

Abstract
Background
The circadian clock plays a crucial role in regulating physiology and is important for maintaining immune homeostasis and responses to inflammatory stimuli. Inflammatory arthritis often shows diurnal variation in disease symptoms and disease markers, and it is now established that cellular clocks regulate joint inflammation. The clock gene Bmal1 is critical for maintenance of 24-h rhythms and plays a key role in regulating immune responses, as well as in aging-related processes. Fibroblast-like synoviocytes (FLS) are circadian rhythmic joint mesenchymal cells which are important for maintenance of joint health and play a crucial role in the development of inflammatory arthritis. The aim of this study was to investigate the importance of the joint mesenchymal cell circadian clock in health and disease.
Methods
Mice were generated which lack Bmal1 in Col6a1-expressing cells, targeting mesenchymal cells in the ankle joints. Joints of these animals were assessed by X-ray imaging, whole-mount staining and histology, and the composition of the synovium was assessed by flow cytometry. Arthritis was induced using collagen antibodies.
Results
Bmal1 deletion in joint mesenchymal cells rendered the FLS and articular cartilage cells arrhythmic. Targeted mice exhibited significant changes in the architecture of the joints, including chondroid metaplasia (suggesting a switch of connective tissue stem cells towards a chondroid phenotype), reductions in resident synovial macrophages and changes in the basal pro-inflammatory activity of FLS. Loss of Bmal1 in FLS rendered these resident immune cells more pro-inflammatory in response to challenge, leading to increased paw swelling, localised infiltration of mononuclear cells and enhanced cytokine production in a model of arthritis.
Conclusions
This study demonstrates the importance of Bmal1 in joint mesenchymal cells in regulating FLS and chondrocyte development. Additionally, we have identified a role for this core clock component for restraining local responses to inflammation and highlight a role for the circadian clock in regulating inflammatory arthritis.
Electronic supplementary material
The online version of this article (10.1186/s13075-018-1770-1) contains supplementary material, which is available to authorized users.
Keywords: BMAL1, Circadian, Fibroblast-like synoviocytes, Chondrocyte, Arthritis, Macrophage, Synovium
Background
The circadian clock enables organisms to regulate their physiology in synchrony with the changing 24-h environment. This biological timer regulates a wide range of physiological processes, including metabolism and immunity [1, 2]. Multiple different cell types possess the clockwork machinery, a network of genes forming a transcriptional-translational feedback loop. In mammals the core clock genes include Bmal1, Clock, Cryptochrome (Cry) and Period (Per), where Bmal1 is the only non-redundant gene. Mice lacking Bmal1 from pre-natal development are behaviourally arrhythmic in the absence of an entraining light/dark cycle, and show loss of rhythmic physiology [3]. In addition to co-ordinating circadian rhythms, Bmal1 regulates other physiological functions, and these global knockout mice have reduced lifespan and fertility and exhibit pathologies affecting the eyes, brain and bone [4, 5]. This includes a progressive non-inflammatory arthropathy resulting in joint ankylosis [5]. Interestingly, this phenotype persists if Bmal1 is rescued in brain or muscle [6], but is absent if Bmal1 is only deleted after birth [7].
In the present study, we explored the contribution of Bmal1 in fibroblast-like synoviocytes (FLS) to joint architecture. FLS are found within the lining of the synovium, the thin organised membrane located between the joint cavity and joint capsule. They are stromal cells of mesenchymal origin, producing a range of extracellular matrix components and secreted factors essential to maintaining the normal environment of the synovial fluid and articular surface [8]. FLS play a critical role in the pathogenesis of inflammatory arthritis, producing inflammatory mediators which contribute to the recruitment and activation of leucocytes, cartilage breakdown and joint remodelling [9]. It is well established that the core clock proteins (PERIOD1/2, BMAL1 and CLOCK) are expressed by FLS [10, 11], and we and others have shown that these immunoregulatory cells are circadian rhythmic [11–13]. Intriguingly, there is mounting evidence that under chronic inflammatory conditions, such as rheumatoid arthritis, these intrinsic timers are disrupted [10, 11, 13–15].
By deleting Bmal1 in Col6a1-expressing cells we rendered joint mesenchymal cells (FLS and articular chondrocytes) arrhythmic. This targeted deletion had profound effects on joint architecture, homeostasis and inflammatory joint disease, highlighting the critical importance of the joint mesenchymal cell clock in health and disease.
Methods
Mice
B6.Cg-Tg(Col6a1-cre)1Gkl/Flmg mice, referred to hereinafter as Col6a1cre/+ mice, were purchased from the European Mutant Mouse Archive repository as frozen embryos and re-derived in-house. These mice, generated by Prof. G. Kollias [16], express Cre recombinase under the control of a collagen VI promoter cassette known to drive gene expression in mesenchymal cells in the ankle joints, mainly fibroblast-like cells but also articular chondrocytes [16, 17]. Bmal1flox/flox PER2::luc mice (as described previously [18]) were bred with Col6a1Cre/+ mice to produce Bmal1flox/flox PER2::luc Col6a1 Cre+/− mice (Col6a1-Bmal1−/−) and Bmal1flox/flox PER2::luc Col6a1 Cre−/− mice (wild-type counterparts). Global Bmal1−/− mice [19] were bred as heterozygous pairs to produce Bmal1−/− and wild-type littermates. All animal procedures were carried out in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986 and were subject to local ethical review by the University of Manchester Animals Welfare and Ethical Review Board. Mice were maintained under standard 12-h/12-h light/dark lighting with ad libitum access to standard chow. Unless stated otherwise, male mice aged 8–20 weeks were used.
Murine FLS cultures
Mouse FLS were cultured as described previously [12]. In brief, mice were killed, and their skin was removed from the hind paws, which were then cut 1 mm above the ankle joint and placed into Hanks’ balanced salt solution. After a wash, the paws were dissected between the toe and ankle joints using a scalpel blade, and the dissected tissue was placed into DMEM (containing 1% penicillin-streptomycin and 10% FBS) with 10 mg/ml collagenase (from Clostridium histolyticum type IV; Sigma-Aldrich, Gillingham, UK) and placed into a shaking incubator for 1.5 h at 37 °C. Cells (and some tissue debris) were pelleted by centrifugation, re-suspended in DMEM and plated out. Cells were cultured in DMEM to passage 3 before further experimental use, in order to obtain a purified population of FLS.
Bead sorting of FLS
Cultured mouse FLS (passage 3) were purified further by labelling them with a phycoerythrin (PE)-CD90.2 antibody (clone 30-H12) and positively selecting PE-labelled cells (EasySep PE Positive Selection Kit, as per kit instructions; STEMCELL Technologies, Vancouver, BC, Canada) for further culture. Bead-sorted FLS were used for Western blotting of the BMAL1 protein, as well as for qPCR analysis and in tumour necrosis factor (TNF)-α stimulation experiments (see below).
