

Abstract
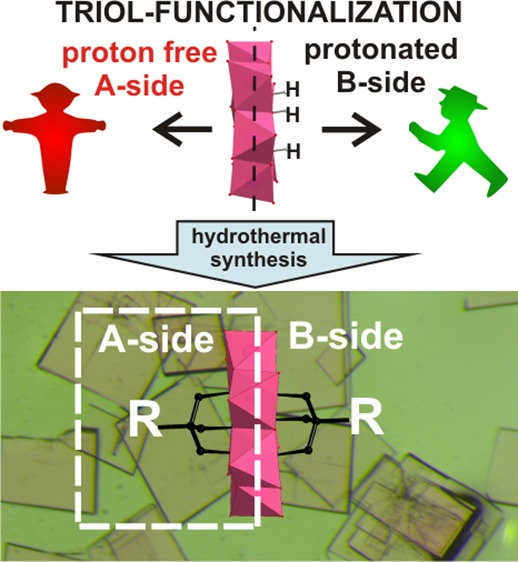
Since the first successful triol-functionalization of the Anderson polyoxometalates, the six protons of their central octahedron X(OH)6 (X—heteroatom, p- or d-element) have been considered as a prerequisite for their functionalization with tripodal alcohols, and therefore, the functionalization of Anderson structures from the unprotonated sides have never been reported. Here, we describe the triol-functionalization of [Cr(OH)3W6O21]6– leading to the single-side grafted anions [Cr(OCH2)3CRW6O21]6– (CrW6-tris-R, R = −C2H5, −NH2, −CH2OH) and the unprecedented double-side functionalized anion [Cr((OCH2)3CC2H5)2W6O18]3– (CrW6-(tris-C2H5)2), despite the lack of protons in the parent anion in the solid state. CrW6-(tris-C2H5)2 demonstrates the first example of double-side functionalized Anderson POT with the partially one-side protonated corresponding parent anion. The new heteropolytungstates were characterized by single-crystal X-ray diffraction, elemental analysis, Fourier-transform infrared spectroscopy, thermal gravimetric analysis, cyclic voltammetry, and electrospray ionization mass spectrometry. Density functional theory calculations were performed to investigate and compare the stability among the different isomers of the parent anion [Cr(OH)3W6O21]6–.
Short abstract
Since the first successful triol-functionalization of the Anderson polyoxometalates, the protons of the central octahedron X(OH)6 (X—heteroatom) have been considered as a prerequisite for their functionalization and therefore the functionalization of Anderson structures from the unprotonated sides have never been reported. Herein, for the first time, we organically functionalized the mixed-type Anderson polyoxometalate with real-time observation of hybrid anion formation.
Introduction
Anderson polyoxometalates (POMs) are a class of transformable discrete polynuclear metal-oxo clusters,1 which contain six edge-sharing pseudo-octahedrally coordinated MO6 units (M = Mo or W) surrounding a central heteroatom X (p- or d-element).2 The polyoxomolybdate (POMo) XMo6 system incorporates a greater variety of heteroatoms than the respective polyoxotungstate (POT) XW6 system, despite both being structurally identical. The physical and chemical properties of Anderson POMos and POTs are highly versatile and depend on the kind of the heteroatom, countercation, and its organic functionalization. The Anderson POMs are divided into two types according to the protonation state of the six triple-bridged oxygen atoms, μ3-O, connecting the heteroatom with the addenda atoms (Mo and W, respectively). The general formula of the non-protonated A-type is [Xn+M6O24](12–n)– with the heteroatom exhibiting its highest oxidation state (e.g., I7+,3 Sb5+,4 Te6+5), whereas the fully protonated B-type has the general formula [Xn+(OH)6M6O18](6–n)– with the heteroatom exhibiting lower oxidation states (e.g., Ni2+,6 Co2+,7 Fe3+,8 Al3+9).1 For a long time, the partial protonation (number of protons less than 6) of B-type Anderson POMos and POTs was known only for PtIV-centered anions, which usually do not possess an integer number of protons and tend to form dimeric structures via hydrogen bonds.10 Recently, the mixed type anion [Cr(OH)3W6O21]6– was reported by Kortz11 and our group;12 however, the existence of a heteropolyanion with a molar ratio Cr/W = 1:6 was predicted back in the 1970s.13 X-ray analysis and bond valence sum calculations showed that in solid state [Cr(OH)3W6O21]6– contains an unprotonated “A-side” and a protonated “B-side” (three protonated μ3-O atoms).11 Using an alternative synthetic procedure with a different component ratio (see the Supporting Information), Na6[Cr(OH)3W6O21] has been obtained with the same protonation state (Tables S10 and S11).
Among the various synthetic strategies for the organic functionalization of POMs,14 alkoxylation has gained much attention because of the diversity and tunability of alkoxyl ligands, especially when using the disk-shaped Anderson type anions with a wide spectrum of central heteroatoms.15 The resulting hybrid materials showed interesting application in the synthesis of bioinorganic hybrid materials,16a,16b supramolecular self-assembly,16c charge storage,16d metal–organic frameworks,16e nanostructured materials16f and photochemistry.16g Since the first tris-alkoxo functionalized [RC(CH2OH)3, R = e.g., −NH2, −OH, −CH2OH, etc.] Anderson-type hybrid POMos were published,17 there was a clear conviction that only B-type (protonated) POMs can be tris-functionalized as the incorporation of tris-ligands represents a nominal dehydrative condensation reaction,17 where the protons of the POM’s hydroxo groups are putatively replaced by carbon atoms of the organic ligand. Moreover, even in the case of the formation of a χ Anderson isomer, in which the ligand is anchored on two μ3-O atoms and one μ2-O atom, the addition of a stoichiometric amount of protons for the protonation of the μ2-O atom was considered necessary.18 In contrast to the B-type clusters, tris-functionalized A-type anions have never been reported, but when the [IMo6O24]5– POMo is refluxed under acidic conditions, it becomes protonated and can be methylated, yielding the methoxyl-derivative [IMo6O23(μ2-OMe)]4–.19 Interestingly, the protonation in [IMo6O24]5– occurs rather at the μ2-O atoms than at the μ3-O sites, despite the former being usually less basic. The potential reactivity of the unprotonated μ3-O atoms of A-type Anderson POTs to undergo functionalization was shown during the crystallization of aurone synthase from Coreopsis grandiflora (cgAUS1)20 using [TeW6O24]6– (TEW) as additive because the POT formed a covalent bond with a glutamic acid via two of its μ3-O atoms, yielding the first proteinogenically functionalized [TeW6O24O2(Glu)]7– anion.21
Tris-functionalized Anderson POMos, which make up a lion’s share in alkoxylation chemistry of Anderson-type POMs, are mainly synthesized from octamolybdate [Mo8O26]4– in organic solvent or from the parent Anderson anion in aqueous solution under reflux.2,14,22 Interestingly, the role of the protons is considered important for both synthetic routes, but their importance is even emphasized in the latter case, namely, during the self-assembly reaction starting from octamolybdate in the presence of the heteroatom and tris-ligand.17 Hydrothermal reaction, which has been proven as an effective method for the preparation of POMs is rarely utilized for alkoxylation but was successfully applied for the tris-modification of the fully protonated B-type POMo [Cr(OH)6Mo6O18]3–.23 Depending on the stoichiometric ratio between the parent anion and the triol-ligand, both single- and double-side POMos were obtained.23
Attempts to obtain organically functionalized Anderson-type POTs were unsuccessful for a long period because of the inertness of the bridging oxygen atoms in the {WO6} octahedra. However, in 2016, we demonstrated that the B-type anion [Ni(OH)6W6O18]4– can be covalently modified from one side with pentaerythritol CH2OH–C(CH2OH)3 using an acidified aqueous solution with excess of triol-ligand.12 More recently, Li and Wei extended the family of single-side functionalized Anderson-type POTs by AlIII-, CrIII-, and CoIII-containing structures, which were obtained via direct assembly of the preformed heteroatom-tris-ligand complex and ortho- or ditungstate in aqueous solution.24 Despite using a fourfold excess of the triol-ligand, the authors claim that the X(H2O)3(RC(CH2O)3) (X = Al3+, Co3+, Cr3+; R = −CH2OH, CH3) complexes are formed, which then react with the tungstates. The parent inorganic Anderson structures containing AlIII and CoIII are not reported yet; however, the authors made a conclusion based on the functionalization mode that these structures should be three-protonated from one side as the [Cr(OH)3W6O21]6– anion. Note, that all four existing functionalized Anderson POTs were decorated from one side by aqueous one-pot reactions.
