

Abstract
Purpose:
Pancreatic ductal adenocarcinoma (PDAC) is one of the most deadly cancers with a 5-year survival rate of less than 10%. Physicians often rely on biopsy or CT to guide treatment decisions, but these techniques fail to reliably measure the actions of therapeutic agents in PDAC. KRAS mutations are present in >90% of PDAC and are connected to many signaling pathways through its oncogenic cascade, including extracellular regulated kinase (ERK) and MYC. A key downstream event of MYC is transferrin receptor (TfR), which has been identified as a biomarker for cancer therapeutics and imaging.
Experimental Design:
In this study, we aimed to test if zirconium-89 transferrin ([89Zr]Zr-Tf) could measure changes in MYC depending on KRAS status of PDAC, and assess target engagement of anti-MYC and anti-ERK targeted therapies.
Results:
Mice bearing iKras*p53* tumors showed significantly higher (P < 0.05) uptake of [89Zr]Zr-Tf in mice withdrawn from inducible oncogenic KRAS. A therapy study with JQ1 showed a statistically significant decrease (P < 0.05) of [89Zr]Zr-Tf uptake in drug vs. vehicle-treated mice bearing Capan-2 and Suit-2 xenografts. IHC analysis of resected PDAC tumors reflects the data observed via PET imaging and radiotracer biodistribution.
Conclusions:
Our study demonstrates that [89Zr]Zr-Tf is a valuable tool to noninvasively assess oncogene status and target engagement of small molecule inhibitors downstream of oncogenic KRAS, allowing a quantitative assessment of drug delivery.
Keywords: transferrin, MYC, KRAS, ERK, zirconium-89, PET
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most challenging malignancies with a 5-year survival rate of less than 10% (1). PDAC remains to be one of the most difficult public health issues due poor prognosis, with very limited diagnostic and treatment options (1). Currently, while surgery is the single curative option to combat PDAC, only ~20% of patients are candidates for this procedure due to the typically late diagnosis of PDAC, with advanced and metastatic disease eliminating this as a treatment option (2). With chemotherapy and/or targeted therapy as the remaining alternative, evaluating success of said therapy can be difficult to predict due to the high interstitial fluid pressure and stromal barrier within PDAC tumors, along with the inevitable heterogeneity that plagues many cancers, especially those of the pancreas (3). These shortcomings embody a critically unmet clinical need to identify new biomarkers and/or develop innovative tools capable of more specifically identifying tumors that respond to systemic and targeted therapies.
KRAS mutations have been identified in greater than 90% of PDAC (4), yet targeted therapies to modulate the protein have not yet reached the clinic. One of the more recently explored and promising areas of this research includes targeting downstream signaling as a viable approach to affect KRAS (5). Recent evidence connects the RAS-RAF-MEK-ERK mitogen-activated protein kinase (MAPK) cascade to downstream of signal transduction pathways, including those closely related to MYC (6–8). Kortlever and coworkers have established cooperation between KRAS and MYC through the immune suppression and tumor microenvironment (9), which suggests that dysregulation of downstream pathways may also contribute to PDAC. Small molecule inhibitors that target BRD4 and ERK have been shown to indirectly target MYC in KRAS mutant PDAC (10, 11). MEK inhibitors have anti-tumorigenic activity in MAPK-dependent cancers with RAS mutations (12), but their resistance associated with pathway alterations results in phospho-ERK (pERK) reactivation (13–15). A newer ERK inhibitor, SCH772984, has shown promise in KRAS mutant cancers (5, 11, 16, 17). BRD4 inhibitors have been explored as a promising option for PDAC as both single agents and combination therapies (6, 18, 19), have been explored as stromal regulators in pancreatic tumors (20, 21), and have been implemented in clinical trials to treat KRAS mutant tumors (22).
Fully understanding protein relationships is critical to the success of experimental targeted therapies, as evidenced by the trials with vemurafenib (15, 23, 24), a drug that is now FDA-approved for BRAF-mutated (V600E) melanoma. Despite the presence of the V600E mutation in other cancer subtypes, this therapy is not always efficacious. Similarly, while it has been established that oncogenic KRAS signaling is a major driver of PDAC progression, dysregulation of KRAS signaling in pancreatic cancer is not necessarily associated with KRAS mutations and outcome (25, 26), and as a result may not be the best prognostic factor or molecular target for therapy. Many inhibitors that target aberrant proteins in PDAC along the KRAS cascade (e.g. MEK and EGFR) have also failed in clinical trials due to modest antitumor activity (27), but warrant further studies in molecularly enriched patient subsets (28). Sophisticated molecular imaging strategies can achieve this unmet need to assess target engagement and stratify respondents (29) by taking advantage of pathway intersection of downstream proteins such as MYC and TfR.
A key downstream event of MYC upregulation is amplification of the transferrin receptor (TfR), which has been implicated as a target for cancer therapeutics and imaging (30–34). Cancer cells express higher levels of TfR to accommodate the persistent need for Fe3+, which is utilized in many biological processes including those that involve cell proliferation (35). Transferrin (Tf), an 80 kDa serum protein that binds Fe3+, was labeled with zirconium-89 (89Zr) by our group to generate a PET tracer to annotate MYC status and other signaling pathways in cancer. Our radiotracer [89Zr]Zr-Tf has been used successfully to measure MYC and upstream inputs like mTORC1 and PI3K in multiple cancer subtypes (31, 35–38). On this basis, we chose to investigate this radiotracer further in the context of KRAS and pancreatic cancer, to facilitate a noninvasive method of evaluating oncogene status and therapy response through target engagement.
Materials and Methods
Cell culture and small molecule inhibitors.
All tissue culture manipulations were performed using sterile techniques, and all cells were grown at 37 °C and 5% CO2 in a humidified atmosphere. Suit-2 cells were a kind gift from the Tuveson Lab (Cold Spring Harbor) and were cultured in RPMI 1640 + 10% FBS and 1% Penicillin/Streptomycin cocktail (PenStrep). Doxycycline-induced KRAS mutant cells (iKras*p53*, denoted as such in text and figures) were a kind gift from the Pasca di Magliano Lab (University of Michigan) and were cultured in RPMI 1640 + 10% FBS 1% PenStrep, supplemented with 1 μg/mL doxycycline to facilitate constitutive KRAS mutation. Human PDAC cell lines (MIA PaCa-2, Capan-2, and BxPC-3) were obtained from the American Type Culture Collection, where they are authenticated via short tandem repeat (STR) DNA profiling. Stocks from the original thaw of each cell lines were used for all experiments between passage 5 and 15. Suit-2 and murine iKras*p53* cells (4292, 4668, and 9805) were not authenticated and used for experiments between passage 5 and 15. All of the cell lines discussed in this manuscript (human and murine) thawed from the same original stocks were tested and found to be free of mycoplasma contamination in July 2018. A table of cell lines used in this project, including culture conditions and in vivo inoculation procedures can be found in Supplemental Table 1. Experimental details regarding flow cytometry and western blot are described in the Supplemental Data.
Small molecule inhibitors.
BRD4 inhibitors were purchased from ApexBio (JQ1) and Cayman Chemical (OTX015) and used at a concentration of 1 μM. ERK inhibitor SCH772984 was purchased from Active Biochem and used at a concentration range of 250 nM-1 μM. MEK inhibitor trametinib was purchased from Selleckchem and used at a concentration range of 25–100 nM. All drugs were reconstituted in sterile, dry dimethyl sulfoxide, aliquotted, and store at −80 °C until further use.
