

Abstract
Conventional methods for detecting small quantities of nucleic acids require amplification by the polymerase chain reaction (PCR), which necessitates prior purification and introduces copying errors. While amplification-free methods do not have these shortcomings, they are generally orders of magnitude less sensitive and specific than PCR-based methods. In this review, we provide a practical guide to a novel amplification-free method, single-molecule recognition through equilibrium Poisson sampling (SiMREPS), that provides both single-molecule sensitivity and single-bas e selectivity by monitoring the repetitive interactions of fluorescent probes to immobilized targets. We demonstrate how this kinetic fingerprinting filters out background arising from the inevitable nonspecific binding of probes, yielding virtually zero background signal. As practical applications of this digital detection methodology, we present the quantification of microRNA miR-16 and the detection of the mutation EGFR L858R with an apparent single-base discrimination factor of over 3 million.
Keywords: Single-molecule Fluorescence Microscopy, Kinetic Fingerprinting, Amplification-free, MicroRNA, DNA, Mutation
1. Introduction
1.1. Conventional high-sensitivity analysis of nucleic acids requires amplification
The detection of nucleic acid sequences with high specificity plays an important role in both basic biological research and diagnostics due to the fundamental roles of genetics, epigenetics, and gene expression in both normal physiology and pathology. For instance, specific mutations [1] and aberrant methylation patterns of DNA [2] have been linked to various types of cancer, showing promise for early detection of disease, monitoring of treatment response and relapse, and indicating whether a cancer is likely to respond to a given course of treatment [3–6]. Expression levels of specific microRNAs (miRNAs) [7–10] and long non-coding RNAs (lncRNAs) [11–13] are strongly correlated to cell differentiation states and thus of interest as biomarkers of disease.
To ensure adequate sensitivity for most nucleic acid analyses, samples must be amplified by procedures such as the polymerase chain reaction (PCR) for adequate specificity, sometimes following generation of cDNA by reverse transcriptase and/or the ligation of adapter sequences. However, such preparative procedures introduce several challenges for the quantitative analysis of nucleic acids. First, DNA polymerases and thermal cycling can both introduce artifactual sequence changes such as base substitutions during the amplification process [14,15], which may result in false positives when attempting to detect rare single-base mutations (e.g., for liquid biopsy of cancer). Second, reverse transcriptases and ligases exhibit significant sequence biases, introducing significant artifacts [16] such as spurious differences in expression levels and even the complete absence of certain sequences. Third, polymerases and ligases can be susceptible to inhibition by contaminants such as heparin and heme [17,18], necessitating additional purification steps prior to amplification. Finally, many classes of analytes are simply not amenable to direct amplification, including epigenetic modifications, short or fragmented nucleic acids, or non-nucleic acid analytes.
1.2. Advantages and shortcomings of amplification-free detection methods
Due to these challenges, several amplification-free methods [19–21] have been pursued for the analysis of nucleic acids and other biomolecules, in some cases permitting the direct capture and quantitation of analytes from biological matrices without prior purification. However, these amplification-free approaches typically suffer from a different set of challenges. First, since they lack the geometric amplification of PCR, these methods are generally limited by finite thermodynamic discrimination factors between closely related sequences [22]. This thermodynamic specificity limit is embodied by the parameter , where ΔΔGo is the difference in the Gibbs free energy of hybridization of a detection probe to a target sequence and of the same probe to a related but spurious target sequence; in practice, this translates to Qmax,therm values ranging from about 20 to 20,000 for singlenucleotide variants [22]. In most cases, the actual single-base specificity realized is only 90–99% [20, 22]. Second, since many amplification-free assays are surface-based, true single-molecule sensitivity becomes challenging due to the inability to completely suppress nonspecific binding of probes to the detection surfaces.
1.3. Single-molecule kinetic fingerprinting enables amplification-free detection with arbitrarily high specificity
To realize amplification-free biomolecule detection without being bound by thermodynamic limits of specificity, we developed an approach based on time-resolved measurement of the interaction kinetics between fluorescent probes and single immobilized analyte molecules [23]. This approach, termed SiMREPS (single-molecule recognition through equilibrium Poisson sampling), exploits repeated observations of transient probe interactions with each surface-bound copy of the analyte to create a “kinetic fingerprint” that is highly characteristic of that particular analyte molecule when detected at the single molecule level (Fig. 1A), and is significantly perturbed by even small alterations such as singlebase substitutions. As a result, nonspecific binding of probes to the surface and to closely related sequences can be confidently screened out due to their distinct kinetics (Fig. 1B-C), yielding essentially background-free detection of single analyte molecules after applying appropriate filters for signal-to-noise, intensity, and probe binding and dissociation kinetics (Fig. 1D). To facilitate the observation of repeated fluorescent probes binding to the same copy of analyte, the analyte is typically immobilized to a biotin-functionalized surface via a streptavidin bridge and a biotin-labeled capture probe (Fig. 1A). While DNA oligonucleotides have been successfully employed as capture probes for SiMREPS, several locked nucleic acid (LNA) modifications are usually incorporated when the analyte is a short nucleic acid such as a miRNA to permit high-affinity capture while leaving several unpaired nucleobases to interact with the fluorescent probe.
Fig. 1.
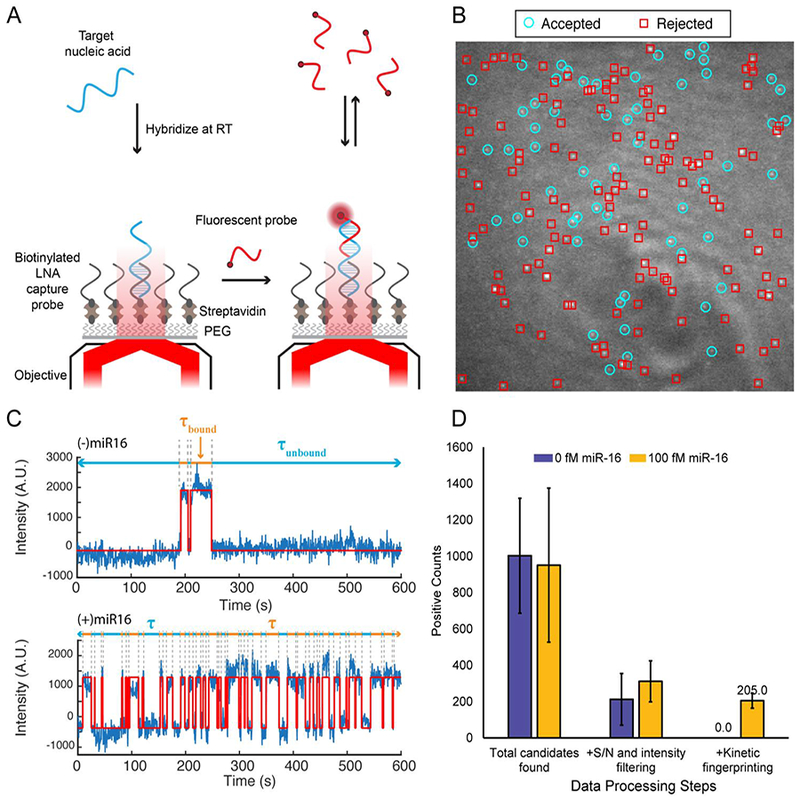
Overview of the SiMREPS technique for low-background, high-specificity detection of single molecules. (A) Schematic illustrating the experimental principles of SiMREPS. A target analyte is captured at the surface of a coverslip via a biotinylated capture probe. Then, using TIRF microscopy, each copy of surface-bound analyte is detecting by monitoring the repeated transient binding of a fluorescent probe, which yields a distinctive kinetic fingerprint. (B) Single movie frame from a representative field of view from SiMREPS using objective-type TIRF microscopy. Red squares indicate positions of binding events that were rejected as likely background binding by kinetic fingerprinting, and the cyan circles indicate positions of repeated binding events with kinetics that suggest the presence of the analyte (C) Representative fluorescence-versus-time traces observed in the presence and absence of a miRNA target, hsa-miR-16. The kinetics of transitions between FP-bound and FP-unbound states are analyzed to distinguish between true and false positives at the single-molecule level. (D) Number of spots counted in positive and negative control experiments for miR-16 before (‘total counts’) and after (‘accepted counts’) kinetic filtering. While filtering based on intensity and signal-to-noise (S/N) alone does not yield a significant difference between positive and negative controls (due to background binding of the probe), the application of kinetic filtering criteria (see section 2.7.4) reduces accepted counts in the negative control to essentially zero.
