

Abstract
Background
Leaf mold, one of the major diseases of tomato caused by Cladosporium fulvum (C. fulvum), can dramatically reduce the yield and cause multimillion dollar losses annually worldwide. Mapping the resistance genes (R genes) of C. fulvum and devising MAS based strategies for breeding new cultivars is an effective approach to improve the resistance in tomato. Up to now, many C. fulvum genes or QTLs have been mapped using different genetic materials, but few studies focused on Cf-10 gene positioning.
Results
In this study, we investigated the genetic rules for Cf-10 and used a novel combinatorial strategy to rapidly map the Cf-10 gene. Initially, the performance of F1, F2 and BC1F1 individuals after infection, demonstrated that the resistance against C. fulvum was controlled by a single dominant gene. Two pools of resistant and susceptible individuals from F2 population were investigated, using mapping by sequencing approach and Cf-10 was found to be localized to 3.35 Mb and 3.74 Mb on chromosome 1, employing SNP/InDel index methods, respectively. After accounting for overlapping regions, these two algorithms yielded a total length of 3.29 Mb, narrowing down the target region. We further developed five serviceable KASP markers for this region based on sequencing data and conducted local QTL mapping using individuals from the F2 population, except for mapping by sequencing as mentioned above. Finally Cf-10 gene was mapped spanning a region of 790 kb, where only one gene (Solyc01g007130.3) was annotated as probable receptor protein kinase TMK1 with a LRR motif, a common R gene characteristic. The RT-qPCR analysis further confirmed the localization and the relative expression of Solyc01g007130.3 in Ontario 792 and was found to be significantly higher than that in Moneymaker at 9 dpi and 12 dpi, respectively.
Conclusion
This study proposed a novel combinatorial strategy by combining SNP-index, InDel-index analyses and local QTL mapping using KASP genotyping approach to rapidly map genes responsible for specific traits and provided a robust base for cloning the Cf-10 gene. Furthermore, these analyses suggest that Solyc01g007130.3 is a potential candidate to be regarded as Cf-10 gene.
Electronic supplementary material
The online version of this article (10.1186/s12870-018-1616-7) contains supplementary material, which is available to authorized users.
Keywords: Tomato, Cladosporium fulvum, Cf-10 gene, QTL mapping, SNP-index, InDel-index, KASP
Background
Cloning genes associated with important agronomic traits is one of the major components of plant functional genomics research. Map-based cloning is an important and reliable method, and many genes with vital functions from economically important crops [1–5] and fruit trees [6, 7] have been cloned, primarily using simple sequence repeat (SSR), sequence-characterized amplified region (SCAR) markers and/or cleaved amplified polymorphic sequence (CAPS) based approaches. A number of genes or quantitative trait loci (QTLs) such as ER4.1 [8], dl [9], Ty-2 [10], ms10 [11], fw2.2 [12] and fw11.3 [13] were fine mapped and isolated from tomato using SSR/InDel makers. However, the markers and CAPS derived from single nucleotide polymorphisms (SNPs) to clone these genes often results in low resolution and are time-consuming. Mapping and cloning of genes quickly is a major challenge in the post-genomic era. However, next-generation sequencing (NGS) technologies provide an effective solution to the problem and a number of genes were fine mapped and cloned by employing high-density genetic map based approaches. A high-density genetic map constructed by using Illumina-based whole genome re-sequencing of soybean genome, a 29.7 kb QTL containing two candidate genes involved in resistance against southern root-knot nematode (RKN) was mapped on chromosome 10 [14]. Another research highlighted the mapping of QTL-U5 responsible for fruit colour in cucumber spanning a region of 313.2 kb located on chromosome 5 containing 320 genes, of which 39 were synonymous [15]. Mapping by sequencing or bulked segregant analysis (BSA) is another method for rapid gene mapping [16, 17]. Pandey et al. [18] identified a region of 3.06 Mb on pseudomolecule A03 responsible for rust and late leaf spot (LLS) resistance using whole-genome re-sequencing QTL-seq approach. The study further identified that about 25 candidate genes were affected by 30 non-synonymous SNPs besides other nine candidate genes affected by three synonymous SNPs responsible for rust and LLS resistance, respectively. The RNA Seq-BSA was also adopted to identify SNPs associated with Yr15, involved in resistance against yellow rust in wheat [19]. Several other studies used similar methods to fine map genes for different economically important traits [20–22].
Plants have evolved a plethora of resistance strategies in response to the invasion of pathogens [23–26], and most prominent among them encompasses R gene induced recognition, exploiting nucleotide-binding sites and leucine-rich repeat (NBS-LRR) domains to trigger the disease resistance response [27, 28].
C. fulvum series genes are special R genes that play important roles in resisting tomato leaf disease caused by Cladosporium fulvum, which can cause serious damage to tomato yield and quality. To date, many C. fulvum genes have been mapped in different genomic regions, but most of these C. fulvum genes are located at the Milky Way site of chromosome 1 such as, Cf-4, Cf-9, Hcr9-4E, Hcr9-9B, Cf-ECP1, Cf-ECP2, Cf-ECP3, Cf-ECP4, Cf-ECP5 and Cf-19 genes [29–38]. Whereas, other C. fulvum genes, such as Cf-2, Cf-5 and Cf-6, were shown to be closely associated with chromosome 6 [39, 40]. Among all these genes, only Cf-2, Cf-4, Cf-5, Cf-9 and Cf-19 have been cloned and well characterized. All of these cloned Cf genes belong to one of the two multigene families designated as homologues of Cladosporium fulvum resistance genes (Hcr). The Cf-9 and Cf-2 genes consisting of 28 and 39 LRRs respectively, codes for transmembrane proteins [41, 42] while Cf-5 gene has been predicted to encode a largely extracytoplasmic protein containing 32 LRRs. On the other hand, Cf-4 encodes a membrane-anchored extracellular glycol-protein possessing only two LRRs which is much less than the LLRs encoded by Cf-9 gene [31]. The candidate gene Cf-19 (Solyc01g006550.2.1) encodes 30 LRRs, which is different from other Hcr genes. The C terminal of the protein encoded by Cf-4 gene was similar to those of Cf-9 and Cf-19 genes [38, 39]. These genes seem to be evolutionarily conserved although, they have shown a certain degree of structural differences [38].
Previously, our research team had developed four molecular markers associated with Cf-10 gene [43], but this is far from gene positioning or cloning. Recently, we investigated the expression patterns of differentially expressed genes (DEGs) in Ontario 792 harbouring Cf-10 gene after infection [44]. In the current study, we explored the genetic characteristics of the Cf-10 gene by developing a large F2 population and a BC1F1 population. By combining the SNP/InDel-index sequencing methods, we preliminarily mapped the Cf-10 gene and then narrowed this key region by developing kompetitive allele-specific PCR (KASP) markers for the primary mapping region to genotype the remaining individuals of F2 population. The RT-qPCR was used to validate the expression of candidate genes. Our results provide a novel, rapid and labour-saving approach for mapping genes in general and form the basis of cloning and deciphering molecular dynamics of Cf-10 gene in particular.
