

Abstract
Glycogen synthase kinase-3 (GSK3) is a constitutively active serine threonine kinase with 1) two isoforms (GSK3A and GSK3B) that have unique and overlapping functions, 2) multiple molecular intracellular mechanisms that involve phosphorylation of diverse substrates, and 3) implications in pathogenesis of many diseases. Insulin causes phosphorylation and inactivation of GSK3 and mammalian oocytes have a functional insulin-signaling pathway whereby prolonged elevated insulin during follicle/oocyte development causes GSK3 hyperphosphorylation, reduced GSK3 activity, and altered oocyte chromatin remodeling. Periconceptional diabetes and chronic hyperinsulinemia are associated with congenital malformations and onset of adult diseases of cardiovascular origin. Objectives were to produce transgenic mice with individual or concomitant loss of GSK3A and/or GSK3B and investigate the in vivo role of oocyte GSK3 on fertility, fetal development, and offspring health. Wild-type males bred to females with individual or concomitant loss of oocyte GSK3 isoforms did not have reduced fertility. However, concomitant loss of GSK3A and GSK3B in the oocyte significantly increased neonatal death rate due to congestive heart failure secondary to ventricular hyperplasia. Individual loss of oocyte GSK3A or GSK3B did not induce this lethal phenotype. In conclusion, absence of oocyte GSK3 in the periconceptional period does not alter fertility yet causes offspring cardiac hyperplasia, cardiovascular defects, and significant neonatal death. These results support a developmental mechanism by which periconceptional hyperinsulinemia associated with maternal metabolic syndrome, obesity, and/or diabetes can act on the oocyte and affect offspring cardiovascular development, function, and congenital heart malformation.
Keywords: congenital heart malformation, glycogen synthase kinase 3, mouse gene knockout, oocyte, periconception
Introduction
Diabetes mellitus type II, schizophrenia, Alzheimer disease, bipolar disorder and some types of cancer have a common denominator: glycogen synthase kinase 3 (GSK3) [1–8]. Glycogen synthase kinase 3 is a constitutively active serine threonine kinase comprising two isoforms referred to as GSK3A (51 kDa) and GSK3B (47 kDa) [9]. Isoforms of GSK3 regulate several physiologic processes, and their dysregulation are proposed to be involved with pathogenesis of numerous diseases through divergent mechanisms of actions regulating the phosphorylation of substrates such as glycogen synthase [10], beta-catenin [11, 12], neuronal kinesin [13], tau protein [14, 15], and activity of DNA methyltransferases (DNMTs) [16–18].
The prevalence of type-2 diabetes and obesity has currently reached alarming levels in both men and women. Accordingly, increasing numbers of reproductive age women are either diabetic and/or obese before conception (preconception), during conception (periconception, including the oocyte growth and preimplantation embryo development periods), and/or during gestation. Maternal nutritional status, body composition, and diabetic conditions during the preconception, periconception, and gestational period can increase the lifelong risk of numerous chronic diseases in offspring and represents an example of the developmental origin of health and diseases paradigm, incorporating the Barker hypothesis [19, 20]. Diabetes, during preconception or periconception, increases spontaneous abortions [21] and offspring teratogenicity, including congenital cardiac defects [22]. Maternal diabetes manifestations of increased congenital abnormalities, and increased disease risk in offspring, are not thought to be caused by genetic aberrations, yet are likely due to dysregulated intracellular signal transduction and/or epigenetic modifications [20].
Insulin receptors are present in Caenorhabditis elegans [23], xenopus [24–26], mouse [27], rat [28], pig [29], cow [30], and human [31] oocytes. Insulin binding to its receptor initiates a signal transduction cascade involving insulin-dependent activation of tyrosine kinase receptor, resulting in phosphorylation of insulin receptor substrates and subsequent activation of the phosphatidylinositol pathway [32]. This pathway includes activation of the phosphoinositide-3 kinase (PI3K) signaling through second messenger molecules, including phosphorylated 3-phosphoinositide-dependent protein kinase-1 (PDPK1), which, in turn, phosphorylates its substrate thymoma viral proto-oncogene 1 (AKT1)/protein kinase B (PKB) [33, 34]. Phosphorylated/activated AKT1/PKB is the primary regulator of the terminal enzyme in insulin signaling, GSK3A and B, and AKT/PKB-mediated phosphorylation of GSK3A at serine 21 or GSK3B at serine 9 results in GSK3 inactivation [35–39]. Components of this insulin-signaling pathway have been demonstrated in oocytes [27, 40, 41]. More importantly, this insulin-signaling pathway is functional in oocytes, supported by studies where follicle culture for 10 days, in the presence of insulin, resulted in hyperphosphorylation and inactivation of oocyte GSK3B [27]. Finally, the inhibition of oocyte GSK3 during oocyte growth and/or meiosis, with insulin and pharmacological inhibitors of GSK3, resulted in changes in histone modifications and chromatin remodeling [27, 41].
Detailed investigations on the in vivo impact and consequences of decreased oocyte GSK3 activity during the preconception/periconception period on fertility, fetal development, and developmental origin of health and disease are wanting. Therefore, our objectives were to produce transgenic mice with individual or concomitant loss of oocyte GSK3A and/or GSK3B and investigate the in vivo role of oocyte GSK3 on fertility, fetal development, and offspring health.
Materials and Methods
Generating Transgenic Mice
The University of Michigan Animal Care and Use Committee approved all uses and procedures with animals reported in this study. Constitutive Gsk3a knockout and Gsk3b wild-type/flox heterozygous FVB/NJ males (Gsk3a+/−b+/f) were generously provided by Dr. Jim R. Woodgett of the Lunenfeld-Tanenbaum Research Institute, Mount Sinai Hospital, Toronto, Canada, which founded the breeding colony. Briefly, founder males were bred to C57Bl/6J-ZP3Cre+ female (The Jackson Laboratory) and heterozygous males (Gsk3a+/−b+/f Cre+) and females (Gsk3a+/−b+/f or Gsk3a−/−b+/f) were selected and constituted the breeding colony for the production of females yielding oocytes with loss of GSK3A (Gsk3a−/−bf/f), loss of GSK3B (oGsk3a+/+b−/−), or concomitant loss of GSK3A and GSK3B (oGsk3a−/−b−/−) (Fig. 1). Constitutive GSK3A knockout males were excluded from the breeding scheme due to subfertility.
Fig. 1.
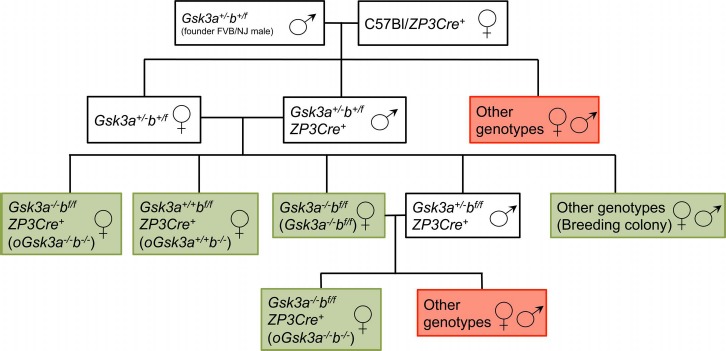
Graphic representation of breeding scheme to produce Gsk3a−/−bf/f, oGsk3a+/+b−/−, oGsk3a−/−b−/−, and breeding colony females. Animals/genotypes within white boxes were used in intermediate breeding for production of target transgenic animals. Green boxes indicate females used for terminal experiments, and red boxes indicate animals/genotypes that were discarded.
Ear punches were collected for genomic DNA extraction using the HotSHOT method for preparation of genomic DNA [42]. Mouse genotyping was performed at 21 days of age with primers to identify wild-type Gsk3a and b, exon 2 excised alleles of Gsk3a, loxP flanked alleles of Gsk3b (Supplemental Table S1; Supplemental Data are available online at www.biolreprod.org), and ZP3-Cre recombinase knock in (003651; The Jackson Laboratory, multiplex PCR, detailed in Fig. 2A). Production of oocytes with absent/reduced activity of Gsk3a and/or b was assessed with RT-PCR and Western blot analysis for mRNA and protein, respectively.
Fig. 2.
