

Abstract
Telomeres and the shelterin complex cap and protect the ends of chromosomes. Telomeres are flanked by the subtelomeric sequences that have also been implicated in telomere regulation, although their role is not well defined. Here, we show that, in Schizosaccharomyces pombe, the telomere‐associated sequences (TAS) present on most subtelomeres are hyper‐recombinogenic, have metastable nucleosomes, and unusual low levels of H3K9 methylation. Ccq1, a subunit of shelterin, protects TAS from nucleosome loss by recruiting the heterochromatic repressor complexes CLRC and SHREC, thereby linking nucleosome stability to gene silencing. Nucleosome instability at TAS is independent of telomeric repeats and can be transmitted to an intrachromosomal locus containing an ectopic TAS fragment, indicating that this is an intrinsic property of the underlying DNA sequence. When telomerase recruitment is compromised in cells lacking Ccq1, DNA sequences present in the TAS promote recombination between chromosomal ends, independent of nucleosome abundance, implying an active function of these sequences in telomere maintenance. We propose that Ccq1 and fragile subtelomeres co‐evolved to regulate telomere plasticity by controlling nucleosome occupancy and genome stability.
Keywords: genome stability, heterochromatin, nucleosomes, shelterin, telomeres
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics
Introduction
The chromatin of eukaryotic cells is organized into structural and functional domains that are crucial for genome stability. For example, centromeres and telomeres are composed of large repetitive DNA elements assembled into heterochromatin, which mediate the faithful inheritance and integrity of chromosomes. Telomeres are composed of G/T‐rich tandem DNA repeats that provide docking sites for telomere‐binding proteins to protect the chromosomal ends from erosion and fusion. In humans and Schizosaccharomyces pombe, telomere protection is mediated by the shelterin complex, which consists of six subunits (Fig 1A): Pot1 (hPOT1); Tpz1 (hTPP1); Poz1 (hTIN2); Rap1; Taz1 (hTRF1 and hTRF2) 1, 2, 3, 4. Fission yeast has an additional subunit Ccq1, which associates with shelterin via Tpz1 and is essential for maintaining telomere length through recruiting telomerase 2, 5, 6, 7, 8, 9, 10. When telomeres shorten to a critical size (after about 100 generations), cells undergo telomere crisis resulting in cell cycle arrest and cellular senescence 11. However, survivors can arise through telomerase‐independent maintenance mechanisms. Cells without telomerase mainly survive through the formation of circular intrachromosomal fusions, whereas mutants lacking Ccq1 maintain linear chromosomes through a homologous recombination (HR) pathway 8.
Figure 1. CLRC promotes the shelterin–chromatin association.
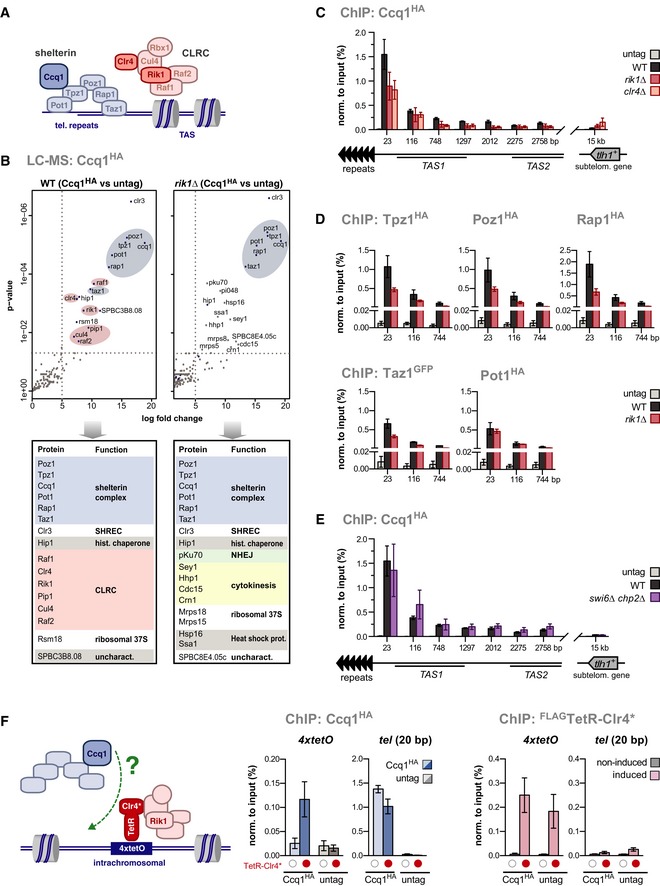
-
AScheme of the shelterin complex and CLRC at telomeres.
-
BMass spectrometry analysis of proteins co‐purified with Ccq1HA. Shown is a volcano plot for proteins significantly enriched in Ccq1HA relative to untagged Ccq1 (left panel: WT cells; right panel: rik1∆ cells). Members of the CLRC complex and shelterin are highlighted in red and blue, respectively. Bottom panel displays proteins enriched (log2 ≥ 5 or P ≤ 0.01).
-
C–EChIP‐qPCR analysis of epitope‐tagged shelterin components in WT and mutant strains as indicated (negative control: untagged strain). Positions on x‐axis denote distances relative to telomeric repeats (telomere‐proximal PCR primer) (n = 3 independent experiments).
-
FChIP analysis of Ccq1HA and FLAG‐TetR‐Clr4* (see scheme). Expression and tethering of FLAG‐TetR‐Clr4* are controlled by thiamine/AHT addition (red dots indicate induced/tethered FLAG‐TetR‐Clr4*; empty circles indicate non‐induced/non‐bound controls) (n = 3 independent experiments).
Subtelomeres often comprise homologous sequence elements that differ from the telomeric repeats, yet their function is not well defined. The subtelomeric homology (SH) sequences in S. pombe contain loosely repetitive elements known as telomere‐associated sequences (TAS), which are enriched in poly[dA:dT] tracts and found telomere‐proximal on nearly every chromosomal arm. Whereas SH sequences are dispensable for viability and telomere length, their absence causes harmful interchromosomal fusions in cells lacking telomerase 12, suggesting that they promote chromosome circularization upon telomere attrition. In addition, the subtelomeres may be involved in telomerase‐independent, HR‐mediated maintenance of linear chromosomes 1, 13; however, whether these mechanisms require specific SH sequences remains unknown.
Schizosaccharomyces pombe subtelomeres harbor heterochromatic domains telomere‐distal from the TAS, which are marked by high levels of di‐ and trimethylated lysine‐9 of histone H3 (H3K9me2/3) deposited by the sole histone methyltransferase, Clr4 14. Clr4 associates with Rik1 to form a complex (CLRC) that contains the ubiquitin E3 ligase Cul4‐Rik1Raf1/2. This complex is essential for H3K9me establishment, but its ubiquitylation substrate remains unknown (Fig 1A) 15, 16, 17, 18, 19. H3K9me is recognized by members of the heterochromatin protein 1 (HP1) family, Swi6 and Chp2. HP1 proteins mediate spreading and the assembly into a heterochromatic platform by recruiting other factors, like SHREC (Snf2‐like/HDAC‐containing repressor complex) 20, 21, which consists of Mit1, Clr3, Clr2, and Clr1 21, 22. In addition, SHREC is targeted to subtelomeres via its interaction with Ccq1 9, 21. Ccq1 also recruits CLRC and promotes silencing of reporter genes inserted next to telomeric repeats 9, 23. However, unlike Ccq1, heterochromatin factors are not required for telomere maintenance 24. Telomere‐adjacent (TAS) and telomere‐distal subtelomeres differ in their requirements for heterochromatin silencing 25, and the heterochromatin structure of the former is largely unknown.
Here, we describe a role of Ccq1 in nucleosome maintenance at telomere‐adjacent subtelomeres. We identified Ccq1 as a main interactor and recruiter of CLRC, being critical for H3K9me deposition near telomeres, in agreement with two previous reports 23, 26. However, we further demonstrate that the major role of Ccq1 in heterochromatic silencing arises from maintaining critical levels of nucleosomes at the subtelomeric TAS. This chromosomal region displays unusually low nucleosome abundance, which is independent of the proximity of the telomeric repeats but intrinsic to the DNA sequence of the TAS. Deletion of ccq1 + results in a near complete loss of histones at the TAS. This unexpected function of Ccq1 in nucleosome maintenance is mediated by CLRC and SHREC, which are recruited by Ccq1 and act epistatically with Ccq1 in heterochromatic silencing. Thus, in contrast to other heterochromatic domains, gene silencing at the TAS appears to be regulated at the level of nucleosome stability. Furthermore, we show that recombination in the absence of Ccq1 is promoted by homologous sequences present in the TAS, implying that these sequences are actively implicated in telomere maintenance by a mechanism resembling ALT (alternative telomere lengthening).
Results
CLRC promotes the shelterin–chromatin association
To identify physical interactors of CLRC, we used a proteomic approach employing an epitope fusion with Raf1, the putative receptor of the Cul4‐Rik1 E3 ubiquitin ligase. LC‐MS analysis retrieved all members of CLRC as well as the shelterin complex (Fig EV1A–C; Table EV1). This association required the presence of the adaptor protein Rik1 and was confirmed by co‐immunoprecipitation experiments for the shelterin subunits Ccq1 and Tpz1 (Fig EV1D). We further validated the Rik1‐dependent interaction between CLRC and shelterin by a reciprocal approach expressing Ccq1HA in WT and rik1Δ cells (Fig 1A and B). In the absence of Rik1, the interaction pattern of Ccq1 was significantly altered, resulting in the loss of the interaction with CLRC and most of the other Ccq1‐associated proteins (except the SHREC subunit Clr3 and the histone chaperone Hip1; Fig 1B, right panel). Interestingly, we found several new interactions with factors involved in protein folding, cytokinesis, and non‐homologous end‐joining (NHEJ). Together, our proteomics data show that the shelterin complex is a main interactor of CLRC, confirming two recent reports 23, 26. We further demonstrate that the interaction profile of Ccq1 is affected by the presence of CLRC, suggesting that this association confines the cooperation of shelterin with other factors.
Figure EV1. CLRC interacts with shelterin without affecting ubiquitylation.
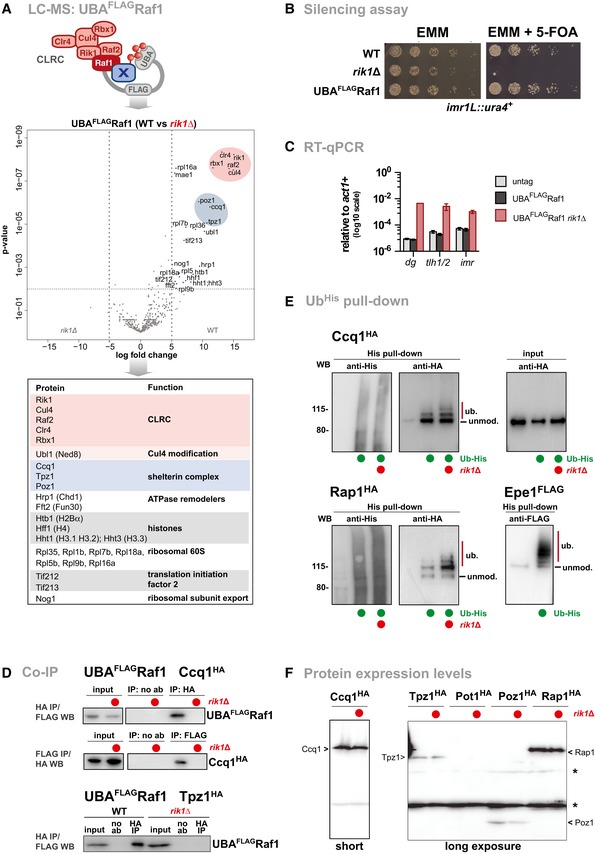
- Mass spectrometry analysis of proteins co‐purified with Raf1 fused to the ubiquitin‐associated (UBA) domain of the ubiquitin receptor Dsk2 from S. cerevisiae (UBAFLAGRaf1). This method is based on the Ubiquitin Ligase Trapping method originally developed in S. cerevisiae 57 , which stabilizes the binding of substrates to the ubiquitin ligase. The volcano plot depicts proteins enriched in UBAFLAGRaf1 WT cells relative to UBAFLAGRaf1 rik1Δ. Members of the CLRC and shelterin complexes are highlighted in red and blue, respectively. Bottom panel displays nuclear proteins enriched (log2 ≥ 5 or P ≤ 0.01).