Because the binding of fluorescent probes to a single analyte molecule can be modeled as a Poisson process, the number of probe binding and dissociation events observed for each analyte molecule (Nb+d) will increase linearly over time, with a coefficient of variation (C.V.) that decreases as [23]. This decrease in C.V. with increasing observation time permits the kinetic fingerprint resulting from a single analyte molecule to be separated from the signals resulting from nonspecific binding to an arbitrarily high degree. Similarly, the lifetimes of the analyte in the probe-bound (τbound) and probe-unbound (τunbound) states become better separated from the background binding as an increasing number of probe-binding events to each analyte is observed. This increased confidence in the source of a given kinetic fingerprint is the core feature of SiMREPS, and means that probes with finite thermodynamic discrimination can be used to detect an analyte with arbitrarily high specificity, given an adequate number of binding events. In other words, the specific time evolution of the detection signal becomes a heretofore untapped observation parameter that serves to enhance the accuracy of analyte identification, in concept similar to the revolution conventional fluorescence microscopy experienced upon introduction of super-resolution approaches that observe a time series of sparse signals from single molecules to determine their cellular localization more accurately [24].
As a proof of concept, we show that miRNAs such as miR-16 [23] can be detected using SiMREPS with essentially zero background signal from surface binding of fluorescent probes if kinetic fingerprints from single molecules are filtered by Nb+d and τbound (Fig. 1D). In this article, we discuss practical considerations for the use of SiMREPS to detect short nucleic acids such as miRNAs and DNA fragments, including guidelines for instrumentation and assay design. In addition, we demonstrate the high specificity of the technique through proof-of-concept measurement of the cancer point mutation EGFR L858R with an apparent discrimination factor of > 1,000,000.
2. Materials and Methods
2.1. Instrumentation and sample cell design
Since SiMREPS in its current implementation requires the presence of an excess of fluorescent probe in binding equilibrium with the surface-immobilized analyte, a microscope capable of total internal reflection fluorescence (TIRF) illumination is required to reject background signal from the majority of freely diffusing (non-surface-bound) fluorescent probes. Most commonly, TIRF measurements are carried out using either a prism-type (P-TIRF) or objective-type (O-TIRF) illumination geometry (Fig. 2A-B). Excitation light is provided by a laser of appropriate wavelength (e.g., 640 nm for probes labeled with Cy5) and output power (typically 10–100 mW) and undergoes total internal reflection at the interface between the coverslip and the aqueous solution containing the fluorescent probe. To reliably detect single fluorescent probes with satisfactory signal-to-noise, an illumination intensity of ~50 W/cm2 is typically used, and the TIRF angle adjusted to achieve a calculated penetration depth of ~80–110 nm of the evanescent field. Emission light from surface- or analyte-bound fluorescent probes is collected through a microscope objective lens, passed through dichroic mirrors and/or chromatic filters to remove the majority of the excitation light, and detected by a high-sensitivity camera such as an ICCD, EMCCD, or sCMOS. In our study, an EMCCD camera is used in O-TIRF and an ICCD camera is used in P-TIRF. In SiMREPS imaging, the signal integration time (exposure time) per frame is typically 500 ms, and typically 1200 movie frames are acquired per field of view (FOV).
Fig. 2.
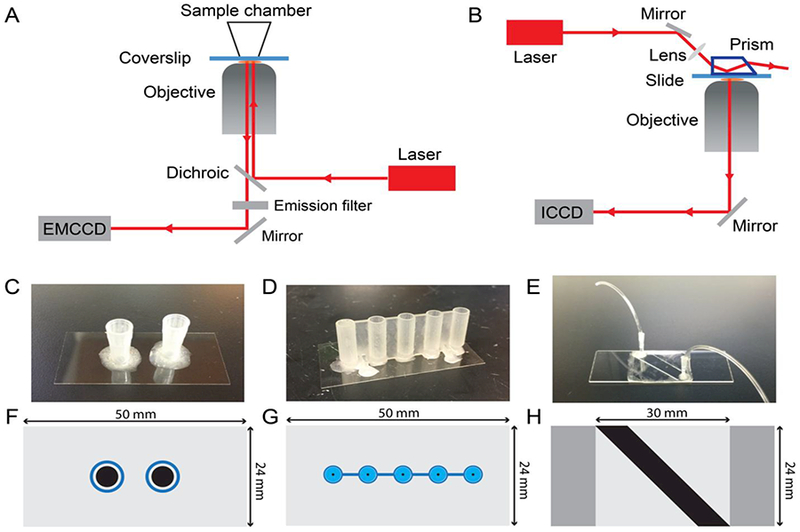
Overview of instrumentation and sample cells. (A) Objective-type TIRF microscope. (B) Prism-type TIRF microscope. (C) Pipet tip chamber sample cell. (D) 3D-printed sample cell with cylindrical reservoir and tapered conical base. (E) Sandwich-type sample cell for prism-TIRF measurements. (F)-(G) Scale drawings showing a top view of each sample cell type shown in (C)-(E). The black-shaded region in each panel represents the surface area available for target capture and imaging on the coverslip or slide. Blue-shaded regions in (F) and (G) represent the plastic walls of the sample wells.
SiMREPS is compatible with a variety of sample cell types (Fig. 2C-H). Because the sample cell must be positioned between the prism and objective, P-TIRF requires thin flow cells that are typically constructed by sandwiching two pieces of double-sided tape between a coverslip and a biotinfunctionalized microscope slide, with optional plastic tubing added for ease of sample injection (Fig. 2E). However, with O-TIRF taller sample cells constructed from cut pipet tips (Fig. 2C) or 3D-printed plastic parts (Fig. 2D) attached to a biotinylated coverslip may also be used. These taller sample cells permit the immobilization of analyte on the imaging surface at higher densities, providing g reater sensitivity than thin flow cells. Thus, for high-sensitivity measurements (LOD < 1 pM) O-TIRF is preferred over PTIRF for SiMREPS. However, due to their open-top geometry, measurements that take a long time (>1 h) or using fluorescent probes with slow-off rates (< 2 min−1) may benefit from filling the sample chamber to the top with imaging solution and sealing it with parafilm to slow the influx of atmospheric oxygen. All the data presented here were collected by O-TIRF using sample cells constructed from cut pipet tips. Recently, other instrumentation has been introduced for super-resolution studies, including spinning disk confocal microscopes (CSU-W1, Yokogawa Electric) [25] and the Oxford Nanoimager [26]; these may provide other options for SiMREPS measurements in the future.
