast cancer, across all subtypes and patient cohorts (30,31). Specifically, we examined the expression of cyclin E, by IHC, in 2,494 breast cancer patients of all subtypes from four different patient cohorts. In multivariable analysis, compared with other prognostic factors (e.g., estrogen receptor status, progesterone receptor status, HER2 status, Ki67 status), cyclin E staining was associated with the greatest risk of recurrence. These studies suggest that cyclin E is likely to identify patients with the highest likelihood of recurrence consistently across different patient cohorts and breast cancer subtypes. In the current study, we propose that cyclin E may also be used as a potential predictive biomarker for AZD1775 based therapies. Using the guidelines set forth by the Reporting Recommendation for Tumor Marker Prognostic Studies (REMARK) (59), we evaluated the utility of cyclin E both as a prognostic and a potential predictive marker (table S11). While the role of cyclin E as a prognostic marker meets the REMARK guidelines, the criteria for the use of cyclin E as a predictive biomarker of response to AZD1775 have only been partially met, since the studies presented here are the pre-clinical evaluation of cyclin E expression as a biomarker of response to AZD1775 in cell lines and PDX models. However, these pre-clinical studies provided the needed rationale for a biomarker driven clinical trial (NCT03253679) entitled “Wee1 inhibitor AZD1775 in treating patients with advanced refractory solid tumors with CCNE1 amplification”. This new phase II trial will enroll 35 patients with the histologically advanced solid tumors harboring cyclin E amplification. The patients enrolled in this trial will receive AZD1775 on days 1–5 and 8–12, which will repeat every 21 days in the absence of disease progression or unacceptable toxicity. Hence, linking the functionality of cyclin E amplification or overexpression to Wee1 kinase in individual tumors is critical for patient selection for AZD1775 therapy.
The present study also provides mechanistic evidence in preclinical models to support the use of cyclin E to predict the response to AZD1775 in TNBC. We show that the replication stress pathway is activated upon cyclin E overexpression, which is biochemically functional and CDK2 dependent and results in sensitivity to Wee1 inhibition. The replicative stress pathway (fig. 6H) as measured by the changes in the ATR-CHK1 axis, RPA foci–positive cells, and DNA replication progression are all modulated by either knock out or overexpression of cyclin E or CDK2 levels in TNBC cells.
Several previous studies have examined the association of genes involved in DNA repair (such as TP53, SIRT1, and RAD18), DNA synthesis (such as SETD2, RRM2, and Polκ), and G1/S phase regulation (such as SKP2, CUL1, and CDK2) with sensitivity to Wee1 kinase inhibition (12,38,60,61), however, these findings have not been readily translated into the clinic. For example, while preclinical studies have shown that cancer cells with TP53 mutation are more sensitive to Wee1 kinase inhibition than those with wild-type TP53 (10,15,16), none of the patients with only TP53 mutation responded to AZD1775 treatment in a recent phase I trial (NCT01748825) (20). In the same trial, two patients carrying both BRCA and TP53 mutations achieved only partial response. These data indicate that owing to the complexity of cell cycle checkpoint regulation, additional biomarkers are required to determine sensitivity to Wee1 kinase inhibitors.
Our study also identified an alternate treatment strategy in cyclin E low TNBC tumors. We found that certain chemotherapeutic (i.e. platinum) or targeted (i.e. dinaciclib) agents are capable of transiently inducing the expression of cyclin E. Once cyclin E levels are induced, the cells and tumor models are then responsive to AZD1775 in both in vitro and in vivo models. As such we propose that treatment strategies combining a platinum agent first, followed by AZD1775 in sequence may have activity in cancers with low cyclin E levels. In a recent study combination treatment effect of three agents; roscovitine, AZD1775 and AZD6738 (ATR inhibitor) (62) in MDA231, revealed that inhibition of the three pathways being targeted may be antagonistic, however the AZD1775 dose response studies as well as the short term growth inhibition assay system were not consistent with other published studies (38,63).