To investigate the possibility of triol-ligand attachment to the mixed-type anion [Cr(OH)3W6O21]6–, which in a solid state demonstrates protonated and proton-free sides, we explored hydrothermal synthesis for the alkoxylation of POTs. Herein, we report for the first time the successful double-sided tris-functionalization of an Anderson-type POM, which is not fully protonated in the pure inorganic parent form, yielding [Cr((OCH2)3CC2H5)2W6O18]3– (CrW6-(tris-C2H5)2). Furthermore, the proposed controlled hydrothermal synthesis allows the synthesis of the single-side functionalized CrIII-centered anions [Cr(OCH2)3CC2H5W6O21]6– (CrW6-tris-C2H5), [Cr(OCH2)3CNH2W6O21]6– (CrW6-tris-NH2), and [Cr(OCH2)3CCH2OHW6O21]6– (CrW6-tris-CH2OH). Among the five reported functionalized Anderson POTs [Ni(OH)3W6O18(OCH2)3CCH2OH]4–,12 [XO3W6O18(OCH2)3CCH2OH]6– (X = Al3+, Co3+, Cr3+),24 and [CoO3W6O18(OCH2)3CCH3]6–,24CrW6-(tris-C2H5)2 represents the first example of a double-side functionalized Anderson POT, which is usually more stable and could therefore be an excellent building block candidate for the design of novel metal oxide-based materials.
Experimental Section
Physical Methods
Infrared spectra (4000–400 cm–1) of all samples were recorded on a Bruker Vertex 70 IR Spectrometer equipped with a single-reflection diamond-ATR unit. Elemental analysis was performed with an iCAP 6500 series inductively coupled plasma-optical emission spectrometry (ICP-OES) spectrometer (Thermo Scientific, USA). The ICP-OES was equipped with a standard sample introduction system consisting of a concentric nebulizer and a cyclonic spray chamber. Transportation of sample solutions was performed by the peristaltic pump of the iCAP 6500 coupled to an ASX-520 auto sampler (Cetac, USA). Per element two sensitive and noninterfered emission lines were used, the first line for measurement and the second line for quality control. The water content was determined by thermal gravimetric analysis (TGA) with a Mettler SDTA851e thermogravimetric analyzer under nitrogen flow with a heating rate of 5 K min–1 in the region 298–1023 K. Mass spectrometry (MS) was performed with an electrospray ionization (ESI)-Qq-oaRTOF supplied by Bruker Daltonics Ltd. Bruker Daltonics Data Analysis software was used to analyze the results. The measurement was carried out in a 1:1 mixture of water/MeCN, collected in negative ion mode and with the spectrometer calibrated with the standard tune-mix to give an accuracy of ca. 5 ppm in the region of m/z 300–3000. The cyclic voltammetry measurements were carried out using a HEKA PG 390 potentiostat at room temperature. A conventional three-electrode glass cell of 3 mL capacity was used. A 2 mm diameter glassy carbon disk electrode was used as working electrode (GCE). A platinum wire served as the counter electrode and a normal hydrogen electrode served as the reference electrode. All solutions were deoxygenated using argon gas for 10–15 min prior to electrochemical experiments. X-ray powder diffraction was performed on a Bruker D8 ADVANCE diffractometer, Cu Kα radiation, λ = 1.54056 Å, LYNXEYE silicon strip detector and SolX energy dispersive detector, variable slit aperture with 12 mm, 8° ≤ 2θ ≤ 50°.
X-ray Diffraction on Single Crystals
The X-ray data were measured on a Bruker D8 VENTURE equipped with a multilayer monochromator, Mo Kα Incoatec Microfocus sealed tube, and Kryoflex cooling device. The structure was solved by direct methods and refined by full-matrix least-squares. Nonhydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were inserted at calculated positions and refined with riding coordinates. The following software was used for the structure-solving procedure: frame integration, Bruker SAINT software package25 using a narrow-frame algorithm (absorption correction), SADABS26 (structure solution), SHELXS-201327 (refinement), SHELXL-2013,27 OLEX2,28 and SHELXLE29 (molecular diagrams). Experimental data and CCDC-codes can be found in Table S10.
Computational Methods
All density functional theory (DFT) calculations were carried out using Gaussian (G16 rev. B01).30 The geometries and vibrational modes of all molecules were computed using unrestricted DFT using the hybrid exchange–correlation functional PBE0,31,32 the def2-TZVP33 basis set, and implicit aqueous solvation through application of the polarizable continuum model,34 as this level of theory has been demonstrated to work well in predicting bond distances, nuclear magnetic resonance shifts, and relative protonation energies.35,36 An ultrafine integration grid with extra-tight convergence criteria was used in all computations. In addition, DFT calculations for the three isomers of [Cr(OH)3W6O21]6– were also performed using PBE37 and B3LYP38,39 as exchange–correlation functionals to gauge the influence of the functional, and the relative enthalpies between the three different methodologies differ by less than 1 kcal/mol (see Table S13). For all Cr(III) species, a multiplicity of 4 was assumed. Spin contamination was found to be less than 10% for all species. Energies and optimized geometries are provided as the Supporting Information.
Syntheses
All materials were purchased from Sigma-Aldrich and used without further purification.