2.2. Analyte Scope
Since it does not require any nucleic acid-specific enzymes such as ligases or polymerases, SiMREPS is in principle capable of detecting any analyte that can (1) be immobilized at a surface, preferably via a specific interaction, and (2) remain free to transiently recruit fluorescent probes from solution while bound to the surface. It thus has a much broader scope than amplification-based approaches. To date, SiMREPS has been successfully applied to the identification and counting of short nucleic acids such as miRNAs (miR-16, miR-21, let-7a, let-7c, miR-141, cel-miR-139) [23] and ~22–160 bp fragments of single-stranded or double-stranded DNA [27–29] such as cancer-related EFFR mutations (see Results). Since the assay is typically performed at ambient room temperature, to ensure maximal sensitivity for double-stranded or highly structured analytes, care must be taken to fully denature and sequester any interfering secondary structure that might interfere with surface capture or fluorescent probe binding, e.g., by brief heating in an excess (e.g., 1–2 μM) of a carrier oligonucleotide or sequence-specific oligonucleotides that prevent the formation of interfering secondary structure. In contrast, short nucleic acids that are difficult to detect with amplification-based approaches are readily detected by SiMREPS and are thus particularly strong candidates for the technique. Finally, owing to its high specificity, SiMREPS is capable of discriminating single-nucleotide variants such as let-7a and let-70 [23].
2.3. Probe design
2.3.1. Capture probes
For sequence-specific capture of analytes, terminally biotin-labeled capture probes (CPs) are immobilized on a streptavidin-coated coverslip or microscope slide surface. The CPs may, in principle, comprise any type of nucleotide or modified nucleotide, including DNA, RNA, and other non-natural nucleic acids such as LNAs, peptide nucleic acids (PNAs), or unlocked nucleic acids (UNAs) [30]. When using SiMREPS for miRNA detection, it is important to leave ~10 nucleotides unpaired for interaction with the fluorescent probe, necessitating the use of a relatively short capture probe (10–12 nucleotides). We therefore typically employ CPs comprising mostly DNA nucleotides, but also incorporating several (4–5) LNA nucleotides to maintain a high melting temperature (Tm) despite the short length. Compared to natural DNA and RNA oligonucleotides, an LNA oligonucleotide offers substantially increased affinity for its complementary strand, which makes it an ideal capture probe of short RNA and DNA targets. The positions of LNA modifications are determined semi-empirically using the online Tm and self-structure prediction tools available from Exiqon [31]; the goals are to achieve a predicted Tm (under standard conditions) > 60 °C and a self-structure score as low as possible, preferably < 25 °C. LNA capture probes used in our study were purchased from Exiqon (now distributed by Qiagen) with HPLC purification. For instance, the melting temperature of the capture probe for miR16 increases from 47 °C to 79 °C when replacing 4 out of 10 DNA nucleotides with the corresponding LNA nucleotides. For longer targets such as genomic DNA fragments, long DNA capture probes with suitably large Tm values (> 60 °C) have also been employed with success; these have the advantage of higher capture specificity than short CPs. Regardless of the type of CP used, the choice of capture region should be chosen such that it minimizes any interfering secondary structure in the CP and target. Such optimization can be carried out using prediction tools such as Exiqon’s OligoAnalyzer, Integrated DNA Technology’s Tm prediction tool, or NUPACK [27–29].
2.3.2. Fluorescent probes
To permit kinetic fingerprinting of single molecules by SiMREPS, reversible binding is required to allow for many cycles of binding and dissociation of the fluorescence probe (FP) to each copy of the target. Typically, this means that the Tm of the interaction between the FP and target should be comparable to the temperature at which the assay is conducted (usually room temperature, 20–25 °C). The lifetime of bound state should be longer than the camera exposure time (in our case, 500 ms) but not so long as to impede the observation of enough binding events to separate the positive signal from background binding within a convenient sample imaging time frame (in our case, typically ~10 minutes). At constant temperature and ionic strength, the dissociation kinetics of a short oligonucleotide probe are exponentially dependent upon the length of the probe [35], making the choice of FP length a particularly important parameter [23]. In the high-ionic strength buffers typically used in SiMREPS measurements of nucleic acids (see section 2.6) and for observations near room temperature, the optimal length of FPs with ~50% GC content is typically ~9 nucleotides for RNA targets, and ~8 nucleotides for DNA targets. Probes against sequences with high GC content can be designed with one or more intentional mismatches to achieve appropriate kinetics; alternatively, denaturants such as 5–30% formamide can be added to mildly destabilize the FP-target interaction. Formamide lowers the (Tm) of DNAs linearly by 2.4–2.9°C/mole of formamide depending on the (GC) composition and state of hydration [36]. Higher observation temperatures (e.g., using a heated microscope objective and/or stage) can be contemplated as another way of destabilizing FP-target interactions. If denaturants or higher temperatures are used, the stability of the CP-target interaction should be verified under the new conditions, e.g., by performing SiMREPS measurements after variable incubation times and determining whether there is a systematic decrease in detected target molecules over time.
When choosing the binding register of the FP on the target sequence, the followin g criteria should be observed for optimal performance:
GC content of the FP-target interaction should be ≤_50%, if possible, to ensure rapid binding and dissociation kinetics;
There should be at least 1–2 unpaired nucleotides between the binding sites of the CP and FP on the target in order to avoid stacking interactions between adjacently binding probes that will tend to lengthen the bound-state lifetime of the FP;
It is preferable to position the fluorophore distally on the FP relative to the CP, to reduce the likelihood of stacking interactions between the fluorophore and the CP; alternatively, an additional 1–2 unpaired bases between the FP and CP can accommodate a proximally positioned fluorophore;
If single-base discrimination is desired, note that the selectivity is higher when the mismatched nucleotide is near the middle of the FP than it is when positioned near the 3’- or 5’-end of the probe-target duplex [33–35]. While mismatches near the end of the duplex can also provide adequate discrimination by SiMREPS [23], longer observation times may be necessary to achieve perfect kinetic discrimination.
Notably, the use of fluorescent probes with only 8–9 nucleotides will not provide sufficient specificity to uniquely identify a sequence against a background of genomic DNA or RNA. Additional specificity is provided by the capture probe (~10 nucleotides), which can be engineered to be as specific as needed, for example, by lengthening it upon removal of LNA moieties or increase of the assay temperature or formamide concentration, and the slide surface should be well passivated against nonspecific binding of nucleic acids. Furthermore, addition of a second fluorescent probe to create a FRET pair has been employed in super-resolution imaging with DNA-PAINT [40] and could provide additional specificity by requiring the proximity of two short (e.g., 8–10 nucleotide) sequences to observe a positive kinetic fingerprint. The addition of a second fluorescent probe will slightly increase the footprint of the assay (from ~20 to ~30 nucleotides), but this footprint will still be comparable to, or shorter than, that required by the majority of other nucleic acid assays based on PCR or thermodynamic binding, while also providing extremely high single-base discrimination power without any purification or enzymatic processing. Since SiMREPS has notably fewer required components than enzymatic assays, the choice of both probes and buffer conditions is particularly flexible and can be adjusted to match most specificity requirements imposed by a particular sample matrix.
2.3.3. Auxiliary oligonucleotides
While SiMREPS can often be performed using only the CP and FP, other oligonucleotides may be helpful in preventing re-hybridization of double-stranded targets, in preventing secondary structures in the target that could interfere with FP binding, or in reducing off-target binding of the FP to the CP or spurious target sequences.
Carrier oligonucleotide: 1–5 μM of a polythymidine oligonucleotide such as (dT)10 can reduce sample loss due to adsorption as well as prevent re-hybridization of double-stranded DNA targets after denaturation.
CP blocker: some combinations of CP and FP sequences will result in a large amount of transient FP binding to the CP, which can lead to false positives or false negatives; in such cases, a short oligonucleotide probe complementary to the CP can be added to the imaging solution at a sufficient concentration (e.g., > 10 nM) to saturate any non-target-bound CPs at the imaging surface.