In summary, the strengths of the current study are 1) establishing the prognostic value of cyclin E for TNBC patients; 2) providing the needed evidence to design and implement a biomarker-driven clinical trials with AZD1775 as a single agent for cyclin E–high tumors (NCT03253679) and in combination with a platinum agent for cyclin E–low tumors. However, our study has some limitations. Since the prediction of response to the AZD1775 based treatments were investigated based on cell-based and mouse models, they do not meet the REMARK criteria. Therefore, further clinical studies are required to establish cyclin E as a predictive biomarker for the mono- and combination AZD1775 based treatment strategies. The expression of cyclin E will need to be examined in all patients to stratify them into cyclin E low and cyclin high cohorts. We propose that the cyclin E high cohort are likely to respond to AZD1775 mono-therapy, while the cyclin E low cohort are more suited to the sequential carboplatin followed by AZD1775 treatment. Furthermore, the levels of cyclin E should also be measured following carboplatin administration and prior to AZD1775 in a window-trial setting to monitor if carboplatin leads to the induction of cyclin E protein. Sequential cyclin E–inducing chemotherapy with carboplatin followed by AZD1775 may also prove to be less toxic than the combination administered concomitantly since carboplatin is used as a biological modulator (of cyclin E) rather than as a cytotoxic chemotherapy. As such, carboplatin can be dosed at a non-therapeutic dose at lower doses that would lead just to the induction of cyclin E. Our in vitro and in vivo studies suggest that the dose needed for inducing cyclin E is lower (50mg/kg) than those used for therapeutic response in other studies (85–120mg/kg) (64,65). Such dosing of carboplatin could be translated clinically to help reduce the significant adverse events currently associated with carboplatin therapy (66). These proposed clinical trials could have significant therapeutic implications for cyclin E–dependent cancers for which no targeted therapy is currently available.
Supplementary Material
Acknowledgments
Financial support: Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award (P30CA016672) to The University of Texas MD Anderson Cancer Center, R01 grants CA1522218, CA223772 and Cancer Prevention and Research Institute of Texas (CPRIT) RP170079 grants to K. Keyomarsi, the Susan G. Komen for the Cure grant KG100521 to K.K. Hunt, the Susan G. Komen post-doctoral fellowship grant PDF14302675 to J.P.W. Carey and the CPRIT RP170067 training grant to S.Vijayaraghavan, the NCI Experimental Therapeutics Clinical Trials Network grant 3UM1CA186688-03S2 (to F. Meric-Bernstam).
Footnotes
Conflict of interest: The authors have no conflicts.
References
- 1.Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer 2007;109(9):1721–8 doi 10.1002/cncr.22618. [DOI] [PubMed] [Google Scholar]
- 2.Morris GJ, Naidu S, Topham AK, Guiles F, Xu Y, McCue P, et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: a single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer 2007;110(4):876–84 doi 10.1002/cncr.22836. [DOI] [PubMed] [Google Scholar]
- 3.Oakman C, Viale G, Di Leo A. Management of triple negative breast cancer. Breast 2010;19(5):312–21 doi 10.1016/j.breast.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 4.Dumay A, Feugeas JP, Wittmer E, Lehmann-Che J, Bertheau P, Espie M, et al. Distinct tumor protein p53 mutants in breast cancer subgroups. Int J Cancer 2013;132(5):1227–31 doi 10.1002/ijc.27767. [DOI] [PubMed] [Google Scholar]
- 5.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012;486(7403):395–9 doi 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner N, Moretti E, Siclari O, Migliaccio I, Santarpia L, D’Incalci M, et al. Targeting triple negative breast cancer: is p53 the answer? Cancer Treat Rev 2013;39(5):541–50 doi 10.1016/j.ctrv.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature 1993;366(6456):701–4. [DOI] [PubMed] [Google Scholar]
- 8.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998;282(5393):1497–501. [DOI] [PubMed] [Google Scholar]
- 9.Russell P, Nurse P. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell 1987;49(4):559–67. [DOI] [PubMed] [Google Scholar]