Synthesis of Na3[Cr((OCH2)3CC2H5)2W6O18]·13H2O (Na3CrW6-(tris-C2H5)2)
Na2WO4·2H2O (6.6 g, 20 mmol) was dissolved in 80 mL of water before adding 20 mL of HCl (1.0 M). Cr(NO3)3·9H2O (1.33 g, 3.3 mmol) was dissolved in water (10 mL) and added dropwise to the acidified solution of Na2WO4. The molar ratio of Cr3+/WO42–/H+ in the aqueous solution was 1:6:6 (Cr/W/H+) corresponding to the maximum amount of [Cr(OH)3W6O21]6– (3.33 mmol). The reaction mixture of green color was heated for 1 h at 80 °C, filtered, and kept at room temperature for 5 days. The pH of filtered solution was 5.7. 1,1,1-Tris(hydroxymethyl)propane (tris-C2H5) (0.27 g, 2 mmol) was added to 20 mL of the initial solution (max 0.67 mmol of [Cr(OH)3W6O21]6–) and was sealed in a 50 mL stainless steel reactor with a Teflon liner and heated at 160 °C for 48 h. After the reaction mixture was cooled to room temperature, the pink crystals of Na3CrW6-(tris-C2H5)2 appeared within 3 weeks. Yield: 0.93 g (69% based on W). Elemental analysis found (calculated) for C12H48CrNa3O37W6: Na 4.22 (3.43), Cr 2.62 (2.59), W 55.30 (54.92). IR (see Figure S1 in the Supporting Information): 3399 (b), 2934 (w), 2872 (w), 1652 (m), 1455 (m), 1357 (m), 1110 (m), 1058 (w), 1007 (m), 957 (m), 936 (m), 903 (s), 889 (s), 835 (w), 771 (w), 668 (s), 559 (m), 527 (m), 470 (m), 428 (m) cm–1.
Synthesis of Na3K3[Cr((OCH2)3CC2H5)W6O21]·17H2O (Na3K3CrW6-tris-C2H5)
The synthesis of Na3K3CrW6-C2H5 is similar to that of Na3CrW6-(tris-C2H5)2, with the exception that the reaction mixture was heated at 140 °C for 48 h. The blue-green crystals appeared in 1 week after the addition of KCl solution (1.0 M). Elemental analysis found (calculated) for Na3K3CrW6O48C6H45: Na 3.26 (3.10), K 5.56 (5.27), Cr 2.46 (2.34), W 52.16 (49.54), O 31.02 (34.49). Yield: 0.62 g (57% based on W). IR (see Figure S1 in the Supporting Information): 3333 (b), 1613 (m), 1480 (m), 1375 (m), 1113 (m), 1045 (w), 933 (m), 869 (s), 765 (w), 659 (s), 513 (m), 435 (s) cm–1.
Synthesis of Na5[Cr((OCH2)3CNH3)W6O21]·17H2O (Na5CrW6-tris-NH3)
The synthesis of Na5CrW6-tris-NH3 is similar to that of Na3K3CrW6-tris-C2H5 with the exception that tris(hydroxymethyl)aminomethane (tris-NH2) was used instead of 1,1,1-tris(hydroxymethyl)propane. The pH of the reaction mixture after tris-NH2 addition increased to 7.7 and after reaction was 7.3. Yield: 0.72 g (65% based on W). Elemental analysis found (calculated) for Na5CrW6O41C4NH44: Na, 6.30 (5.66); Cr, 2.51 (2.56); W, 59.1 (54.27); O, 29.53 (32.28). IR (see Figure S1 in the Supporting Information): 3333 (b), 3035 (w), 1634 (m), 1120 (m), 1072 (w), 1032 (w), 933 (m), 865 (s), 644 (s), 521 (m), 439 (s) cm–1.
Synthesis of Na4(TMA)2[Cr((OCH2)3CCH2OH)W6O21]·19H2O (Na4(TMA)2CrW6-tris-CH2OH, TMA = Tetramethylammonium)
The synthesis of Na4(TMA)2CrW6-tris-CH2OH is similar to that of Na3K3CrW6-tris-C2H5, with the exception that pentaerythritol (tris-CH2OH) was used instead of 1,1,1-tris(hydroxymethyl)propane. After the reaction, tetramethylammonium chloride (0.3 g, 2.5 mmol) was added to induce crystallization. Colorless crystals of [TMA]6[W12O38(OH)2]·9H2O appeared within 2 days, exhibiting the following cell parameters a = 12.9872(5) Å, b = 21.0354(9) Å, c = 13.1053(5) Å, α = γ = 90°, and β = 91.6841(2)°. Evaporation of the green-blue filtrate at air led to the formation of blue block-shaped crystals within 2–3 weeks. Yield: 0.68 g (61% based on W). Elemental analysis found (calculated) for Na4CrW6O44N2C13H71: Na, 4.32 (4.17); Cr, 2.44 (2.36); W, 51.77 (49.99); O, 32.29 (31.90). IR (see Figure S1 in the Supporting Information): 3322 (b), 3025 (w), 2873 (w), 1636 (m), 1486 (m), 1118 (m), 1067 (w), 1031 (w), 932 (m), 862 (s), 646 (s), 522 (m), 438 (s) cm–1.
Results and Discussion
Synthesis and Structures
Initially, an aqueous solution of Cr3+–WO42– (C = 0.2 M)–H+ was prepared at a molar ratio of 1:6:6 (Cr/W/H+) and a final pH of 5.7. The reaction mixture was kept for 3 days under ambient conditions to achieve equilibrium in the solution. Because the initial solution containing Cr3+–WO42––H+ had a lower pH compared with the values reported for the synthesis of the parent anion [Cr(OH)3W6O21]6– (pH ≈ 8),11,12 ESI-MS was used to aid elucidation of the speciation in solution. The ESI-MS spectra (see Figure S11, Table S8, Supporting Information) of the solution with Cr3+–WO42––H+ measured after 1, 3, and 10 days at pH 5.7 are the same and are dominated by peaks which can be assigned to isopolytungstate fragments ([WmO3m+1]2–, H[WmO3m+1]− or Na[WmO3m+1]−, where m = 1–6). Decomposition fragments of [Cr(OH)3W6O21]6– (e.g., [CrW2O8]−, [CrW3O11]−, Na[CrW4O15]2–, [Cr(OH)3W4O16]−, [CrW5O18]3–, and NanH1–n[CrW5O18]2–; n = 0 or 1) are also present in the spectra. It should be noted that the relatively low pH of the initial solution does not lead to the formation of the elusive fully protonated B-type hexatungstochromate, and none of the spectra contains signals at m/z = 515.19, 773.29, or 784.27, where the fully protonated species [Cr(OH)6W6O18]3–, H[Cr(OH)6W6O18]2–, and Na[Cr(OH)6W6O18]2– would have occurred.