Results
The variation in disease resistance and test crosses
To investigate the genetic basis of Cf-10, we evaluated the disease resistance potential of Ontario 792, Moneymaker, F1, F2 and BC1F1 populations. Initially, we tested the resistance response of the two parents to ten different races (Table 1). Moneymaker demonstrated significantly higher susceptibility to all these races, while Ontario 792 was immune to race 1.2.4 and 1.4 besides being resistant to other eight races, indicating that Ontario 792 had broad-spectrum resistance against C. fulvum. Then we adopted the predominant physiological race 1.2.3.4 in Heilongjiang Province to test the performance of F1 and F2 individuals. To simplify the phenotype, plants with a disease score between 0 and 3 points were marked as resistant, while those with scores ≥5 points were marked as susceptible. All F1 plants showed resistance against the disease (Table 2), 391 individuals out of 529 from F2 population showed disease resistance while remaining 138 were found to be susceptible with a 3:1 segregation ratio as revealed by Chi-square test results (Fig. 1c), suggesting that Cf-10 was controlled by a single dominant gene. This hypothesis was further supported by the BC1F1 population presenting a 1:1 Mendelian ratio (Fig. 1c; Table 2; χ2 = 0.011).
Table 1.
The disease response of Moneymaker and Ontario 792 to different physiological races
| Physiological races | Moneymaker | Ontario 792 | ||
|---|---|---|---|---|
| Disease index | Resistance level | Disease index | Resistance level | |
| 1.2 | 65.4 | HS | 15.7 | R |
| 1.2.3 | 50.1 | MS | 20.1 | R |
| 1.2.3.4 | 55.5 | HS | 11.4 | R |
| 1.2.4 | 76.1 | HS | 0.0 | I |
| 1.3.4 | 58.4 | HS | 12.5 | R |
| 2.3 | 70.7 | HS | 22.6 | R |
| 1.3 | 59.9 | HS | 26.8 | R |
| 1.4 | 55.2 | HS | 0.0 | I |
| 1.2.3.4.5 | 56.1 | HS | 20.4 | R |
| 1.2.3.4.9 | 62.3 | HS | 15.3 | R |
I immune, HR highly resistant, R resistant, MR moderately resistant, MS moderate susceptibility, HS highly susceptible
Table 2.
Genetic analysis of the Cf-10 resistance gene in different populations
| Generation | Total No. of plants | No. of resistant plants | No. of susceptible plants | The segregation ratio of R:S | χ2 |
|---|---|---|---|---|---|
| Ontario 792 | 50 | 50 | 0 | – | – |
| Moneymaker | 50 | 0 | 50 | – | – |
| F1 | 20 | 20 | 0 | – | – |
| F2 | 529 | 391 | 138 | 2.83:1 | 0.082 |
| BC1F1 | 45 | 23 | 22 | 1.045:1 | 0.011 |
χ2 0.05, 1 = 3.84
Fig. 1.

The phenotype and segregation patterns of tomato. a The performance of Ontario 792 (left) and Moneymaker (right) after inoculation at 21 dpi. b The performance of F2 individuals after inoculation at 21 dpi. The left leaf is susceptible, while the right one is resistant. c The distribution of resistant and susceptible plants in the F2 and BC1F1 populations
QTL-seq analysis combining SNP-index and InDel-index
A total of 116.30 Gb clean data were obtained by Illumina sequencing, including 62.61 Gb from the parents and 53.69 Gb from the two mixed pools, all of high quality (94.33% > Q30 > 91.25%) and with a stable GC content (36.77% > GC > 35.69%) (Table 3). The average sequencing depths for the two parents and the two F2 pools were 33.50 × and 29 ×, respectively. These high-quality data lay a robust foundation for subsequent analysis.
Table 3.
An overview of the sequencing results
| Sample | Clean Reads | Clean Bases (Gb) | Q30 (%) | GC (%) | Properly mapped (%) |
|---|---|---|---|---|---|
| R01 | 89,945,037 | 26.94 | 93.89 | 36.77 | 89.45 |
| R02 | 119,076,889 | 35.67 | 91.25 | 35.69 | 89.53 |
| R03 | 96,599,912 | 28.93 | 94.27 | 36.18 | 89.80 |
| R04 | 82,672,515 | 24.76 | 94.33 | 36.30 | 90.47 |
These reads were mapped onto the reference genome of Solanum lycopersicum. The average mapping rate was 96.61% for these four samples. A total of 1,465,985 SNPs were obtained from the parents, of which 10,293 SNPs were non-synonymous. A total of 202,784 SNPs were obtained from the two mixed pools. Notably, we detected 214,921 small InDels amongst the parents and 51,348 small InDels from the mixed pools. Venn diagrams were used to demonstrate the relationships between SNP and InDels among the parents and the mixed pools. As shown in the Venn diagrams, these four samples share 159,740 SNPs and 74,988 InDels respectively (Fig. 2a, b).
Fig. 2.

SNP statistics and BSA analysis. a Venn diagram of SNP in the four pools. b Venn diagram of InDel in the four pools. c SNP-index algorithm to map Cf-10 gene. d InDel-index algorithm to map Cf-10 gene
We used two different methods to map the QTLs responsible for Cf-10 resistance. As shown in Fig. 2, only one QTL was identified and both the SNP-index and InDel-index association algorithms mapped this QTL to chromosome 1. More specifically, this QTL was located in the region 0–3,350,000 bp (3.35 Mb) using SNP-index method while InDel-index method revealed its presence in the region spanning 60,000–3,800,000 bp (3.74 Mb). By taking overlapping regions into account, these two methods yielded a total length of 3.29 Mb on chromosome 1. A total of 408 genes were annotated in this associated region, including 73 non-synonymous genes, 16 frameshifted genes and 16 genes with an extracellular LRR (eLRR) domain.
Further mapping of the Cf-10 gene
Since the 3.29-Mb region still contained a large number of genes, we developed 16 KASP markers, and five valid KASP markers in this region were used for genotyping an additional set of 147 individuals. The KASP genotyping results obtained from Ontario 792, Moneymaker and F1 individuals were consistent with our sequencing results, indicating that the sequencing and SNP calling results were reliable (Additional file 1: Table S5). Furthermore, we developed genetic map and also performed QTL mapping of this region so that the position of Cf-10 resistance QTL was narrowed down and was located between SNP 1 and SNP 2, a 790-kb region (Fig. 3; Additional file 2: Figure S1). Fortunately, only the Solyc01g007130.3 gene was annotated as probable receptor protein kinase TMK1 with a LRR motif, a common R gene characteristic suggesting that Solyc01g007130.3 is a potential candidate to be regarded as Cf-10 gene. Nevertheless, no SNPs or InDels were called in Solyc01g007130.3 from Ontario 792 and Moneymaker according to our re-sequencing data. We further used Sanger sequencing to compare Solyc01g007130.3 variation between Ontario 792 and Moneymaker and that no apparent differences were observed between them (data not shown).