Murine macrophages
Mice were killed, and the peritoneal cavity was lavaged with two washes of 5 ml of RPMI (containing 1% penicillin-streptomycin and 10% FBS). The recovered media was centrifuged (432 × g for 10 min). For RNA analysis, the resultant pellet of peritoneal exudate cells was lysed immediately, and RNA extraction was carried out (as described below). For photomultiplier tube recordings, the pellet was re-suspended in RPMI, and cells were plated out into a 35-mm dish. After 2-h incubation at 37 °C, non-adherent cells were removed via three washes in warmed RPMI. For bone marrow-derived macrophages, mice were killed and the femur and tibia were removed. The bone marrow was flushed out with DMEM (containing 1% penicillin-streptomycin and 10% FBS), and the effluent was centrifuged (432 × g for 10 min). The pelleted cells were re-suspended in media containing 50 ng/ml macrophage colony-stimulating factor (M-CSF) (eBioscience, San Diego, CA, USA). The media were replaced after 3 days, and on day 6, the bone marrow-derived macrophages were scraped from the flask and lysed to extract protein for Western blot analysis (described below).
Bioluminescence recordings
FLS (passage 3) and peritoneal macrophages were plated out on 35-mm dishes. Primary murine lung fibroblasts were cultured as described elsewhere [20] and plated out on 35-mm dishes. Femoral head tissue was placed directly onto tissue culture inserts within the dishes. Cells and tissues were synchronised (200 nM dexamethasone, 1 h), and the media were replaced with recording media containing luciferin [21]. Dishes were sealed over with a glass coverslip using vacuum grease [22]. Bioluminescence from FLS, femoral head tissue and lung fibroblasts was recorded every minute using photomultiplier tubes. Bioluminescence data from peritoneal macrophages were collected and analysed using a LumiCycle (ActiMetrics, Wilmette, IL, USA). Data were plotted using Prism software (GraphPad Software, La Jolla, CA, USA).
Western blot analysis
Cultured FLS and bone marrow-derived macrophages were lysed using a radioimmunoprecipitation assay buffer (Sigma-Aldrich) containing cOmplete Mini, EDTA-free Protease Inhibitor Cocktail Tablets (Roche Diagnostics, Mannheim, Germany). Protein concentrations of cell lysates were measured with a bicinchoninic acid protein quantification kit (Merck, Kenilworth, NJ, USA), and an equal amount of protein was separated on SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Thermo Fisher Scientific, Waltham, MA, USA). Membranes were incubated with primary antibody against BMAL1 (D2L7G; Cell Signaling Technology, Danvers, MA, USA) and β-ACTIN (ab8226; Abcam, Cambridge, UK), followed by horseradish peroxidase-conjugated secondary antibody, and imaged using enhanced chemiluminescence (Clarity; Bio-Rad Laboratories, Hercules, CA, USA).
TNF-α stimulation of FLS
Bead-sorted FLS were stimulated with 10 ng/ml murine TNF-α (Sigma-Aldrich). Eight hours later the supernatant was removed. Cytokine levels in supernatants were quantified using the Bio-Plex Pro Mouse Cytokine 23-plex assay (Bio-Rad Laboratories) as per kit instructions, and run on a Bio-Plex 200 System. CXCL5 and M-CSF levels in supernatant were quantified separately using DuoSet enzyme-linked immunosorbent assay (ELISA) kits as per kit instructions (R&D Systems, Minneapolis, MN, USA).
Collagen antibody-induced arthritis
Male mice (aged 10 weeks) were administered a cocktail of collagen antibodies (ArthritoMab; MD Biosciences, Egg, Switzerland) intravenously (4 mg/mouse) on day 0, and lipopolysaccharide (100 μg/mouse) intraperitoneally on day 3. Both treatments were administered during the mid-light phase (Zeitgeber time [ZT] 6). Each paw was scored for disease severity using a 4-point scale (1 = inflammation of one digit; 2 = inflammation of more than one digit; 3 = inflammation and swelling of the footpad with or without involvement of one or more digits; 4 = severe inflammation and swelling of the footpad and ankle with or without involvement of one or more digits). Each paw score was totalled to produce an animal score. Paw thickness was measured in the hind limbs using a thickness gauge. Mice were killed on day 7, when serum was harvested along with the left hind paw for RNA analysis and the right hind paw was harvested for flow cytometry. ELISAs for interleukin (IL)-6 in mouse serum were performed using DuoSet ELISAs (R&D Systems) as per kit instructions.
qPCR
RNA was extracted from snap-frozen limbs and Peyer’s patches using TRIzol reagent (Life Technologies, Carlsbad, CA, USA). Frozen limbs were first pulverised using a pestle and mortar containing liquid nitrogen, and the resulting material was transferred to a Lysing Matrix D tube (MP Biomedicals, Santa Ana, CA, USA) and homogenised in TRIzol reagent using a FastPrep-24 machine (MP Biomedicals). Peyer’s patches were added directly to the Lysing Matrix D tubes containing TRIzol reagent for homogenisation. RNA was extracted using the standard TRIzol procedure. RNA was extracted from cultured primary cells using RNeasy kits (Qiagen, Hilden, Germany). After DNase treatment (DNase I, Thermo Fisher Scientific), RNA was converted to complementary DNA (cDNA) using the High-Capacity RNA-to-cDNA Kit (Thermo Fisher Scientific). qPCR was performed using a StepOne Real Time PCR system (Thermo Fisher Scientific) with TaqMan primers (see Additional file 1: Table S1). The housekeeping gene Gapdh was used to normalise data.
Flow cytometry
Whole limbs were dissected at the joints, and the tissue was digested in collagenase (as described earlier). Isolated cells were counted using a nucleocounter NC-250 (ChemoMetec, Allerod, Denmark). A sample (5 μl) of the cell suspension was diluted in PBS (1:4), and a solution of acridine orange and 4′,6-diamidino-2-phenylindole (Solution 18; ChemoMetec) was added to provide counts of total and dead cells, respectively. For flow cytometric analysis, the remaining cells were subjected to live/dead staining (LIVE/DEAD Fixable Blue Dead Cell Stain Kit; Thermo Fisher Scientific) for 20 min. ArC Amine Reactive Compensation Beads (Thermo Fisher Scientific) were used to obtain compensation controls for live/dead staining. After washing off the live/dead stain, an Fc block (CD16/CD32) was applied for 15 min, and then cells were stained with a cocktail of antibodies (Additional file 1: Tables S2 and S3). Cells were analysed on a BD LSR II flow cytometer (BD Biosciences, San Jose, CA, USA) after compensation set-up with OneComp eBeads (Thermo Fisher Scientific). Gating strategies are outlined in Additional file 1: Figures S4 and S5. Data were analysed using FlowJo software (FlowJo, Ashland, OR, USA). In some experiments FLS (CD45−CD90.2+) were sorted from these samples (BD Influx cell sorter; BD Biosciences) into lysis buffer, and RNA was prepared using RNeasy Plus Micro kits (Qiagen).
Histology
The skin was removed from hind limbs, which were then placed into formalin overnight. The next day, tissue was transferred to OSTEOSOFT (VWR, Radnor, PA, USA) for 2 weeks to allow de-calcification. De-calcified tissue was processed and embedded into paraffin wax. Sections (5 μm) were cut and mounted onto glass slides. After rehydration, slides were subjected to H&E staining or Safranin O staining using standard protocols. Slides were imaged on a Leica DM2000 microscope, and images were captured with a Leica DFC295 camera (Leica Microsystems, Wetzlar, Germany).
