

Abstract
RNA metabolism impacts different steps of mRNA life cycle including splicing, polyadenylation, nucleo-cytoplasmic export, translation, and decay. Growing evidence indicates that defects in any of these steps leads to devastating diseases in humans. This chapter reviews the various RNA metabolic mechanisms that are disrupted in Myotonic Dystrophy—a trinucleotide repeat expansion disease—due to dysregulation of RNA Binding Proteins. We also compare Myotonic Dystrophy to other microsatellite expansion disorders and describe how some of these mechanisms commonly exert direct versus indirect effects towards disease pathologies.
Keywords: Microsatellite repeat expansions, Post-transcriptional gene regulation, RNA toxicity, Alternative splicing and polyadenylation, RNA binding proteins
1. Introduction
Gene expression is a highly coordinated multistep process, which allows organisms to integrate intrinsic and environmental information to exert appropriate cellular functions. The expression of most genes can be regulated at distinct stages of RNA metabolism including synthesis or transcription, post-transcriptional processing or maturation, nucleo-cytoplasmic export, translation, as well as degradation at a rate that is often dictated by transcript- and cell-type-specific cues. Although transcription is a general point of control, many co- and post-transcriptional pre-mRNA processing events add substantial capacity to tune overall gene expression [1]. The typical pre-mRNA processing events comprise 5’ capping, splicing, and 3’ polyadenylation, which are directly linked to the nucleo-cytoplasmic export and eventual fate of mRNAs. RNA Binding Proteins (RBPs) are essential in carrying out these processing events in both the nucleus and cytoplasm by interacting with RNA sequence or structural elements and forming distinct mRNA-protein (mRNP) complexes [2]. Disruption of RBP function(s), therefore, frequently results in deleterious RNA metabolism defects that in some cases become pathogenic [3, 4].
Neurodegenerative diseases are a heterogeneous group of neurological disorders characterized by progressive degeneration of structure and function of the central or peripheral nervous systems. Aberrant RNA metabolism is increasingly implicated in neurodegenerative diseases, a subset of which are caused by the expansion of short repetitive elements (microsatellites) within particular genes [5]. The causative repeat expansion mutation for this group of disorders is unstable because the repeat size changes through generations and even within an individual, as different tissues have cell populations with variable repeat length and in some cases the repeat length varies within the same tissue [6]. The severity of a repeat expansion disease is dependent on numerous variables, including the length of the repeat, its sequence context, and the native function of the protein-coding gene with which the repeats are associated. A typical pathogenic feature of these diseases is the accumulation of repeat-containing transcripts into aberrant RNA foci, which can sequester RBPs and prevent them from performing their normal functions [7–9]. Interestingly, once the expansions cross a critical number, the repeat-containing RNAs can undergo phase separation—partitioning into granules due to multivalent base-pairing between repeat RNAs—or spontaneous gelation to form RNA foci, explaining why disease symptoms appear to be triggered after the expansions have reached a particular threshold number [10].
2. Toxicity of coding and non-coding microsatellite repeat expansions
Over 25 human genes with tandem repeat expansions have been identified to date, and these disease-causing repeats can occur in the coding or non-coding regions [6] (Figure 1 and Table 1). Majority of the microsatellites arise due to the expansion of trinucleotide repeats. However, expanded tetranucleotide, pentanucleotide, and hexanucleotide repeats are also detected. In the early nineties, two microsatellites were discovered providing the first evidence that simple repeat expansions are linked to human disease. Fragile X Syndrome (FXS)—an X-linked disorder caused by CGG repeat expansions in the 5′ untranslated region (UTR) of the FMR1 gene—is the most prevalent form of inherited cognitive impairment and mental retardation [11–16]. The repeat expansion in FXS causes loss of FMR1 gene product FMRP, a polyribosome-associated RBP that binds ~4% of brain mRNAs and regulates their expression—either enhancing or suppressing translation through unknown mechanisms [17–20].
Figure 1. Origin and expansion of microsatellite repeats in human disease.

Schematic of the gene location for various disease-associated repeat expansions. Types of repeat expansions are indicated within the parentheses along with the range of expanded repeat numbers.
Table: 1.
Summary of the tissue specific symptoms of the repeat expansion diseases with the disease-associated gene
| Defected mRNA region | Disease | Defected Gene | Tissue-specific clinical symptoms | |
|---|---|---|---|---|
| Neuronal Tissues |
Other Tissues |
|||
| 5’UTR | FXTAS | FMR1 [158] | Ataxia [159], brain atrophy, white matter lesions [160, 161], cognitive decline, parkinsonism [160], peripheral neuropathy, autonomic dysfunction and short-term memory loss [162]. | Premature ovarian failure, hypothyroidism in female [159], limb proximal muscle weakness [160] |
| FXS | FMR1 [14] | Autism [163], mental retardation, developmental delay and increased susceptibility to seizures [15]. | Macroorchidism [15], cardiac murmur [164], hyperflexible joints, hernias, flat feet [165] | |
| SCA12 | PPP2R2B [166] | Ataxia, cerebral and/or cerebellar atrophy [167], seizures [22]. | Dysarthria, action tremors in upper limbs [167]. | |
| Intron | DM2 | ZNF9 [168] | Cognitive impairment [169], intellectual disability, sleepiness and fatigue [170], brain atrophy, white and grey matter abnormalities [171, 172]. | Myotonia, muscle dysfunction, cardiac arrhythmia [40, 173], hypertrophy calf muscles [174]. |
| ALS | C9orf72 [28, 29] | Motor neuron degeneration, frontotemporal lobar dysfunction, dementia and cognitive impairment [175]. | Progressive spasticity, muscle wasting, weakness and muscle atrophy [28] | |
| FTD | C9orf72 [28, 29] | Frontotemporal lobar dysfunction, motor neuron dysfunction [176], changes in personality, behavior, and language ability, dementia [175]. | Fasciculation, muscle atrophy, weakness [177]. | |
| Coding Region | Polyglutamine (PolyQ) diseases | |||
| SBMA | AR [21] | Lower motor neuron degeneration [178], androgen insensitivity [22]. | Muscle weakness, gynecomastia and reduced fertility [22, 178] | |
| HD | HTT [179] | Cognitive decline and dementia [22], dystonia [180]. | Chorea [22], movement disorder [181]. | |
| DRPLA | ATN1 [182–184] | White matter lesion, neural loss, ataxia, seizures, choreoathetosis, dementia [22, 185], myoclonus, epilepsy [184]. | Chorea, incoordination [185] | |
| SCA 1,2,3,6,7,17 | Ataxin 1 [186, 187], Ataxin 2 [188–190], Ataxin 3 [191], CACNA1A [192], Ataxin 7 [193], TBP [194] | Ataxia, tremor, and dysarthria, parkinsonism (SCA3), retinal dystrophy (SCA7), seizures (SCA17) [22]. | Slurred speech (SCA1); Hyporeflexia (SCA2) Cardiac dysfunction (SCA7) [22]. | |
| Poly Alanine (Poly A) diseases | ||||
| OPMD (OPMD) | PABPN1 [195] | -(no data) | Eyelid ptosis and dysphagia [195], involuntary muscle weakness [196]. | |
| XLMR | ARX [197] | Cognitive impairment [198], mental retardation [199], dysarthria [200]. | Involuntary hand movements (MRXS), growth abnormality [200]. | |
| 3’UTR | DM1 | DMPK [37] | Neuropsychiatric disturbances, cognitive defeats, sleepiness and fatigue; brain atrophy [169], white and grey matter abnormalities [201], mood disorder, emotion problem and memory problem. | Myotonia, muscle wasting, cardiac arrhythmias, insulin resistance, gastrointestinal dysfunctions, posterior iridescent cataracts [54] |
| SCA8 | ATXN8 [202] | Cerebellar atrophy[203], progressive ataxia[204]. | Limb ataxia, dysarthria, nystagmus, spasticity [22]. | |
Abbreviations: Fragile X associated tremor/ataxia syndrome (FXTAS); Fragile X Syndrome (FXS); Spinocerebellar ataxia type 12 (SCA12); Myotonic Dystrophy type 2 (DM2); Amyotrophic lateral sclerosis (ALS); Frontotemporal degeneration (FTD); Spinal and Bulbar Muscular Atrophy (SBMA); Huntington disease (HD); Dentatorubral pallidoluysian Atrophy (DRPLA); Spinocerebellar Ataxias (SCA); Polyalanine (PolyA) diseases; Oculopharyngeal muscular dystrophy (OPMD); Syndromic and non-syndromic X-linked mental retardation (XLMR); Myotonic Dystrophy type 1 (DM1)
Spinal and bulbar muscular atrophy (SBMA)—the other microsatellite disease discovered along with FXS—arises due to a CAG repeat expansion in the coding region of the X chromosome-linked androgen receptor (AR) gene [21]. The discovery of SBMA was soon followed by the elucidation of a similar mutation as the basis for a group of disorders now known as the polyglutamine (polyQ) neurodegenerative diseases (Table 1). Along with SBMA, the polyQ diseases include Huntington disease (HD), dentatorubral-pallidoluysian atrophy, and six spinocerebellar ataxias (SCA) 1, 2, 3, 6, 7, and 17 [22]. As a group, these nine diseases are amongst the more common forms of inherited neurodegeneration. The translation of exons containing CAG repeats gives rise to elongated stretches of polyQs in mutant proteins, which aggregate into nuclear or cytoplasmic inclusions in the diseased brain [23–25]. Several observations indicate that the CAG repeat-containing RNAs, in the absence of coding for a protein, may also be a source of toxicity in polyQ diseases [26, 27]. GGGGCC hexanucleotide repeat expansion in the C9orf72 gene has gained much attention in the past few years and is now considered the most frequent inherited cause of Amyotrophic lateral sclerosis (ALS) and Frontotemporal dementia (FTD) [28, 29]. Pathology occurs due to the toxicity of expanded repeats, which are transcribed in both sense and antisense directions and give rise to distinct sets of intracellular RNA and protein aggregates [30–33].