Fig. 3.

Molecular mapping of the Cf-10 gene using SNP/InDel combinatorial approach
Screening of expression pattern of the candidate gene
We conducted a RT-qPCR assay to determine the expression response of Solyc01g007130.3 in Ontario 792 and Moneymaker after inoculation. The RT-qPCR results demonstrated that the expression of Solyc01g007130.3 was significantly induced in Ontario 792, except at 16 dpi (Fig. 4). No significant differences were observed between Ontario 792 and Moneymaker at 0 dpi, but at 9 dpi and 12 dpi, the relative expression of Solyc01g007130.3 in Ontario 792 was significantly higher than that in Moneymaker. At 5 dpi and 16 dpi, the difference in expression was higher in Ontario 792 than in Moneymaker but was of lesser statistical significance. Considering these results together, we concluded that Solyc01g007130.3 is a possible candidate for the gene underlying the QTL for Cf-10 resistance.
Fig. 4.
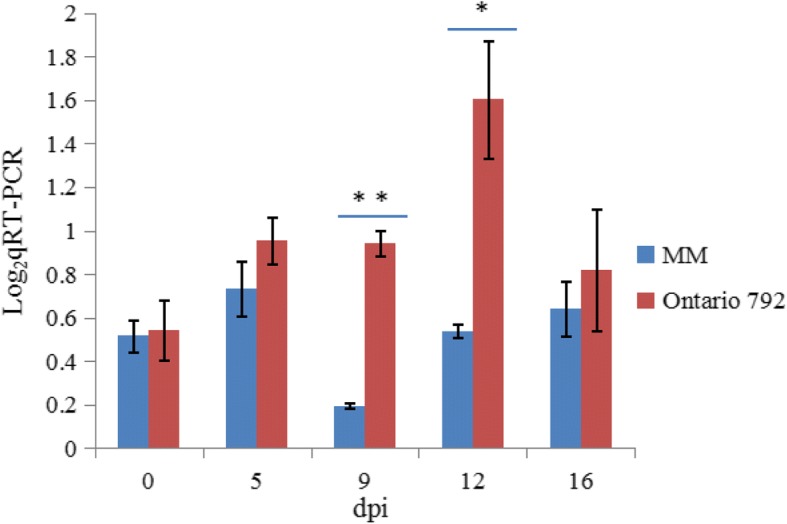
RT-qPCR analysis of Solyc01g007130.3 in the QTL region on chromosome 1 in response to C. fulvum inoculation. Both Ontario 792 and Moneymaker were inoculated with C. fulvum for 5, 9, 12, and 16 d. The blue and red bars represent Moneymaker and Ontario 792, respectively. The results were statistically analysed using Student’s t-test (*, P < 0.05; **, P < 0.01) and the asterisks indicate that the difference in gene expression in Ontario 792 and Moneymaker plants was highly significant
Discussion
Rapid gene cloning from a large F2 population
Traditional map-based cloning is an efficient approach to isolate genes/QTLs responsible for desired agronomic traits [45–47]. Usually, a genetic map of an F2, double-haploid (DH) or recombinant inbred line (RIL) population based on hundreds of SSR or InDel markers is used to make a primary map, and then a near-isogenic line (NIL) is developed based on marker-assisted selection (MAS; newly discovered in the primary mapping region) to narrow down the region of interest to a sufficient size to screen for a few candidate genes. Unfortunately, this workflow always takes a relatively long time [48, 49]. Compared with genetic maps, the mapping by sequencing using NGS is a faster and reliable method for mapping QTLs [50]. Nevertheless, one mixed pool typically contains approximately 20–100 individuals and generally maps the target region at a Mb-level interval [19, 51–54] because of insufficient meiotic recombination events. We still have to perform fine mapping or use omics methods such as RNA-seq to further screen the candidate genes [55, 56].
KASP is one of the uniplex SNP genotyping platforms [57] and has been successfully used to genotype and map QTLs in bi-parental populations [58, 59]. Recently, Xu et al. [60] developed KASP markers spanning a 303 kb region mapped by a 50 + 50 mapping sequence strategy and genotyped 2274 F2 individuals. Finally, the region was narrowed down to 36.1 kb, and they identified a candidate gene, ARN6.1, responsible for waterlogging tolerance.
In the present study, we used mapping by sequencing by combining SNP-index and InDel-index analyses to primarily position the Cf-10 gene to a 3.29 Mb region. Five KASP markers developed in this region helped us to narrow the Cf-10 gene to 790 kb region. We thus put forward an approach that could rapidly fine map QTLs using only a large F2 population, especially for those traits governed by single nuclear-encoded genes or some threshold traits (Fig. 5). Developing a large F2 population, mapping by sequencing analysis and KASP genotyping for QTL mapping could be completed in a short time, and a progeny test could ensure accurate phenotype identification and helps in achieving the desired objectives rapidly and accurately. We believe that this approach can be adopted for quick QTL fine mapping in the future.
Fig. 5.
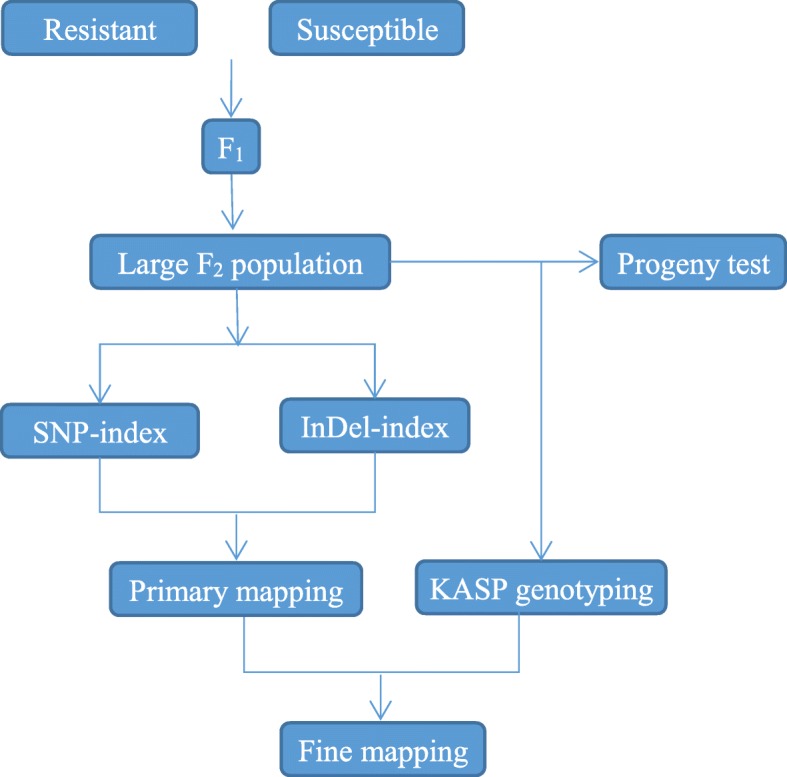
A novel combinatorial strategy to rapidly fine map genes
The characteristics of the potential candidate gene Solyc01g007130.3
We used ProtComp 9.0 for the sub-cellular localization of Solyc01g007130.3 and found that the encoded product was located in the plasma membrane (Additional file 3: Table S6), in agreement with other Cf genes. The CDD online protocol (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) predicted that Solyc01g007130.3 encodes a member of the STKc_IRAK-like superfamily and the PKc-like superfamily with 10 LRRs associated with disease resistance [61]. To further investigate the relationship between Solyc01g007130.3 and other Cf genes, we used MEGA 5.05 to perform multiple DNA sequence alignment analyses. A long evolutionary distance was observed between Solyc01g007130.3 and other Cf genes (Hcr2, Cf-2, Cf-4, Cf-5, Cf-9) (Additional file 4: Figure S2, A), strongly suggesting that comparatively different resistance mechanisms underlie Cf-10 gene. Amino acid sequence clustering also support these results. More follow-up experimental investigation is required to further understand the molecular function of Solyc01g007130.3.
