

Abstract
The μ opioid receptor (μOR) is a G protein-coupled receptor (GPCR) that is the target of most clinically and recreationally used opioids. The induced positive effects of analgesia and euphoria are mediated by μOR signaling through the adenylyl cyclase-inhibiting heterotrimeric G protein Gi. We present the 3.5-Å resolution cryo-electron microscopy (cryo-EM) structure of the μOR bound to the agonist peptide DAMGO and nucleotide-free Gi. DAMGO occupies the morphinan ligand pocket, with its N-terminus interacting with conserved receptor residues while its C-terminus engages regions important for opioid ligand selectivity. Comparison of the μOR-Gi complex to previously determined structures of other GPCRs bound to the stimulatory G protein Gs reveals differences in the movement of transmembrane receptor helix 6 and in the interactions between the G protein α subunit and the receptor core. Together, these results shed light into the structural features that contribute to the Gi protein coupling specificity of the μOR.
The μOR is the primary target of morphine and many clinically used opioid analgesics1. Opioid binding to the μOR leads to clinically desired analgesic and antitussive actions but also important negative side effects including addiction and potentially lethal respiratory suppression. Opioids have become the most prescribed class of medication in the United States2, leading to a national epidemic of addiction and an unprecedented level of drug overdose deaths.
Like other GPCRs, the μOR achieves many of its physiological actions by stimulating signaling via a heterotrimeric G protein. While other GPCRs have been shown to signal through more than one G protein subtype, the μOR signals almost exclusively through the adenylyl-cyclase inhibitory family of G proteins (Gi/o)3. The analgesic activity of opioids is driven by G protein activation4, but activated μOR can also interact with β-arrestins, whose recruitment has been associated with the respiratory depression induced by many opioids5,6. Recently developed molecules that favor Gi signaling over arrestin recruitment display analgesic efficacy with reduced side effects, suggesting that different signaling pathways can be selectively targeted to yield unique physiological outcomes7,8. Though a framework for GPCR interactions with the stimulatory G protein Gs has recently been enabled by X-ray crystallography9 and cryo-electron microscopy (cryo-EM)10,11, the structural basis for GPCR signaling through other G protein subtypes remains undefined. To better understand the mechanism of selective activation of Gi by the μOR, we sought to determine the structure of the μOR-Gi complex.
3.5-Å cryo-EM map of a μOR-Gi complex
DAMGO (H-Tyr-D-Ala-Gly-N-MePhe-Gly-OH) is a μOR-selective synthetic analog of the natural peptide agonist enkephalin. DAMGO-bound μOR was incubated with Gi1 heterotrimer and the complex was treated with the nucleotide hydrolase apyrase to remove GDP. The resulting nucleotide-free complex was further stabilized by a single-chain variable fragment (scFv16) that binds to heterotrimeric Gi (Extended Data Figure 1) and prevents GTPγS mediated dissociation of nucleotide-free complexes. We applied single-particle cryo-EM to initially obtain a three-dimensional map of the μOR-DAMGO-Gi-scFv16 complex at an indicated nominal resolution of 3.6 Å (Extended Data Fig. 2–3, Extended Data Table 1). Notably, scFv16 binds a composite interface comprised of the αN helix of Gαi and the β propeller of Gβ, a site that is more than 20 Å distal to the μOR-Gαi interface and does not perturb the interface between Gα and Gβ subunits (Extended Data Figure 1). Subtraction of the scFv16 signal from raw particle images led to an improved map with an indicated global resolution of 3.5 Å. This map displayed enhanced features particularly in the receptor transmembrane core (Extended Data Figures 2, 3, 4), enabling the high resolution visualization of the μOR-Gi interface and ligand binding. Accordingly, we employed this improved 3.5-Å map to examine interactions between μOR and DAMGO, and between μOR and Gi (Fig. 1a, b).
Figure 1. Cryo-EM structure of the μOR-Gi complex.
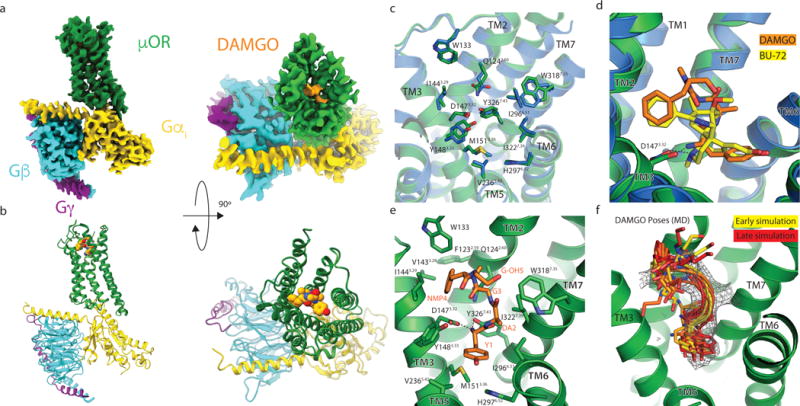
a, Orthogonal views of the cryo-EM density map of the μOR-Gi heterotrimer complex colored by subunit (μOR in green, DAMGO in orange, G⍺s Ras-like domain in gold, Gβ in cyan, Gγ in purple). b, Model of the μOR-Gi complex in the same views and color scheme as shown in a. c, Residues that line the μOR orthosteric binding pocket are shown as sticks for the μOR-Gi complex (green) and the μOR-Nb39 complex (PDB 5C1M; blue). The binding pocket residues of DAMGO and BU-72 occupied μOR show nearly identical conformations, despite differences in ligand structure. d, Comparison of BU-72 (yellow carbons) in the orthosteric pocket of the μOR-Nb39 complex (blue) with DAMGO (orange carbons) in the orthosteric pocket of the μOR-Gi complex (green). e, view of DAMGO in the orthosteric binding pocket with critical residues shown. f, A frame from every 100 ns of a 1 μs MD simulation (yellow for t = 0 fading to red for t = 1 μs) shows that the first 4 residues of DAMGO (bottom) are stable, whereas the C-terminal Gly-ol (top) is dynamic but frequently returns to the modeled pose.
Activation of μOR by a peptide agonist
We previously determined the active-state crystal structure of μOR bound to the morphinan agonist BU72 and an active-state stabilizing nanobody (Nb39) at a resolution of 2.2 Å12. Like other small molecule morphinans, BU72 is rigidified by a complex ring system, in contrast to flexible opioid peptides like DAMGO that have multiple rotatable bonds. The cryo-EM map includes well-defined features for most amino acids forming the orthosteric binding pocket (Extended Data Fig. 4a). Despite differences in agonist structure, the conformation of the active-state binding pocket and the orientation of the amino acids that interact with the agonist are highly similar for the μOR bound to BU72 or DAMGO (Fig. 1c), suggesting that the μOR recognizes structurally distinct agonists in a stereotypic manner.
Although DAMGO is a flexible ligand, we observe density for the entire peptide bound to the receptor (Fig. 1a, Extended Data Fig. 3, 4). The DAMGO N-terminus occupies a similar position in the binding pocket as BU72. In contrast, the C-terminus of DAMGO extends ~8 Å further towards the extracellular loops compared to BU72 (Fig. 1d, e). To identify stable atomic-level interactions between DAMGO and the binding pocket, we performed molecular (MD) dynamics simulations. In over 1μs of simulation, DAMGO remained close to its initially modeled pose, with the amino-terminal portion largely remaining confined to the experimentally determined EM density (Fig. 1f, Extended Data Fig 5). The DAMGO N-terminus maintained a persistent salt bridge with D1473.32, a feature previously observed in structures of morphinans bound to opioid receptors (Fig. 1e; superscripts indicate Ballesteros-Weinstein numbering for GPCRs13). The same amine group also often formed a hydrogen bond with Y3267.43. More generally, the amino-terminal Tyr of DAMGO overlaps the phenolic group of other small molecule opioids characterized previously by X-ray crystallography14–17.
MD simulations also revealed a water-mediated hydrogen bonding network that closely overlaps with the water network observed in the high-resolution crystal structure of μOR12 (Extended Data Fig. 6). In particular, the simulations revealed a stable, water-mediated interaction formed between the DAMGO phenol and H2976.52. Though the crystal structure of the μOR bound to BU-72 shows two water molecules bridging the DAMGO phenol and H2976.52, simulations of μOR bound to DAMGO and other phenolic ligands8,12 suggest that one of these waters rapidly dissociates and that a single water is required for stable ligand binding. This interaction is a hallmark of opioid recognition that has been observed for morphinans in complex with the μOR12,14 as well as other small molecule and peptide-mimetic agonists for the homologous δ and κ opioid receptors (κOR)15,16,18.
DAMGO is more than 500-fold selective for the μOR over the δOR and κOR19. As elucidated in prior structures, ligand interactions with the extracellular loops encode ligand subtype specificity among closely related opioid receptors15. Indeed, DAMGO selectivity for μOR over δOR has been shown to result from residues in ECL1 while selectivity over κOR results from differences in ECL320. The map density for the carboxy-terminal residues of DAMGO is slightly weaker than for the amino-terminus, consistent with increased mobility of this region in simulations (Fig. 1f, Extended Data Fig 5). In our model, the DAMGO N-Me-Phe side chain occupies a conserved hydrophobic pocket near ECL1 and the Gly-OH group folds back over the ligand (Fig. 1e). This model is consistent with the high affinity μOR binding of cyclized enkephalins that bridge the +2 and +5 positions of the peptide21.
Structure of Gi-Stabilized Active μOR
The overall structure of Gi-bound μOR is similar to the active conformation of the BU72-bound μOR stabilized by Nb39 (root mean square deviation of 1 Å)12 with a predominant outward displacement of TM6 from the heptahelical bundle relative to the inactive state (Fig. 2a, b). A number of highly conserved residues in the GPCR family have been shown to be important for receptor activation, including the D3.49R3.50Y3.51, the N7.49P7.50xxY7.53, and conserved core triad (I3.40, P5.50, and F6.44) motifs. The conformation of each of these regions in the μOR-Gi complex is virtually identical to the active state observed in complex with Nb39 (Fig. 2c). The structural similarity of μOR between Nb39 and Gi-coupled states indicate that these changes underlie ligand-mediated activation and are not specific to a particular intracellular binder. Indeed, Nb39 and Gi promote a similar increase in agonist affinity12, which supports a common mechanism of allosteric communication between the intracellular G protein coupling domain and the ligand binding pocket12.