- 10.Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer discovery 2012;2(6):524–39 doi 10.1158/2159-8290.CD-11-0320. [DOI] [PubMed] [Google Scholar]
- 11.Mir SE, De Witt Hamer PC, Krawczyk PM, Balaj L, Claes A, Niers JM, et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell 2010;18(3):244–57 doi 10.1016/j.ccr.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pfister SX, Markkanen E, Jiang Y, Sarkar S, Woodcock M, Orlando G, et al. Inhibiting WEE1 selectively kills histone H3K36me3-deficient cancers by dNTP starvation. Cancer Cell 2015;28(5):557–68 doi 10.1016/j.ccell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Connor MJ. Targeting the DNA Damage Response in Cancer. Mol Cell 2015;60(4):547–60 doi 10.1016/j.molcel.2015.10.040. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Dai Q, Park D, Deng X. Targeting DNA replication stress for cancer therapy. Genes (Basel) 2016;7(8) doi 10.3390/genes7080051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther 2009;8(11):2992–3000 doi 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- 16.Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, Buchholz TA, et al. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res 2011;17(17):5638–48 doi 10.1158/1078-0432.CCR-11-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mueller S, Hashizume R, Yang X, Kolkowitz I, Olow AK, Phillips J, et al. Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro Oncol 2014;16(3):352–60 doi 10.1093/neuonc/not220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Indovina P, Marcelli E, Di Marzo D, Casini N, Forte IM, Giorgi F, et al. Abrogating G(2)/M checkpoint through WEE1 inhibition in combination with chemotherapy as a promising therapeutic approach for mesothelioma. Cancer Biol Ther 2014;15(4):380–8 doi 10.4161/cbt.27623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kato S, Schwaederle M, Daniels GA, Piccioni D, Kesari S, Bazhenova L, et al. Cyclin-dependent kinase pathway aberrations in diverse malignancies: clinical and molecular characteristics. Cell Cycle 2015;14(8):1252–9 doi 10.1080/15384101.2015.1014149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol 2015;33(30):3409–15 doi 10.1200/JCO.2014.60.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ekholm-Reed S, Mendez J, Tedesco D, Zetterberg A, Stillman B, Reed SI. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J Cell Biol 2004;165(6):789–800 doi 10.1083/jcb.200404092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mailand N, Diffley JF. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 2005;122(6):915–26 doi 10.1016/j.cell.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 23.Ekholm SV, Reed SI. Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr Opin Cell Biol 2000;12(6):676–84. [DOI] [PubMed] [Google Scholar]
- 24.Keyomarsi K, Pardee AB. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc Natl Acad Sci U S A 1993;90(3):1112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keyomarsi K, Conte D Jr., Toyofuku W, Fox MP. Deregulation of cyclin E in breast cancer. Oncogene 1995;11(5):941–50. [PubMed] [Google Scholar]
- 26.Eder AM, Sui X, Rosen DG, Nolden LK, Cheng KW, Lahad JP, et al. Atypical PKCiota contributes to poor prognosis through loss of apical-basal polarity and cyclin E overexpression in ovarian cancer. Proc Natl Acad Sci U S A 2005;102(35):12519–24 doi 10.1073/pnas.0505641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richter J, Wagner U, Kononen J, Fijan A, Bruderer J, Schmid U, et al. High-throughput tissue microarray analysis of cyclin E gene amplification and overexpression in urinary bladder cancer. Am J Pathol 2000;157(3):787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fukuse T, Hirata T, Naiki H, Hitomi S, Wada H. Prognostic significance of cyclin E overexpression in resected non-small cell lung cancer. Cancer research 2000;60(2):242–4. [PubMed] [Google Scholar]
- 29.Keyomarsi K, Tucker SL, Buchholz TA, Callister M, Ding Y, Hortobagyi GN, et al. Cyclin E and survival in patients with breast cancer. N Engl J Med 2002;347(20):1566–75 doi 10.1056/NEJMoa021153. [DOI] [PubMed] [Google Scholar]
