

Abstract
Protein structures are essential in modern biology yet experimental methods are far from being able to catch up with the rapid increase in available genomic data. Computational protein structure prediction methods aim to fill the gap while the role of protein structure refinement is to take approximate initial template-based models and bring them closer to the true native structure. Current methods for computational structure refinement rely on molecular dynamics simulations, related sampling methods, or iterative structure optimization protocols. The best methods are able to achieve moderate degrees of refinement but consistent refinement that can reach near-experimental accuracy remains elusive. Key issues revolve around the accuracy of the energy function, the inability to reliably rank multiple models, and the use of restraints that keep sampling close to the native state but also limit the degree of possible refinement. A different aspect is the question of what exactly the target of high-resolution refinement should be as experimental structures are affected by experimental conditions and different biological questions require varying levels of accuracy. While improvement of the global protein structure is a difficult problem, high-resolution refinement methods that improves local structural quality such as favorable stereochemistry and the avoidance of atomic clashes are much more successful.
Graphical Abstract
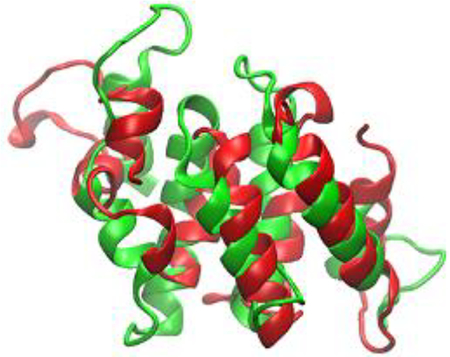
Protein structure refinement to improve a template-based model (green) towards the native structure (red) continues to be a challenging computational problem.
Introduction
When the first protein structures were solved via X-ray crystallography1, 2, it became instantly clear that detailed structural information at the atomistic level is the key to a full mechanistic understanding of biological processes. The sequence-structure-function paradigm is now an essential feature of modern molecular and cellular biology3. Accurate protein structures are necessary as starting points for rational drug design4, protein engineering applications5, and to understand functional implications of mutations associated with genomic variations within populations6. We have reached an age where structural information is available for most types of proteins7. Nevertheless, the number of known genes continues to greatly outnumber the number of available protein structures by orders of magnitude as experimental protein structure determination remains a slow and tedious process. This has motivated computational efforts to predict protein structures soon after the first protein structures became available8, 9. Since early on, ab initio methods have aimed at structure prediction via physics-motivated models with the idea that, starting from extended chains, the native structure could be found via sampling or optimization as the lowest (free) energy state10, 11. To overcome computational limitations, these approaches have often invoked simplified models of proteins such as polymers decorated with patterns of polar and hydrophobic side chains12–14. While much has been learned at the fundamental level about protein structures and the folding process13, pure ab initio methods that do not use any information from known structures largely fail to accurately and reliably predict the structures of all but very small proteins15, 16. This remains true today even as more sophisticated models can be sampled extensively on today’s computers. In contrast, using knowledge from known structures to predict new structures, has always been a far more successful strategy17–20. Homology modelling, which assumes that similar amino acid sequences lead to similar structures, is the standard method for predicting protein structures21. Furthermore, advanced methods that assemble models based on structural fragments taken from known structures are often able to generate useful models for sequences where homologous proteins with known structures cannot be found22–24.
Predicted structural models generated with today’s methods can be impressively accurate25, 26, but they still often do not reach experimental accuracy (considered to be <1Å root mean square deviation (RMSD) for all heavy atoms). The reliance on structural templates inherently limits the ability to capture subtle variations in protein structures as a result of minor differences in amino acid sequence. Template-based modelling is also problematic when interaction partners such as other proteins, nucleic acids or ligands, perturb the structure and when templates that include such interactions are not available. This has created the need for structure refinement methods to improve initial models towards experimental accuracy27–36. Structure refinement methods are widely used already to derive atomistic structures from nuclear magnetic resonance (NMR) restraints37, 38 and during crystallographic structure determination39–41 either de novo or via molecular replacement. The use of lower-resolution data, such as from electron microscopy (EM)42, small-angle X-ray scattering (SAXS)43, 44, or cross-linking information45 can also be very effective in guiding structure refinement when combined with template-based modelling. In the absence of experimental data, structure refinement is tasked with improving initial template-based models using just computation. As template-based models often come within 2–5 Å Cα RMSD from an experimentally determined native structure46, the goal of structure refinement is a seemingly moderate degree of improvement in accuracy by just a few Å. While an accurate overall fold of the native structure, based on the backbone Cα atoms, is often the main target of structure prediction, high-resolution refinement also comes with the expectation of generating accurate side chain orientations47 and a high stereochemical quality that is comparable to experimental structures48–50.
Template-based models already incorporate knowledge from existing structures. Therefore, structure refinement has to rely on alternative strategies51. A common idea is to use atomistic force fields in conjunction with energy minimization or more extensive sampling methods, in particular molecular dynamics (MD) simulations52. The resolution of atomistic force fields matches the target resolution of structure refinement. The hope is that conformational sampling can reach the native state as the state with the lowest free energy with no or few kinetic barriers since the initial model is already very close to the correct structure. Other strategies involve the targeted optimization of certain aspects of a given model53–55. Such an approach would be especially effective when there is knowledge about which parts of a given structure are least accurate and would benefit most from refinement. Highly successful structure refinement has been documented in anecdotal cases for a while28, 34, 36, 52, 56–60, but broadly useful strategies for structure refinement have only recently begun to emerge61–66. What seemed to be a relatively straightforward task, turned out to be exceedingly challenging. The objective of this review is to present the current state of the art and outline the significant challenges that remain.
The goal of structure refinement: How close is close enough?
Template-based models often correctly capture the overall fold of a given protein structure. On the other hand, effective atomic coordinate precisions as high as 0.2 Å provided by most crystal structures67 are not needed, for example, to determine which residues to mutate in biochemistry experiments that investigate a proposed biochemical mechanism. Therefore, it has to be established first what level of accuracy is necessary to obviate experimental structures before discussing how to reach such accuracy.
Mechanistic analyses
A primary use of high-resolution structures is to provide mechanistic insight that may require 1 Å resolution or better, especially when reaction mechanisms involving quantum mechanics would be applied68. Mechanistic analyses likely also require the accurate modelling of side chains and cofactors such as ions or ligands. However, as such questions are often focused on certain parts of a given structure, an accuracy better than 1 Å RMSD for all heavy atoms may not be needed across an entire structure for a model to be useful. Fortunately, the functionally most important regions of a given protein are also typically most conserved among homologues with the same function. Therefore, initial template-based models may already be highly accurate at mechanistic key sites and refinement methods may not need to improve those regions by much in order to reach experimental accuracy.
Prediction of binding partners
Protein structures serve as the starting point for a variety of further computational analyses. An important application is the identification of potential binding partners, such as proteins, ligands, and potential drug molecules69, 70. This type of analysis may be important for functional predictions but also during rational drug design applications. Previous studies have shown that homology models are often inferior to experimental structures for reliably predicting protein-protein interfaces71 and for generating docked conformations with small-molecule ligands72, 73. The docking of small molecules is expected to require high structural accuracy near the binding site and preferable models of holo structures since docking to apo structures may not be successful if, for example, binding sites are occluded74. Protein-protein interactions are sensitive to shape complementary over larger parts of a given protein surface. This may necessitate the accurate modelling of dynamic regions on the surface such as loops as they can alter the overall shape substantially if modelled incorrectly, even if such parts may otherwise not be functionally important75–77.
Biological relevance of experimental reference structures
The ultimate goal of structural prediction is to generate high-resolution models of proteins in their biologically most relevant conformation. In contrast, much of the assessment of structure prediction methods is driven by comparisons against experimental reference structures from X-ray crystallography or NMR, e.g. in the context of the biannual Critical Assesment of protein Structure Prediction (CASP) competitions25. Crystal structures may or may not exactly correspond to the biologically most relevant due to crystal contacts78 and other peculiarities of the artificial crystal environment that could have a significant effect on protein structures79. While it may be possible to consider the crystal environment during computational structure refinement, focusing refinement methods too much on exactly reproducing X-ray structures distracts from capturing what is biologically most relevant. Another related concern is that although crystallography has promoted a largely static view of structural biology, in reality, protein structures exhibit significant dynamics, especially at physiological temperatures80. Therefore, the most accurate representation of protein structures would be an ensemble spanning the native state with, for example, alternate conformations for flexible loops81. Computational refinement methods could in principle deliver such ensembles and comparisons could be made against crystallographic B-factors or NMR-derived ensembles. But a broader validation of how accurate computer-generated ensembles are could bemore challenging.