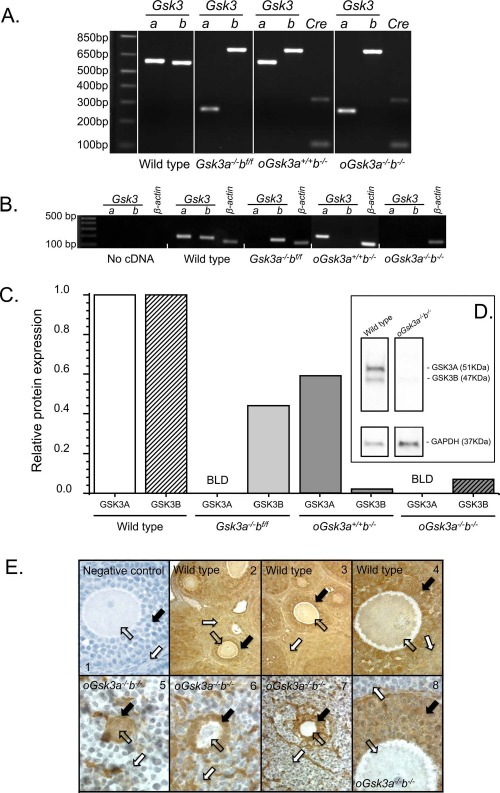
Characterization of constitutive Gsk3a (Gsk3a−/−bf/f), oocyte-specific Gsk3b (oGsk3a+/+b−/−), and concomitant constitutive Gsk3a and oocyte-specific Gsk3b (oGsk3a−/−b−/−) knockout females, ovaries, and oocytes. A) Genomic DNA from wild-type and oGsk3a+/+b−/− animals used in PCR genotyping presented an ∼600 base pairs (bp) amplicon of Gsk3a, while the excision of exon 2 of Gsk3a in Gsk3a−/−bf/f and oGsk3a−/−b−/− females produced an ∼250 bp Gsk3a amplicon. Flanking Gsk3b exon 2 with lox excision sites produced a super shift in the amplicon (∼700 bp) after electrophoresis in Gsk3a−/−bf/f, oGsk3a+/+b−/−, and oGsk3a−/−b−/− females in comparison to wild type (∼550 bp). The ZP3-driven Cre-recombinase knock-in was detected with a multiplex PCR. Image of gel lane loaded with the product of multiplex PCR (not shown for wild type and Gsk3a−/−bf/f) displayed an amplification control targeting Mus musculus strain C57BL/6J chromosome 3, GRCm38.p3 C57BL/6J sequence (NCBI reference sequence: NC_000069.6) of ∼324 bp present for all animals and a 100 bp amplicon corresponding to the Cre-recombinase knock-in observed only in oGsk3a+/+b−/− and oGsk3a−/−b−/− females. B) Individual or concomitant knockout of Gsk3 isoforms produced oocytes lacking transcripts for Gsk3a (Gsk3a−/−bf/f and oGsk3a−/−b−/−) and/or Gsk3b (oGsk3a+/+b−/− and oGsk3a−/−b−/−). C) By Western blot analysis with band density quantification (one oocyte pool/genotype), wild-type oocytes presented the two isoforms of GSK3 that were individually normalized to GAPDH. GSK3A was below limits of detection (BLD) in oocytes from Gsk3a−/−bf/f and oGsk3a−/−b−/−. Expression of GSK3B was greatly decreased in oocytes from oGsk3a+/+b−/− and oGsk3a−/−b−/− in comparison to wild-type oocytes, as demonstrated in this Western blot image (D). E) GSK3 was observed with immunohistochemistry in oocytes (open arrows; E2–E4), granulosa (black arrows; E2–E4) and theca/interstitial cells (white arrows; E2–E4) of wild-type animals. Image E1 shows an immunohistochemistry negative control in which primary antibody was not applied. GSK3 was detected in oocytes of primordial follicle in oGsk3a−/−b−/− females (open arrow; E5). Oocytes contained in primary and secondary follicles of oGsk3a−/−b−/− females lacked detectable GSK3 (open arrows; E6–E8); nevertheless, GSK3 protein was observed in granulosa (black arrows; E5–E8) and theca/interstitial (white arrows; E5–E8) cells in follicles of all stages of development in oGsk3a−/−b−/− ovaries. Original magnification ×400 (E1, E4, E5, E6, E8) and ×100 (E2, E3, E7).
Assessment of Gsk3 Expression in Oocytes and Ovaries
Meiotically competent germinal vesicle-intact (GVI) oocytes enclosed in cumulus cells were collected from ovaries of 8- to 9-wk-old wild type, Gsk3a−/−bf/f, oGsk3a+/+b−/−, and oGsk3a−/−b−/− females following ovarian stimulation with injection of 5 international units of equine chorionic gonadotropin (eCG) (Sigma) and manual antral ovarian follicle rupture at 44–46 h post-eCG administration. Mechanically denuded GVI oocytes were triple washed in human tubal fluid-HEPES (Irvine Scientific) supplemented with 0.3% bovine serum albumin, observed microscopically to confirm complete cumulus cell removal, and snap frozen prior to RT-PCR and Western blot analysis.
Pools of 20 oocytes per genotype were submitted to total RNA extraction with Picopure RNA Isolation kit (ABI Systems) following the manufacturer's instruction and used as a template for cDNA synthesis in RT-PCR with MultiScribeTM reverse transcriptase (ABI Systems). Polymerase chain reaction for Gsk3a and Gsk3b (Supplemental Table S2) with cDNA equivalent to three oocytes was carried out in duplicate with a Bio-Rad MyCycler thermal cycler using Platinum Taq enzyme system (Invitrogen) for 30 cycles.
Western blots were performed using oocyte lysates. Briefly, 198 denuded GVI oocytes pooled from four females with each genotype were lysed in Laemmli sample buffer (Bio-Rad) with 5% 2-mercaptoethanol, vortexed, and placed on ice for 15 min. Following sonication on ice for 10 sec, samples were denatured at 90°C for 10 min and loaded for electrophoresis. Proteins on the gel were then transferred to polyvinylidene fluoride membrane (Amersham) in a semidry electrophoretic transfer cell at 20 V for 50 min (Bio-Rad). Blots were blocked in 5% nonfat dry milk in 50 mM Tris, 150 mM NaCl, and 0.1% Tween (TBST) at room temperature for 1 h and incubated with the appropriate primary antibody diluted in TBST with 5% bovine serum albumin overnight at 4°C with agitation. The antibodies included anti-GSK3A/B (1:1000 dilution, 44-610; Invitrogen) and GAPDH (1:1000 dilution, NB300-326; Novus). After complete washing in TBST, blots were incubated with peroxidase-conjugated immunoglobulin G secondary antibody (Amersham) at room temperature for 2 h, washed in TBST, and developed with SuperSignal ECL reagents (Thermo) according to the manufacturer's instructions. Membranes were reprobed using different primary antibodies after stripping at 50°C for 30 min in stripping buffer (62.5 mM Tris-HCl, pH 6.7, 2% SDS, and 100 mM of 2-mercaptoethanol).
Ovarian paraffin sections (5 μm) from wild-type and oGsk3a−/−b−/− mice were deparaffinized with Histoclear (National Diagnostics), rehydrated with decreasing concentrations of ethanol in deionized water, and then boiled in 10 mM sodium citrate (pH 6.0) for 10 min to unmask antigens. Sections were then treated in 3% H2O2 for 5 min and washed with PBS for 5 min. Immunohistochemistry was performed using Vectastain ABC kit (Vector Laboratories, Inc.) following the manufacturer's instructions. The primary antibody (GSK3A/B, Cell signaling) was diluted (1:200) in PBS supplemented with 2.5% normal goat serum and incubated on sections overnight at 4°C. The ImmPACT DAB kit (Vector Laboratories, Inc.) was used for colorimetric reaction following the manufacturer's instructions.
Assessment of Fertility and Offspring Development
The 8-wk-old Gsk3a−/−bf/f, oGsk3a+/+b−/−, and oGsk3a−/−b−/− females were mated to FVB-NJ males of known fertility. Female age at first litter and litter size were recorded and compared to age-matched colony breeders. In addition, Gsk3a−/−bf/f, oGsk3a+/+b−/−, and oGsk3a−/−b−/− females were evaluated for estrous cycle length [43]. Peripartum death rate (defined as percentage of dead offspring observed within 4 h after deliver) and cumulative 24 h death rate (defined as percentage of dead offspring in the initial 24 h after birth) were recorded for comparison between Gsk3a−/−bf/f, oGsk3a+/+b−/−, and oGsk3a−/−b−/− female-derived litters as well as litters from breeding colony females.
Offspring Histopathology and Heart Assessment
Litters were observed hourly during the first 24 h after birth for collection and fixation of newly dead offspring to avoid autolysis. Specimens were fixed in 4% formalin and processed for histology, or hearts were dissected for Western blot analysis of GSK3 protein expression.