Figure 2. Structural changes in the μOR stabilized by nucleotide-free Gi.
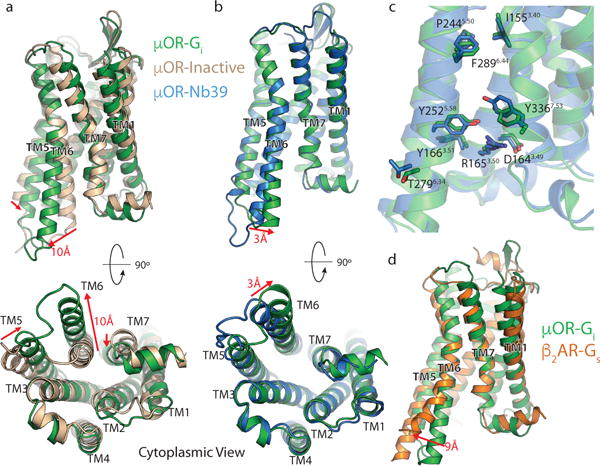
a, Comparison of inactive μOR (brown) and the Gi stabilized active state of μOR (green). b, Comparison of Nb39 and Gi stabilized active states of the μOR (blue and green, respectively). The structures are nearly identical except for a slight shift of TM6 towards TM7 in the Gi -bound state. c, Residues important for activation of the μOR show nearly identical conformations despite the difference in ligands. d, Comparison of Gs-stabilized β2AR (orange) and Gi-stabilized μOR (green). While most transmembrane helices align well between the two receptors, TM6 is kinked further outward by 9Å in the β2AR. Distance calculated between Cα of residue 6.29 (Ballesteros-Weinstein numbering) in TM6.
Two differences between Nb39 and Gi stabilized active-states of μOR are particularly notable. First, compared with the nanobody-stabilized active-state μOR, TM6 in the μOR-Gi complex is further displaced by 3 Å towards TM7 (Fig. 2b). Second, the conformation of intracellular loop 3 (ICL3) is different in the two structures (Fig. 2a). It is likely that the specific ICL3 conformation of μOR stabilized by Nb39 reflects interactions that are unique to the nanobody rather than a general feature of receptor activation prior to G protein coupling. A similar difference in ICL3 conformation was previously observed for the β2-adrenergic receptor (β2AR) between nanobody (Nb80)22 and Gs-coupled states. The comparison of the G protein bound states of both receptors shows that the β2AR TM6 is displaced outward by another 9 Å compared to the μOR (Fig. 2d).
Structural Changes in Gi
The quality of the cryo-EM map enabled accurate modeling of Gi in its nucleotide-free state, providing insight into the structural changes that underlie nucleotide release. The changes we observe are similar to those observed in nucleotide-free Gs in complex with other GPCRs. The most striking difference between the GDP bound23 and nucleotide free heterotrimer in complex with μOR involves the separation of the α-helical domain (AHD) from the Ras-like domain in the alpha subunit of Gi (Gαi) (Fig. 3a). Due to its relative flexibility, we excluded the AHD density from the high-resolution map refinement. The dynamic character of the AHD has been observed previously by spectroscopic and structural studies in complexes between receptors and both Gs9–11 and Gi24,25. Displacement of the AHD disrupts several contacts with GDP and is necessary, but not sufficient for nucleotide release, a process that involves breaking additional contacts with the Ras domain24.
Figure 3. Changes in Gi upon coupling to the μOR.
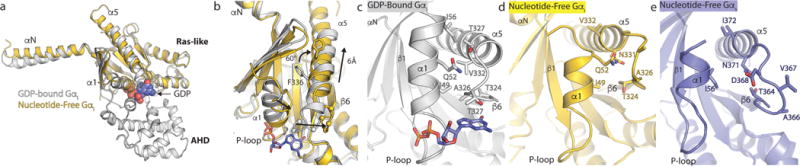
a, b, Comparison of GDP-bound Gαi (PDB 1GP2, grey) and nucleotide-free Gαi from the μOR-Gi complex (gold). GDP is shown as blue spheres in panel a and sticks in panel b. The primary differences between these two structures are the opening and outward movement of the alpha helical domain (AHD), and an upward shift of the α5 helix by 6Å to engage the receptor core. The α-carbons of the TCAT motif are represented as spheres in panel b. The TCAT motif coordinates the guanosine base of GDP. The upward shift of the α5 helix and repositioning of the TCAT motif leads to nucleotide release. c, d, e, The interface between the α1 helix and the N-terminal end of the α5 helix and TCAT motif for GDP-bound Gαi (c), nucleotide free Gαi (d), and nucleotide free Gs from the β2AR -Gs complex (e). The upward movement of the α5 helix disrupts the interaction between the α1 and α5 helices leading to changes in the P-loop that coordinates the phosphates of GDP.
Gi coupling to the μOR also involves a 6-Å translation as well as a 60° rotation of the Gαi α5 helix into the receptor core (Fig. 3b). This movement has been shown to be essential for nucleotide release in Gi24. In particular, the motion of α5 leads to a change in the position of the β6-α5 loop containing the conserved TCAT motif that forms direct interactions with the guanine base of GDP. This displacement disrupts key contacts between the G protein and nucleotide. Furthermore, the observed translation and rotation of the α5 helix requires the displacement of the fully conserved F336 away from the hydrophobic pocket formed by residues in the β2/β3 strands and the α1 helix26 (Fig. 3b). Movement of the α5 helix is also propagated to the phosphate binding P-loop connecting the β1 strand and the α1 helix by disruption of a hydrophobic network between the α1 and α5 helices (Fig. 3b–d). Correspondingly, upon transition of Gi to the nucleotide-free state, we observe a 4-Å shift of α1 towards the α5 helix in Gi whereby the hydrophobic contacts are replaced by polar interactions with the β6-α5 loop as it is released from its guanine binding position (Fig. 3c, d). These changes contrast those observed in structures of Gs-coupled complexes, in which α1 not only becomes more unstructured, but also tends to lose interactions with the α5 helix (Fig. 3e). Our structure is consistent with previous studies suggesting that engagement of a GPCR with the α5 helix and αN- β1 loop leads to concerted changes in the α1 helix and P loop that destabilize contacts with the guanine nucleotide leading to its release27.
Structural insights into Gi coupling specificity of the μOR
Although the μOR couples exclusively to Gi/o3, many GPCRs can couple to multiple G protein subtypes; a well-studied example is the β2AR, which couples to both Gs and Gi/o. Prior sequence-level analyses have failed to identify a linear GPCR epitope that determines G protein coupling specificity, suggesting that it is likely determined by a more complex three-dimensional network of interactions. Globally, the structure of the μOR-Gi complex is similar to the β2AR-Gs, likely reflecting a similarity in the conformation of nucleotide-free states of Family A GPCR-G protein complexes. The primary interaction sites in both complexes occur between ICL2, ICL3 and TMs 3,5, and 6 on the receptor and the αN, αN- β1 loop, and α5 helix on the Gα subunit of the G protein (Fig. 4). The most striking difference between the β2AR-Gs and μOR-Gi complex is in the relative position of the α5 helix of both G proteins, as well as the corresponding shift in the position of TM6 of the receptor. The α5 helix of Gαi is rotated ~21° relative to the α5 helix of Gαs, leading to a 5 Å displacement of the extreme C-terminus of the Gαi helix α5 toward TM7 of the μOR (Fig. 4a). This difference in α5 positioning is associated with a smaller outward displacement of the μOR TM6. The C-terminal residues of α5 that interact with TMs 5 and 6 of the receptor are bulkier in Gs than in Gi, with Y and E compared to C and G at positions −4 and −3 from the C-terminus, respectively. Accordingly, substitution of these two amino acids of Gs into Gi would lead to steric clashes with TM3 and the TM7-Helix 8 loop (Extended Data Fig. 7). In the Gs-coupled Family B calcitonin11 and GLP-110 receptors, G protein coupling is associated with a large kink of TM6 at the conserved PxxG motif, which produces an even larger outward displacement of TM6 than what observed in the β2AR-Gs complex.
Figure 4. Comparison of the receptor-G protein binding interfaces of the μOR-Gi and β2AR-Gs complexes.
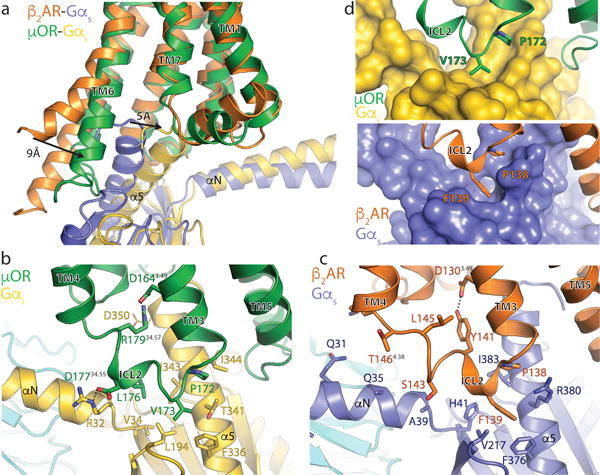
a, Comparison of the conformation of the α5 helix of Gα and receptor TM6 in β2AR-Gs and μOR-Gi complexes after alignment on the receptor. b, Interactions between ICL2 of the μOR (green) and Gαi (gold). Asp 350 of Gαi is depicted with narrow lines to indicate uncertainty in its conformation due to poor cryo-EM density for its side chain. c, Interactions between ICL2 of the β2AR (orange) and Gαs (blue). d, Surface view of the hydrophobic pockets in Gαi (top panel) and Gαs (bottom panel) that interact with a non-polar amino acid in ICL2 of the μOR and β2AR, respectively.