Structure refinement via minimization and molecular dynamics simulations
The use of force-field based energy minimization or MD simulation has long been a popular strategy for structure refinement following homology model generation82–90. The immediate benefit of force-field based refinement is the resolution of atomic clashes, deviations from standard bonding geometries, and other gross pathologies that may have been introduced as a result of using coarse-grained representations91 or the combination of templates from multiple structures92 during homology modeling. Distorted bond geometries and unfavorable atomic interactions correspond to high-energy states with atomistic force fields that can be quickly relieved when subjected to short optimization55, 83, 91, 93–95. On the other hand, the use of longer MD simulations for protein structure refinement (see Fig. 1, left side) has appeared as an obvious choice to achieve more extensive refinement without requiring any further knowledge or assumptions86, 96. Confidence in the ability of MD simulations to deliver on the promise of being able to refine approximate models stems from ample evidence that modern atomistic simulations typically maintain native conformations in close correspondence with experimental data97, 98 while also being able to fold peptides and small proteins via MD simulations given sufficient sampling16, 99–103. This suggests that MD simulations should in principle be able to accomplish refinement by sampling conformations on a downhill energy landscape towards the native state, at least to within the 1–3 A Cα RMSD deviations that are typically seen in MD simulations that are started from experimental structures104. The two main challenges in MD-based structure refinement are the accuracy of the model to ensure that the native state is indeed at the global minimum105 and the amount of conformational sampling that is needed to reach the native state from an initial model32. Refinement may be further complicated by a lack of a clear downhill funnel from near-native conformations to the native basin as a result of roughness in the energy landscape and/or force field inaccuracies when atomistic models are applied87, 106. Another challenge is the selection of a refined model from the conformational ensemble generated in MD simulations where the last conformation in a given run may not necessarily be the most native-like structure62. All of these issues are discussed in more detail in the following.
Figure 1:

Typical refinement protocols via MD-based sampling (left) and iterative structure optimization (right). Grey colors indicate optional elements. KB: Knowledge-based.
Force fields and solvation models
An accurate energy description is essential to distinguish the native state from similar, but less accurate initial models that may have been generated by template-based modeling. The first choice may be atomistic force fields that have improved significantly in accuracy over the last decade97, 98, 100, 107–111. Backbone torsion potentials, a key ingredient to correctly reproduce secondary structure propensities, have been fine-tuned to balance ab initio data with experimental data on small peptides and torsional distributions in structures from the Protein Data Bank (PDB)97, 107, 109. Other improvements have focused on side chain rotamer sampling and a better balance of solvation and salt-bridge interactions of charged and polar residues107, 112, 113. Most recently, the attention has shifted to the sampling of disordered regions as most available force fields have a tendency to over-stabilize ordered and compact states107, 114–117. While the sampling of disordered peptides is less relevant in the context of structure refinement, which generally targets well-folded proteins, a better balance between folded and disordered states may benefit the sampling of more dynamic regions such as longer loops that are part of many folded proteins. The most recent sets of force fields perform very well when folding model peptides and smaller proteins and there is evidence that force field improvements translate into better accuracy during structure refinement61, 87, 107, 118, 119. In addition to physics-based force fields, statistical potentials have also been used in MD- or minimization-based refinement methods56, 83, 120. Other efforts aim at specifically training potentials to deepen the energy of protein native states87, 119 and/or to penalize excursions to non-native states88.
As the inclusion of solvent is generally important in modeling biological systems, it is also key for successful refinement121. Atomistic force fields are typically meant to be combined with explicit water to account for solvation effects. However, the high cost of explicit solvent and requirements for extensive sampling demand significant computational resources. Therefore, implicit solvent methods have been used as a more efficient alternative in MD simulations122–125. Generalized Born models are especially attractive and have led to success in protein folding and refinement34, 35, 94, 120, 121, 126–136, but even simpler models such as a distant-dependent dielectric can be applicable during refinement85. However, implicit solvent models may have artifacts such as an overstabilization of salt-bridges and secondary structure elements that may require specially optimized force fields to be effective133, 137–140.
Coarse-grained models offer advantages over fully atomistic force fields141, 142 by reducing computational costs, and providing smoother energy landscapes that are helpful in navigating the transition from a slightly incorrect but already well-folded initial model to the native state. Although very coarse models, such as Cα-based protein models are problematic when applied to high-resolution structure refinement, moderately coarse-grained models such as UNRES143, 144 or PRIMO145, 146 can be suitable alternatives to the more expensive all-atom standard force field treatments during refinement85.
Conformational sampling strategies
In addition to the accuracy of the energy function, conformational sampling is essential to overcome the kinetic barriers during the transition from an initial model to the native state during refinement. Initial attempts at MD-based structure refinement quickly established that sub-nanosecond time scales are not sufficient52. Instead, much longer time scales on the order of 100 ns or beyond may be required to achieve any significant refinement at all36. Furthermore, it has been found that unrestrained sampling that is started from an initial template-based model often deviates quickly and sometimes quite significantly away from the native state rather than towards it58, 82. Even simulations on 100 μs time scales, when unrestrained, generally do not come back towards the native state32. Force field issues have been suggested as an obvious culprit32. Another argument is that the initial template-based models are overly compact so that the only way to refine further would be to first expand the structure away from the native state92. More generally, the idea is that homology models may not lie exactly on the folding funnel exhibiting and local defects such as mispacked side chains or incorrect secondary structures may only be resolvable on low free energy paths via partial unfolding and eventual refolding147. Since unfolding and refolding may take a very long time, this has led to a general practice of applying weak restraints with respect to a given initial model during refinement so that large structural drifts are prevented while still allowing some refinement towards the native state32, 34, 56, 61, 62, 89, 90, 95. Restraints are commonly implemented based on Cα RMSD but may alternatively encode pairwise distances within a given reference structure126. The application of restraints can be effective when the initial model is close to the native state, but this practice opposes larger conformational arrangements that are needed to refine models that are further away from the native state. In some refinement protocols, restraints are applied based on selected fragments rather than the entire structure106 or are limited to regions deemed least reliable59, 60, either based on previous knowledge59, 60 or quality assessment criteria95. There are also methods that combine multiple, possibly conflicting sets of restraints based on homologs89, 90 or other knowledge to maintain the conformational sampling close to the native state148.
MD simulations are often combined with enhanced sampling methods such as replica exchange (REXMD)149 or other related techniques150. In protein folding simulations, enhanced sampling techniques can accelerate in silico folding by several orders of magnitude151–156. Consequently, some of these methods have also been used in the context of refinement37, 58, 59, 120, 127, 128, 147, 157. Other strategies for navigating the high-dimensional space of atomistic models during refinement include normal mode (NMA) based sampling31, 41, 158, torsional dynamics58, 84, 126, 159, or multi-scale methods where coarse-grained and atomistic models are mixed85, 132, 160–165. Other methods sample conformations either via the direct replacement of fragments from a library, often via Monte Carlo (MC) sampling,53, 158, 166 or by guiding MD sampling by constraints from such fragments106. Thus, traditional force field based methods are effectively combined with knowledge-based (KB) approaches. In all cases, the general idea is to focus sampling on lower-resolution space to more efficiently overcome the major barriers during refinement. However, refinement either via directly enhancing sampling of atomistic models or by confining sampling to lower-resolution space has so far not quite provided the breakthrough one would hope for. The reason is probably that the accelerated sampling does not prevent structures drifting away from the native state if restraints are not applied157. On the other hand, the use of restraints largely cancels the advantage of enhanced sampling so that the effective performance becomes similar to what can be achieved with regular MD simulations.
Selecting refined structures from MD ensembles
MD simulations generate trajectories that follow the free energy landscape for a given system according to the force field used in the simulation. In the ideal case where the native structure indeed corresponds to the free energy minimum and the sampling is long enough to reach full convergence, the largest population of structures in the ensemble generated by MD would be expected to correspond to the native state. However, it is not guaranteed that any snapshot, and in particular the final structure from a long MD run, corresponds to the native state as there may be excursions to non-native states with energies similar to the native state. In practical scenarios, where sampling is more limited and/or the native state is only a metastable state with a given force field, there may be larger excursions for much of a given trajectory. Therefore, simply taking the final structure from an MD run is rarely the best choice for obtaining improved structures. One common approach to overcome this challenge is to treat the structural ensemble as a set of decoys and apply clustering and/or scoring methods to find the most native-like structures35, 94, 105, 126, 167–172. The generation of decoys followed by scoring is a widely applied strategy in structure prediction. However, during refinement, where the generated models are very similar to each other and to the native state, the limited accuracy and noise in typical scoring functions may cause high false positive rates35, 170, 173, 174. As a consequence, it is often very difficult to identify the most refined models, or even distinguish refined models from models that have become worse in snapshots from MD simulations31, 62. An interesting alternative to traditional scoring is to use the stability in MD simulations as a scoring function118. In a similar spirit, one may also score based on the distance by which models deviate from an initial model during refinement62 as structures that remain closer to an initial model generally appear to be closer to the native state.