Preparation and radiolabeling of [89Zr]Zr-transferrin.
Apo-transferrin (Tf) was functionalized with p-isothiocyanatobenzyl-desferrioxamine (DFO-Bn-NCS; Macrocyclics, Inc.) as previously described (38). 89Zr was produced through proton beam bombardment of yttrium foil and isolated in high purity as [89Zr]Zr-oxalate at Memorial Sloan Kettering Cancer Center according to a previously published procedure (39). Further details of radiolabeling procedures are described in the Supplemental Data.
Small animal models.
All animal studies were conducted in accordance with the guidelines set by the Institutional Animal Care and Use Committee at MSKCC. Female athymic nude (NU/NU) mice (Charles River Laboratories, 6–8 weeks, 20–22 g) were inoculated with PDAC tumors. Cells were implanted subcutaneously (details for conditions are listed in Supplemental Table 1) in the lower right flank in 200 μL of 1:1 media:Matrigel (BD Biosciences) and grown to a tumor volume ~100–150 mm3 before use for JQ1 therapy, and ~250–300 mm3 before SCH772984 therapy. Tumors were measured with a Peira TM900 tumor measurement device before the start of therapy and randomized into groups, and were measured once more at the end of the therapy.
iKras*p53* Animal Models.
Mice bearing subcutaneous murine PDAC xenografts (iKras*p53*) were placed on special husbandry to facilitate tumor growth through KRAS dependency. Mice were provided water ab libitum that was supplemented with 0.2 mg/mL doxycycline and 5% m/v sucrose. Mice were removed from special husbandry for one week before KRAS-off experiments commenced.
In vivo therapy.
Mice bearing subcutaneous human PDAC xenografts were treated via intraperitoneal injection of either vehicle (5% DMSO v/v in 10% m/v 2-hydroxypropyl-β-cyclodextrin) or BRD4 inhibitor JQ1 (50 mg/kg in 5% DMSO v/v in a 10% m/v 2-hydroxypropyl-β-cyclodextrin) administered 12 h apart, for a total of 12 doses. The same dosing schedule and amount administered was used for ERK inhibitor SCH772984 (prepared in 5% DMSO v/v, 10% PEG-300 v/v, in 10% m/v 2-hydroxypropyl-β-cyclodextrin). A solution with the same concentration of DMSO and PEG-300 in 10% m/v 2-hydroxypropyl-β-cyclodextrin was prepared as the vehicle control for the SCH772984 therapy.
In vivo imaging.
Mice were anesthetized with 1.5%−2% isoflurane (Baxter Healthcare) in oxygen. PET imaging experiments were accomplished with a microPET Focus 120 (Concorde Microsystems). Mice were administered [89Zr]Zr-labeled Tf (9–11 MBq, 60–70 μg) in ~200 μL of 1× PBS formulations via lateral tail vein injections. PET whole-body acquisitions were recorded for mice at 48 h post-injection while anesthetized as described above. Images were acquired until 20 million coincidence events were achieved, and images underwent 3D reconstruction. PET images were analyzed using ASIPro VM software (Concorde Microsystems).
Immunohistochemistry.
Immunohistochemical staining of TfR and c-MYC were carried out on paraffin-embedded tumors using antibodies targeting proteins of interest, along with TfR blocking peptide as a control (anti-c-MYC [Y69] ab32072, 4 μg/mL; anti-TfR [ab84036], 4 μg/mL dilution; human transferrin receptor peptide [ab101219], 15× excess). Sequential sections were stained with hematoxylin and eosin (H&E) stain. Images for all immunohistochemical staining were prepared using Pannoramic Viewer.
Statistical considerations.
Tumor uptake was determined from biodistribution studies as previously described (31, 35–38) and are further described in the Supplemental Data. All data was analyzed by the unpaired, two-tailed Student’s t-test and differences at the 95% confidence level (P < 0.05) was considered statistically significant. Positive and negative controls were included through all experiments whenever possible to ensure rigor and reproducibility throughout experimental design.
Results
KRAS dependency shows differences to BRD4 inhibition in PDAC cells at the protein level.
We began our study with assessing the pharmacological effects of BRD4 inhibitors JQ1 and OTX015 on proliferation and the proteins of interest (MYC and TfR) in doxycycline-inducible iKras*p53* murine PDAC cells (denoted as 4292*, 9805*, and 4668* when supplemented with doxycycline and KRAS mutant, and 4292, 9805, and 4668 without doxycycline supplement, and therefore KRAS WT) (40). The 4292* cells were seeded in two medias, one with (4292*) and one without (4292) doxycycline-supplemented media, allowing a subset of cells to return to a normal (rather than oncogenic) KRAS state. This was done to compare the effects of these drugs on KRAS mutant vs. cells no longer dependent on KRAS mutations (with all other transcriptional components normalized). The results show that both subsets murine PDAC cells were responsive BRD4 inhibitors at the proliferation level, regardless of KRAS status, with a significant decrease (P < 0.05 – 0.001) of cells in the G0/G1 phase and S phase upon drug treatment (Figure 1A). However, a selective decrease in surface TfR expression (Figure 1C) was observed by flow cytometry at 48 h in the doxycycline-dependent (KRAS mutant) vs. withdrawn (KRAS-WT) cells. This same trend with regards to MYC can be seen in the western blot series in Supplemental Figure 2D, where KRAS mutant 4292* and 9805* and WT 4292 and 9805 cells were drugged with BRD4 inhibitors for 48 h. There is a clear decrease in MYC protein levels in the KRAS mutant (4292* and 9805*) samples upon drug treatment, with little to no effect on the KRAS-WT cells (4292 and 9805). These results are in part supported by multiple studies that show success with BRD4 inhibitors in KRAS mutant cells, including PDAC (20, 21), and how each of these therapies directly affects MYC protein expression. With a head-to-head comparison of this therapy in cells of different KRAS dependency, we observe a potential preferential sensitivity to KRAS mutant cells that is currently being further explored with regards to our proteins of interest.
FIGURE 1:
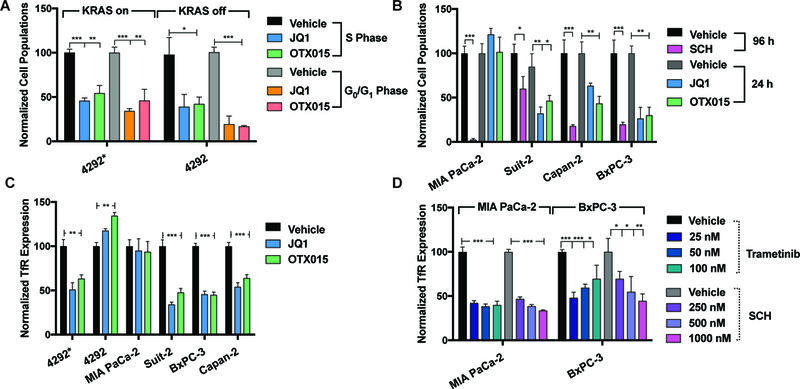
Cell cycle and protein analysis of drug treatment with BRD4, ERK, and MEK inhibitors in human PDAC and doxycycline-inducible KRAS mutant murine PDAC cells. (A) % of cells in S-phase and G0/G1 24 h post-treatment with BRD4 inhibitors in doxycycline-inducible KRAS mutant murine cells. 4292* indicates iKras*p53* mutant upon doxycycline induction, and 4292 indicates cells that are not doxycycline supplemented or KRAS mutant. (B) % of cells in S-phase post-treatment with BRD4 inhibitors and ERK inhibitor (1 μM of drug administered) in human PDAC cell lines. Timeline of analysis is indicated in the figure legend. (C) A drug-dependent decrease of surface TfR protein expression upon BRD4 inhibition in murine and human PDAC cells 48 h post-treatment. (D) A drug-dependent decrease of surface TfR protein expression in MIA PaCa-2 and BxPC-3 cells upon ERK inhibition with SCH772984 and MEK inhibition with trametinib. For statistical analysis: *P < 0.05, **P < 0.01, ***P < 0.001.