Surprisingly, the structure of μOR-Gi shows substantial similarity to an active-state structure of the visual pigment rhodopsin (Meta II) in complex with a modified peptide derived from the 11 C-terminal residues of the α subunit of the visual G protein transducin (GαtCT2) (Extended Data Fig. 8)28. Despite the lack of the remainder of the heterotrimeric G protein in the MetaII- GαtCT2 structure, the conformation of TM6 of MetaII is highly similar to that of the μOR, while the location of the GαtCT2 peptide is almost identical to the C-terminus of Gi in complex with μOR. This finding is consistent with observations that substitution of the last five amino acids of the Gα α5 helix is sufficient to change G protein coupling specificity29.
In Extended Data Table 2, we list amino acids in the μOR that interact with the cytoplasmic surface of Gi. The μOR ICL2 primarily forms interactions with the αN and α5 helices of Gαi, including a key ionic interaction between the μOR D17734.55 [G Protein Coupled Receptor Data Base (GPCRDB) numbering30] in ICL2 and R32 in the αN- β1 loop of Gαi (Fig. 4b). Although D34.55 in ICL2 is conserved in all opioid receptors with available sequences (GPCRDB31), it is variable in most other Gi coupled receptors. Another notable interaction involves R17934.57 in μOR ICL2, which simultaneously coordinates the highly conserved D1643.49 in the DRY motif and potentially forms an additional interaction with D350 in the Gαi α5 helix (−5 position) (Fig. 4b). This arginine is essential for μOR induced Gi signaling, as the polymorphic variant R179C abolishes signaling in vitro32 and leads to insensitivity to morphine in patients homozygous for the mutation33. The potential role of this interaction network in G protein coupling is supported by the preponderance of basic residues (arginine and lysine) at this position in most Gi coupled receptors, whereas Gs-coupled receptors employ alternative residues (Extended Data Table 2).
A further group of contacts occurs between P17234.50 and V17334.51 of μOR and a hydrophobic patch on Gαi comprised of residues F336, I343, I344, and T340 on the α5 helix and L194 on the β2- β3 loop (Fig. 4b, d). In the GDP bound state, these α5 helix residues are buried by the adjacent β2 and β3 loops. Coupling to a receptor involves an upward shift of the α5 helix and exposes these residues to form a shallow hydrophobic pocket that interacts with μOR V17334.51 in ICL2 (Fig. 4b, d). In the case of Gs, a deeper hydrophobic pocket in this region engages the bulky aromatic F13934.51 in ICL2 of the β2AR (Fig. 4c, d).
In the μOR, ICL3 stabilizes the interface between receptor and G protein through two sets of interactions: one set involves multiple contacts with a hydrophobic patch on the α5 helix of Gαi, while another engages the β6 strand of Gαi through a network of charged residues (Fig. 5a, b). The hydrophobic interface formed by ICL3 is similar in both the μOR and β2AR; in the β2AR, TM5 is helically extended to form a larger hydrophobic interaction around nonpolar residues in the α5 helix of Gαs (Fig. 5c, d). While the shorter ICL3 of the μOR does not form a similar helical extension, it nevertheless fulfills the same role. Residues V2625.68, M264 and L265 fold back to form a hydrophobic patch that interacts with hydrophobic residues on the α5 helix of Gαi (Figure 5a).
Figure 5. Comparison of the receptor-G protein binding interfaces of the μOR-Gi and β2AR-Gs complexes.
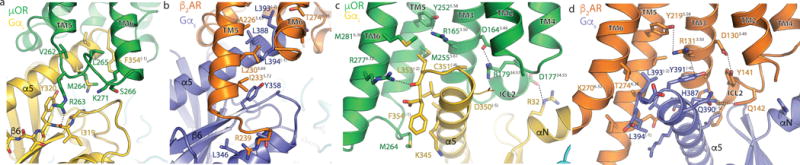
Top panels show interactions between ICL3 of μOR and Gαi (a) and between the cytosolic ends of TMs 3,5,6, of the μOR and the α5 helix of Gi (b). Asp 350 of Gαi is depicted with narrow lines to indicate uncertainty in its position due to poor cryo-EM density for its side chain. Bottom panels show these same interfaces between β2AR and Gs (c,d).
The second set of polar contacts involves μOR R263 and a backbone carbonyl to I319 on the β6 strand of Gαi (Fig. 5a). Mutations of R263 reduce, but do not abolish, Gi signaling34, which is consistent with the potential importance of stabilizing the Gαi β6 strand in the observed conformation. A similar interaction is absent in the β2AR-Gs complex (Fig. 5c). This additional recognition interface may be necessary for efficient μOR-Gi coupling due to the higher affinity for GDP to Gi relative to Gs. Compared to Gs-coupled receptors, additional interactions with the β6 strand in Gi-coupled receptors may be required to disrupt interactions between the Ras domain and GDP for efficient nucleotide exchange.
The cytosolic ends of μOR TMs 3,5 and 6 further stabilize the nucleotide-free conformation of the α5 helix by interacting with highly conserved residues in the distal C-terminus of Gαi (Figure 5b). In particular, C351 (−4 position) of Gαi is in close proximity to the cytosolic end of μOR TM3. This cysteine residue has previously been identified as the site of pertussis toxin-mediated inhibition of Gi/o family proteins by enzymatic ADP-ribosylation35. The close apposition of C351 to the μOR cytoplasmic surface highlights how the addition of a bulky modification at this position can completely inhibit receptor coupling and nucleotide exchange35. In addition to this interaction, μOR residues M2555.61, I2786.33, M2816.36, and V2826.37 form a hydrophobic pocket that engages the absolutely conserved Gαi residue L353 (−2 position) in the α5 helix. Methionines M2555.61 and M2816.36 have previously been observed in NMR experiments to respond to activation by DAMGO36, suggesting that this region undergoes conformational changes prior to G protein coupling. Further stabilization, however, is likely provided by a hydrogen bond between R2776.32 and the backbone carbonyl of L353 (Fig. 5b). Notably, interactions between the C-terminus of the α5 helix and the receptor core are entirely different in the in the β2AR-Gs complex (Fig. 5c).
Our findings provide structural insights into the inability of the μOR to couple to Gs, but do not explain the mechanism of G protein coupling specificity across all GPCRs. It is possible that coupling specificity is determined at an intermediate step in the formation of a GPCR-G protein complex, such as the initial interactions between the GDP-bound G protein and the agonist-bound receptor. Recent single molecule fluorescence studies provide evidence for a transient intermediate complex between GDP-bound Gs and the β2AR that is associated with a smaller outward movement of TM637. Previous studies suggest that amino acids C-terminal to helix 8 confer coupling specificity for Gq in the M3 muscarinic receptor (M3R)38. Given that there are no interactions between the C-terminus of the β2AR or μOR with their respective G proteins in the nucleotide-free complexes, we postulate that engagement of Gq and the M3R C-terminus may occur at an earlier stage in complex formation. Thus, the nucleotide-free GPCR-G protein complex may be preceded by one or more GDP-bound intermediates characterized by dynamic low affinity interactions with the receptor. Such initial encounter complexes may have larger energetic differences for interactions with various G protein subtypes than the nucleotide-free state, and would thereby contribute more critically to coupling specificity. The transient nature of such interactions, however, currently poses challenges for structure determination by both crystallography and cryo-EM.
METHODS
Online Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Purification of μ-opioid receptor
These studies utilized a previously described mouse μOR construct with cleavable amino and carboxy terminal domains12. Briefly, the receptor was expressed in Spodoptera frugiperda Sf9 insect cells using the baculovirus method (Expression Systems), extracted from insect cell membranes with n-dodecyl-β-D-maltoside (DDM, Anatrace), and purified by nickel-chelating sepharose chromatography. The Ni-NTA eluate was loaded onto M1 anti-FLAG immunoaffinity resin and washed with progressively lower concentrations of the antagonist naloxone. The μOR was then eluted in a buffer consisting of 20 mM Hepes pH 7.5, 100 mM NaCl, 0.1% DDM, 0.01% cholesterol hemisuccinate (CHS) supplemented with 50 nM naloxone, FLAG peptide and 5 mM EDTA. The monomeric fraction was purified by size exclusion chromatography on a Superdex 200 10/300 gel filtration column (GE) in 20 mM Hepes pH 7.5, 100 mM NaCl, 0.1% DDM, 0.01% CHS, and 1 μM DAMGO. A further 2-fold molar excess of DAMGO was added to the preparation and the resulting agonist-bound μOR preparation was concentrated to ~100 μM.
Expression and purification of heterotrimeric Gi
Heterotrimeric Gi was expressed and purified as previously described24. Briefly, Trichuplusia ni Hi5 insect cells were coinfected with two viruses, one encoding the wild-type human Gαi subunit and another encoding the wild-type human β1γ2 subunits with an octahistidine tag inserted at the amino terminus of the β1 subunit. Cultures were harvested 48 hours post infection. Cells were lysed in hypotonic buffer and lipid-modified heterotrimeric Gi was extracted in a buffer containing 1% sodium cholate. The soluble fraction was purified using Ni-NTA chromatography, and the detergent was exchanged from cholate to DDM on column. After elution, the protein was dialyzed against a buffer containing 20 mM Hepes pH 7.5, 100 mM NaCl, 0.015% DDM, 100 μM TCEP, 10 μM GDP, and concentrated to ~20 mg/mL for further complexing with the μOR.
Generation of scFv16
6–8 week old female Balb/c mice were immunized with a purified rhodopsin-Gi complex39. Hybridoma cells were prepared using splenocytes of immunized mice using standard methods in combination with PAI myeloma cells. Clones that showed a positive reaction to purified rhodopsin(N2C/D282C/M257Y)/Gi1 complex in an ELISA assay and by immunoprecipitation were further characterized as monoclonal antibodies or Fab fragments. Fab-16 was selected from the initial pool of clones because it prevented dissociation of the rhodopsin (N2C/D282C/M257Y)/Gi1 complex by GTPγS, and therefore acted as a stabilizing chaperone in the same manner as Nb35 for Gs. The full sequence of constructs used is listed in Supplemental Figure 1. All animal studies were performed at Roche Innovation Center Basel according to ethical guidelines. All cell lines were obtained from manufacturer and tested for contamination.