A threefold excess of the tris-ligand was added to 20 mL of the respective stock solution prior to the hydrothermal reaction. The single-side functionalization of the [Cr(OH)3W6O21]6– is possible under hydrothermal conditions at a synthesis temperature of 140 °C leading to the formation of CrW6-tris-R (R = −C2H5, −NH2, −CH2OH). At 150 °C, the crystallization of a mixture of single- and double-side decorated anions is observed, whereas at 160 °C, the crystallization of solely the double-functionalized product CrW6-(tris-C2H5)2 occurs (Scheme 1). A further increase in the synthesis temperature leads to the decomposition of the ligand. This synthetic route indicates that the temperature is the key factor in the formation of the double-side product. All hybrid POTs were crystallized as their alkali salts, namely, Na3CrW6-(tris-C2H5)2·13H2O (CCDC 1842741), K3Na3CrW6-tris-C2H5·17H2O (CCDC 1842742), Na5CrW6-tris-NH3·17H2O (CCDC 1842739), and Na4(C4H12N)2CrW6-tris-CH2OH·19H2O (CCDC 1842740). The successful double-sided functionalization can be followed visually by the color of the crystals, as single-side alkoxo-decorated compounds are of pine green color, while the double-side decorated Na3CrW6-(tris-C2H5)2·13H2O exhibits a pink color, which is characteristic for anhydrous chromium(III) salts indicating a different Cr3+ coordination sphere compared with CrW6-tris-R.
Scheme 1. Synthesis of CrW6-(tris-C2H5)2 and CrW6-tris-R (R = −C2H5, −CH2OH, −NH2); HT = Hydrothermal Synthesis; Color Code: WO6, Pink Octahedra; Cr, Green; O, Red; C, Black; H, White.
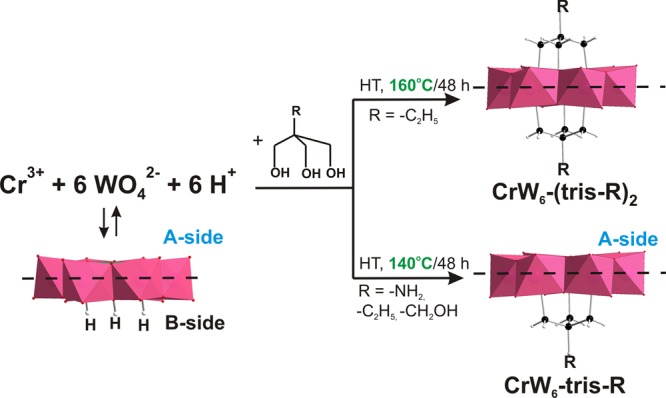
All structures are showed based on the single-crystal X-ray analysis (CCDC for CrW6-tris-C2H5 1842742; CrW6-tris-CH2OH 1842740, CrW6-tris-NH2 1842739, CrW6-(tris-C2H5)2 1842741, CSD for [Cr(OH)3W6O21]6– 1870211) and ESI-MS.
Applying the proposed method, [Cr(OH)3W6O21]6– can be asymmetrically functionalized in two steps, where two different organic groups are selectively grafted on each side. The reaction of CrW6-tris-C2H5 with tris-CH2OH at 160 °C leads to the formation of the asymmetric product CH2OH-tris-CrW6-tris-C2H5 that was proven with ESI-MS (Figure S9, Table S6). The possibility of the asymmetric step by step functionalization of [Cr(OH)3W6O21]6– proves the crucial role of the synthesis temperature.
Because the introduced organic ligands present active functional groups, Anderson-type hybrids can be utilized as precursors for further post-functionalization via imine, amide, or ester bond formation. The post-functionalization of CrW6-tris-NH2 with cinnamic acid in acetonitrile was performed using a well-established protocol.2 The successful formation of a new hybrid can be seen in the ESI-MS spectrum of the reaction mixture (Figure S10, Table S7).
Both double-side and single-side decorated compounds show the characteristic Anderson-type structure with six edge-shared WO6 arranged hexagonally around the central CrO6. The Cr–O bond lengths are in the range of 1.947(6)–1.969(5) Å for CrW6-(tris-C2H5)2, 1.964(7)–1.977(8) Å for CrW6-tris-C2H5, 1.956(4)–1.993(4) Å for CrW6-tris-NH2, and 1.960(1)–1.997(9) Å for CrW6-tris-CH2OH, which is in good agreement with the corresponding bond lengths of the parent Anderson-anion [Cr(OH)3W6O21]6–.11 Structural analysis indicates that the bond lengths of all three types of W–O bonds (W−μ2-O, W−μ3-O, and W=Oterminal) are also close to those of the parent species [Cr(OH)3W6O21]6–11 and corresponding POT analogues.24
Mechanistic Proposal
For real-time observation of the self-assembly of CrW6-(tris-C2H5)2 under hydrothermal condition, aliquots for ESI-MS of the reaction solution were taken after 12, 24, and 48 h from the moment the reaction had been started. The measured spectra for all time intervals contain the same peaks with slightly different intensities, which can be assigned to the target compound CrW6-(tris-C2H5)2 (m/z = 568.6 for triple-charge, m/z = 853.4 and 864.4 for double-charged), the single-side decorated CrW6-tris-C2H5 anions, their decomposition fragments, and isopolytungstate fragments with lower nuclearity ([WmO3m+1]2–, H[WmO3m+1]− or Na[WmO3m+1]−, where m = 1–4) (see Figure S12, Table S9, Supporting Information). The absence of signals at m/z = 515.19, 773.29, or 784.27 is assigned to [Cr(OH)6W6O18]3–, H[Cr(OH)6W6O18]2–, and Na[Cr(OH)6W6O18]2–. According to the ESI-MS spectra recorded from the reaction solution, all the Cr-containing anions are coordinated to triol-ligand, pointing toward the alkoxylation mechanism proposed by Li and Wei for single-side functionalized structures,24 according to which free Cr3+ ions and ligand molecules first formed the complex {Cr((OCH2)3CR)2} before further reacting to the functionalized anion (Scheme 2A).
Scheme 2. Proposed Mechanism for the Formation of CrW6-(tris-C2H5)2: (A) via the Formation of {Cr(RC(CH2O)3)2} Complex; (B) via the Protonation of μ3-O Atoms; Color Code: WO6, Pink Octahedra; CrO6, Green Octahedron; Cr, Green; O, Red; C, Black; H, White.
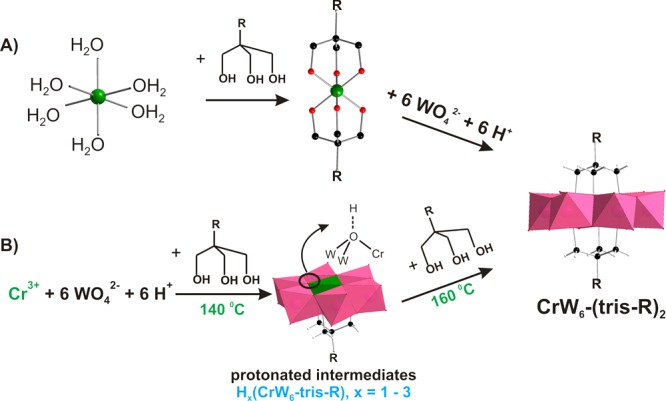
All structures are shown based on the single-crystal X-ray analysis (CCDC for CrW6-tris-C2H5 1842742; CrW6-tris-CH2OH 1842740, CrW6-tris-NH2 1842739, CrW6-(tris-C2H5)2 1842741, CSD for [Cr(OH)3W6O21]6– 1870211) and ESI-MS.