- Silencing reporter assay at imr1L::ura4 +. Fivefold serial dilution on non‐selective (EMM) and selective media (EMM + 5‐FOA) of WT, rik∆, and UBAFLAGRaf1 cells. The N‐terminal fusion does not interfere with the function of CLRC in heterochromatin formation. The rik1∆ strain was used as negative control.
- RT–qPCR analysis in strains expressing untagged Raf1, UBAFLAGRaf1, and UBAFLAGRaf1 rik1∆ (n = 3 independent experiments). Data are normalized to input and represented as mean ± SEM.
- Co‐immunoprecipitation of Raf1 with Ccq1 (top and middle panel) and Tpz1 (bottom panel) in presence and absence of Rik1. Strains expressing endogenous levels of UBAFLAGRaf1 and Ccq1HA or Tpz1HA were subjected to anti‐HA immunoprecipitation in the presence of Benzonase (indicating that the interactions are independent of DNA or RNA; negative control: no antibody, noAb). Input and immunoprecipitated material were analyzed by anti‐FLAG immunoblots. For Ccq1HA, the reciprocal experiment is also shown.
- Ubiquitin pull‐down experiments. Top panels shows in vivo ubiquitylation of Ccq1HA in WT and rik1∆ cells expressing 6His‐ubiquitin. Left panels: precipitated 6His‐ubiquitin conjugates (20%) analyzed by anti‐His and anti‐HA immunoblots. Right panel: input fraction (0.1%) analyzed by anti‐HA immunoblot. The bottom panels show 6His‐ubiquitin pull‐down assays for Rap1HA in WT and rik1∆ cells (left) and Epe1FLAG in WT cells (right). Epe1 is shown as an example for poly‐ubiquitylation. WT cells expressing untagged ubiquitin are used as negative control.
- Anti‐HA immunoblot of epitope‐tagged shelterin proteins expressed from their endogenous locus in WT and rik1∆ cells. The asterisks denote unspecific signals derived from cross‐reactions with the anti‐HA antibody. Note that Ccq1 is expressed at higher levels compared to other shelterin subunits; endogenous levels of Pot1 are low and not detected by the immunoblot. Estimated sizes (kDa) of the tagged proteins: Ccq1 (86 kDa), Tpz1 (61 kDa), Pot1 (67 kDa), Poz1 (34 kDa), and Rap1 (84 kDa).
Although CLRC contains a ubiquitin ligase subcomplex (Cul4‐Rik1Raf1/Raf2), our results show that CLRC does not regulate shelterin through ubiquitylation (Fig EV1E–F). Ccq1 and other shelterin subunits are enriched at telomeric repeats and the telomere‐adjacent TAS. We therefore tested whether CLRC regulates the abundance of Ccq1 on subtelomeres and analyzed several loci next to the telomeric repeats known to bind the shelterin complex (Fig 1C; see also Appendix Fig S1A and B) 7, 8, 21. Lack of Rik1 or Clr4 causes a significant decrease in the binding of Ccq1, particularly next to the telomeric repeats (Fig 1C). Similarly, association of all other shelterin components with chromatin is reduced in rik1∆ cells (Fig 1D), except for Pot1. Since the latter binds the ssDNA overhang of the telomeric 3′ end in the semi‐open configuration 2, we speculate that binding to the G‐tail overhang is less sensitive to perturbations caused by the absence of Rik1. CLRC promotes heterochromatin assembly through the recruitment of HP1; however, no decrease in Ccq1 binding was observed in a double mutant lacking both Swi6 and Chp2 (Fig 1E). This suggests that CLRC‐mediated stable binding of shelterin is not a direct function of the heterochromatin structure. To study whether CLRC can promote Ccq1 binding to chromatin independently of telomeres, we expressed Ccq1HA in a strain in which the TetRoff‐Clr4* fusion protein can be tethered via a 4xtetO array to an ectopic chromatin site 27. Ccq1 associates with this ectopic site in a manner that strictly depends on the expression and binding of TetRoff‐Clr4* to the tetO array (Fig 1F). Thus, CLRC promotes shelterin association with chromatin in a ubiquitylation‐ and HP1‐independent manner.
Ccq1 promotes nucleosome stability at native subtelomeres
In agreement with two previous reports 23, 26, we found that both Rik1 and Raf1 associate with telomere‐adjacent chromatin in a Ccq1‐dependent manner, indicating that shelterin also recruits CLRC to chromatin (Appendix Fig S2A and B). We also observed that H3K9me2 is strongly reduced in ccq1∆ cells at TAS (Fig 2A and B, left panel, and Appendix Fig S2C). This H3K9me2 loss was also seen for the m23::ura4 + reporter gene placed next to telomeric repeats and for an intrachromosomal Taz1‐binding site (isl15) known to recruit the shelterin complex (Fig 2A and B, middle and right panel), confirming results of the previous studies 23, 26. Nonetheless, we noticed that the chromatin distribution of CLRC markedly differs from the subtelomeric H3K9me2 profile. Whereas Rik1 and Raf1 are mostly abundant at the telomeric end and decline with increasing distance, H3K9me2 is mostly enriched at the heterochromatic tlh1 + gene located 15 kb away of the telomeric repeats (compare Appendix Fig S2A and B with Fig 2). We further observed that H3K9me2 levels at the telomere‐proximal TAS are unusually low, even in WT cells, reaching barely 10% of the levels found at tlh1 + (Fig 2B, left panel). This low H3K9me2 level at TAS contrasts the levels seen at m23::ura4 + and the heterochromatin island isl15 (Fig 2B; middle and right panel). Hence, we wondered whether TAS are restrictive for H3K9 methylation or whether this chromatin region has lower nucleosome occupancy. ChIP for total histone H3 revealed that WT cells have indeed lower nucleosome levels at the telomere‐proximal TAS1 region (0.3‐fold relative to euchromatin, EC) compared to the telomere‐distal tlh1 + locus (1.25‐fold relative to EC; Fig 2C). In the absence of Ccq1, H3 levels are even further decreased (0.1‐fold at TAS1 relative to EC; Fig 2C), but this marked decrease was not seen for tlh1 + or other heterochromatic loci (m23::ura4 +, isl15; Fig 2C; middle and right panel). By performing several control experiments (Appendix Fig S2E and F), we excluded the possibility that the H3 loss at TAS is caused by progressive telomere shortening, a phenotype often seen for cells upon deletion of ccq1 + after continuous growth 8. When determining the relative decrease, i.e., WT:mutant ratios for H3 and H3K9me2, we found a strong correlation for the telomere‐proximal TAS, suggesting that the loss of H3 largely explains the loss of H3K9me2 at this subtelomeric region (Fig 2D). Nonetheless, H3K9me2 levels are still significantly lower than H3 levels even in WT cells; thus, additional mechanisms likely contribute to constrain this repressive mark at TAS. In conclusion, our findings demonstrate that the chromatin structure at native subtelomeres differs substantially from other heterochromatin domains and reveals an unexpected role of Ccq1 in nucleosome maintenance.
Figure 2. Ccq1 promotes nucleosome stability at native subtelomeres.
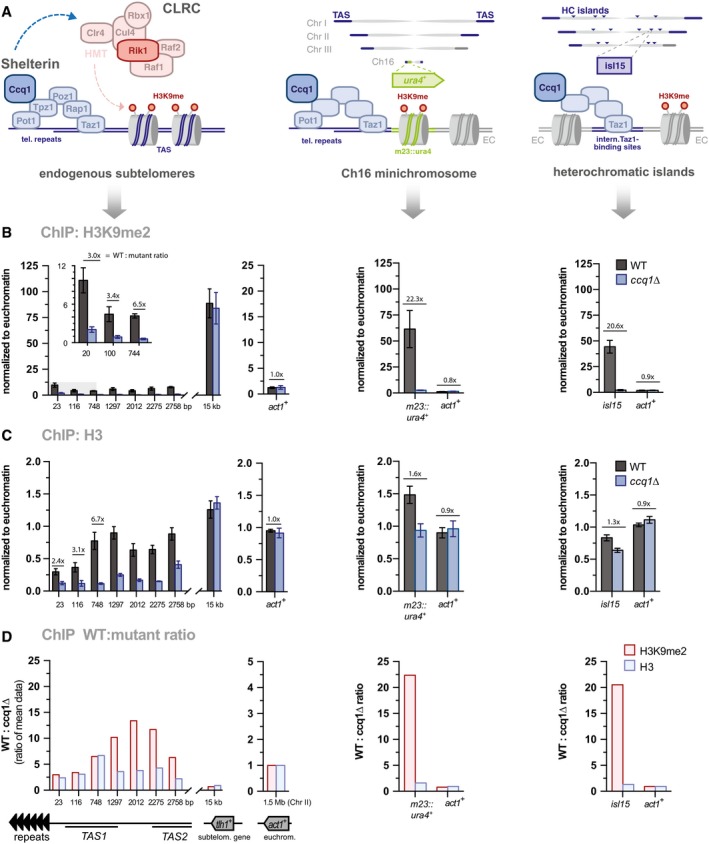
-
ALeft: Schemes of CLRC and the shelterin complex. Middle and right: Scheme depicting the position of the ura4 + reporter gene and heterochromatin islands relative to telomeric repeats and TAS regions; note that the minichromosome Ch16 does not contain TAS.
-
B, CChIP‐qPCR analyses of H3K9me2 (B) and H3 (C) in WT and ccq1Δ cells (n = 3 independent experiments). Data are normalized to input and to the average of three euchromatic loci (EC) as internal control (see Materials and Methods) and represented as mean ± SEM.
-
DFold change of WT over ccq1Δ for H3K9me2 and H3 levels (see Materials and Methods).
Ccq1 maintains nucleosome stability by recruiting CLRC and SHREC
Ccq1 may contribute to nucleosome stability through the recruitment of CLRC, which promotes H3K9 methylation and in turn prevents transcription‐dependent histone turnover by RNA polymerase II (reviewed in ref. 28). To test whether CLRC acts downstream of Ccq1 in nucleosome maintenance, we analyzed H3 levels in mutants of CLRC (Fig 3A). H3 levels are reduced to similar levels in single and double mutants of rik1 + and ccq1 +, implying that the loss of CLRC alone is sufficient to produce this nucleosome loss phenotype (Fig 3B, upper panel). Similar phenotypes were observed for clr4∆ cells and, albeit to lesser extent, for the swi6∆ chp2∆ double mutant (Fig 3B, middle and lower panel). The latter is consistent with the fact that HP1 proteins provide a binding platform for CLRC (and SHREC, see below) and likely act redundantly with Ccq1 at the TAS. In agreement with the findings for ccq1∆, the loss of H3 in mutants of CLRC is specific for TAS and not seen at tlh1 + (Fig 3B).