Myotonic Dystrophy (DM) is part of a group of diseases characterized by repeat expansions in non-coding regions of genes. DM is defined in two clinical and molecular forms: myotonic dystrophy type 1 (DM1), and type 2 (DM2), both of which are inherited in autosomal dominant fashion. The combined worldwide incidence of DM is approximately 1 in 8000 [34, 35]. DM1 is the most prevalent form of adult onset muscular dystrophy [36] and is caused by a CTG repeat expansion in the 3′ UTR of Dystrophia Myotonica Protein Kinase (DMPK) gene [37, 38]. DM2, on the other hand, is caused by a CCTG repeat expansion in an intron of Zinc Finger Protein 9 (ZNF9) gene [39]. While 5–37 repeats are considered normal, DM1 patients can have up to several thousand CTG repeats, which can reduce expression of DMPK [40] (Figure 2a). DMPK is expressed in multiple tissues, and the major symptoms of the disease include muscle hyperexcitability (myotonia), progressive muscle wasting, cardiac defects, insulin resistance and neuropsychiatric disturbances [41–44]. Table 1 provides further description of tissue-specific symptoms observed in DM and other microsatellite expansion disorders.
Figure 2. Schematic showing different pathological mechanisms for Myotonic Dystrophy type 1 (DM1) and 2 (DM2).
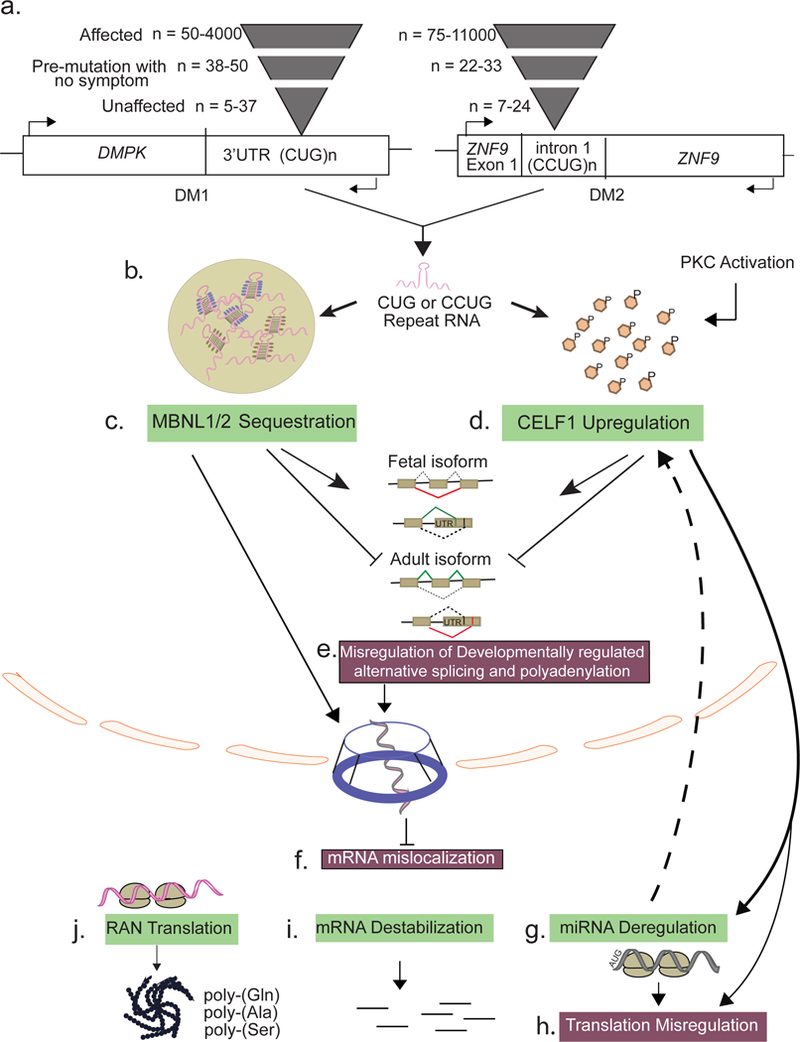
(a) Causative mutation for DM1 is CUG repeat expansion in 3’UTR of DMPK gene and for DM2 is CCUG repeat expansion in intron 1 of Znf9 gene. The severity of the disease is dependent on the number of repeats. Although these mutations are in two different genes, the disease mechanisms for both diseases are surprisingly similar. Most of the pathology is consistent with the toxic RNA gain-of-function mechanism and affects general RNA metabolism both in the nucleus and cytoplasm. (b) After transcription, the repeat containing transcripts form stable hairpin loop comprising secondary structures (pink), which aggregate to form ribonuclear foci. (c) Members of the Mbnl family of RNA binding proteins (RBPs) MBNL1/2 (purple) bind the CUG or CCUG repeats and are sequestered in the ribonuclear foci. (d) Hyperphosphorylation by PKC stabilizes another RBP, CELF1, resulting in its gain-of-function. (e) Both MBNL and CELF proteins regulate various aspects of RNA metabolism during normal development. Alterations in their functional levels due to toxic repeat RNA causes adult-to-fetal reversion of splicing and polyadenylation for many pre-mRNAs in the nucleus. (f) MBNL depletion also leads to cellular mislocalization of many mRNAs. CELF1 gain-of-function further affects (g) miRNA metabolism and (h) mRNA translation. (i) Deregulation of MBNL and CELF activity in the cytoplasm also affects mRNA stability through various mechanisms. (j) Both sense and antisense CUG/CCUG containing transcripts are subject to RAN translation in all three frames giving rise to homopolymeric polypeptides that accumulate in the cytoplasm and form pathological intracellular aggregates.
3. RNA metabolism defects in Myotonic Dystrophy
Closely after the discovery of repeats, DMPK haploinsufficiency model was put forward to explain the DM1 pathology. However, the removal of Dmpk gene in mice failed to recapitulate the major neuromuscular symptoms of DM1 [45, 46]. A separate hypothesis proposed that expanded CTG repeats might affect the expression of nearby genes. Although the adjacent gene, SIX5, exhibits reduced expression in DM1 patients [47], Six5 knockout mice also do not reproduce DM1 muscle pathology [48]. Instead, the CTG repeats alone, regardless of the gene context, are sufficient to induce pathogenic features of DM1 [49, 50]. The predominant pathology of DM1 actually stems from the toxic effects of expanded CUG RNA, which disrupts the normal activity of certain RBPs. Further support for the RNA toxicity model comes from the finding that although the repeat expansion in DM2 is on an entirely different gene, both diseases exhibit similar symptoms.
In both DM1 and DM2, the RNAs with expanded repeats (CUG in DM1; and CCUG in DM2) fold into stable hairpin loops that accumulate as ribonuclear foci in the nuclei of affected tissues [9] (Figure 2b). These expanded RNA transcripts directly trap RBPs such as muscleblind-like proteins (MBNLs) and cause deregulation of CUG-binding protein 1 (CELF1) family of alternative splicing factors [51–54], which results in aberrant splicing of many transcripts and a broad, multi-systemic phenotype (Figures 2c and 2d). Alternative pre-mRNA splicing generates much of the transcriptome diversity in higher eukaryotes as it enables the production of multiple transcripts with potentially different functions from individual genes [55]. Alternative splicing decisions are generally influenced by cis-acting regulatory elements within pre-mRNAs that promote or inhibit exon recognition, as well as expression/activity of trans-acting factors (e.g., MBNL and CELF proteins) that bind to these cis elements and regulate the accessibility of the spliceosome to splice sites [56]. The misregulated splicing events in DM are usually developmentally regulated and exhibit an adult-to-embryonic switch in splicing patterns (Figure 2e). Some of these embryonic isoforms fail to meet the adult tissue requirements and thus directly contribute to the overall disease pathology [54].
3a. Misregulation of mRNA processing
MBNL loss-of-function in DM1 and DM2 is a prominent example of RBP sequestration by disease-associated microsatellite expansion RNAs. The MBNL proteins were initially identified in Drosophila melanogaster for their requirement in muscle development and eye differentiation [57], and they were later shown as direct regulators of alternative splicing [58]. There are three MBNL paralogues in mammals, named MBNL1–3. MBNL1 and MBNL2 are widely expressed across many tissues, including brain, heart, muscle, and liver, whereas MBNL3 expression is restricted to the placenta [59]. In a majority of tissues, MBNL1 and MBNL2 mRNA levels rise during differentiation [60, 61]. Besides their roles in pre-mRNA processing, MBNLs also influence gene expression by regulating cellular mRNA transport, stability as well as microRNA biogenesis [62–67]. The high expression of MBNL1 in the heart and skeletal muscle is consistent with the most severe DM phenotypes in these tissues. For instance, independent of the repeat expansion, Mbnl1 deletion in mice reproduces many of the cardinal symptoms of DM1 such as myotonia, myopathy, cataracts, and misregulation of developmentally regulated RNA processing [63, 68].
The expanded repeat-containing RNAs in DM sequester MBNL1, 2 and 3 in nuclear RNA foci [69–71], and this protein redistribution explains the inhibition of their normal functions predominantly in alternative splicing and polyadenylation, microRNA processing, and mRNA localization [58, 62, 67, 72–75]. The MBNL loss-of-function hypothesis is further supported by studies on Mbnl single- and compound-knockout mice, which recapitulate many of the DM phenotypes [68, 76–78]. The extent of symptoms, however, varies depending on the tissue context, relative concentrations of MBNL paralogues, and the degree to which they are sequestered [78]. For instance, compared to skeletal muscle, only few splicing defects are observed in the brains of Mbnl1 knockout mice [63, 79]. Alternatively, Mbnl2 knockout mice exhibit a number of DM-related central nervous system abnormalities including irregular REM sleep propensity and deficits in spatial memory [76], which is consistent with the observation that MBNL2 expression in the brain is higher than MBNL1 [59]. MBNL2 is directly sequestered by repeat expansions in the brain tissue of human DM patients resulting in misregulation of alternative splicing and polyadenylation of its normal RNA targets [80]. One of the most misspliced mRNA due to loss of MBNL2 is human microtubule-associated protein tau (MAPT) in the DM1 frontal cortex [80]. RNA toxicity mediated through MBNL2 sequestration leads to abnormal expression of tau isoforms & the progressive appearance of neurofibrillary tangles composed of intraneuronal aggregates of hyper-phosphorylated tau protein [81].