TfR is a downstream marker for ERK, MEK, and MYC inhibition in human PDAC cells.
To further our in vitro exploration, BRD4 inhibitors JQ1 and OTX015, ERK inhibitor SCH772984, which is also known to result in MYC degradation upon long-term treatment (5), along with FDA-approved MEK inhibitor trametinib, were explored in a panel of human PDAC cells (MIA PaCa-2, Suit-2, Capan-2: KRAS mutant and BxPC-3: KRAS WT). Differential responses to BRD4 inhibitors were observed in the human cell lines studied with regards to proliferation (Figure 1B) and surface TfR expression (Figure 1C). Human PDAC cells showed a marked antiproliferative response to JQ1 and OTX015 in Suit-2, BxPC-3, and Capan-2 cells (Figure 1B), as the % cells in S-phase was significantly decreased compared to vehicle control in responsive cell lines, confirming cell cycle arrest at the G0/G1 phase at 24 h for BRD4 inhibitors and 96 h for SCH772984 (P < 0.05 – 0.001). MIA PaCa-2 cells were not responsive to BRD4 inhibitors either through proliferation or protein analysis (Figures 1B and 1C). This result is consistent with findings of Sherman et al. (21), where widespread transcriptome analysis upon JQ1 treatment in this cell line did not show any effect on MYC in these cells upon drug treatment. All of the other human PDAC cell lines that showed a positive response to BRD4 inhibitors at the proliferation level (Suit-2, Capan-2, and BxPC-3) also showed a decrease MYC protein levels via western blot and partial response in whole TfR expression (Supplemental Figure 2C). BxPC-3 and Suit-2 cells showed a distinct decrease in MYC with both BRD4 inhibitors, but only JQ1 showed significant response in Capan-2 cells with MYC, establishing the rationale for using this drug in vivo across each of these models moving forward. All of the human PDAC lines tested were sensitive to SCH772984 (P < 0.05 – 0.001) at the proliferation and protein level (Figure 1B and Supplemental Figure 2A and B), including MIA PaCa-2 cells, and is consistent with results described by Hayes and coworkers (5). Earlier time points (4 h and 24 h) were used to assess pERK expression levels, respectively, upon SCH772984 treatment prior to known rebound effects of pERK reactivation (Supplemental Figure 2A and 2B). Since the most dramatic decrease was observed in BxPC-3 and MIA PaCa-2 cells, a dose dependent drugging experiment was performed to observe surface levels of TfR upon SCH772984 and trametinib treatment in these cells. A drug-dependent decrease in surface TfR 48 h post-SCH772984 treatment (Figure 1D) was observed upon SCH772984 treatment in BxPC-3 and MIA PaCa-2 cells (P < 0.05 – 0.001). The highest dose (1000 nM) significantly reduced surface TfR expression levels in BxPC-3 cells compared to the two lower doses (250 nM and 500 nM, respectively). Treatment with trametinib did not show a significant dose dependency with regards to surface TfR at 48 h in either cell line. In fact, a slight rebound effect was observed in the BxPC-3 cells at the highest dose, with no significant differences observed between each of the three doses in MIA PaCa-2 cells.
KRAS independent tumors rebound MYC expression and have increased uptake of [89Zr]Zr-Tf.
To explore the KRAS dependency and its correlation with MYC in an in vivo setting, we inoculated iKras*p53* tumors (4292*, 9805*, and 4668*) into female athymic nude mice to assess their uptake with [89Zr]Zr-Tf in vivo. Figure 2 represents the PET imaging hypothesis of this work with regard to uptake of the [89Zr]Zr-Tf radiotracer, with oncogenic MYC leading to an overexpression of TfR on the surface of cancer cells. To achieve KRAS-WT murine tumors, a cohort of mice from each group were removed from doxycycline water husbandry and tumor growth was measured over time (Figure 3D, H, and L). PET imaging with [89Zr]Zr-Tf in both KRAS dependent and doxycycline-withdrawn mice showed uptake between 3–5 % ID/g (Figure 3A, E, and I). Uptake of [89Zr]Zr-Tf in the doxycycline-withdrawn tumors was significantly higher (P < 0.05) than the KRAS-mutant tumors, as quantified by biodistribution studies (Figure 3B, F, and J). Full biodistribution studies can be found in Supplemental Table 2. Flow cytometry was performed with all three sets of KRAS-inducible cells (4292, 9805, and 4668). Each of these cell lines were plated either with (or without) doxycycline in the media to observe changes in protein expression (TfR and MYC) upon differences in KRAS dependency. Across each of these cell lines, MYC and TfR were significantly upregulated in the doxycycline-withdrawn cells (Figure 3C, G, and K). This result was supported by Collins et al., which showed a similar results upon immunohistochemical analysis of MYC in these tumors after withdrawing them from doxycycline and awaiting their growth rebound (40). This study describes a loss-of doxycycline dependence in the transgene, evidenced by elevated constitutive KRAS* expression, indicating a selective pressure, but not acquired independence from KRAS*. Determining the origin and mechanism of this phenomenon is currently under further study, but we show that we are able to use [89Zr]Zr-Tf PET to delineate MYC and TfR in these models, and how constitutive MYC expression may be a consequence of KRAS selective pressure in vivo. We confirmed this result with our own in-house ex vivo immunohistochemistry (IHC), which also shows a clear increase in MYC and TfR protein expression in the doxycycline-withdrawn tumor tissue (Figure 4 shows zoomed in tissue at 100 μm and Supplemental Figure 3 for whole tissue at a 2000 μm scale). We posit that the rebound elevation of MYC upon loss of KRAS mutant status is significant to these results, with constitutive expression of MYC as a potential mechanism of acquired resistance to these inhibitors at the protein level, as shown in our in vitro results. While this remains unproven, we were able to noninvasively image changes in MYC in mice of contrasting KRAS status, and can successfully use TfR as our downstream target to assess oncogene changes over time.