DFT calculations were carried out to determine the most likely protonation sites and proton distribution of [CrW6O24]9– (CrW6) in aqueous solution. The non-protonated anion CrW6 was optimized, and the most nucleophilic O atoms according to their molecular electrostatic potential were found to be the μ3-O atoms (see Figure S14). Moreover, the enthalpies of the [Cr(OH)1W6O23]8– isomers were computed for the states when the proton is localized on a triply-bridging oxygen (μ3-O), doubly-bridging oxygen (μ2-O), or terminal oxygen (Ot) atom. The relative enthalpies (see Table S12) show that the triply-bridging (μ3-O) sites are the most probable ones to be protonated, in agreement with the existing experimental data. To investigate and compare the formation energies of the different protonation states in [Cr(OH)xW6O24–x](9–x)– (x = 0–6), the different protonation states and isomers of protonation states were optimized by DFT and enthalpies computed through normal mode analysis. The different isomers of the each protonation state of [Cr(OH)xW6O24–x](9–x)– (x = 0–6) were structurally optimized and the relative energies computed as protonation enthalpies (Tables S14–S16). The only isolated11,12 in solid-state isomer [Cr(OH)3W6O21]6– has three possible different positional isomers, namely, two fac isomers (one with all protons on the same side (fac1) and one with a proton on the opposite side (fac2) of the anion) and one mer isomer (mer) (Figure 1). The relative enthalpies of these isomers are shown in Figure 1 and reveal that the fac1-[Cr(OH)3W6O21]6– (three protonated μ3-O atoms from one side) is lower in energy than the other two isomers by 4 and 8 kcal/mol, respectively, suggesting that at the very least the fac1-isomer is the dominant form of the tri-protonated molecule.
Figure 1.
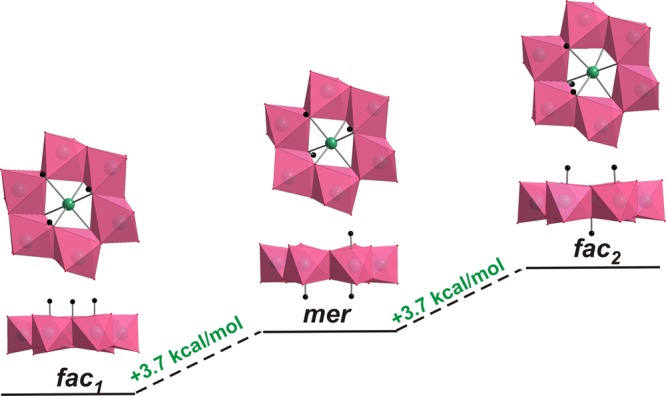
Relative enthalpies (ΔHrel) of three positional isomers of [Cr(OH)3W6O21]6– with respect to the fac1 isomer. Color code: WO6, pink octahedra; Cr, green; O, red; H, black.
The computed bond distances in the optimized fac1-[Cr(OH)3W6O21]6– structure agree well with those observed experimentally (see Supporting Information Figure S12, Table S10 and CSD 1870211) in square brackets for [Cr(OH)3W6O21]6–, with W=Ot 1.740–1.752 Å [1.723–1.765 Å], W−μ2-O 1.931–1.951 Å [1.919–1.965 Å], W−μ3-O 2.109–2.399 Å [2.094–2.303 Å], Cr–O 1.967–2.008 Å [1.964–2.003 Å] and W···W 3.194–3.435 Å [3.149–3.375 Å].
The estimated enthalpies of protonation (Table 1) were calculated based on a reaction with aqueous H+, using the absolute aqueous enthalpy of the proton as determined by Coe and co-workers40
| 1 |
Table 1. Enthalpies of Stepwise Protonation of [CrW6O24]9– According to Eq 1a.
| reactant | product | ΔH (kcal/mol) |
|---|---|---|
| [CrW6O24]9– | [Cr(OH)1W6O23]8– | –49.5 |
| [Cr(OH)1W6O23]8– | [Cr(OH)2W6O22]7– | –43.4 |
| [Cr(OH)2W6O22]7– | [Cr(OH)3W6O21]6– | –36.9 |
| [Cr(OH)3W6O21]6– | [Cr(OH)4W6O20]5– | –22.8 |
| [Cr(OH)4W6O20]5– | [Cr(OH)5W6O19]4– | –15.4 |
| [Cr(OH)5W6O19]4– | [Cr(OH)6W6O18]3– | –7.01 |
In the cases where several protonated isomers exist, only the enthalpy of the most stable isomer of each protonated species is given.
The enthalpy of protonation decreases by 40 kcal/mol when proceeding from the unprotonated to the penta-protonated anion, reflecting at least in part the decreasing charge of the cluster ion. The estimation with sufficient accuracy of the absolute pKa’s of the different isomers using computed free energies of protonation is often difficult, even for organic molecules and is not attempted here. It is, however, interesting to note that while the protonation enthalpy changes by ca. +6–7 kcal/mol for each added proton in most cases, the addition of a proton to the tri-protonated species is +14 kcal/mol larger than the addition of a proton to the di-protonated species, which may well give an indication as to why in particular the tri-protonated species is generally observed experimentally.
Mizuno et al.41 proposed a mechanism for the alkoxylation of lacunary Keggin anions [A-α-XW9O34]4– (X = Si, Ge) involving the instantaneous protonation of oxygen atoms followed by a dehydrative condensation reaction, which could also be a possible mechanism for the formation of CrW6-(tris-C2H5)2 (Scheme 2B) considering the fact that mono- and di-protonated CrW6-tris-C2H5 were present in the ESI-MS spectra (m/z = 835.3 for Na2H2[Cr(OCH2)3CC2H5W6O21]2– and 845.8 for Na3H[Cr(OCH2)3CC2H5W6O21]2–) (see Figure S12, Table S9, Supporting Information) and that protonation of [Cr(OH)xW6O24–x](9–x)– (x = 0–6) is likely to be exothermic under most conditions according to DFT protonation enthalpies (Table 1). Both suggested mechanisms overcome the putative impossibility of functionalizing A-type Anderson POMs.