Hematoxylin and eosin-stained sections were obtained for histopathology analysis of hearts, livers, kidneys, lungs, and brains. Offspring of oGsk3a−/−b−/− females were compared to offspring from the breeding colony euthanized at 25 h after birth. Heart images were acquired at 25× magnification for atrial and ventricular chamber width and length measurements using ImageJ (National Institutes of Health).
Heart sections were also stained with wheat-germ agglutinin (WGA) conjugated to AlexaFluor 488 (Life Technologies) and counterstained with Hoechst 33342. Briefly, samples were deparaffinized and rehydrated as previously described. Sections were exposed to WGA dissolved in Hank's Balanced Salt Solution with 0.2% of Triton-X 100 (5 μl/ml) for 10 min and washed three times (5 min each) with deionized water and covered with Hoechst 33342 solution (5 μg/ml). After removal of excess Hoechst, slides were mounted with 25 μl of Vectashield hard-set mount media (Vector Laboratories, Inc.) and stored overnight at 4°C protected from light for hardening. Slides were observed under fluorescence microscopy (400× magnification) to obtain digital images of cardiomyocytes in comparable transverse orientation. Cell margins observed with WGA-Alexa Fluor 488 were used to quantify cell area using ImageJ software. Fifty cells in four different heart sections were measured for each animal, and the mean cardiomyocyte cross-sectional area of each animal was used for comparison of means of each group. Additionally, images of nuclei stained with Hoechst 33342 from heart apexes were obtained at 400× magnification for determination of number of nuclei/100 μm2. Two fields from two different heart sections were counted for each animal.
Expression of neonatal heart GSK3 isoforms were assessed with Western blot analysis. Briefly, hearts were dissected under stereomicroscope and frozen prior to protein extraction. For protein extraction, hearts were mechanically homogenized in lysis buffer (150 mM NaCl, 50 mM Tris and 1% Triton X-100; pH 8.0) with protease inhibitors (cOmplete; 04693116001, Roche). and protein concentrations were determined by Bradford analysis. Protein separation (20 μg of protein loaded per well) was performed with electrophoresis in a 4%–20% gradient polyacrylamide gel. After electrophoretic separation and transfer to polyvinylidene fluoride membranes, Western blots were treated as previously described above.
Statistical Analysis
Normal distribution and homoscedasticity were assessed with Kolmogorov-Smirnov test and f test for variances, respectively. Comparisons between groups were performed with Student t-test, Kruskal-Wallis test followed by Mann-Whitney U-test, Z-test for one proportion or ANOVA followed by Tukey test for means where appropriate. Data were expressed as mean ± SEM. Significance was attained at P < 0.05.
Results
Transgenic Mice with Individual or Concomitant Loss of Oocyte GSK3A and GSK3B
Constitutive Gsk3a knockout female mice were reported to be viable and fertile [44–48]; however, constitutive Gsk3b knockout mice have a lethal phenotype of controversial causes [45, 49]. Therefore, to investigate the role of GSK3 isoforms in the developing oocyte, constitutive Gsk3a knockout females (Gsk3a−/−bf/f), oocyte-specific Gsk3b knockout females (oGsk3a+/+b−/−), and constitutive Gsk3a knockout and oocyte-specific Gsk3b knockout females (oGsk3a−/−b−/−; Fig. 2A) were obtained through genotype-oriented mating.
Transcription of Gsk3 isoforms was investigated in GVI oocytes harvested from wild-type, Gsk3a−/−bf/f, oGsk3a+/+b−/−, and oGsk3a−/−b−/− animals with RT-PCR. Both Gsk3 isoforms were transcribed in GVI oocytes from wild-type animals (Fig. 2B). Constitutive Gsk3a knockout females (Gsk3a−/−bf/f) and oGsk3a−/−b−/− produced oocytes lacking Gsk3a transcripts, and Gsk3b transcripts were absent in GVI from oGsk3a+/+b−/− and oGsk3a−/−b−/− (Fig. 2B). When transcripts were identified, amplicons were sequenced and demonstrated >95% homology to anticipated Gsk3 isoforms.
Isoforms of GSK3 share 85% homology [9], and commercially available antibodies failed to distinguish GSK3 isoforms in immunohistochemistry; however, GSK3 isoforms A and B can be discerned in Western blots because of different molecular masses. Extracts of 198 pooled oocytes from each female group (wild type, Gsk3a−/−bf/f, oGsk3a+/+b−/−, and oGsk3a−/−b−/−) were assessed by Western blot analysis (Fig. 2C). Wild-type oocytes contained both GSK3A and GSK3B at the anticipated molecular masses 51 and 47 kDa, respectively, and with greater levels of GSK3A compared to GSK3B as previously reported (Fig. 2D) [27]. Gsk3a−/−bf/f oocytes had no detectable GSK3A (below the level of detection), yet had GSK3B at 47 kDa (Supplemental Fig. S1). The oGsk3a+/+b−/− oocytes had GSK3A at the appropriate mass and levels of GSK3B slightly above the level of detection (Supplemental Fig. S1), yet markedly reduced compared to GSK3B in wild type (approximately 2% of wild type) and Gsk3a−/−bf/f oocytes. Finally, oGsk3a−/−b−/− oocytes (Fig. 2D) had no detectable GSK3A, and GSK3B was barely visible or quantifiable and was reduced to approximately 7% of wild-type oocytes. Therefore, we were able to produce oocytes with loss/reduction of individual GSK3 isoforms and with concomitant loss of GSK3A and GSK3B (Fig. 2, C and D).
In addition to Western blot analysis, ovary sections of oGsk3a−/−b−/− were assessed with immunohistochemistry against GSK3 isozymes to determine if GSK3 was detectable in oocytes from follicles of progressive developmental stages. The primary antibody used recognizes both GSK3A and B. Ovaries from wild-type females showed extensive GSK3A/B localization in theca/interstitial tissue, granulosa cells, and oocytes of primordial, primary, secondary (Fig. 2E2–E4), and antral follicles (data not shown), in agreement with past reports of ovarian GSK3 [50]. Ovaries from Gsk3a−/−bf/f and oGsk3a+/+b−/− females were not evaluated because oocyte staining would be noninformative due to primary antibody isoform cross-reactivity. Deletion of floxed Gsk3b alleles was obtained with expression of Cre-recombinase driven by ZP3 promoter at the primary follicles stage transition [51]. Consequently, GSK3 immunostaining in oGsk3a−/−b−/− female ovaries was predicted to be present in oocytes from primordial follicles, but not from those enclosed in latter stages of folliculogenesis. Ovary sections from oGsk3a−/−b−/− females presented GSK3 staining in oocytes of primordial follicles (few squamous granulosa cells; Fig. 2E5), but not in oocytes of primary (cuboidal to columnar granulosa cells; Fig. 2E6), secondary (Fig. 2, E7 and E8), or antral follicles (data not shown). Western blot analysis for detection of GSK3 isoforms in oGsk3a−/−b−/− indicated that only GSK3B is expressed in somatic cells (brain, liver, and heart; Supplemental Fig. S2). This information led us to a secondary observation in the GSK3 ovarian immunohistochemistry of oGsk3a−/−b−/−: the assessment of the presence and differential GSK3B staining of granulosa and theca/interstitial cells of primordial, primary, and secondary follicles. While squamous, cuboidal, and columnar granulosa cells stained positive for GSK3B (Fig. 2E5 and E6), not all granulosa cells of secondary follicles displayed GSK3B staining (Fig. 2E7). In addition, this evaluation also demonstrated that GSK3B staining of theca/interstitial is not ubiquitous, yet there are distinct groupings of cells with and without GSK3B staining in this region of the ovary.
Loss of Oocyte Individual and Concomitant GSK3 Isoforms and Fertility
To investigate the loss of oocyte individual and/or concomitant GSK3 isoforms on female fertility, females were mated to wild-type FVB-NJ males. This design allowed the study of oocyte GSK3 isoforms without potential interference of an embryonic lethal because following fertilization, the male gamete would contribute one copy of Gsk3a and Gsk3b. In large-scale breeding colonies generating Gsk3a−/+ and Gsk3b−/+ embryos, offspring are born and survive at an anticipated Mendelian genetic ratio [48].