A carboxy-terminal hexahistidine-tagged single chain construct of Fab16 (scFv16) was cloned into a modified pVL1392 vector containing a GP67 secretion signal immediately prior to the amino terminus of the scFv, expressed in secreted form from Trichuplusia ni Hi5 insect cells using the baculoviral method, and purified by Ni-NTA chromatography. Supernatant from baculoviral infected cells was pH balanced by addition of Tris pH 8.0. Chelating agents were quenched by addition of 1 mM nickel chloride and 5 mM calcium chloride and incubation with stirring for 1 hr at 25 °C. Resulting precipitates were removed by centrifugation and the supernatant was loaded onto Ni-NTA resin. The column was washed with 20 mM Hepes pH 7.5, 500 mM NaCl, and 10 mM imidazole followed by a low salt wash comprised of the same buffer substituted with 100 mM NaCl. Following elution with the same buffer supplemented with 250 mM imidazole, the carboxy-terminal hexahistidine tag was cleaved by incubation with human rhinovirus 3C protease, and the protein was dialyzed into a buffer consisting of 20mM Hepes pH 7.5 and 100 mM NaCl. Cleaved scFv16 was further purified by reloading over Ni-NTA resin. The flow-through was collected and purified over gel filtration chromatography using a Superdex 200 16/60 column. Monomeric fractions were pooled, concentrated, and flash frozen in liquid nitrogen until further use.
Formation and purification of the μOR-Gi-scFv16 complex
Purified DAMGO-bound μOR was mixed with a 1.2 molar excess of Gi heterotrimer. The coupling reaction was allowed to proceed at 24 °C for 1 hour and was followed by addition of apyrase to catalyze hydrolysis of unbound GDP, which destabilizes the nucleotide-free complex40. After one more hour at 25 °C, a 4-fold volume of 20 mM Hepes pH 7.5, 100 mM NaCl, 1% lauryl maltose neopentyl glycol (L-MNG), 0.1% CHS was added to the complexing reaction to initiate detergent exchange. After one hour incubation at 25 °C to allow micelle exchange, 1 mM MnCl2 and lambda phosphatase (New England Biolabs) were added to dephosphorylate the preparation. This reaction was further incubated at 4 °C for 2 hours. To remove excess G protein and residual DDM, the complexing mixture was purified by M1 anti-FLAG affinity chromatography. Bound complex was first washed in a buffer containing 1% L-MNG, followed by washes in gradually decreasing L-MNG concentrations. The complex was then eluted in 20mM Hepes pH 7.5, 100mM NaCl, 0.01% MNG/0.001% CHS, 300 nM DAMGO, 5 mM EDTA, and FLAG peptide. The eluted complex was supplemented with 100 μM TCEP to provide a reducing environment. The tobacco etch virus (TEV) protease and human rhinovirus 3C protease were added to cleave the flexible μOR amino- and carboxy- termini. Finally, a 1.2 molar excess of scFv16 was added to the preparation. Once cleavage of the termini was confirmed by SDS-PAGE, the μOR-Gi-scFv16 complex was purified by size exclusion chromatography on a Superdex 200 10/300 column in 20mM Hepes pH 7.5, 100mM NaCl, 300 nM DAMGO, 0.00075% MNG and 000025% GDN. Peak fractions were concentrated to ~7 mg/mL for electron microscopy studies.
Cryo-electron microscopy of μOR-Gi-scFv16 complex
3.0 μL of purified μOR-Gi-scFv16 complex was applied to glow-discharged 200 mesh grids (Quantifoil R1.2/1.3) and subsequently vitrified using a Vitrobot Mark IV (Thermo Fischer Scientific). Cryo-EM imaging was performed on a Titan Krios operated at 300 kV at a nominal magnification of 130,000x using a Gatan K2 Summit direct electron camera in counted mode, corresponding to a pixel size of 1.04 Å. A total of 2642 image stacks were obtained with a defocus range of −0.8 to −2.6 μm. Each stack movie was recorded for a total of 8 seconds with 0.1s per frame. The dose rate was 5 e/Å2/s, resulting in an accumulated dose of 40 electrons per Å2.
Dose fractionated image stacks were subjected to beam-induced motion correction using MotionCor241. A sum of all frames, filtered according to exposure dose, in each image stack was used for further processing. CTF parameters for each micrograph were determined by Gctf v1.0642. Particle selection, two-dimensional and three-dimensional classification, and 3D reconstruction were performed using RELION2.143, apart from the last round of local refinement and reconstruction that was performed with Frealign44. Semi-automated selected 893,426 particle projections were subjected to reference-free two-dimensional classification and averaging using a binned data set with a pixel size of 2.08 Å. 379,373 particles belonging to well-defined averages were subjected to further processing. An ab initio map generated by VIPER45 was used as initial reference model for maximum-likelihood-based three-dimensional classification, which, however did not produce classes with notable differences. Thus, all 379,373 particle projections were subjected to 3D refinement, producing a map at 4.3 Å resolution. The dataset was further reduced by removing particle projections from micrographs with resolution lower than 4.5 Å, resulting in a data set of 359,406 particles that were subjected to refinement and reconstruction after subtracting densities for the mobile Gα α-helical domain and the detergent micelle11. Particle projection assignments from RELION were imported into Frealign46 for a final round of local refinement and reconstruction. To prevent overfitting, the resolution limit for every alignment iteration never exceeded the 0.9 value of the Frealign calculated FSC. The map was further improved map after additionally subtracting densities corresponding to the ScFv from the raw particle projections11. The indicated resolution, using Phenix “gold standard” FSC47, of the final reconstruction is 3.5 Å and 3.6 Å at FSC 0.143 for the ScFv subtracted map and the ScFv including map, respectively. Local resolution was determined using the Bsoft package48 with unfiltered half-maps as input.
Model Building and Refinement
The building of a full atomic model for the μOR-Gi complex was aided by the quality and resolution of our map, as well as the existence of high-resolution crystal structures of each of the components that make up the complex. A composite model was formed by rigid body fitting of the active-state μOR (PDB ID: 5C1M)12 with nanobody removed, as well as the Ras domain and βγ subunits of GDP-bound Gi (PDB ID: 1GP2)23. The α5 helix of Gαi was removed and manually fit to the density, and the final 8 residues missing from the extreme C-terminus of the 1GP2 structure were manually built in coot49. This starting model was then subjected to iterative rounds of automated refinement in Rosetta50 and Phenix real space refine47, and manual building in Coot49. In the regions of the model for which side chain density was too weak to unambiguously assign a conformation, we stubbed residues to their C βposition, while preserving sequence information (Supplemental Figure 2,3). The final model was visually inspected for general fit to the map, and geometry was further evaluated using Molprobity51 as part of the Phenix suite of software. Initial restraints for DAMGO were generated using the PRODRG server52. To further refine the pose of DAMGO, we chose a pose from molecular dynamics simulation consistent with our map and then performed a refinement using Phenix. This involved manually editing the residue and atom names from a CHARMM parameter file to match the 3-letter codes and atom names from the rcsb. In particular, DAL for D-alanine, MEA for N-methyl phenylalanine, and ETA for Gly-ol C terminus. Additional, custom, restraints were generated to keep planarity of the final peptide bond between MEA and ETA as a supplement to the natural library of phenix amino acid restraints. Model overfitting was evaluated through its refinement against one cryo-EM half map after randomly displacing all atoms by 0.2 Å. FSC curves were calculated between the resulting model and the half map used for refinement (green curve, Extended Data Fig. 2b, c), as well as between the resulting model and the other half map for cross- validation (blue curve, Extended Data Fig. 2b, c), and also against the full map (red curve, Extended Data Fig. 2b, c). The final refinement statistics for both models are provided in Extended Data Table 1.
System setup for molecular dynamics simulations
Molecular dynamics simulations were initiated from an earlier refinement of the structure reported in this study after removing the G protein and ScFv fragment. Prior to beginning simulations, Schrödinger Glide53 was used to relax DAMGO to an energetically favorable conformation. The initial DAMGO pose is depicted in Extended Data Figure 3. We performed five independent simulations, for each of which initial atom velocities were assigned randomly and independently. Prime (Schrödinger, Inc.) was used to model missing side chains, and neutral acetyl and methylamide groups were added to cap protein termini. Titratable residues remained in their dominant protonation state at pH 7, as determined using PropKa, except for D2.50 and D3.49 which were protonated. Our simulations incorporated the waters from the 5C1M crystal structure.
The prepared protein structures were aligned to the Orientation of Proteins in Membranes (OPM) structure for PDB entry 5C1M54. The aligned structures were then inserted into a pre-equilibrated palmitoyl-oleoyl-phosphatidylcholine (POPC) bilayer using Dabble, a simulation preparation software55. Sodium and chloride ions were added to neutralize each system at a concentration of approximately 150 mM. Bilayer dimensions were chosen to maintain at least a 30 Å buffer between protein images in the x-y plane and a 20 Å buffer between protein images in the z direction. Final system dimensions were approximately 80 × 75 × 90 Å3. Simulation times for each replicate were approximately 1 μs.
Molecular dynamics simulation protocols
We used the CHARMM36m force field for proteins, lipids, and ions and the TIP3P model for waters56–60. Parameters for the non-canonical residues in DAMGO were determined by analogy to N-methyl glycine for assigning N-methyl parameters to N-methyl phenylalanine (residue 4) and by analogy to serine to assign parameters to the Gly-ol capping group (residue 5). CMAP terms for D-alanine were inverted from those for L-Alanine to account for the inverted chirality of the residue.