Figure 3. Ccq1 maintains nucleosome stability through recruiting CLRC and SHREC .
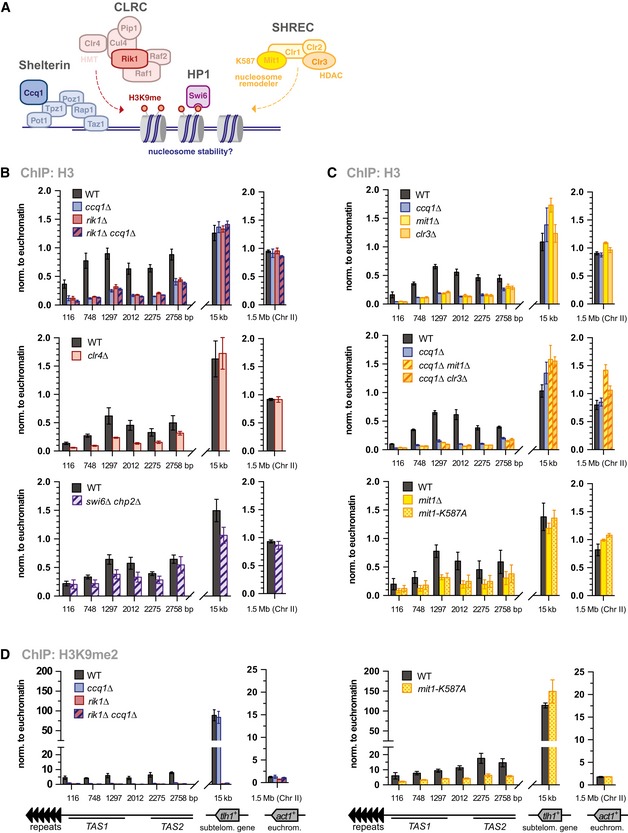
-
AScheme of shelterin, CLRC and SHREC complexes, and Swi6HP1 bound to H3K9me2.
-
B–DChIP‐qPCR analyses of H3 and H3K9me2 in WT and mutants as indicated (n = 3 independent experiments). Data are represented as mean ± SEM (normalization as in Fig 2B and C).
Ccq1 was further shown to interact with the HDAC Clr3, which is part of the repressor complex SHREC that also contains the Snf2‐like nucleosome remodeler Mit1 (Fig 3A) 9, 21, 22, 29. Mit1 and Clr3 have been associated with nucleosome positioning and turnover and prevent the formation of nucleosome‐depleted regions within heterochromatin 21, 29, 30, 31. Similar to mutants of CLRC, we found that H3 is strongly decreased in mit1∆ or clr3∆ mutants at TAS, while the corresponding double mutants with ccq1Δ displayed an epistatic phenotype (Fig 3C, upper and middle panel). To test whether the remodeler activity of Mit1 is required for nucleosome stability at native subtelomeres, we examined telomeric chromatin in a strain expressing an ATPase‐dead mutant, mit1‐K587A 21. H3 levels in this point mutant are similar to the ones observed in mit1∆ (Fig 3C, lower panel). This decrease in H3 at TAS is also reflected by reduced H3K9me2 in the mit1‐K587A mutant, largely recapitulating the phenotype of ccq1∆ but differing from rik1∆, which lacks H3K9me2 at tlh1 + (Fig 3D). This finding further corroborates the notion that low levels of nucleosomes, or their improper positioning, result in fragile chromatin, precluding the establishment of H3K9me2 at TAS. In conclusion, Ccq1 maintains nucleosomes at telomere‐adjacent subtelomeres through the recruitment of CLRC and SHREC.
The subtelomeric DNA sequence causes nucleosome instability
It has been proposed that subtelomeric sequences in S. pombe contain low affinity nucleosome‐binding sites forming nucleosome‐free regions 21, 29, 31, 32; however, these studies focused primarily on heterochromatin regions excluding the TAS regions. Employing ChIP‐seq for H3, we find that telomere‐proximal H3 levels in WT cells are low across the first 10 kb including the TAS, but rise with increasing distance from the telomeric ends (Fig 4A). TAS2 and TAS3 have above‐average A/T content (i.e., more than 70%) and a high percentage of exclusive poly[dA:dT] tracks of 5‐mers and longer (Fig 4B). Such DNA properties are absent at tlh1 + and further telomere‐distal subtelomeric regions, which all display high levels of H3. Using an in silico nucleosome prediction algorithm 33, we found a strong correlation between our experimental H3 data and predicted nucleosome occupancy (Fig 4C). Moreover, when analyzing mutants lacking Ccq1, Rik1, or Mit1, we found H3 levels to be further reduced throughout the TAS but to be mostly unaffected at the tlh1 + locus (Fig EV2A–C).
Figure 4. Nucleosome abundance is low throughout the telomere‐proximal chromosomal region.
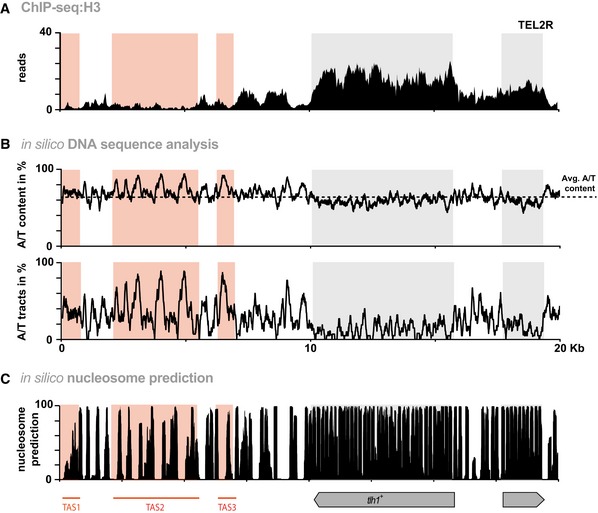
- ChIP‐seq reads mapped to TEL2R (shown in reverse orientation for consistency).
- In silico analyses of TEL2R DNA sequence. Black line in top panel shows A/T content. Dotted line represents average A/T content in S. pombe (www.pombase.org). Bottom panel shows the percentage of poly[dA:dT] tracts (defined as a sequence of 5 or more nucleotides consisting only of A or T). Red and gray shaded areas show TAS regions and subtelomeric genes, respectively.
- Nucleosome prediction for TEL2R using prediction algorithm 33.
Figure EV2. Subtelomeric H3 profiles in WT cells and mutants deficient in Ccq1, CLRC, and SHREC .
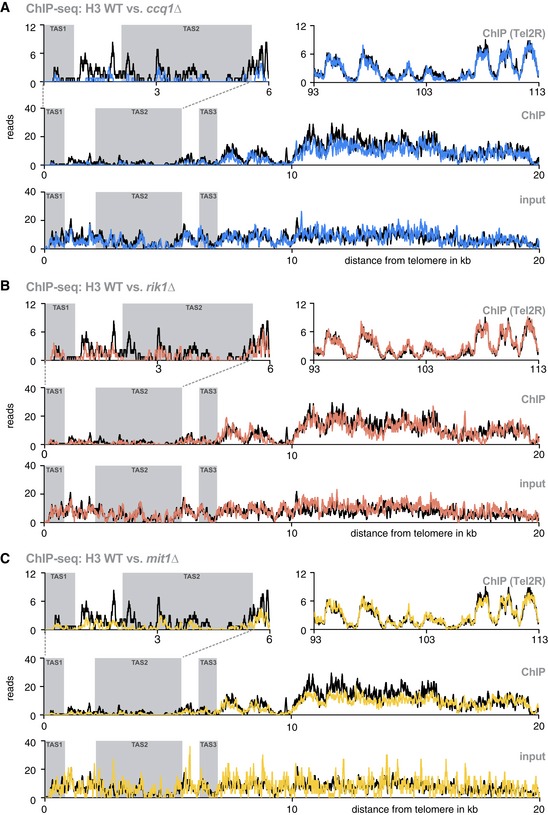
-
A–CChIP‐seq reads mapped to TEL2R (shown in reverse orientation for consistency) for WT (black) vs. ccq1Δ (blue) (A), rik1Δ (red) (B), and mit1Δ (yellow) cells (C). Shown are reads of ChIP data for TAS1 and TAS2 (top left panel), a nucleosome‐free region reported by Tashiro et al 12 (top right panel), and the subtelomeres including the TAS1‐3 and tlh1 + (middle panel). The bottom panel shows input samples for the same region. Gray shaded areas are TAS regions.
To examine whether the low nucleosome occupancy at the TAS is an intrinsic feature of the underlying DNA sequence, we integrated a 790‐bp fragment comprising a partial sequence of TAS1 (115–905 bp) into the intrachromosomal leu1 + locus using a plasmid‐insertion strategy. Since TAS sequences are present at most of the chromosomal arms, distinguishing between subtelomeric and ectopic TAS sequences is difficult. We therefore integrated this TAS fragment in a strain that lacks all subtelomeric homologous (SH) sequences, including the TAS on chromosome I + II and the left arm on chromosome III 12. ChIP‐qPCR revealed that H3 levels are low at the ectopic TAS1 fragment, recapitulating the H3 levels and distribution of the endogenous TAS1 region (Fig 5A). Notably, H3 levels were further reduced at the ectopic TAS fragment when ccq1 + was deleted in this strain, similar to the endogenous TAS, suggesting that shelterin can bind to this fragment even in the absence of telomeric repeats (Fig 5A). Together, these results indicate that the DNA sequence of this TAS fragment is sufficient to mediate nucleosome instability and regulation by Ccq1.
Figure 5. Ectopic insertion of TAS fragment is sufficient to cause nucleosome instability.
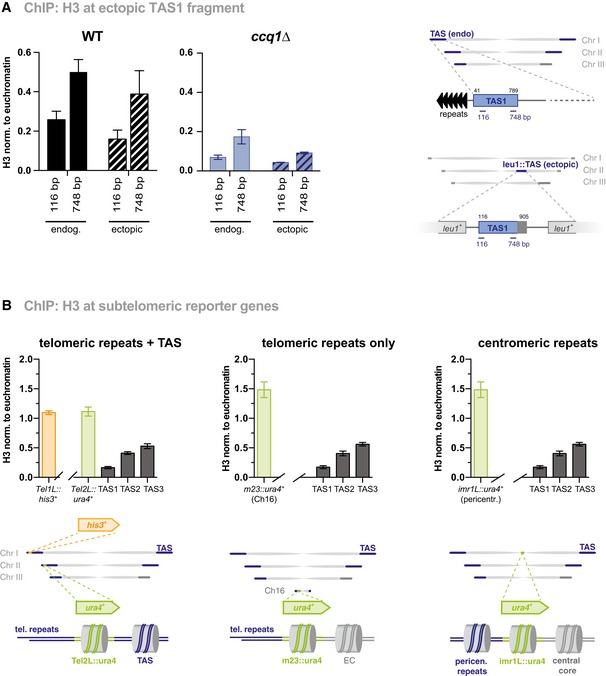
- ChIP‐qPCR analysis of H3 at endogenous TAS and a fragment spanning the TAS region from 115 to 905 bp (relative to telomeric repeat) inserted into the leu1 + locus (see scheme). The TAS fragment was inserted into a strain that lacks endogenous TAS (see text) 12. Shown are ChIP analyses for WT (left) and ccq1∆ (right; note different the scale of the y‐axis) (n = 9–10 independent experiments except for ectopic TAS in ccq1∆ strain where n = 3).