Loss of oocyte individual or concomitant GSK3 isoforms did not significantly alter the age of the first litter when compared to the mouse breeding colony or between GSK3 isoform transgenic animal groupings: breeding colony, 63 ± 10 days; Gsk3a−/−bf/f, 75 ± 12 days; oGsk3a+/+b−/−, 85 ± 17 days; and oGsk3a−/−b−/−, 74 ± 10 days (P = 0.3; Fig. 3A). Females presenting loss of oocyte individual or concomitant GSK3 isoforms had comparable estrous cycles duration—Gsk3a−/−bf/f, 6 ± 0.5 days; oGsk3a+/+b−/−, 5 ± 0.6 days; and oGsk3a−/−b−/−, 6 ± 0.3 days (P = 0.6)—that were within the reported range of this mouse strain [52].
Fig. 3.
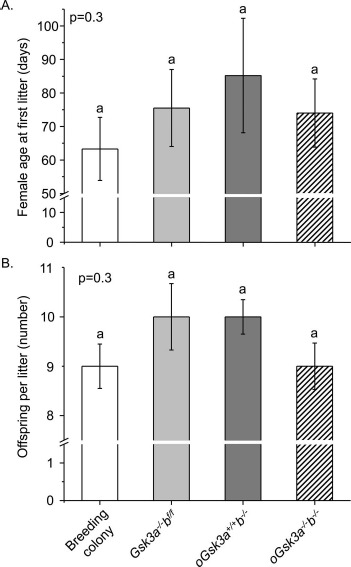
Characterization of Gsk3a−/−bf/f, oGsk3a+/+b−/−, and oGsk3a−/−b−/− females fertility in comparison to fertility of breeding colony females. A) Age at first conception of Gsk3a−/−bf/f (n = 4), oGsk3a+/+b−/− (n = 5), and oGsk3a−/−b−/− (n = 6) females were similar between transgenic groups and females in the mouse breeding colony (n = 15). Values represent mean ± SEM. B) The number of offspring per litter of transgenic females (Gsk3a−/−bf/f (n = 9), oGsk3a+/+b−/− (n = 10), and oGsk3a−/−b−/− (n = 13)) bred to FVB/NJ males were similar between transgenic groups and females in the mouse breeding colony (n = 29). Values represent mean ± SEM.
Inbred C57BL/6J and FVB/NJ mouse strains produce litters with 8 ± 2 and 9 ± 2 offspring, respectively [53]. Similarly, our mouse breeding colony produced 9 ± 0.5 offspring/litter. Loss of oocyte GSK3A, GSK3B, or concomitant loss of GSK3 isoforms did not significantly affect litter sizes in comparison to the breeding colony: Gsk3a−/−bf/f, 10 ± 0.7; oGsk3a+/+b−/−, 10 ± 0.4; and oGsk3a−/−b−/−, 10 ± 0.5 offspring/litter (P = 0.3; Fig. 3B). In summary, individual or concomitant loss of GSK3 isoforms in the oocyte did not cause infertility.
Loss of Oocyte Individual and Concomitant GSK3 Isoforms and Offspring Survival
Constitutive heterozygous knockout of GSK3 isoforms (Gsk3a+/−b+/−) produced normal and viable animals [48] within the appropriate Mendelian ratios. In our breeding colony controls, the cumulative death rate of offspring within 24 h of birth was 9% ± 3% and was comparable to previous reports for C57/B16 dams (5% ± 6%) [53]. Similarly, low offspring 24 h death rate was observed when dams were Gsk3a−/−bf/f (4% ± 2%) or oGsk3a+/+b−/− (5% ± 2%). However, significantly more offspring from oGsk3a−/−b−/− dams died within 24 h of birth (63% ± 8%; P < 0.0001; Fig. 4). While a significant percentage of offspring from oGsk3a−/−b−/− dams died within the first 4 h after birth (25% ± 7%), the majority of death occurred between 4 to 24 h after birth (39% ± 9%). In the following 24 h of life (48 h postparturition) only 1% ± 1% of offspring from oGsk3a−/−b−/− dams died, and this incidence was similar to other dam groupings and did not increase through the subsequent preweaning period.
Fig. 4.
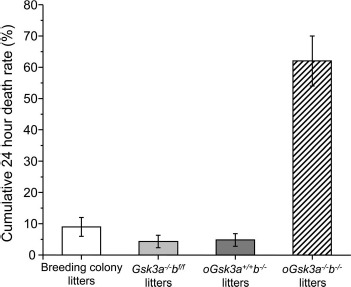
Comparison of 24 h cumulative death rate in offspring from Gsk3a−/−bf/f, oGsk3a+/+b−/−, oGsk3a−/−b−/−, and breeding colony dams. Loss of oocyte GSK3A (Gsk3a−/−bf/f, n = 9 litters) or GSK3B (oGsk3a+/+b−/−, n = 10 litters) in dams did not increase offspring 24 h cumulative death rate in comparison to each other, nor to litters from the mouse breeding colony (n = 29 litters). However, concomitant loss of oocyte GSK3A and GSK3B (oGsk3a−/−b−/−, n = 13 litters) in dams significantly increased offspring 24 h cumulative death rate in comparison to litters from the mouse breeding colony, Gsk3a−/−bf/f, and oGsk3a+/+b−/− dams (P < 0.01). Values represent means ± SEM.
Concomitant Loss of Oocyte GSK3 Isoforms Induce Congestive Heart Failure Secondary to Ventricular Hyperplasia
In a subset of experiments, offspring derived from the breeding colony, Gsk3a−/−bf/f dams, and oGsk3a+/+b−/− dams were euthanized at 25 h after birth and prepared for histology and comparison to oGsk3a−/−b−/− dam-derived offspring that died within 24 h of birth. Offspring derived from the mouse breeding colony, Gsk3a−/−bf/f, and oGsk3a+/+b−/− females had normal heart, kidney, liver, lung, and brain histology (Fig. 5A–G).
Fig. 5.
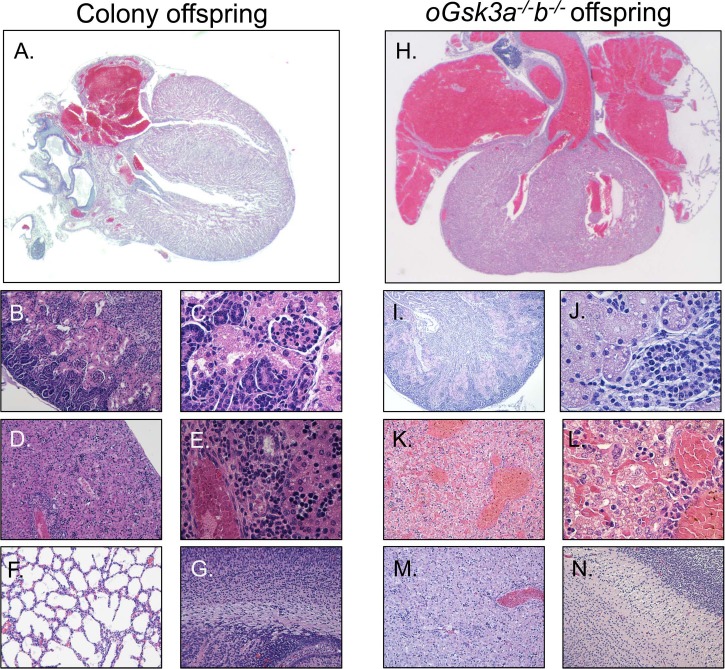
Histology analysis determined that offspring derived from the mouse breeding colony had normal hearts (A), kidneys (B, C), liver (D, E), lungs (F), and brains (G). However, animals derived from oocytes lacking both isoforms of GSK3 (oGsk3a−/−b−/− offspring) presented with great vessel and atrial dilation (H), ischemic nephrosis (I, J), severe liver congestion (K, L), and lung atelectasis (M); however, the brain had a normal appearance (N). Original magnification ×25 (A, H), ×100 (B, D, F, G, I, K, M, N), or ×400 (C, E, J, L).
Compared to the offspring from the mouse breeding colony, Gsk3a−/−bf/f, and oGsk3a+/+b−/− females, offspring derived from oGsk3a−/−b−/− females presented with severe atrial and great pulmonary vessels dilation (Fig. 5H). These cardiovascular alterations in oGsk3a−/−b−/− dam-derived offspring copresented with ischemic nephrosis (Fig. 5, I and J), liver congestion (Fig. 5, K and L), and atelectasis (Fig. 5M). Brains of oGsk3a−/−b−/− female-derived offspring were morphology normal (Fig. 5N). Histopathology findings suggested that oGsk3a−/−b−/− female-derived pups had congestive cardiac failure.