X-ray imaging
Mice were killed and X-rayed on a Faxitron MX-20 (32 kV for 55 s) (Faxitron Bioptics, Tucson, AZ, USA). Scanned X-ray films were analysed using ImageJ software. Using these images, pixel intensity was quantified in a standardised size ROI superior to the calcaneus or between vertebral disks. This value was adjusted for background intensity.
Whole-mount staining
Whole-mount staining was performed as described elsewhere [23]. In brief, skin and adipose tissue were removed from the hind limbs, and limbs were detached at the pelvis. Limbs were fixed in 95% ethanol for 48 h, then placed into 100% acetone (2 days). They were stained with Alcian blue solution for 3 days (0.03% wt/vol Alcian blue, 80% ethanol, 20% glacial acetic acid). After a brief wash (70% ethanol) they were de-stained overnight in 95% ethanol. Limbs were then placed in potassium hydroxide (1% wt/vol) for 4 h before staining with Alizarin red solution (0.005% wt/vol Alizarin red in 1% wt/vol KOH) for 4–5 days. After a wash in distilled water they were placed in 1% KOH for 2 days to begin clearing. Clearing solution was changed weekly, with decreasing amounts of KOH and increasing amounts of glycerol, until after 3 weeks limbs were placed in 100% glycerol for imaging and storage.
Statistical analysis
Flow cytometry data were analysed using FlowJo version 10 software. All other data were analysed using Prism 7 software. Data are presented as mean ± SEM. Where statistical tests were used, statistical significance is annotated as follows: *P < 0.05, **P < 0.01, ***P < 0.005.
Results
Bmal1 deletion renders FLS arrhythmic
Mice were generated which lack the core clock component Bmal1 in Col6a1-expressing cells (Fig. 1a). To confirm efficient gene targeting, FLS were cultured to passage 3 (61% purity) and were then further purified by selecting CD90.2+ cells (96% purity) (Fig. 1b). These cells were then subjected to qPCR and Western blot analysis. Results showed significantly reduced expression of the targeted gene (exon 8) (Fig. 1c) and reduced BMAL1 protein (Fig. 1d).
Fig. 1.

Targeting Bmal1 for deletion in joint mesenchymal cells. a The basic helix-loop-helix–encoding exon of Bmal1 was targeted for deletion in cells expressing Col6a1. b Fibroblast-like synoviocytes (FLS) were cultured from mouse hind limbs to passage 3, and assessed by flow cytometry for the absence of leukocytes (CD45−) and presence of the FLS marker CD90.2. CD90.2+ cells were then selected for further culture. c qPCR confirmed significant reduction in Bmal1 transcript in targeted cells (n = 3; unpaired t test), which corresponded with (d) loss of BMAL1 protein
Breeding these mice onto a PER2::luc background [21] permitted bioluminescence monitoring of cells and tissue from these mice as a readout of clock activity. Real-time luciferase recording under photomultiplier tubes confirmed Bmal1 deletion rendered FLS arrhythmic (Fig. 2a). Conversely, lung fibroblasts remained rhythmic (Additional file 1: Figure S1a). The femoral head, containing chondrocytes, also showed dampened PER2::luc rhythms (Fig. 2a). In further studies, cultured and purified FLS (as described above) were sampled every 4 h. As expected, these showed reduction in Bmal1 expression, loss of circadian oscillation in Dbp and Rev-erbα, and increased, non-oscillatory expression of Cry1 and Per2 (both repressed by BMAL1) (Fig. 2b). Finally, to confirm that the targeting rendered these cells arrhythmic in vivo, FLS were sorted by fluorescence-activated cell sorting (CD45−CD90.2+) from naïve limbs at opposing times of day (ZT6, mid-light versus ZT18, mid-dark). qPCR analysis revealed loss of rhythmic Per2 and impaired rhythmicity of Cry1 and Rev-erbα (Fig. 2c), confirming results from in vitro approaches.
Fig. 2.
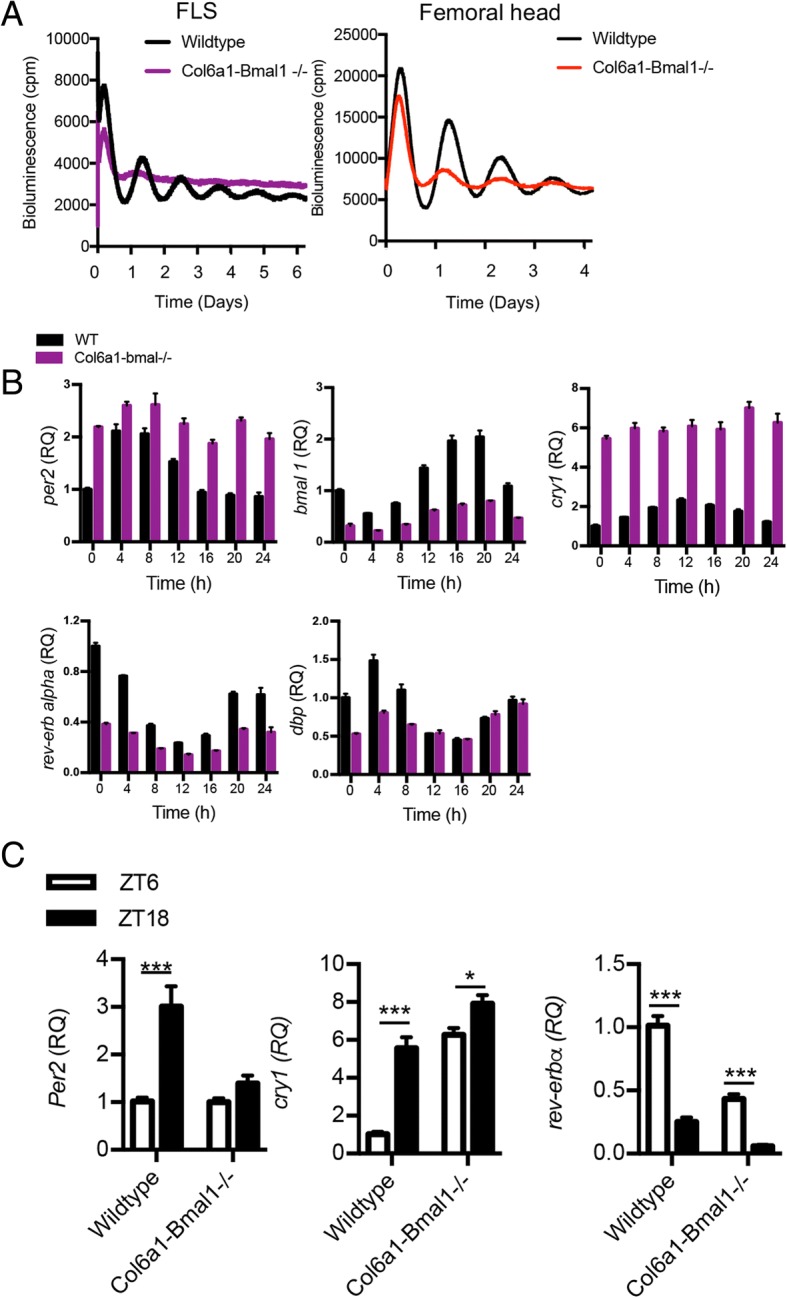
Deletion of Bmal1 in joint mesenchymal cells renders fibroblast-like synoviocytes (FLS) and chondrocytes arrhythmic. a Cultured and purified FLS and femoral head tissue from Col6a1-Bmal1−/− and wild-type animals (on a PER2::luc background) were placed under photomultiplier tubes to record bioluminescence (representative of n = 3/genotype). b Cultured FLS were harvested every 4 h for RNA extraction, and clock gene expression was profiled. Values are made relative to expression in wild-type cells at time point 0. c Clock gene expression in FLS (CD45−CD90.2+) sorted from the limbs of male and female mice (9–13 weeks) at ZT6 and ZT18 (n = 4–6). Two-way analysis of variance and post hoc Bonferroni correction were performed
Loss of Bmal1 in FLS significantly affects joint structure
Col6a1-Bmal1−/− animals showed thickening of the hind footpad (Fig. 3a), and this was evident in both sexes (Additional file 1: Figure S2a). X-ray imaging (Fig. 3b and Additional file 1: Figure S2b) and whole-mount staining (Fig. 3c) revealed density changes around the tarsocrural joint. There was also evidence of calcification of intervertebral disk space within the spine and tail (Additional file 1: Figure S2c and d), in keeping with observations in global Bmal1-/- animals [5] and mice lacking Bmal1 in cells of the intervertebral disk (including chondrocytes and cells of the annulus fibrosus, which are morphologically similar to fibroblasts) [24, 25]. By 9 months, there were signs that the phenotype could affect mobility and general well-being; consequently mice were not maintained beyond this age. Quantitative analysis of X-ray images of the hind limbs confirmed increased bone density in a defined region above the calcaneus in Col6a1-Bmal1−/− hind limbs (Fig. 3d). There was an increase in calcified tissue in the joint spaces between the metatarsal and phalangeal bones and within the ankles. A calcaneal spur was evident from 6 months. Similar observations had been reported in global