FIGURE 2:
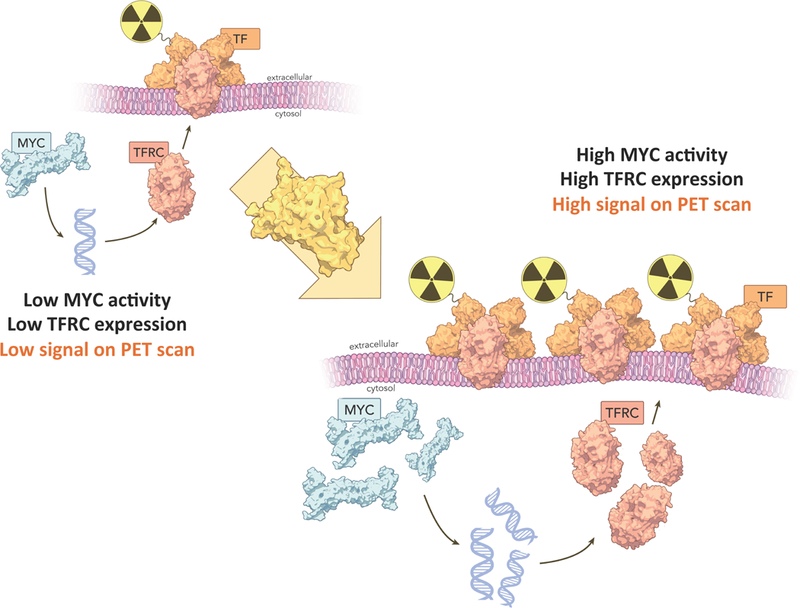
Schematic representation of the PET imaging hypothesis connecting the protein cascade from oncogenic MYC expression to surface transferrin receptor expression (TfR) in pancreatic cancer. High MYC activity, especially in the context of oncogenesis, will directly result in amplified expression of TfR, subsequently formulating an imaging target for radiolabeled transferrin. This correlation is represented across multiple cohorts as [89Zr]Zr-Transferrin has increased uptake in tumors with higher protein expression of both MYC and TfR, and is observed in preclinical models of controlled KRAS dependency and via target engagement along the KRAS oncogenic cascade.
FIGURE 3:
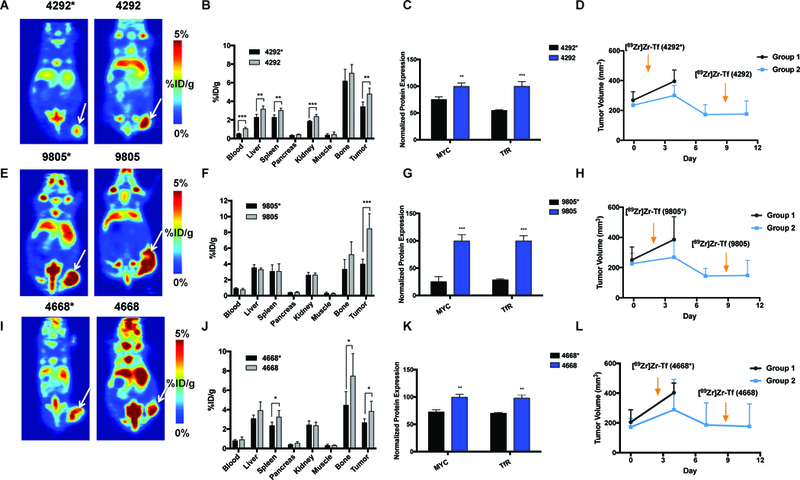
PET imaging of [89Zr]Zr-Transferrin uptake in doxycycline-inducible KRAS-mutant murine xenografts, with corresponding biodistribution at 48 h, in vitro protein analysis of MYC and TfR levels, and tumor growth measurement and imaging timeline. Tumors were inoculated with mice on doxycycline water, and measured and randomized 1 week after inoculation (shown as day 0 in far right panel). A cohort of animals were removed from doxycycline water at day 0 (n = 5 /group). Tumors were followed for each group until endpoint (day 11 across each cohort). Arrows indicate the times of injection of [89Zr]Zr-Tf for each group (dox-on* and dox-off). The nomenclature 4292*, 9805*, and 4668* denote tumors and cells that are doxycycline dependent (KRAS mutant). 4292, 9805, and 4668 (without the asterisk) denote tumors and cells that have been withdrawn from doxycycline and are therefore KRAS independent, which were imaged one week later. (A) Coronal slices of iKras*p53* 4292 tumors and (B) corresponding biodistribution at 48 h. Tumors had significantly more tumor uptake of [89Zr]Zr-Tf in doxycycline-withdrawn mice (*P < 0.05). (C) Protein levels of MYC and TfR in doxycycline-on (4292*) and doxycycline-off cells (4292), showing a significant increase in protein levels in KRAS-WT cells (*P < 0.01–0.001). (D) Timeline of inoculation, imaging, and rebound of tumor size (4292) after doxycycline withdrawal. (E) Coronal slices or iKras*p53* 9805 tumors and (F) and corresponding biodistribution at 48 h. (G) Protein levels of MYC and TfR in doxycycline-on (9805*) and doxycycline-off cells (9805), showing a significant increase in protein levels in KRAS-WT cells (P < 0.001). (H) Timeline of inoculation, imaging, and rebound of tumor size (9805) after doxycycline withdrawal. (I) Coronal slices or iKras*p53* 4668 tumors and (J) and corresponding biodistribution at 48 h. Tumors had significantly more tumor uptake of [89Zr]Zr-Tf in doxycycline-withdrawn mice (*P < 0.05). (K) Protein levels of MYC and TfR in doxycycline-on (4668*) and doxycycline-off cells (4668), showing a significant increase in protein levels in KRAS-WT cells (P < 0.01). (L) Timeline of inoculation, imaging, and rebound of tumor size (4668) after doxycycline withdrawal. *P < 0.05, **P < 0.01, ***P < 0.001.
FIGURE 4:
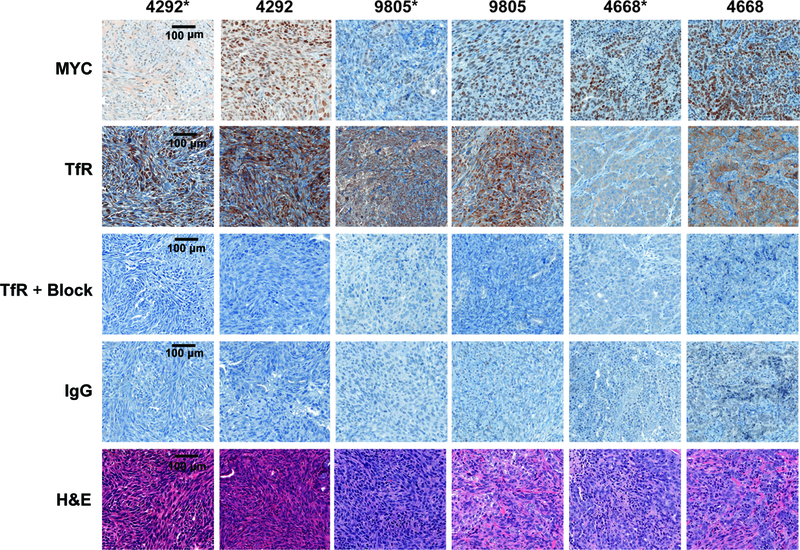
Ex vivo immunohistochemistry of resected murine iKras*p53* tumors. Expression of MYC and TfR is more abundant in KRAS-WT (4292, 9805, and 4668) tumors, with less amplification of MYC and TfR observed in KRAS-mutant tumors (4292*, 9805*, and 4668*). Specific TfR uptake is demonstrated by en excess of blocking peptide and IgG controls on subsequent sections. For each staining selection, n = 3 tumors, and 2–3 levels each were analyzed, and representative images from the same level of tumor from subsequent sections were chosen for figure representation. Images are shown at 20× magnification with 100 μm scale and is represented in the left panel only for brevity.
[89Zr]Zr-Tf PET measures target engagement of pharmacologically induced changes in MYC signaling in KRAS-mutant human PDAC xenografts.
