

Abstract
Nineteen metabolites with diverse structures, including the rare pyrroloindoline alkaloid verrupyrroloindoline (1), the unprecedented highly fused benzosesquiterpenoid verrubenzospirolactone (2), the new asteriscane-type sesquiterpenoid 10-deoxocapillosanane D (3), and the two new cyclopentenone derivatives (4S*,5S*)-4-hydroxy-5-(hydroxymethyl)-2,3-dimethyl-4-pentylcyclopent-2-en-1-one (4) and (S)-4-hydroxy-5-methylene-2,3-dimethyl-4-pentylcyclopent-2-en-1-one (5), were isolated from a South China Sea collection of the soft coral Sinularia verruca. Eleven previously described marine metabolites (7–15, 18, and 19) were also obtained as well as three new EtOH-adduct artifacts (6, 16, and 17). The structures of the new compounds were elucidated by extensive spectroscopic analysis and by comparison with previously reported data. Compounds 4, 5, and 16 showed protection against the cytopathic effects of HIV-1 infection with EC50 values of 5.8–34 μM, and 4, 6, and 16 exhibited inhibition against LPS-induced NO production with IC50 values of 24–28 μM.
Graphical Abstract
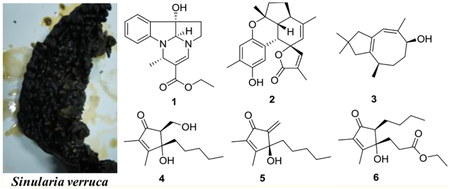
Soft corals of the genus Sinularia (family Alcyoniidae) are commonly distributed in the tropical coral reef systems of the world and are well known for being a rich source of biologically active secondary metabolites. In the past several decades, more than 50 Sinularia species have been chemically investigated, resulting in the discovery of hundreds of secondary metabolites including sesquiterpenoids, diterpenoids, polyhydroxylated steroids, cyclopentenones, and butenolides.1 Many of these metabolites are structurally novel and display a wide range of significant ecological and pharmacological activities, such as antifouling, antifeedant, allelopathy, cytotoxic, antimicrobial, and anti-inflammatory properties.2–8 In the course of our ongoing research on bioactive natural products from marine invertebrates of the South China Sea,9–11 a previously unstudied soft coral, Sinularia verruca van Ofwegen, 2008, was collected. Detailed chemical examination of this specimen led to the isolation of 19 compounds with diverse structural scaffolds, including a rare pyrroloindoline alkaloid (1), a novel benzosesquiterpenoid (2), a new asteriscane-type sesquiterpenoid (3), and two new cyclopentenone derivatives (4 and 5). Eleven known compounds were also isolated from the chemically rich extract as well as three new EtOH-adduct artifacts (6, 16, and 17). All compounds were evaluated for in vitro antiviral activity against human immunodeficiency virus type 1 (HIV-1) and for inhibitory effects on lipopolysaccharide (LPS)-induced nitric oxide (NO) production in mouse peritoneal macrophages (PEMΦ). This paper reports details of the isolation, structure elucidation, and bioactivity assess-ment of these compounds.
RESULTS AND DISCUSSION
Compound 1 had a molecular formula of C17H20N2O3 according to its HREASIMS data, implying nine degrees of unsaturation. The IR spectrum showed absorption bands attributed to hydroxy (3437 cm−1), carbonyl (1677 cm−1), and aromatic ring (1606 and 1466 cm−1) functionalities. The presence of an ortho-disubstituted benzene ring was revealed by 1H NMR signals at δH 7.31 (dd, J = 7.2, 1.2 Hz, H-3), 7.24 (td, J = 7.8, 1.2 Hz, H-5), 6.85 (t, J = 7.2 Hz, H-4), and 6.69 (d, J = 7.8 Hz, H-6) (Table 1) and 13C NMR signals at δC 150.9 (C, C-1), 131.1 (C, C-2), 131.0 (CH, C-5), 124.1 (CH, C-3), 120.3 (CH, C-4), and 111.1 (CH, C-6) (Table 2), as well as COSY correlations between H-3/H-4, H-4/H-5, and H-5/H-6 and HMBC correlations from H-3 to C-1 and from H-6 to C-2 (Figure 1). A disubstituted ethyl isopentenoate unit was established by HMBC correlations from methyl doublet H3- 14 (δH 1.41 d, J = 6.6 Hz) to methine carbon C-11 (δC 46.3) and nonprotonated olefinic carbon C-12 (δc 101.7), from H-11 (δH 4.53 d, J = 6.6 Hz) to carbonyl carbon (3–15 (δC 167.4) and olefinic methine carbon C-13 (δC 142.6), from olefinic proton H-13 (δH 7.13 s) to C-15 and C-11, and from the oxymethylene protons at δH 4.13 (q, J = 7.2 Hz) to the carbonyl carbon C-15, in combination with COSY couplings between H3-14 and H-11 and between a methyl triplet (δH 1.25 t, J = 7.2 Hz) and the oxymethylene protons mentioned above. On the basis of the remaining degrees of unsaturation, 1 had to be tetracyclic. HMBC correlations from H-3 to nonprotonated carbon C-7 (δC 84.1), from H-10 (δH 4.75 s) to C-1, C-2, and C-9 (δC 49.8, CH2), from H2-8 (δH 2.61 m) to C-2, and from H2-9 to C-7, and a COSY correlation between H2-8 and H2-9, in combination with the chemical shifts of C-1, C-7, C-9, C-10, H-9, and H-10, allowed us to establish a 2,3,8,8a-tetrahydropyrrolo[2,3-b]indol-3a(1H)-ol moiety. Moreover, HMBC correlations from H-11 to C-1 and C-10 and from H-13 to C-9 and C-10 revealed that the ethyl isopentenoate unit was connected to the two N atoms of the fused indolin-3-ol and pyrrolidine moieties via C-11 and C-13, respectively. The relative configuration of 7S*, 10S*, and 11S* was assigned based on NOESY correlations between H-6/H-11, H3-14/H-10, and H2-8/H-3 (Figure 2). Compound 1 was given the name verrupyrroloindoline. The ethyl ester is possibly an artifact because EtOH was used during the extraction process; however no evidence of a nonesterified precursor of 1 was observed during the isolation procedure. In either case, the free acid or ethyl ester form of 1 represents a rare natural alkaloid class with a 2a, 5a-diazacyclopenta[jk]fluorene skeleton.12
Table 1.
1H NMR Data for Compounds 1−6 (CDCl3, 600 MHz)a
| no. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 3.24, d (4.8) | 2.28, d (15.6) | ||||
| 2 | 1.80, dd (12.0, 4.8) | 1.99, d (15.6) | ||||
| 3 | 7.31, dd (7.2, 1.2) | 2.22, m | ||||
| 4 | 6.85, t (7.2) | 2.10, m 1.93, m |
1.78, m 1.43, m | |||
| 5 | 7.24, td (7.8, 1.2) | 1.99, m 1.37, m |
1.78, m 1.59, m | 2.71, t (7.2) | 2.34, t (6.6) | |
| 6 | 6.69, d (7.8) | 2.56, m | 4.83, br s | 4.05, dd (10.8, 7.8) | 6.05, s 5.53, s |
1.81, m 1.39, m |
| 3.88, br t (8.4) | ||||||
| 7 | 1.72, m 1.70, m |
1.87, m 1.82, m |
1.62, m 1.51, m |
|||
| 8 | 2.61, m | 4.88, s | 5.65, s | 0.87, m | 0.96, m 0.87, m |
1.39, m |
| 9 | 3.73, m 2.88, dd (18.6, 9.6) | 1.24, m | 1.22, m | 0.94, t (7.2) | ||
| 10 | 4.75, s | 7.11, s | 2.21, d (15.0) 2.01, d (15.0) |
1.24, m | 1.23, m | 2.05, m 1.93, m |
| 11 | 4.53, q (6.6) | 0.85, t (6.6) | 0.83, t (6.6) | 2.04, m | ||
| 12 | 1.04, s | 1.73, s | 1.81, s | |||
| 13 | 7.13, s | 1.96, s | 1.03, s | 1.99, s | 2.00, s | 1.70, s |
| 14 | 1.41, d (6.6) | 1.68, s | 1.73, s | 1.98, s | ||
| 15 | 1.33, s | 0.99, d (7.2) | ||||
| 3′ | 7.29, s | |||||
| 6′ | 6.54, s | |||||
| 7′ | 2.15, s | |||||
| OEt | 4.13, q (7.2) | 4.11, q (6.6) | ||||
| 1.25, t (7.2) | 1.24, t (6.6) |
The coupling constants (J) are in parentheses and reported in Hz; chemical shifts are given in ppm.
Table 2.