A different strategy to selecting one or few structures is to consider larger subsets of conformations from an MD-generated ensemble and obtain a refined model via conformational averaging of those conformations61, 62, 66, 158. The subset may be determined via scoring as well but applying scoring functions as a filter is more robust than selecting one or few structures. In addition, the conformational averaging further reduces sensitivity to noise in the scoring function175. Conformational averaging of a structural ensemble generated via MD is also conceptually more consistent with how MD ensembles should be compared to experimental structures since experiments implicitly involve extensive ensemble- and time-averaging176. Indeed, subset averaging of MD simulations has contributed to the most robust success in structure refinement to date62, 175.
Structure refinement via structure optimization
While MD-based refinement typically relies on a physics-based force field to move an initial model towards the native state, an alternative approach is the targeted optimization of specific aspects of a given structure (see Fig. 1, right side), often via iterative protocols. In particular, the hydrogen bond network that is key for secondary structure formation may be examined and re-optimized55, 93, 106 or structural elements of a given model such as secondary structure elements or loops may be resampled extensively to find better conformations28, 58, 95, 118. The resampling of structural fragments using a fragment library as already mentioned above in the context of MD simulations106, 158, 166 would also fall into this category as such methods have the potential to re-optimize larger sections at once. While most optimization protocols involve conventional sampling or minimization using a suitable energy function, an alternative approach is the use of a constraint-based geometric technique where parts of a structure are iteratively pulled open and reannealed to mimic chaperon-induced unfolding and refolding147. As in the application of restraints, selective refinement could target only certain parts of a structure based on quality assessment criteria to determine which parts are most likely in need of refinement. Targeted optimization often involves the generation of decoys followed by subsequent scoring which would be subject to same challenges and uncertainties as the scoring of snapshots generated via full MD simulations118.
Certain structure quality criteria can be targeted via optimization without requiring knowledge of the native structure. An example is the MolProbity score177 that combines various stereochemical quality criteria such as the avoidance of atomic clashes, bond, angle, and torsion distributions consistent with statistics from known structures, favorable hydrogen bond patterns and other generic features of protein structures. Most optimization protocols focus at least in part on structure quality by improving side chain orientations, hydrogen bonding, and/or general stereochemical accuracy49, 83, 178. Clashes and major bond distortions are easily removed with standard force fields whereas hydrogen bonding geometries of donors and acceptors in close proximity can be optimized with KB functions106, 179. Since the local structure quality and overall accuracy of the fold, measured based on Cα coordinates, are only weakly correlated in typical template-based models49, the local quality can often be improved without altering the Cα coordinates49 and therefore quality optimization can be employed as a final step for example after MD-based refinement that would focus more on improving the overall fold49.
Performance of refinement methods
Until recently, successful structure refinement without the use of experimental data was limited to selected cases while overall consistency was lacking. Up until CASP8, the overall most consistent structure refinement method was essentially not to attempt refinement at all65. In CASP9, refinement methods started to eke out, on average, very slight improvements64. Beginning with CASP10, MD-based refinement became successful as a result of force field improvements, extensive sampling, and the application of ensemble averaging46, 63. The current state of the art is that almost any model can now be refined by a modest amount (on average by 1–3 units of the Global Distance Test (GDT) score187), with very few models becoming worse while some models may become significantly better (up to 10 GDT units; see the example for TR872 in Fig. 2). GDT captures the number of residues where Cα coordinates can be superimposed within four different RMSD cutoffs (1, 2, 4, 8 Å for GDT_TS and 0.5, 1, 2, 4 Å for GDT_HA) and essentially reflects how much of a given structure is accurate to within a few Å while being insensitive to incorrectly modeled parts. Improvements in GDT scores are consistent with the refinement of structural regions that are already fairly good in the initial model. In terms of RMSD, reliable refinement methods rarely exceed improvements by more than 1 Å. Since the overall RMSD is more sensitive to parts of a model that deviate significantly from the native structure, this suggests that consistent refinement of those regions is more difficult. The most successful refinement protocols (see Table 1) largely use MD or MC-type sampling methods and/or energy minimization with a combination of atomistic force fields and/or KB scoring terms, but alternative methods focusing on structure optimization can also perform similarly well. The use of restraints during the MD simulations is common but it limits more significant refinement, especially in more dynamic regions such as loops. Less conservative approaches that sample more aggressively and do not use restraints continue to result in remarkable successes (see for example the best predictions for the TR868 and TR870 CASP12 targets from the Wu and Baker groups in Fig. 2)29, 158. As these methods also generate many deteriorated models for other targets, a lack of consistency hinders practical applications, though. In particular, it remains difficult, despite much effort, to discriminate successful from unsuccessful cases158. Therefore, less conservative methods are only useful in cases where high failure rates can be tolerated, for example to improve success with molecular replacement during crystallographic refinement64, 188.
Figure 2:

Examples of successful protein structure refinement based on CASP12 targets (from CASP12 web site: http://www.predictioncenter.org/casp12/index.cgi). Experimental, initial, and refined structures are shown in red, green, and orange, respectively. RMSD values refer to Cα atom deviations. The GDT_HA score is explained in the text.
Table 1:
Selected Refinement Protocols and Web Services
| Name Reference | Method | Selection | Web server |
|---|---|---|---|
| KoBaMIN Chopra et al.83 | Minimization w/ KB force field | Minimized structure | http://csb.stanford.edu/kobamin/ |
| Carlsen & Røgen88 | Optimization with iteratively refined KB potential | Minimized structure | N/A |
| 3DRefineCheng et al.55, 180 | H-Bond Optimization Minimization w/physics & KB force field (MESHI178) | Minimized structure | http://sysbio.rnet.missouri.edu/3Drefine/ |
| Anton Shaw et al.32 | Very long MD (100 μs) | Force field/Cluster size | N/A |
| PREFMD Feig et al.61, 62, 66 | MD (μs scale) with restraints | Averaging over subset based on DFIRE181, RW+182, iRMSD62 | http://feig.bch.msu.edu/web/services/prefmd |
| Schröder et al.89, 90 | MD (ns scale) with restraints from coupled homologs | Final structure | N/A |
| Wu et al.29 | (REX)MD (μs scale) with optimized force field | Best RMSD | N/A |
| Honig & Mark120 | REXMD | Scoring with RAPDF183/DFIRE181 | N/A |
| Olson & Lee157 | REXMD | Scoring with force field, GOAP184, dDFIRE185 | N/A |
| GNEIMO Vaidehi et al.58, 84 | Torsional space REXMD | Force field | N/A |
| FG-MD Zhang et al.106 | Fragment-guided MD | KB scoring function | http://zhanglab.ccmb.med.umich.edu/FG-MD/ |
| GalaxyRefine Seok et al.47, 186 | Sidechain repacking Iterative MD (ps scale) | Final structure | http://galaxy.seoklab.org/refine |
| TIGRESS Floudas et al.126 | Constraint based sampling (torsion MD, Rosetta24, MD) | Selection based on machine learning | http://atlas.princeton.edu/refinement |
| Kosztin et al.118 | Targeted optimization (RosettaRelax24) and MD | MD-based stability | N/A |
| ROSETTA Baker et al.158 | Iterative relaxation MD-based sampling NMA-based sampling Fragment rebuilding | Clustering and averaging Scoring with GOAP184 | N/A |
| FRODA Ozkan et al.147 | Selective unfolding with geometric method, refolding with REXMD | Clustering and scoring with DFIRE181 | N/A |
| Skolnick et al.87 | REXMC (A-TASSER119) with optimized force field | Force field energy | N/A |
| locPREFMD Feig et al.49 | Iterative optimization targeting local geometry | MolProbity117 score | http://feig.bch.msu.edu/web/services/locprefmd |
Closer inspection of Fig. 2 reveals a general trend that refinement is most successful in regions where only minor rearrangements are necessary. Correct reorientations of helices appear to be especially successful. On the other hand, larger structural errors remain largely uncorrected or structures may even become worse in those areas after refinement. In other words, the most problematic parts of a given model that would benefit most from refinement remain also the hardest parts to improve. How to improve those parts of an initial model will be the main challenge going forward towards achieving near-experimental accuracy.
Structure refinement via MD can incur significant computational costs as the best current protocols apply sampling from tens of nanoseconds to microsecond scales. Although very long MD simulations do not necessarily lead to better refinement32, even moderate-length MD simulations require significant resources. This has limited broad adoption of MD-based refinement as part of standard structure prediction pipelines. Although refinement web servers have become available, they tend to offer only a limited amount of refinement55, 83, 180, 186. It will require additional efforts and resources to focus on optimizing computational efficiency for MD-based methods so that more significant refinement via web-based services can benefit the broader community.