Our compelling results in murine PDAC gave us the opportunity to use TfR as an imaging target to assess MYC status, so we moved forward to assess the capacity of [89Zr]Zr-Tf to observe target engagement and therapy response in human PDAC xenografts. A BRD4 therapy study with JQ1 was launched in preclinical models of human PDAC, using the cell lines that responded to the drug therapy in vitro (Suit-2, Capan-2, and BxPC-3) as xenograft models. Suit-2, Capan-2, and BxPC-3 xenografts were inoculated in mice when tumors were grown to a size of ~100–150 mm3, animals were administered JQ1 twice daily (50 mg/kg) for a total of 12 doses. On the fifth day of treatment, animals were administered [89Zr]Zr-Tf, and drug treatment continued for an additional 3 doses the following two days. Mice were imaged at 48 h, a previously determined optimal time point for delineated uptake of disease for [89Zr]Zr-Tf (31, 35, 36, 38), to assess therapy response. We did not expect this later time point to be a conflicting factor in receptor changes, as the therapy continued post-radiotracer administration. Supplemental Figure 4 highlights the dosing regimen and timeline of radiotracer administration. PET imaging shows a modest but statistically significant decrease in [89Zr]Zr-Tf uptake in drug-treated vs. vehicle-treated Suit-2 and Capan-2 xenografts, respectively, yet no response in BxPC-3 mice, which can be seen in both the coronal and transverse slices in Figures 5A and5B. Quantitative biodistribution studies confirm that the decrease in the tumor uptake of [89Zr]Zr-Tf was significant (P < 0.05) in Suit-2 and Capan-2 xenografts, but not significant in BxPC-3 mice (Figure 5C). Tumor measurement at experimental start and end is represented in Figure 5D, showing that we were able to detect changes in biology [89Zr]Zr-Tf PET before any significant decrease in tumor size. Ex vivo IHC with vehicle vs. JQ1-treated tumors shows an abundance of MYC and TfR in control (vehicle-treated) animals, with a discernable decrease in MYC and TfR expression in the Suit-2 and Capan-2 tumors that were treated with JQ1 (Figure 6 shows zoomed in tissue at 100 μm and Supplemental Figure 5 for whole tissue at 2000 μm scale), which is consistent with the biodistribution data. Full biodistribution studies for these experiments can be found in Supplemental Table 3. This study highlights successful target engagement with [89Zr]Zr-Tf PET, but also that TfR and/or MYC may not be the optimal target for biomarker and/or drug discovery within each subtype of PDAC. IHC (20× enhanced in Figure 6) analysis showed a strong presence of desmoplastic stroma in the BxPC-3 tumors, which is a common characteristic of pancreatic tumors and often confers resistance to therapy (3, 41). We believe the presence of this desmoplasia is a contributing factor for why these xenografts failed to respond in vivo despite the encouraging results observed in vitro.
FIGURE 5:
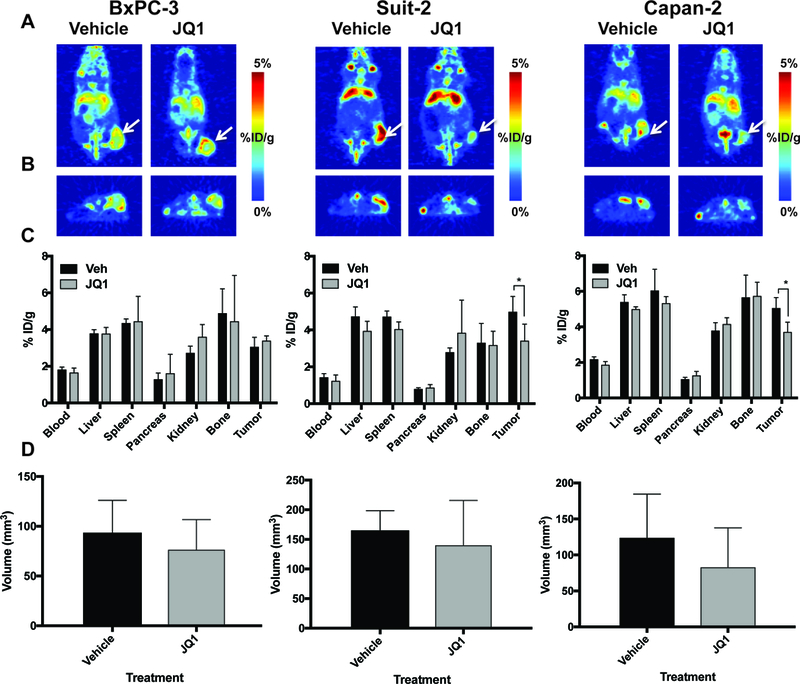
PET imaging of [89Zr]Zr-Transferrin uptake in vehicle vs. JQ1-treated animals bearing human PDAC xenografts. Mice were treated with Vehicle or JQ1 for 6 days at 50 mg/kg, twice daily. Tumors were measured and randomized at the start of each experiment. No significant difference was noted in tumor size at the end of the experiment between vehicle and drug treatment. (A) Coronal PET slices of 89Zr-Tf uptake in vehicle and JQ1-treated animals bearing Suit-2, BxPC-3, and Capan-2 human PDAC xenografts. (B) Transverse slices of vehicle vs. JQ1-treated animals in each group, giving an additional perspective of differences in tracer uptake in vehicle vs. drug-treated tumors. Corresponding biodistribution at 48 h (C) and tumor measurement (D) at experimental end of vehicle vs. JQ1-treated animals, showing a significant decrease in radiotracer uptake in drug-treated animals bearing Suit-2 And Capan-2 (KRAS-mutant) tumors, with no significance in BxPC-3 (KRAS WT). *P < 0.05, n = 5/group.
FIGURE 6:
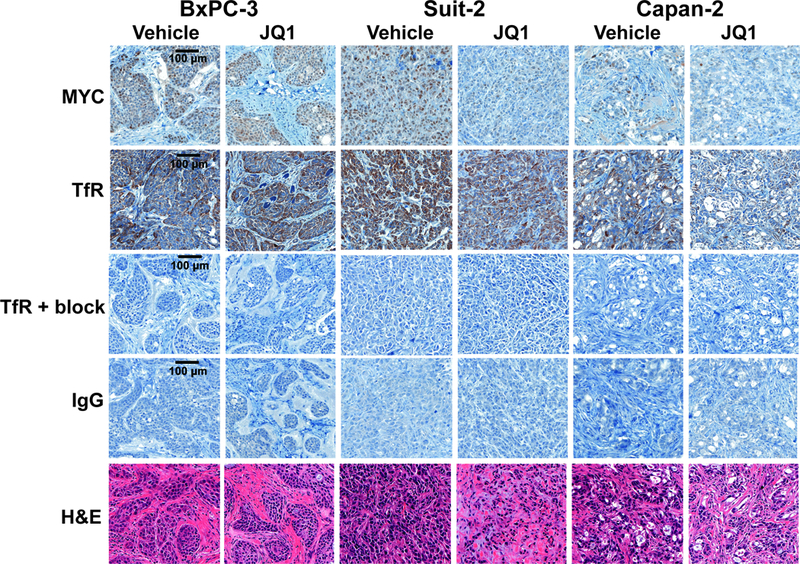
Ex vivo immunohistochemistry of resected human PDAC tumors pre and post-treatment with JQ1. Differences in MYC and TfR protein expression can be observed in tumors that responded to JQ1 treatment with regards to [89Zr]Zr-transferrin uptake in vivo. Specific TfR uptake is demonstrated by en excess of blocking peptide and IgG controls on subsequent sections. For each staining selection, n = 3 tumors were analyzed, with 2–3 levels each, and representative images from the same level of tumor in each group were chosen for figure display. Images are shown at 20× magnification with 100 μm scale and is represented in the left panel only for brevity.