13C NMR Data for Compounds 1−6 (CDCl3, 150 MHz)a
| no. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 150.9, C | 36.6, CH | 36.5, CH2 | 206.1, C | 194.1, C | 204.9, C |
| 2 | 131.1, C | 50.1, CH | 142.4, C | 136.3, C | 139.4, C | 136.6, C |
| 3 | 124.1, CH | 82.0, C | 36.9, CH | 169.3, C | 166.3, C | 166.7, C |
| 4 | 120.3, CH | 39.0, CH2 | 31.0, CH2 | 81.0, C | 78.7, C | 81.5, C |
| 5 | 131.0, CH | 25.1, CH2 | 33.3, CH2 | 59.7, CH | 148.8, C | 58.9, CH |
| 6 | 111.1, CH | 40.7, CH | 71.5, CH | 59.4, CH2 | 114.7, CH2 | 24.3, CH2 |
| 7 | 84.1, C | 143.1, C | 136.7, C | 36.2, CH2 | 37.0, CH2 | 30.9, CH2 |
| 8 | 33.9, CH2 | 119.9, CH | 122.8, CH | 25.1, CH2 | 24.1, CH2 | 23.0, CH2 |
| 9 | 49.8, CH2 | 89.0, C | 129.3, C | 32.0, CH2 | 31.9, CH2 | 13.9, CH3 |
| 10 | 78.4, CH | 153.5, CH | 53.0, CH2 | 22.4, CH2 | 22.4, CH2 | 30.1, CH2 |
| 11 | 46.3, CH | 128.0, C | 29.2, C | 14.0, CH3 | 13.9, CH3 | 31.0, CH2 |
| 12 | 101.7, C | 174.0, C | 29.6, CH3 | 7.7, CH3 | 8.2, CH3 | 173.1, C |
| 13 | 142.6, CH | 10.4, CH3 | 29.6, CH3 | 11.3, CH3 | 10.7, CH3 | 8.0, CH3 |
| 14 | 21.7, CH3 | 20.2, CH3 | 17.2, CH3 | 11.3, CH3 | ||
| 15 | 167.4, C | 23.8, CH3 | 22.7, CH3 | |||
| 1′ | 146.4, C | |||||
| 2′ | 116.8, C | |||||
| 3′ | 115.4, CH | |||||
| 4′ | 147.4, C | |||||
| 5′ | 124.5, C | |||||
| 6′ | 119.7, CH | |||||
| 7′ | 15.6, CH3 | |||||
| OEt | 14.5, CH3 | 14.2, CH3 | ||||
| 59.2, CH2 | 60.9, CH2 |
The assignments were based on HMQC, HMBC, and COSY spectra.
Figure 1.

Key COSY and HMBC correlations for 1−6.
Figure 2.

Key NOE correlations and computer-generated models for 1, 2, 4, and 6. Structures 1, 2, 4a, and 6 were generated using MM2 force field calculations, while 4b was produced by MD simulation and represents a higher energy conformer of the alternative 4S*,5R*-diastereomer that is not supported by the NOE data.
Compound 2 had a molecular formula of C22H24O4 as determined by HRESIMS data, which implied 11 degrees of unsaturation. IR absorptions indicated the presence of hydroxy (3444 cm−1), carbonyl (1743 cm−1), and aromatic ring (1610, 1512, and 1450 cm−1) functionalities. The 13C NMR spectrum exhibited 22 carbon signals including an ester carbonyl (δC and 10 additional sp2 carbons, which indicated that 2 was pentacyclic. Signals for two oxygenated aliphatic carbons, four methyls, two methylenes, and three aliphatic methines were also present in the 13C NMR spectrum. A 1,2,4,5-tetrasubstituted benzene ring was established by HMBC correlations from H3-7′ (δH 2.15 s) to C-4′ (δC 147.4, C), C-5′ (δC 124.5, C), and C-6′ (δC 119.7, CH), from H-3′ (δh 7.29 s) to C-1′ (δC 146.4, C) and C-5′, and from H-6′ (δH 6.54 s) to C-2′ (δC 116.8, C) and C-4′, while an α,β-unsaturated-γ-lactone moiety was recognized by HMBC correlations from H3-13 (δH 1.96 s) to the carbonyl carbon C-12 (δC 174.0) and two olefinic carbons C-10 (δC 153.5, CH) and C-11 (δC 128.0, C) and from H-10 (δH 7.11, s) to the oxygenated nonprotonated carbon C-9 (δC 89.0) (Figure 1). COSY correlations between H-1 (δH 3.24 d, J = 4.8 Hz)/H-2 (δH 1.80 dd, J = 12.0, 4.8 Hz), H-2/H-6 (δH 2.56 m), H-6/H-5 (δH 1.99 m; 1.37 m), and H-5/H-4 (δH 2.10 m; 1.93 m) defined