Bmal1−/− mice, but the cell type responsible had not been determined [5]. In keeping with this, we observed that global Bmal1 deletion [19] also resulted in increased paw thickness, abnormalities between the metatarsal and phalanges, and a calcaneal spur (Additional file 1: Figure S3). Histological assessment of Col6a1-Bmal1−/− ankle joints in 14- to 17-week-old mice (Fig. 3e, f) revealed three distinctive changes restricted to the tarsocrural joint: (1) a thickening of the synovial subintima, in the absence of an increase in the number of surface synoviocytes; (2) erosion and exuberant chondro-osseous repair of the entheses at the condyles of the distal tibia with the formation of a spur; and (3) nodular chondroid metaplasia at the interface between the synovium, the ankle joint capsule and the damaged/healing entheses. Morphologically, there was minimal neutrophilic or lymphocytic inflammation either in the thickened synovium or at the site of the enthesopathy. The thickened synovium was generally fibrotic, but at its junction with bone and capsule consisted of adipose tissue. It contained a mixture of round and spindle-shaped connective tissue cells and infrequent cells resembling histiocytes. The disordered cartilage was at various stages of maturation, with many cells large and hypertrophic (a phenotype associated with endochondral ossification). These changes reflect observations by Bunger et al. in the knees of global Bmal1−/− mice, describing areas of calcification and ossification around tendons and ligaments closely associated with bone insertion sites [5].
Fig. 3.

Effect of Bmal1 deletion in joint mesenchymal cells on joint development. a Hind paw thickness (in mm) plotted against animal weight (in g) measured in male (7 wild-type, 8 Col6a1-Bmal1−/−) and female (18 wild-type, 7 Col6a1-Bmal1−/−) animals. b X-ray images at 9 months. c Whole-mount staining of the hind limbs from 9-month-old wild-type and Col6a1-Bmal1−/− mice. d Quantification of pixel intensity from x-rays of 9-month-old mice in an ROI superior to the calcaneus (n = 4–6 limbs). Analysis was by unpaired t test. e Safranin O staining of ankle joints from 14- to 17-week-old mice where cartilage is stained red. Scale bar represents 500 μm. f H&E staining of ankle joints from 14- to 17 week old mice. Scale bar represents 500 μm. Col6a1-Bmal1−/− mice show thickened synovium (S); the enthesis (N) is eroded (dashed line delimits the interface between eroded bone and repair tissue), and the erosion is filled with exuberant chondro-osseous repair tissue (C) with the formation of a spur (P); there is chondroid metaplasia (M) at the junction of the synovium, joint capsule and enthesis repair tissue. The chondroid metaplasia differs from the chondro-osseous repair tissue in its morphology and tinctorial reaction with Safranin O. The synovium is fibrotic (B) and contains a mixture of spindle and round connective tissue cells. At its interfaces with connective tissues other than metaplastic chondroid tissue, there is a marked increase in adipocytes (A)
To further investigate these histological changes, cells were collagenase-digested from limbs for flow cytometric analysis. Strikingly, numbers of FLS were markedly reduced in Col6a1-Bmal1−/− joints, supporting histological evidence for a phenotypic switch from FLS to chondrocytes (Fig. 4a). Neutrophil numbers were consistent between genotypes. In keeping with others [26] we identified two populations of macrophages within the joints based on differential expression of major histocompatibility complex (MHC) class II. Numbers of MHC II− macrophages, which make up the majority of the macrophages found within the synovial lining and are considered true tissue-resident macrophages, were reduced in Col6a1-Bmal1−/− mice. Numbers of MHC II+ macrophages, considered to originate in the bone marrow, although reduced, were not significantly altered in Col6a1-Bmal1−/− animals. qPCR analysis of whole hind limbs revealed that chondrocyte-associated genes were significantly upregulated in Col6a1-Bmal1−/− animals. Expression of Col2a1, aggrecan, proteoglycan 4 and Indian hedgehog were significantly increased within the hind paws in the absence of mesenchymal cell Bmal1, further indicating increased numbers of active chondrocytes (Fig. 4b).
Fig. 4.

Bmal1 deletion alters the balance of fibroblasts and chondrocytes within joints. a Flow cytometric analysis of the cellular composition of the joints from wild-type (n = 9) and Col6a1-Bmal1−/− (n = 12) mice (aged 9–18 weeks) after exclusion of doublets and dead cells. Analysis by unpaired t test. b Expression of chondrocyte-associated genes in the hind limbs of mice, aged 10 weeks (n = 4). Analysis by unpaired t test
Bmal1 in FLS represses inflammatory arthritis
To investigate the role of Bmal1 in joint mesenchymal cells under inflammatory conditions, arthritis was induced using a cocktail of collagen antibodies. Disease was evident 5 days after administration. There was no genotypic difference in disease score; however, swelling of affected paws was greater in Col6a1-Bmal1−/− animals than in wild-type animals (Fig. 5a). Serum IL-6 concentrations were also significantly higher in Col6a1-Bmal1−/− animals (Fig. 5b), in keeping with a more aggressive inflammatory arthritis. Infiltrating neutrophils and Ly6Chi (but not Ly6Clo) monocytes were increased in arthritic Col6a1-Bmal1−/− limbs (Fig. 5c and d). In keeping with this, pro-inflammatory cytokine transcripts (Il6, Cxcl1, Ccl2, Cxcl5) and receptor activator of nuclear factor κ ligand (Rankl) transcript levels were significantly higher. There were no discernible genotypic differences in cytokines that have been attributed to an anti-inflammatory role in inflammatory arthritis (Il10, Ifnγ and Il13) [27–29] (Fig. 5e).