More recently, MBNL proteins were found to serve essential roles in poly (A) site selection for many transcripts. By integrating HITS-CLIP and RNA-seq from MBNL knockout cells and transgenic DM1 mouse model, along with minigene reporter studies, Swanson and colleagues demonstrated that MBNL proteins directly suppress or activate polyadenylation for thousands of pre-mRNAs [75, 80]. Thus, MBNL proteins coordinate multiple pre-mRNA processing steps and their sequestration in DM depletes them from their normal RNA targets.
Besides MBNL loss-of-function, there is accumulation and aberrant sub-cellular distribution of another splicing factor CELF1 in DM. CELF proteins are normally downregulated during postnatal striated muscle development, which facilitates fetal-to adult splicing transitions in hundreds of muscle transcripts [61, 82]. CELF1 actually does not colocalize with RNA foci [83], and its upregulation in DM1 occurs through two separate mechanisms. First, CELF1 protein is stabilized through its hyper-phosphorylation [84]; and second, reduced levels of microRNAs in DM1 derepress CELF1 protein translation [85, 86]. The situation is less clear in DM2, with conflicting reports of normal [87, 88] and increasing CELF1 protein levels [89] in patient tissues and cells. It is interesting to note that for many pre-mRNAs whose splicing is disrupted in DM1, CELF1 and MBNL1 regulate them in an antagonistic manner [58, 61, 90–92]. The antagonism, however, is not due to direct competition for the binding site as both CELF1 and MBNL1 bind and regulate splicing independently via distinct cis-acting RNA motifs.
In addition to MBNL and CELF proteins, other RNA splicing factors are implicated in DM. For instance, hnRNP H binds to DMPK-derived CUG expanded RNAs in vitro and increased hnRNP H levels may also contribute towards DM pathogenesis [93]. hnRNP H forms a repressor complex with MBNL1 and 9 other proteins (hnRNP H2, H3, F, A2/B1, K, L, DDX5, DDX17, and DHX9) in normal myoblast extracts but elevated hnRNP H levels in DM1 disrupt the stoichiometry of these complexes which affects splicing of specific pre-mRNAs [94, 95]. Since expanded CUG repeat RNAs fold into hairpin structures [96], the partial recruitment and co-localization of the RNA helicase DDX5/p68 with RNA foci may also have a contributing role in splicing deregulation. Moreover, p68/DDX5 can modulate MBNL1 binding activity, and its co-localization with nuclear RNA foci can further stimulate MBNL1 binding to repeat RNAs [97].
3b. Misregulation of mRNA localization and stability
Following transcription, newly synthesized and fully processed mRNAs are bound by specific RBPs to form export-competent mRNPs, which help their transport through the nuclear pore complex (NPC). Some pre-mRNAs are processed at the speckle periphery before being exported and repeat-containing nuclear foci can co-localize at the periphery of nuclear speckles, a non-membrane bound nuclear assembly of macromolecules including splicing factors. The presence of expanded CUG repeats may, therefore, prevent entry of other RNAs into the nuclear speckle [98, 99]. However, in DM2, the mutant ZNF9 mRNA is exported normally as the expanded CCUG repeats are removed during splicing. The nuclear foci formed by DM2 intronic repeats are widely dispersed in the nucleoplasm and not associated with nuclear speckles. Also, it is not yet clear whether the DM1 and/or DM2 nuclear foci contain partially degraded fragments of CUG or CCUG repeats or larger intact RNAs respectively.
As discussed above, CELF1 upregulation and MBNL sequestration by the CUG repeats in DM cause misprocessing of hundreds of transcripts. Aberrant processing results in nucleocytoplasmic export defects for many of these transcripts. Furthermore, MBNL proteins are localized both in the nucleus and cytoplasm and several studies have demonstrated their direct roles in mRNA localization [62, 100] (Figure 2f). For instance, by interacting with the 3′-UTR of Integrin α3, MBNL2 moves it to the plasma membrane for its local translation [64]. Similarly, MBNL1 also plays major roles in mRNA localization and membrane-associated translation. Transcriptome-wide analyses of subcellular compartments from mouse myoblasts showed widespread defects in mRNA localization upon combined depletion of MBNL1 and MBNL2 [62]. Many of the mislocalized mRNAs encode for secreted proteins, extracellular matrix components, and proteins involved in cell–cell communication. MBNL depletion in DM can thus have a significant impact on mRNA localization potentially affecting proper neuromuscular junction formation.
In the cytoplasm, MBNLs also regulate mRNA stability [101] (Figure 2i). MBNL1 specifically recognizes YGCY-containing motifs within the 3’-UTR regions and destabilizes the target mRNAs through unknown mechanisms [65, 92]. CELF1, on the other hand, induces mRNA decay of short-lived transcripts through interactions with GU-rich elements (GREs) in their 3′-UTR and possibly recruitment of poly(A) specific ribonuclease, which promotes deadenylation of target transcripts [102–104]. Many of the CELF mRNA targets with GREs encode proteins essential for muscle cell development and function [105–108]. Interestingly, CELF1 binds to the mRNAs coding for SRP protein subunits and promotes their decay [109]. Signal recognition particle (SRP) is a cytoplasmic ribonucleoprotein complex, which regulates the translation of secreted and membrane-associated proteins. It is likely that the CELF1 overexpression contributes to the faster turnover of SRP mRNAs and the reduced SRP levels thereby attenuate the protein secretory pathway in DM1 [109].
3c. Misregulation of mRNA translation
CELF1 is additionally involved in regulation of mRNA translation [106, 110–112] (Figure 2h). The affinity of CELF1 towards its mRNA targets can be modulated through its phosphorylation status [113]. For instance, phosphorylated CELF1 interacts with a subunit of initiation factor eIF2, leading to the recruitment of translational machinery to target mRNAs [106]. In myoblasts, AKT phosphorylates CELF1 and increases its affinity for CCND1 mRNA. During myoblast-to-myotube differentiation, cyclinD3-cdk4/6 phosphorylates CELF1, which increases CELF1 interaction with 5’-UTR of p21 mRNA (a cell cycle inhibitor) and enhances its translation. Myoblasts from DM1 patients show an increased interaction between CELF1 and AKT and have reduced cyclinD3-CDK4/6 levels during differentiation [105]. Moreover, DM1 myoblasts during differentiation show a reduced ability to withdraw from cell cycle, which may be due to altered translation of P21 or the myogenic transcription factor MEF2A by CELF1 [111, 112].
mRNA translation in DM1 is also affected due to microRNA deregulation (Figure 2g). A subset of developmentally regulated microRNAs associated with cardiac arrhythmias is downregulated in the hearts of DM1 patients and mice [67, 86]. Downregulation of these microRNAs recapitulate particular gene expression deficits seen in DM1 hearts including enhanced protein levels of miR-1 targets CX43 and Cav1.2 as well as miR-23a/b target CELF1 [67, 86]. In DM1 and DM2 skeletal muscle biopsies, both the levels and cellular distribution of several evolutionarily conserved microRNAs are altered affecting their downstream targets [114–117]. Furthermore, specific microRNAs are differentially detected in peripheral blood plasma of DM1 patients, which inversely correlate with skeletal muscle strength and may serve as non-invasive biomarkers [118]. More recently, reduced expression of miR-200c/141 tumor suppressor family was correlated with increased oncologic risk in women with DM1 especially for gynecologic, brain, and thyroid cancer [119].
Besides altering cellular translation through misregulation of RBPs and microRNAs, the microsatellite expansions also promote unconventional translation of repeats in multiple reading frames producing homopolymeric peptides that aggregate both in the nucleus and cytoplasm [120] (Figure 2j). Designated as Repeat Associated Non-AUG Translation (RAN translation), it was first described for the expanded CAG and CTG repeats that cause spinocerebellar ataxia 8 (SCA8) and DM1, respectively [120]. Interestingly, the efficiency of RAN translation increases with the size of repeats and when RNA forms hairpin-like structures [121]. Additionally, the cells making the toxic RAN protein products are prone to apoptosis and are detected in tissues of affected patients, indicating a role in pathogenicity. In addition to DM1, Zu et al. recently demonstrated that in DM2 the tetranucleotide expansion repeats are bidirectionally transcribed, and the resulting transcripts are RAN translated, producing tetrapeptide expansion proteins with Leu-Pro-Ala-Cys (LPAC) from the sense strand or Gln-Ala-Gly-Arg (QAGR) repeats from the antisense strand [122]. These RAN proteins were readily detected in the DM2 patient brains; however, the specific roles of these RAN proteins regarding toxicity, mechanism of action and their regulation are yet to be determined.
Since their original discovery, RAN translation has now been observed in many other repeat-expansion diseases, including ALS/FTD, FXTAS and Huntington’s disease [52, 123]. However, the exact mechanisms initiating translation from these repeats likely differ across diverse sequence contexts [124]. For instance, in case of FMR1, expanded CGG repeats in the 5’-UTR initiate CAP-dependent RAN translation upstream of the canonical AUG start codon, producing FMRpolyGlycine and FMRpolyAlanine in FXTAS [123, 125]. In contrast to FXTAS, the expanded repeats in DM1 exist within the 3′ UTR of DMPK mRNA, which is not in the normal path of ribosome scanning; thus, unconventional ribosome interactions must contribute in their translation. For HTT in Huntington’s disease, the CAG repeats are in the ORF, and canonical translation starts at the native AUG codon upstream of the repeats. But in some instances, HTTpolySerine and HTTpolyAlanine proteins are also produced due to RAN-translation and frame shifting from the normal HTTpolyGlutamine frame of the repeats [126]. Finally, in case of ALS/FTD, the GGGGCC repeats are within C9ORF72 intron, and the RAN-translation generates polyGlycine-Alanine, polyGlycine-Arginine, and polyGlycine-Proline dipeptide products [31, 127]. The RAN translation in this case, however, may occur from the intron retained transcript, spliced lariat, or a 3′ truncated RNA generated due to stalled transcription [124, 128].