Related to the development of computationally efficient refinement protocols is the question of how to tell when sufficient refinement has been achieved. In principle, successful refinement methods should continue refinement until the native structure is reached and at that point there should be no further structural changes. However, the lack of structural variation during continued application of a given refinement protocol could also simply reflect being stuck in a non-native metastable state. How to distinguish those two is so far an unresolved problem.
The practical utility of refinement methods would benefit from knowledge about what kind of initial models are most likely to improve upon refinement. One question is how refinement success depends on the initial model, both in terms of distance from the native structure and how it was generated. Analysis from recent rounds of CASP suggests that models that are closer to the native structure may be generally more amenable to refinement46, 63, 66. This may be useful information since the quality of initial models can be assessed with some degree of certainty189. Another question is how the method used for initial model generation affects refinement. Analysis based on CASP results has offered some insights66, but as prediction methods used in CASP often involve convoluted pipelines and may include refinement already as part of their modeling protocol, it is difficult to arrive at clear conclusions.
While the refinement of the overall protein fold remains a significant challenge, at least the improvement of the local stereochemical quality has turned out to be more straightforward49, 83, 106. Generally, the optimization of force field and/or KB terms with iterative protocols that target a reduction in quality scores such as MolProbity177 are sufficient and perform well. For example, the locPREFMD method is able to improve MolProbity scores for almost any starting structure to values below 1–1.5, which is comparable to experimental structures. Although locPREFMD uses MD simulations as part of the protocol, simulations are short and the resulting stereochemical refinement can be completed on moderate resources within minutes49.
Conclusions
Computational protein structure refinement aims at turning approximate initial models, which can be generated today for many genes where no experimental structures exist, into structures with accuracy and quality that is comparable to experimental structures. While that remains a significant challenge, recent successes, especially with protocols based on MD simulations that can deliver modest but consistent refinement, are beginning to point in the right direction. Such MD-based methods may become part of standard prediction pipelines to improve template-based models. Moreover, methods that target local structural quality have become available to apply finishing touches to generate models that satisfy most quality criteria expected from experimental structures. However, as the overall degree of refinement with the best methods still remains modest on average, the refinement problem is far from solved.
Going forward, it appears that the ever-recurring issues of force field and scoring function accuracy on one side and sampling insufficiency on the other side still need to be fully addressed. Current force fields are much improved, but they are far from perfect and the most successful refinement protocols combine standard atomistic force fields with KB functions in some form or fashion. One underappreciated aspect involves polarization effects that are difficult to capture with fixed charge force fields and that have limited for example the accurate description of hydrogen bonding179, 190. It will, therefore, be interesting to see how much can be gained with polarizable force fields191 in the context of structure refinement. However, sampling is probably still the more significant challenge. Having to use restraints to obtain consistent refinement is a major impediment towards more significant refinement and the key to developing more effective refinement methods will be how to prevent initial models from deviating too far from initial models while still allowing refinement by several Å RMSD. One idea to overcome this conundrum may be the simultaneous use of multiple restraints148, 192 and/or adaptive restraints15, 89, 90 to limit sampling while still being able to reach the native state. Another successful path may be to continue to explore strategies that explicitly follow an unfolding/refolding scheme147 as that more likely mimics what nature would do to reach the native state from misfolded states via chaperones193.
As refinement methods become better at reaching experimental accuracy, the exact reproduction of experimental conditions will become increasingly important. Current methods generally do not fully consider ionization equilibria of titratable amino acid side chains, the potential effect of ligands and ions, and the structural constraints imposed by crystal packing for structures solved by crystallography. There is a danger, however, to overemphasize single structures under very specific, possibly artificial conditions, whereas the ultimate goal should be to produce structural ensembles that are representative of the biological context including the crowded conditions of the cellular environment194, 195.
Finally, another underdeveloped direction is the prediction and refinement of membrane protein structures196–198. It may be that similar strategies as for soluble proteins will be successful but limited assessment, during CASP for example, has effectively discouraged efforts by the community to date. As a consequence, it is much less clear, for example, how well scoring functions work for membrane protein structures199, 200 and how the sampling challenges present for soluble proteins translate to membrane proteins201.
Footnotes
The author declares no competing interests.
References
- 1.Perutz MF, Rossmann MG, Cullis AF, Muirhead H, Will G, North ACT. Structure of Haemoglobin - 3-dimensional Fourier Synthesis at 5.5-Å Resolution, Obtained by X-ray Analysis. Nature 1960, 185:416–422. [DOI] [PubMed] [Google Scholar]
- 2.Kendrew JC, Bodo G, Dintzis HM, Parrish RG, Wyckoff H, Phillips DC. 3-dimensional Model of the Myoglobin Molecule Obtained by X-ray Analysis. Nature 1958, 181:662–666. [DOI] [PubMed] [Google Scholar]
- 3.Whisstock JC, Lesk AM. Prediction of protein function from protein sequence and structure. Q. Rev. Biophys. 2003, 36:307–340. [DOI] [PubMed] [Google Scholar]
- 4.Gane PJ, Dean PM. Recent Advances in structure-based rational drug design. Curr. Opin. Struct. Biol. 2000, 10:401–404. [DOI] [PubMed] [Google Scholar]
- 5.Lutz S Beyond directed evolution - semi-rational protein engineering and design. Curr Opin Biotech 2010, 21:734–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durbin RM. A map of human genome variation from population-scale sequencing. Nature 2010, 467:1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rose PW, Prlić A, Altunkaya A, Bi C, Bradley AR, Christie CH, Costanzo LD, Duarte JM, Dutta S, Feng Z, et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2016, 45:D271–D281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levitt M Protein Conformation, Dynamics, and Folding by Computer-Simulation. Annu. Rev. Biophys. Bioeng. 1982, 11: 251–271. [DOI] [PubMed] [Google Scholar]
- 9.Garnier J Protein-Structure Prediction. Biochimie 1990, 72:513–524. [DOI] [PubMed] [Google Scholar]
- 10.Liwo A, Lee J, Ripoll DR, Pillardy J, Scheraga HA. Protein structure prediction by global optimization of a potential energy function. Proc. Natl. Acad. Sci. U.S.A 1999, 96:5482–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardin C, Pogorelov TV, Luthey-Schulten Z. Ab initio protein structure prediction. Curr. Opin. Struct. Biol 2002, 12:176–181. [DOI] [PubMed] [Google Scholar]
- 12.Levitt M Simplified Representation of Protein Conformations for Rapid Simulation of Protein Folding. J. Mol. Biol 1976, 104:59–107. [DOI] [PubMed] [Google Scholar]
- 13.Dill KA, Bromberg S, Yue KZ, Fiebig KM, Yee DP, Thomas PD, Chan HS. Principles of Protein-Folding - a Perspective from Simple Exact Models. Protein Sci. 1995, 4:561–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shakhnovich E Protein folding thermodynamics and dynamics: Where physics, chemistry, and biology meet. Chem. Rev 2006, 106:1559–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonneau R, Baker D. Ab Initio Protein Structure Prediction: Progress and Prospects. Annu. Rev. Biophys. Biomol. Struct 2001, 30:173–189. [DOI] [PubMed] [Google Scholar]
- 16.Simmerling C, Strockbine B, Roitberg AE. All-atom structure prediction and folding simulations of a stable protein. J. Am. Chem. Soc 2002, 124:11258–11259. [DOI] [PubMed] [Google Scholar]