The casual variation in Cf-10 responsible for disease resistance
Most plant disease resistance genes (R genes) cloned so far encode NBS-LRR proteins [27, 28]. Therefore, when we located the Cf-10 gene in the 790-kb interval, we only screened for genes containing the LRR domain. Fortunately, we only found one gene containing this domain (Solyc01g007130.3), and RT-qPCR analysis showed that significant or highly significant expression differences existed at 9 dpi and 12 dpi. However, Solyc01g007130.3 was not included in the set of DEGs at 16 dpi in our previous transcriptome research [44]. This phenomenon may be explained by the fact that Solyc01g007130.3 expression was significantly higher in the early stage of infection and that, in the later stage, downstream regulation played a leading role, but the hypothesis requires validation.
We also compared the parental re-sequencing data and found that no SNP or InDel variation was observed between the two parents. To better understand this step, we used Sanger sequencing to compare Solyc01g007130.3 between the parents and found that no difference in this gene existed between the parents (data not shown). We did not rule out the possibility of a methylation modification difference between the parents [62] and also compared the 5 kb region upstream of this gene. Unfortunately, this region could not be cloned by traditional PCR. Using reference genome information available for tomato (ftp://ftp.solgenomics.net/tomato_genome/assembly/build_3.00/), we found that the GC content of this 5 kb region upstream of Solyc01g007130.3 was 24.14%, which was much lower than the average value (35.20%) of the entire genome. This could be the possible reason hindering PCR based cloning of the region. In future research, we plan to establish the bacterial artificial chromosome (BAC) library for Ontario 792 to solve this problem of cloning the 5 kb sequence upstream of Solyc01g007130.3. Recently, a new method for cloning R genes that utilizes a three-step method (MutRenSeq) combining chemical mutagenesis with exome capture and sequencing to clone genes with specific domains may solve the problems associated with cloning the Cf-10 gene [63].
Conclusion
We used two steps of bioinformatic analysis named SNP-index and InDel-index to co-location and narrow down the Cf-10 gene region to 3.29 Mb. Then QTL mapping analysis adopting a total of five KASP markers developed by SNPs in this region further mapped Cf-10 gene to 790 Kb. This combinatorial approach can rapidly map or fine map genes responsible for specific traits just utilizing a big F2 population. Furthermore, we screened possible candidate genes according to their annotation information and found that Solyc01g007130.3 harbored a receptor protein kinase TMK1 with a LRR motif. Different expression by RT-qPCR validated that Solyc01g007130.3 was a candidate gene for Cf-10. The location and candidate gene screening of Cf-10 could lay a robust foundation for later cloning the Cf-10 gene and applications in MAS selection programs.
Methods
Plant materials and breeding strategy
In order to map the Cf-10 gene in tomato using map-based strategy, we crossed accession Ontario 792 (Institute of Vegetable and Flowers, Chinese Academy of Agricultural Science) harbouring the Cf-10 resistant gene (R gene) with the susceptible accession Moneymaker (Tomato Genetic Resource Center, LA2706) to harvest F1 and an F2 population subsequently containing 529 individuals, generated by selfing F1 population. At the same time, a BC1F1 population was also generated by back-crossing the F1 plants with Moneymaker to test the genetic basis of the Cf-10 gene. All plants were grown at the Horticultural Experimental Station of Northeast Agricultural University by applying normal water and fertilizer practices while weeds were managed manually.
Cladosporium fulvum inoculation and resistance evaluation
Ten physiological races (1.2, 1.2.3, 1.2.3.4, 1.2.4, 1.3.4, 2.3, 1.3, 1.4, 1.2.3.4.5, 1.2.3.4.9) provided by tomato research group of Horticulture College, Northeast Agricultural University were used to test the resistance potential of Ontario792 and Moneymaker. The disease score (0–9 points) for the severity of symptoms was assessed according to Zhao et al.’s method [38]. The disease index and the classification criteria were judged as per Li et al. [64] recommendations.
At five-to-six leaf stage, the seedling of Ontario 792 (50 pure lines), Moneymaker (50 pure lines), F1, F2 and BC1F1 plants were inoculated with C. fulvum race 1.2.3.4, which is a predominant physiological race in Heilongjiang Province, China. To ensure that the leaves of each individual were successfully inoculated, we sprayed the C. fulvum suspension at 1 × 107 sporangia per mL in the morning, afternoon and evening of a sunny day as mentioned by Liu et al. [44]. The plants were assessed for disease severity as described by Wang et al. [65]. The disease severity of the plants was converted to a disease score of 0–9 points according to the method of Zhao et al. [38]. Scoring in the range of 0–3 points was demarcated as resistant and a score ≥ 5 points was defined as susceptible. For the subsequent linkage analysis, the resistant and susceptible phenotypes were assigned as 1 and 0, respectively. Chi-square analysis was performed to test the segregation ratio of the resistant and susceptible individuals of F2 population and BC1F1 population.
DNA extraction, library construction and whole-genome re-sequencing
Using a minor modified cetyltrimethylammonium bromide (CTAB) method [66], all young leaves of 529 F2 individuals were harvested separately for total genomic DNA extraction. Of these, 20 resistant and 20 susceptible plants from the F2 population and the two parents were chosen for library construction and whole-genome re-sequencing. To simplify the description in later parts of this paper, we abbreviate Ontario 792 as R01, Moneymaker as R02, resistant pool as R03 and susceptible pool as R04. The isolated DNA was quantified using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Fremont, CA, USA). All 20 resistant and 20 susceptible plants were precisely quantified on Qubit® 2.0 Fluorometer (Life Technologies, CA, USA). Equal amounts of DNA from the resistant and susceptible plants were mixed to prepare R03 and R04. These two samples along with R01 and R02 were sonicated to generate 350-bp fragments using S2/E210 Ultrasonicator (Covaris, Woburn, MA, USA); which were subsequently end repaired and nucleotide (A) overhangs were generated. Afterwards, sequencing adapters were ligated using T4 DNA ligase and PCR was performed. The PCR products were then purified and loaded onto an Illumina sequencing platform (Illumina, Inc., San Diego, CA, USA) for paired-end sequencing according to the manufacturer’s recommendations.