Electrospray Ionization Mass Spectrometry
Further evidence for the existence of the intact CrW6-(tris-C2H5)2 (Figure 1) and CrW6-tris-R (R = −C2H5, −NH2, −CH2OH) (see the Supporting Information) in solution and their exact stoichiometric composition was obtained from ESI-MS. The most intense peak at m/z = 568.6 (calcd m/z = 568.6) in the ESI-MS spectrum of CrW6-(tris-C2H5)2·13H2O (Figure 2) corresponds to the triply charged anion [Cr((OCH2)3CC2H5)2W6O18]3– (CrW6-(tris-C2H5)2). The peaks of the doubly charged H[Cr((OCH2)3CC2H5)2W6O18]2– and Na[Cr((OCH2)3CC2H5)2W6O18]2– appeared at m/z = 853.4 (calcd m/z = 853.4) and m/z = 864.4 (calcd m/z = 864.4), respectively. The double-side capping effect enhances the stability of the complex and leads to an almost complete absence of decomposition fragments, which are usually unavoidable in POM MS spectra.42 The ESI-MS spectra of the CrW6-tris-R structures represent a more complex character exhibiting a peak envelope, which can be unambiguously assigned to the doubly-charged anions Na4–xHxCrW6-tris-R2– (x = 1–3; see Figures S6–S8, Tables S3–S5, Supporting Information) indicating the presence of the intact one-side grafted clusters in solution. A series of peak envelopes in the rest of the spectra could be assigned to different common POTs fragments (HWO4–, W2O72–, HW2O7–, NaW2O7–, W3O102–, HW3O102–, etc.) and some more specific decomposition products, of which the most intensive are [CrW4O13(OCH2)3CR]2–, [CrW5O16(OCH2)3CR]2–, and H[CrW3O10(OCH2)3CR]− (Figures S6–S8, Tables S2–S4, Supporting Information).
Figure 2.
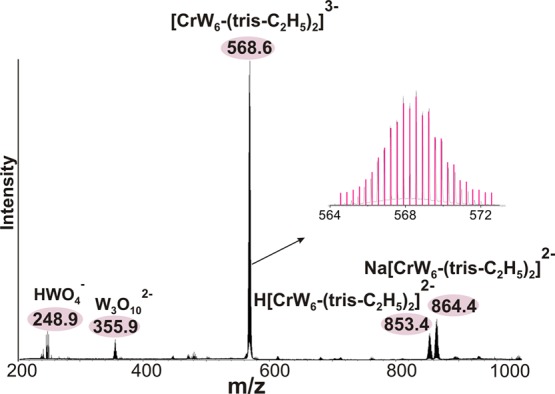
ESI mass spectrum of Na3CrW6-(tris-C2H5)2 in mixed CH3CN/MeOH/H2O using the negative mode. The envelope of CrW6-(tris-C2H5)2 is shown in black with the corresponding simulated spectra shown in pink.
Electrochemistry
The electrochemical behaviors of the single-side functionalized CrW6-tris-C2H5 and double-side functionalized CrW6-(tris-C2H5)2 have been studied by cyclic voltammetry (Figure S13). For CrW6-tris-C2H5, one irreversible oxidation wave is observed at 0.480 V and attributed to the CrVI/III couple.43 In the cathodic domain, two successive reductions at −0.259 and −0.455 V have been measured (Figure S13A), corresponding to the reduction of the central Cr ion (CrIII/II and CrII/0 couples).43 The cyclic voltammogram of CrW6-(tris-C2H5)2 does not exhibit either oxidative or reduction processes (Figure S13B). The more concealed position of Cr in the double-side decorated anion most likely affects the absence of redox waves in the case of CrW6-(tris-C2H5)2. The absence of reduction processes for tungsten in both cases is caused by the fact that Anderson anions represent type II POMs44,45 with two terminal oxo ligands per W atom, which lacks a nonbonding orbital for electrons acceptance.
IR Spectroscopy
IR spectroscopy was applied to investigate the anion in Na3CrW6-(tris-C2H5)2·13H2O, K3Na3CrW6-tris-C2H5·17H2O, Na5HCrW6-tris-NH2·17H2O, and Na4(C4H12N)2CrW6-tris-CH2OH·30H2O (see Figure S1, Table S1, Supporting Information). The characteristic peaks of the core structure in CrW6-(tris-C2H5)2 and CrW6-tris-R (R = −C2H5, −NH2, −CH2OH) are all in agreement with the peaks observed in the spectrum of Na6[Cr(OH)3W6O21]·22H2O.11 The stretching vibrations of the terminal W=O units appears within the range of 936–932 cm–1. The peaks at approximately 865 cm–1 and in the region of 420–800 cm–1 correspond to the antisymmetric and symmetric deformation vibrations of W–O–W and W–O–Cr bridging fragments. The peaks appearing at ∼1100 and 1050 cm–1 could be assigned to C–O stretching vibrations, indicating the successful grafting of tris-ligands. The spectrum of CrW6-(tris-C2H5)2 is slightly different from that of the single-functionalized products, which is associated with their lower symmetry.
Conclusions
In conclusion, for the first time, we organically functionalized the Anderson POT from two sides and showed that the double-side functionalization is possible even in the case when the parent inorganic anion state exhibits only three protons in the solid. It was generally accepted that it is not possible to graft triol ligands onto the Anderson anions, which represent unprotonated μ3-O atoms in their structures as confirmed by single crystal X-ray analysis. By varying the synthesis temperature, both the double- and single-side tris-modified Cr-centered Anderson POTs have been obtained in good yields (>60%). DFT studies revealed the fac1-[Cr(OH)3W6O21]6–-isomer (three protons attached from one side) as the most stable form of all triply protonated isomers, which is therefore likely to dominate over the other two possible isomers (mer and fac2) in solution. This finding supports the fact that only the three-protonated [Cr(OH)3W6O21]6– and the unprotonated [CrW6O21(OCH2)3CR]6–CrW6-tris-R can be isolated in solid state. DFT in itself does not claim to conclusively predict that the degree of protonation is going to be the major one under the synthetic conditions but it provides general insights into possible protonation states of CrW6 anion. The DFT studies together with the real-time ESI-MS experiments suggest two principal mechanisms of CrW6-(tris-C2H5)2 formation, which include either potential {triol-Cr3+} complexes or protonated HxCrW6-tris-C2H5 (x = 1–3). The new compound CrW6-(tris-C2H5)2 can be seen as a “game changer” for alkoxylation of A-type Anderson POTs, exhibiting new properties that differ from those of the already existing hybrids based on the protonated Anderson archetype. This can be expected because the physical and chemical properties of Anderson POMs highly depend on the nature of the heteroatom, which for A-type Anderson POTs and POMos are highly charged p- and d-elements. Applying the proposed synthetic way, mixed-type Anderson POMs can be asymmetrically functionalized in two steps, where two different organic groups are selectively grafted on each side. The asymmetric product formation was proven by ESI-MS. In the future, asymmetrically functionalized Anderson derivatives can be introduced into oligomers in a controlled manner by, for example, applying click-chemistry reactions.
Acknowledgments
The research was funded by the Austrian Science Fund (FWF): M2203 (N.I.G.) and P27534 (A.R.) and the University of Vienna. The authors are grateful to Mag. A. Ronacher and Mag. A. Fabisikova for the support with the ESI-MS measurements at the MS Centre, Univ. of Vienna. We thank Ao.Uni.-Prof. C. L. Lengauer for TGA measurements at the Institut für Mineralogie und Kristallographie, Univ. of Vienna and Assoc.-Prof. Dr. Andreas Limbeck for ICP-OES measurements at the Institute of Chemical Technology and Analytics, Vienna Univ. of Technology. Last, the authors wish to thank Dr. A. Bijelic, E. Al-Sayed, MSc, E. Tanuhadi, MSc, and Mag. A. Dobrov for valuable discussions regarding this work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.8b01740.