an extended proton spin system. HMBC correlations from H3-14 (δH 1.68 s) to the aliphatic methine carbon C-6 (δC 40.7) and two olefinic carbons C-7 (δC 143.1, C) and C-8 (δC 119.9, CH), from H3-15 (δH 1.33 s) to the other oxygenated nonprotonated carbon C-3 (δC 82.0), an aliphatic methine carbon C-2 (δC 50.1), and a methylene carbon C-4 (δC 39.0), and from H-1 to C-8 and two carbons (C-9 and C-10) of the α,β-unsaturated-γ-lactone moiety allowed us to establish the 5,6-membered ring system C/D, in which C-3 and C-7 were substituted by methyls, C-7/C-8 composed a double bond, and C-9 was a spiro carbon that was also a constituent of the α,β-unsaturated-γ-lactone. Direct linkage of the substituted benzene ring to C-1 was indicated by the chemical shift of H-1 and confirmed by HMBC correlations from H-1 to C-1′, C-2′, and C-3′. An ether linkage between C-3 and C-1′ was supported by their chemical shifts (δC 82.0 and 146.4, respectively) and the remaining unsaturation equivalent, which thus required C-4′ to be hydroxylated. The relative configurations of the stereogenic centers in 2 were assigned on the basis of coupling constants and NOESY analysis. The coupling constant values JH-1/H-2 (4.8 Hz) and JH-2/H-6 (12.0 Hz) uggested the cis and trans fusions of rings B/D and C/D, respectively, while NOESY correlations between H3-15/H-1, H-1/H-2, and H-1/H-10 indicated that H3-15, H-1, H-2, and H-10 had the same facial orientation (Figure 2). Thus, compound 2 was established to be a highly fused benzosesquiterpenoid lactone, and it was named verrubenzo-spirolactone. It is interesting to note that 2 represents a novel skeleton that could be derived from capillobenzopyranol 1213 via a series of oxidation and cyclization reactions.
Compound 3 was assigned a molecular formula of C15H24O based on its HRESIMS data, requiring four degrees of unsaturation. The 13C NMR spectrum showed 15 carbon signals including four olefinic carbons, indicating that 3 must be bicyclic. A 4,4-dimethylcyclopent-1-ene moiety was established by HMBC correlations from H3-12 (δH 1.04 s) and H3-13 (δH 1.03 s) to C-1 (δC 36.5, CH2), C-10 (δC 53.0, CH2), and C-11 (δC 29.2, C) and from H2-1 (δH 2.28 d, J = 15.6 Hz; 1.99 d, J = Hz) and H2-10 (δH 2.21 d, J = 15.0 Hz; 2.01 d, J = 15.0 Hz) to two nonprotonated olefinic carbons C-2 (δC 142.4) and C-9 (δC 129.3). COSY correlations established a spin system from H-3 to H-6 and extended to H3-15. Additional HMBC correlations from H3-14 (δH 1.73 s) to oxygenated carbon C-6 (δC 71.5, CH) and two olefinic carbons C-7 (δC 136.7, C) and C-8 (δC 122.8, CH), from H-8 (δH 5.65 s) to C-10 and C-2, and from H3-15 (δH 0.99 d, J = 7.2 Hz) to C-2 allowed connection of the structural subunits to define an asteriscanetype sesquiterpenoid. Accordingly, the planar structure of 3 was established as 6-hydroxyasterisca-2(9),7-diene. The relative configurations at C-3 and C-6 and the Z configuration of the C-7/C-8 double bond in 3 were suggested to be the same as those of the known analogues capillosananes B—D14 from Sinularia capillosa based on their very similar NMR data. As such, compound 3 was assigned as 10-deoxocapillosanane D.