Competitor oligonucleotides: to block transient binding to closely related sequences, short unlabeled oligonucleotides may be included in the imaging solution. For instance, in the detection of EGFR L858R presented in this work, an 8-nucleotide probe complementary to the wild-type (WT) sequence - a so-called WT competitor - is used to reduce binding of the FP to the WT EGFR sequence.
Secondary structure blockers: short (10–14 nucleotide) oligonucleotide probes complementary to the regions of the target that are directly adjacent to the CP and/or FP binding region can be useful in improving both capture efficiency and accessibility of the target to the FP. These may be added either prior to surface capture or in the imaging buffer.
2.4. Slide and sample cell preparation
2.4.1. Surface functionalization
The objectives of surface functionalization are twofold: first, to passivate the imaging surface against excessive nonspecific binding of the FP and other components; and second, to provide an affinity tag, usually biotin, that can be used for subsequent immobilization of the CP. Whether glass coverslips or microscope slides are used as the imaging surface, a typical surface functionalization is performed as follows, based on a published protocol [41].
First, the slides or coverslips (hereafter referred to as “slides”) are placed in a slide staining jar (Coplin-type) and sonicated for 10 min in 1M KOH. The KOH is removed, and the slides are washed at least three times with deionized water. Next, the slides are immersed for 20 min in an aqueous “base piranha” solution consisting of 14.3% v/v ammonium hydroxide and 14.3% v/v hydrogen peroxide that is heated to 60–70 °C. The slides are rinsed at least three times with deionized water (optionally, if fused silica slides are being re-used, they may be heated for ~1 min with a propane torch at this step to burn off any residual microscopic contaminants). The slides are then rinsed once with acetone (HPLC purity or higher).
Next, the slides are immersed in a 2% v/v solution of (3-aminopropyl) triethoxysilane (ATPES) in acetone for 10 min, sonicated for 1 min, and incubated for another 10 min. The APTES/acetone solution is discarded and the slides are immediately rinsed 3–5 times with deionized water, then dried completely under nitrogen flow. The slides are now functionalized with surface amines for further reaction with N-hydroxysuccinimidyl esters of polyethylene glycol (PEG) and biotin-PEG.
To functionalize the slides with biotin-PEG and PEG, a 1:10 mixture of biotin-PEG-succinimidyl valerate (biotin-PEG-SVA, MW ~5000, Laysan Bio, Inc.) and methoxy-PEG-succinimidylvalerate (mPEG-SVA, MW ~5000, Laysan Bio, Inc.) is dissolved in freshly prepared 0.1 M NaHCO3 to a final total PEG concentration of 21.6% w/v. The mixture is briefly centrifuged (1 min at 10,000 rpm in a benchtop Eppendorf microcentrifuge) to remove any suspended air bubbles, and 70–80 μL of the PEG solution is immediately sandwiched between two slides, making sure to exclude air bubbles. The slide sandwiches are kept in a humidified environment in the dark at room temperature for 2–3 h. The slides are then carefully disassembled, placed in a slide staining jar (keeping track of the orientation of the coated side) and rinsed at least three times with deionized water, then dried completely under nitrogen flow.
Remaining surface amines are quenched with disulfosuccinimidyltartrate (sulfo-DST, Soltec Ventures) to reduce nonspecific binding of nucleic acids to the surface, as follows. A 10-mg portion of sulfo-DST is dissolved in 350 μL of 1 M aqueous NaHCO3, briefly centrifuged (1 min at 10,000 rpm in a benchtop Eppendorf microcentrifuge), and 70–80 μL of the solution is immediately sandwiched between two slides with the PEG-functionalized surfaces pointing inward towards the sulfo-DST solution. The slide sandwiches are incubated in a humidified chamber for 30 min at room temperature, then rinsed thoroughly with deionized water and dried completely with nitrogen. The slides are stored in the dark under air for up to 2 weeks, or in a desiccator (preferably under inert gas or vacuum) for several weeks.
2.4.2. Sample cells
For prism-type TIRF microscopy experiments, fluidic sample cells are constructed using two pieces of double-sided tape sandwiched between a quartz slide and glass coverslip as previously described[42] (Fig. 2E). Optional drilling of holes in the backing slide and attachment of Tygon tubing permits convenient buffer exchange, while use of quartz microscope slides permits them to be cleaned with detergent and re-used [42], though cheaper borosilicate glass slides may also be used. After use, these slides can be disassembled and re-cleaned as follows: immerse in boiling water for 30 min; carefully peel off any tape and adhesive with a razor blade; rub slide thoroughly with a thick paste of an abrasive detergent such as Alconox; then rinse thoroughly with deionized water and subject to the cleaning protocol in section 2.4.1. Note that no visible residue of adhesive should remain on the slide prior to beginning the protocol of section 2.4.1.
For objective-type TIRF microscopy measurements, sample cells are constructed by fixing a cut 1cm length of a pipet tip (e.g., Eppendorf brand) to a coverslip using epoxy adhesive (Double Bubble, Hardman Adhesives; Fig. 2C). We have also successfully employed 3D-printed sample cells (Fig. 2D) that have a smaller area of contact with the coverslip (~0.2 mm2) and a tapered base that permits the use of as little as 5–10 μL of analyte solution without sacrificing sensitivity. The custom design was prepared in Autodesk Fusion 360 and printed on a ProJet 3500 using the M3 Crystal resin at the highest print resolution of 16 μm per layer. As with the pipet tip sample cells, the 3D-printed sample cells are attached to coverslips with epoxy adhesive, but in this case the attachment is performed with the aid of an electronics vise (e.g., PanaVise) to firmly hold the 3D-printed wells against the coverslip during the application of epoxy to prevent the adhesive from seeping in and clogging the small aperture between the interior of the sample well and the coverslip. While the sandwich-type flow cell can be used on objective TIRF as well, the sample cells constructed from pipet tips or tall 3D-printed wells provide higher sensitivity because of a higher ratio between the volume of analyte solution and the contact area with the coverslip; that is, a larger fraction of the analyte may be captured in a small region of the imaging surface, yielding more detectable molecules per field of view. One drawback of the sample cells constructed from pipet tips and 3D-printed wells is they are both for one-time use only. Regardless of type, the completed sample cells may be stored in a dry, inert, dark environment for several weeks prior to use in SiMREPS.
2.5. Surface capture of the target analyte
The following protocol applies to all sample cell types with biotin-PEG-functionalized surfaces, but for the sake of clarity all solution volumes apply specifically to sample wells constructed from cut pipet tips, which were used to collect all data presented in this study. Before imaging, the slide surface is briefly washed with 100 μL T50 buffer (10 mM Tris-HCl, 50 mM NaCl, pH 8.0) followed by the addition of 40 μL of 1 mg/ml streptavidin to the sample well. After 10 min, the streptavidin solution is removed and the surface is washed three times with 100 μL of 1× PBS. The surface is then incubated with 40 μL of a solution containing 100 nM of the appropriate biotinylated LNA capture probe in 1× PBS buffer for 10 min. The solution is removed and then the sample cell is washed three times with 100 μL of 1× PBS. Finally, a 100-μl portion of sample containing the target RNA or DNA and 2 μM carrier oligonucleotide is introduced into the sample chamber and incubated for 1 h to capture the analyte at the imaging surface. Note that double-stranded DNA samples must first be denatured by, for example, heating to 95 °C in the presence of 2 μM carrier oligonucleotide, then cooling to room temperature in a water bath for 5 min before adding to the sample cell. For direct capture of analytes from crude biofluids such as cell extract or serum, a pre-incubation step in ~2% (w/v) sodium dodecyl sulfate (SDS) and 0.16 U/μL of proteinase K (New England BioLabs, Inc.) is used to liberate nucleic acids from any protein binding partners as well as to inactivate any nucleases present in the sample [23]. After the 1-h capture incubation, the sample solution is removed, and 1× PBS buffer is added to the sample cell until the imaging buffer (see section 2.6) is added. Note that, while analytes can be captured from crude biofluids [23], the imaging should still be performed in a standard imaging buffer to ensure reproducible probe binding and dissociation kinetics.