4. Disrupted function of RBPs in other microsatellite expansion disorders
Recent paradigm-shifting advances have established that defective RNA processing through disrupted function of RBPs is central to many other repeat expansion diseases (Table 2). For instance, RBP defects occur in both familial and sporadic cases of ALS/FTD [129, 130]. Mutations in TDP-43 and FUS/TLS genes result in abnormal aggregation of these proteins in neurons and are considered pathogenic for ALS/FTD. TDP-43 and FUS/TLS are RNA/DNA-binding proteins, with noticeable structural and functional similarities.
Table 2:
Common postulated pathological mechanisms and associated RNA Binding Proteins (RBPs) for disease-associated microsatellite repeat expansions
| Diseases Name | RBPs | Pathological Mechanism | |
|---|---|---|---|
| a. | FXTAS, FXS | FMRP [11–14] Pur α and hnRNP A2/B1 [205, 206], CELF1 [207], Sam68 [208]. | mRNP transport and translation [209–213] Nuclear Foci and RBP Sequestration leads to changes in expression and cellular distribution of several proteins [214, 215], RNA Splicing [208, 216]. |
| b. | DM1/2 | MBNL1/2/3 [49, 68, 72–74]; and CELF [39, 83, 84, 90, 91, 111, 217], HnRNP H [93], p68/DDX5 [97] | Nuclear Foci and RBP Sequestration, RNA splicing [58, 61, 62, 82, 218, 219] and polyadenylation misregulation [75, 80], miRNA biogenesis [67, 86, 115], Translation and cellular localization disruption [62, 99], Intracellular aggregation by non-canonical RAN translation [122] |
| c. | ALS/FTD | TDP-43[220, 221] FUS[222, 223], TAF15 [141, 142], EWSR1 [143, 144] hnRNPA1 and hnRNPA2B1 [146], Ataxin 2 [145], TIA1 [224] | Nuclear foci [225], Splicing misregulation [132, 134, 226], translation, and RNA transport [227], impaired cytoplasmic localization [154, 228, 229], mutated LCD domain mediated cytoplasmic inclusions [146, 230–233] |
| d. | SCA 8 | MBNL/CELF [234], Staufen [235] | RNA Splicing [234], RAN Translation [120] |
Abbreviations: Fragile X associated tremor/ataxia syndrome (FXTAS); Fragile X Syndrome (FXS); Myotonic Dystrophy type 1/2 (DM1/2); Amyotrophic lateral sclerosis (ALS); Frontotemporal degeneration (FTD); Spinocerebellar Ataxias (SCA); fragile X mental retardation protein (FMRP), CUGBP Elav-Like Family Member (CELF); Muscleblind Like Splicing Regulator (mbnl); heterogeneous nuclear ribonucleoprotein (hnRNPs); TATA box–binding protein–associated factor 15 (TAF15), Ewing sarcoma breakpoint region 1 (EWSR1), T cell intracytoplasmic antigen (TIA1).
TDP-43 functions in multiple RNA processing steps including pre-mRNA splicing [131–134], RNA stability [135–137] and transport [138]. Similar to TDP-43, FUS interacts with serine-arginine (SR) proteins that serve diverse roles in splicing [139] and regulates transcription by recruiting other RBPs through non-coding RNAs [140]. Hence, the association of TDP43 and FUS/TLS with ALS and FTD is redirecting research efforts toward identifying additional RBPs that are mutated in neurological diseases, defining their normal RNA substrates and determining the misprocessed RNAs that underlie particular disease symptoms. In fact, mutations in several other RBPs that are functionally and structurally similar to FUS/TLS such as TAF15 [141, 142] and EWSR1 [143, 144], as well as the less closely related RBPs—ataxin 2 [145], hnRNPA2B1 [146], hnRNPA1[146], and matrin3 [147] were recently identified. Among these RBPs, TDP-43, FUS and hnRNPA1 harbor low complexity domains (LCDs), which, can polymerize and drive phase separation to form dynamic membrane-less organelles or liquid droplets. For instance, a 57-residue segment within the FUS-LCD was recently shown to assemble into a fibril core that promotes phase-separation and hydrogel formation. Interestingly, phosphorylation of the core forming residues by DNA-dependent protein kinase dissolves the FUS-LCD liquid droplets providing a molecular basis for the dynamics of LCD polymerization and phase separation [148].
Disease-associated mutations within LCDs of RBPs also enhance prion-like properties and accelerate the shift from liquid to solid phase disturbing proper ribonucleoprotein (RNP) formation [127, 149, 150]. These mutations likely trigger protein aggregation due to aberrant self-assembly of LCDs. The cytoplasmic aggregation of RBPs not only affects their typical functions in RNA metabolism but also diminishes general nucleocytoplasmic trafficking, a common consequence of ALS-initiating mutations [151–153]. While the exact reasons impeding nuclear/cytoplasmic transport in ALS are not yet fully established, multiple independent and overlapping mechanisms have been proposed. For example, nucleocytoplasmic trafficking defects can arise due to proteotoxicity caused by cytoplasmic β-sheet containing protein aggregates [154], direct interactions between repeat RNAs and nuclear import factors [153], or inhibition by RAN translation-products of repeat RNAs [151]. Curiously, arginine-containing dipeptide repeats produced from RAN translation of hexanucleotide GGGGCC expansions in ALS interact with LCDs of RBPs, which disrupts the dynamics and functions of membrane-less organelle formation by LCDs [155, 156]. Furthermore, subsets of these arginine-containing dipeptides frequently bind to the LCDs encoded by the nuclear pore proteins blocking the transport of macromolecules into and out of the nucleus [157]. Thus, interaction of RAN translation products with LCDs is a yet another pathogenic mechanism that interferes with the normal function of RBPs in microsatellite expansion disorders.
5. Conclusions
The past decade has seen remarkable progress in our understanding of the molecular pathogenesis of microsatellite repeat expansion disorders. Although the repeats may vary in terms of their length and location within the gene or the multiple ways through which they cause disease, one commonality of microsatellite expansions is the production of toxic RNA species containing repeats. Mechanistically, the pathology arises either due to loss-of-function of the affected gene, or gain-of-function of the repeat-containing RNAs. Regarding loss-of-function, the repeats can induce transcriptional silencing of the affected gene through epigenetic modifications or produce a non-functional protein that contains a long stretch of homopolymeric amino acids. In case of gain-of-function, the RNAs with expanded repeats often sequester RBPs and thus disrupt their normal activities. Alternatively, the translated protein with a repetitive stretch of homopolymeric peptide sequence can misfold, aggregate, and trap critical cellular proteins causing nucleo-cytoplasmic export defects and further proteotoxicity. For a number of repeat expansion disorders, there is an intricate overlap of such loss- and gain-of-function mechanisms resulting in complex molecular pathologies. We envision that for many repeat expansions, the future investigations will be geared towards determining the unique versus overlapping disease mechanisms, dissecting direct versus indirect RNA metabolism defects, and finally, understanding whether alterations in RNA metabolism occur early or during late stages of the disease.
Acknowledgements
A.K. is supported by grants from the US National Institute of Health (R01HL126845), Muscular Dystrophy Association (MDA514335), and the Center for Advanced Study at the University of Illinois. C.M. is supported by the American Heart Association post-doctoral fellowship (16POST29950018).