QTL-seq and linkage mapping analysis
The raw sequencing data were filtered [reads with more than 50% of bases having Q-value ≤10 or an ambiguous sequence content (“N”) exceeding 10%] using an in-house Perl script available with Biomarker Technologies Co. Ltd. (Beijing, China). Then, these high-quality data were mapped onto the Solanum lycopersicum genome sequence (ftp://ftp.solgenomics.net/tomato_genome/assembly/build_3.00/) using a Burrows-Wheeler Aligner (Li et al. 2009). Picard software (https://sourceforge.net/projects/picard/) was used to mark duplicates. The SNP and InDel (1–5 bp) calling were realized by GATK [67] using default settings. In order to obtain highly accurate SNP and InDel set, a range of filters were also employed [68]. Both SNP-index [69] and InDel-index [17] methods were used for the association analysis. The SNP-index dot loess was obtained by regression fitting as described by Abe et al. [69]. The threshold value to map Cf-10 resistant gene controlled by a dominant locus was expected to be 0.667 for an F2 population.
Using information from the QTL-seq analysis on the SNPs in the QTL region, a total of 16 KASP markers were designed using Primer 5.0 (Additional file 5: Table S1) to genotype 147 individuals, including 141 from F2 and two from each of F1, Ontario 792 and Moneymaker individuals respectively. The KASP assays were performed in a 1536-well plate format following the protocol of LGC Genomics (LGC, Middlesex, UK). The KASP reaction mixture system is shown in Additional file 6: Table S2. A S1000TM Thermal Cycler PCR (Bio-Rad, Hercules, CA) was used with the following settings: thermal activation at 95 °C for 15 min, denaturation at 95 °C for 20 s, primer annealing at 65 °C for 60 s (decreasing by 1 °C per cycle, 10 cycles totally) and finally 30 cycles of amplification (95 °C for 10 s; 57 °C for 60 s). The Synergy H1 full-function microplate reader (FLUOstar Omega, BMG Labtech, Germany) was used to read the fluorescence signal upon completion of the reaction.
Real time quantitative PCR (RT-qPCR) analysis
The RT-qPCR assay was carried out in order to explore the expression pattern of candidate genes in Ontario 792 and Moneymaker at 0, 5, 9, 12 and 16 days post-infection (dpi). The leaves from Ontario 792 and Moneymaker individuals were sampled independently for total RNA extraction using Trizol (Invitrogen) as per manufacturer’s recommendations. The primers for candidate genes were designed using primer 5.0 software. Actin-EFα1 was used for internal normalization. The primer sequences and reaction system are shown in Additional files 7 and 8: Tables S3 and S4, respectively and the RT-qPCR was performed in an iQ5 system (Bio-Rad, USA). A 20 μL reaction mixture, including 2 μL of cDNA (1:10 dilution), 10 μL of 2× TransStart Top Green qPCR SuperMix (TransGen, China), 0.5 μL of each primer (10 μmol/μL) and ddH2O, was prepared. Reaction conditions were as follows: 95 °C for 7 min; 40 cycles at 95 °C for 10 s, 58 °C for 30 s, and 72 °C for 20 s and then 71 cycles at 95 °C for 10 s, 60.5 °C for 10 s, and 95 °C for 10 s. The relative gene expression levels were calculated using the 2−ΔΔCt method [70].
Additional files
Table S5. The KASP genotyping results. (XLSX 16 kb)
Figure S1. QTL mapping analysis by Icimapping software. (PDF 36 kb)
Table S6. The sub-cellular location prediction of Cf-10. (XLSX 14 kb)
Figure S2. Cluster analysis of candidate and other genes. A Cluster analysis on account of DNA sequence. B Cluster analysis on account of amino acid sequence. (PDF 51 kb)
Table S1. The primer information of KASP markers. (XLSX 11 kb)
Table S2. The KASP reaction system. (XLSX 10 kb)
Table S3. The primer information of EFα1 and candidate gene. (XLSX 10 kb)
Table S4. The qRT-PCR reaction system. (XLSX 10 kb)
Acknowledgements
We thank Yuhui Xu, Lingxue Xu and Dongmei Yin from the Biomarker Limited Company for helping the data processing of this manuscript.
Funding
The publication costs of the article was funded by China Agriculture Research System (CARS-23-A-16). The funders have no role in the study design, data analysis and interpretation, and manuscript writing, but just provide the financial. The design of this study was funded by the Natural Science Foundation of Heilongjiang Province (C2017024)590048, and the China postdoctoral science foundation grant (2018 M630333). The analysis of the study was funded by the University Nursing Program for Young Scholars with Creative Talents in Heilongjiang Province (UNPYSCT-2018169).
Availability of data and materials
All the sequencing clean data were uploaded to the SRA database of NCBI (SRR6914778, SRR6914779, SRR6914776, SRR6914777). However, due to patent application, the data will be made public on 1 April 2019. However, before 1 April 2019, the datasets are available from the corresponding author on reasonable request.
Abbreviations
- BSA
bulked segregant analysis
- CAPS
cleaved amplified polymorphic sequence
- DEGs
differentially expressed genes
- DH
double-haploid
- InDel
insertion-deletion
- KASP
kompetitive allele-specific PCR
- MAS
marker-assisted selection
- NBs-LRR
nucleotide-binding sites and leucine-rich repeat
- NGS
next-generation sequencing
- NIL
near-isogenic line
- QTL
quantitative trait locus
- RIL
recombinant inbred line
- RT-qPCR
real time quantitative polymerase chain reaction
- SCAR
sequence-characterized amplified region
- SNPs
single nucleotide polymorphisms
- SSR
simple sequence repeat
Authors’ contributions
XX and JL conceived and designed the experiments; JJ and XY performed the RNA isolation, RT-qPCR experiments and the genetic analysis of the plant populations; GL and TZ performed the data analysis and wrote the manuscript. JJ and XY revised the manuscript. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The authors declared that a patent on a SNP marker tightly linked with Cf-10 resistant gene has been applied in connection with the work submitted.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Guan Liu, Email: liuguan.1989@163.com.
Tingting Zhao, Email: 411370676@qq.com.
Xiaoqing You, Email: 424252981@qq.com.
Jingbin Jiang, Email: 71040584@qq.com.
Jingfu Li, Email: lijf_2005@126.com.
Xiangyang Xu, Email: lijingfuls@163.com.