[89Zr]Zr-Tf PET measures target engagement of pharmacologically induced changes in ERK signaling in KRAS WT human xenografts.
We set out to explore this response assessment further by using a second inhibitor of interest in BxPC-3 tumors higher up the oncogenic cascade. BxPC-3 cells, although KRAS WT, harbor a plethora of additional mutations that contribute to their oncogenic properties, including BRAF and TP53, allowing them to circumvent the KRAS pathway by upregulation of MAPK and PI3K (42). For the second drug, we chose ERK inhibitor SCH772984, as it has been shown to respond in both KRAS WT and KRAS mutant cells, does not have a preference to either subtype, and has been shown to induce MYC degradation over time (5). With this, we expected this drug to work both in vitro and in vivo, regardless of KRAS status. Supporting our hypothesis, were able to utilize [89Zr]Zr-Tf to successfully assess target engagement of SCH772984 in animals bearing BxPC-3 xenografts using PET imaging (Supplemental Figure 6A), as significantly less [89Zr]Zr-Tf was taken up in drug vs. vehicle-treated tumors (Supplemental Figure 6B), but with no significant decrease in tumor growth (Supplemental Figure 6C). Although we did not observe any specific toxicity in these animals in this cohort, we noted a significant increase uptake of [89Zr]Zr-Tf in the blood pool and pancreas in SCH772984- vs. vehicle-treated animals, indicating potential inflammation. Our data still encourages translation of this tracer, as it may be a useful tool to interrogate target engagement and but also detect potentially unfavorable side effects in patients receiving targeted therapy. Full biodistribution for these studies can be found in Supplemental Table 4.
Discussion
All of the data herein supports that [89Zr]Zr-Tf is an excellent radiotracer to assess oncogene status and target engagement in preclinical PDAC. We were able to use [89Zr]Zr-Tf to assess oncogene status over time as changes in MYC were induced by withdrawal of the KRAS oncogenic state. We observed significant effects of both BRD4 and ERK inhibition on PDAC cell proliferation, coupled to robust effects on MYC, TfR, and pERK protein expression upon drug treatment. Much of the in vitro data described is corroborated with in vivo experiments, as we were able to successfully interrogate target engagement of both BRD4 and ERK inhibitors in preclinical PDAC using [89Zr]Zr-Tf PET. With all of this data taken together, we were able to confirm that TfR is a useful biomarker to interrogate target engagement for drugs aimed to inhibit protein function downstream of the KRAS hierarchy.
TfR has been validated as a clinically relevant target for PET imaging in models of prostate cancer, glioblastoma, lymphoma, and triple negative breast cancer (31, 35–37), leading to the ongoing translation of [89Zr]Zr-Tf into the clinic. The research described herein validates TfR as a successful biomarker for PET imaging in multiple models of PDAC, allows us to investigate treatment response to small molecule targeted therapy both in vitro and in vivo, and furthers our understanding of the relationship between multiple oncogenic proteins that are key to solving the PDAC dilemma. While we maintain that dysregulation of KRAS signaling in pancreatic cancer is not necessarily associated with KRAS mutations and outcome (25), there remains a critical need to investigate new therapies along this cascade paired with noninvasive techniques to have a successful readout to assess response and target engagement.
Establishing biomarkers in pancreatic cancer is key to improve the prognosis of this challenging malignancy. The ability to predict effects of KRAS oncogenic signaling, whether it is through PET imaging or assessing downstream proteins such as ERK, MYC, and TfR, vastly improves the diagnostic options. Previous work done by Galvez et al. did not identify a significant link between MAPK inhibition and TfR through siRNA screen of human signaling proteome (43). However, these studies were only done in HeLa cells, and the experiments we describe herein were performed with multiple PDAC cell lines, directly tied to the effects at the protein level, and are robust and reproducible. We are able to clearly highlight that pharmacological modulation of proteins downstream of MAPK signaling can indeed affect TfR. The specificity of this relationship is yet to be determined, but we have the ability to take advantage of it to push for improvements in the clinical readouts of trials that include these types of therapies.
Clinical trials for KRAS and MEK inhibitors have classically failed or had conflicting results across patient cohorts, and reasons for this are still not well understood (15, 27, 44). Current criteria for clinical trials (including RECIST) do not always have the ability to assess target engagement of experimental therapies, and often lack a specific imaging component that can successfully assess therapy response. Additionally, other groups have found that BRD4 inhibitors such as JQ1 may affect stromal remodeling independent of MYC (20, 21). As such, it is possible that we would not be able to detect such changes in with our [89Zr]Zr-Tf PET radiotracer in tumors that do have the presence of the desmoplastic stroma as noted by the BxPC-3 slices in Figure 6. However, Hayes et al. describes long-term ERK inhibition having significant and specific effects on MYC, along with lack of preference of KRAS-mutant vs. KRAS WT to drug responsiveness to SCH772984 (5). We suspect the dynamic actions and unexplored mechanisms of this class of drugs supports our data for the differential response in our cohorts, especially in BxPC-3 mice, and speaks to the heterogeneity of PDAC tumors. [89Zr]Zr-Tf PET has the potential to shed light on both MYC-dependent and independent mechanisms, and we are using this data to spearhead additional applications for both these drugs and radiotracer. We aim to improve the current clinical standards and encourage molecular imaging as a critical tool for precision medicine.
The usual standard for imaging PDAC in the clinic is typically computed tomography (CT) or magnetic resonance imaging (MRI) (45). While these techniques can be sufficient for diagnostic purposes, CT and MRI do not offer all of the advantages that PET does in terms of noninvasive and quantitative measurement of biology. Currently, PET imaging in PDAC is slowly improving, and one promising field of study includes multiple clinical trials with CA19.9-targeted antibody 5B1 for both imaging and radioimmunotherapy (46). Additionally, Sugyo and coworkers have published work with anti-transferrin receptor antibody imaging and radioimmunotherapy in PDAC, giving promise to the future of imaging and therapy with our target of interest in mind (47–49). We believe in the immediate clinical relevance of utilizing an endogenous system (with [89Zr]Zr-Tf PET) for interrogating biology, as it may avoid issues of potential immunogenicity and can follow clinically translatable pharmacological effects.
Assessing drug pharmacodynamics is a huge challenge in the oncology field. Repeat biopsies are rare, particularly for patients with widespread metastatic disease, and do not always yield evaluable tissue. In addition to this, sampling issues, invasiveness, and patient compliance builds a wall between a reliable diagnosis and appropriating treatment. Metabolic radiotracers such as 18F-labeled FDG have a reputation for being non-specific, and are not always sufficient in assessing therapy response (50). [89Zr]Zr-Tf has the ability to assess target engagement downstream of a critical oncogenic cascade in PDAC, and using MYC signaling through oncogene dependence.