REFERENCES
- 1.Arif W, Datar G, Kalsotra A: Intersections of post-transcriptional gene regulatory mechanisms with intermediary metabolism. Biochimica et biophysica acta 2017, 1860(3):349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lewis CJ, Pan T, Kalsotra A: RNA modifications and structures cooperate to guide RNA-protein interactions. Nature reviews Molecular cell biology 2017, 18(3):202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scotti MM, Swanson MS: RNA mis-splicing in disease. Nature reviews Genetics 2016, 17(1):19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brinegar AE, Cooper TA: Roles for RNA-binding proteins in development and disease. Brain research 2016, 1647:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mirkin SM: Expandable DNA repeats and human disease. Nature 2007, 447(7147):932–940. [DOI] [PubMed] [Google Scholar]
- 6.La Spada AR, Taylor JP: Repeat expansion disease: progress and puzzles in disease pathogenesis. Nature reviews Genetics 2010, 11(4):247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Renoux AJ, Todd PK: Neurodegeneration the RNA way. Progress in neurobiology 2012, 97(2):173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Rourke JR, Swanson MS: Mechanisms of RNA-mediated disease. The Journal of biological chemistry 2009, 284(12):7419–7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang N, Ashizawa T: RNA toxicity and foci formation in microsatellite expansion diseases. Current opinion in genetics & development 2017, 44:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain A, Vale RD: RNA phase transitions in repeat expansion disorders. Nature 2017, 546(7657):243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heitz D, Rousseau F, Devys D, Saccone S, Abderrahim H, Le Paslier D, Cohen D, Vincent A, Toniolo D, Della Valle G et al. : Isolation of sequences that span the fragile X and identification of a fragile X-related CpG island. Science 1991, 251(4998):1236–1239. [DOI] [PubMed] [Google Scholar]
- 12.Kremer EJ, Pritchard M, Lynch M, Yu S, Holman K, Baker E, Warren ST, Schlessinger D, Sutherland GR, Richards RI: Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 1991, 252(5013):1711–1714. [DOI] [PubMed] [Google Scholar]
- 13.Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas MF, Mandel JL: Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 1991, 252(5009):1097–1102. [DOI] [PubMed] [Google Scholar]
- 14.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP et al. : Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65(5):905–914. [DOI] [PubMed] [Google Scholar]
- 15.Bhakar AL, Dolen G, Bear MF: The pathophysiology of fragile X (and what it teaches us about synapses). Annual review of neuroscience 2012, 35:417–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santoro MR, Bray SM, Warren ST: Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annual review of pathology 2012, 7:219–245. [DOI] [PubMed] [Google Scholar]
- 17.Ashley CT Jr., Wilkinson KD, Reines D, Warren ST: FMR1 protein: conserved RNP family domains and selective RNA binding. Science 1993, 262(5133):563–566. [DOI] [PubMed] [Google Scholar]
- 18.Brown V, Small K, Lakkis L, Feng Y, Gunter C, Wilkinson KD, Warren ST: Purified recombinant Fmrp exhibits selective RNA binding as an intrinsic property of the fragile X mental retardation protein. The Journal of biological chemistry 1998, 273(25):15521–15527. [DOI] [PubMed] [Google Scholar]
- 19.Siomi H, Choi M, Siomi MC, Nussbaum RL, Dreyfuss G: Essential role for KH domains in RNA binding: impaired RNA binding by a mutation in the KH domain of FMR1 that causes fragile X syndrome. Cell 1994, 77(1):33–39. [DOI] [PubMed] [Google Scholar]
- 20.Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G: The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 1993, 74(2):291–298. [DOI] [PubMed] [Google Scholar]
- 21.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH: Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352(6330):77–79. [DOI] [PubMed] [Google Scholar]
- 22.Orr HT, Zoghbi HY: Trinucleotide repeat disorders. Annual review of neuroscience 2007, 30:575–621. [DOI] [PubMed] [Google Scholar]
- 23.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP: Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90(3):537–548. [DOI] [PubMed] [Google Scholar]
- 24.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N: Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277(5334):1990–1993. [DOI] [PubMed] [Google Scholar]
- 25.Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN: Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 1997, 19(2):333–344. [DOI] [PubMed] [Google Scholar]
- 26.Li LB, Bonini NM: Roles of trinucleotide-repeat RNA in neurological disease and degeneration. Trends in neurosciences 2010, 33(6):292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gatchel JR, Zoghbi HY: Diseases of unstable repeat expansion: mechanisms and common principles. Nature reviews Genetics 2005, 6(10):743–755. [DOI] [PubMed] [Google Scholar]
- 28.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J et al. : Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72(2):245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L et al. : A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72(2):257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW 3rd, Rademakers R et al. : Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013, 77(4):639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C et al. : The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339(6125):1335–1338. [DOI] [PubMed] [Google Scholar]
- 32.Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS et al. : Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America 2013, 110(19):7778–7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH et al. : RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proceedings of the National Academy of Sciences of the United States of America 2013, 110(51):E4968–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faustino NA, Cooper TA: Pre-mRNA splicing and human disease. Genes & development 2003, 17(4):419–437. [DOI] [PubMed] [Google Scholar]
- 35.Wheeler TM: Myotonic dystrophy: therapeutic strategies for the future. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics 2008, 5(4):592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harper P: Myotonic Dystrophy London: W.B. Saunders; 2001. [Google Scholar]
- 37.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T et al. : Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell 1992, 68(4):799–808. [DOI] [PubMed] [Google Scholar]
- 38.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O’Hoy K et al. : Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene. Science 1992, 255(5049):1253–1255. [DOI] [PubMed] [Google Scholar]
- 39.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP: Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293(5531):864–867. [DOI] [PubMed] [Google Scholar]
- 40.Yum K, Wang ET, Kalsotra A: Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Current opinion in genetics & development 2017, 44:30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Groh WJ, Groh MR, Saha C, Kincaid JC, Simmons Z, Ciafaloni E, Pourmand R, Otten RF, Bhakta D, Nair GV et al. : Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. The New England journal of medicine 2008, 358(25):2688–2697. [DOI] [PubMed] [Google Scholar]
- 42.Heatwole C, Bode R, Johnson N, Quinn C, Martens W, McDermott MP, Rothrock N, Thornton C, Vickrey B, Victorson D et al. : Patient-reported impact of symptoms in myotonic dystrophy type 1 (PRISM-1). Neurology 2012, 79(4):348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phillips MF, Harper PS: Cardiac disease in myotonic dystrophy. Cardiovasc Res 1997, 33(1):13–22. [DOI] [PubMed] [Google Scholar]
- 44.Salehi LB, Bonifazi E, Stasio ED, Gennarelli M, Botta A, Vallo L, Iraci R, Massa R, Antonini G, Angelini C et al. : Risk prediction for clinical phenotype in myotonic dystrophy type 1: data from 2,650 patients. Genet Test 2007, 11(1):84–90. [DOI] [PubMed] [Google Scholar]
- 45.Jansen G, Groenen PJ, Bachner D, Jap PH, Coerwinkel M, Oerlemans F, van den Broek W, Gohlsch B, Pette D, Plomp JJ et al. : Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nature genetics 1996, 13(3):316–324. [DOI] [PubMed] [Google Scholar]
- 46.Berul CI, Maguire CT, Aronovitz MJ, Greenwood J, Miller C, Gehrmann J, Housman D, Mendelsohn ME, Reddy S: DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. The Journal of clinical investigation 1999, 103(4):R1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thornton CA, Wymer JP, Simmons Z, McClain C, Moxley RT, 3rd: Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nature genetics 1997, 16(4):407–409. [DOI] [PubMed] [Google Scholar]
- 48.Klesert TR, Cho DH, Clark JI, Maylie J, Adelman J, Snider L, Yuen EC, Soriano P, Tapscott SJ: Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nature genetics 2000, 25(1):105–109. [DOI] [PubMed] [Google Scholar]
- 49.Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA: Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289(5485):1769–1773. [DOI] [PubMed] [Google Scholar]
- 50.Gomes-Pereira M, Cooper TA, Gourdon G: Myotonic dystrophy mouse models: towards rational therapy development. Trends in molecular medicine 2011, 17(9):506–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Echeverria GV, Cooper TA: RNA-binding proteins in microsatellite expansion disorders: mediators of RNA toxicity. Brain research 2012, 1462:100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cleary JD, Ranum LP: Repeat associated non-ATG (RAN) translation: new starts in microsatellite expansion disorders. Current opinion in genetics & development 2014, 26:6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mohan A, Goodwin M, Swanson MS: RNA-protein interactions in unstable microsatellite diseases. Brain research 2014, 1584:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chau A, Kalsotra A: Developmental insights into the pathology of and therapeutic strategies for DM1: Back to the basics. Developmental dynamics : an official publication of the American Association of Anatomists 2015, 244(3):377–390. [DOI] [PubMed] [Google Scholar]
- 55.Nilsen TW, Graveley BR: Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463(7280):457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kalsotra A, Cooper TA: Functional consequences of developmentally regulated alternative splicing. Nature reviews Genetics 2011, 12(10):715–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Begemann G, Paricio N, Artero R, Kiss I, Perez-Alonso M, Mlodzik M: muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins. Development 1997, 124(21):4321–4331. [DOI] [PubMed] [Google Scholar]
- 58.Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA: Muscleblind proteins regulate alternative splicing. The EMBO journal 2004, 23(15):3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kanadia RN, Urbinati CR, Crusselle VJ, Luo D, Lee YJ, Harrison JK, Oh SP, Swanson MS: Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene expression patterns : GEP 2003, 3(4):459–462. [DOI] [PubMed] [Google Scholar]
- 60.Konieczny P, Stepniak-Konieczna E, Sobczak K: MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic acids research 2014, 42(17):10873–10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA: A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proceedings of the National Academy of Sciences of the United States of America 2008, 105(51):20333–20338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S et al. : Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 2012, 150(4):710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, Hall MP, Shiue L, Swanson MS, Thornton CA et al. : Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nature structural & molecular biology 2010, 17(2):187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adereth Y, Dammai V, Kose N, Li R, Hsu T: RNA-dependent integrin alpha3 protein localization regulated by the Muscleblind-like protein MLP1. Nature cell biology 2005, 7(12):1240–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, Ohno K: CUGBP1 and MBNL1 preferentially bind to 3’ UTRs and facilitate mRNA decay. Scientific reports 2012, 2:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Osborne RJ, Lin X, Welle S, Sobczak K, O’Rourke JR, Swanson MS, Thornton CA: Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Human molecular genetics 2009, 18(8):1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D et al. : Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nature structural & molecular biology 2011, 18(7):840–845. [DOI] [PubMed] [Google Scholar]