References
- 1.Liu J, Hua W, Hu Z, Yang H, Zhang L, Li RJ, et al. Natural variation in ARF18 gene simultaneously affects seed weight and silique length in polyploid rapeseed. Proc Natl Acad Sci. 2015;112:E5123–E5132. doi: 10.1073/pnas.1502160112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu C, Li X, Meng D, Jin WW, Yan JB, Chen SJ. A 4-bp insertion at ZmPLA1 encoding a putative phospholipase a generates haploid induction in maize. Mol Plant. 2017;10:520–522. doi: 10.1016/j.molp.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 3.Lu S, Zhao X, Hu Y, Liu H, Nan H, Li X, et al. Natural variation at the soybean J locus improves adaptation to the tropics and enhances yield. Nat Genet. 2017;49:773–779. doi: 10.1038/ng.3819. [DOI] [PubMed] [Google Scholar]
- 4.Tucker EJ, Baumann U, Kouidri A, Suchecki R, Baes M, Garcia M, et al. Molecular identification of the wheat male fertility gene Ms1 and its prospects for hybrid breeding. Nat Commun. 2017;8:869. doi: 10.1038/s41467-017-00945-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurokawa Y, Nagai K, Huan PD, Shimazaki K, Qu HQ, Mori Y, et al. Rice leaf hydrophobicity and gas films are conferred by a wax synthesis gene (LGF 1) and contribute to flood tolerance. New Phytol. 2018;218:1558–1569. doi: 10.1111/nph.15070. [DOI] [PubMed] [Google Scholar]
- 6.Yao G, Ming M, Allan AC, Gu C, Li L, Wu X, et al. Map-based cloning of the pear gene MYB114 identifies an interaction with other transcription factors to coordinately regulate fruit anthocyanin biosynthesis. Plant J. 2017;92:437–451. doi: 10.1111/tpj.13666. [DOI] [PubMed] [Google Scholar]
- 7.Han Z, Hu Y, Lv Y, Rose JKC, Sun Y, Shen F, et al. Natural variation underlies differences in ETHYLENE RESPONSE FACTOR 17 activity in fruit peel degreening. Plant Physiol. 2018. 10.1104/pp.17.01320. [DOI] [PMC free article] [PubMed]
- 8.Cui L, Qiu Z, Wang Z, Gao JC, Guo YM, Huang ZJ, et al. Fine mapping of a gene (ER4. 1) that causes epidermal reticulation of tomato fruit and characterization of the associated transcriptome. Front Plant Sci. 2017;8:1254. doi: 10.3389/fpls.2017.01254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang J, Yu T, Gao S, Xiong C, Xie QM, Li HX, et al. Fine mapping of the dialytic gene that controls multicellular trichome formation and stamen development in tomato. Theor Appl Genet. 2016;129:1531–1539. doi: 10.1007/s00122-016-2722-2. [DOI] [PubMed] [Google Scholar]
- 10.Yang X, Caro M, Hutton SF, Scott JW, Guo YM, Wang XX, et al. Fine mapping of the tomato yellow leaf curl virus resistance gene ty-2 on chromosome 11 of tomato. Mol Breed. 2014;34:749–760. doi: 10.1007/s11032-014-0072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Huang Z, Wang X, Gao JC, Guo YM, Du YC, et al. Fine mapping and molecular marker development of anthocyanin absent, a seedling morphological marker for the selection of male sterile 10 in tomato. Mol Breed. 2016;36:107. doi: 10.1007/s11032-016-0531-6. [DOI] [Google Scholar]
- 12.Frary A, Nesbitt TC, Frary A, Knaap E, Cong B, Liu J, et al. fw2. 2: a quantitative trait locus key to the evolution of tomato fruit size. Science. 2000;289:85–88. doi: 10.1126/science.289.5476.85. [DOI] [PubMed] [Google Scholar]
- 13.Mu Q, Huang Z, Chakrabarti M, Illa-Berenguer E, Liu X, Wang Y, et al. Fruit weight is controlled by cell size regulator encoding a novel protein that is expressed in maturing tomato fruits. PLoS Genet. 2017;13:e1006930. doi: 10.1371/journal.pgen.1006930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu X, Zeng L, Tao Y, Vuong T, Wan J, Boerma R, et al. Pinpointing genes underlying the quantitative trait loci for root-knot nematode resistance in palaeopolyploid soybean by whole genome resequencing. Proc Natl Acad Sci. 2013;110:13469–13474. doi: 10.1073/pnas.1222368110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Q, Miao H, Li S, Zhang SP, Wang Y, Weng YQ, et al. A sequencing-based linkage map of cucumber. Mol Plant. 2015;8:961–963. doi: 10.1016/j.molp.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 16.Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C, et al. QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013;74:174–183. doi: 10.1111/tpj.12105. [DOI] [PubMed] [Google Scholar]
- 17.Singh VK, Khan AW, Saxena RK, Sinha P, Kale SM, Parupalli S, et al. Indel-seq: a fast-forward genetics approach for identification of trait-associated putative candidate genomic regions and its application in pigeonpea (Cajanus cajan) Plant Biotechnol J. 2017;15:906–914. doi: 10.1111/pbi.12685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pandey MK, Khan AW, Singh VK, Vishwakarma MK, Shasidhar Y, Kumar V, et al. QTL-seq approach identified genomic regions and diagnostic markers for rust and late leaf spot resistance in groundnut (Arachis hypogaea L.) Plant Biotechnol J. 2017;15:927–941. doi: 10.1111/pbi.12686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramirez-Gonzalez RH, Segovia V, Bird N, Fenwick P, Holdgate S, Berry S, et al. RNA-Seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotechnol J. 2015;13:613–624. doi: 10.1111/pbi.12281. [DOI] [PubMed] [Google Scholar]
- 20.Lu H, Lin T, Klein J, Wang SH, Qi JJ, Zhou Q, et al. QTL-seq identifies an early flowering QTL located near Flowering Locus T in cucumber. Theor Appl Genet. 2014;127:1491–1499. doi: 10.1007/s00122-014-2313-z. [DOI] [PubMed] [Google Scholar]
- 21.Xu X, Lu L, Zhu B, Xu Q, Qi XH, Chen XH. QTL mapping of cucumber fruit flesh thickness by SLAF-seq. Sci Rep. 2015;5:15829. doi: 10.1038/srep15829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Q, Song J, Du WP, Xu LY, Jiang Y, Zhang J, et al. Identification, mapping, and molecular marker development for Rgsr8. 1: a new quantitative trait locus conferring resistance to gibberella stalk rot in maize (Zea mays L.) Front Plant Sci. 2017;8:1355. doi: 10.3389/fpls.2017.01355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng Y, Zhai K, Xie Z, Yang D, Zhu X, Liu J, et al. Epigenetic regulation of antagonistic receptors confers rice blast resistance with yield balance. Science. 2017;355:962–965. doi: 10.1126/science.aai8898. [DOI] [PubMed] [Google Scholar]