Despite KRAS being an “undruggable” target, mechanisms that drive the resistance to these inhibitors are consistently of interest to the biomedical field. Drugs that target KRAS and downstream proteins in the oncogenic cascade are constantly being synthesized and tested preclinically and in clinical trials. The exciting finding of the study is the evaluation of a single PET tracer to interrogate target engagement of downstream proteins involved in (and directly related to) KRAS dependency, which may allow us to noninvasively study the actions of drugs to confer less resistance and increase successes and patient longevity. With our PET imaging strategy, physicians could identify target engagement vs. efficacy to better stratify patients for selected therapies in KRAS-driven PDAC.
Conclusion
There is a critical need for noninvasive tools to assess metabolic effects, predict treatment response, and demarcate biology within PDAC. Pancreatic cancer carries a very unfavorable prognosis, and there is a clinically unmet necessity to establish biomarkers to predict which patients will favorably respond to newly targeted therapies. We have found that our radiotracer [89Zr]Zr-Tf is a valuable tool in preclinical PDAC to assess oncogene status and target engagement of MYC- and ERK-targeted therapy. Molecular imaging tools that can quantify oncogene activity could significantly impact the diagnosis of pancreatic cancer, provide the ability predict therapy response and assess tumor biology post-treatment, and reveal future successes within this devastating disease. Noninvasive [89Zr]Zr-Tf PET demonstrates the importance of TfR as a marker for target engagement in pancreatic cancer, establishes a potential link of KRAS dependency to therapy response, and highlights the relevance of this tracer for clinical translation moving forward.
Supplementary Material
Translational Relevance:
Currently, there is a critical need for specific, clinically translatable, and noninvasive tools to assess oncogene status, predict treatment response, and demarcate biology within PDAC. KRAS mutations have been identified in greater than 90% of PDAC, yet targeted therapies to modulate this protein have not been cleared for clinical use. Our radiotracer [89Zr]Zr-transferrin shows promise as a tool for interrogating oncogene signaling and target engagement of small molecule inhibitors for proteins downstream of oncogenic KRAS (e.g. ERK and MYC). This strategy can be used to noninvasively detect early action of targeted therapies along the KRAS cascade, which is a clinically unmet need within the realm of pancreatic cancer.
Acknowledgements
The authors thankfully acknowledge the MSKCC Small Animal Imaging Core Facility, the Radiochemistry & Molecular Imaging Probe Core, the Molecular Cytology Core Facility, and the Flow Cytometry Core Facility, all of which are supported in part by the National Institutes of Health (P30 CA08748). The authors gratefully acknowledge the Tuveson Lab (Cold Spring Harbor) for providing the Suit-2 cell line, and the Pasca di Magliano Lab (University of Michigan) for providing the iKras*p53* cell lines (4292, 9805, and 4668). JSL and KEH gratefully acknowledge Kimberly Kelly (University of Virginia) for critical reading of the manuscript and helpful discussions. This work was funded by the National Institutes of Health (R01CA17661: JSL, MJE) and the MSKCC Center for Molecular Imaging and Nanotechnology Tow Fellowship (KEH). MJE was supported by the 2013 David H. Koch Young Investigator Award from the Prostate Cancer Foundation, the National Institutes of Health (R00CA172695), a Department of Defense Idea Development Award (PC140107), the UCSF Academic Senate, an American Cancer Society Research Scholar Grant (130635-RSG-17–005-01-CCE), and GE Healthcare.
Financial Support: This work was funded by the National Institutes of Health (R01CA17661, JSL, MJE) and the MSKCC Center for Molecular Imaging and Nanotechnology Tow Fellowship (KEH). The MSKCC Small Animal Imaging Core Facility, Radiochemistry & Molecular Imaging Probe Core, Molecular Cytology Core Facility, and Flow Cytometry Core Facility are supported in part by the National Institutes of Health (P30CA08748). MJE was supported by the 2013 David H. Koch Young Investigator Award from the Prostate Cancer Foundation, the National Institutes of Health (R00CA172695), a Department of Defense Idea Development Award (PC140107), an American Cancer Society Research Scholar Grant (130635-RSG-17–005-01-CCE), the UCSF Academic Senate, and GE Healthcare.
Abbreviations
- Tf
Transferrin
- TfR
Transferrin receptor
- PET
positron emission tomography
- PDAC
pancreatic ductal adenocarcinoma
- BRD4
bromodomain-containing protein 4
- ERK
extracellular regulated kinase
- DFO
desferrioxamine
- 89Zr
zirconium-89
Footnotes
Conflicts of Interest: The authors have declared no conflicts of interest.
REFERENCES
- 1.Dimastromatteo J, Houghton JL, Lewis JS, Kelly KA. Challenges of Pancreatic Cancer. Cancer J 2015;21:188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cameron JL, Riall TS, Coleman J, Belcher KA. One Thousand Consecutive Pancreaticoduodenectomies. Ann Surg 2006;244:10–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dimou A, Syrigos KN, Saif MW. Overcoming the stromal barrier: technologies to optimize drug delivery in pancreatic cancer. Therap Adv Med Onc 2012;4:271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeitouni D, Pylayeva-Gupta Y, Der CJ, Bryant KL. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers 2016;8:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayes Tikvah K, Neel Nicole F, Hu C, Gautam P, Chenard M, Long B, et al. Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell 2016;29:75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia PL, Miller AL, Kreitzburg KM, Council LN, Gamblin TL, Christein JD, et al. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene 2016;35:833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ischenko I, Zhi J, Hayman MJ, Petrenko O. KRAS-dependent suppression of MYC enhances the sensitivity of cancer cells to cytotoxic agents. Oncotarget 2017;8:17995–8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaub FX, Dhankani V, Berger AC, Trivedi M, Richardson AB, Shaw R, et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Systems 2018;6:282–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017;171:1301–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N Engl J Med 2011;364:2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014;15:323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jha S, Morris EJ, Hruza A, Mansueto MS, Schroeder GK, Arbanas J, et al. Dissecting Therapeutic Resistance to ERK Inhibition. Mol Cancer Therap 2016;15:548–59. [DOI] [PubMed] [Google Scholar]
- 13.Lemstrova R, Brynychova V, Hughes DJ, Hlavac V, Dvorak P, Doherty JE, et al. Dysregulation of KRAS signaling in pancreatic cancer is not associated with KRAS mutations and outcome. Oncology Lett 2017;14:5980–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins MA, Bednar F, Zhang Y, Brisset J-C, Galbán S, Galbán CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012;122:639–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ko AH, Bekaii-Saab T, Van Ziffle J, Mirzoeva OM, Joseph NM, Talasaz A, et al. A Multicenter, Open-Label Phase II Clinical Trial of Combined MEK plus EGFR Inhibition for Chemotherapy-Refractory Advanced Pancreatic Adenocarcinoma. Clin Cancer Res 2016;22:61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko AH. Progress in the Treatment of Metastatic Pancreatic Cancer and the Search for Next Opportunities . J Clin Oncol 2015;33:1779–86. [DOI] [PubMed] [Google Scholar]