To better understand the congestive cardiac failure phenotype, dimensions of hearts collected from offspring of the mouse breeding colony and oGsk3a−/−b−/− females affected with congestive heart failure were compared (Fig. 6, A and B). Atrial width of oGsk3a−/−b−/− female-derived offspring (3.8 ± 0.1 mm) was significantly greater than atrial widths of breeding colony offspring (2.3 ± 0.1 mm, P < 0.05; Fig. 6C). Additionally, atrial length of oGsk3a−/−b−/− female-derived offspring (2.5 ± 0.2 mm) was greater than in offspring obtained from the breeding colony (1.4 ± 0.2 mm, P < 0.01; Fig. 6D). Ventricular width was greater in oGsk3a−/−b−/− female-derived animals (2.7 ± 0.1 mm) than in offspring from the breeding colony (2.3 ± 0.05 mm, P < 0.05; Fig. 6E); however, the length of ventricles of oGsk3a−/−b−/− female-derived offspring (2.4 ± 0.03 mm) was similar to heart ventricular length of breeding colony offspring (2.2 ± 0.1 mm, P = 0.2; Fig. 6F). Because ventricular dimensions were altered, cardiomyocyte area and number were assessed in heart apexes.
Fig. 6.
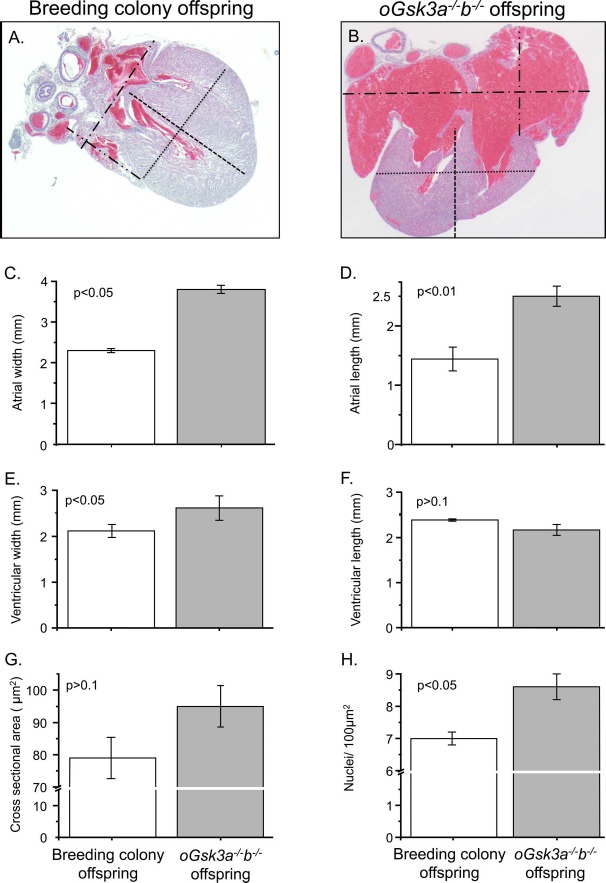
Comparison of the breeding colony and oGsk3a−/−b−/− offspring heart phenotypes. A and B) Representative micrographs used to quantify atrial and ventricular dimensions of hearts from mouse breeding colony and oGsk3a−/−b−/− female-derived offspring (×25 magnification): atrial width, dash-dotted lines; atrial length, dash-dot-dotted lines; ventricular width, dotted lines; and ventricular length, dashed lines. B) Atria from oGsk3a−/−b−/− female-derived pups were visually increased in relation to atria from mouse breeding colony offspring. C and D) Atrial width and length were significantly greater in oGsk3a−/−b−/− offspring hearts (n = 6) in comparison to atrial dimensions of mouse breeding colony offspring hearts (n = 3). E) Ventricular width was greater in hearts from oGsk3a−/−b−/− offspring (n = 6) in relation to mouse breeding colony offspring hearts (n = 3); however, the ventricular length was similar between groups (F). G) Cross-sectional area of cardiomyocytes in heart ventricles from mouse breeding colony (n = 5) and oGsk3a−/−b−/− (n = 5) offspring were similar, but apexes from oGsk3a−/−b−/− female-derived animals had significantly greater cell density than in mouse breeding colony offspring hearts (H). Values in column graphs are means ± SEM.
Ventricular apex cardiomyocyte cross-sectional area was similar between offspring derived from the breeding colony (79 ± 6.4 μm2) and oGsk3a−/−b−/− female-derived offspring (95 ± 6.4 μm2, P > 0.1; Fig. 6G). Offspring derived from oGsk3a−/−b−/− females had a greater number of cardiomyocytes in the ventricular apex (8.6 ± 0.4 nuclei/100 μm2) than in offspring from the breeding colony (7.0 ± 0.2 nuclei/100 μm2, P < 0.05; Fig. 6H). In summary, concomitant loss of oocyte GSK3 isozymes during oocyte growth and the periconception period resulted in offspring death due to congestive heart failure secondary to ventricular cardiomyocyte hyperplasia.
Heart protein expression levels of GSK3 isoforms were compared by Western blot analysis in offspring from the breeding colony euthanized 25 h after birth and oGsk3a−/−b−/− female-derived animals that died within 24 h after birth to investigate if ventricular cardiomyocyte hyperplasia was associated to postnatal GSK3 haploinsufficiency. Expression of GSK3 isoforms were detected in offspring from the breeding colony and oGsk3a−/−b−/− (Fig. 7A). Relative expression of heart GSK3A normalized to GAPDH was comparable between offspring from the breeding colony (1 ± 0.2) and oGsk3a−/−b−/− (1.9 ± 0.6; P > 0.1) females (Fig. 7B). Similarly, the relative expression of heart GSK3B normalized to GAPDH did not differ between groups (breeding colony offspring = 1 ± 0.16 and oGsk3a−/−b−/− offspring = 1.17 ± 0.4; Fig. 7C).
Fig. 7.
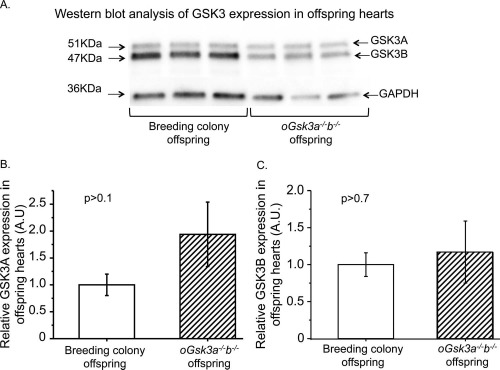
Comparison of GSK3 isoforms protean expression in hearts from breeding colony and oGsk3a−/−b−/− female-derived offspring. A) Representative micrographs of GSK3A, GSK3B, and GAPDH protein detection in hearts from breeding colony and oGsk3a−/−b−/− female-derived offspring. B) Levels of GSK3A in breeding colony offspring hearts (n = 11) were comparable to those observed in hearts from oGsk3a−/−b−/− female-derived offspring (n = 7). C) Heart GSK3B levels in breeding colony offspring (n = 11) were similar to those observed in hearts from oGsk3a−/−b−/− female-derived offspring (n = 7). Values in column graphs are means ± SEM.
Discussion
GSK3 has pleiotropic effects [10–15] and is involved in the pathogenesis of several diseases [1–8]. Cell treatments with small molecules such as lithium chloride, alsterpaullone [54–56], 6-bromoindirubin-3′-oxime, and insulin [27, 40, 50, 57] are common pharmacological and physiological means of inhibiting GSK3 and are also used to study its role in different cell types. However, GSK3 inhibitors frequently compete with ATP, have limited specificity, and can cause inhibition of other kinases [58]. Knockout models serve as an alternative approach to investigate the role of GSK3 in different tissues circumventing unwanted effects of GSK3 inhibitors. Yet, caveats exist in this approach, especially if both GSK3 isoforms are coexpressed in the cell/tissue of interest [48, 59], as is the case of mammalian oocytes. Constitutive knockout of Gsk3a did not alter survival or female reproduction in mice [44–48]; however, attempts to produce constitutive knockout of Gsk3b resulted in embryonic/neonatal lethality [45, 49]. Consequently, any study addressing the role of GSK3B with knockout models requires the generation of a conditional Gsk3b knockout. In addition, GSK3 isozymes have tissue/cell type-specific actions [48, 60, 61], functional redundancy, and potential compensatory actions [48, 59]. For this reason, knocking out Gsk3a, oocyte Gsk3b, and both isoforms concomitantly was required to study the in vivo relevance of oocyte GSK3 in female reproductive function and offspring health. Therefore, creation of mice lacking GSK3 isoforms in oocytes was attained through a series of genotype-oriented matings of Gsk3a+/−b+/f males to animals carrying a ZP3-Cre knock-in. The success of this strategy in providing female mice with oocyte lacking individual GSK3A or GSK3B, or concomitant loss of GSK3A and GSK3B, was documented with RT-PCR for amplification of exon 2 of Gsk3a and Gsk3b and Western blot detection of GSK3 isoforms in oocytes from wild-type, Gsk3a−/−bf/f, oGsk3a+/+b−/−, and oGsk3a−/−b−/− animals.