We performed the simulations using the Compute Unified Device Architecture (CUDA) version of Particle-Mesh Ewald Molecular Dynamics (PMEMD) in AMBER on one or two graphical processing units (GPUs)61. Simulations were performed using the AMBER1662 software. Three rounds of minimization were performed, each consisting of 500 iterations of steepest descent minimization, followed by 500 iterations of conjugate gradient descent minimization, with harmonic restraints of 10.0, 5.0, and 1.0∙kcal∙mol−1∙Å−2 placed on the protein and lipids. Systems were heated from 0K to 100K in the NVT ensemble over 12.5 ps and then from 100K to 310K in the NPT ensemble over 125 ps, using 10.0∙kcal∙mol−1∙Å−2 harmonic restraints applied to lipid and protein heavy atoms. Systems were then equilibrated at 310 K in the NPT ensemble at 1 bar, with harmonic restraints on all protein heavy atoms tapered off by 1.0 kcal∙mol−1∙Å−2 starting at 5.0∙kcal∙mol−1∙Å−2 in a stepwise fashion every 2 ns for 10 ns and then by 0.1 kcal∙mol−1∙Å−2 in a stepwise fashion every 2 ns for 20 ns. Production simulations were performed in the NPT ensemble at 310K and 1 bar, using a Langevin thermostat for temperature coupling and a Monte Carlo barostat for pressure coupling. These simulations used a 4 fs time step with hydrogen mass repartitioning63. Bond lengths to hydrogen atoms were constrained using SHAKE. Simulations used periodic boundary conditions. Non-bonded interactions were cut off at 9.0 Å, and long-range electrostatic interactions were computed using Particle Mesh Ewald (PME) with an Ewald coefficient of approximately 0.31 Å and an interpolation order of 4. The FFT grid size was chosen such that the width of a grid cell was approximately 1 Å.
During production simulations, all residues within 5 Å of the G protein interface were restrained to the initial structure using 5.0 kcal∙mol−1∙Å−2 harmonic restraints applied to non-hydrogen atoms. Using such restraints reduces the overall system size, enabling more simulation, while ensuring that the receptor maintains an active conformation throughout the simulation.
Analysis protocols for MD simulation
Trajectory snapshots were saved every 200 ps during production simulations. The AmberTools17 CPPTRAJ package was used to reimage and center trajectories64. Simulations were visualized and analyzed using Visual Molecular Dynamics (VMD)65. In two simulations, DAMGO was trapped in an unstable binding pose, wherein the water-mediated interaction between the DAMGO Tyr residue and His297 failed to form during equilibration, and instead a direct hydrogen bond between these residues was formed. Our analysis is based on the other three simulations, in which DAMGO’s pose was consistent with the EM density. Water occupancy maps were generated using AmberTools17 GIST66,67. Frames from every 1 ns of simulation, excluding the first 400 ns, aligned to the initial structure, were used as input. The grid size was set to 0.25 Å. The resulting map was smoothed using a Gaussian filter with a standard deviation of 2 grid cells.
Extended Data
Extended Data Figure 1. scFv binding characteristics.
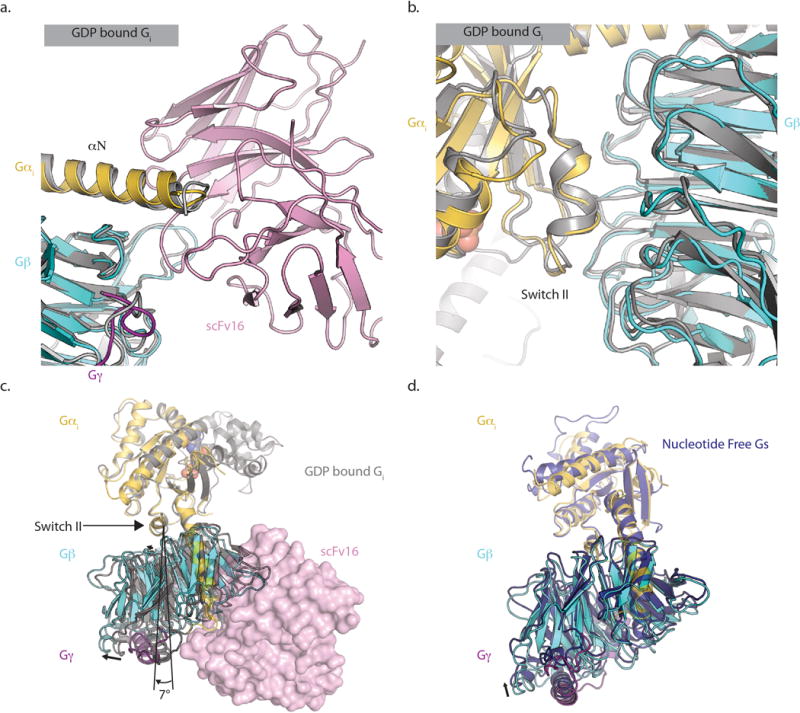
scFv 16 does not perturb the interfaces between Gα and Gβ at a) its binding epitope or b) the Switch II region located ~40A away. Our structure is colored by chain, while the structure of GDP-bound Gi1 heterotrimer (PDB 1GP2) is colored grey. c) In the nucleotide-free state, there is a ~7˚ rotation of G βγ relative to the Gαs Switch II domain when compared to the GDP-bound form. This rotated conformation is similar to that observed in nucleotide-free Gs coupled to the β2AR (PDB ID 3SN6) as shown in panel d).
Extended Data Figure 2. Cryo-EM data processing.
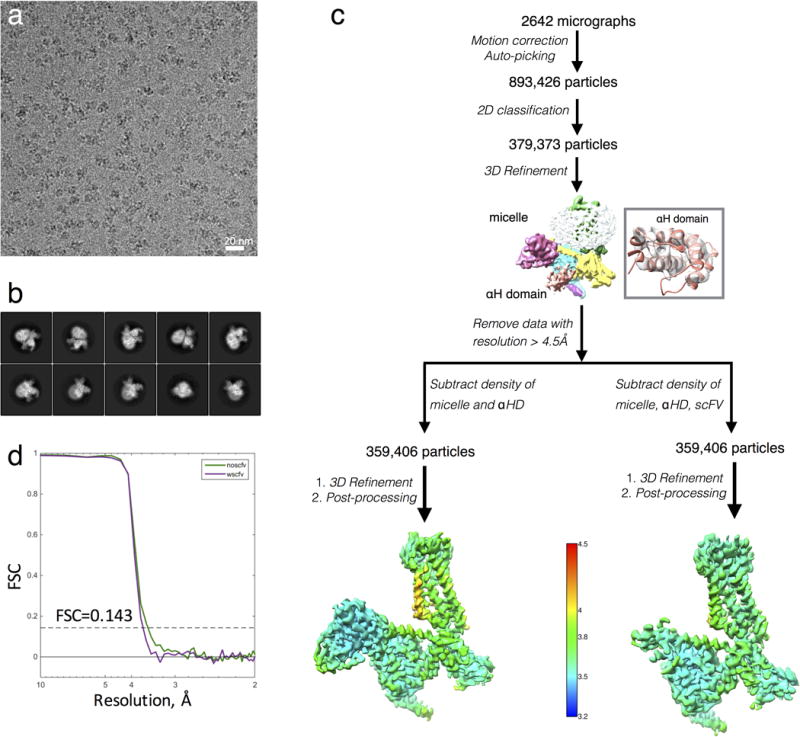
a, Representative cryo-EM micrograph of the μOR-Gi complex. Scale bar, 20nm.
b, Representative two-dimensional averages showing distinct secondary structure features from different views of the complex.
c, Flow chart of cryo-EM data processing. The unmasked map in the middle of the chart has been colored by subunit. The inset shows the fit of the crystal structure of the α-helical domain in the corresponding density of the unmasked reconstruction. Three-dimensional density maps colored according to local resolution.
d, “Gold standard” Fourier shell correlation (FSC) curves from Phenix indicates overall nominal resolutions of 3.5 Å and 3.6 Å using the FSC=0.143 criterion for the scFv-subtracted map (green curve) and scFv-retained maps (purple curve), respectively.
Extended Data Figure 3. Cryo-EM map vs. refined structure.
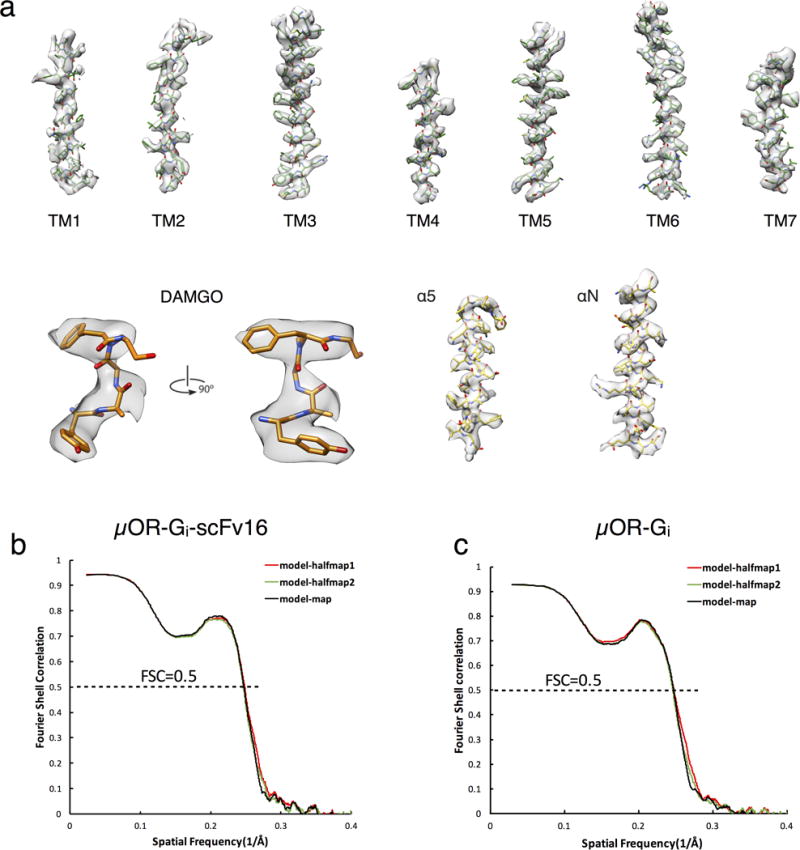
a) EM density map (scFv subtracted) and model are shown for all seven transmembrane α-helices of the μOR, DAMGO, and Gα helices α5 and αN.
b,c) Cross-validation of model to EM density map. The model was refined against one half map after displacement of atoms by 0.2A, and FSC curves were calculated between this model and the final cryo-EM map (full dataset, black), of the outcome of model refinement with a half map versus the same map (red), and of the outcome of model refinement with a half map versus the other half map (green). The results of the scFv-retained model vs. map and of scFv subtracted model vs. map are shown in b) and c), respectively.