- 17.Wirth M, Schneider G. MYC: A Stratification Marker for Pancreatic Cancer Therapy. Trends in Cancer 2016;2:1–3. [DOI] [PubMed] [Google Scholar]
- 18.Dang CV. MYC, Metabolism, Cell Growth, and Tumorigenesis. Cold Spring Harbor Perspect Med. 2013;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doran MG, Carnazza KE, Steckler JM, Spratt DE, Truillet C, Wongvipat J, et al. Applying 89Zr-Transferrin To Study the Pharmacology of Inhibitors to BET Bromodomain Containing Proteins. Mol Pharm 2016;13:683–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryschich E, Huszty G, Knaebel HP, Hartel M, Büchler MW, Schmidt J. Transferrin receptor is a marker of malignant phenotype in human pancreatic cancer and in neuroendocrine carcinoma of the pancreas. Eur J Cancer 2004;40:1418–22. [DOI] [PubMed] [Google Scholar]
- 21.Daniels TR, Bernabeu E, Rodríguez JA, Patel S, Kozman M, Chiappetta DA, et al. Transferrin receptors and the targeted delivery of therapeutic agents against cancer. Biochim Biophys Acta 2012;1820:291–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Donnell KA, Yu D, Zeller KI, Kim J-w, Racke F, Thomas-Tikhonenko A, et al. Activation of Transferrin Receptor 1 by c-Myc Enhances Cellular Proliferation and Tumorigenesis. Mol Cell Biol 2006;26:2373–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holland JP, Evans MJ, Rice SL, Wongvipat J, Sawyers CL, Lewis JS. Annotating MYC oncogene status with (89)Zr-transferrin imaging. Nat Med 2012;18:1586–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans MJ, Holland JP, Rice SL, Doran MG, Cheal SM, Campos C, et al. Imaging Tumor Burden in the Brain with (89)Zr-Transferrin. J Nucl Med 2013;54:90–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Truillet C, Cunningham am JT, Parker MFL, Huynh LT, Conn CS, Ruggero D, et al. Non-invasive measurement of mTORC1 signaling with 89Zr-transferrin. Clin Cancer Res 2017;23:3045–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henry KE, Dilling TR, Abdel-Atti D, Edwards KJ, Evans MJ, Lewis JS. Non-invasive 89Zr-Transferrin PET Shows Improved Tumor Targeting Compared to 18F-FDG PET in MYC-overexpressing Human Triple Negative Breast Cancer. J Nucl Med 2018;59:51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rembielak AI, Jain P, Jackson AS, Green MM, Santorelli GR, Whitfield GA, et al. Phase II Trial of Cetuximab and Conformal Radiotherapy Only in Locally Advanced Pancreatic Cancer with Concurrent Tissue Sampling Feasibility Study. Transl Oncol 2014;7:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manchado E, Weissmueller S, Morris JP, Chen C-C, Wullenkord R, Lujambio A, et al. A combinatorial strategy for treating KRAS mutant lung cancer. Nature. 2016;534:647–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oikonomou E, Koustas E, Goulielmaki M, Pintzas A. BRAF vs RAS oncogenes: are mutations of the same pathway equal? differential signalling and therapeutic implications. Oncotarget. 2014;5:11752–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goetz EM, Ghandi M, Treacy DJ, Wagle N, Garraway LA. ERK Mutations Confer Resistance to Mitogen-Activated Protein Kinase Pathway Inhibitors. Cancer Res 2014;74:7079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N Engl J Med. 2012;367:1694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, et al. Discovery of a Novel ERK Inhibitor with Activity in Models of Acquired Resistance to BRAF and MEK Inhibitors. Cancer Discov 2013;3:742–50. [DOI] [PubMed] [Google Scholar]
- 33.Hatzivassiliou G, Liu B, O’Brien C, Spoerke JM, Hoeflich KP, Haverty PM, et al. ERK Inhibition Overcomes Acquired Resistance to MEK Inhibitors. Mol Cancer Therap 2012;11:1143–54. [DOI] [PubMed] [Google Scholar]
- 34.Sahai V, Redig AJ, Collier KA, Eckerdt FD, Munshi HG. Targeting BET bromodomain proteins in solid tumors. Oncotarget 2016;7:53997–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sanchez-Rivera FJ, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21:1163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamamoto K, Tateishi K, Kudo Y, Hoshikawa M, Tanaka M, Nakatsuka T, et al. Stromal remodeling by the BET bromodomain inhibitor JQ1 suppresses the progression of human pancreatic cancer. Oncotarget. 2016;7:61469–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sherman MH, Yu RT, Tseng TW, Sousa CM, Liu S, Truitt ML, et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc Natl Acad Sci 2017;114:1129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riveiro ME, Astorgues-Xerri L, Vazquez R, Frapolli R, Kwee I, Rinaldi A, et al. OTX015 (MK-8628), a novel BET inhibitor, exhibits antitumor activity in non-small cell and small cell lung cancer models harboring different oncogenic mutations. Oncotarget 2016;7:84675–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holland JP, Sheh Y, Lewis JS. Standardized methods for the production of high specific-activity zirconium-89. Nuclear medicine and biology. 2009;36:729–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collins MA, Brisset J-C, Zhang Y, Bednar F, Pierre J, Heist KA, et al. Metastatic Pancreatic Cancer Is Dependent on Oncogenic Kras in Mice. PLoS ONE 2012;7:e49707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watt J, Kocher HM. The desmoplastic stroma of pancreatic cancer is a barrier to immune cell infiltration. Oncoimmunology 2013;2:e26788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deer EL, Gonzalez-Hernandez J, Coursen JD, Shea JE, Ngatia J, Scaife CL, et al. Phenotype and Genotype of Pancreatic Cancer Cell Lines. Pancreas 2010;39:425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galvez T, Teruel MN, Heo WD, Jones JT, Kim ML, Liou J, et al. siRNA screen of the human signaling proteome identifies the PtdIns(3,4,5)P(3)-mTOR signaling pathway as a primary regulator of transferrin uptake. Genome Biol 2007;8:R142-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brauswetter D, Gurbi B, Varga A, Várkondi E, Schwab R, Bánhegyi G, et al. Molecular subtype specific efficacy of MEK inhibitors in pancreatic cancers. PLOS ONE. 2017;12:e0185687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee ES, Lee JM. Imaging diagnosis of pancreatic cancer: A state-of-the-art review. World J Gastroenterol 2014;20:7864–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viola-Villegas NT, Rice SL, Carlin S, Wu X, Evans MJ, Sevak KK, et al. Applying PET to Broaden the Diagnostic Utility of the Clinically Validated CA19.9 Serum Biomarker for Oncology. J Nucl Med 2013;54:1876–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sugyo A, Tsuji AB, Sudo H, Nagatsu K, Koizumi M, Ukai Y, et al. Preclinical evaluation of 89Zr-labeled human antitransferrin receptor monoclonal antibody as a PET probe using a pancreatic cancer mouse model. Nucl Med Commun 2015;36:286–94. [DOI] [PubMed] [Google Scholar]
- 48.Sugyo A, Tsuji AB, Sudo H, Okada M, Koizumi M, Satoh H, et al. Evaluation of Efficacy of Radioimmunotherapy with (90)Y-Labeled Fully Human Anti-Transferrin Receptor Monoclonal Antibody in Pancreatic Cancer Mouse Models. PLoS ONE. 2015;10:e0123761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sugyo A, Tsuji AB, Sudo H, Nomura F, Satoh H, Koizumi M, et al. Uptake of 111In-labeled fully human monoclonal antibody TSP-A18 reflects transferrin receptor expression in normal organs and tissues of mice. Oncol Rep 2017;37:1529–36. [DOI] [PubMed] [Google Scholar]
- 50.Long NM, Smith CS. Causes and imaging features of false positives and false negatives on (18)F-PET/CT in oncologic imaging. Insights Into Imaging. 2011;2:679–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.