Compound 4 had a molecular formula of C13H22O3 as determined by HRESIMS data, requiring three degrees of unsaturation. IR absorptions at 3393, 1693, and 1647 cm−1 indicated the presence of hydroxy, carbonyl, and olefinic functionalities. The 13C NMR spectrum showed 13 carbon signals including a conjugated ketone carbonyl (δC 206.1) and two nonprotonated olefinic carbons at δC 169.3 and 136.3 (Table 2), which accounted for two of the three degrees of unsaturation; therefore 4 was monocyclic. The 1H NMR spectrum exhibited three methyl signals including two olefinic methyl singlets (δH 1.99 s, H3-13; 1.73 s, H3-12) and a methyl triplet (δH 0.85 t, J = 6.6 Hz, H3-11) (Table 1). In addition, a tertiary oxygen-bearing carbon (δC 81.0, C-4), a secondary alcohol (δC 59.4, C-6; δH 4.05 dd, J = 10.8, 7.8 Hz, H-6a; 3.88 br t, J = 8.4 Hz, H-6b), an aliphatic methine (δC 59.7, C-5; δH 2.71 t, J = 7.2 Hz, H-5), anad four aliphatic methylenes were recognized from the 1H and 13C NMR data. An n-pentyl unit was established by COSY correlations between H3-11/H2-10 (δh 1.24 m), H2-7 (δh 1.72 m; 1.70 m)/H2-8 (δH 0.87 m), and H2-8/H2-9 (δH 1.24 m) and HMBC correlations (Figure 1) from H3-11 and H2-7 to the aliphatic methylene carbon C-9 (δC). Finally, the gross structure of 4 was established as 4-hydroxy-5-(hydroxymethyl)-2,3-dimethyl-4-pentylcyclopent-2-en-1-one based on HMBC correlations from H3-12 to the ketone carbonyl C-1 and the two olefinic carbons C-2 and C-3, from H3-13 to C-2, C-3, and the oxygenated nonprotonated carbon C-4, from H2-7 to C-3, C-4, and the methine carbon C-5, and from the oxymethylene H2-6 to C-1, C-5, and C-4. COSY correlations between H-5 and H2-6 were observed, and an NOE correlation between H2-6 and H2-7 could establish their cis relationship (4a in Figure 2) in the cyclopent-2-enone nucleus, which is much more rigid than a typical five-membered ring system, such as a cyclopentane ring. In order to support this assignment, a set of 500 conformers of the trans isomer 4b (Figure 2) were modeled, and the distance between H2-6 and H2-7 was determined to be 3.6–4.8 Å, which is greater than the typical limit for seeing NOE correlations. Compound 4 was found to be closely related to the previously reported metabolite (+)-sinularone J, which has a methoxy in place of the hydroxy at C-6.15
The closely related analogue 16 was also isolated and assigned a molecular formula of C15H26O3 based on HRESIMS data. It provided NMR data (Tables 1 and 2) that were very similar to those of 4, except there were additional signals for an ethoxy group (δC 66.8 (CH2), 15.2 (CH3); δH 3.54 2H, q, J = 6.6 Hz; 1.22 3H, t, J = 6.6 Hz). Linkage of the ethoxy group to C-6 in 16 was indicated by the deshielded chemical shift of C-6 (δC 66.8) in comparison with that of 4 (C-6, δC 59.4) and further confirmed by HMBC correlations from H2-6 (δH 3.96 dd, J = 9.6, 4.0 Hz; 3.52 dd, J = 10.2, 9.6 Hz) to the ketone carbon C-1 (δC 202.7) and to the oxymethylene carbon of the ethoxy group (Figure 1). The configurations at C-4 and C-5 in 16 were tentatively determined to be the same as those in 4 since it showed similar NOE correlations between H2-6 and H2- 7 (δH 1.86 ddd, J = 13.2, 12.0, 4.2 Hz; 1.78 ddd, J = 13.2, 12.6, 5.4 Hz) and it had the same sign of specific rotation (, MeOH). The co-occurrence of 16 and 4 in the extract and the fact that EtOH was used during the extraction process suggest that compound 16 is likely a 6-O-ethyl artifact of 4.
The molecular formula of compound 5 was determined to be C13H20O2 by HRESIMS, indicating four degrees of unsaturation. NMR data analyses revealed that 5 had a structure closely related to that of 4. The only difference was loss of signals for an aliphatic methine and a secondary alcohol and the appearance of signals for an exocyclic double bond (δC 148.8 (C), 114.7 (CH2); δH 6.05 s, 5.53 s). HMBC correlations from the exomethylene protons to a ketone (δC 194.1, C-1) and a hydroxylated nonprotonated carbon (δC 78.7, C-4) revealed the exocyclic double bond was located at C-5/C-6. Thus, compound 5 was elucidated as 4-hydroxy-5-methylene-2,3-dimethyl-4-pentylcyclopent-2-en-1-one. The measured ECD spectrum of 5 was similar to the calculated ECD spectrum for the S-enantiomer, allowing the tentative assignment of the 4S configuration for 5 (Figure 3).