- 17.Baker D, Sali A. Protein structure prediction and structural genomics. Science 2001, 294:93–96. [DOI] [PubMed] [Google Scholar]
- 18.Al-Lazikani B, Jung J, Xiang ZX, Honig B. Protein structure prediction. Curr. Opin. Chem. Biol 2001, 5:51–56. [DOI] [PubMed] [Google Scholar]
- 19.Blundell TL, Sibanda BL, Sternberg MJE, Thornton JM. Knowledge-Based Prediction of Protein Structures and the Design of Novel Molecules. Nature 1987, 326:347–352. [DOI] [PubMed] [Google Scholar]
- 20.Dudek MJ, Ramnarayan K, Ponder JW. Protein Structure Prediction Using a Combination of Sequence Homology and Global Energy Minimization: II. Energy Functions. J. Comput. Chem 1998, 19:548–573. [Google Scholar]
- 21.Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol 1993, 234:779–815. [DOI] [PubMed] [Google Scholar]
- 22.Xu D, Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 2012, 80:1715–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Arakaki AK, Skolnick JR. TASSER: An automated method for the prediction of protein tertiary structures in CASP6. Proteins 2005, 61: 91–98. [DOI] [PubMed] [Google Scholar]
- 24.Rohl CA, Strauss CE, Misura KM, Baker D. Protein Structure Prediction Using Rosetta. Methods Enzymol 2004, 383:66–93. [DOI] [PubMed] [Google Scholar]
- 25.Kryshtafovych A, Fidelis K, Moult J. CASP10 results compared to those of previous CASP experiments. Proteins 2014, 82:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moult J, Fidelis K, Kryshtafovych A, Schwede T, Tramontano A. Critical assessment of methods of protein structure prediction: Progress and new directions in round XI. Proteins 2016, 84 (Suppl 1):4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Zhang W, He B, Walker SE, Zhang H, Govindarajoo B, Virtanen J, Xue Z, Shen H-B, Zhang Y. Template-based protein structure prediction in CASP11 and retrospect of I-TASSER in the last decade. Proteins 2016, 84 (Suppl. 1):233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu J, Xie L, Honig B. Structural Refinement of Protein Segments Containing Secondary Structure Elements: Local Sampling, Knowledge-Based Potentials, and Clustering . Proteins 2006, 65: 463–479. [DOI] [PubMed] [Google Scholar]
- 29.Xun S, Jiang F, Wu Y-D. Significant Refinement of Protein Structure Models Using a Residue-Specific Force Field. J. Chem. Theory Comput 2015, 11:1949–1956. [DOI] [PubMed] [Google Scholar]
- 30.Verma A, Wenzel W. Protein structure prediction by all-atom free-energy refinement. BMC Struct. Biol 2007, 7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stumpff-Kane AW, Maksimiak K, Lee MS, Feig M. Sampling of Near-Native Protein Conformations during Protein Structure Refinement Using a Coarse-Grained Model, Normal Modes, and Molecular Dynamics Simulations. Proteins 2008, 70:1345–1356. [DOI] [PubMed] [Google Scholar]
- 32.Raval A, Piana S, Eastwood MP, Dror RO, Shaw DE. Refinement of protein structure homology models via long, all-atom molecular dynamics simulations. Proteins 2012, 80:2071–2079. [DOI] [PubMed] [Google Scholar]
- 33.Misura KMS, Baker D. Progress and Challenges in High-Resolution Refinement of Protein Structure Models. Proteins 2005, 2005:15–29. [DOI] [PubMed] [Google Scholar]
- 34.Chen J, Brooks CL III Can Molecular Dynamics Simulations Provide High-Resolution Refinement of Protein Structure? Proteins 2007, 67:922–930. [DOI] [PubMed] [Google Scholar]
- 35.Lee MS, Olson MA. Assessment of detection and refinement strategies for de novo protein structures using force field and statistical potentials. J. Chem. Theory Comput 2007, 3:312–324. [DOI] [PubMed] [Google Scholar]
- 36.Fan H, Mark AE. Refinement of homology-based protein structures by molecular dynamics simulation techniques. Protein Sci. 2004, 13:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen JH, Im W, Brooks CL. Refinement of NMR structures using implicit solvent and advanced sampling techniques. J. Am. Chem. Soc 2004, 126:16038–16047. [DOI] [PubMed] [Google Scholar]
- 38.Lee SY, Zhang Y, Skolnick J. TASSER-based refinement of NMR structures. Proteins 2006, 63:451–456. [DOI] [PubMed] [Google Scholar]
- 39.Raves ML, Doreleijers JF, Vis H, Vorgias CE, Wilson KS, Kaptein R. Joint refinement as a tool for thorough comparison between NMR and X-ray data and structures of HU protein. J. Biomol. NMR 2001, 21:235–248. [DOI] [PubMed] [Google Scholar]
- 40.Adams PD, Pannu NS, Brünger AT. Cross-Validated Maximum Likelihood Enhances Crystallographic Simulated Annealing Refinement. Proc. Natl. Acad. Sci. U.S.A 1997, 94:5018–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kidera A, Go N. Refinement of Protein Dynamic Structure - Normal Mode Refinement. Proc. Natl. Acad. Sci. U.S.A 1990, 87:3718–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Topf M, Baker ML, Marti-Renom MA, Chiu W, Sali A. Refinement of protein structures by iterative comparative modeling and cryoEM density fitting. J. Mol. Biol 2006, 357:1655–1668. [DOI] [PubMed] [Google Scholar]
- 43.Grishaev A, Ying J, Canny MD, Pardi A, Bax A. Solution structure of tRNAVal from refinement of homology model against residual dipolar coupling and SAXS data. J. Biomol. NMR 2008, 42:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rozycki B, Kim YC, Hummer G. SAXS Ensemble Refinement of ESCRT-III CHMP3 Conformational Transitions. Structure 2010, 19:109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rozbesky D, Man P, Kavan D, Chmelik J, Cerny J, Bezouska K, Novak P. Chemical Cross-Linking and H/D Exchange for Fast Refinement of Protein Crystal Structure. Anal. Chem 2012, 84:867–870. [DOI] [PubMed] [Google Scholar]
- 46.Modi V, Dunbrack RLJ. Assessment of refinement of template-based models in CASP11. Proteins 2016, 84 (Suppl. 1):260–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heo L, Park H, Seok C. GalaxyRefine: Protein Structure Refinement Driven by Side-Chain Repacking. Nucleic Acids Res. 2013, 41:W384–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Headd JJ, Immormino RM, Keedy DA, Emsley P, Richardson DC, Richardson JS. Autofix for Backward-Fit Sidechains: Using MolProbity and Real-Space Refinement to Put Misfits in Their Place. J. Struct. Funct. Genomics 2009, 10:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feig M Local Protein Structure Refinement via Molecular Dynamics Simulation with locPREFMD. J. Chem. Inf. Model 2016, 56:1304–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Cryst 1993, 26:283–291. [Google Scholar]
- 51.Gront D, Kmiecik S, Blaszczyk M, Ekonomiuk D, Kolinski A. Optimization of protein models. Wiley Interdiscp. Rev.: Comput. Mol. Sci 2012, 2:479–493. [Google Scholar]
- 52.Lee MR, Tsai J, Baker D, Kollman PA. Molecular Dynamics in the Endgame of Protein Structure Prediction. J. Mol. Biol 2001, 2001:417–430. [DOI] [PubMed] [Google Scholar]
- 53.Park H, Seok C. Refinement of unreliable local regions in template-based protein models. Proteins 2012, 80:1974–1986. [DOI] [PubMed] [Google Scholar]
- 54.Park H, Ko J, Joo K, Lee J, Seok C, Lee J. Refinement of protein termini in template-based modeling using conformational space annealing. Proteins 2011, 79:2725–2734. [DOI] [PubMed] [Google Scholar]
- 55.Bhattacharya D, Cheng JL. 3Drefine: Consistent protein structure refinement by optimizing hydrogen bonding network and atomic-level energy minimization. Proteins 2013, 81:119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu H, Skolnick J. Application of statistical potentials to protein structure refinement from low resolution Ab initio models. Biopolymers 2003, 70:575–584. [DOI] [PubMed] [Google Scholar]
- 57.Jonassen I, Klose D, Taylor WR. Protein model refinement using structural fragment tessellation. Comp. Biol. Chem 2006, 30:360–366. [DOI] [PubMed] [Google Scholar]
- 58.Park IH, Gangupomu V, Wagner J, Jain A, Vaidehi N. Structure Refinement of Protein Low Resolution Models Using the GNEIMO Constrained Dynamics Method. J. Phys. Chem. B 2012, 116:2365–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cao W, Terada T, Nakamura S, Shimizu K. Refinement of Comparative-Modeling Structures by Multicanonical Molecular Dynamics. Genome Inform. 2003, 14:484–485. [Google Scholar]
- 60.Ishitani R, Terada T, Shimizu K. Refinement of comparative models of protein structure by using multicanonical molecular dynamics simulations. Mol. Simul 2008, 34:327–336. [Google Scholar]