Accession Codes
CCDC 1842739–1842742 and 1870211 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Dedication
Dedicated to Professor Dr. Dr. h. c. mult. Achim Müller on the occasion of his 80th birthday.
Supplementary Material
References
- Pope M. T.Inorganic Chemistry Concepts. Heteropoly and Isopoly Oxometalates; Springer: Berlin, 1983; Vol. 8. [Google Scholar]
- Blazevic A.; Rompel A. The Anderson-Evans polyoxometalate: From inorganic building blocks via hybrid organic-inorganic structures to tomorrows “Bio-POM”. Coord. Chem. Rev. 2016, 307, 42–64. 10.1016/j.ccr.2015.07.001. [DOI] [Google Scholar]
- Kondo H.; Kobayashi A.; Sasaki Y. The structure of the hexamolybdoperiodate anion in its potassium salt. Acta Crystallogr., Sect. B: Struct. Sci. 1980, 36, 661–664. 10.1107/s0567740880004037. [DOI] [Google Scholar]
- Naruke H.; Yamase T. Structure of a photoluminescent polyoxotungstoantimonate. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1992, 48, 597–599. 10.1107/s0108270191010478. [DOI] [Google Scholar]
- Schmidt K. J.; Schrobilgen G. J.; Sawyer J. F. Hexasodium hexatungstotellurate(VI) 22-hydrate. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1986, 42, 1115–1118. 10.1107/s0108270186093204. [DOI] [Google Scholar]
- Rozantsev G. M.; Radio S. V.; Gumerova N. I.; Baumer V. N.; Shishkin O. B. Phase formation in the Ni2+-WO42–-H+-H2O system (Z = 1.00). Crystal structure and properties of sodium heteropolyhexatunsten nickelate(2+) Na4[Ni(OH)6W6O18]·16H2O. J. Struct. Chem. 2009, 50, 296–305. 10.1007/s10947-009-0041-z. [DOI] [Google Scholar]
- Lee U.; Joo H. C. Tripotassium hexahydrogenhexamolybdocobaltate(III) potassium nitrate tetrahydrate, K3[H6CoMo6O24]·KNO3·4H2O. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2000, 56, e423–e424. 10.1107/s0108270100012154. [DOI] [Google Scholar]
- Wu C.; Lin X.; Yu R.; Yang W.; Lu C.; Zhuang H. [Na4(H2O)7][Fe(OH)6Mo6O18]: A new [12] metallacrown-6 structure with an octahedrally coordinated iron at the center. Sci. China, Ser. B 2001, 44, 49–54. 10.1007/bf02879735. [DOI] [Google Scholar]
- Manikumari S.; Shivaiah V.; Das S. K. Identification of a Near-Linear Supramolecular Water Dimer, (H2O)2, in the Channel of an Inorganic Framework Material. Inorg. Chem. 2002, 41, 6953–6955. 10.1021/ic025803x. [DOI] [PubMed] [Google Scholar]
- a Lee U.; Joo H.-C. A novel protonated hexatungstoplatinate(IV), Na5.5[H2.5PtW6O24]·17H2O. Acta Crystallogr., Sect. E: Struct. Rep. Online 2004, 60, i33–i36. 10.1107/s1600536804002302. [DOI] [Google Scholar]; b Day V. W.; Goloboy J. C.; Klemperer W. G. Synthesis and Solid State Structures of the Hydrogen-Bonded Hexamolybdoplatinate(IV) Tetramer [(PtMo6O24)4H23]9– and the Hexamolybdoplatinate(IV) Trimers [(PtMo6O24)3H16]8– and [(PtMo6O24)3H14]10–. Eur. J. Inorg. Chem. 2009, 5079–5087. 10.1002/ejic.200900873. [DOI] [Google Scholar]
- Liu W.; Lin Z.; Bassil B. S.; Al-Oweini R.; Kortz U. Synthesis and Structure of Hexatungstochromate(III), [H3CrIIIW6O24]6–. Chimia 2015, 69, 537–540. 10.2533/chimia.2015.537. [DOI] [PubMed] [Google Scholar]
- Gumerova N. I.; Roller A.; Rompel A. [Ni(OH)3W6O18(OCH2)3CCH2OH]4–: the first tris-functionalized Anderson-type heteropolytungstate. Chem. Commun. 2016, 52, 9263–9266. 10.1039/c6cc04326g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzyn V. I.; Kolli I. D.; Evchenko T. I.; Lunk H.-J. Investigation of chromium polytungstates. Russ. J. Inorg. Chem. 1973, 18, 2176–2179. [Google Scholar]
- Dolbecq A.; Dumas E.; Mayer C. R.; Mialane P. Hybrid organic–inorganic polyoxometalate compounds: from structural diversity to applications. Chem. Rev. 2010, 110, 6009–6048. 10.1021/cr1000578. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Huang Y.; Li G.; Wei Y. Recent advances in alkoxylation chemistry of polyoxometalates: From synthetic strategies, structural overviews to functional applications. Coord. Chem. Rev. 2019, 378, 395–414. 10.1016/j.ccr.2017.10.025. [DOI] [Google Scholar]
- a Yvon C.; Surman A. J.; Hutin M.; Alex J.; Smith B. O.; Long D.-L.; Cronin L. Polyoxometalate clusters integrated into peptide chains and as inorganic amino acids: solution- and solid-phase approaches. Angew. Chem., Int. Ed. 2014, 53, 3336–3341. 10.1002/anie.201311135. [DOI] [PubMed] [Google Scholar]; b Yang H.-K.; Cheng Y.-X.; Su M.-M.; Xiao Y.; Hu M.-B. Polyoxometalate–biomolecule conjugates: A new approach to create hybrid drugs for cancer therapeutics. Bioorg. Med. Chem. Lett. 2013, 23, 1462–1466. 10.1016/j.bmcl.2012.12.081. [DOI] [PubMed] [Google Scholar]; c Macdonell A.; Johnson N. A. B.; Surman A. J.; Cronin L. Configurable nanosized metal oxide oligomers via precise “click” coupling control of hybrid polyoxometalates. J. Am. Chem. Soc. 2015, 137, 5662–5665. 10.1021/jacs.5b02466. [DOI] [PubMed] [Google Scholar]; d Ji Y.; Hu J.; Huang L.; Chen W.; Streb C.; Song Y.-F. Covalent attachment of Anderson-type polyoxometalates to single-walled carbon nanotubes gives enhanced performance electrodes for lithium ion batteries. Chem.—Eur. J. 2015, 21, 6469–6474. 10.1002/chem.201500218. [DOI] [PubMed] [Google Scholar]; e Li X.-X.; Wang Y.-X.; Wang R.-H.; Cui C.-Y.; Tian C.-B.; Yang G.-Y. Designed assembly of heterometallic cluster organic frameworks based on Anderson-type polyoxometalate clusters. Angew. Chem., Int. Ed. 2016, 55, 6462–6466. 10.1002/anie.201602087. [DOI] [PubMed] [Google Scholar]; f Song Y.-F.; McMillan N.; Long D.-L.; Kane S.; Malm J.; Riehle M. O.; Pradeep C. P.; Gadegaard N.; Cronin L. Micropatterned surfaces with covalently grafted unsymmetrical polyoxometalate-hybrid clusters lead to selective cell adhesion. J. Am. Chem. Soc. 2009, 131, 1340–1341. 10.1021/ja807091v. [DOI] [PubMed] [Google Scholar]; g Schaming D.; Allain C.; Farha R.; Goldmann M.; Lobstein S.; Giraudeau A.; Hasenknopf B.; Ruhlmann L. Synthesis and photocatalytic properties of mixed polyoxometalate–porphyrin copolymers obtained from anderson-type polyoxomolybdates. Langmuir 2010, 26, 5101–5109. 10.1021/la903564d. [DOI] [PubMed] [Google Scholar]