- 68.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS: A muscleblind knockout model for myotonic dystrophy. Science 2003, 302(5652):1978–1980. [DOI] [PubMed] [Google Scholar]
- 69.Kino Y, Mori D, Oma Y, Takeshita Y, Sasagawa N, Ishiura S: Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Human molecular genetics 2004, 13(5):495–507. [DOI] [PubMed] [Google Scholar]
- 70.Warf MB, Berglund JA: MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. Rna 2007, 13(12):2238–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yuan Y, Compton SA, Sobczak K, Stenberg MG, Thornton CA, Griffith JD, Swanson MS: Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic acids research 2007, 35(16):5474–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, Brook JD: Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Human molecular genetics 2002, 11(7):805–814. [DOI] [PubMed] [Google Scholar]
- 73.Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA: Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Human molecular genetics 2004, 13(24):3079–3088. [DOI] [PubMed] [Google Scholar]
- 74.Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS: Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. The EMBO journal 2000, 19(17):4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, Goodwin M, Zhang C, Sobczak K, Thornton CA et al. : Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Molecular cell 2014, 56(2):311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G et al. : Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 2012, 75(3):437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Poulos MG, Batra R, Li M, Yuan Y, Zhang C, Darnell RB, Swanson MS: Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice. Human molecular genetics 2013, 22(17):3547–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee KY, Li M, Manchanda M, Batra R, Charizanis K, Mohan A, Warren SA, Chamberlain CM, Finn D, Hong H et al. : Compound loss of muscleblind-like function in myotonic dystrophy. EMBO molecular medicine 2013, 5(12):1887–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Suenaga K, Lee KY, Nakamori M, Tatsumi Y, Takahashi MP, Fujimura H, Jinnai K, Yoshikawa H, Du H, Ares M Jr. et al. : Muscleblind-like 1 knockout mice reveal novel splicing defects in the myotonic dystrophy brain. PloS one 2012, 7(3):e33218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goodwin M, Mohan A, Batra R, Lee KY, Charizanis K, Fernandez Gomez FJ, Eddarkaoui S, Sergeant N, Buee L, Kimura T et al. : MBNL Sequestration by Toxic RNAs and RNA Misprocessing in the Myotonic Dystrophy Brain. Cell reports 2015, 12(7):1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sergeant N, Sablonniere B, Schraen-Maschke S, Ghestem A, Maurage CA, Wattez A, Vermersch P, Delacourte A: Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Human molecular genetics 2001, 10(19):2143–2155. [DOI] [PubMed] [Google Scholar]
- 82.Ladd AN, Charlet N, Cooper TA: The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Molecular and cellular biology 2001, 21(4):1285–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fardaei M, Larkin K, Brook JD, Hamshere MG: In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts. Nucleic acids research 2001, 29(13):2766–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kuyumcu-Martinez NM, Wang GS, Cooper TA: Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Molecular cell 2007, 28(1):68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kalsotra A, Wang K, Li PF, Cooper TA: MicroRNAs coordinate an alternative splicing network during mouse postnatal heart development. Genes & development 2010, 24(7):653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kalsotra A, Singh RK, Gurha P, Ward AJ, Creighton CJ, Cooper TA: The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell reports 2014, 6(2):336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA: Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Human molecular genetics 2006, 15(13):2087–2097. [DOI] [PubMed] [Google Scholar]
- 88.Pelletier R, Hamel F, Beaulieu D, Patry L, Haineault C, Tarnopolsky M, Schoser B, Puymirat J: Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiology of disease 2009, 36(1):181–190. [DOI] [PubMed] [Google Scholar]
- 89.Salisbury E, Schoser B, Schneider-Gold C, Wang GL, Huichalaf C, Jin B, Sirito M, Sarkar P, Krahe R, Timchenko NA et al. : Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. The American journal of pathology 2009, 175(2):748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Savkur RS, Philips AV, Cooper TA: Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nature genetics 2001, 29(1):40–47. [DOI] [PubMed] [Google Scholar]
- 91.Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA: Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Molecular cell 2002, 10(1):45–53. [DOI] [PubMed] [Google Scholar]
- 92.Wang ET, Ward AJ, Cherone JM, Giudice J, Wang TT, Treacy DJ, Lambert NJ, Freese P, Saxena T, Cooper TA et al. : Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome research 2015, 25(6):858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim DH, Langlois MA, Lee KB, Riggs AD, Puymirat J, Rossi JJ: HnRNP H inhibits nuclear export of mRNA containing expanded CUG repeats and a distal branch point sequence. Nucleic acids research 2005, 33(12):3866–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Paul S, Dansithong W, Kim D, Rossi J, Webster NJ, Comai L, Reddy S: Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. The EMBO journal 2006, 25(18):4271–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Paul S, Dansithong W, Jog SP, Holt I, Mittal S, Brook JD, Morris GE, Comai L, Reddy S: Expanded CUG repeats Dysregulate RNA splicing by altering the stoichiometry of the muscleblind 1 complex. The Journal of biological chemistry 2011, 286(44):38427–38438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Krzyzosiak WJ, Sobczak K, Wojciechowska M, Fiszer A, Mykowska A, Kozlowski P: Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic acids research 2012, 40(1):11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Laurent FX, Sureau A, Klein AF, Trouslard F, Gasnier E, Furling D, Marie J: New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats. Nucleic acids research 2012, 40(7):3159–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Holt I, Mittal S, Furling D, Butler-Browne GS, Brook JD, Morris GE: Defective mRNA in myotonic dystrophy accumulates at the periphery of nuclear splicing speckles. Genes to cells : devoted to molecular & cellular mechanisms 2007, 12(9):1035–1048. [DOI] [PubMed] [Google Scholar]
- 99.Smith KP, Byron M, Johnson C, Xing Y, Lawrence JB: Defining early steps in mRNA transport: mutant mRNA in myotonic dystrophy type I is blocked at entry into SC-35 domains. The Journal of cell biology 2007, 178(6):951–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Taliaferro JM, Vidaki M, Oliveira R, Olson S, Zhan L, Saxena T, Wang ET, Graveley BR, Gertler FB, Swanson MS et al. : Distal Alternative Last Exons Localize mRNAs to Neural Projections. Molecular cell 2016, 61(6):821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang ET, Taliaferro JM, Lee JA, Sudhakaran IP, Rossoll W, Gross C, Moss KR, Bassell GJ: Dysregulation of mRNA Localization and Translation in Genetic Disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 2016, 36(45):11418–11426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vlasova IA, Tahoe NM, Fan D, Larsson O, Rattenbacher B, Sternjohn JR, Vasdewani J, Karypis G, Reilly CS, Bitterman PB et al. : Conserved GU-rich elements mediate mRNA decay by binding to CUG-binding protein 1. Molecular cell 2008, 29(2):263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee JE, Lee JY, Wilusz J, Tian B, Wilusz CJ: Systematic analysis of cis-elements in unstable mRNAs demonstrates that CUGBP1 is a key regulator of mRNA decay in muscle cells. PloS one 2010, 5(6):e11201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Moraes KC, Wilusz CJ, Wilusz J: CUG-BP binds to RNA substrates and recruits PARN deadenylase. Rna 2006, 12(6):1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Timchenko L: Molecular mechanisms of muscle atrophy in myotonic dystrophies. The international journal of biochemistry & cell biology 2013, 45(10):2280–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dasgupta T, Ladd AN: The importance of CELF control: molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins. Wiley interdisciplinary reviews RNA 2012, 3(1):104–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rattenbacher B, Beisang D, Wiesner DL, Jeschke JC, von Hohenberg M, St Louis-Vlasova IA, Bohjanen PR: Analysis of CUGBP1 targets identifies GU-repeat sequences that mediate rapid mRNA decay. Molecular and cellular biology 2010, 30(16):3970–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang L, Lee JE, Wilusz J, Wilusz CJ: The RNA-binding protein CUGBP1 regulates stability of tumor necrosis factor mRNA in muscle cells: implications for myotonic dystrophy. The Journal of biological chemistry 2008, 283(33):22457–22463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Russo J, Lee JE, Lopez CM, Anderson J, Nguyen TP, Heck AM, Wilusz J, Wilusz CJ: The CELF1 RNA-Binding Protein Regulates Decay of Signal Recognition Particle mRNAs and Limits Secretion in Mouse Myoblasts. PloS one 2017, 12(1):e0170680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vlasova-St Louis I, Dickson AM, Bohjanen PR, Wilusz CJ: CELFish ways to modulate mRNA decay. Biochimica et biophysica acta 2013, 1829(6–7):695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Timchenko NA, Iakova P, Cai ZJ, Smith JR, Timchenko LT: Molecular basis for impaired muscle differentiation in myotonic dystrophy. Molecular and cellular biology 2001, 21(20):6927–6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Timchenko NA, Patel R, Iakova P, Cai ZJ, Quan L, Timchenko LT: Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. The Journal of biological chemistry 2004, 279(13):13129–13139. [DOI] [PubMed] [Google Scholar]
- 113.Salisbury E, Sakai K, Schoser B, Huichalaf C, Schneider-Gold C, Nguyen H, Wang GL, Albrecht JH, Timchenko LT: Ectopic expression of cyclin D3 corrects differentiation of DM1 myoblasts through activation of RNA CUG-binding protein, CUGBP1. Experimental cell research 2008, 314(11–12):2266–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gambardella S, Rinaldi F, Lepore SM, Viola A, Loro E, Angelini C, Vergani L, Novelli G, Botta A: Overexpression of microRNA-206 in the skeletal muscle from myotonic dystrophy type 1 patients. Journal of translational medicine 2010, 8:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Perbellini R, Greco S, Sarra-Ferraris G, Cardani R, Capogrossi MC, Meola G, Martelli F: Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscular disorders : NMD 2011, 21(2):81–88. [DOI] [PubMed] [Google Scholar]
- 116.Fernandez-Costa JM, Garcia-Lopez A, Zuniga S, Fernandez-Pedrosa V, Felipo-Benavent A, Mata M, Jaka O, Aiastui A, Hernandez-Torres F, Aguado B et al. : Expanded CTG repeats trigger miRNA alterations in Drosophila that are conserved in myotonic dystrophy type 1 patients. Human molecular genetics 2013, 22(4):704–716. [DOI] [PubMed] [Google Scholar]
- 117.Greco S, Perfetti A, Fasanaro P, Cardani R, Capogrossi MC, Meola G, Martelli F: Deregulated microRNAs in myotonic dystrophy type 2. PloS one 2012, 7(6):e39732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Perfetti A, Greco S, Bugiardini E, Cardani R, Gaia P, Gaetano C, Meola G, Martelli F: Plasma microRNAs as biomarkers for myotonic dystrophy type 1. Neuromuscular disorders : NMD 2014, 24(6):509–515. [DOI] [PubMed] [Google Scholar]
- 119.Fernandez-Torron R, Garcia-Puga M, Emparanza JI, Maneiro M, Cobo AM, Poza JJ, Espinal JB, Zulaica M, Ruiz I, Martorell L et al. : Cancer risk in DM1 is sex-related and linked to miRNA-200/141 downregulation. Neurology 2016, 87(12):1250–1257. [DOI] [PubMed] [Google Scholar]