Extended Data Figure 4. Selected cryo-EM densities of μOR-Gi Complex.
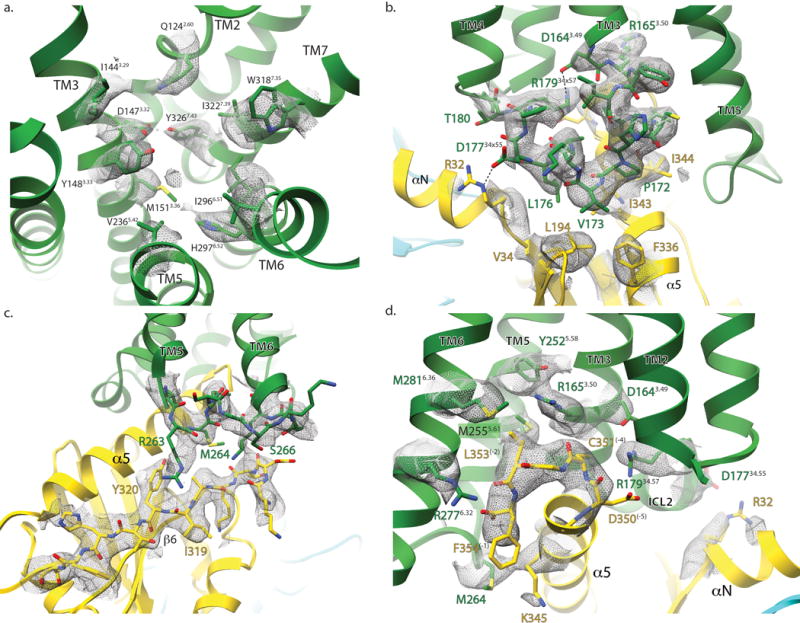
Cryo-EM density (displayed as mesh) surrounding residues involved in a) DAMGO binding, b) μOR-Gαi interaction around ICL2, c) ICL3, and d) cytoplasmic ends of the μOR transmembrane helices. These figures accompany the models shown in figures 1e, 4b, 5a, and 5b respectively.
Extended Data Figure 5. Stability of DAMGO in MD Simulations.
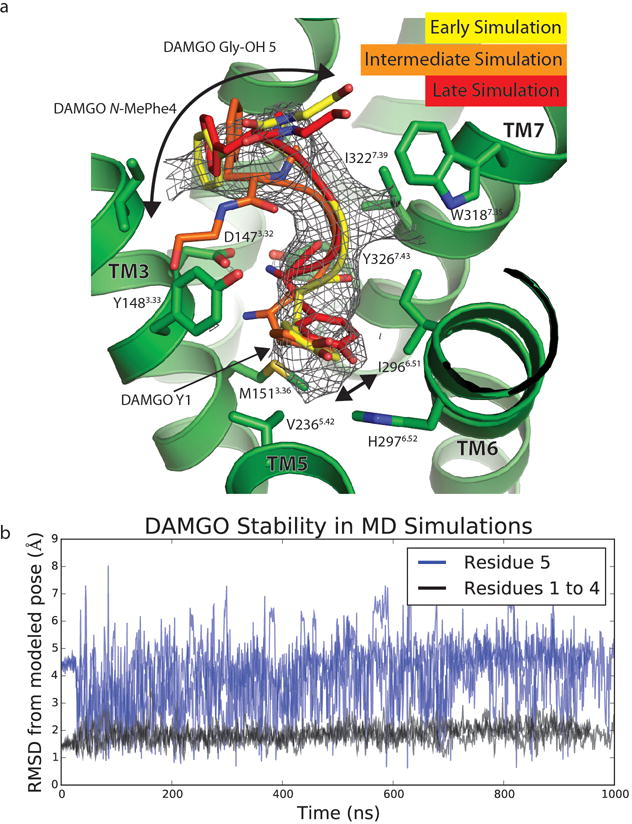
a. Over the course of MD simulations, the positions of the first 4 residues of DAMGO do not significantly change, while the 5th residue (Gly-ol) shows significant variability in position. Frames from the first and last 100 ns are shown with an intermediate to highlight both the relative stability of the first 4 amino acids, as well as the flexibility of the fifth. Arrows show the extent of motion in the N- and C-terminal residues over the course of simulation. Cryo-EM density for DAMGO is shown as mesh.
b. Root mean standard deviations (RMSDs) from the modeled pose of DAMGO to the pose during MD simulations. The RMSD calculations include heavy atoms on the peptide backbone. Data from three independent simulations are plotted. The RMSDs for residues 1 to 4 (black) and the C-terminal Gly-ol (blue) are plotted separately to highlight their stability and mobility, respectfully.
Extended Data Figure 6. Water occupancy in orthosteric binding site.
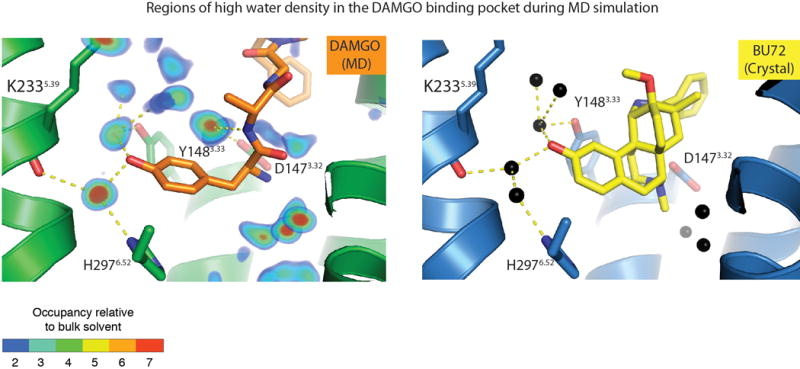
Left panel, water occupancy in MD simulations of DAMGO-bound μOR overlaid with a representative conformation from MD simulations. ‘Occupancy relative to bulk solvent’ is the ratio of the rate at which water is observed in a given volume to the rate at which water is expected to be observed in an equivalent volume in the bulk solvent. For example, blue regions (occupancy ratio = 2) are occupied by water twice as often as an equivalent region in the bulk solvent. Right panel, crystallographic waters in the BU72-bound μOR binding pocket (PDB ID: 5C1M). Waters are shown as black spheres, BU72 is shown as yellow sticks, and hydrogen bonds are shown as dashed lines.
Extended Data Figure 7. Comparison of the C-termini of Gαs and Gαi.
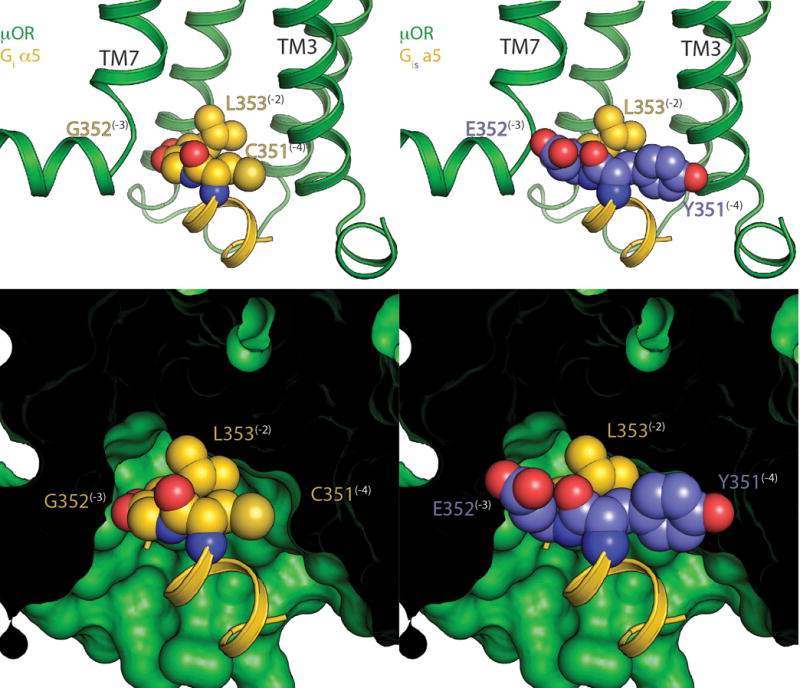
The C-terminus of Gαs is bulkier than that of Gαi due to substitution of small amino acids C (−4 position) and G (−3 position) in Gαi to Y and E respectively in Gαs. This leads to steric clashes with TMs 3 and 7 of the μOR.
Top - ribbon view of μOR (green) with WT Gαi (gold, left) and a Gαis model (right) created by substituting C and G for Y and E based on the β2AR-Gs crystal structure. Substituted positions are colored in light purple. The −4 to −2 positions have their side chains shown as spheres, and the rest as a ribbon.
Bottom - space filling view of the μOR showing the steric clashes that result from these substitutions.
Extended Data Figure 8. Comparison of Gai C terminal peptide binding modes.
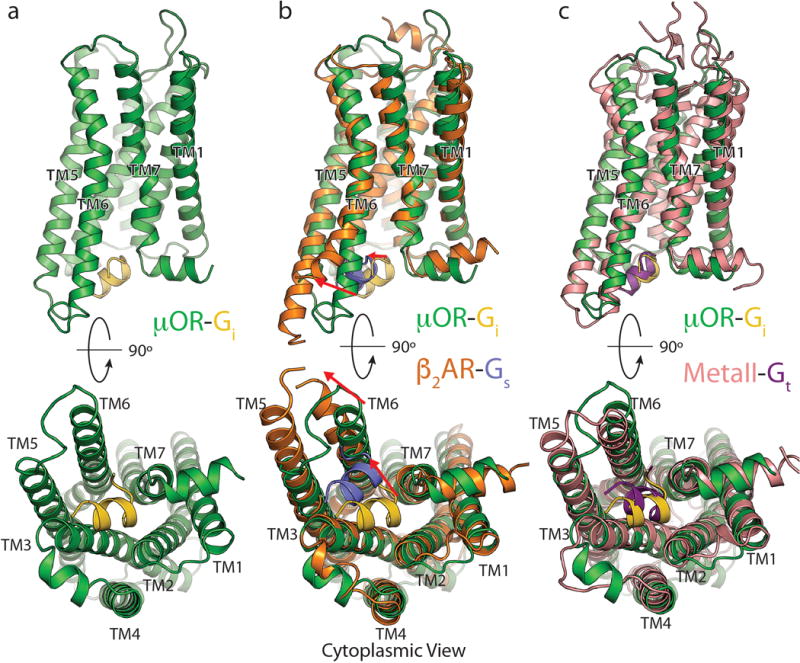
Side (top half), and cytoplasmic (bottom half) views of a) the μOR (green) with the last 11 residues of Gαi (gold) alone, b) compared to the β2AR(orange) with the last 11 residues of Gαs (light purple) (PDB ID 3SN6), or c) compared to MetaII Rhodopsin (pink) in complex with an 11 residue Gtransducin (Gt) C-terminal peptide (dark purple) (PDB ID 3PQR). The mOR-Gi complex aligns best with the MetaII-Gt complex both in terms of TM6 displacement as well as position of the α5 peptide.