- 30.Karakas C, Biernacka A, Bui T, Sahin AA, Yi M, Akli S, et al. Cytoplasmic Cyclin E and Phospho-Cyclin-Dependent Kinase 2 Are Biomarkers of Aggressive Breast Cancer. Am J Pathol 2016;186(7):1900–12 doi 10.1016/j.ajpath.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hunt KK, Karakas C, Ha MJ, Biernacka A, Yi M, Sahin AA, et al. Cytoplasmic Cyclin E Predicts Recurrence in Patients with Breast Cancer. Clin Cancer Res 2017;23(12):2991–3002 doi 10.1158/1078-0432.CCR-16-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Molecular and cellular biology 1994;14(3):1669–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bagheri-Yarmand R, Biernacka A, Hunt KK, Keyomarsi K. Low molecular weight cyclin E overexpression shortens mitosis, leading to chromosome missegregation and centrosome amplification. Cancer Res 2010;70(12):5074–84 doi 10.1158/0008-5472.CAN-09-4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bagheri-Yarmand R, Nanos-Webb A, Biernacka A, Bui T, Keyomarsi K. Cyclin E deregulation impairs mitotic progression through premature activation of Cdc25C. Cancer Res 2010;70(12):5085–95 doi 10.1158/0008-5472.CAN-09-4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keck JM, Summers MK, Tedesco D, Ekholm-Reed S, Chuang LC, Jackson PK, et al. Cyclin E overexpression impairs progression through mitosis by inhibiting APC(Cdh1). J Cell Biol 2007;178(3):371–85 doi 10.1083/jcb.200703202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neelsen KJ, Zanini IM, Herrador R, Lopes M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol 2013;200(6):699–708 doi 10.1083/jcb.201212058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hills SA, Diffley JF. DNA replication and oncogene-induced replicative stress. Curr Biol 2014;24(10):R435–44 doi 10.1016/j.cub.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 38.Heijink AM, Blomen VA, Bisteau X, Degener F, Matsushita FY, Kaldis P, et al. A haploid genetic screen identifies the G1/S regulatory machinery as a determinant of Wee1 inhibitor sensitivity. Proceedings of the National Academy of Sciences of the United States of America 2015;112(49):15160–5 doi 10.1073/pnas.1505283112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duong MT, Akli S, Wei C, Wingate HF, Liu W, Lu Y, et al. LMW-E/CDK2 deregulates acinar morphogenesis, induces tumorigenesis, and associates with the activated b-Raf-ERK1/2-mTOR pathway in breast cancer patients. PLoS Genet 2012;8(3):e1002538 doi 10.1371/journal.pgen.1002538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jabbour-Leung NA, Chen X, Bui T, Jiang Y, Yang D, Vijayaraghavan S, et al. Sequential Combination Therapy of CDK Inhibition and Doxorubicin Is Synthetically Lethal in p53-Mutant Triple-Negative Breast Cancer. Mol Cancer Ther 2016;15(4):593–607 doi 10.1158/1535-7163.MCT-15-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evans KW, Yuca E, Akcakanat A, Scott SM, Arango NP, Zheng X, et al. A population of heterogeneous breast cancer patient-derived xenografts demonstrate broad activity of PARP inhibitor in BRCA1/2 wild-type tumors. Clin Cancer Res 2017;23(21):6468–77 doi 10.1158/1078-0432.CCR-17-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nanos-Webb A, Jabbour NA, Multani AS, Wingate H, Oumata N, Galons H, et al. Targeting low molecular weight cyclin E (LMW-E) in breast cancer. Breast Cancer Res Treat 2012;132(2):575–88 doi 10.1007/s10549-011-1638-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015;163(2):506–19 doi 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486(7403):346–52 doi 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teixeira LK, Wang X, Li Y, Ekholm-Reed S, Wu X, Wang P, et al. Cyclin E deregulation promotes loss of specific genomic regions. Curr Biol 2015;25(10):1327–33 doi 10.1016/j.cub.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hughes BT, Sidorova J, Swanger J, Monnat RJ Jr., Clurman BE. Essential role for Cdk2 inhibitory phosphorylation during replication stress revealed by a human Cdk2 knockin mutation. Proc Natl Acad Sci U S A 2013;110(22):8954–9 doi 10.1073/pnas.1302927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones RM, Mortusewicz O, Afzal I, Lorvellec M, Garcia P, Helleday T, et al. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 2013;32(32):3744–53 doi 10.1038/onc.2012.387. [DOI] [PubMed] [Google Scholar]