- 120.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA et al. : Non-ATG-initiated translation directed by microsatellite expansions. Proceedings of the National Academy of Sciences of the United States of America 2011, 108(1):260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cleary JD, Ranum LP: Repeat-associated non-ATG (RAN) translation in neurological disease. Human molecular genetics 2013, 22(R1):R45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zu T, Cleary JD, Liu Y, Banez-Coronel M, Bubenik JL, Ayhan F, Ashizawa T, Xia G, Clark HB, Yachnis AT et al. : RAN Translation Regulated by Muscleblind Proteins in Myotonic Dystrophy Type 2. Neuron 2017, 95(6):1292–1305 e1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kearse MG, Todd PK: Repeat-associated non-AUG translation and its impact in neurodegenerative disease. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics 2014, 11(4):721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Green KM, Linsalata AE, Todd PK: RAN translation-What makes it run? Brain research 2016, 1647:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V et al. : CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 2013, 78(3):440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Banez-Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK, Pletnikova O, Borchelt DR, Ross CA, Margolis RL et al. : RAN Translation in Huntington Disease. Neuron 2015, 88(4):667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Harrison AF, Shorter J: RNA-binding proteins with prion-like domains in health and disease. The Biochemical journal 2017, 474(8):1417–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tran H, Almeida S, Moore J, Gendron TF, Chalasani U, Lu Y, Du X, Nickerson JA, Petrucelli L, Weng Z et al. : Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron 2015, 87(6):1207–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu EY, Cali CP, Lee EB: RNA metabolism in neurodegenerative disease. Disease models & mechanisms 2017, 10(5):509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ling SC, Polymenidou M, Cleveland DW: Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 2013, 79(3):416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE: Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. The EMBO journal 2001, 20(7):1774–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ling JP, Pletnikova O, Troncoso JC, Wong PC: TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 2015, 349(6248):650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shiga A, Ishihara T, Miyashita A, Kuwabara M, Kato T, Watanabe N, Yamahira A, Kondo C, Yokoseki A, Takahashi M et al. : Alteration of POLDIP3 splicing associated with loss of function of TDP-43 in tissues affected with ALS. PloS one 2012, 7(8):e43120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V et al. : Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nature neuroscience 2011, 14(4):452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Costessi L, Porro F, Iaconcig A, Muro AF: TDP-43 regulates beta-adducin (Add2) transcript stability. RNA biology 2014, 11(10):1280–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liu X, Li D, Zhang W, Guo M, Zhan Q: Long non-coding RNA gadd7 interacts with TDP-43 and regulates Cdk6 mRNA decay. The EMBO journal 2012, 31(23):4415–4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra-Lantz C, Shoesmith C: TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Molecular and cellular neurosciences 2007, 35(2):320–327. [DOI] [PubMed] [Google Scholar]
- 138.Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A et al. : Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014, 81(3):536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yang L, Embree LJ, Tsai S, Hickstein DD: Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. The Journal of biological chemistry 1998, 273(43):27761–27764. [DOI] [PubMed] [Google Scholar]
- 140.Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R: Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454(7200):126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, Erion R, Oristano R, Liu AX, Ramos D, Jethava N, Hosangadi D et al. : A yeast functional screen predicts new candidate ALS disease genes. Proceedings of the National Academy of Sciences of the United States of America 2011, 108(52):20881–20890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ticozzi N, Vance C, Leclerc AL, Keagle P, Glass JD, McKenna-Yasek D, Sapp PC, Silani V, Bosco DA, Shaw CE et al. : Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 2011, 156B(3):285–290. [DOI] [PubMed] [Google Scholar]
- 143.Neumann M, Bentmann E, Dormann D, Jawaid A, DeJesus-Hernandez M, Ansorge O, Roeber S, Kretzschmar HA, Munoz DG, Kusaka H et al. : FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain : a journal of neurology 2011, 134(Pt 9):2595–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, Mojsilovic-Petrovic J, Panossian S et al. : Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Human molecular genetics 2012, 21(13):2899–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM et al. : Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466(7310):1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A et al. : Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495(7442):467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, Pliner HA, Abramzon Y, Marangi G, Winborn BJ et al. : Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nature neuroscience 2014, 17(5):664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Murray DT, Kato M, Lin Y, Thurber KR, Hung I, McKnight SL, Tycko R: Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell 2017, 171(3):615–627 e616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FT, Michel CH et al. : ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88(4):678–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Taylor JP, Brown RH Jr., Cleveland DW: Decoding ALS: from genes to mechanism. Nature 2016, 539(7628):197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC et al. : GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525(7567):129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW 3rd, Sun S, Herdy JR, Bieri G et al. : Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nature neuroscience 2015, 18(9):1226–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S et al. : The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525(7567):56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU et al. : Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 2016, 351(6269):173–176. [DOI] [PubMed] [Google Scholar]
- 155.Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A et al. : C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167(3):774–788 e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lin Y, Mori E, Kato M, Xiang S, Wu L, Kwon I, McKnight SL: Toxic PR Poly-Dipeptides Encoded by the C9orf72 Repeat Expansion Target LC Domain Polymers. Cell 2016, 167(3):789–802 e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Shi KY, Mori E, Nizami ZF, Lin Y, Kato M, Xiang S, Wu LC, Ding M, Yu Y, Gall JG et al. : Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proceedings of the National Academy of Sciences of the United States of America 2017, 114(7):E1111–E1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hagerman RJ, Hull CE, Safanda JF, Carpenter I, Staley LW, O’Connor RA, Seydel C, Mazzocco MM, Snow K, Thibodeau SN et al. : High functioning fragile X males: demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. American journal of medical genetics 1994, 51(4):298–308. [DOI] [PubMed] [Google Scholar]
- 159.Coffey SM, Cook K, Tartaglia N, Tassone F, Nguyen DV, Pan R, Bronsky HE, Yuhas J, Borodyanskaya M, Grigsby J et al. : Expanded clinical phenotype of women with the FMR1 premutation. American journal of medical genetics Part A 2008, 146A(8):1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, Brunberg JA, Greco C, Des Portes V, Jardini T, Levine R et al. : Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. American journal of human genetics 2003, 72(4):869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, Jacquemont S, Leehey M, Hagerman PJ: Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain : a journal of neurology 2002, 125(Pt 8):1760–1771. [DOI] [PubMed] [Google Scholar]
- 162.Leehey MA: Fragile X-associated tremor/ataxia syndrome: clinical phenotype, diagnosis, and treatment. Journal of investigative medicine : the official publication of the American Federation for Clinical Research 2009, 57(8):830–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Penagarikano O, Mulle JG, Warren ST: The pathophysiology of fragile x syndrome. Annual review of genomics and human genetics 2007, 8:109–129. [DOI] [PubMed] [Google Scholar]
- 164.Sreeram N, Wren C, Bhate M, Robertson P, Hunter S: Cardiac abnormalities in the fragile X syndrome. British heart journal 1989, 61(3):289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lozano R, Azarang A, Wilaisakditipakorn T, Hagerman RJ: Fragile X syndrome: A review of clinical management. Intractable & rare diseases research 2016, 5(3):145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Holmes SE, O’Hearn EE, McInnis MG, Gorelick-Feldman DA, Kleiderlein JJ, Callahan C, Kwak NG, Ingersoll-Ashworth RG, Sherr M, Sumner AJ et al. : Expansion of a novel CAG trinucleotide repeat in the 5’ region of PPP2R2B is associated with SCA12. Nature genetics 1999, 23(4):391–392. [DOI] [PubMed] [Google Scholar]
- 167.O’Hearn E, Holmes SE, Calvert PC, Ross CA, Margolis RL: SCA-12: Tremor with cerebellar and cortical atrophy is associated with a CAG repeat expansion. Neurology 2001, 56(3):299–303. [DOI] [PubMed] [Google Scholar]
- 168.Ranum LP, Rasmussen PF, Benzow KA, Koob MD, Day JW: Genetic mapping of a second myotonic dystrophy locus. Nature genetics 1998, 19(2):196–198. [DOI] [PubMed] [Google Scholar]
- 169.Peric S, Rakocevic Stojanovic V, Mandic Stojmenovic G, Ilic V, Kovacevic M, Parojcic A, Pesovic J, Mijajlovic M, Savic-Pavicevic D, Meola G: Clusters of cognitive impairment among different phenotypes of myotonic dystrophy type 1 and type 2. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology 2017, 38(3):415–423. [DOI] [PubMed] [Google Scholar]
- 170.Tieleman AA, Knoop H, van de Logt AE, Bleijenberg G, van Engelen BG, Overeem S: Poor sleep quality and fatigue but no excessive daytime sleepiness in myotonic dystrophy type 2. Journal of neurology, neurosurgery, and psychiatry 2010, 81(9):963–967. [DOI] [PubMed] [Google Scholar]
- 171.Hund E, Jansen O, Koch MC, Ricker K, Fogel W, Niedermaier N, Otto M, Kuhn E, Meinck HM: Proximal myotonic myopathy with MRI white matter abnormalities of the brain. Neurology 1997, 48(1):33–37. [DOI] [PubMed] [Google Scholar]
- 172.Schneider-Gold C, Bellenberg B, Prehn C, Krogias C, Schneider R, Klein J, Gold R, Lukas C: Cortical and Subcortical Grey and White Matter Atrophy in Myotonic Dystrophies Type 1 and 2 Is Associated with Cognitive Impairment, Depression and Daytime Sleepiness. PloS one 2015, 10(6):e0130352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Meola G, Sansone V, Perani D, Scarone S, Cappa S, Dragoni C, Cattaneo E, Cotelli M, Gobbo C, Fazio F et al. : Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM-1) and in proximal myotonic myopathy (PROMM/DM-2). Neuromuscular disorders : NMD 2003, 13(10):813–821. [DOI] [PubMed] [Google Scholar]
- 174.Meola G, Cardani R: Myotonic Dystrophy Type 2: An Update on Clinical Aspects, Genetic and Pathomolecular Mechanism. Journal of neuromuscular diseases 2015, 2(s2):S59–S71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Giordana MT, Ferrero P, Grifoni S, Pellerino A, Naldi A, Montuschi A: Dementia and cognitive impairment in amyotrophic lateral sclerosis: a review. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology 2011, 32(1):9–16. [DOI] [PubMed] [Google Scholar]
- 176.Graff-Radford NR, Woodruff BK: Frontotemporal dementia. Seminars in neurology 2007, 27(1):48–57. [DOI] [PubMed] [Google Scholar]
- 177.Kirshner HS: Frontotemporal dementia and primary progressive aphasia, a review. Neuropsychiatric disease and treatment 2014, 10:1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Thomas PS Jr., Fraley GS, Damian V, Woodke LB, Zapata F, Sopher BL, Plymate SR, La Spada AR: Loss of endogenous androgen receptor protein accelerates motor neuron degeneration and accentuates androgen insensitivity in a mouse model of X-linked spinal and bulbar muscular atrophy. Human molecular genetics 2006, 15(14):2225–2238. [DOI] [PubMed] [Google Scholar]