- 24.Li W, Zhu Z, Chern M, Yin JJ, Yang C, Ran L, et al. A natural allele of a transcription factor in rice confers broad-spectrum blast resistance. Cell. 2017;170:114–126. e15. doi: 10.1016/j.cell.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 25.Ma Z, Zhu L, Song T, Wang Y, Zhang Q, Xia Y, et al. A paralogous decoy protects Phytophthora sojae apoplastic effector PsXEG1 from a host inhibitor. Science. 2017;355:710–714. doi: 10.1126/science.aai7919. [DOI] [PubMed] [Google Scholar]
- 26.Bacete L, Mélida H, Miedes E, Molina A. Plant cell wall-mediated immunity: cell wall changes trigger disease resistance responses. Plant J. 2018;93:614–636. doi: 10.1111/tpj.13807. [DOI] [PubMed] [Google Scholar]
- 27.Jones JDG, Dangl JL. The plant immune system. Nature. 2006;444:323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 28.Jones JDG, Vance RE, Dangl JL. Intracellular innate immune surveillance devices in plants and animals. Science. 2016;354:aaf6395. doi: 10.1126/science.aaf6395. [DOI] [PubMed] [Google Scholar]
- 29.Balint-Kurti PJ, Dixon MS, Jones DA, NorcottJ KA, Jones DJ. RFLP linkage analysis of the cf-4 and cf-9 genes for resistance to Cladosporium fulvum in tomato. Theor Appl Genet. 1994;88:691–700. doi: 10.1007/BF01253972. [DOI] [PubMed] [Google Scholar]
- 30.Jones DA, Thomas CM, Hammond-Kosack KE, Balint-Kurti PJ, Jones DJ. Isolation of the tomato cf-9 gene for resistance to Cladosporium fulvum by transposon tagging. Science. 1994;266:789–793. doi: 10.1126/science.7973631. [DOI] [PubMed] [Google Scholar]
- 31.Thomas CM, Jones DA, Parniske M, Harrison K, Balint-Kurti PJ, Hatzixanthis K, et al. Characterization of the tomato Cf-4 gene for resistance to Cladosporium fulvum identifies sequences that determine recognitional specificity in Cf-4 and Cf-9. Plant Cell. 1997;9:2209–2224. doi: 10.1105/tpc.9.12.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takken FLW, Schipper D, Nijkamp HJJ, Hille J. Identification and ds-tagged isolation of a new gene at the Cf-4 locus of tomato involved in disease resistance to Cladosporium fulvum race 5. Plant J. 1998;14:401–411. doi: 10.1046/j.1365-313X.1998.00135.x. [DOI] [PubMed] [Google Scholar]
- 33.Haanstra JPW, Laugé R, Meijer-Dekens F, Bonnema G, de Wit PJGM, Lindhout P. The Cf-ECP2 gene is linked to, but not part of, the Cf-4/Cf-9 cluster on the short arm of chromosome 1 in tomato. Mol Gen Genet MGG. 1999;262(4–5):839–845. doi: 10.1007/s004380051148. [DOI] [PubMed] [Google Scholar]
- 34.Haanstra JPW, Meijer-Dekens F, Lauge R, Seetanah DC, Joosten MHAJ, de Wit PJGM, et al. Mapping strategy for resistance genes against Cladosporium fulvum on the short arm of chromosome 1 of tomato: Cf-ECP5 near the Hcr9 milky way cluster. Theor Appl Genet. 2000;101:661–668. doi: 10.1007/s001220051528. [DOI] [Google Scholar]
- 35.Panter SN, Hammond-Kosack SN, Harrison K, Jones DJ, Jones DA. Developmental control of promoter activity is not responsible for mature onset of cf-9B-mediated resistance to leaf mold in tomato. Mol Plant-Microbe Interact. 2002;15:1099–1107. doi: 10.1094/MPMI.2002.15.11.1099. [DOI] [PubMed] [Google Scholar]
- 36.Yuan Y, Haanstra J, Lindhout P, Bonnema G. The cladosporium fulvum resistance gene cf-ecp3 is part of the orion cluster on the short arm of tomato chromosome 1. Mol Breed. 2002;10(1–2):45–50. doi: 10.1023/a:1020328827442. [DOI] [Google Scholar]
- 37.Soumpourou E, Iakovidis M, Chartrain L, Lyall V, Thomas CM. The Solanum pimpinellifolium cf-ECP1 and cf-ECP4 genes for resistance to Cladosporium fulvum are located at the Milky Way locus on the short arm of chromosome 1. Theor Appl Genet. 2007;115:1127–1136. doi: 10.1007/s00122-007-0638-6. [DOI] [PubMed] [Google Scholar]
- 38.Zhao T, Jiang J, Liu G, He SS, Zhang H, Chen XL, et al. Mapping and candidate gene screening of tomato Cladosporium fulvum-resistant gene Cf-19, based on high-throughput sequencing technology. BMC Plant Biol. 2016;16:51. doi: 10.1186/s12870-016-0737-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dixon MS, Jones DA, Keddie JS, Thomas CM, Harrison K, Jonathan DG, et al. The tomato Cf-2 disease resistance locus comprises two functional genes encoding leucine-rich repeat proteins. Cell. 1996;84:451–459. doi: 10.1016/S0092-8674(00)81290-8. [DOI] [PubMed] [Google Scholar]
- 40.Dixon MS, Hatzixanthis K, Jones DA, Harrison K, Jones JD. The tomato cf-5 disease resistance gene and six homologs show pronounced allelic variation in leucine-rich repeat copy number. Plant Cell. 1998;10:1915–1925. doi: 10.1105/tpc.10.11.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ellis J, Dodds P, Pryor T. The generation of plant disease resistance gene specificities. Trends Plant Sci. 2000;5:373–379. doi: 10.1016/S1360-1385(00)01694-0. [DOI] [PubMed] [Google Scholar]
- 42.Takken FLW, Joosten MHAJ. Plant resistance genes: their structure, function and evolution. Eur J Plant Pathol. 2000;106:699–713. doi: 10.1023/A:1026571130477. [DOI] [Google Scholar]
- 43.Li N, Jiang JB, Li JF, Xu XY. Development of molecular marker linked to Cf-10 gene using SSR and AFLP method in tomato. J Northeast Agricult Univ (English Edition) 2012;19:30–36. doi: 10.1016/S1006-8104(13)60047-1. [DOI] [Google Scholar]
- 44.Liu G, Liu J, Zhang C, You XQ, Zhao TT, Jiang JB, et al. Physiological and RNA-seq analyses provide insights into the response mechanism of the Cf-10-mediated resistance to Cladosporium fulvum infection in tomato. Plant Mol Biol. 2018;96:403–416. doi: 10.1007/s11103-018-0706-0. [DOI] [PubMed] [Google Scholar]
- 45.Zuo W, Chao Q, Zhang N, Ye JR, Tan GQ, Li BL, et al. A maize wall-associated kinase confers quantitative resistance to head smut. Nat Genet. 2015;47:151–157. doi: 10.1038/ng.3170. [DOI] [PubMed] [Google Scholar]
- 46.Lu X, Xiong Q, Cheng T, Li QT, Liu XL, Bi YD, et al. A PP2C-1 allele underlying a quantitative trait locus enhances soybean 100-seed weight. Mol Plant. 2017;10:670–684. doi: 10.1016/j.molp.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Z, Li J, Pan Y, Li J, Zhou L, Shi H, et al. Natural variation in CTB4a enhances rice adaptation to cold habitats. Nat Commun. 2017;8:14788. doi: 10.1038/ncomms14788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu Z, Tan L, Fu Y, Liu FX, Cai HW, Xie DX, et al. Genetic control of inflorescence architecture during rice domestication. Nat Commun. 2013;4:1345–1346. doi: 10.1038/ncomms3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan Y, Yang J, Mathioni SM, Yu J, Shen J, Yang X, et al. PMS1T, producing phased small-interfering RNAs, regulates photoperiod-sensitive male sterility in rice. Proc Natl Acad Sci. 2016;113:15144–15149. doi: 10.1073/pnas.1619159114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schneeberger K. Using next-generation sequencing to isolate mutant genes from forward genetic screens. Nat Rev Genet. 2014;15:662–676. doi: 10.1038/nrg3745. [DOI] [PubMed] [Google Scholar]