2.6. Imaging
All data discussed in this paper were collected using an Olympus IX-81 objective-type TIRF microscope equipped with a 60× oil-immersion objective (APON 60×OTIRF, 1.49 NA) with both Cell^TIRF and z-drift control (ZDC2) modules, and an EMCCD camera (IXon 897, And or, EM gain 300). Cy5 excitation was provided by a 640-nm red laser (Coherent CUBE 640–100C, 100 mW) and Cy3 excitation by a 532-nm green laser (CrystaLaser CL532–150mW-L). To delay the photobleaching of fluorophores and thus obtain more accurate measurements of the bound-state lifetime of the FP, a 25 nM solution of the FP is prepared in an imaging buffer containing 4× PBS, 2.5 mM 3,4-dihydroxybenzoate, 25 nM protocatechuate dioxygenase, 1 mM Trolox (oxygen scavenger system, OSS [43]), and added to the sample chamber for SiMREPS imaging. The imaging solution for EGFR L858R mutant and wild-type discrimination in this study also includes 100 nM of a WT competitor sequence to block FP binding to the WT EGFR sequence. Usually 3–5 minutes are allowed for the OSS to achieve a low steady-state oxygen concentration before imaging. The transient binding of FP to captured target molecules is monitored for 10 min under TIRF illumination, with a movie acquisition rate of 2 Hz and an EM gain setting of 150. All imaging is performed at a darkened room at an environmentally controlled temperature of 20 ± 3 °C.
The high ionic strength of the imaging buffer promotes rapid binding of the FP to the target [35], allowing for many cycles of FP binding and dissociation within the 10-min observation period for well-optimized FP sequences. The concentration of FP in the imaging buffer may be adjusted, but typically is optimal in the range of 25–50 nM; lower concentrations will reduce the frequency of FP binding, while much higher concentrations will result in prohibitively high levels of background fluorescence from freely diffusing probes during imaging. If dissociation kinetics of the FP are relatively slow, for instance due to a longer or more GC-rich FP sequence, denaturants such as 10–30% formamide can be used to decrease the duration of the bound state, albeit at greater risk of target dissociating from the CP during the experiment.
The length of the observation period for each field of view is a particularly important parameter, since enough time must be allowed for multiple (e.g., >10) cycles of binding and dissociation to each surface-bound analyte molecule, thus permitting adequate separation between specific and nonspecific binding signatures for zero-background measurements. The exact imaging time required is dependent on the kinetics of specific and nonspecific binding, as well as the degree of separation between signal and background peaks that is desired. A useful guideline for selecting a minimum observation time is embodied in the following relationship [23]:
| (1) |
where t is the observation time, s is the desired number of standard deviations separating the signal and background peaks, k’bind is the pseudo-first order binding rate constant for the query probe to the target, kdiss is the first-order dissociation rate constant of the query probe from the target, and is the ratio between the average number of nonspecific binding and dissociation events observed per trace () and the average number of specific binding and dissociation events observed per trace ((Nb+d)specific). For example, if a separation of s = 3 standard deviations is desired between signal and background, and with k’bind = 5 min−1, kdiss = 5 min−1, and , the minimum observation time is 3.9 min. The sampling interval (exposure time per frame) should be significantly less than the smaller of Tboundand and Tunbound¦ in the above example, significantly less than 0.2 min, e.g., ~1 s per frame (sampling frequency of ~1 Hz).
2.7. Data analysis for kinetic fingerprinting
All MATLAB scripts for SiMREPS data analysis will be deposited in a publicly available github repository (https://github.com/ajohnsonbuck/simreps-2018-08). A typical analysis of movies from SiMREPS experiments consists of the following steps: 1) identification of “candidate” regions of interest (ROIs) within the image exhibiting greater frame-to-frame intensity fluctuations than their surrounding pixels (Fig. 3A-B); 2) calculating the frame-by-frame fluorescence intensity of each ROI (Fig. 3C), 3) hidden Markov modelling (HMM) to calculate FP binding and dissociation kinetics for single-molecule kinetic fingerprinting (Fig. 3D); and 4) application of filters to distinguish nonspecific from specific binding based on signal-to-noise, intensity, and FP kinetics (Fig. 3E). Prior to step 1), a software-based drift correction may be applied to compensate for lateral stage drift during the experiment, though this is often not necessary if the microscope system is sufficiently stable (e.g., < 3 pixels of drift during the 10-min movie). candidate region. Upon request, MATLAB scripts for all the necessary processing steps below can be provided.
Fig. 3.
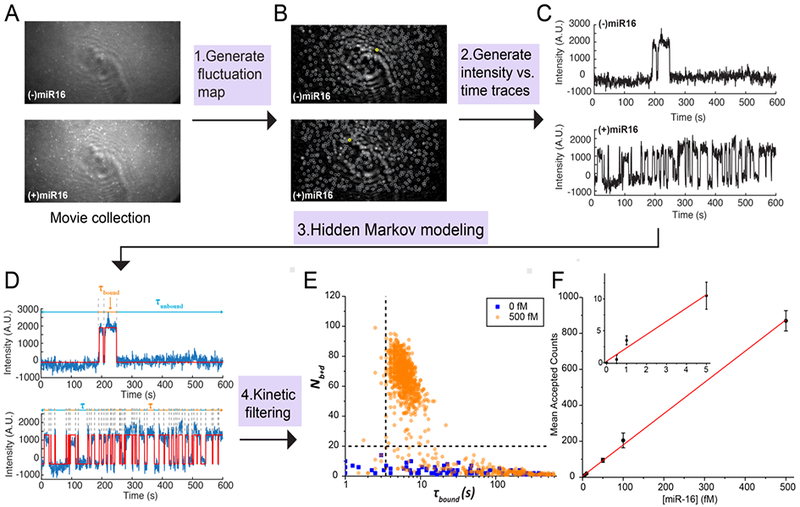
Data analysis pipeline. (A) Single-frame images of representative fields of view from TIRF microscopy. (B) Intensity fluctuation maps of the fields of view shown in (A). Grey circles indicate positions of local maxima in the fluctuation map, from which candidate ROIs are identified for further analysis by generation of intensity vs. time traces. (C) Representative intensity vs. time traces generated from the ROIs identified in (B), circled in yellow. (D) HMM idealization (red lines) for each intensity vs. time trace. Bound and unbound-state dwell times (τbound and τunbound, respectively) are indicated by the orange and blue horizontal line segments above the idealization. (E) Candidates in the positive (orange circles) and negative (blue squares) controls for miR-16 are well separated by thresholds of Nb+d > 20 and τbound > 2.5 s (black dashed lines), permitting discrimination of specific and nonspecific binding at the single-molecule level. Data are pre-filtered for signal-to-noise > 2.5 and intensity > 1000. (F) miR-16 standard curve. n = 3 replicates for blank, 2 replicates for other measurements. Error bars represent 1 standard deviation.