- ChIP‐qPCR analysis of H3 at reporter genes (ura4 + and his3 +) at various chromosomal locations (see schemes) in WT cells. TAS1, TAS2, and TAS3 correspond to position 116, 2,851, and 6,291 bp (relative to telomeric repeats), respectively (n = 3 independent experiments).
To investigate whether the proximity to telomeres also contributes to nucleosome instability, we examined H3 levels at the ura4 + gene inserted between the telomeric repeats and TAS on the left arm of chromosome II (Tel2L::ura4 +; scheme see Fig 5B, left) 34. In analogy to ura4 + inserted at the m23 locus on the Ch16 minichromosome 35, Tel2L::ura4 + is marked by H3K9me2 and efficiently silenced 34. Yet in contrast to TAS, Tel2L::ura4 + and m23::ura4 + display high H3 levels in WT cells, which are comparable to H3 levels of ura4 + inserted at the pericentromeric imr1L locus (Fig 5B). These high H3 levels at m23::ura4 + and Tel2L::ura4 + are mostly maintained in ccq1∆ cells (Figs 2C and EV3A). We made similar observations for the Tel1L::his3 + reporter gene present on the left arm on chromosome I (Fig 5B). Based on these findings, we conclude that the presence of the telomeric repeats is not sufficient to cause nucleosome instability. We also tested whether the shelterin complex competes with nucleosomes for DNA binding by analyzing H3 levels in cells lacking Taz1, which recruits shelterin to dsDNA 1, 2. However, deleting taz1 + resulted in reduced H3 levels similar to ccq1∆ or mit1∆. Consistent with this finding, nucleosome levels were not recovered in taz1∆ double mutants lacking Ccq1 or Mit1 (Fig EV3B), indicating that removing shelterin from subtelomeres is not sufficient to restore nucleosome levels, neither to levels seen WT cells nor to levels found at other genomic regions. Together, these findings demonstrate that the specific chromatin context of the underlying DNA sequences, or the DNA sequence itself, but not the chromosomal positioning, determines nucleosome stability at TAS.
Figure EV3. Low nucleosome occupancy at TAS is not caused by proximity of telomeric repeats or shelterin binding, RSC .
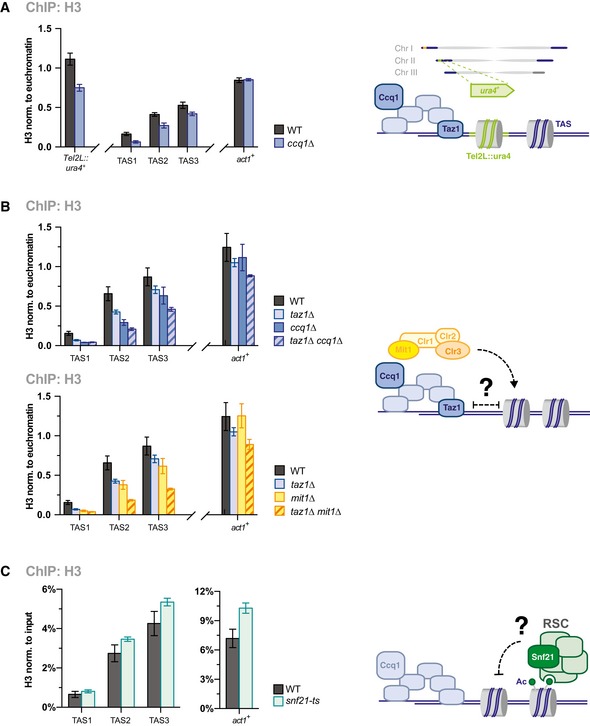
- ChIP‐qPCR analysis of H3 at reporter gene (ura4 +) and TAS regions WT and ccq1Δ strains (see scheme) (n = 3 independent experiments).
- ChIP‐qPCR analysis of H3 in WT, ccq1Δ, and taz1Δ (top) or mit1Δ (bottom) and corresponding double mutants (see scheme) (n = 3 independent experiments).
- ChIP‐qPCR analysis of H3 in WT and snf21‐ts cells (see scheme) (n = 3 independent experiments each derived from 2 to 3 parallel ChIP samples).
Subtelomeric nucleosome stability is linked to heterochromatic silencing
To understand the impact of nucleosome stability on telomere function, we analyzed subtelomeric silencing by examining the expression of endogenous subtelomeric non‐coding RNAs (here dubbed as TERRA, telomeric repeat‐containing non‐coding RNA, for simplicity) from telomere‐adjacent TAS regions (Fig EV4A) 25, 36. Mutants of ccq1 and mit1 cause comparable levels of TERRA derepression and display an epistatic genetic interaction (Fig 6A, left panel), similar to that observed for nucleosome stability. Loss of Clr3 and Rik1 resulted in a slightly larger increase of TERRA and showed partially additive effects with ccq1∆, suggesting that they can also act independently in TERRA silencing (Fig 6A, left and right panels). Similar observations were made for the binding of RNA polymerase II to chromatin regions associated with TERRA transcription (Fig EV4B). However, in contrast to the mostly non‐additive derepression of TERRA, we found that ccq1∆ double mutants also deficient in SHREC or CLRC display strong synthetic silencing defects at tlh1 + (Fig 6B). As a control region, we used the pericentromeric cen‐dg repeats, which are not affected by Ccq1 (Fig EV4C). This led us to conclude that Ccq1 contributes to silencing by different mechanisms: At telomere‐adjacent chromatin, Ccq1 acts to maintain nucleosome stability mainly through the combined actions of SHREC and CLRC, whereas at tlh1 +, Ccq1 has a different function that is independent of SHREC and CLRC.
Figure EV4. Expression of telomeric transcripts in the absence of CLRC and SHREC .
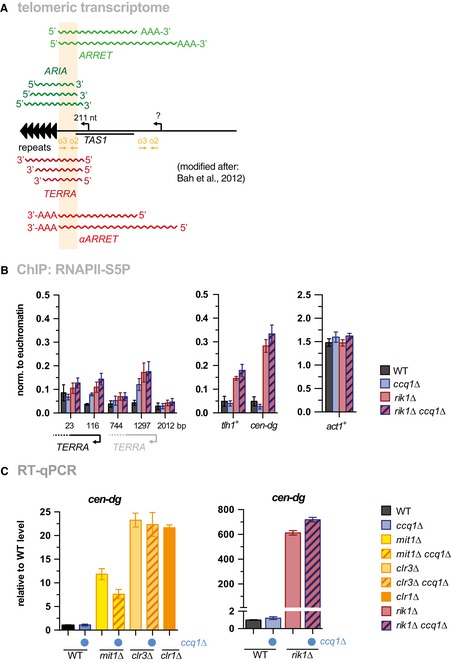
- Scheme of telomeric transcripts (modified after 36). Whereas ARRET and αARRET RNAs have a poly‐A tail, only a small percentage of TERRA and ARIA transcripts are poly‐adenylated. The amplicon (primers o2 + o3) used for RT–qPCR and ChIP‐qPCR analysis anneals to all telomeric transcripts without discriminating strand specificity or shorter species that lack transcribed parts from the telomeric repeats. For simplicity, we refer to these transcripts as “TERRA”. An identical sequence of the o2/o3 amplicon is present in a telomere‐distal region, but it is unknown whether this region also contains transcription start sites.
- ChIP‐qPCR analysis of RNAPII‐S5P in WT, ccq1Δ, rik1Δ, and the corresponding double mutant (n = 3 independent experiments).
- Ccq1 is not involved in silencing of pericentromeric heterochromatin. RT–qPCR analysis of transcript levels of cen‐dg in WT, ccq1Δ, mit1Δ, clr3Δ, rik1Δ, double mutants with ccq1Δ (indicated by blue dot), and clr1Δ cells (n = 3 independent experiments)
Figure 6. Nucleosome stability at subtelomeres is linked to heterochromatic silencing.
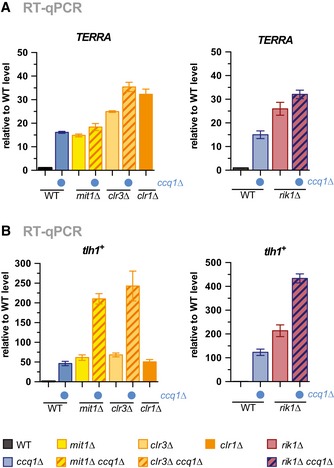
-
A, BRT–qPCR analysis of transcript levels of TERRA (A) and tlh1 + (B) in WT strain and mutants as indicated (double mutants with ccq1Δ are indicated by blue dot). Data are represented as mean ± SEM from three independent experiments and shown relative to WT level (see Materials and Methods).
TAS promote subtelomeric DNA recombination in the absence of Ccq1
Our findings indicate that the intrinsic properties of the TAS sequence impede nucleosome binding, raising the question about the benefit of maintaining these sequences at subtelomeres. Deletion of Ccq1 was previously shown to cause genomic rearrangements of subtelomeres, which likely contribute to telomere maintenance in the absence of telomerase 8. Thus, we wondered whether TAS are critical for this hyper‐recombinogenic phenotype of ccq1∆. While we did not observe changes in the overall abundance of TAS under our experimental conditions (Appendix Fig S2F), recombination events between individual subtelomeres (e.g., gene conversion or break‐induced replication) would be difficult to detect due to the identical DNA sequences on most chromosomal arms. To overcome this technical challenge, we used reporter genes inserted into telomeric regions that served as unique barcodes. As such, we used ura4 + placed next to telomeric repeats on the minichromosome Ch16 (m23::ura4 +), which lacks any TAS sequences 35, and compared its stability with his3 + and ura4 + inserted between the endogenous telomeric repeats and TAS1 (Tel1L::his3 + Tel2L::ura4 +) 34. Cultures of WT cells untreated or freshly disrupted for ccq1 + were grown continuously for about 40 generations, and genomic copy numbers of the reporter gene and TAS were assessed every six to seven generations (see scheme, Fig 7A).
Figure 7. Subtelomeric DNA promotes recombination in the absence of Ccq1.