Extended Data Table 1.
Cryo-EM data collection, refinement and validation statistics
| μOR-Gi Complex (EMDB-xxxx) (PDB xxxx) | μOR-Gi-scFvl6 Complex (EMDB-yyyy) (PDB yyyy) | |
|---|---|---|
| Data collection and processing | ||
| Magnification | 48,076 | 48,076 |
| Voltage (kV) | 300 | 300 |
| Electron exposure (e–/Å2) | 40 | 40 |
| Defocus range (μm) | −0.8 ~ −2.6 | −0.8 ~ −2.6 |
| Pixel size (Å) | 1.04 | 1.04 |
| Symmetry imposed | C1 | C1 |
| Initial particle images (no.) | 893,426 | 893,426 |
| Final particle images (no.) | 359,406 | 359,406 |
| Map resolution (Å) | 3.5 Å | 3.6 Å |
| FSC threshold | (0.143) | (0.143) |
| Map resolution range (Å) | 3.3-4.5 | 3.3-4.5 |
| Refinement | ||
| Initial model used (PDB code) | 5C1M 1GP2 |
5C1M 1GP2 |
| Model resolution (Å) | 3.5 | 3.6 |
| Model resolution range (Å) | 3.3-4.5 | 3.3-4.5 |
| Map sharpening B factor (Å2) | Pre −90, post −60 | Pre −90, post −60 |
| Model composition | ||
| Non-hydrogen atoms | 6986 | 8731 |
| Protein residues | 886 residues (6949 atoms) | 1119 residues (8694 atoms) |
| Ligands | 1 (37 atoms) | 1 (37 atoms) |
| B factors (Å2) | ||
| Protein | 33.23 | 60.55 |
| Ligand | 31.27 | 79.99 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.007 | 0.007 |
| Bond angles (°) | 1.311 | 1.015 |
| Validation | ||
| MolProbity score | 1.89 | 1.89 |
| Clashscore | 7.02 | 8.16 |
| Poor rotamers (%) | 0.72 | 0.92 |
| Ramachandran plot | ||
| Favored (%) | 91.54 | 92.93 |
| Allowed (%) | 8.35 | 6.98 |
| Disallowed (%) | 0.11% | 0.09% |
Extended Data Table 2.
Sequence alignment of residues that form the interaction interface between μOR and Gi. Receptors from different branches of the GPCR family with different coupling specificity were selected for analysis. Sequences and alignment were performed using GPCRDB (gpcrdb.org)
| Coupling | Branch | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mOR Residue | T103 | V169 | P172 | V173 | D177 | R179 | T180 | M255 | K271 | R277 | I278 | ||
| 2.39 | 3.54 | 34.50 | 34.51 | 34.55 | 34.57 | 4.38 | 5.61 | 6.26 | 6.32 | 6.33 | |||
| [Human] 5-HT1A receptor | Gi | α | A | I | P | I | N | R | T | I | A | K | T |
| [Human] 5-HT1B receptor | Gi | α | A | I | A | V | A | R | T | I | M | K | A |
| [Human] M2 receptor | Gi | α | N | V | P | L | V | R | T | I | P | K | V |
| [Human] M4 receptor | Gi | α | N | V | P | L | A | R | T | I | M | K | V |
| [Human] alpha2A-adrenoceptor | Gi | α | Q | I | A | I | L | R | T | I | R | R | F |
| [Human] FPR1 | Gi | γ | T | V | P | V | N | R | T | I | – | R | P |
| [Human] FPR2/ALX | Gi | γ | T | V | P | V | N | R | T | I | – | R | P |
| [Human] GAL1 receptor | Gi | γ | T | I | S | R | S | R | V | V | – | K | T |
| [Human] GAL3 receptor | Gi | γ | T | V | P | L | A | R | T | T | R | R | A |
| [Human] δ receptor | Gi | γ | T | V | P | V | D | R | T | M | K | R | I |
| [Human] κ receptor | Gi | γ | T | V | P | V | D | R | T | M | K | R | I |
| [Human] μ receptor | Gi | γ | T | V | P | V | D | R | T | M | K | R | I |
| [Human] NOP receptor | Gi | γ | T | I | P | I | D | R | T | M | K | R | I |
| [Human] SST1 receptor | Gi | γ | T | V | P | I | R | R | R | I | R | K | I |
| [Human] SST2 receptor | Gi | γ | T | V | P | I | K | R | R | I | R | K | V |
| [Human] SST3 receptor | Gi | γ | T | V | P | T | R | R | T | I | R | R | V |
| [Human] SST4 receptor | Gi | γ | T | V | P | L | T | R | R | I | R | K | I |
| [Human] SST5 receptor | Gi | γ | T | V | P | L | R | R | R | I | – | K | V |
| [Human] CCR1 | Gi | γ | T | I | A | V | R | R | T | I | – | K | A |
| [Human] CCR4 | Gi | γ | T | I | A | V | R | R | T | I | – | K | A |
| [Human] CXCR4 | Gi | γ | T | I | A | T | R | R | K | I | – | K | A |
| [Human] A1 receptor | Gi | α | T | V | P | L | M | V | T | V | Y | K | I |
| [Human] beta1-adrenoceptor | Gs | α | T | I | P | F | S | L | T | V | V | K | A |
| [Human] beta2-adrenoceptor | Gs | α | T | I | P | F | S | L | T | V | F | K | A |
| [Human] MC1 receptor | Gs | α | M | I | A | L | S | V | T | G | – | K | G |
| [Human] MC2 receptor | Gs | α | M | I | A | L | S | V | T | K | – | K | G |
| [Human] MC4 receptor | Gs | α | M | I | A | L | N | M | T | R | – | K | G |
| [Human] A2A receptor | Gs | α | T | I | P | L | G | V | T | I | T | H | A |
| [Human] H2 receptor | Gs | α | T | V | P | L | V | V | T | I | A | K | A |
| [Human] TA1 receptor | Gs | α | T | V | P | L | A | M | N | I | S | K | A |
| [Human] RXFP1 | Gs | δ | Y | I | P | F | R | – | G | M | Q | I | L |
| [Human] RXFP2 | Gs | δ | H | I | P | F | R | – | G | M | C | A | V |
| [Human] V2 receptor | Gs | β | I | I | P | M | R | G | S | I | V | K | T |
| [Human] 5-HT2A receptor | Gq | α | T | I | P | I | R | N | S | T | S | K | A |
| [Human] 5-HT2B receptor | Gq | α | T | I | P | I | Q | N | S | T | T | R | A |
| [Human] M1 receptor | Gq | α | N | V | P | L | A | R | T | I | S | K | A |
| [Human] M3 receptor | Gq | α | N | I | P | L | A | R | T | I | S | K | A |
| [Human] M5 receptor | Gq | α | N | I | P | L | A | R | T | I | V | K | A |
| [Human] alpha1A-adrenoceptor | Gq | α | T | V | P | L | T | V | T | V | K | K | A |
| [Human] GAL2 receptor | Gq | γ | T | I | P | L | E | R | T | T | A | K | V |
| [Human] OX1 receptor | Gq | β | T | I | P | L | – | – | T | I | Q | K | T |
| [Human] OX2 receptor | Gq | β | T | I | P | L | – | – | T | I | Q | K | T |
| [Human] NK1 receptor | Gq | β | T | I | – | – | – | – | S | V | Q | K | V |
Supplementary Material
Acknowledgments
We thank Jean-Philippe Carralot (F. Hoffmann-La Roche Ltd) for help in antibody generation, Craig Yoshioka and Claudia Lopez for assistance with data collection, Martin Siegrist, Georg Schmid, Bernard Rutten, Doris Zulauf, Stephanie Kueng (Roche Non-Clinical Biorepository) and Ralf Thoma for technical assistance for biomass and cell line generation. We also acknowledge Nigel Moriarty (Lawrence Berkeley National Laboratories) for help with generation of parameters for DAMGO and general advice for refinement of our model. The work is supported by NIH grant R37DA036246 (B.K.K, S.G. and G.S.) and NIH grant R01GM083118 (B.K.K.). Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number T32GM007276 (A.K.). The content is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health. Shoji Maeda was supported by the Roche Postdoctoral Fellowship (RPF ID: 113). Gebhard F.X. Schertler acknowledges the Swiss National Science Foundation for grants 310030_153145 and 310030B_17335 (to G.F.X.S.) and long-term financial support from the Paul Scherrer Institute. Brian Kobilka is a Chan-Zuckerberg Biohub Investigator.
Footnotes
Author Contributions
A.K. prepared the μOR-Gi complex and refined the structure from cryo-EM density maps. H.H. obtained and processed cryo-EM data with the assistance of Y.Z. and Q.Q. S.M. identified and prepared scFv16 with assistance from R.D. and H.M and under supervision of G.F.X.S. A.M., D.H. S.G. and A.K. developed the procedure for forming the μOR-Gi complex. N.R.L and J.M.P performed molecular dynamics simulations under supervision of R.O.D. W.I.W. aided in map interpretation and model refinement. A.K., A.M. B.K.K. and G.S. wrote the manuscript. A.M., G.S. and B.K.K. supervised the project.
Author Information
The authors declare one competing interest: Brian Kobilka is a founder of and consultant for ConfometRx, Inc. Readers are welcome to comment on the online version of the paper.