- 61.Mirjalili V, Noyes K, Feig M. Physics-Based Protein Structure Refinement through Multiple Molecular Dynamics Trajectories and Structure Averaging. Proteins 2014, 82:196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mirjalili V, Feig M. Protein Structure Refinement through Structure Selection and Averaging from Molecular Dynamics Ensembles. J. Chem. Theory Comput 2013, 9:1294–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nugent T, Cozzetto D, Jones DT. Evaluation of predictions in the CASP10 model refinement category. Proteins 2014, 82:98–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.MacCallum JL, Perez A, Schnieders MJ, Hua L, Jacobson MP, Dill KA. Assessment of protein structure refinement in CASP9. Proteins 2011, 79:74–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.MacCallum JL, Hua L, Schnieders MJ, Pande VS, Jacobson MP, Dill KA. Assessment of the protein-structure refinement category in CASP8. Proteins 2009, 77:66–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feig M, Mirjalili V. Protein Structure Refinement via Molecular-Dynamics Simulations: What Works and What Does Not? Proteins 2016, 84 (Suppl. 1):282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cruickshank DWJ. Remarks about protein structure precision. Acta Crystallogr. D 1999, 55:583–601. [DOI] [PubMed] [Google Scholar]
- 68.Himo F Quantum chemical modeling of enzyme active sites and reaction mechanisms. Theor. Chem. Acc 2006, 116:232–240. [Google Scholar]
- 69.Cavasotto CN. Homology Models in Docking and High-Throughput Docking. Curr. Top. Med. Chem 2011, 11:1528–1534. [DOI] [PubMed] [Google Scholar]
- 70.Capriotti E Comparative modeling and structure prediction: application to drug discovery In: Lill MA, ed. In Silico Drug Discovery and Design. London: Future Medicine; 2013, 34–48. [Google Scholar]
- 71.Vajda S, Kozakov D. Convergence and combination of methods in protein-protein docking. Curr. Opin. Struct. Biol 2009, 19:164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wieman H, Tondel K, Anderssen E, Drablos F. Homology-based modelling of targets for rational drug design. Mini. Rev. Med. Chem 2004, 4:793–804. [PubMed] [Google Scholar]
- 73.Bordogna A, Pandini A, Bonati L. Predicting the accuracy of protein-ligand docking on homology models. J. Comput. Chem 2011, 32:81–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Du H, Brender JR, Zhang J, Zhang Y. Protein structure prediction provides comparable performance to crystallographic structures in docking-based virtual screening. Methods 2015, 71:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang C, Bradley P, Baker D. Protein-Protein Docking with Backbone Flexibility. J. Mol. Biol 2007, 373:503–519. [DOI] [PubMed] [Google Scholar]
- 76.Bonvin AMJJ. Flexible protein-protein docking. Curr. Opin. Struct. Biol 2006, 16:194–200. [DOI] [PubMed] [Google Scholar]
- 77.Bastard K, Prévost C, Zacharias M. Accounting for loop flexibility during protein-protein docking. Proteins 2006, 62:956–969. [DOI] [PubMed] [Google Scholar]
- 78.Janin J, Rodier F. Protein-protein interaction at crystal contacts. Proteins 1995, 23:580–587. [DOI] [PubMed] [Google Scholar]
- 79.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol 2002, 320:597–608. [DOI] [PubMed] [Google Scholar]
- 80.Kundu S, Melton JS, Sorensen DC, Phillips Jr. GN. Dynamics of Proteins in Crystals: Comparison of Experiment with Simple Models. Biophys. J 2002, 83:723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Burnley BT, Afonine PV, Adams PD, Gros P. Modelling dynamics in protein crystal structures by ensemble refinement. eLife 2012, 1:e00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Summa CM, Levitt M. Near-native structure refinement using in vacuo energy minimization. Proc. Natl. Acad. Sci. U.S.A 2007, 104:3177–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rodrigues JPGLM, Levitt M, Chopra G. KoBaMIN: a knowledge-based minimization web server for protein structure refinement. Nucleic Acids Res. 2012, 40:W323–W328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Larsen AB, Wagner JR, Jain A, Vaidehi N. Protein Structure Refinement of CASP Target Proteins Using GNEIMO Torsional Dynamics Method. J. Chem. Inf. Model 2014, 54:508–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Feig M, Gopal SM, Vadivel K, Stumpff-Kane AW. Conformational sampling in structure prediction and refinement with atomistic and coarse-grained models In: Kolinski A, ed. Multiscale approaches to protein modeling: Structure prediction, dynamics, thermodynamics and macromolecular assemblies. New York City: Springer; 2010. [Google Scholar]
- 86.Engh RA, Huber R. Accurate Bond and Angle Parameters for X-Ray Protein-Structure Refinement. Acta Crystallogr. A 1991, 47:392–400. [Google Scholar]
- 87.Jagielska A, Wroblewska L, Skolnick J. Protein model refinement using an optimized physics-based all-atom force field. Proc. Natl. Acad. Sci. U.S.A 2008, 105:8268–8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carlsen M, Rogen P. Protein structure refinement by optimization. Proteins 2015, 83:1616–1624. [DOI] [PubMed] [Google Scholar]
- 89.Della Corte D, Wildberg A, Schröder GF. Protein structure refinement with adaptively restrained homologous replicas. Proteins 2016, 84 (Suppl. 1):302–313. [DOI] [PubMed] [Google Scholar]
- 90.Wildberg A, Della Corte D, Schroder GF. Coupling an Ensemble of Homologues Improves Refinement of Protein Homology Models. J. Chem. Theory Comput 2015, 11:5578–5582. [DOI] [PubMed] [Google Scholar]
- 91.Kmiecik S, Gront D, Kolinski A. Towards the high-resolution protein structure prediction. Fast refinement of reduced models with all-atom force field. BMC Struct. Biol 2007, 7:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Y Protein structure prediction: when is it useful? Curr. Opin. Struct. Biol 2009, 19:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bhattacharya D, Cheng JL. i3Drefine Software for Protein 3D Structure Refinement and Its Assessment in CASP10. Plos One 2013, 8:e69648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lin MS, Head-Gordon T. Reliable Protein Structure Refinement Using a Physical Energy Function. J. Comput. Chem 2011, 32:709–717. [DOI] [PubMed] [Google Scholar]
- 95.Lee GR, Heo L, Seok C. Effective protein model structure refinement by loop modeling and overall relaxation. Proteins 2016, 84 (Suppl. 1):293–301. [DOI] [PubMed] [Google Scholar]
- 96.Hospital A, Goñi JR, Orozco M, Gelpi JL. Molecular dynamics simulations: advances and applications. Adv. Appl. Bioinform. Chem 2015, 8:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Best RB, Zhu X, Shim J, Lopes P, Mittal J, Feig M, MacKerell AD Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput 2012, 8:3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lindorff-Larsen K, Maragakis P, Piana S, Eastwood MP, Dror RO, Shaw DE. Systematic Validation of Protein Force Fields against Experimental Data. Plos One 2012, 7:e32131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. How Fast-Folding Proteins Fold. Science 2011, 334:517–520. [DOI] [PubMed] [Google Scholar]
- 100.Best RB, Mittal J, Feig M, MacKerell AD. Inclusion of Many-Body Effects in the Additive CHARMM Protein CMAP Potential Results in Enhanced Cooperativity of α-Helix and β-Hairpin Formation. Biophys. J 2012, 103:1045–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lane TJ, Shukla D, Beauchamp KA, Pande VS. To milliseconds and beyond: challenges in the simulation of protein folding. Curr. Opin. Struct. Biol 2013, 23:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Perez A, Morrone JA, Simmerling C, Dill KA. Advances in free-energy-based simulations of protein folding and ligand binding. Curr. Opin. Struct. Biol 2016, 36:25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee MR, Baker D, Kollman PA. 2.1 and 1.8 Å Average Cα RMSD Structure Predictions on Two Small Proteins, HP-36 and S15. J. Am. Chem. Soc 2001, 123:1040–1046. [DOI] [PubMed] [Google Scholar]
- 104.van der Kamp MW, Schaeffer RD, Jonsson AL, Scouras AD, Simms AM, Toofanny RD, Benson NC, Anderson PC, Merkley ED, Rysavy S, et al. Dynameomics: A Comprehensive Database of Protein Dynamics. Structure 2010, 18:423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wroblewska L, Skolnick J. Can a physics-based, all-atom potential find a protein’s native structure among misfolded structures? I. Large scale AMBER benchmarking. J. Comput. Chem 2007, 28:2059–2066. [DOI] [PubMed] [Google Scholar]
- 106.Zhang J, Liang Y, Zhang Y. Atomic-Level Protein Structure Refinement Using Fragment-Guided Molecular Dynamics Conformation Sampling. Structure 2011, 19:1784–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, MacKerell AD Jr. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14:71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cino EA, Choy WY, Karttunen M. Comparison of Secondary Structure Formation Using 10 Different Force Fields in Microsecond Molecular Dynamics Simulations. J. Chem. Theory Comput 2012, 8:2725–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11:3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou CY, Jiang F, Wu YD. Residue-Specific Force Field Based on Protein Coil Library. RSFF2: Modification of AMBER ff99SB. J. Phys. Chem. B 2015, 119:1035–1047. [DOI] [PubMed] [Google Scholar]