- 51.Haase NJ, Beissinger T, Hirsch CN, Vaillancourt B, Deshpande S, Barry K, et al. Shared genomic regions between derivatives of a large segregating population of maize identified using bulked segregant analysis sequencing and traditional linkage analysis. G3: genes, genomes. Genetics. 2015;5:1593–1602. doi: 10.1534/g3.115.017665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng W, Wang Y, Wang L, Ma ZB, Zhao JM, Wang P, et al. Genetic mapping and molecular marker development for Pi65 (t), a novel broad-spectrum resistance gene to rice blast using next-generation sequencing. Theor Appl Genet. 2016;129:1035–1044. doi: 10.1007/s00122-016-2681-7. [DOI] [PubMed] [Google Scholar]
- 53.Geng X, Jiang C, Yang J, Wang L, Wu X, Wei W. Rapid identification of candidate genes for seed weight using the SLAF-Seq method in Brassica napus. PLoS One. 2016;11:e0147580. doi: 10.1371/journal.pone.0147580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kayam G, Brand Y, Faigenboim-Doron A, Patil A, Hedvat I, Hovav R. Fine-mapping the branching habit trait in cultivated peanut by combining bulked segregant analysis and high-throughput sequencing. Front Plant Sci. 2017;8:467. doi: 10.3389/fpls.2017.00467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song J, Li Z, Liu Z, Guo Y, Qiu LJ. Next-generation sequencing from bulked-Segregant analysis accelerates the simultaneous identification of TWO qualitative genes in soybean. Front Plant Sci. 2017;8:919. doi: 10.3389/fpls.2017.00919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gu AX, Meng C, Chen YQ, Wei L, Dong H, Yin L, et al. Coupling Seq-BSA and RNA-Seq analyses reveal the molecular pathway and genes associated with leaf head pattern formation at top region in Chinese cabbage. Front Genet. 2017;8:176. doi: 10.3389/fgene.2017.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Semagn K, Babu R, Hearne S, Olsen M. Single nucleotide polymorphism genotyping using Kompetitive allele specific PCR (KASP): overview of the technology and its application in crop improvement. Mol Breed. 2014;33:1–14. doi: 10.1007/s11032-013-9917-x. [DOI] [Google Scholar]
- 58.Patterson EL, Fleming MB, Kessler KC, Nissen S, Gaines T. A KASP genotyping method to identify northern watermilfoil, eurasian watermilfoil, and their interspecific hybrids. Front Plant Sci. 2017;8:752. doi: 10.3389/fpls.2017.00752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qureshi N, Bariana HS, Zhang P, McIntosh R, Bansal K, Wong D. Genetic relationship of stripe rust resistance genes Yr34 and Yr48 in wheat and identification of linked KASP markers. Plant Dis. 2018;102:413–420. doi: 10.1094/PDIS-08-17-1144-RE. [DOI] [PubMed] [Google Scholar]
- 60.Xu X, Ji J, Xu Q, Qi X, Weng Y, Chen X. The major-effect QTL CsARN6.1 encodes an AAA-ATPase domain-containing protein that is associated with waterlogging stress tolerance through promoting adventitious root formation. Plant J. 2018;93. 10.1111/tpj.13819. [DOI] [PubMed]
- 61.Wu P, Xie J, Hu J, Qiu D, Liu Z, Li J, et al. Development of molecular markers linked to powdery mildew resistance gene Pm4b by combining SNP discovery from transcriptome sequencing data with bulked segregant analysis (BSR-Seq) in wheat. Front Plant Sci. 2018;9:95. doi: 10.3389/fpls.2018.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giovannoni JJ. Fruit ripening mutants yield insights into ripening control. Curr Opin Plant Biol. 2007;10:283–289. doi: 10.1016/j.pbi.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 63.Steuernagel B, Periyannan SK, Hernández-Pinzón I, Witek K, Rouse M, Yu G, et al. Rapid cloning of disease-resistance genes in plants using mutagenesis and sequence capture. Nat Biotechnol. 2016;34:652. doi: 10.1038/nbt.3543. [DOI] [PubMed] [Google Scholar]
- 64.Li GY, Li JF, Li YH, Go QX, Wang F. Preliminary study on physiological races differentiation of tomato leaf mold in China's three northeastern provinces. J Northeast Agric Univ. 1994;25:122–125. [Google Scholar]
- 65.Wang A, Meng F, Xu X, Wang J, Li J. Development of molecular markers linked to Cladosporium fulvum resistant gene Cf-6 in tomato by RAPD and SSR methods. Hortic Sci. 2007;42:11–15. [Google Scholar]
- 66.Fulton TM, Chunwongse J, Tanksley SD. Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol Biol Report. 1995;13:207–209. doi: 10.1007/BF02670897. [DOI] [Google Scholar]
- 67.Mckenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a map reduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reumers J, De Rijk P, Liekens A, Smeets D, Cleary J, et al. Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat Biotechnol. 2012;30:61–68. doi: 10.1038/nbt.2053. [DOI] [PubMed] [Google Scholar]
- 69.Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol. 2012;30:174–178. doi: 10.1038/nbt.2095. [DOI] [PubMed] [Google Scholar]
- 70.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S5. The KASP genotyping results. (XLSX 16 kb)
Figure S1. QTL mapping analysis by Icimapping software. (PDF 36 kb)
Table S6. The sub-cellular location prediction of Cf-10. (XLSX 14 kb)
Figure S2. Cluster analysis of candidate and other genes. A Cluster analysis on account of DNA sequence. B Cluster analysis on account of amino acid sequence. (PDF 51 kb)
Table S1. The primer information of KASP markers. (XLSX 11 kb)
Table S2. The KASP reaction system. (XLSX 10 kb)
Table S3. The primer information of EFα1 and candidate gene. (XLSX 10 kb)
Table S4. The qRT-PCR reaction system. (XLSX 10 kb)
Data Availability Statement
All the sequencing clean data were uploaded to the SRA database of NCBI (SRR6914778, SRR6914779, SRR6914776, SRR6914777). However, due to patent application, the data will be made public on 1 April 2019. However, before 1 April 2019, the datasets are available from the corresponding author on reasonable request.