2.7.1. Identifying candidate ROIs
For optional drift correction, a custom routine written in Matlab (available upon request) based on the subpixel correlation between consecutive recorded images can be used to compensate for any x-y stage drift that would interfere with subsequent intensity-versus-time analysis of candidate ROIs. After this optional step, candidate ROIs—generally 5-pixel×5-pixel regions with significant frame-to-frame intensity fluctuations—are identified as follows. Each of the N movie frames is subtracted from the previous frame and the absolute value taken to generate a new image of the same dimensions as the original, but in which each pixel value represents the absolute value of the intensity change from the previous frame to the current frame. This is repeated for all movie frames, resulting in a new image stack with (N-1) frames. Finally, the value of each pixel in this image stack is averaged, resulting in a single image called a “fluctuation map” containing the average frame-to-frame change in intensity for each pixel. Pixels representing local maxima within this image are selected to serve as the center pixel of each candidate ROI for further processing.
2.7.2. Calculation of intensity-versus-time traces
The intensity-versus-time trace for each candidate ROI identified from the fluctuation map is generated as follows. Within the first frame of the original movie file, the intensity of all 25 pixels within the 5-pixel-×5-pixel ROI is summed to create a single fluorescence value, and the median intensity value of the 2-pixel-wide region surrounding the ROI is subtracted to find the background-subtracted intensity of this ROI within the first frame. This process is repeated for each frame of the movie, and the list of intensity values combined to create an intensity-versus-time trajectory for this ROI. The process is repeated for each ROI identified from the fluctuation map, and the intensity-versus-time trajectories are exported as an ASCII file for import into the HMM software QuB [44].
2.7.3. Hidden Markov modeling
The traces are imported into the HMM software QuB and fit using a two-state model. Proper parametrization is essential for convergence of HMM fitting; that is, the amplitudes, standard deviations, and kinetics should be as close as possible to the expected behavior of the FP binding to the target, and ideally within ~1 order of magnitude. It is important to use the same model to fit all datasets that are to be compared. The HMM fitting results table from QuB is exported for further analysis of the intensity and kinetics in MATLAB
2.7.4. Filtering specific from nonspecific binding
A binary classification is performed on each candidate ROI based on whether its intensity-versus-time trace satisfies certain criteria. The criteria are established by an empirical evaluation of traces collected in negative and positive control experiments—e.g., in the absence and presence of 500 fM synthetic target nucleic acid—and chosen so as to reject essentially all traces in the negative controls while accepting as many traces as possible in the positive controls. Since nonspecific binding of the probe to the surface can vary somewhat between coverslip or slide preparations, it is generally advisable to establish these criteria based on several independent technical replicates, preferably on different days. While the specific criteria will vary depending on factors such as the target, FP sequence and concentration, imaging buffer, and acquisition temperature, in this study a candidate ROI is considered to contain a true positive signature of the analyte if it satisfies the following c riteria:
Intensity difference between bound state and unbound state (ΔI) > 1000 counts for detection of miR-16, > 500 counts for detection of EGFR L858R
Signal-to-noise (ΔI/σ, where σ is the standard deviation of the intensity in the FP-unbound state) > 2.5 for miR-16, > 2 for EGFR L858R
Number of FP binding and dissociation events per observation period, Nb+d ≥ 20
Median lifetime in the FP-bound state, Tbound,median > 4 s for miR-16, > 5 s and < 20 s for EGFR L858R
Median lifetime in the FP-unbound state, Tunboundmedian > 0 for miR-16, > 20 s and < 50 s for EGFR L858R
All traces satisfying these criteria are counted as true positives, and those that do not are considered to show insufficient evidence to be counted as true positives. Of the above criteria, the most critical for rejecting false positives (as determined from negative control measurements) tend to be Nb+d and Tbound, median.
3. Results and Discussion
In negative control measurements with imaging buffer containing the FP, but in the absence of the target analyte, a considerable number of FP binding events were always observed—typically numbering in the hundreds—suggesting that transient or long-lasting interactions between the FP and the imaging surface were difficult to suppress entirely (Fig. 1D). In a conventional analysis without kinetic fingerprinting, it would be necessary to subtract these counts from all measurements as background; however, the large standard deviation of this background (Fig. 1D) would impose a limit of detection (LOD) of hundreds of captured target molecules per FOV.
In contrast, by applying the kinetic filtering criteria as outlined in section 2.7, essentially all of these background counts were filtered out in the negative control experiments (Fig. 1D), permitting the confident identification and counting of even single-digit numbers of target molecules per FOV. This is because, through repeated sampling of the same surface-immobilized target molecules through multiple cycles of FP binding, a progressively better estimate of kinetic parameters such as Nb+d, Tbound,median, and Tunboundmedian was obtained for each candidate ROI, and it became easier to resolve true and false positives by a binary classification based on the kinetic criteria outlined in section 2.7 (Fig 3E). The number of accepted counts (candidate ROIs that pass kinetic filtering) was linear within the range of approximately 1–800 molecules per FOV, as shown by the standard curve for miR-16 (Fig. 3F). Due to the essentially zero background, even 0.5 fM miR-16 yielded significant counts above the negative control, resulting in an LOD that was mainly limited by the capture efficiency of analyte on the imaging surface rather than on background binding of the FP or autofluorescence of the imaging surface. In terms of absolute concentration [45], the calculated limit of blank (LOB) of this assay is 0 (since no blank counts were detected), and the estimated LOD is 0.4 fM.
If more than ~500 molecules are present in a FOV, the diffraction-limited analysis presented here will result in a sub-linear increase and eventually a decrease in the accepted counts due to the inability to resolve closely spaced molecules. If it is desired to extend the dynamic range beyond this ~2.5 orders of magnitude into the range of thousands of molecules per FOV or more, it will likely be necessary to switch to a more conventional quantification scheme based on fluorescence intensity, or to implement super-resolution methods to analyze the kinetics of FP binding with sub-pixel accuracy [29]. Indeed, one recent paper describes the use of super-resolution imaging and kinetic analysis of dissociation kinetics to discriminate single-nucleotide variants in DNA with 95% accuracy [46].
We also tested the ability of SiMREPS to distinguish between closely related sequences, using as a model the point mutation EGFR L858R (c.2573T>G), a common driver mutation in non-small cell lung carcinoma. Note that the high GC content surrounding this mutation necessitated two design choices for the FP: the intentional introduction of a G-T wobble mismatch in the FP-target interaction, and the positioning of the mutation towards one end of the FP-target duplex to reduce the GC content slightly (Fig. 4A). While considerable FP binding was observed in the presence of the wild-type (WT) sequence, the traces in the WT-only experiment could be distinguished from the mutant (MUT) traces on the basis of the median bound-state lifetime (τboundtmedian), which was longer for some traces in the presence of the MUT (Fig. 4B,C). Indeed, the number of accepted traces in the presence of 100 nM WT was > 30-fold lower than in the presence of only 1 pM MUT, despite the fact that the WT was present at a 100,000× higher concentration. The apparent discrimination factor of this assay is thus approximately 100,000 × 32.5 / 1, or 3.25 million (Fig. 4D). This is far greater than the theoretical maximum for thermodynamic binding assays of any point mutation (Qmax,therm), and demonstrates the power of SiMREPS to discriminate between very closely related analytes, entirely without amplification.
Fig. 4.