- 111.Mackerell AD. Empirical force fields for biological macromolecules: Overview and issues. J. Comput. Chem 2004, 25:1584–1604. [DOI] [PubMed] [Google Scholar]
- 112.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78:1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Oostenbrink C, Villa A, Mark AE, van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem 2004, 25:1656–1676. [DOI] [PubMed] [Google Scholar]
- 114.Fluitt AM, de Pablo JJ. An Analysis of Biomolecular Force Fields for Simulations of Polyglutamine in Solution. Biophys. J 2015, 109:1009–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lindorff-Larsen K, Trbovic N, Maragakis P, Piana S, Shaw DE. Structure and Dynamics of an Unfolded Protein Examined by Molecular Dynamics Simulation. J. Am. Chem. Soc 2012, 134:3787–3791. [DOI] [PubMed] [Google Scholar]
- 116.Best RB, Zheng W, Mittal J. Balanced Protein-Water Interactions Improve Properties of Disorderd Proteins and Non-Specific Protein Association. J. Chem. Theory Comput 2014, 10:5113–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Piana S, Donchev AG, Robustelli P, Shaw DE. Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119:5113–5123. [DOI] [PubMed] [Google Scholar]
- 118.Zhang J, Barz B, Zhang JF, Xu D, Kosztin I. Selective refinement and selection of near-native models in protein structure prediction. Proteins 2015, 83:1823–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wroblewska L, Jagielska A, Skolnick J. Development of a physics-based force field for the scoring and refinement of protein models. Biophys. J 2008, 94:3227–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhu J, Fan H, Periole X, Honig B, Mark AE. Refining homology models by combining replica-exchange molecular dynamics and statistical potentials. Proteins 2008, 72:1171–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chopra G, Summa CM, Levitt M. Solvent dramatically affects protein structure refinement. Proc. Natl. Acad. Sci. U.S.A 2008, 105:20239–20244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Krol M Comparison of various implicit solvent models in molecular dynamics simulations of immunoglobulin G light chain dimer. J. Comput. Chem 2003, 24:531–546. [DOI] [PubMed] [Google Scholar]
- 123.Roux B, Simonson T. Implicit Solvent Models. Biophys. Chem 1999, 78:1–20. [DOI] [PubMed] [Google Scholar]
- 124.Feig M, Brooks CL III. Recent advances in the development and application of implicit solvent models in biomolecule simulations. Curr. Opin. Struct. Biol 2004, 14:217–224. [DOI] [PubMed] [Google Scholar]
- 125.Feig M, Chocholousova J, Tanizaki S. Extending the horizon: towards the efficient modeling of large biomolecular complexes in atomic detail. Theor. Chem. Acc 2006, 116:194–205. [Google Scholar]
- 126.Khoury GA, Tamamis P, Pinnaduwage N, Smadbeck J, Kieslich CA, Floudas CA. Princeton_TIGRESS: Protein geometry refinement using simulations and support vector machines. Proteins 2014, 82:794–814. [DOI] [PubMed] [Google Scholar]
- 127.Olson MA, Lee MS. Structure refinement of protein model decoys requires accurate side-chain placement. Proteins 2013, 81:469–478. [DOI] [PubMed] [Google Scholar]
- 128.Olson MA, Chaudhury S, Lee MS. Comparison Between Self-Guided Langevin Dynamics and Molecular Dynamics Simulations for Structure Refinement of Protein Loop Conformations. J. Comput. Chem 2011, 32:3014–3022. [DOI] [PubMed] [Google Scholar]
- 129.Lee MS, Olson MA. Protein Folding Simulations Combining Self-Guided Langevin Dynamics and Temperature-Based Replica Exchange. J. Chem. Theory Comput 2010, 6:2477–2487. [DOI] [PubMed] [Google Scholar]
- 130.Vitalis A, Pappu RV. ABSINTH: A New Continuum Solvation Model for Simulations of Polypeptides in Aqueous Solutions. J. Comput. Chem 2009, 30:673–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Felts AK, Gallicchio E, Chekmarev D, Paris KA, Friesner RA, Levy RM. Prediction of protein loop conformations using the AGBNP implicit solvent model and torsion angle sampling. J. Chem. Theory Comput 2008, 4:855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Olson MA, Feig M, Brooks CL. Prediction of protein loop conformations using multiscale Modeling methods with physical energy scoring functions. J. Comput. Chem 2008, 29:820–831. [DOI] [PubMed] [Google Scholar]
- 133.Zhu J, Alexov E, Honig B. Comparative study of generalized Born models: Born radii and peptide folding. J. Phys. Chem. B 2005, 109:3008–3022. [DOI] [PubMed] [Google Scholar]
- 134.Nymeyer H, Garcia AE. Simulation of the folding equilibrium of α-helical peptides: A comparison of the generalized Born approximation with explicit solvent. Proc. Natl. Acad. Sci. U.S.A 2003, 100:13934–13939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhou RH. Free energy landscape of protein folding in water: Explicit vs. implicit solvent. Proteins 2003, 53:148–161. [DOI] [PubMed] [Google Scholar]
- 136.Ozkan SB, Wu GA, Chodera JD, Dill KA. Protein folding by zipping and assembly. Proc. Natl. Acad. Sci. U.S.A 2007, 104:11987–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen JH, Brooks CL, Khandogin J. Recent advances in implicit solvent-based methods for biomolecular simulations. Curr. Opin. Struct. Biol 2008, 18:140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chen JH, Im WP, Brooks CL. Balancing solvation and intramolecular interactions: Toward a consistent generalized born force field. J. Am. Chem. Soc 2006, 128:3728–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55:383–394. [DOI] [PubMed] [Google Scholar]
- 140. Roe DR, Okur A, Wickstrom L, Hornak V, Simmerling C. Secondary structure bias in generalized born solvent models: Comparison of conformational ensembles and free energy of solvent polarization from explicit and implicit solvation. J. Phys. Chem. B 2007, 111:1846–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tozzini V Coarse-grained models for proteins. Curr. Opin. Struct. Biol 2005, 15:144–150. [DOI] [PubMed] [Google Scholar]
- 142.Kar P, Feig M. Recent Advances in Transferable Coarse-Grained Modeling of Proteins. Adv. Prot. Chem. Struct. Biol 2014, 96:143–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Krupa P, Mozolewska MA, Wisniewska M, Yin YP, He Y, Sieradzan AK, Ganzynkowicz R, Lipska AG, Karczynska A, Slusarz M, et al. Performance of protein-structure predictions with the physics-based UNRES force field in CASP11. Bioinformatics 2016, 32: 3270–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lee J, Liwo A, Scheraga HA. Energy-Based de novo Protein Folding by Conformational Space Annealing and an Off-Lattice United-Residue Force Field: Application to the 10–55 Fragment of Staphylococcal Protein A and to apo calbindin D9K. Proc. Natl. Acad. Sci. U.S.A 1999, 96:2055–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gopal SM, Mukherjee S, Cheng Y-M, Feig M. PRIMO/PRIMONA: A coarse-grained model for proteins and nucleic acids that preserves near-atomistic accuracy. Proteins 2010, 78:1266–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kar P, Gopal SM, Cheng YM, Predeus AV, Feig M. PRIMO: A Transferable Coarse-Grained Force Field for Proteins. J. Chem. Theory Comput 2013, 9:3769–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kumar A, Campitelli P, Thorpe MF, Ozkan SB. Partial unfolding and refolding for structure refinement: A unified approach of geometric simulations and molecular dynamics. Proteins 2015, 83:2279–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Perez A, Morrone JA, Brini E, MacCallum JL, Dill KA. Blind protein structure prediction using accelerated free-energy simulations. Sci. Adv 2016, 2:e1601274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314:141–151. [Google Scholar]
- 150.Abrams C, Bussi G. Enhanced Sampling in Molecular Dynamics Using Metadynamics, Replica-Exchange, and Temperature-Acceleration. Entropy 2014, 16:163–199. [Google Scholar]
- 151.Rao F, Caflisch A. Replica exchange molecular dynamics simulations of reversible folding. J. Chem. Phys. 2003, 119:4035–4042. [DOI] [PubMed] [Google Scholar]
- 152.Rhee YM, Pande VS. Multiplexed-Replica Exchange Molecular Dynamics Method for Protein Folding Simulation. Biophys. J 2003, 84:775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Paschek D, Gnanakaran S, Garcia AE. Simulations of the pressure and temperature unfolding of an alpha-helical peptide. Proc. Natl. Acad. Sci. U.S.A 2005, 102:6765–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Baumketner A, Shea JE. The thermodynamics of folding of a beta hairpin peptide probed through replica exchange molecular dynamics simulations. Theor. Chem. Acc 2006, 116:262–273. [Google Scholar]
- 155.Paschek D, Nymeyer H, Garcia AE. Replica exchange simulation of reversible folding/unfolding of the Trp-cage miniprotein in explicit solvent: On the structure and possible role of internal water. J. Struct. Biol. 2007, 157:524–533. [DOI] [PubMed] [Google Scholar]