Figure 3.

Experimental ECD and calculated ECD spectra for 5.
A molecular formula of C16H26O4 was established for compound 6 by HRESIMS data, which required four unsaturation equivalents. The 2,3-dimethylcyclopent-2-enone nucleus was recognized from diagnostic NMR signals for conjugated ketone carbonyl C-1 (δC 204.9), two non-protonated olefinic carbons C-2 (δC 136.6) and C-3 (δC 166.7), and two olefinic methyls (δC 11.3, 8.0; δH 1.98, 1.70). An n-butyl group was established by COSY correlations between H3-9 (δH 0.94 t, J = 7.2 Hz)/H2-8 (δH 1.39 m), H2-8/H2-7 (δH 1.62 m; 1.51, m), and H2-7/H2-6 (δH 1.81 m; 1.39 m). In addition, an ethylpropanoate unit was constructed by a COSY correlation between two vicinal methylenes H2-10 (δH 2.05 m; 1.93 m) and H2-11 (δH 2.04 m) and the coupling between an oxymethylene (δH 4.11 q, J = 6.6 Hz) and the second methyl triplet (δH 1.24 t, J = 6.6 Hz), in combination with HMBC correlations from H2-10, H2-11, and the oxymethylene protons to ester carbonyl C-12 (δC 173.1). HMBC correlations from H2-10 to olefinic carbon C-3, oxygenated nonprotonated carbon C-4 (δC 81.5), and methine C-5 (δC 58.9), along with a COSY correlation between H2-6 and the methine proton H-5 (δH 2.34 t, J = 6.6 Hz), allowed connection of the ethylpropanoate and n-butyl units to C-4 and C-5, respectively. This indicated that C-4 was hydroxylated. Thus, compound 6 was established as 4-hydroxy-4-(ethyl-propanoate)-2,3-dimethyl-5-butylcyclopent-2-en-1-one, probably an ethanolysis artifact formed from the co-occurring analogue sinularone D (8).16 The absolute configurations at C-4 and C-5 were tentatively assigned to be identical to those in 8 based on biogenetic considerations and similar NOE correlations between H2-6 and H2-10 (Figure 2).
Eleven known compounds were also isolated from the S. verruca extract and identified as sinularones A (7),16 D (8),16 G (9),16 and I (10),16 (S)-5-hydroxy-3,4-dimethyl-5-pentylfuran-2(5H)-one (11),17 capillobenzopyranol (12),13 flexibilisqui-none (13),18 5-(6-hydroxy-2,5,7,8-tetramethylchroman-2-yl)-2-methylpentanoic acid methyl ester (14),19 capillosanane I (15),14 (Z)-N-(4-hydroxyphenethyl)-3-methyldodec-2-enamide,20 and (Z)-N-(3-hydroxy-4-methoxyphenethyl)-3-methyl-dodec-2-enamide,20 by comparison of their 1H and 13C NMR and MS spectroscopic data, as well as specific rotations, with those reported in the literature.
A final compound, 17, was isolated, and its molecular formula determined to be C21H36O5 on the basis of HRESIMS data. Therefore, compound 17 was isomeric with compound 10, and its NMR data also corresponded very closely with the NMR data for 10. HMBC correlations from H3-18 to the carbonyl carbon C-1 and two olefinic carbons C-2 and C-3 and from H3-19 to the two olefinic carbons and the acetal carbon C-4 disclosed the same α,β-unsaturated-2,3-dimethyl-γ-lactone unit as found in butenolide 10. An ethoxy group [δC 58.3 (CH2), 15.2 (CH3); δH 3.40 and 3.14 (both 1H, dq, J = 12.0, 6.6 Hz), 1.17 (3H, t, J = 6.6 Hz)] and 12 aliphatic methylenes were also similar to those seen in the artifact sinularone I (10).16 An HMBC correlation from the oxymethylene protons to the acetal carbon C-4 revealed the location of the ethoxy group at C-4 in 17, instead of forming an ethyltridecanoate unit as in 10. Thus, compound 17 was defined as 13-(2-ethoxy-3,4-dimethyl-5-oxo-2,5-dihydrofuran-2-yl)tridecanoic acid, which has not previously been reported in the chemical literature, and it was named isosinularone I. The absolute configuration at C-4 was tentatively assigned to be the same as that of 10 based on its similar specific rotation , MeOH).