In addition to Western blot analysis, immunohistochemistry analysis of oGsk3a−/−b−/− ovary sections, with an antibody that recognizes both GSK3 isoforms, showed that GSK3B was expressed in primordial follicle oocytes, and the expected expression of Cre-recombinase by Zp3 promoter at the primordial to primary follicle developmental transition [51] led to loss of GSK3B in oocytes from more developed follicles. Interestingly, wild-type ovaries presented GSK3 immunostaining throughout all the ovarian cell types, whilst oGsk3a−/−b−/− ovaries presented differential expression of GSK3B in granulosa and theca/interstitial cells of follicles in different developmental stages. This indicates that GSK3B is expressed in different granulosa cell subpopulations, as wells as having spatial differential expression in theca/interstitial cells, during follicle growth. This differential expression, and its relationship to cell type and/or function, requires further investigation.
Inhibition of GSK3 activity in ovarian follicles and oocytes with small molecules altered chromatin condensation [27, 50] during meiosis with the potential of causing meiotic disjunction, aberrant meiotic chromatin segregation, and chaotic aneuploidy in the resulting embryos. Treatment of bovine cumulus-oophorus-complexes with the GSK3 inhibitor 6-bromoindirubin-3′-oxime caused chromosomal misalignment and disruption of meiotic progression [40]. These observations prompted the hypothesis that inhibition of oocyte GSK3 would result in infertility. However, individual or concomitant knock out of GSK3A and GSK3B in mouse oocytes did not interfere with age at first conception or estrous cycle length. In addition, litter sizes of transgenic females (Gsk3a−/−bf/f, oGsk3a+/+b−/−, and oGsk3a−/−b−/−) were within the normal range of litter sizes from the mouse breeding colony and litter sizes reported for FVB/NJ and C57/Bl6 mouse strains. Discordance between the results observed with GSK3 knockouts with no fertility affect in this study and pharmacological GSK3 inhibition studies and perturbed oocyte meiosis [27, 40, 41, 62] may reflect unintended inhibition of other kinases involved in meiotic progression [63] by pharmacological inhibitors.
Individual or concomitant loss of GSK3A and GSK3B did not cause infertility; however, offspring 24 h cumulative death rate was approximately 10 times higher in litters originated from oocytes with concomitant loss of GSK3A and GSK3B (oGsk3a−/−b−/− offspring) than in litters generated within the mouse breeding colony or with oocytes lacking individual isoforms of GSK3 (Gsk3a−/−bf/f and oGsk3a+/+b−/− female-derived litters). Transgenic females were mated with wild-type FVB/NJ males, consequently zygotes generated from oocytes with concomitant loss of GSK3A and GSK3B had one wild-type copy of each GSK3 isoform. Mice with constitutive loss of GSK3A and only one functional allele of GSK3B (Gsk3a−/−b+/−) were previously reported by Itoh and coworkers [48] to be viable and survive as offspring, despite impaired skeletal development. In the same study, animals with only one functional allele of each GSK3 isoforms (Gsk3a+/−b+/−) were reported to be normal [48] and were produced in the expected Mendelian ratio ([48]; S. Itoh, personal communication; J. Woodgett, personal communication). Evidence that Gsk3a+/−b+/− mouse are viable, and thrive, indicates that loss of a single functional allele of each GSK3 isoform during embryogenesis is insufficient to impair animal viability and development. Collectively, these data support the conclusion that the lack of offspring viability observed in the present study are caused by the concomitant loss of both GSK3 isoforms within the oocyte during oogenesis.
The cause of death in animals with concomitant loss of GSK3 isoforms (oGsk3a−/−b−/−) was congestive heart failure secondary to ventricular hyperplasia. Constitutive knockout of Gsk3b, with normal GSK3A, is a lethal phenotype due to cardiac malformation. Despite similarities between the histology findings of Kerkela and colleagues [45] and the present study, the mechanism underpinning these cardiac malformations are different because neither offspring from Gsk3a−/−bf/f or oGsk3 a+/+b−/− females had perinatal death, cardiac dysfunction, or cardiac malformation. Knocking out GSK3B in every cell during embryogenesis disrupts normal function of the Wnt-signaling pathway required for specification of cardiac mesoderm and cardiogenesis [45, 47, 64]. However, heart malformations observed in this study occurred despite the presence of embryo monoallelic Gsk3 and only after knockout of both isoforms of Gsk3 in the oocyte. Additionally, in the present study, offspring produced from oocytes lacking GSK3 isoforms and wild-type sperm that presented the lethal phenotype had heart GSK3 levels comparable to healthy offspring from the breeding colony, suggesting that postnatal heart GSK3 levels have no causal relation to the lethal cardiac phenotype.
A mechanism by which concomitant loss of oocyte GSK3A and GSK3B impairs offspring cardiogenesis may involve epigenetically regulated genes important for normal fetal and cardiac development and neonatal survival [65]. During oocyte growth and acquisition of meiotic competence, oocyte chromatin is remodeled with the addition of de novo methyl groups to cytosines located in cytosine-guanine (CG) dinucleotides and non-CG cytosine [66] regions of imprinted genes. This oocyte methylation process is mediated by DNMTs [66]. GSK3 alters the expression/activity of DNMTs in tissue- or cell-specific manners [16–18]. Most relevant to the role of insulin signaling and GSK3 in the oocyte is the report that the PIK3-mediated activation of Akt can mediate DNA methylation in mouse embryonic stem cells [67]. Additionally, inhibition of GSK3 in embryonic stem cells caused reduced expression of Dnmt3a2 and misexpression of imprinted genes [67]. Over the last two decades, it has been appreciated that abnormal expression of differentially imprinted genes in gametes and/or preimplantation embryos can affect fetal development [68–70]. Loss of proper DNA methylation and altered maternally imprinted gene expression may be underlying the lethal cardiac phenotype observed when concomitant GSK3 activity is absent during late stages of oogenesis.
Besides providing new information on GSK3 isoform functional specificity in the oocyte, and their concomitant requirement for proper development of healthy offspring, the results from this study suggests that changes occurring during the periconceptional period can affect progeny development, survival and long-term/late onset health effects as previously proposed [71–73]. The present study is of particular translational importance because it suggests that oocyte GSK3 during the preconception and periconception periods may represent an intermediate in mechanisms that link maternal hyperinsulinemia/insulin resistance present in individuals with metabolic syndrome, diabetes, and/or increased body mass index and offspring congenital birth defects and/or failure to thrive [74–77]. In summary, loss of oocyte GSK3 in the periconceptional period does not alter fertility yet causes offspring cardiac hyperplasia, cardiovascular defects, and significantly increased death. These results support a developmental mechanism by which periconceptional hyperinsulinemia associated with maternal metabolic syndrome, obesity, and/or diabetes can act on the oocyte and affect offspring cardiovascular development, function, and congenital heart malformation.
Supplementary Material
Acknowledgment
We would like to acknowledge Dr. Jim R. Woodgett from the Lunenfeld-Tanenbaum Research Institute in Toronto, Canada, for providing founder males used to establish our mouse colony. We also appreciate the critical review of this manuscript and comments by Drs. Carol Elias and Vijayaraghavan Srinivasan.