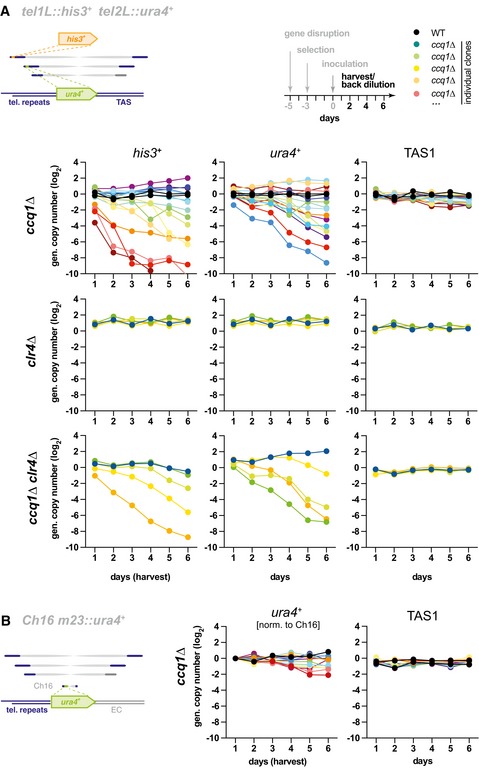
- Genomic copy number of his3 +, ura4 +, and TAS1 in indicated strains harboring the reporter genes tel1L::his3 + and tel2L::ura4 +. Cultures from individual WT and freshly generated knockout clones (ccq1Δ, n = 16; clr4Δ, n = 5; ccq1Δ clr4Δ, n = 5) were pre‐grown on selective media for several days and inoculated at day 0 to grow in liquid media with regular back‐dilution (every 24 h, approximately 7 generations). Samples were taken at indicated harvest times, and relative copy numbers of genomic regions were assessed by qPCR (normalization against intrachromosomal loci). Black and rainbow color lines indicate WT strains and individual ccq1 + deletion mutants (clones), respectively.
- qPCR analysis as in (A) but with strains harboring the minichromosome Ch16 m23::ura4 + (ccq1Δ, n = 14). Since Ch16 is unstable in ccq1Δ cells (see Appendix Fig S3A and B and 37, m23::ura4 + has been normalized to minichromosome levels (details on normalization, see Materials and Methods). Note that Ch16 does not harbor TAS sequences.
In contrast to WT cells, we found that both Tel1L::his3 + and Tel2L::ura4 + were highly unstable in ccq1∆ cells, resulting in either additional copies or their gradual loss, but largely independently of each other (Figs 7A and EV5). Moreover, whereas most isolates displayed a partial or near complete loss of the reporter gene, those with a copy number gain showed a maximum of 4–5 copies of either his3 + or ura4 +, suggesting that most TAS‐containing subtelomeres acquired an additional copy of the reporter gene in these cultures. Since we did not observe major changes for TAS (Fig 7A), this implies that the rearrangements of subtelomeric his3 + and ura4 + were largely caused by recombination but not telomere shortening. In contrast to the TAS‐flanked reporters, we found that the m23::ura4 + reporter gene is relatively stable in ccq1∆ cells (Fig 7B and Appendix Figure S3), particularly when taking into account the general instability of the Ch16 minichromosome in this mutant reported previously 37.
Figure EV5. Subtelomeres do not undergo recombination in the absence of CLRC and SHREC .
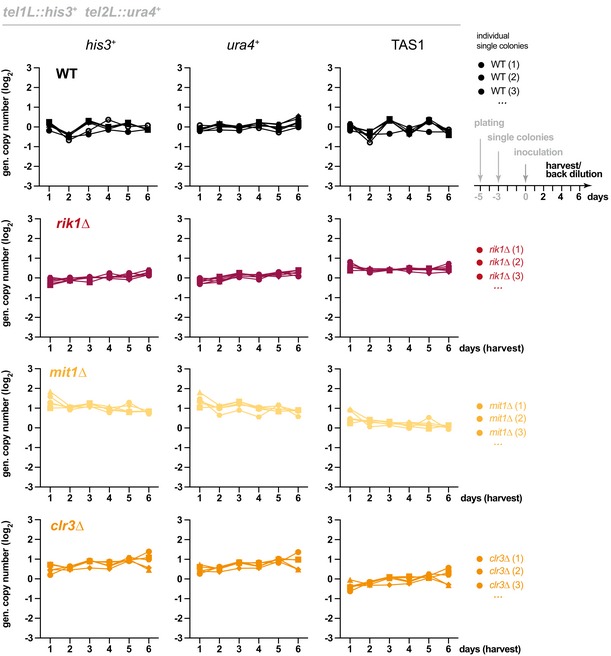
Genomic copy number of his3 +, ura4 +, and TAS1 in indicated strains harboring the reporter genes tel1L::his3 + and tel2L::ura4 + cells. Cultures from individual WT and knockout clones (WT, n = 6; rik1Δ, n = 6; mit1Δ, n = 5; clr3Δ, n = 5) were inoculated at day 0 to grow in liquid media with regular back‐dilution (every 24 h, approximately 7 generations). Samples were taken at indicated harvest times, and relative copy numbers of genomic regions were assessed by qPCR (normalization against intrachromosomal loci).
To dissect whether TAS‐mediated genome instability is linked to nucleosome loss or relates to other functions of Ccq1, we repeated these experiments in strains deficient in CLRC or SHREC. Examining reporter gene stability in single mutants of clr4 + (Fig 7A, middle panel) or rik1 + , mit1 + and clr3 + (Fig EV5) revealed no significant difference to WT cells. This implies that failed recruitment of CLRC and SHREC, and as consequence nucleosome loss, is not sufficient or involved to cause this phenotype. We further tested whether CLRC is required for recombination, in analogy to HAATI (heterochromatin amplification‐mediated and telomerase‐independent), an alternative telomere maintenance mechanism 38. Deleting clr4 + did not prevent the hyper‐recombinogenic phenotype of ccq1∆ (Fig 7A, lower panel), indicating that the requirements for HR involving DNA sequences present within TAS are different from HAATI (which predominantly involves rDNA). In conclusion, our findings imply that subtelomeric sequences are intrinsically prone to recombination, which is prevented by Ccq1 through a mechanism independent of its role in nucleosome maintenance.
Discussion
Here, we demonstrate that the subtelomeric TAS comprise a unique chromatin structure that is controlled by the shelterin subunit Ccq1. This chromosomal region displays very low nucleosome abundance, which is sequence‐dependent but position‐independent. Neither transcription, nor the presence of telomeric repeats, nor competition with shelterin explains these low levels (Figs 5 and 6, and EV3). Rather, the presence of exclusive poly[dA:dT] tracts strongly correlates with the low nucleosome occupancy (Fig 4), and integration of a TAS fragment at an ectopic locus is sufficient to cause nucleosome instability (Fig 5). Consistently, the role of Ccq1 in nucleosome stability, mediated by CLRC and SHREC, is strictly confined to TAS but not found at telomere‐distal subtelomeres, which have normal nucleosome abundance. Interestingly, those telomere‐distal regions still display Ccq1‐dependent silencing, yet in a CLRC‐ and SHREC‐independent manner, suggesting that Ccq1 contributes to heterochromatin maintenance at distinct regions by different mechanisms. Hence, it is an attractive hypothesis that Ccq1's property to interact with CLRC and SHREC has co‐evolved as a “built‐in function” with the appearance of subtelomeric nucleosome‐refractory sites to accommodate the need of maintaining appropriate histone levels.
The low nucleosome occupancy at TAS may result from less well‐positioned nucleosomes. This is in agreement with a recent study that describes a non‐canonical chromatin structure at chromosomal ends consisting of irregularly spaced nucleosomes and telomere‐binding proteins (“telosomes”) 39. However, while this structure appears to be restricted to the ~1.5 kb telomere‐proximal region and requires the presence of chromosomal ends, we find that the low nucleosome occupancy and instability at TAS extends to chromosomal regions more distal (Fig 4) and is independent of telomeric repeats (Fig 5). This suggests that TAS act autonomously in controlling nucleosome binding, which appears to be encoded in the DNA sequence. The A/T‐rich sequence may disfavor nucleosome binding 29 or TAS may recruit in a sequence‐dependent manner other factors that compete with nucleosome binding. For example, Taz1 binds DNA beyond the telomeric repeats 40, which may be facilitated by low nucleosome occupancy. However, deleting taz1 + is not sufficient to restore nucleosome levels (Fig EV3B), suggesting that additional or other mechanisms are involved. We tested a potential role by the Remodels Structure of Chromatin complex (RSC), which promotes nucleosome eviction and has been proposed to antagonize Mit1 29, 32. While TAS2 and TAS3 displayed a modest H3 increase in a temperature‐sensitive mutant of RSC, this increase was even stronger at euchromatic genes, and overall nucleosome occupancy remained low at TAS compared to other genomic regions (Fig EV3C). Hence, we conclude that RSC does not play a specific role in the nucleosome instability at TAS. Interestingly, origins of replication correlate with low nucleosome occupancy 41 and the origin recognition complex (ORC) binds adenine stretches via its Orc4 subunit, which contains several AT‐hook subdomains 42, 43. Thus, ORC may be a potential factor that competes with nucleosomes for binding to TAS.
Since nucleosomes are the major obstacle for transcribing RNA polymerase II, the absence of Ccq1, or its downstream partners CLRC and SHREC, likely has a direct consequence on gene expression. We find that the defect in maintaining H3 levels in these mutants correlates with the derepression of ncRNAs expressed from TAS (Fig 6). The role of Ccq1 in repressing transcription could be directly mediated through the nucleosome remodeler activity of Mit1, which may help keeping nucleosomes at refractory binding sites, in agreement with previous reports 21, 29, 31, 32. The activities of Clr3 and CLRC may additionally contribute to nucleosome stability through preventing open chromatin structure and access to RNA polymerase II, which would be consistent with the role of Clr3 in restricting histone turnover at other heterochromatic domains 30. However, even when fully derepressed, those other heterochromatin domains maintain high nucleosome levels, whereas perturbing any of these mechanisms at TAS causes a substantial decrease of H3 with nearly a complete loss at some sites (Fig 3). Thus, we conclude that transcription alone is not sufficient to cause the nucleosome loss at TAS, underscoring the fragile nature of this subtelomeric region, which makes it necessary to provide multiple mechanisms to maintain a critical level of nucleosomes and repression of TERRA (see model, Fig 8A). On the other hand, since telomere elongation is promoted by the expression of TERRA 44, the metastable state of nucleosomes may also serve as a cue to dynamically control telomere length. This is further in line with the negative regulation of telomere elongation by SHREC, which in addition displaces telomerase from shelterin 45.
Figure 8. Working models for Ccq1‐mediated nucleosome stability and telomere maintenance.
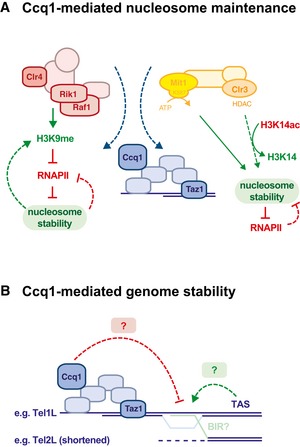
- Ccq1 recruits CLRC and SHREC to subtelomeric chromatin, which counteracts nucleosome instability conferred by the DNA sequence of TAS. CLRC may contribute to nucleosome stability indirectly by depositing H3K9me, which prevents access to RNAPII and, thus, nucleosome eviction through transcription. Conversely, stabilized nucleosomes will contribute to maintaining critical levels of H3K9me at TAS. SHREC may contribute to nucleosome stability directly through nucleosome positioning via its ATP‐dependent remodeler activity conferred by Mit1; indirectly through histone deacetylation via the HDAC moiety Clr3, thereby preventing open chromatin structure and access to RNAPII.
- TAS or other DNA sequences may represent fragile sites causing DNA breaks through replication fork collapse or the recruitment of unknown factors causing genome instability. Ccq1 may prevent genomic instability by binding to subtelomeric chromatin, thereby stabilizing fragile sites and/or competing with the binding of destabilizing factors. Homologous sequences present in the TAS may promote recombination by BIR (break‐induced replication) or similar mechanisms.