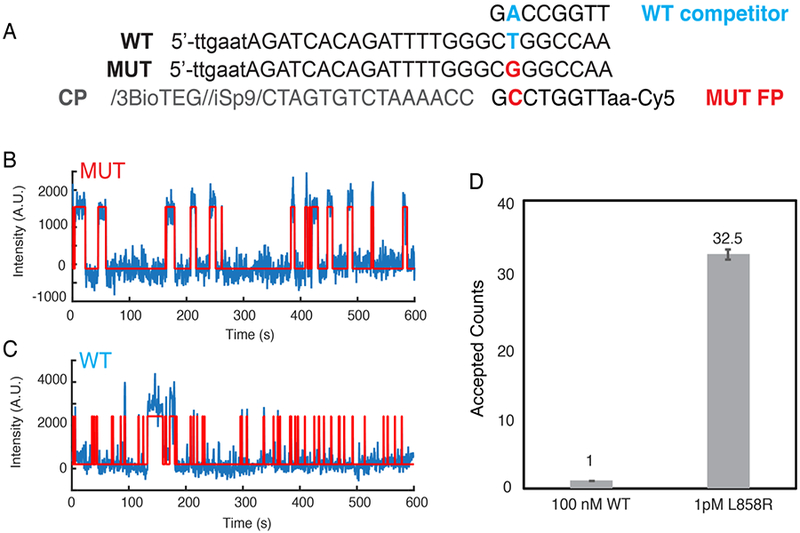
Single-base selectivity of SiMREPS. (A) Sequences of WT and L858R MUT targets, as well as the capture probe (CP), MUT fluorescent probe (FP) and WT competitor. (B) Representative intensity vs. time trace from MUT-only positive control. (C) Representative intensity vs. time trace from WT-only control. (D) The accepted counts after kinetic filtering of traces collected in the presence of 100 nM EGFR WT or 1 pM L858R MUT. The apparent discrimination factor between MUT and WT is 3.25 million.
4. Conclusions
We here have presented a workflow for the detection of nucleic acid targets by single-molecule kinetic fingerprinting through SiMREPS, and shown that this method affords detection of single analyte molecules with essentially no background (0–1 counts per FOV) in negative controls, even when challenged with a large concentration of closely related sequence. We further show that the single-base selectivity of the technique is sufficient to detect a mutation as subtle as a single T-to-G substitution with an apparent discrimination factor > 1 million, far in excess of any other amplification-free technique and comparable to the best available methods (i.e., droplet digital PCR). The ability of SiMREPS to accommodate very short (< 25 nt) analyte sequences, and those captured from crude biofluids with minimal processing, are unique advantages relative to most amplification-based methods. To make the technique more widely applicable and convenient, future improvements may include the use of techniques to improve mass transfer of analytes to the surface in order to increase the density of captured analyte, thus increasing sensitivity; modified probe or assay designs to permit more rapid cycling between bound and unbound states to shorten the imaging time needed to reach any desired level of specificity; and/or automated signal detection and counting algorithms. For instance, while published data here and elsewhere [23] indicate typical limits of detection of ~1 fM for passive analyte capture in our standard pipet-tip sample cells (Fig. 2c), further exploratory work suggests that attomolar detection limits may be achievable in the near future (data not shown); furthermore, in theory, even single-digit copy numbers could be detected with sufficiently high capture efficiency. Furthermore, there is no fundamental limit to the type of analyte that can accurately be detected and quantified using SiMREPS, making it a universal platform that - with further refinements - may transform biomarker detection just as super-resolution has conventional fluorescence microscopy.
ACKNOWLEDGEMENTS
The authors acknowledge support from a Michigan Economic Development Corporation MTRAC for Life Sciences grant to M.T., N.G.W., and A.J-B.; pilot grants from the University of the Michigan MCubed 2.0 program, the James Selleck Bower Permanently Endowed Innovative Promise Funds for Cancer Research of the University of Michigan Rogel Cancer Center, and the Fast Forward GI Innovation Fund to N.G.W. and M.T.; and NIH grant R21 CA204560 to N.G.W. and M.T. The authors also wish to thank X. Su for acquisition of the miR-16 data used in Fig. 3; S.L. Hayward and P.A. Lund for technical discussions regarding DNA detection as well as contributions to SiMREPS analysis code; and the Single Molecule Analysis in Real-Time (SMART) Center of the University of Michigan, seeded by NSF MRI-R2- ID award DBI-0959823 to N.G.W., as well as J.D. Hoff for training, technical advice and use of the objective-type TIRF microscope.
Abbreviations:
- SiMREPS
single-molecule recognition through equilibrium Poisson sampling
- ROI
region of interest
- FOV
field of view
- CP
capture probe
- FP
fluorescent probe
- LNA
locked nucleic acid
- PNA
peptide nucleic acid
- MUT
mutant
- WT
wild-type
- APTES
(3-aminopropyl)triethoxysilane
- OSS
oxygen scavenger system
- HMM
hidden Markov modeling
- sulfo-DST
sulfo-disuccinimidyltartrate
- Nb+d
number of binding and dissociation events
- P-TIRF
prism-type total internal reflection fluorescence
- OTIRF
objective-type total internal reflection fluorescence
- ICCD
intensified charge-coupled device
- EMCCD
electron-multiplying charge-coupled device
- sCMOS
scientific complementary metal oxide semiconductor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
The University of Michigan has filed patent applications related to the single-molecule kinetic fingerprinting approach described herein, on which A.J.B, M.T. and N.G.W. are co-inventors. A.J.B., M.T., and N.G.W. are co-founders of a startup company, Alight Sciences LLC, which seeks to commercialize this technology.
References
- [1].Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jäger N, Jones DTW, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdés-Mas R, van Buuren MM, van ‘t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Initiative APCG, Consortium IBC, Consortium IM-S, PedBrain I, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, and Stratton MR, Signatures of mutational processes in human cancer, Nature 500 (2013) 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Li M, Chen W, Papadopoulos N, Goodman SN, Bjerregaard NC, Laurberg S, Levin B, Juhl H, Arber N, Moinova H, Durkee K, Schmidt K, He Y, Diehl F, Velculescu VE, Zhou S, D. LA Jr, Kinzler KW, Markowitz SD, and Vogelstein B, Sensitive digital quantification of DNA methylation in clinical samples, Nat. Biotechnol 27 (2009) 858–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Murtaza M, Dawson S-J, Tsui DWY, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin S-F, Kingsbury Z, Wong ASC, Marass F, Humphray S, Hadfield J, Bentley D, Chin TM, Brenton JD, Caldas C, and Rosenfeld N, Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA, Nature 497 (2013) 108–112. [DOI] [PubMed] [Google Scholar]
- [4].Crowley E, Nicolantonio FD, Loupakis F, and Bardelli A, Liquid biopsy: monitoring cancer-genetics in the blood, Nat. Rev. Clin. Oncol 10 (2013) 472–484. [DOI] [PubMed] [Google Scholar]
- [5].Ansari J, Yun JW, Kompelli AR, Moufarrej YE, Alexander JS, Herrera GA, and Shackelford RE, The liquid biopsy in lung cancer, Genes Cancer 7 (2016) 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gazdar AF, Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors, Oncogene 28 (2009) S24–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, and Cui Q, An Analysis of Human MicroRNA and Disease Associations, PLOS ONE 3 (2008) e3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Schwarzenbach H, Hoon DSB, and Pantel K, Cell-free nucleic acids as biomarkers in cancer patients, Nat. Rev. Cancer 11 (2011) 426–437. [DOI] [PubMed] [Google Scholar]