- 48.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005;434(7035):864–70 doi 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 49.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011;145(3):435–46 doi 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beck H, Nahse-Kumpf V, Larsen MS, O’Hanlon KA, Patzke S, Holmberg C, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol 2012;32(20):4226–36 doi 10.1128/MCB.00412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bauer TM, Jones SF, Greenlees C, Cook C, Jewsbury PJ, Mugundu G, et al. A phase 1b, open-label, multi-center study to assess the safety, tolerability, pharmacokinetics and anti-tumor activity of AZD1775 monotherapy in patients with advanced solid tumors: expansion cohorts. J Clin Oncol 2016;34(suppl; abstr TPS2608). [Google Scholar]
- 52.Wingate H, Puskas A, Duong M, Bui T, Richardson D, Liu Y, et al. Low molecular weight cyclin E is specific in breast cancer and is associated with mechanisms of tumor progression. Cell Cycle 2009;8(7):1062–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shoaib M, Sorensen CS. Epigenetic deficiencies and replicative stress: driving cancer cells to an early grave. Cancer Cell 2015;28(5):545–7 doi 10.1016/j.ccell.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 54.Piwko W, Mlejnkova LJ, Mutreja K, Ranjha L, Stafa D, Smirnov A, et al. The MMS22L-TONSL heterodimer directly promotes RAD51-dependent recombination upon replication stress. EMBO J 2016;35(23):2584–601 doi 10.15252/embj.201593132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ouyang KJ, Yagle MK, Matunis MJ, Ellis NA. BLM SUMOylation regulates ssDNA accumulation at stalled replication forks. Front Genet 2013;4:167 doi 10.3389/fgene.2013.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 2010;107(50):21931–6 doi 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dominguez-Kelly R, Martin Y, Koundrioukoff S, Tanenbaum ME, Smits VA, Medema RH, et al. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol 2011;194(4):567–79 doi 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stover EH, Konstantinopoulos PA, Matulonis UA, Swisher EM. Biomarkers of response and resistance to DNA repair targeted therapies. Clin Cancer Res 2016;22(23):5651–60 doi 10.1158/1078-0432.CCR-16-0247. [DOI] [PubMed] [Google Scholar]
- 59.McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM, et al. Reporting recommendations for tumor marker prognostic studies (REMARK). J Natl Cancer Inst 2005;97(16):1180–4 doi 10.1093/jnci/dji237. [DOI] [PubMed] [Google Scholar]
- 60.Chen G, Zhang B, Xu H, Sun Y, Shi Y, Luo Y, et al. Suppression of Sirt1 sensitizes lung cancer cells to WEE1 inhibitor MK-1775-induced DNA damage and apoptosis. Oncogene 2017. doi 10.1038/onc.2017.297. [DOI] [PubMed] [Google Scholar]
- 61.Yang Y, Gao Y, Mutter-Rottmayer L, Zlatanou A, Durando M, Ding W, et al. DNA repair factor RAD18 and DNA polymerase Polkappa confer tolerance of oncogenic DNA replication stress. J Cell Biol 2017;216(10):3097–115 doi 10.1083/jcb.201702006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jin J, Fang H, Yang F, Ji W, Guan N, Sun Z, et al. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia 2018;20(5):478–88 doi 10.1016/j.neo.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, et al. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol Cancer Ther 2013;12(8):1442–52 doi 10.1158/1535-7163.MCT-13-0025. [DOI] [PubMed] [Google Scholar]
- 64.Jandial DD, Messer K, Farshchi-Heydari S, Pu M, Howell SB. Tumor platinum concentration following intraperitoneal administration of cisplatin versus carboplatin in an ovarian cancer model. Gynecol Oncol 2009;115(3):362–6 doi 10.1016/j.ygyno.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aharinejad S, Fink M, Abri H, Nedwed S, Schlag MG, Macfelda K, et al. Efficient carboplatin single therapy in a mouse model of human testicular nonseminomatous germ cell tumor. J Urol 2002;167(1):368–74. [PubMed] [Google Scholar]
- 66.Fujiwara K, Sakuragi N, Suzuki S, Yoshida N, Maehata K, Nishiya M, et al. First-line intraperitoneal carboplatin-based chemotherapy for 165 patients with epithelial ovarian carcinoma: results of long-term follow-up. Gynecol Oncol 2003;90(3):637–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.