Fig. 5.
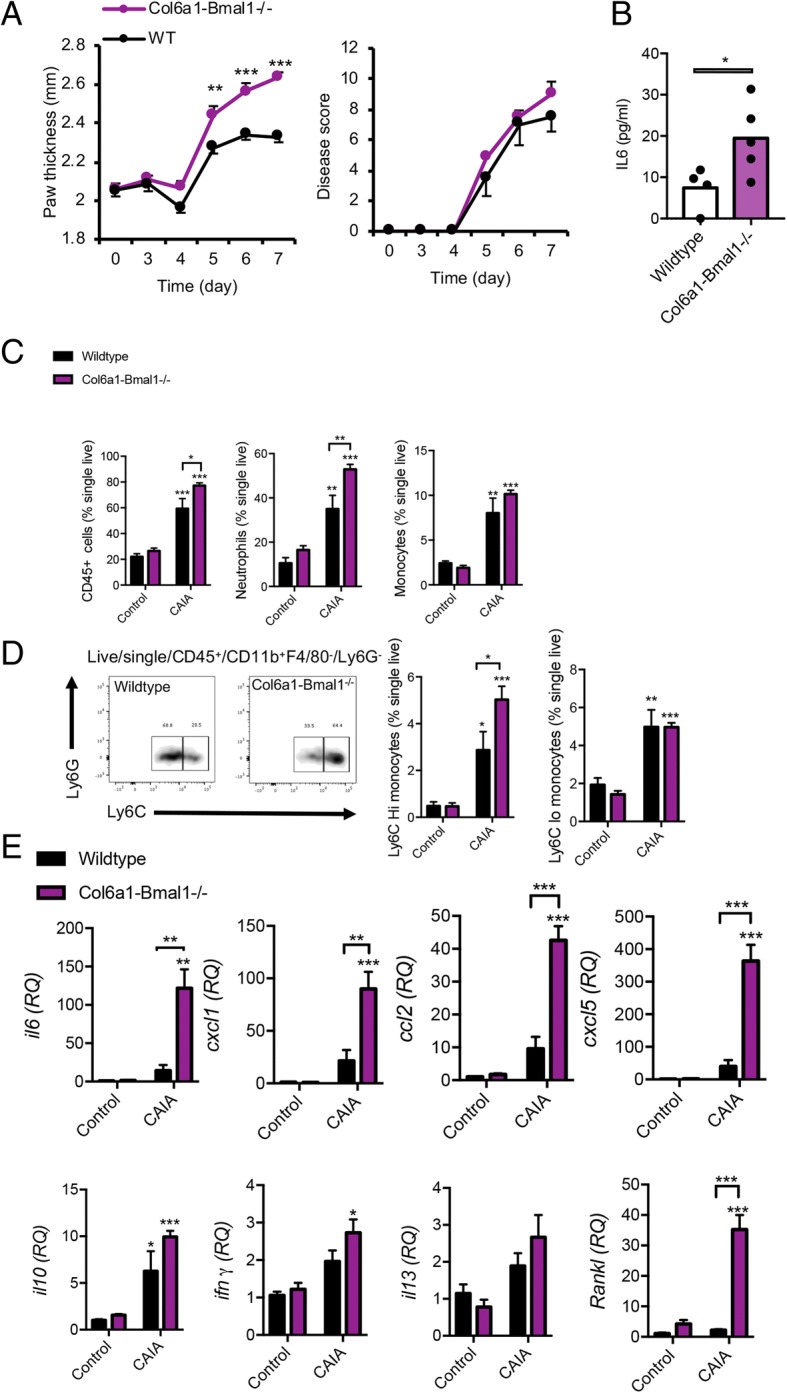
Enhanced response of Col6a1-Bma1−/− mice to collagen antibody-induced arthritis. a Hind paw thickness and disease score after initiation of collagen antibody-induced arthritis (CAIA) in wild-type (n = 4) and Col6a1-Bmal1−/− (n = 5) mice. One-way analysis of variance (ANOVA) and Bonferroni multiple comparisons. b Serum IL-6 levels in CAIA animals at the time they were killed on day 7. Analysis by t test. c Quantification of leukocytes within hind paws of arthritic (n = 4–5/group) and non-arthritic (n = 4/group) animals. Two-way ANOVA and post hoc Bonferroni correction. Stars directly above CAIA bars indicate significant difference compared with control counterparts. d Representative plots showing increased presence of LycChi monocytes in inflamed limbs of Col6a1-Bmal1−/− mice, and quantification of Ly6Chi and Ly6Clo monocytes within hind paws of arthritic (n = 4–5/group) and non-arthritic (n = 4/group) animals. Two-way ANOVA and post hoc Bonferroni correction. Stars directly above CAIA bars indicate significant differences compared with control counterparts. e Transcripts of inflammatory cytokines in hind limbs harvested from control (n = 4/group) and CAIA mice (n = 4–5/group). Two-way ANOVA and post hoc Bonferroni correction. Stars directly above CAIA bars indicate significant difference compared with control counterparts
To assess how FLS contribute to this enhanced inflammatory response, cultured, purified FLS were stimulated with the pro-inflammatory cytokine TNF-α, and the supernatant was analysed for cytokines by performing a Bio-Plex assay (Fig. 6 and Additional file 1: Table S4). After stimulation, Bmal1−/− FLS secreted higher levels of IL-6, CXCL1, CCL2 and CXCL5, and levels of cytokines with known anti-inflammatory properties in inflammatory arthritis (IL-10, interferon [IFN]-γ and IL-13) were dampened. Interestingly, under basal conditions, Bmal1 loss results in increased secretion of several pro-inflammatory cytokines (e.g., CCL2 and CXCL5), but this did not reach statistical significance. Together these data suggest disruption of the basal inflammatory state in FLS in the absence of Bmal1, as well as an enhanced pro-inflammatory response in a stimulatory environment.
Fig. 6.

Altered response of fibroblast-like synoviocytes (FLS) lacking Bma1−/− to inflammatory stimuli. FLS were cultured from Col6a1-Bmal1−/− and wild-type littermates to passage 3 and purified for CD90.2 expression before stimulation for 8 h with tumour necrosis factor (TNF)-α. Expression of inflammatory cytokines in cell supernatants was assessed by Bioplex and enzyme-linked immunosorbent assay (CXCL5 only) (n = 5–6). One-way analysis of variance and post hoc Bonferroni correction. Stars directly above TNF-α bars indicate significant difference compared with control counterparts
Discussion
This study addresses the role of the core clock component Bmal1 in defining musculoskeletal structure. By deleting Bmal1 in a targeted cell population, we were able to identify the cell lineage responsible for a previously recognised joint arthropathy. Bmal1 deletion in joint mesenchymal cells rendered FLS and articular cartilage arrhythmic and led to disruption of ankle joint architecture. This was evidenced by focal chondroid metaplasia, decreased resident synovial macrophage numbers, and altered basal secretion of a subset of cytokines by FLS in the native state.