- [9].Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O’Briant KC, Allen A, Lin DW, Urban N, Drescher CW, Knudsen BS, Stirewalt DL, Gentleman R, Vessella RL, Nelson PS, Martin DB, and Tewari M, Circulating microRNAs as stable blood-based markers for cancer detection, Proc. Natl. Acad. Sci 105 (2008) 10513–10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Iorio MV and Croce CM, MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review, EMBO Mol. Med 4 (2012) 143–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, Baker JC, Grützner F, and Kaessmann H, The evolution of lncRNA repertoires and expression patterns in tetrapods, Nature 505(2014)635–640. [DOI] [PubMed] [Google Scholar]
- [12].Prensner JR and Chinnaiyan AM, The Emergence of lncRNAs in Cancer Biology, Cancer Discov.1 (2011) 391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lee JT, Epigenetic Regulation by Long Noncoding RNAs, Science 338 (2012) 1435–1439. [DOI] [PubMed] [Google Scholar]
- [14].Potapov V and Ong JL, Examining Sources of Error in PCR by Single-Molecule Sequencing, PLoS ONE 12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen G, Mosier S, Gocke CD, Lin M-T, and Eshleman JR, Cytosine Deamination Is a Major Cause of Baseline Noise in Next-Generation Sequencing, Mol. Diagn. Ther 18 (2014) 587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, Nusbaum C, and Jaffe DB, Characterizing and measuring bias in sequence data, Genome Biol. 14 (2013) R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Akane A, Matsubara K, Nakamura H, Takahashi S, and Kimura K, Identification of the Heme Compound Copurified with Deoxyribonucleic Acid (DNA) from Bloodstains, a Major Inhibitor of Polymerase Chain Reaction (PCR) Amplification, J. Forensic Sci 39 (1994) 13607J. [PubMed] [Google Scholar]
- [18].Satsangi J, Jewell DP, Welsh K, Bunce M, and Bell JI, Effect of heparin on polymerase chain reaction, The Lancet 343 (1994) 1509–1510. [DOI] [PubMed] [Google Scholar]
- [19].Jin Z, Geißler D, Qiu X, Wegner KD, and Hildebrandt N, A Rapid, Amplification-Free, and Sensitive Diagnostic Assay for Single-Step Multiplexed Fluorescence Detection of MicroRNA, Angew. Chem. Int. Ed 54 (2015) 10024–10029. [DOI] [PubMed] [Google Scholar]
- [20].Kim K, Oh J-W, Lee YK, Son J, and Nam J-M, Associating and Dissociating Nanodimer Analysis for Quantifying Ultrasmall Amounts of DNA, Angew. Chem. Int. Ed 56 (2017) 9877–9880. [DOI] [PubMed] [Google Scholar]
- [21].Cohen L, Hartman MR, Amardey-Wellington A, and Walt DR, Digital direct detection of microRNAs using single molecule arrays, Nucleic Acids Res. 45 (2017) e137-e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang DY, Chen SX, and Yin P, Optimizing the specificity of nucleic acid hybridization, Nat. Chem 4 (2012) 208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Johnson-Buck A, Su X, Giraldez MD, Zhao M, Tewari M, and Walter NG, Kinetic fingerprinting to identify and count single nucleic acids, Nat. Biotechnol 33 (2015) 730–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sahl SJ, Hell SW, and Jakobs S, Fluorescence nanoscopy in cell biology, Nat. Rev. Mol. Cell Biol 18 (2017) 685–701. [DOI] [PubMed] [Google Scholar]
- [25].Wide Field of View | Yokogawa Electric Corporation. https://www.yokogawa.com/solutions/products-platforms/life-science/spinning-disk-confocal/csu-w1-confocal-scanner-unit/, 2018. (accessed: 16 May 2018).
- [26].Home - Oxford Nanoimaging. https://www.oxfordni.com/, 2018. (accessed: 16 May 2018).
- [27].Su X, Li Z, Yan X, Wang L, Zhou X, Wei L, Xiao L, and Yu C, Telomerase Activity Detection with Amplification-Free Single Molecule Stochastic Binding Assay, Anal. Chem 89 (2017) 3576–3582. [DOI] [PubMed] [Google Scholar]
- [28].Su X, Li L, Wang S, Hao D, Wang L, and Yu C, Single-Molecule Counting of Point Mutations by Transient DNA Binding, Sci. Rep 7 (2017) 43824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hayward SL, Lund PE, Kang Q, Johnson-Buck A, Tewari M, Walter NG, Ultra-specific and amplification-free quantification of mutant DNA by single-molecule kinetic fingerprinting, under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nucleic acid analogues. https://www.atdbio.com/content/12/Nucleic-acid-analogues, 2018. (accessed: 16 May 2018).
- [31].TM Prediction Tool. https://www.exiqon.com/ls/Pages/ExiqonTMPredictionTool.aspx, 2018. (accessed: 16 May 2018).
- [32].NUPACK: Analysis input. http://www.nupack.org/partition/new, 2018. (accessed: 16 May 2018).
- [33].OligoAnalyzer 3.1 | IDT. https://www.idtdna.com/calc/analyzer, 2018. (accessed: 16 May 2018).
- [34].Oligo Optimizer Tool. https://www.exiqon.com/ls/Pages/ExiqonOligoOptimizerTool.aspx, 2018. (accessed: 16 May 2018).
- [35].Dupuis NF, Holmstrom ED, and Nesbitt DJ, Single-Molecule Kinetics Reveal Cation-Promoted DNA Duplex Formation Through Ordering of Single-Stranded Helices, Biophys. J 105 (2013) 756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Blake RD and Delcourt SG, Thermodynamic effects of formamide on DNA stability., Nucleic Acids Res. 24 (1996) 2095–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cisse II, Kim H, and Ha T, A rule of seven in Watson-Crick base-pairing of mismatched sequences, Nat. Struct. Mol. Biol 19 (2012) 623–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dahse R, Berndt A, and Kosmehl H, PCR-based testing for therapy-related EGFR mutations in patients with non-small cell lung cancer, Anticancer Res. 28 (2008) 2265–2270. [PubMed] [Google Scholar]
- [39].Sanromán-Iglesias M, Lawrie CH, Liz-Marzán LM, and Grzelczak M, Nanoparticle-Based Discrimination of Single-Nucleotide Polymorphism in Long DNA Sequences, Bioconjug. Chem 28 (2017) 903–906. [DOI] [PubMed] [Google Scholar]
- [40].Auer A, Strauss MT, Schlichthaerle T, and Jungmann R, Fast, Background-Free DNA-PAINT Imaging Using FRET-Based Probes, Nano Lett. 17 (2017) 6428–6434. [DOI] [PubMed] [Google Scholar]
- [41].Abelson J, Blanco M, Ditzler MA, Fuller F, Aravamudhan P, Wood M, Villa T, Ryan DE, Pleiss JA, Maeder C, Guthrie C, and Walter NG, Conformational dynamics of single pre-mRNA molecules during in vitro splicing, Nat. Struct. Mol. Biol 17 (2010) 504–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Michelotti N, de Silva C, Johnson-Buck AE, Manzo AJ, and Walter NG, Chapter Six - A Bird’s Eye View: Tracking Slow Nanometer-Scale Movements of Single Molecular Nano-assemblies, in Methods in Enzymology, 475, Walter NG, Ed. Academic Press, 2010, 121–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Aitken CE, Marshall RA, and Puglisi JD, An Oxygen Scavenging System for Improvement of Dye Stability in Single-Molecule Fluorescence Experiments, Biophys. J 94 (2008) 1826–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].QUB - Markov Analysis. https://qub.mandelics.com/, 2018. (accessed: 16 May 2018).
- [45].Armbruster DA and Pry T, Limit of Blank, Limit of Detection and Limit of Quantitation, Clin. Biochem. Rev 29 (2008) S49–S52. [PMC free article] [PubMed] [Google Scholar]
- [46].Peterson EM and Harris JM, Identification of Individual Immobilized DNA Molecules by Their Hybridization Kinetics Using Single-Molecule Fluorescence Imaging, Anal. Chem 90 (2018) 5007–5014. [DOI] [PubMed] [Google Scholar]