Taz1 and other shelterin proteins are known to be critical for H3K9 methylation 23, 26, 40, 46. The recruitment of CLRC by Ccq1 has therefore been proposed to be essential for robust heterochromatin assembly and the initiation of heterochromatin spreading from telomeric repeats to telomere‐distal subtelomeres 23. However, our results suggest that the decrease in H3K9me2 in ccq1∆, at least at telomere‐adjacent subtelomeres, is less a cause but rather a consequence of nucleosome loss. This becomes very obvious in cells lacking Mit1, which results in a similar loss of H3K9me2 at TAS yet does not affect H3K9me2 maintenance at other heterochromatin domains (Fig 3), consistent with previous findings 29. Furthermore, TAS recruitment of CLRC by Ccq1 results only in low H3K9me levels in WT cells (Fig 2), raising the question of whether these low H3K9me levels are sufficient to promote HP1‐dependent spreading. These apparent discrepancies between our work and previous studies can likely be explained by H3K9 methylation and silencing having been primarily examined in the context of reporter genes 23, 40, 46. In contrast to native subtelomeres, these euchromatic genes exhibit normal nucleosome abundance and allow the establishment of high levels of H3K9me when placed next to telomeric repeats (Fig 2). We made similar observations for nucleosome occupancy at intrachromosomal Taz1‐binding sites (Fig 2), which promote Ccq1‐dependent assembly of facultative heterochromatin 26. Thus, Ccq1 appears to fulfill distinct functions, promoting nucleosome maintenance and heterochromatin establishment, largely depending on the chromatin environment. From this, we predict that heterochromatin at TAS is primarily controlled by nucleosome stability, in contrast to other heterochromatin regions that display high levels of H3K9me and depend on HP1‐mediated spreading. Finally, the physical interaction between Ccq1 and CLRC may also impact other processes. In the absence of telomerase, linear chromosomes can be maintained through HAATI, which depends on Ccq1 and heterochromatic factors 38, 47. It has been proposed that Ccq1 could be recruited to heterochromatin via its interaction with Clr3 38. Since we find that the association of Ccq1 with telomeres or an ectopic intrachromosomal locus is also controlled by CLRC (Fig 1), this may suggest that CLRC also plays an active role in shelterin regulation and HAATI recruitment.
The persistent challenge of maintaining proper nucleosome levels raises the question of what is the advantage of keeping these nucleosome‐binding refractory sequences at subtelomeres. Whereas SH sequences, which include TAS, are dispensable for mitotic and meiotic growth, their relevance for telomere maintenance becomes evident when Trt1 (the catalytic subunit of telomerase) is absent. Under this condition, loss of SH sequences shifts the balance among trt1∆ survivors from intra‐ to interchromosomal fusions absent 12. The conclusion that has been drawn from this observation is that the SH sequences promote circularization upon telomere erosion. However, in contrast to trt1∆ cells, early ccq1∆ mutants do not circularize, but instead maintain linear chromosomes, via gross genomic rearrangements, suggesting an additional function of subtelomeres. This pathway involves ATR (Ataxia telangiectasia and Rad3 related)‐dependent checkpoint activation and an unusual HR mechanism that depends on Rad51 but not Rad52 8. We confirmed this hyper‐recombinogenic phenotype of ccq1∆ and demonstrated that recombination depends on the presence of the TAS, suggesting that the homologous sequences of the TAS, or specific DNA sequences within, are directly involved in the telomere maintenance mechanism that becomes active upon loss of Ccq1. As we observe that donor sequences often get amplified in ccq1∆ resulting in loss of heterozygosity, we hypothesize that recombination occurs through break‐induced replication (BIR), resembling other telomere maintenance mechanisms in S. cerevisiae 48 or in human tumor cells commonly referred to as ALT (alternative lengthening of telomeres) 49.
Several mechanisms could contribute to the genomic instability at TAS. Formally, we cannot exclude the possibility that any homologous donor sequence would be sufficient to sustain a high recombination rate at subtelomeres. However, while the presence of homologous donor sequences likely affects the mode of repair (HR versus NHEJ), DNA damage at those hyper‐recombinogenic sites may be due to additional intrinsic properties of the chromatin region. Thus, we favor the hypothesis that the genomic instability is specifically caused by the DNA sequence of the TAS (see model, Fig 8B). For instance, recombination may be facilitated by the sequence properties of the TAS (e.g., poly[dA:dT] tracts), which may be more prone to double‐strand breaks (DSBs) 50. This idea would be consistent with our observation that TAS trigger recombination of telomere‐proximal reporter genes, implying that DSBs frequently occur telomere‐distal of the reporters within subtelomeric sequences when Ccq1 is not present. A similar role has been proposed for intrachromosomal Taz1‐dependent heterochromatin islands, which replicate late (a feature linked to fragile sites) and map to meiotic DSB sites 26. In addition, the mouse homolog of Taz1, TRF1, was shown to bind to a common fragile site containing an internal telomeric repeat and to stabilize this locus in a sequence‐ but position‐independent manner 51. Thus, the absence of Taz1 (TRF1), or shelterin in general, would trigger genome instability at those fragile sites. Conversely, a role for telomerase in DNA damage repair has been proposed in budding yeast 52. According to this model, telomerase may promote repair upon replication stress by elongating broken telomeres or the G‐rich single strand following replication fork resection, while its absence causes recombination by alternative pathways. These recombination pathways are not restricted to telomeres but also include translocations between subtelomeric X and Y’ elements through pairing of interstitial telomeric sequences (ITS) 53. While this model of telomerase alleviating replication stress is also attractive in light of Ccq1's role of suppressing recombination and recruitment of Trt1, further work needs to be done to examine a potential role of telomerase in DNA break repair in fission yeast.
Alternative to accumulation of stochastic damage within the TAS, sequence‐specific elements within the DNA sequence may mediate the recruitment of specific factors promoting HR, as this has been observed in human ALT cells. Here, rearrangements of telomere repeats are driven by the nuclear hormone receptor (NHR) family members NR2C/F. These receptors recognize variant telomere repeat motifs and are thought to dimerize and “bridge” the canonical and variant repeats, resulting in their clustering and recombination 54. Finally, ncRNAs derived from the subtelomeric TAS, such as TERRA, could also contribute to genome stability through the formation of RNA:DNA hybrids, as discussed for ALT 49. However, although we find that TERRA transcription is upregulated in ccq1∆ and mutants of CLRC (Figs 2, 3, 4), the absence of CLRC or SHREC alone is not sufficient to induce recombination (Fig EV5). This is consistent with the minor role of heterochromatin in inhibiting related telomere recombination pathways 55 and implies that other downstream functions controlled by Ccq1 are critical in this process.
Many species harbor subtelomeres with mosaic homologous sequence elements, and there is increasing evidence that they play a role in telomere length control independent of telomerase, as seen in yeast, flies, and humans 56. Our findings demonstrate that the TAS in S. pombe differ from other chromatin regions through their nucleosome instability and high recombinogenic potential. Further studies of the phenomena may provide crucial insights into the still opaque functions of subtelomeres and, specifically, how telomere maintenance mechanisms are encoded in the subtelomeric DNA sequence.
Materials and Methods
Yeast techniques, plasmids and strains
Standard media and genome engineering methods were used (Fission Yeast. A Laboratory Manual. Hagan, Carr, Grallert and Nurse. Cold Spring Harbor Press. New York 2016). 5‐FOA media contained 1 g/l 5‐fluoroorotic acid. EMM‐leu media were used for growing strains harboring pREP1 plasmids. PMG medium supplemented with 10 μM anhydrotetracycline (AHT) and 15 μM thiamine was used for experiments with 4xTetO strains expressing FLAG‐TetR‐Clr4*. The plasmid used for generating the UBA trap fusion strains (3xFLAG‐DSK2) was described previously 57. The plasmids pBS‐3xFLAG:kanMX6, pFA6a–HA:kanMX4, and pREP‐nmt1p‐His‐ubi‐LEU2 were provided by M. Smolle (LMU Munich), A. Ladurner (LMU Munich), and T. Toda (Hiroshima University), respectively. The ectopic TAS strain was generated by amplifying 790 bp of TAS1 region using genomic DNA from the strain PSB0017 as a template and oligonucleotides Sg3182 and Sg3183. The PCR product was cloned into the plasmid pJK148. For integration into the leu1‐32 locus, the resulting plasmid was linearized using NdeI (NEB R0111S) and transformed into S. pombe strain ST3479 that lack endogenous TAS (gift by J. Kanoh, Osaka University). Strains used in this study are listed in Appendix Table S1.
Large‐scale Immunoprecipitation (IP)
Cell extracts were prepared with 20–30 g of cells. Liquid cultures were harvested by centrifugation, washed with STOP Buffer (NaCl 150 mM, EDTA 10 mM pH 7.4, 2 mg/ml sodium fluoride, 0.065 mg/ml sodium azide), and resuspended in 3.5–5 ml of lysis buffer (25 mM HEPES‐KOH, 150 mM KOAc, 10 mM MgCl2, 5 mM CaCl2, 20% glycerol, 4 mM AEBSF [Sigma, 11585916001], 1 Roche Complete EDTA‐free protease inhibitor tablet [Roche, 05056489001] per 10 ml, and 0.01 mg/ml MG132 [Sigma, C2211]); cell droplets were flash‐frozen by pipetting into liquid nitrogen. Frozen droplet cells were broken during 9 cycles (3 min ON/2 min OFF, power 9) with a freezer mill [SPEX, 6970EFM‐50 ml adaptor]. Cells were diluted up to 0.6 g/ml and treated with 174 U/ml of Benzonase for 1 h at 4°C. Cell debris was discarded (17,530 g, 15 min at 4°C), and the soluble fraction was clarified by centrifuging at 40 K for 45 min at 4°C. After a pre‐clearing step with Dynabeads Prot G [Life Technologies, 10009D], the supernatant was incubated with the Dynabeads Prot G beads coupled to the respective antibody for 3–12 h. The bound material was washed two times with lysis buffer and four times with 50 mM NH4HCO3. Next, beads were incubated with 10 ng/μl trypsin in 1 M urea 50 mM NH4HCO3 for 30 min and washed once with 50 mM NH4HCO3. Subsequently, the supernatant was digested in the presence of 1 mM DTT over night. Digested peptides were alkylated and desalted prior to LC‐MS analysis.
Mass spectrometry
For the mass spectrometry analysis, the desalted peptides were separated in a 15 cm analytical column C18 micro column (75 μm ID homepacked with ReproSil‐Pur C18‐AQ 2.4 μm from Dr. Maisch) with a 40‐min gradient from 5 to 60% acetonitrile in 0.1% formic acid (Ultimate 3000 RSLCnano system). The effluent from the HPLC was directly electro‐sprayed into a LTQ‐Orbitrap mass spectrometer (Thermo Fisher Scientific). The MS instrument was operated in data‐dependent mode to automatically switch between full scan MS and MS/MS acquisition. Survey full scan MS spectra (from m/z 300–2,000) were acquired in the Orbitrap with resolution R = 60,000 at m/z 400 (after accumulation to a “target value” of 500,000 in the linear ion trap). The six most intense peptide ions with charge states between 2 and 4 were sequentially isolated to a target value of 10,000 and fragmented in the linear ion trap by collision‐induced dissociation (CID). All fragmentation spectra were recorded in the LTQ part of the instrument. For all measurements with the Orbitrap detector, three lock‐mass ions from ambient air were used for internal calibration as described before 58. Typical MS conditions were as follows: spray voltage, 1.5 kV; no sheath and auxiliary gas flow; heated capillary temperature, 200°C; normalized CID energy 35%; activation q = 0.25; activation time = 30 ms. MaxQuant 1.5.2.8 was used to identify proteins and quantify by iBAQ. Maxquant conditions were as follows: Database, Uniprot_Spombe_150114; MS tol, 10 ppm; MS/MS tol, 0.5 Da; Peptide FDR, 0.1; Protein FDR, 0.01 min. peptide Length, 5; Variable modifications, Oxidation (M); Fixed modifications, Carbamidomethyl (C); Peptides for protein quantitation, razor and unique; Min. peptides, 1; Min. ratio count, 2.