Global Bmal1-knockout mice show a progressive ectopic mineralisation with new bone formation at tendon and ligament insertion sites [5]. Our findings suggest this ossification may be a consequence of a change in mesenchymal stem cell differentiation towards a bone-depositing chondroid phenotype. These pathological chondrocytes drive calcification of the tendon sheath to form a calcaneal spur. Global deletion of Bmal1 in adult mice does not result in this abnormal calcification [7]; therefore, it is likely a consequence of the absence of Bmal1 in mesenchymal cells during embryogenesis/early post-natal development. Articular chondrocytes and synovial fibroblasts are derived from common joint progenitor cells, with the action of different factors driving them towards a chondrogenic or anti-chondrogenic lineage [30]. Furthermore, Col6a1 is present in synovial membrane and articular cartilage in 16.5-day murine embryos [31]. We suggest that BMAL1 expression in joint mesenchymal cells may be essential for regulating the balance between FLS and chondrocytes of the developing embryo. In support of this, higher expression levels of chondrocyte-associated genes were detected in the hind limbs of Col6a1-Bmal1−/− mice. This includes genes encoding for type II collagen and aggrecan (major structural components of cartilage), proteoglycan 4 (synthesised by chondrocytes) and Indian hedgehog (associated with chondrocyte differentiation, proliferation and maturation). Intriguingly, others have shown that targeted ablation of Bmal1 in mouse chondrocytes causes progressive degeneration of articular cartilage within the knee, with loss of chondrocytes and extracellular matrix evident from 3 months of age [32]. Taken together, it is clear that Bmal1 within mesenchymal cells is critical for joint health throughout development and later life.
Col6a1-Bmal1−/− animals demonstrated further alterations in the cellular composition of the synovial tissue. Tissue-resident MHC II− macrophages were significantly reduced within the joints. Resident synovial macrophages play critical roles in immune surveillance, maintenance of tissue integrity and limiting inflammation. It should be noted that human and murine macrophages express Col6a1 and secrete collagen VI protein [33, 34]. Macrophages derived from Col6a1-Bmal1−/− mice express low levels of Col6a1 and Cre (data not shown). However, Cre expression was insufficient to drive recombination and did not affect expression of the targeted exon of Bmal1 (exon 8) within the macrophage (Additional file 1: Figure S6a). In keeping with this, expression of BMAL1 protein was unchanged (Additional file 1: Figure S6b). Furthermore, Col6a1-Bmal1−/− macrophages showed robust circadian rhythms in PER2::luc bioluminescence (Additional file 1: Figure S6c). (Previous work demonstrated that efficient deletion of Bmal1 renders these cells arrhythmic [18].) Thus we suggest that the observed change in numbers of MHC II− macrophages within the joints is a consequence of loss of BMAL1 in the mesenchymal cell population. The differentiation and function of resident synovial macrophages is governed by (as yet undetermined) tissue-specific cues most likely derived from FLS [35]. Our data suggest that Bmal1 deletion in mesenchymal joint cells indirectly affects the joint macrophage population by disrupting signalling between FLS and synovial macrophages. Indeed, we saw that in stimulated FLS the expression of granulocyte-macrophage colony-stimulating factor, which regulates the proliferation and differentiation of macrophages, was significantly decreased in the absence of Bmal1 (Additional file 1: Table S4). Conversely, M-CSF levels released from FLS lacking Bmal1 were heightened. Taken together, this suggests that Bmal1 is important for the regulation of cytokine signals from FLS which facilitate the proliferation, differentiation and survival of macrophages within the synovium. The disordered macrophage population may contribute to the destructive joint phenotype, and may also affect the propagation and resolution of inflammatory arthritis.
FLS are key players in inflammatory arthritis, releasing a range of pro-inflammatory mediators. Chondrocytes respond to these mediators (e.g., TNF-α and IL-1) by releasing matrix-degrading proteinases, resulting in joint damage. Chondrocytes do also have the capacity to release pro-inflammatory cytokines in response to stimulation [36], and so may also contribute as a cellular source of inflammatory mediators which damage the joint [37]. Col6a1-Bmal1−/− mice showed enhanced localised inflammation after induction of arthritis. In comparison to wild-type counterparts, the affected limbs exhibited enhanced swelling, increased expression of pro-inflammatory cytokines (e.g., Ccl2, Cxcl1 and Cxcl5) and increased infiltration of neutrophils and Ly6Chi monocytes. Both Ly6Chi and Ly6Clo monocytes were recruited to the joints during collagen antibody-induced arthritis. Interestingly, we did not see significant differences in numbers of infiltrating Ly6Clo monocytes between the two genotypes, only Ly6Chi. Recent work has established that Ly6Clo monocytes have an increased potential to differentiate into osteoclasts and inflammatory macrophages, which play a key role in the destruction of articular bone during arthritis [26, 38]. Furthermore, given the increased levels of Rankl (which plays a role in bone erosion) in the arthritic joints of Col6a1-Bmal1−/− animals, it would be interesting to determine, in future studies, the effects of loss of Bmal1 in joint mesenchymal cells on bone erosion. CCL2 drives monocyte recruitment, whilst CXCL1 and CXCL5 are neutrophil chemoattractants and thus likely explain the observed increase in monocytes and neutrophils. This more severe inflammatory phenotype extended to a systemic response, because arthritic Col6a1-Bmal1−/− mice exhibited increased levels of circulating IL-6. Some stromal cells within secondary lymphoid organs (Peyer’s patches and isolated lymphoid follicles) express Col6a1 [39]. In control experiments, qPCR analysis confirmed the expression of Col6a1 in Peyer’s patches of wild-type and Col6a1-Bmal1−/− animals (Additional file 1: Figure S1b) and Cre expression in Col6a1-Bmal1−/− animals only (data not shown). However, this did not affect expression of Bmal1 (Additional file 1: Figure S1b). Consequently, this increase in systemic IL-6 is most likely a consequence of targeting of joint-resident stroma rather than an extra-articular source.
In vitro studies demonstrated that loss of Bmal1 in FLS promotes enhanced secretion of pro-inflammatory cytokines and reduced secretion of anti-inflammatory cytokines after stimulation, suggesting that Bmal1 within FLS is critical for restraining joint inflammation. Cultured Col6a1-Bmal1−/− FLS demonstrated elevated release of a subset of pro-inflammatory cytokines under naïve conditions. This reflects observations in pulmonary Club cells [40], macrophages and monocytes [41], where BMAL1 regulates recruitment of the glucocorticoid receptor (Club cells) and the polycomb repressive complex (macrophages/monocytes) to target genes, resulting in repression of Cxcl5 and Ccl2 transcription, respectively. Given the role of Bmal1 in restraining joint inflammation, it is important to consider the effects of the chronic inflammatory environment on Bmal1 expression. It is reported that TNF-α induces the expression of Bmal1 in human synovial fibroblasts via calcium-dependent pathways [11, 15]. Similarly, studies have shown higher levels of BMAL1 protein in synovium from patients with rheumatoid arthritis than in synovium from patients with osteoarthritis [13]. In light of our findings, we suggest that this enhanced BMAL1 expression in rheumatoid arthritis may in fact act to dampen inflammation.