- 179.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72(6):971–983. [DOI] [PubMed] [Google Scholar]
- 180.Louis ED, Lee P, Quinn L, Marder K: Dystonia in Huntington’s disease: prevalence and clinical characteristics. Movement disorders : official journal of the Movement Disorder Society 1999, 14(1):95–101. [DOI] [PubMed] [Google Scholar]
- 181.Kim SD, Fung VS: An update on Huntington’s disease: from the gene to the clinic. Current opinion in neurology 2014, 27(4):477–483. [DOI] [PubMed] [Google Scholar]
- 182.Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, Takahashi H, Kondo R, Ishikawa A, Hayashi T et al. : Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nature genetics 1994, 6(1):9–13. [DOI] [PubMed] [Google Scholar]
- 183.Nagafuchi S, Yanagisawa H, Sato K, Shirayama T, Ohsaki E, Bundo M, Takeda T, Tadokoro K, Kondo I, Murayama N et al. : Dentatorubral and pallidoluysian atrophy expansion of an unstable CAG trinucleotide on chromosome 12p. Nature genetics 1994, 6(1):14–18. [DOI] [PubMed] [Google Scholar]
- 184.Ikeuchi T, Koide R, Tanaka H, Onodera O, Igarashi S, Takahashi H, Kondo R, Ishikawa A, Tomoda A, Miike T et al. : Dentatorubral-pallidoluysian atrophy: clinical features are closely related to unstable expansions of trinucleotide (CAG) repeat. Annals of neurology 1995, 37(6):769–775. [DOI] [PubMed] [Google Scholar]
- 185.Nucifora FC Jr., Ellerby LM, Wellington CL, Wood JD, Herring WJ, Sawa A, Hayden MR, Dawson VL, Dawson TM, Ross CA: Nuclear localization of a non-caspase truncation product of atrophin-1, with an expanded polyglutamine repeat, increases cellular toxicity. The Journal of biological chemistry 2003, 278(15):13047–13055. [DOI] [PubMed] [Google Scholar]
- 186.Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT: Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 1998, 95(1):41–53. [DOI] [PubMed] [Google Scholar]
- 187.Skinner PJ, Vierra-Green CA, Emamian E, Zoghbi HY, Orr HT: Amino acids in a region of ataxin-1 outside of the polyglutamine tract influence the course of disease in SCA1 transgenic mice. Neuromolecular medicine 2002, 1(1):33–42. [DOI] [PubMed] [Google Scholar]
- 188.Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, Wakisaka A, Tashiro K, Ishida Y, Ikeuchi T et al. : Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nature genetics 1996, 14(3):277–284. [DOI] [PubMed] [Google Scholar]
- 189.Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I, Pearlman S, Starkman S, Orozco-Diaz G, Lunkes A et al. : Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nature genetics 1996, 14(3):269–276. [DOI] [PubMed] [Google Scholar]
- 190.Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier JM, Weber C, Mandel JL, Cancel G, Abbas N et al. : Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nature genetics 1996, 14(3):285–291. [DOI] [PubMed] [Google Scholar]
- 191.Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Akiguchi I et al. : CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nature genetics 1994, 8(3):221–228. [DOI] [PubMed] [Google Scholar]
- 192.Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC: Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nature genetics 1997, 15(1):62–69. [DOI] [PubMed] [Google Scholar]
- 193.David G, Abbas N, Stevanin G, Durr A, Yvert G, Cancel G, Weber C, Imbert G, Saudou F, Antoniou E et al. : Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nature genetics 1997, 17(1):65–70. [DOI] [PubMed] [Google Scholar]
- 194.Koide R, Kobayashi S, Shimohata T, Ikeuchi T, Maruyama M, Saito M, Yamada M, Takahashi H, Tsuji S: A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Human molecular genetics 1999, 8(11):2047–2053. [DOI] [PubMed] [Google Scholar]
- 195.Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM, Lafreniere RG, Rommens JM, Uyama E, Nohira O et al. : Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nature genetics 1998, 18(2):164–167. [DOI] [PubMed] [Google Scholar]
- 196.Anvar SY, Raz Y, Verway N, van der Sluijs B, Venema A, Goeman JJ, Vissing J, van der Maarel SM, t Hoen PA, van Engelen BG et al. : A decline in PABPN1 induces progressive muscle weakness in oculopharyngeal muscle dystrophy and in muscle aging. Aging 2013, 5(6):412–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Stromme P, Mangelsdorf ME, Shaw MA, Lower KM, Lewis SM, Bruyere H, Lutcherath V, Gedeon AK, Wallace RH, Scheffer IE et al. : Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nature genetics 2002, 30(4):441–445. [DOI] [PubMed] [Google Scholar]
- 198.Ropers HH, Hamel BC: X-linked mental retardation. Nature reviews Genetics 2005, 6(1):46–57. [DOI] [PubMed] [Google Scholar]
- 199.Nawara M, Szczaluba K, Poirier K, Chrzanowska K, Pilch J, Bal J, Chelly J, Mazurczak T: The ARX mutations: a frequent cause of X-linked mental retardation. American journal of medical genetics Part A 2006, 140(7):727–732. [DOI] [PubMed] [Google Scholar]
- 200.Messaed C, Rouleau GA: Molecular mechanisms underlying polyalanine diseases. Neurobiology of disease 2009, 34(3):397–405. [DOI] [PubMed] [Google Scholar]
- 201.Winblad S, Lindberg C, Hansen S: Cognitive deficits and CTG repeat expansion size in classical myotonic dystrophy type 1 (DM1). Behavioral and brain functions : BBF 2006, 2:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, Ranum LP: An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nature genetics 1999, 21(4):379–384. [DOI] [PubMed] [Google Scholar]
- 203.Ikeda Y, Dalton JC, Moseley ML, Gardner KL, Bird TD, Ashizawa T, Seltzer WK, Pandolfo M, Milunsky A, Potter NT et al. : Spinocerebellar ataxia type 8: molecular genetic comparisons and haplotype analysis of 37 families with ataxia. American journal of human genetics 2004, 75(1):3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Paulson HL: The spinocerebellar ataxias. Journal of neuro-ophthalmology : the official journal of the North American Neuro-Ophthalmology Society 2009, 29(3):227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST: Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 2007, 55(4):556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J: RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 2007, 55(4):565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Timchenko LT, Miller JW, Timchenko NA, DeVore DR, Datar KV, Lin L, Roberts R, Caskey CT, Swanson MS: Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic acids research 1996, 24(22):4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ et al. : Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. The EMBO journal 2010, 29(7):1248–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL: Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66(4):817–822. [DOI] [PubMed] [Google Scholar]
- 210.Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM: Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. The Journal of neuroscience : the official journal of the Society for Neuroscience 1997, 17(5):1539–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Khandjian EW, Huot ME, Tremblay S, Davidovic L, Mazroui R, Bardoni B: Biochemical evidence for the association of fragile X mental retardation protein with brain polyribosomal ribonucleoparticles. Proceedings of the National Academy of Sciences of the United States of America 2004, 101(36):13357–13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Stefani G, Fraser CE, Darnell JC, Darnell RB: Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. The Journal of neuroscience : the official journal of the Society for Neuroscience 2004, 24(33):7272–7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Greenough WT, Klintsova AY, Irwin SA, Galvez R, Bates KE, Weiler IJ: Synaptic regulation of protein synthesis and the fragile X protein. Proceedings of the National Academy of Sciences of the United States of America 2001, 98(13):7101–7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hokkanen S, Feldmann HM, Ding H, Jung CK, Bojarski L, Renner-Muller I, Schuller U, Kretzschmar H, Wolf E, Herms J: Lack of Pur-alpha alters postnatal brain development and causes megalencephaly. Human molecular genetics 2012, 21(3):473–484. [DOI] [PubMed] [Google Scholar]
- 215.Qurashi A, Li W, Zhou JY, Peng J, Jin P: Nuclear accumulation of stress response mRNAs contributes to the neurodegeneration caused by Fragile X premutation rCGG repeats. PLoS genetics 2011, 7(6):e1002102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Li Y, Jin P: RNA-mediated neurodegeneration in fragile X-associated tremor/ataxia syndrome. Brain research 2012, 1462:112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wang GS, Kearney DL, De Biasi M, Taffet G, Cooper TA: Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. The Journal of clinical investigation 2007, 117(10):2802–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ho TH, Savkur RS, Poulos MG, Mancini MA, Swanson MS, Cooper TA: Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. Journal of cell science 2005, 118(Pt 13):2923–2933. [DOI] [PubMed] [Google Scholar]
- 219.Ladd AN, Stenberg MG, Swanson MS, Cooper TA: Dynamic balance between activation and repression regulates pre-mRNA alternative splicing during heart development. Developmental dynamics : an official publication of the American Association of Anatomists 2005, 233(3):783–793. [DOI] [PubMed] [Google Scholar]
- 220.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM et al. : Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314(5796):130–133. [DOI] [PubMed] [Google Scholar]
- 221.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y et al. : TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochemical and biophysical research communications 2006, 351(3):602–611. [DOI] [PubMed] [Google Scholar]
- 222.Kwiatkowski TJ Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T et al. : Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323(5918):1205–1208. [DOI] [PubMed] [Google Scholar]
- 223.Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P et al. : Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323(5918):1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A et al. : TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 2017, 95(4):808–816 e809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Todd PK: Making sense of the antisense transcripts in C9FTD/ALS. Acta neuropathologica 2013, 126(6):785–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C et al. : Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nature neuroscience 2011, 14(4):459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ito D, Suzuki N: Conjoint pathologic cascades mediated by ALS/FTLD-U linked RNA-binding proteins TDP-43 and FUS. Neurology 2011, 77(17):1636–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N: Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Annals of neurology 2011, 69(1):152–162. [DOI] [PubMed] [Google Scholar]
- 229.Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B et al. : ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. The EMBO journal 2010, 29(16):2841–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.King OD, Gitler AD, Shorter J: The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain research 2012, 1462:61–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J et al. : Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149(4):753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Lim L, Wei Y, Lu Y, Song J: ALS-Causing Mutations Significantly Perturb the Self-Assembly and Interaction with Nucleic Acid of the Intrinsically Disordered Prion-Like Domain of TDP-43. PLoS biology 2016, 14(1):e1002338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP: Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163(1):123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Daughters RS, Tuttle DL, Gao W, Ikeda Y, Moseley ML, Ebner TJ, Swanson MS, Ranum LP: RNA gain-of-function in spinocerebellar ataxia type 8. PLoS genetics 2009, 5(8):e1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Mutsuddi M, Marshall CM, Benzow KA, Koob MD, Rebay I: The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with staufen in Drosophila. Current biology : CB 2004, 14(4):302–308. [DOI] [PubMed] [Google Scholar]