Data availability
All data generated or analyzed during this study are included in this published article and its Supplementary Information. Sequences of constructs used in this study are listed in Supplementary Figure 1. The cryo-EM density maps for the μOR-Gi complex with, and without, scFv16 have been deposited in the Electron Microscopy Data Bank under accession codes EMD-XXXX and EMD-YYYY, respectively. The coordinates for the models of μOR-Gi with, and without scFv-16 have been deposited in the Protein Data Bank under accession numbers XXXX and YYYY respectively.
References
- 1.Matthes HW, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- 2.Barnett ML, Olenski AR, Jena AB. Opioid-Prescribing Patterns of Emergency Physicians and Risk of Long-Term Use. N Engl J Med. 2017;376:663–673. doi: 10.1056/NEJMsa1610524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Connor M, Christie MD. Opioid receptor signalling mechanisms. Clin Exp Pharmacol Physiol. 1999;26:493–499. doi: 10.1046/j.1440-1681.1999.03049.x. [DOI] [PubMed] [Google Scholar]
- 4.Raffa RB, Martinez RP, Connelly CD. G-protein antisense oligodeoxyribonucleotides and mu-opioid supraspinal antinociception. Eur J Pharmacol. 1994;258:R5–7. doi: 10.1016/0014-2999(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 5.Raehal KM, Walker JKL, Bohn LM. Morphine side effects in beta-arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005;314:1195–1201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- 6.Schmid CL, et al. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell. 2017;171:1165–1175.e13. doi: 10.1016/j.cell.2017.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeWire SM, et al. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 8.Manglik A, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasmussen SGF, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature. 2017;546:248–253. doi: 10.1038/nature22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang YL, et al. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature. 2017;546:118–123. doi: 10.1038/nature22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang W, et al. Structural insights into μ-opioid receptor activation. Nature. 2015;524:315–321. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ballesteros JA, Weinstein H. Receptor Molecular Biology. Vol. 25. Elsevier; 1995. pp. 366–428. [Google Scholar]
- 14.Manglik A, et al. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Granier S, et al. Structure of the delta opioid receptor bound to naltrindole. 2012 doi: 10.2210/pdb4ej4/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu H, et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Che T, et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell. 2018;172:55–67.e15. doi: 10.1016/j.cell.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fenalti G, et al. Structural basis for bifunctional peptide recognition at human δ-opioid receptor. Nat Struct Mol Biol. 2015;22:265–268. doi: 10.1038/nsmb.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emmerson PJ, Liu MR, Woods JH, Medzihradsky F. Binding affinity and selectivity of opioids at mu, delta and kappa receptors in monkey brain membranes. J Pharmacol Exp Ther. 1994;271:1630–1637. [PubMed] [Google Scholar]
- 20.Minami M, et al. DAMGO, a mu-opioid receptor selective ligand, distinguishes between mu-and kappa-opioid receptors at a different region from that for the distinction between mu- and delta-opioid receptors. FEBS Lett. 1995;364:23–27. doi: 10.1016/0014-5793(95)00340-f. [DOI] [PubMed] [Google Scholar]
- 21.DiMaio J, Schiller PW. A cyclic enkephalin analog with high in vitro opiate activity. Proc Natl Acad Sci USA. 1980;77:7162–7166. doi: 10.1073/pnas.77.12.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rasmussen SGF, et al. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wall MA, et al. The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 24.Dror RO, et al. SIGNAL TRANSDUCTION. Structural basis for nucleotide exchange in heterotrimeric G proteins. Science. 2015;348:1361–1365. doi: 10.1126/science.aaa5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Eps N, et al. Interaction of a G protein with an activated receptor opens the interdomain interface in the alpha subunit. Proceedings of the National Academy of Sciences. 2011;108:9420–9424. doi: 10.1073/pnas.1105810108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaya AI, et al. A conserved phenylalanine as a relay between the α5 helix and the GDP binding region of heterotrimeric Gi protein α subunit. J Biol Chem. 2014;289:24475–24487. doi: 10.1074/jbc.M114.572875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung KY, et al. Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature. 2011;477:611–615. doi: 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choe HW, et al. Crystal structure of Metarhodopsin II in complex with a C-terminal peptide derived from the Galpha subunit of transducin. 2011 doi: 10.2210/pdb3pqr/pdb. [DOI] [Google Scholar]
- 29.Conklin BR, Farfel Z, Lustig KD, Julius D, Bourne HR. Substitution of three amino acids switches receptor specificity of Gq alpha to that of Gi alpha. Nature. 1993;363:274–276. doi: 10.1038/363274a0. [DOI] [PubMed] [Google Scholar]
- 30.Isberg V, et al. Generic GPCR residue numbers - aligning topology maps while minding the gaps. Trends Pharmacol Sci. 2015;36:22–31. doi: 10.1016/j.tips.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Isberg V, et al. GPCRDB: an information system for G protein-coupled receptors. Nucleic Acids Res. 2014;42:D422–5. doi: 10.1093/nar/gkt1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ravindranathan A, et al. Functional characterization of human variants of the mu-opioid receptor gene. Proceedings of the National Academy of Sciences. 2009;106:10811–10816. doi: 10.1073/pnas.0904509106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skorpen F, et al. The rare Arg181Cys mutation in the μ opioid receptor can abolish opioid responses. Acta Anaesthesiol Scand. 2016;60:1084–1091. doi: 10.1111/aas.12739. [DOI] [PubMed] [Google Scholar]
- 34.Chaipatikul V, Loh HH, Law PY. Ligand-selective activation of mu-oid receptor: demonstrated with deletion and single amino acid mutations of third intracellular loop domain. J Pharmacol Exp Ther. 2003;305:909–918. doi: 10.1124/jpet.102.046219. [DOI] [PubMed] [Google Scholar]
- 35.West RE, Moss J, Vaughan M, Liu T, Liu TY. Pertussis toxin-catalyzed ADP-ribosylation of transducin. Cysteine 347 is the ADP-ribose acceptor site. J Biol Chem. 1985;260:14428–14430. [PubMed] [Google Scholar]
- 36.Okude J, et al. Identification of a Conformational Equilibrium That Determines the Efficacy and Functional Selectivity of the μ-Opioid Receptor. Angew Chem Int Ed Engl. 2015;54:15771–15776. doi: 10.1002/anie.201508794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gregorio GG, et al. Single-molecule analysis of ligand efficacy in β2AR-G-protein activation. Nature. 2017;547:68–73. doi: 10.1038/nature22354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin K, Dong C, Wu G, Lambert NA. Inactive-state preassembly of G(q)-coupled receptors and G(q) heterotrimers. Nat Chem Biol. 2011;7:740–747. doi: 10.1038/nchembio.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maeda S, et al. Crystallization Scale Preparation of a Stable GPCR Signaling Complex between Constitutively Active Rhodopsin and G-Protein. PLoS ONE. 2014;9:e98714. doi: 10.1371/journal.pone.0098714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Westfield GH, et al. Structural flexibility of the G alpha s alpha-helical domain in the beta2-adrenoceptor Gs complex. Proceedings of the National Academy of Sciences. 2011;108:16086–16091. doi: 10.1073/pnas.1113645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng S, Palovcak E, Armache JP, Cheng Y, Agard D. Anisotropic Correction of Beam-induced Motion for Improved Single-particle Electron Cryo-microscopy. 2016 doi: 10.1101/061960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang K. Gctf: Real-time CTF determination and correction. Journal of Structural Biology. 2016;193:1–12. doi: 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheres SHW. Journal of Structural Biology. Journal of Structural Biology. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grigorieff N. FREALIGN: high-resolution refinement of single particle structures. Journal of Structural Biology. 2007;157:117–125. doi: 10.1016/j.jsb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 45.Penczek PA, Grassucci RA, Frank J. The ribosome at improved resolution: new techniques for merging and orientation refinement in 3D cryo-electron microscopy of biological particles. Ultramicroscopy. 1994;53:251–270. doi: 10.1016/0304-3991(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 46.Grigorieff N. Frealign: An Exploratory Tool for Single-Particle Cryo-EM. Meth Enzymol. 2016;579:191–226. doi: 10.1016/bs.mie.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adams PD, et al. The Phenix software for automated determination of macromolecular structures. Methods. 2011;55:94–106. doi: 10.1016/j.ymeth.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heymann JB, Belnap DM. Bsoft: image processing and molecular modeling for electron microscopy. Journal of Structural Biology. 2007;157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 49.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 50.Wang RYR, et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. eLife. 2016;5:352. doi: 10.7554/eLife.17219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams CJ, et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018;27:293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schüttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 53.Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 1 Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 54.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 55.Betz R. Dabble. 2017 doi: 10.5281/zenodo.836914. [DOI] [Google Scholar]
- 56.Best RB, Mittal J, Feig M, MacKerell AD. Inclusion of many-body effects in the additive CHARMM protein CMAP potential results in enhanced cooperativity of α-helix and β-hairpin formation. Biophys J. 2012;103:1045–1051. doi: 10.1016/j.bpj.2012.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Best RB, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J Chem Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang J, MacKerell AD. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem. 2013;34:2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klauda JB, et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang J, et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Meth. 2016;14:71–73. doi: 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2 Explicit Solvent Particle Mesh Ewald. J Chem Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 62.Case DA, et al. Amber 2017. Available at: (Accessed: 25 April 2018) [Google Scholar]
- 63.Hopkins CW, Le Grand S, Walker RC, Roitberg AE. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J Chem Theory Comput. 2015;11:1864–1874. doi: 10.1021/ct5010406. [DOI] [PubMed] [Google Scholar]
- 64.Roe DR, Cheatham TE., III PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 65.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. Journal of Molecular Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 66.Nguyen CN, Young TK, Gilson MK. Grid inhomogeneous solvation theory: hydration structure and thermodynamics of the miniature receptor cucurbit[7]uril. J Chem Phys. 2012;137:044101. doi: 10.1063/1.4733951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nguyen C, Gilson MK, Young T. Structure and Thermodynamics of Molecular Hydration via Grid Inhomogeneous Solvation Theory. arXiv.org. 2011 doi: 10.1063/1.4733951. q-bio.BM. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.