References
- 1. Latapy C, Rioux V, Guitton MJ, Beaulieu JM.. Selective deletion of forebrain glycogen synthase kinase 3beta reveals a central role in serotonin-sensitive anxiety and social behaviour. Philos Trans R Soc Lond B Biol Sci 2012; 367:2460–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL et al. . Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 2009; 136:1017–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beaulieu JM, Zhang X, Rodriguiz RM, Sotnikova TD, Cools MJ, Wetsel WC, Gainetdinov RR, Caron MG.. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci U S A 2008; 105:1333–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu Y, Tanabe K, Baronnier D, Patel S, Woodgett J, Cras-Meneur C, Permutt MA.. Conditional ablation of Gsk-3beta in islet beta cells results in expanded mass and resistance to fat feeding-induced diabetes in mice. Diabetologia 2010; 53:2600–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu Z, Tanabe K, Bernal-Mizrachi E, Permutt MA.. Mice with beta cell overexpression of glycogen synthase kinase-3beta have reduced beta cell mass and proliferation. Diabetologia 2008; 51:623–631. [DOI] [PubMed] [Google Scholar]
- 6. Tanabe K, Liu Z, Patel S, Doble BW, Li L, Cras-Meneur C, Martinez SC, Welling CM, White MF, Bernal-Mizrachi E, Woodgett JR, Permutt MA.. Genetic deficiency of glycogen synthase kinase-3beta corrects diabetes in mouse models of insulin resistance. PLoS Biol 2008; 6:e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rao R, Hao CM, Redha R, Wasserman DH, McGuinness OP, Breyer MD.. Glycogen synthase kinase 3 inhibition improves insulin-stimulated glucose metabolism but not hypertension in high-fat-fed C57BL/6J mice. Diabetologia 2007; 50:452–460. [DOI] [PubMed] [Google Scholar]
- 8. Lee J, Kim MS.. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res Clin Pract 2007; 77(Suppl 1):S49–S57. [DOI] [PubMed] [Google Scholar]
- 9. Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J 1990; 9:2431–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rylatt DB, Aitken A, Bilham T, Condon GD, Embi N, Cohen P.. Glycogen synthase from rabbit skeletal muscle. Amino acid sequence at the sites phosphorylated by glycogen synthase kinase-3, and extension of the N-terminal sequence containing the site phosphorylated by phosphorylase kinase. Eur J Biochem 1980; 107:529–537. [PubMed] [Google Scholar]
- 11. Hart MJ, de los Santos R, Albert IN, Rubinfeld B, Polakis P.. Downregulation of beta-catenin by human Axin and its association with the APC tumor suppressor, beta-catenin and GSK3 beta. Curr Biol 1998; 8:573–581. [DOI] [PubMed] [Google Scholar]
- 12. Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P.. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science 1996; 272:1023–1026. [DOI] [PubMed] [Google Scholar]
- 13. Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST.. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J 2002; 21:281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ishiguro K, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, Uchida T, Imahori K.. Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett 1993; 325:167–172. [DOI] [PubMed] [Google Scholar]
- 15. Sperber BR, Leight S, Goedert M, Lee VM.. Glycogen synthase kinase-3 beta phosphorylates tau protein at multiple sites in intact cells. Neurosci Lett 1995; 197:149–153. [DOI] [PubMed] [Google Scholar]
- 16. Sun L, Zhao H, Xu Z, Liu Q, Liang Y, Wang L, Cai X, Zhang L, Hu L, Wang G, Zha X.. Phosphatidylinositol 3-kinase/protein kinase B pathway stabilizes DNA methyltransferase I protein and maintains DNA methylation. Cell Signal 2007; 19:2255–2263. [DOI] [PubMed] [Google Scholar]
- 17. Leitch HG, McEwen KR, Turp A, Encheva V, Carroll T, Grabole N, Mansfield W, Nashun B, Knezovich JG, Smith A, Surani MA, Hajkova P.. Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol 2013; 20:311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pyko IV, Nakada M, Sabit H, Teng L, Furuyama N, Hayashi Y, Kawakami K, Minamoto T, Fedulau AS, Hamada J.. Glycogen synthase kinase 3beta inhibition sensitizes human glioblastoma cells to temozolomide by affecting O6-methylguanine DNA methyltransferase promoter methylation via c-Myc signaling. Carcinogenesis 2013; 34:2206–2217. [DOI] [PubMed] [Google Scholar]
- 19. Barker DJ, Osmond C.. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986; 1:1077–1081. [DOI] [PubMed] [Google Scholar]
- 20. Lehnen H, Zechner U, Haaf T.. Epigenetics of gestational diabetes mellitus and offspring health: the time for action is in early stages of life. Mol Hum Reprod 2013; 19:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Greene MF. Spontaneous abortions and major malformations in women with diabetes mellitus. Semin Reprod Endocrinol 1999; 17:127–136. [DOI] [PubMed] [Google Scholar]
- 22. Corrigan N, Brazil DP, McAuliffe F.. Fetal cardiac effects of maternal hyperglycemia during pregnancy. Birth Defects Res A Clin Mol Teratol 2009; 85:523–530. [DOI] [PubMed] [Google Scholar]
- 23. Lopez AL III, , Chen J, Joo HJ, Drake M, Shidate M, Kseib C, Arur S.. DAF-2 and ERK couple nutrient availability to meiotic progression during Caenorhabditis elegans oogenesis. Dev Cell 2013; 27:227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stefanovic D, Erikson E, Pike LJ, Maller JL.. Activation of a ribosomal protein S6 protein kinase in Xenopus oocytes by insulin and insulin-receptor kinase. EMBO J 1986; 5:157–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. El-Etr M, Schorderet-Slatkine S, Baulieu EE.. Meiotic maturation in Xenopus laevis oocytes initiated by insulin. Science 1979; 205:1397–1399. [DOI] [PubMed] [Google Scholar]
- 26. Hainaut P, Kowalski A, Giorgetti S, Baron V, Van Obberghen E.. Insulin and insulin-like-growth-factor-I (IGF-I) receptors in Xenopus laevis oocytes. Comparison with insulin receptors from liver and muscle. Biochem J 1991; 273(Pt 3):673–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Acevedo N, Ding J, Smith GD.. Insulin signaling in mouse oocytes. Biol Reprod 2007; 77:872–879. [DOI] [PubMed] [Google Scholar]
- 28. Zhang X, Kidder GM, Watson AJ, Schultz GA, Armstrong DT.. Possible roles of insulin and insulin-like growth factors in rat preimplantation development: investigation of gene expression by reverse transcription-polymerase chain reaction. J Reprod Fertil 1994; 100:375–380. [DOI] [PubMed] [Google Scholar]
- 29. Quesnel H. Localization of binding sites for IGF-I, insulin and GH in the sow ovary. J Endocrinol 1999; 163:363–372. [DOI] [PubMed] [Google Scholar]
- 30. Watson AJ, Hogan A, Hahnel A, Wiemer KE, Schultz GA.. Expression of growth factor ligand and receptor genes in the preimplantation bovine embryo. Mol Reprod Dev 1992; 31:87–95. [DOI] [PubMed] [Google Scholar]
- 31. Lighten AD, Hardy K, Winston RM, Moore GE.. Expression of mRNA for the insulin-like growth factors and their receptors in human preimplantation embryos. Mol Reprod Dev 1997; 47:134–139. [DOI] [PubMed] [Google Scholar]
- 32. Mendez R, Myers MG Jr, , White MF, Rhoads RE.. Stimulation of protein synthesis, eukaryotic translation initiation factor 4E phosphorylation, and PHAS-I phosphorylation by insulin requires insulin receptor substrate 1 and phosphatidylinositol 3-kinase. Mol Cell Biol 1996; 16:2857–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P.. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 1997; 7:261–269. [DOI] [PubMed] [Google Scholar]
- 34. Toker A, Newton AC.. Cellular signaling: pivoting around PDK-1. Cell 2000; 103:185–188. [DOI] [PubMed] [Google Scholar]
- 35. Sutherland C, Leighton IA, Cohen P.. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J 1993; 296(Pt 1):15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cross DA, Alessi DR, Vandenheede JR, McDowell HE, Hundal HS, Cohen P.. The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. Biochem J 1994; 303(Pt 1):21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA.. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995; 378:785–789. [DOI] [PubMed] [Google Scholar]
- 38. Fang X, Yu SX, Lu Y, Bast RC Jr, , Woodgett JR, Mills GB.. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci U S A 2000; 97:11960–11965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Welsh GI, Proud CG.. Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem J 1993; 294(Pt 3):625–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Uzbekova S, Salhab M, Perreau C, Mermillod P, Dupont J.. Glycogen synthase kinase 3B in bovine oocytes and granulosa cells: possible involvement in meiosis during in vitro maturation. Reproduction 2009; 138:235–246. [DOI] [PubMed] [Google Scholar]