- 156.Su L, Cukier RI. Hamiltonian and distance replica exchange method studies of Met-enkephalin. J. Phys. Chem. B 2007, 111:12310–12321. [DOI] [PubMed] [Google Scholar]
- 157.Olson MA, Lee MS. Evaluation of Unrestrained Replica-Exchange Simulations Using Dynamic Walkers in Temperature Space for Protein Structure Refinement. Plos One 2014, 9:e96638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Park H, DiMaio F, Baker D. CASP11 refinement experiments with ROSETTA. Proteins 2016, 84 (Suppl. 1):314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chen JH, Im W, Brooks CL. Application of torsion angle molecular dynamics for efficient sampling of protein conformations. J. Comput. Chem 2005, 26:1565–1578. [DOI] [PubMed] [Google Scholar]
- 160.Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55:351–367. [DOI] [PubMed] [Google Scholar]
- 161.Chinchio M, Czaplewski C, Oldziej S, Scheraga HA. A Hierarchical Multiscale Approach to Protein Structure Prediction: Production of Low-Resolution Packing Arrangements of Helices and Refinement of the Best Models with a United-Residue Force Field. Multiscale Model. Simul 2006, 5:1175–1195. [Google Scholar]
- 162.Harada R, Kitao A. Exploring the Folding Free Energy Landscape of a beta-Hairpin Miniprotein, Chignolin, Using Multiscale Free Energy Landscape Calculation Method. J. Phys. Chem. B 2011, 115:8806–8812. [DOI] [PubMed] [Google Scholar]
- 163.Feig M, Karanicolas J, Brooks CL III. MMTSB Tool Set: Enhanced Sampling and Multiscale Modeling Methods for Applications in Structural Biology. J. Mol. Graph. Modell 2004, 22:377–395. [DOI] [PubMed] [Google Scholar]
- 164.Zhang W, Chen J. Accelerate Sampling in Atomistic Energy Landscapes Using Topology-Based Coarse-Grained Models. J. Chem. Theory Comput 2014, 10:918–923. [DOI] [PubMed] [Google Scholar]
- 165.Chen Y, Roux B.. Enhanced Sampling of an Atomic Model with Hybrid Nonequilibrium Molecular Dynamics-Monte Carlo Simulations Guided by a Coarse-Grained Model. J. Chem. Theory Comput. 2015, 11:3572–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bhattacharya D, Cheng J. Protein Structure Refinement by Iterative Fragment Exchange. In: International Conference on Bioinformatics, Computational Biology and Biomedical Informatics, BCB’13 Washington, DC; 2013. [Google Scholar]
- 167.Lee MC, Duan Y. Distinguishing protein decoys by using a scoring function based on a new AMBER force field, short molecular dynamics simulations, and the generalized Born solvent model. Proteins 2004, 55:620–634. [DOI] [PubMed] [Google Scholar]
- 168.Stumpff-Kane AW, Feig M. A correlation-based method for the enhancement of scoring functions on funnel-shaped energy landscapes. Proteins 2006, 63:155–164. [DOI] [PubMed] [Google Scholar]
- 169.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15:2507–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Feig M, Brooks CL III. Evaluating CASP4 predictions with physical energy functions. Proteins 2002, 49:232–245. [DOI] [PubMed] [Google Scholar]
- 171.Zhang C, Liu S, Zhou HY, Zhou YQ. An accurate, residue-level, pair potential of mean force for folding and binding based on the distance-scaled, ideal-gas reference state. Protein Sci. 2004, 13:400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhang Y, Skolnick J. SPICKER: A clustering approach to identify near-native protein folds. J. Comput. Chem 2004, 25:865–871. [DOI] [PubMed] [Google Scholar]
- 173.Skolnick J In quest of an empirical potential for protein structure prediction. Curr. Opin. Struct. Biol 2006, 16:166–171. [DOI] [PubMed] [Google Scholar]
- 174.Rykunov D, Fiser A. New statistical potential for quality assessment of protein models and a survey of energy functions. BMC Bioinformatics 2010, 11:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Park H, DiMaio F, Baker D. The Origin of Consistent Protein Structure Refinement from Structural Averaging. Structure 2015, 23:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Cerutti DS, Freddolino PL, Duke RE Jr., Case DA. Simulations of a Protein Crystal with a High Resolution X-ray Structure: Evaluation of Force Fields and Water Models. J. Phys. Chem. B 2010, 114:12811–12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. D 2010, 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kalisman N, Levi A, Maximova T, Reshef D, Zafiri-Lynn S, Gleyzer Y, Kaesar C. MESHI: A new library of Java classes for molecular modeling. Bioinformatics 2005, 21:3931–3932. [DOI] [PubMed] [Google Scholar]
- 179.Kortemme T, Morozov AV, Baker D. An Orientation-dependent Hydrogen Bonding Potential Improves Prediction of Specificity and Structure for Proteins and Protein-Protein Complexes. J. Mol. Biol 2003, 326:1239–1259. [DOI] [PubMed] [Google Scholar]
- 180.Bhattacharya D, Nowotny J, Cao RZ, Cheng JL. 3Drefine: an interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016, 44:W406–W409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhou HY, Zhou YQ. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002, 11:2714–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhang J, Zhang Y. A Novel Side-Chain Orientation Dependent Potential Derived from Random-Walk Reference State for Protein Fold Selection and Structure Prediction. Plos One 2010, 5:e15386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wang K, Fain B, Levitt M, Samudrala R. Improved protein structure selection using decoy-dependent discriminatory functions. BMC Struct. Biol 2004, 4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhou H, Skolnick J. GOAP: A generalized orientation-dependent, all-atom statistical potential for protein structure prediction. Biophys. J 2011, 101:2043–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Yang YD, Zhou YQ. Specific interactions for ab initio folding of protein terminal regions with secondary structures. Proteins 2008, 72:793-–803.. [DOI] [PubMed] [Google Scholar]
- 186.Ko J, Park H, Heo L, Seok C. GalaxyWEB Server for Protein Structure Prediction and Refinement. Nucleic Acids Res 2012, 40:W294–W297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zemla A, Venclovas C, Moult J, Fidelis K. Processing and Analysis of CASP3 Protein Structure Predictions. Proteins 1999, 37 (Supplement 3):22–29. [DOI] [PubMed] [Google Scholar]
- 188.Huang YJP, Mao BC, Aramini JM, Montelione GT. Assessment of template-based protein structure predictions in CASP10. Proteins 2014, 82:43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Liu T, Wang YH, Eickholt J, Wang Z. Benchmarking Deep Networks for Predicting Residue-Specific Quality of Individual Protein Models in CASP11. Sci. Rep.-Uk 2016, 6:19301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Paton RS, Goodman JM. Hydrogen Bonding and π-Stacking: How Reliable are Force Fields? A Critical Evaluation of Force Field Descriptions of Nonbonded Interactions. J. Chem. Inf. Model 2009, 49:944–955. [DOI] [PubMed] [Google Scholar]
- 191.Lopes PEM, Huang J, Shim J, Luo Y, Li H, Roux B, MacKerell AD Jr. Polarizable Force Field for Peptides and Proteins Based on the Classical Drude Oscillator. J. Chem. Theory Comput 2013, 9:5430–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cheng J A multi-template combination algorithm for protein comparative modeling. BMC Struct. Biol 2008, 8:18.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Martin J, Hartl FU. Chaperone-assisted protein folding. Curr. Opin. Struct. Biol 1997, 7:41–52. [DOI] [PubMed] [Google Scholar]
- 194.Yu I, Mori T, Ando T, Harada R, Jung J, Sugita Y, Feig M. Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. eLife 2016, 5:e19274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Feig M, Sugita Y. Reaching new levels of realism in modeling biological macromolecules in cellular environments. J. Mol. Graph. Modell 2013, 45:144–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Barth P, Wallner B, Baker D. Prediction of membrane protein structures with complex topologies using limited constraints. Proc. Natl. Acad. Sci. U.S.A 2009, 106:1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Elofsson A, von Heijne G. Membrane protein structure: Prediction versus reality. Annu. Rev. Biochem 2007, 76:125–140. [DOI] [PubMed] [Google Scholar]
- 198.Leman JK, Umschneider MB, Gray JJ. Computational modeling of membrane proteins. Proteins 2015, 83:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yuzlenko O, Lazaridis T. Membrane protein native state discrimination by implicit membrane models. J. Comput. Chem 2013, 34:731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Gao C, Stern HA. Scoring function accuracy for membrane protein structure prediction. Proteins 2007, 68:67–75. [DOI] [PubMed] [Google Scholar]
- 201.Weiner BE, Woetzel N, Karakas M, Alexander N, Meiler J. BCL::MP-Fold: Folding Membrane Proteins through Assembly of Transmembrane Helices. Structure 2013, 21:1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]