- Hasenknopf B.; Delmont R.; Herson P.; Gouzerh P. Anderson-type heteropolymolybdates containing tris(alkoxo) ligands: synthesis and structural characterization. Eur. J. Inorg. Chem. 2002, 1081–1087. . [DOI] [Google Scholar]
- Zhang J.; Li Q.; Zeng M.; Huang Y.; Zhang J.; Hao J.; Wei Y. The proton-controlled synthesis of unprecedented diol functionalized Anderson-type POMs. Chem. Commun. 2016, 52, 2378–2381. 10.1039/c5cc10071b. [DOI] [PubMed] [Google Scholar]
- Honda D.; Ikegami S.; Inoue T.; Ozeki T.; Yagasaki A. Protonation and Methylation of an Anderson-Type Polyoxoanion [IMo6O24]5-. Inorg. Chem. 2007, 46, 1464–1470. 10.1021/ic061881z. [DOI] [PubMed] [Google Scholar]
- Molitor C.; Mauracher S. G.; Rompel A. Aurone synthase is a catechol oxidase with hydroxylase activity and provides insights into the mechanism of plant polyphenol oxidases. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E1806–E1815. 10.1073/pnas.1523575113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molitor C.; Bijelic A.; Rompel A. In situ formation of the first proteinogenically functionalized [TeW6O24O2(Glu)]7– structure reveals unprecedented chemical and geometrical features of the Anderson-type cluster. Chem. Commun. 2016, 52, 12286–12289. 10.1039/c6cc07004c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sayed E.; Blazevic A.; Roller A.; Rompel A. The synthesis and characterization of aromatic hybrid Anderson–Evans POMs and their serum albumin interactions: the shift from polar to hydrophobic interactions. Chem.—Eur. J. 2015, 21, 17800–17807. 10.1002/chem.201502458. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Blazevic A.; Al-Sayed E.; Roller A.; Giester G.; Rompel A. Tris-functionalized hybrid Anderson polyoxometalates: synthesis, characterization, hydrolytic stability and inversion of protein surface charge. Chem.—Eur. J. 2015, 21, 4762–4771. 10.1002/chem.201405644. [DOI] [PubMed] [Google Scholar]
- Lin C.-G.; Chen W.; Long D.-L.; Cronin L.; Song Y.-F. Step-by-step covalent modification of Cr-templated Anderson-type polyoxometalates. Dalton Trans. 2014, 43, 8587–8590. 10.1039/c4dt00033a. [DOI] [PubMed] [Google Scholar]
- Li Q.; Wei Y. A series of unprecedented triol-stabilized [H3MW6O24]n–: the missing piece between A- and B-type Anderson–Evans polyoxometalates. Chem. Commun. 2018, 54, 1375–1378. 10.1039/c7cc08976g. [DOI] [PubMed] [Google Scholar]
- Bruker SAINT, V8.32B Copyright 2005–2015; Bruker AXS, 2013.
- Sheldrick G. M.SADABS; University of Göttingen: Germany, 1996.
- Sheldrick G. M. A short history of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. 10.1107/s0108767307043930. [DOI] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/s0021889808042726. [DOI] [Google Scholar]
- Hübschle C. B.; Sheldrick G. M.; Dittrich B. ShelXle: a Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. 10.1107/s0021889811043202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Truck G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian G16, Rev. B01; Gaussian, Inc.: Wallingford, CT, 2016.
- Grimme S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. 10.1002/jcc.20078. [DOI] [PubMed] [Google Scholar]
- Ernzerhof M.; Scuseria G. E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. 10.1063/1.478401. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Miertuš S.; Scrocco E.; Tomasi J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. 10.1016/0301-0104(81)85090-2. [DOI] [Google Scholar]
- Ohlin C. A.; Pascual-Borràs M. Protonation and water exchange kinetics in sandwich polyoxometalates. Dalton Trans. 2018, 47, 13602–13607. 10.1039/c8dt02342e. [DOI] [PubMed] [Google Scholar]
- Sharma R.; Zhanga J.; Ohlin C. A. pH-Dependent solution dynamics of a manganese(II) polyoxometalate, [Mn4(H2O)2(P2W15O56)2]16–, and [Mn(H2O)6]2+. Dalton Trans. 2015, 44, 19068–19071. 10.1039/c5dt03138a. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/physrevlett.77.3865. [DOI] [PubMed] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1998, 37, 785–789. 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- Tissandier M. D.; Cowen K. A.; Feng W. Y.; Gundlach E.; Cohen M. H.; Earhart A. D.; Coe J. V.; Tuttle T. R. The Proton’s Absolute Aqueous Enthalpy and Gibbs Free Energy of Solvation from Cluster-Ion Solvation Data. J. Phys. Chem. A 1998, 102, 7787–7794. 10.1021/jp982638r. [DOI] [Google Scholar]
- Minato T.; Suzuki K.; Yamaguchi K.; Mizuno N. Alkoxides of trivacant lacunary polyoxometalates. Chem.—Eur. J. 2017, 23, 14213–14220. 10.1002/chem.201702883. [DOI] [PubMed] [Google Scholar]
- Miras H. N.; Wilson E. F.; Cronin L. Unravelling the complexities of inorganic and supramolecular self-assembly in solution with electrospray and cryospray mass spectrometry. Chem. Commun. 2009, 1297–1311. 10.1039/b819534j. [DOI] [PubMed] [Google Scholar]
- Bard A. J.; Parsons R.; Jordan J.. Standard Potentials in Aqueous Solution; Routledge: New York, 1985. [Google Scholar]
- Pope M. T. Heteropoly and isopoly anions as oxo complexes and their reducibility to mixed-valence blues. Inorg. Chem. 1972, 11, 1973–1974. 10.1021/ic50114a057. [DOI] [Google Scholar]
- Gumerova N. I.; Rompel A. Synthesis, structures and applications of electron-rich polyoxometalates. Nat. Rev. Chem. 2018, 2, 0112. 10.1038/s41570-018-0112. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.