Conclusions
In the present study, we demonstrated the critical importance of Bmal1 in joint mesenchymal cells in regulating FLS and chondrocyte development within the joints. Our data provide a mechanism to explain the abnormal bone phenotype and altered cellular composition of the joint associated with pre-natal deletion of Bmal1. Additionally, we identified a role for Bmal1 in FLS for restraining local responses to inflammation and highlighted a role for the circadian clock in regulating inflammatory arthritis.
Additional file
Supplementary data. (PDF 2885 kb)
Acknowledgements
We thank the Transgenic Core Facilities for re-deriving the Col6a1Cre mice, the Flow Cytometry Core Facility for assistance with fluorescence assisted cell sorting, and the Histology Core Facility for advice on tissue processing and staining. We acknowledge the kind gifts of Col6a1Cre mice from Prof. G. Kollias and global Bmal1-knockout mice from Prof A. Reddy. We thank Dr. M. Dudek and Prof. Q. J. Meng for their assistance with isolating articular cartilage and X-ray imaging, and Dr. James Early for assistance with culturing murine bone marrow-derived macrophages.
Funding
This work was supported by Arthritis Research UK (grant number 20629 awarded to JEG) and the Medical Research Council (grant number MR/L018640/1 awarded to JEG).
Availability of data and materials
The datasets used and analysed during this study are available from the corresponding author on reasonable request.
Abbreviations
- Cry
Cryptochrome
- ELISA
Enzyme-linked immunosorbent assay
- FLS
Fibroblast-like synoviocytes
- IFN
Interferon
- IL
Interleukin
- M-CSF
Macrophage colony-stimulating factor
- MHC
Major histocompatibility complex
- PE
Phycoerythrin
- Per
Period
- RANKL
Receptor activator of nuclear factor κ ligand
- TNF-α
Tumour necrosis factor-α
- ZT
Zeitgeber time
Authors’ contributions
JEG and LEH designed the study. LEH, SHD and JEG carried out the experiments, data collection and analysis. JEG, DWR and AJF prepared the manuscript. JEG supervised all the experiments. All authors read and approved the final manuscript.
Ethics approval
All animal procedures were carried out in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986 and were subject to local ethical review by the University of Manchester Animals Welfare and Ethical Review Board.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Laura E. Hand, Email: laura.hand@manchester.ac.uk
Suzanna H. Dickson, Email: suzanna.dickson@manchester.ac.uk
Anthony J. Freemont, Email: tony.freemont@manchester.ac.uk
David W. Ray, Email: david.w.ray@manchester.ac.uk
Julie E. Gibbs, Phone: +44 (0)161 275 5470, Email: julie.gibbs@manchester.ac.uk
References
- 1.Scheiermann C, Gibbs J, Ince L, Loudon A. Clocking in to immunity. Nat Rev Immunol. 2018;18(7):423–437. doi: 10.1038/s41577-018-0008-4. [DOI] [PubMed] [Google Scholar]
- 2.Dibner C, Schibler U. Circadian timing of metabolism in animal models and humans. J Intern Med. 2015;277(5):513–527. doi: 10.1111/joim.12347. [DOI] [PubMed] [Google Scholar]
- 3.Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103(7):1009–1017. doi: 10.1016/S0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 2006;20(14):1868–1873. doi: 10.1101/gad.1432206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bunger MK, Walisser JA, Sullivan R, Manley PA, Moran SM, Kalscheur VL, Colman RJ, Bradfield CA. Progressive arthropathy in mice with a targeted disruption of the Mop3/Bmal-1 locus. Genesis. 2005;41(3):122–132. doi: 10.1002/gene.20102. [DOI] [PubMed] [Google Scholar]
- 6.McDearmon EL, Patel KN, Ko CH, Walisser JA, Schook AC, Chong JL, Wilsbacher LD, Song EJ, Hong HK, Bradfield CA, et al. Dissecting the functions of the mammalian clock protein BMAL1 by tissue-specific rescue in mice. Science. 2006;314(5803):1304–1308. doi: 10.1126/science.1132430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang G, Chen L, Grant GR, Paschos G, Song WL, Musiek ES, Lee V, McLoughlin SC, Grosser T, Cotsarelis G, et al. Timing of expression of the core clock gene Bmal1 influences its effects on aging and survival. Sci Transl Med. 2016;8(324):324ra316. doi: 10.1126/scitranslmed.aad3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwanaga T, Shikichi M, Kitamura H, Yanase H, Nozawa-Inoue K. Morphology and functional roles of synoviocytes in the joint. Arch Histol Cytol. 2000;63(1):17–31. doi: 10.1679/aohc.63.17. [DOI] [PubMed] [Google Scholar]
- 9.Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233(1):233–255. doi: 10.1111/j.0105-2896.2009.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Becker T, Tohidast-Akrad M, Humpeler S, Gerlag DM, Kiener HP, Zenz P, Steiner G, Ekmekcioglu C. Clock gene expression in different synovial cells of patients with rheumatoid arthritis and osteoarthritis. Acta Histochem. 2014;116(7):1199–1207. doi: 10.1016/j.acthis.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 11.Haas S, Straub RH. Disruption of rhythms of molecular clocks in primary synovial fibroblasts of patients with osteoarthritis and rheumatoid arthritis, role of IL-1β/TNF. Arthritis Res Ther. 2012;14(3):R122. doi: 10.1186/ar3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hand LE, Hopwood TW, Dickson SH, Walker AL, Loudon AS, Ray DW, Bechtold DA, Gibbs JE. The circadian clock regulates inflammatory arthritis. FASEB J. 2016;30(11):3759–3770. doi: 10.1096/fj.201600353R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kouri VP, Olkkonen J, Kaivosoja E, Ainola M, Juhila J, Hovatta I, Konttinen YT, Mandelin J. Circadian timekeeping is disturbed in rheumatoid arthritis at molecular level. PLoS One. 2013;8(1):e54049. doi: 10.1371/journal.pone.0054049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee H, Nah SS, Chang SH, Kim HK, Kwon JT, Lee S, Cho IH, Lee SW, Kim YO, Hong SJ, et al. PER2 is downregulated by the LPS-induced inflammatory response in synoviocytes in rheumatoid arthritis and is implicated in disease susceptibility. Mol Med Rep. 2017;16(1):422–428. doi: 10.3892/mmr.2017.6578. [DOI] [PubMed] [Google Scholar]
- 15.Yoshida K, Nakai A, Kaneshiro K, Hashimoto N, Suzuki K, Uchida K, Hashimoto T, Kawasaki Y, Tateishi K, Nakagawa N, et al. TNF-α induces expression of the circadian clock gene Bmal1 via dual calcium-dependent pathways in rheumatoid synovial cells. Biochem Biophys Res Commun. 2018;495(2):1675–1680. doi: 10.1016/j.bbrc.2017.12.015. [DOI] [PubMed] [Google Scholar]