- 41. Swain JE, Ding J, Brautigan DL, Villa-Moruzzi E, Smith GD.. Proper chromatin condensation and maintenance of histone H3 phosphorylation during mouse oocyte meiosis requires protein phosphatase activity. Biol Reprod 2007; 76:628–638. [DOI] [PubMed] [Google Scholar]
- 42. Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML.. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). BioTechniques 2000; 29:52–54. [DOI] [PubMed] [Google Scholar]
- 43. Byers SL, Wiles MV, Dunn SL, Taft RA.. Mouse estrous cycle identification tool and images. PLoS One 2012; 7:e35538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR.. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab 2007; 6:329–337. [DOI] [PubMed] [Google Scholar]
- 45. Kerkela R, Kockeritz L, Macaulay K, Zhou J, Doble BW, Beahm C, Greytak S, Woulfe K, Trivedi CM, Woodgett JR, Epstein JA, Force T et al. . Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest 2008; 118:3609–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Soutar MP, Kim WY, Williamson R, Peggie M, Hastie CJ, McLauchlan H, Snider WD, Gordon-Weeks PR, Sutherland C.. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J Neurochem 2010; 115:974–983. [DOI] [PubMed] [Google Scholar]
- 47. Zhou J, Lal H, Chen X, Shang X, Song J, Li Y, Kerkela R, Doble BW, MacAulay K, DeCaul M, Koch WJ, Farber J et al. . GSK-3alpha directly regulates beta-adrenergic signaling and the response of the heart to hemodynamic stress in mice. J Clin Invest 2010; 120:2280–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Itoh S, Saito T, Hirata M, Ushita M, Ikeda T, Woodgett JR, Algul H, Schmid RM, Chung UI, Kawaguchi H.. GSK-3alpha and GSK-3beta proteins are involved in early stages of chondrocyte differentiation with functional redundancy through RelA protein phosphorylation. J Biol Chem 2012; 287:29227–29236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR.. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000; 406:86–90. [DOI] [PubMed] [Google Scholar]
- 50. Wang X, Liu XT, Dunn R, Ohl DA, Smith GD.. Glycogen synthase kinase-3 regulates mouse oocyte homologue segregation. Mol Reprod Dev 2003; 64:96–105. [DOI] [PubMed] [Google Scholar]
- 51. Lan ZJ, Xu X, Cooney AJ.. Differential oocyte-specific expression of Cre recombinase activity in GDF-9-iCre, Zp3cre, and Msx2Cre transgenic mice. Biol Reprod 2004; 71:1469–1474. [DOI] [PubMed] [Google Scholar]
- 52. Silver LM. Mouse Genetics: Concepts and Applications. New York:Oxford University Press; 1995. [Google Scholar]
- 53. Murray SA, Morgan JL, Kane C, Sharma Y, Heffner CS, Lake J, Donahue LR.. Mouse gestation length is genetically determined. PLoS One 2010; 5:e12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. de Abreu LA, Calixto C, Waltero CF, Della Noce BP, Githaka NW, Seixas A, Parizi LF, Konnai S. Vaz Ida S, Ohashi K, Logullo C. The conserved role of the AKT/GSK3 axis in cell survival and glycogen metabolism in Rhipicephalus (Boophilus) microplus embryo tick cell line BME26. Biochim Biophys Acta 2013; 1830:2574–2582. [DOI] [PubMed] [Google Scholar]
- 55. Acevedo N, Wang X, Dunn RL, Smith GD.. Glycogen synthase kinase-3 regulation of chromatin segregation and cytokinesis in mouse preimplantation embryos. Mol Reprod Dev 2007; 74:178–188. [DOI] [PubMed] [Google Scholar]
- 56. Leost M, Schultz C, Link A, Wu YZ, Biernat J, Mandelkow EM, Bibb JA, Snyder GL, Greengard P, Zaharevitz DW, Gussio R, Senderowicz AM et al. . Paullones are potent inhibitors of glycogen synthase kinase-3beta and cyclin-dependent kinase 5/p25. Eur J Biochem 2000; 267:5983–5994. [DOI] [PubMed] [Google Scholar]
- 57. Mirakhori F, Zeynali B, Tafreshi AP, Shirmohammadian A.. Lithium induces follicular atresia in rat ovary through a GSK-3beta/beta-catenin dependent mechanism. Mol Reprod Dev 2013; 80:286–296. [DOI] [PubMed] [Google Scholar]
- 58. Eldar-Finkelman H, Licht-Murava A, Pietrokovski S, Eisenstein M.. Substrate competitive GSK-3 inhibitors–strategy and implications. Biochim Biophys Acta 2010; 1804:598–603. [DOI] [PubMed] [Google Scholar]
- 59. Gillespie JR, Ulici V, Dupuis H, Higgs A, Dimattia A, Patel S, Woodgett JR, Beier F.. Deletion of glycogen synthase kinase-3beta in cartilage results in up-regulation of glycogen synthase kinase-3alpha protein expression. Endocrinology 2011; 152:1755–1766. [DOI] [PubMed] [Google Scholar]
- 60. Maurin H, Lechat B, Dewachter I, Ris L, Louis JV, Borghgraef P, Devijver H, Jaworski T, Van Leuven F.. Neurological characterization of mice deficient in GSK3alpha highlight pleiotropic physiological functions in cognition and pathological activity as Tau kinase. Mol Brain 2013; 6:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR.. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell 2007; 12:957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hu Y, Bai Y, Chu Z, Wang J, Wang L, Yu M, Lian Z, Hua J.. GSK3 inhibitor-BIO regulates proliferation of female germline stem cells from the postnatal mouse ovary. Cell Prolif 2012; 45:287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schindler K. Protein kinases and protein phosphatases that regulate meiotic maturation in mouse oocytes. Results Probl Cell Differ 2011; 53:309–341. [DOI] [PubMed] [Google Scholar]
- 64. Tzahor E. Wnt/beta-catenin signaling and cardiogenesis: timing does matter. Dev Cell 2007; 13:10–13. [DOI] [PubMed] [Google Scholar]
- 65. Van Vliet P, Wu SM, Zaffran S, Puceat M.. Early cardiac development: a view from stem cells to embryos. Cardiovasc Res 2012; 96:352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shirane K, Toh H, Kobayashi H, Miura F, Chiba H, Ito T, Kono T, Sasaki H.. Mouse oocyte methylomes at base resolution reveal genome-wide accumulation of non-CpG methylation and role of DNA methyltransferases. PLoS Genet 2013; 9:e1003439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Popkie AP, Zeidner LC, Albrecht AM, D'Ippolito A, Eckardt S, Newsom DE, Groden J, Doble BW, Aronow B, McLaughlin KJ, White P, Phiel CJ.. Phosphatidylinositol 3-kinase (PI3K) signaling via glycogen synthase kinase-3 (Gsk-3) regulates DNA methylation of imprinted loci. J Biol Chem 2010; 285:41337–41347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lau MM, Stewart CE, Liu Z, Bhatt H, Rotwein P, Stewart CL.. Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev 1994; 8:2953–2963. [DOI] [PubMed] [Google Scholar]
- 69. Ogawa O, Eccles MR, Szeto J, McNoe LA, Yun K, Maw MA, Smith PJ, Reeve AE.. Relaxation of insulin-like growth factor II gene imprinting implicated in Wilms' tumour. Nature 1993; 362:749–751. [DOI] [PubMed] [Google Scholar]
- 70. Nystrom A, Hedborg F, Ohlsson R.. Insulin-like growth factor 2 cannot be linked to a familial form of Beckwith-Wiedemann syndrome. Eur J Pediatr 1994; 153:574–580. [DOI] [PubMed] [Google Scholar]
- 71. Steegers-Theunissen RP, Twigt J, Pestinger V, Sinclair KD.. The periconceptional period, reproduction and long-term health of offspring: the importance of one-carbon metabolism. Hum Reprod Update 2013; 19:640–655. [DOI] [PubMed] [Google Scholar]
- 72. Reece EA. Diabetes-induced birth defects: what do we know? What Can We Do? Curr Diab Rep 2012; 12:24–32. [DOI] [PubMed] [Google Scholar]
- 73. Jungheim ES, Moley KH.. Current knowledge of obesity's effects in the pre- and periconceptional periods and avenues for future research. Am J Obstet Gynecol 2010; 203:525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gilboa SM, Correa A, Botto LD, Rasmussen SA, Waller DK, Hobbs CA, Cleves MA, Riehle-Colarusso TJ.. Association between prepregnancy body mass index and congenital heart defects. Am J Obstet Gynecol 2010; 202(51):51.e1-51.e10. [DOI] [PubMed] [Google Scholar]
- 75. Mills JL. Malformations in infants of diabetic mothers. Teratology 1982; 25:385–394. [DOI] [PubMed] [Google Scholar]
- 76. Aberg A, Westbom L, Kallen B.. Congenital malformations among infants whose mothers had gestational diabetes or preexisting diabetes. Early Hum Dev 2001; 61:85–95. [DOI] [PubMed] [Google Scholar]
- 77. Gladman G, McCrindle BW, Boutin C, Smallhorn JF.. Fetal echocardiographic screening of diabetic pregnancies for congenital heart disease. Am J Perinatol 1997; 14:59–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.