Small scale protein analysis
For examining cellular protein levels by immunoblots, extracts were prepared under denaturing conditions 59. Co‐immunoprecipitations were performed with cell extracts prepared under native conditions, similarly to the large‐scale protocol, either from frozen cell droplets using a freezer mill (SPEX, 6970EFM) or from frozen cell pellets using a cell homogenizator (Precellys 24, Peqlab). Ubiquitylation pull‐downs were done under denaturing conditions essentially as described 60, except using Ni‐NTA Magnetic Agarose Beads (Qiagen, 336113). Protein levels were analyzed by immunoblots with anti‐polyHistidine (Sigma, H1029), anti‐FLAG (Sigma, P3165), and anti‐HA (Sigma, H6908) antibodies.
ChIP assays
ChIP experiments were performed essentially as described 61. Cross‐linking was performed with 1% formaldehyde for 10 min at RT. For quantitative ChIP, immunoprecipitations were performed with 2–5 μg of antibody against H3 (or H3 modifications) or epitope‐tagged proteins with lysates corresponding to 15 OD600 and 60 OD600, respectively. The following antibodies were used: anti‐GFP, anti‐phospho RNA pol II (Ser5) (provided by A. Ladurner, LMU), anti‐Flag (Sigma, F3165), anti‐HA (Sigma, H6908), anti‐H3 (Active Motif, 61475), anti‐H3K9me2 (Abcam, ab1220), and anti‐H3K14ac (Abcam, ab52946). Immunoprecipitated DNA was quantified by qPCR using the PowerUp™SYBR Green Master mix (Life Technologies, A25778) and a 7500 Fast real‐time PCR system (Applied Biosystems). Primers are listed in Appendix Table S2. Unless otherwise noted, mean and SEM values were calculated from three independent experiments. qPCR signals were normalized against the input samples for each primer position, which avoids any bias against subtelomeric sequences that might be under‐ or overrepresented due the potential shortening of telomeric ends or recombination in absence of Ccq1. For ChIP experiments with histone proteins, histone modifications, or RNA pol II, input‐normalized qPCR signals were additionally normalized to the mean of 2–3 euchromatic loci (act1 + , ade2 + , tef3 +) as an internal control, which was set to 1 (“EC normalized”).
Internal ChIP normalization:
| (1) |
Using the mean of multiple euchromatic loci (“EC”) instead of single locus (e.g., act1 +) reduces bias coming from variations in ChIP experiments, especially when doing IP experiments with bulk chromatin proteins (see also Appendix Fig S2). For ChIP‐seq, anti‐H3 immunoprecipitated DNA corresponding to a total of 120 OD600 starting material (whole cell lysate) was pooled from independent experiments. The samples were treated with 0.04 mg of RNase A (30 min, 37°C) and 0.04 mg of Proteinase K (1.5 h, 37°C) and purified (Zymo Research, D5201). Samples of at least three different ChIP experiments were combined and prepared for sequencing using NEBnext Ultra II DNA Library prep Kit of Illumina (NEB, E7645S). Single‐end sequencing of libraries, demultiplexing, mapping of Illumina reads, and normalization were performed as previously described 62.
RT–qPCR analyses
RT–qPCR experiments were carried out as previously described 60. cDNA was quantified by qPCR using primers listed in Appendix Table S2. Prior calculation of mean and SEM values from independent experiments, act1 + normalized data sets were standardized to the mean of all samples from each experiment (experimental normalization; Equation (2)). These sample pool‐normalized results were shown relative to the mean value of the sample pool‐normalized wild‐type data from all (n) experiments (equation (3)).
Experimental normalization:
| (2) |
Mean WT normalization:
| (3) |
Using the average from a collection (sample pool) instead of a single strain (e.g., WT) reduces bias, especially when transcripts levels are low in the repressed state and therefore more prone to noise.
Telomere length and genomic stability assays
Telomere‐PCR was performed as previously described 44. Briefly, telomeres of denatured genomic DNA were poly(C)‐tailed. Telomeres were then amplified by PCR using primers that anneal to the poly(C) tail and a downstream region. Telomere length was determined by analyzing PCR products on a 0.8% agarose gel. Primers used for Telomere‐PCR are shown in Appendix Table S2. For the telomere stability assay with ccq1∆ cells, fresh deletion mutants were generated by homologous recombination using standard genome engineering methods. Individual knockout clones grown for 5 days on selective media were used to inoculate 5 ml of media at OD600~0.05. Cultures were grown over night to OD600 = 3–5. Amount of cells corresponding to 5 OD600 was harvested by centrifugation and flash‐frozen. Remaining culture was back diluted to OD600~0.05 to be grown 24 h. This process was repeated to collect samples from 6 days. Genomic DNA was extracted from frozen cell pellets using a yeast DNA extraction kit (Thermo Fisher Scientific, 78870). Relative copy numbers of individual reporter genes or subtelomeric loci were measured by qPCR and normalized to internal reference genes (act1 + , ade2 + , tef3 +) as described above. For quantifying the minichromosome loss of Ch16 m23::ura4 (Appendix Fig S3), the genomic copy number of the ade6 + gene was quantified by qPCR using oligonucleotides that anneal to both the chromosomal ade6‐M210 allele (Chr III) and the ade6‐M216 allele (Ch16), followed by normalization to reference genes (EC: act1 + , ade2 + , tef3 +). Based on the rationale that the level of Ch16 and m23::ura4 + (encoded by the minichromosome) is equal at the start of the experiment, the relative level of ade6‐M216 Ch16 allele was determined by subtracting the copy number of ura4 + at day 1 from the total copy number of ade6 + (i.e., ade6‐M216 Ch16 + ade6‐M210 ChrIII) for each time point. These calculated ade6‐M216 Ch16 values were used for normalizing the m23::ura4 + copy number.
Data availability
ChIP‐seq data have been deposited in Gene Expression Omnibus (GEO) database under the accession number GSE121502 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE121502).
Author contributions
TSE, MF, and SB designed the study. MF, TSE, MC, and SF‐B generated plasmids and yeast strains. MF performed biochemical purifications, pull‐down experiments, and immunoblots. IF designed and performed the LC‐MS analysis under supervision by AI. MF and SF‐B carried out yeast growth assays. The UBA trap method was developed by MS in the laboratory of DT. TSE, MF, SF‐B, ZS, MC, LMC, and SB performed RT–qPCR and ChIP experiments. TSE conducted genome stability and telo‐PCR assays. TSE performed ChIP‐seq together with CB and MH. TSE, MF, IF, CB, ZS, MC, LMC, and SB carried out data analysis. SB, TSE, and MF wrote the manuscript and IF and AI contributed to editing.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figure PDF
Table EV1
Review Process File
Acknowledgements
We thank Bassem Al‐Sady, Ramón Ramos Barrales, and members of our laboratory for critical reading of the manuscript, and Guy Riddihough (Life Science Editors) for editorial assistance. We thank Robin Allshire, Zac Cande, Songtao Jia, Junko Kanoh, Andreas Ladurner, Michaela Smolle, Philipp Korber, and Takashi Toda for strains, plasmids and reagents. We also thank Sebastian Eustermann and the Hopfner Lab (Gene center, LMU) for technical training and sharing equipment, and Scott Wood for initial efforts to identify CLRC interactors. We further thank Songtao Jia for communicating results prior to publication. This work was supported by grants awarded to S.B. from the German Research Foundation (BR 3511/2‐1) and the European Union Network of Excellence EpiGeneSys (HEALTH‐2010‐257082). S.B. is Member of the Collaborative Research Center 1064 funded by the German Research Foundation and acknowledges infrastructure support.