- 16.Armaka M, Apostolaki M, Jacques P, Kontoyiannis DL, Elewaut D, Kollias G. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J Exp Med. 2008;205(2):331–337. doi: 10.1084/jem.20070906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakkou M, Chouvardas P, Ntari L, Prados A, Moreth K, Fuchs H, Gailus-Durner V, Hrabe de Angelis M, Denis MC, Karagianni N, et al. Mesenchymal TNFR2 promotes the development of polyarthritis and comorbid heart valve stenosis. JCI Insight. 2018;3(7):98864. doi: 10.1172/jci.insight.98864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibbs JE, Blaikley J, Beesley S, Matthews L, Simpson KD, Boyce SH, Farrow SN, Else KJ, Singh D, Ray DW, et al. The nuclear receptor REV-ERBα mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc Natl Acad Sci U S A. 2012;109(2):582–587. doi: 10.1073/pnas.1106750109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edgar RS, Stangherlin A, Nagy AD, Nicoll MP, Efstathiou S, O’Neill JS, Reddy AB. Cell autonomous regulation of herpes and influenza virus infection by the circadian clock. Proc Natl Acad Sci U S A. 2016;113(36):10085–10090. doi: 10.1073/pnas.1601895113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meng QJ, McMaster A, Beesley S, Lu WQ, Gibbs J, Parks D, Collins J, Farrow S, Donn R, Ray D, et al. Ligand modulation of REV-ERBα function resets the peripheral circadian clock in a phasic manner. J Cell Sci. 2008;121(Pt 21):3629–3635. doi: 10.1242/jcs.035048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, et al. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A. 2004;101(15):5339–5346. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gibbs JE, Beesley S, Plumb J, Singh D, Farrow S, Ray DW, Loudon AS. Circadian timing in the lung; a specific role for bronchiolar epithelial cells. Endocrinology. 2009;150(1):268–276. doi: 10.1210/en.2008-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rigueur D, Lyons KM. Whole-mount skeletal staining. Methods Mol Biol. 2014;1130:113–121. doi: 10.1007/978-1-62703-989-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dudek M, Yang N, Ruckshanthi JP, Williams J, Borysiewicz E, Wang P, Adamson A, Li J, Bateman JF, White MR, et al. The intervertebral disc contains intrinsic circadian clocks that are regulated by age and cytokines and linked to degeneration. Ann Rheum Dis. 2017;76(3):576–584. doi: 10.1136/annrheumdis-2016-209428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pattappa G, Li Z, Peroglio M, Wismer N, Alini M, Grad S. Diversity of intervertebral disc cells: phenotype and function. J Anat. 2012;221(6):480–496. doi: 10.1111/j.1469-7580.2012.01521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK, 3rd, Berdnikovs S, Filer A, Clark AR, Buckley CD, et al. Nonclassical Ly6C− monocytes drive the development of inflammatory arthritis in mice. Cell Rep. 2014;9(2):591–604. doi: 10.1016/j.celrep.2014.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee SH, Kwon JY, Kim SY, Jung K, Cho ML. Interferon-γ regulates inflammatory cell death by targeting necroptosis in experimental autoimmune arthritis. Sci Rep. 2017;7(1):10133. doi: 10.1038/s41598-017-09767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams AS, Richards PJ, Thomas E, Carty S, Nowell MA, Goodfellow RM, Dent CM, Williams BD, Jones SA, Topley N. Interferon-γ protects against the development of structural damage in experimental arthritis by regulating polymorphonuclear neutrophil influx into diseased joints. Arthritis Rheum. 2007;56(7):2244–2254. doi: 10.1002/art.22732. [DOI] [PubMed] [Google Scholar]
- 29.Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P. Accelerated collagen-induced arthritis in IFN-γ receptor-deficient mice. J Immunol. 1997;158(11):5507–5513. [PubMed] [Google Scholar]
- 30.Koyama E, Shibukawa Y, Nagayama M, Sugito H, Young B, Yuasa T, Okabe T, Ochiai T, Kamiya N, Rountree RB, et al. A distinct cohort of progenitor cells participates in synovial joint and articular cartilage formation during mouse limb skeletogenesis. Dev Biol. 2008;316(1):62–73. doi: 10.1016/j.ydbio.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braghetta P, Fabbro C, Piccolo S, Marvulli D, Bonaldo P, Volpin D, Bressan GM. Distinct regions control transcriptional activation of the α1(VI) collagen promoter in different tissues of transgenic mice. J Cell Biol. 1996;135(4):1163–1177. doi: 10.1083/jcb.135.4.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dudek M, Gossan N, Yang N, Im HJ, Ruckshanthi JP, Yoshitane H, Li X, Jin D, Wang P, Boudiffa M, et al. The chondrocyte clock gene Bmal1 controls cartilage homeostasis and integrity. J Clin Invest. 2016;126(1):365–376. doi: 10.1172/JCI82755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnoor M, Cullen P, Lorkowski J, Stolle K, Robenek H, Troyer D, Rauterberg J, Lorkowski S. Production of type VI collagen by human macrophages: a new dimension in macrophage functional heterogeneity. J Immunol. 2008;180(8):5707–5719. doi: 10.4049/jimmunol.180.8.5707. [DOI] [PubMed] [Google Scholar]
- 34.Chen P, Cescon M, Zuccolotto G, Nobbio L, Colombelli C, Filaferro M, Vitale G, Feltri ML, Bonaldo P. Collagen VI regulates peripheral nerve regeneration by modulating macrophage recruitment and polarization. Acta Neuropathol. 2015;129(1):97–113. doi: 10.1007/s00401-014-1369-9. [DOI] [PubMed] [Google Scholar]
- 35.Kurowska-Stolarska M, Alivernini S. Synovial tissue macrophages: friend or foe? RMD Open. 2017;3(2):e000527. doi: 10.1136/rmdopen-2017-000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aida Y, Maeno M, Suzuki N, Namba A, Motohashi M, Matsumoto M, Makimura M, Matsumura H. The effect of IL-1β on the expression of inflammatory cytokines and their receptors in human chondrocytes. Life Sci. 2006;79(8):764–771. doi: 10.1016/j.lfs.2006.02.038. [DOI] [PubMed] [Google Scholar]
- 37.Otero M, Goldring MB. Cells of the synovium in rheumatoid arthritis. Chondrocytes. Arthritis Res Ther. 2007;9(5):220. doi: 10.1186/ar2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puchner A, Saferding V, Bonelli M, Mikami Y, Hofmann M, Brunner JS, Caldera M, Goncalves-Alves E, Binder NB, Fischer A, et al. Non-classical monocytes as mediators of tissue destruction in arthritis. Ann Rheum Dis. 2018;77(10):1490–1497. doi: 10.1136/annrheumdis-2018-213250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prados A, Kollias G, Koliaraki V. CollagenVI-Cre mice: a new tool to target stromal cells in secondary lymphoid organs. Sci Rep. 2016;6:33027. doi: 10.1038/srep33027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibbs JE, Ince L, Matthews L, Mei J, Bell T, Yang N, Saer B, Begley N, Poolman T, Pariollaud M, Farrow S, Demayo F, Hussell T, Worthen GS, Ray D, Loudon A. An epithelial circadian clock controls pulmonary inflammation, and glucocorticoid action. Nat Med. 2014;20:919–26. [DOI] [PMC free article] [PubMed]
- 41.Nguyen KD, Fentress SJ, Qiu Y, Yun K, Cox JS, Chawla A. Circadian gene Bmal1 regulates diurnal oscillations of Ly6Chi inflammatory monocytes. Science. 2013;341(6153):1483–1488. doi: 10.1126/science.1240636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data. (PDF 2885 kb)
Data Availability Statement
The datasets used and analysed during this study are available from the corresponding author on reasonable request.