EMBO Reports (2019) 20: e47181
References
- 1. Cooper JP, Nimmo ER, Allshire RC, Cech TR (1997) Regulation of telomere length and function by a Myb‐domain protein in fission yeast. Nature 385: 744–747 [DOI] [PubMed] [Google Scholar]
- 2. Miyoshi T, Kanoh J, Saito M, Ishikawa F (2008) Fission yeast Pot1‐Tpp1 protects telomeres and regulates telomere length. Science 320: 1341–1344 [DOI] [PubMed] [Google Scholar]
- 3. Ye JZ‐S, Hockemeyer D, Krutchinsky AN, Loayza D, Hooper SM, Chait BT, de Lange T (2004) POT1‐interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev 18: 1649–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nandakumar J, Cech TR (2013) Finding the end: recruitment of telomerase to telomeres. Nat Rev Mol Cell Biol 14: 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Armstrong CA, Pearson SR, Amelina H, Moiseeva V, Tomita K (2014) Telomerase activation after recruitment in fission yeast. Curr Biol 24: 2006–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Flory MR, Carson AR, Muller EG, Aebersold R (2004) An SMC‐domain protein in fission yeast links telomeres to the meiotic centrosome. Mol Cell 16: 619–630 [DOI] [PubMed] [Google Scholar]
- 7. Harland JL, Chang YT, Moser BA, Nakamura TM (2014) Tpz1‐Ccq1 and Tpz1‐Poz1 interactions within fission yeast shelterin modulate Ccq1 Thr93 phosphorylation and telomerase recruitment. PLoS Genet 10: e1004708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tomita K, Cooper JP (2008) Fission yeast Ccq1 is telomerase recruiter and local checkpoint controller. Genes Dev 22: 3461–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moser BA, Raguimova ON, Nakamura TM (2015) Ccq1‐Tpz1TPP1 interaction facilitates telomerase and SHREC association with telomeres in fission yeast. Mol Biol Cell 26: 3857–3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jun H‐I, Liu J, Jeong H, Kim J‐K, Qiao F (2013) Tpz1 controls a telomerase‐nonextendible telomeric state and coordinates switching to an extendible state via Ccq1. Genes Dev 27: 1917–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dehé P‐M, Cooper JP (2010) Fission yeast telomeres forecast the end of the crisis. FEBS Lett 584: 3725–3733 [DOI] [PubMed] [Google Scholar]
- 12. Tashiro S, Nishihara Y, Kugou K, Ohta K, Kanoh J (2017) Subtelomeres constitute a safeguard for gene expression and chromosome homeostasis. Nucleic Acids Res 45: 10333–10349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakamura TM, Cooper JP, Cech TR (1998) Two modes of survival of fission yeast without telomerase. Science 282: 493–496 [DOI] [PubMed] [Google Scholar]
- 14. Grewal SIS, Jia S (2007) Heterochromatin revisited. Nat Rev Genet 8: 35–46 [DOI] [PubMed] [Google Scholar]
- 15. Hong E‐JE, Villén J, Gerace EL, Gygi SP, Moazed D (2005) A cullin E3 ubiquitin ligase complex associates with Rik1 and the Clr4 histone H3‐K9 methyltransferase and is required for RNAi‐mediated heterochromatin formation. RNA Biol 2: 106–111 [DOI] [PubMed] [Google Scholar]
- 16. Horn PJ, Bastie J‐N, Peterson CL (2005) A Rik1‐associated, cullin‐dependent E3 ubiquitin ligase is essential for heterochromatin formation. Genes Dev 19: 1705–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jia S, Kobayashi R, Grewal SIS (2005) Ubiquitin ligase component Cul4 associates with Clr4 histone methyltransferase to assemble heterochromatin. Nat Cell Biol 7: 1007–1013 [DOI] [PubMed] [Google Scholar]
- 18. Li F, Goto DB, Zaratiegui M, Tang X, Martienssen R, Cande WZ (2005) Two novel proteins, dos1 and dos2, interact with rik1 to regulate heterochromatic RNA interference and histone modification. Curr Biol 15: 1448–1457 [DOI] [PubMed] [Google Scholar]
- 19. Thon G, Hansen KR, Altes SP, Sidhu D, Singh G, Verhein‐Hansen J, Bonaduce MJ, Klar AJS (2005) The Clr7 and Clr8 directionality factors and the Pcu4 cullin mediate heterochromatin formation in the fission yeast Schizosaccharomyces pombe. Genetics 171: 1583–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Motamedi MR, Hong E‐JE, Li X, Gerber S, Denison C, Gygi S, Moazed D (2008) HP1 proteins form distinct complexes and mediate heterochromatic gene silencing by nonoverlapping mechanisms. Mol Cell 32: 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sugiyama T, Cam HP, Sugiyama R, Noma K‐I, Zofall M, Kobayashi R, Grewal SIS (2007) SHREC, an effector complex for heterochromatic transcriptional silencing. Cell 128: 491–504 [DOI] [PubMed] [Google Scholar]
- 22. Job G, Brugger C, Xu T, Lowe BR, Pfister Y, Qu C, Shanker S, Baños Sanz JI, Partridge JF, Schalch T (2016) SHREC silences heterochromatin via distinct remodeling and deacetylation modules. Mol Cell 62: 207–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J, Cohen AL, Letian A, Tadeo X, Moresco JJ, Liu J, Yates JR, Qiao F, Jia S (2016) The proper connection between shelterin components is required for telomeric heterochromatin assembly. Genes Dev 30: 827–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ekwall K, Nimmo ER, Javerzat JP, Borgstrøm B, Egel R, Cranston G, Allshire R (1996) Mutations in the fission yeast silencing factors clr4+ and rik1+ disrupt the localisation of the chromo domain protein Swi6p and impair centromere function. J Cell Sci 109(Pt 11): 2637–2648 [DOI] [PubMed] [Google Scholar]
- 25. Greenwood J, Cooper JP (2012) Non‐coding telomeric and subtelomeric transcripts are differentially regulated by telomeric and heterochromatin assembly factors in fission yeast. Nucleic Acids Res 40: 2956–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zofall M, Smith DR, Mizuguchi T, Dhakshnamoorthy J, Grewal SIS (2016) Taz1‐shelterin promotes facultative heterochromatin assembly at chromosome‐internal sites containing late replication origins. Mol Cell 62: 862–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Audergon P, Catania S, Kagansky A, Tong P, Shukla M, Pidoux A, Allshire RC (2015) Restricted epigenetic inheritance of H3K9 methylation. Science 348: 128–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Workman JL (2006) Nucleosome displacement in transcription. Genes Dev 20: 2009–2017 [DOI] [PubMed] [Google Scholar]
- 29. Creamer KM, Job G, Shanker S, Neale GA, Lin Y‐C, Bartholomew B, Partridge JF (2014) The Mi‐2 homolog Mit1 actively positions nucleosomes within heterochromatin to suppress transcription. Mol Cell Biol 34: 2046–2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aygün O, Mehta S, Grewal SIS (2013) HDAC‐mediated suppression of histone turnover promotes epigenetic stability of heterochromatin. Nat Struct Mol Biol 20: 547–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamane K, Mizuguchi T, Cui B, Zofall M, Noma K‐I, Grewal SIS (2011) Asf1/HIRA facilitate global histone deacetylation and associate with HP1 to promote nucleosome occupancy at heterochromatic loci. Mol Cell 41: 56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garcia JF, Dumesic PA, Hartley PD, El‐Samad H, Madhani HD (2010) Combinatorial, site‐specific requirement for heterochromatic silencing factors in the elimination of nucleosome‐free regions. Genes Dev 24: 1758–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xi L, Fondufe‐Mittendorf Y, Xia L, Flatow J, Widom J, Wang J‐P (2010) Predicting nucleosome positioning using a duration Hidden Markov Model. BMC Bioinformatics 11: 346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nimmo ER, Pidoux AL, Perry PE, Allshire RC (1998) Defective meiosis in telomere‐silencing mutants of Schizosaccharomyces pombe . Nature 392: 825–828 [DOI] [PubMed] [Google Scholar]
- 35. Nimmo ER, Cranston G, Allshire RC (1994) Telomere‐associated chromosome breakage in fission yeast results in variegated expression of adjacent genes. EMBO J 13: 3801–3811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bah A, Wischnewski H, Shchepachev V, Azzalin CM (2012) The telomeric transcriptome of Schizosaccharomyces pombe . Nucleic Acids Res 40: 2995–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Motwani T, Doris R, Holmes SG, Flory MR (2010) Ccq1p and the condensin proteins Cut3p and Cut14p prevent telomere entanglements in the fission yeast Schizosaccharomyces pombe . Eukaryot Cell 9: 1612–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jain D, Hebden AK, Nakamura TM, Miller KM, Cooper JP (2010) HAATI survivors replace canonical telomeres with blocks of generic heterochromatin. Nature 467: 223–227 [DOI] [PubMed] [Google Scholar]
- 39. Greenwood J, Patel H, Cech TR, Cooper JP (2018) Fission yeast telosomes: non‐canonical histone‐containing chromatin structures dependent on shelterin and RNA. Nucleic Acids Res 46: 8865–8875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kanoh J, Sadaie M, Urano T, Ishikawa F (2005) Telomere binding protein Taz1 establishes Swi6 heterochromatin independently of RNAi at telomeres. Curr Biol 15: 1808–1819 [DOI] [PubMed] [Google Scholar]
- 41. Lantermann AB, Straub T, Strålfors A, Yuan G‐C, Ekwall K, Korber P (2010) Schizosaccharomyces pombe genome‐wide nucleosome mapping reveals positioning mechanisms distinct from those of Saccharomyces cerevisiae . Nat Struct Mol Biol 17: 251–257 [DOI] [PubMed] [Google Scholar]
- 42. Chuang RY, Kelly TJ (1999) The fission yeast homologue of Orc4p binds to replication origin DNA via multiple AT‐hooks. Proc Natl Acad Sci USA 96: 2656–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Okuno Y, Satoh H, Sekiguchi M, Masukata H (1999) Clustered adenine/thymine stretches are essential for function of a fission yeast replication origin. Mol Cell Biol 19: 6699–6709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moravec M, Wischnewski H, Bah A, Hu Y, Liu N, Lafranchi L, King MC, Azzalin CM (2016) TERRA promotes telomerase‐mediated telomere elongation in Schizosaccharomyces pombe. EMBO Rep 17: 999–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Armstrong CA, Moiseeva V, Collopy LC, Pearson SR, Ullah TR, Xi ST, Martin J, Subramaniam S, Marelli S, Amelina H et al (2018) Fission yeast Ccq1 is a modulator of telomerase activity. Nucleic Acids Res 46: 704–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tadeo X, Wang J, Kallgren SP, Liu J, Reddy BD, Qiao F, Jia S (2013) Elimination of shelterin components bypasses RNAi for pericentric heterochromatin assembly. Genes Dev 27: 2489–2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Begnis M, Apte MS, Masuda H, Jain D, Wheeler DL, Cooper JP (2018) RNAi drives nonreciprocal translocations at eroding chromosome ends to establish telomere‐free linear chromosomes. Genes Dev 32: 537–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McEachern MJ, Haber JE (2006) Break‐induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem 75: 111–135 [DOI] [PubMed] [Google Scholar]
- 49. Apte MS, Cooper JP (2017) Life and cancer without telomerase: ALT and other strategies for making sure ends (don't) meet. Crit Rev Biochem Mol Biol 52: 57–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schwartz M, Zlotorynski E, Kerem B (2006) The molecular basis of common and rare fragile sites. Cancer Lett 232: 13–26 [DOI] [PubMed] [Google Scholar]
- 51. Bosco N, de Lange T (2012) A TRF1‐controlled common fragile site containing interstitial telomeric sequences. Chromosoma 121: 465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Simon M‐N, Churikov D, Géli V (2016) Replication stress as a source of telomere recombination during replicative senescence in Saccharomyces cerevisiae . FEMS Yeast Res 16: fow085 [DOI] [PubMed] [Google Scholar]
- 53. Churikov D, Charifi F, Simon M‐N, Géli V (2014) Rad59‐facilitated acquisition of Y′ elements by short telomeres delays the onset of senescence. PLoS Genet 10: e1004736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marzec P, Armenise C, Pérot G, Roumelioti F‐M, Basyuk E, Gagos S, Chibon F, Déjardin J (2015) Nuclear‐receptor‐mediated telomere insertion leads to genome instability in ALT cancers. Cell 160: 913–927 [DOI] [PubMed] [Google Scholar]
- 55. Khair L, Subramanian L, Moser BA, Nakamura TM (2010) Roles of heterochromatin and telomere proteins in regulation of fission yeast telomere recombination and telomerase recruitment. J Biol Chem 285: 5327–5337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jain D, Cooper JP (2010) Telomeric strategies: means to an end. Annu Rev Genet 44: 243–269 [DOI] [PubMed] [Google Scholar]
- 57. Mark KG, Simonetta M, Maiolica A, Seller CA, Toczyski DP (2013) Ubiquitin ligase trapping identifies an SCF(Saf1) pathway targeting unprocessed vacuolar/Lysosomal proteins. Mol Cell 53: 148–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Olsen JV, de Godoy LMF, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M (2005) Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C‐trap. Mol Cell Proteomics 4: 2010–2021 [DOI] [PubMed] [Google Scholar]
- 59. Knop M, Siegers K, Pereira G, Zachariae W, Winsor B, Nasmyth K, Schiebel E (1999) Epitope tagging of yeast genes using a PCR‐based strategy: more tags and improved practical routines. Yeast 15: 963–972 [DOI] [PubMed] [Google Scholar]
- 60. Braun S, Garcia JF, Rowley M, Rougemaille M, Shankar S, Madhani HD (2011) The Cul4‐Ddb1Cdt2 ubiquitin ligase inhibits invasion of a boundary‐associated antisilencing factor into heterochromatin. Cell 144: 41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Barrales RR, Forn M, Georgescu PR, Sarkadi Z, Braun S (2016) Control of heterochromatin localization and silencing by the nuclear membrane protein Lem2. Genes Dev 30: 133–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Brönner C, Salvi L, Zocco M, Ugolini I, Halic M (2017) Accumulation of RNA on chromatin disrupts heterochromatic silencing. Genome Res 27: 1174–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figure PDF
Table EV1
Review Process File
Data Availability Statement
ChIP‐seq data have been deposited in Gene Expression Omnibus (GEO) database under the accession number GSE121502 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE121502).