All of the compounds obtained from the S. verruca extract were tested for anti-HIV-1 activity in a cell-based HIV screening assay.21–24 Compounds 4, 5, and 16 were found to be moderately protective against the cytopathic effects of in vitro HIV-1 infection with EC50 values of 34, 5.8, and 30 μM, respectively, and maximum protection rates of 86%, 52%, and 75%, respectively. However, their IC50 values for cytotoxicity against the CEM-SS host cells were determined to be 79, 6.3, and 66 μM, indicating low therapeutic indices. The other compounds tested showed no detectable antiviral activity in this assay.
In addition, all compounds were evaluated for their in vitro anti-inflammatory activities.25 In the primary assay, 4, 6, and 16 showed moderate inhibition against lipopolysaccharide-induced NO production in mouse peritoneal macrophages with IC50 values of 28, 24, and 26 μM, respectively, whereas no inhibitory effect (inhibition <50%) was observed for the other compounds at 30 μM.
EXPERIMENTAL SECTION
General Experimental Procedures.
Optical rotations, UV, and IR spectra were measured using a PoLAAR 3005 digital polarimeter, a TU 1901 spectrometer, and a Bruker Equinox 55 spectrometer, respectively. 1H and 13C NMR spectra were acquired with a Bruker Avance-600FT NMR spectrometer using TMS as an internal standard. HRESIMS data were obtained using a Thermo Scientific Q Exactive hybrid quadrupole-Orbitrap mass spectrometer. Column chromatography was performed using silica gel (200–300 mesh, Qingdao Marine Chemistry Co. Ltd.), Sephadex LH-20 (GE Healthcare Biosciences AB), and ODS (50 μm, YMC). Silica gel GF254 (Qingdao Marine Chemistry Co. Ltd.) was used for TLC analysis. HPLC separation was performed using an Agilent 1100 series instrument equipped with a VWD G1314A detector and a YMC-Pack C18 column (10 μm, 250 × 10 mm).
Animal Material.
Specimens of the soft coral Sinularia verruca van Ofwegen, 2008, were collected from the inner coral reef of Ximao Island, Hainan Province, China, in November 2011, at a depth of 8 m and frozen immediately after collection. The specimen was identified by one of the authors (X.L.). A voucher specimen (HS201102) was deposited at the Laboratory of Marine Natural Products Chemistry, Wenzhou Medical University, China.
Extraction and Isolation.
The frozen soft coral S. verruca (wet weight: 1.20 kg) was homogenized and extracted with 95% EtOH at room temperature (rt). The concentrated extract was partitioned between H2O and EtOAc. Evaporation of EtOAc in vacuo yielded a dark residue of 22.5 g. The EtOAc fraction (22.0 g) was subjected to silica gel vacuum column chromatography, eluting with a gradient of EtOAc/petroleum ether (1:20, 1:10, 1:5, 1:3, and 1:2), to obtain eight fractions (A1–A8). Fraction A3 (700.2 mg) was chromatographed on a Sephadex LH-20 column, eluting with CH2Cl2/MeOH (1:1), to afford four fractions (A3a–A3d). Fraction A3b (100.5 mg) was further separated on an ODS column, eluting with MeOH/H2O (75:25, 80:20), to afford 8 (6.4 mg) and 11 (26.1 mg). Fraction A3d (60.0 mg) was purified by semipreparative HPLC, using MeOH/H2O (75:25) as eluent, to yield 5 (6.0 mg) and 15 (22.5 mg). Fraction A4 (1.1 g) was subjected to a Sephadex LH-20 column, eluting with CH2Cl2/MeOH (1:1), to obtain three fractions (A4a–A4c). Fraction A4c (798.1 mg) was further separated on an ODS column, eluting with MeOH/H2O (70:30, 75:25, 80:20, 85:15, and 90:10), to afford seven fractions (A4c1–A4c7). Fraction A4c2 (40.0 mg) was purified by HPLC (MeOH/H2O, 60:40) to yield 9 (3.5 mg) and 1 (1.5 mg). Fraction A4c3 (19.8 mg) was purified by HPLC (MeOH/H2O, 62:38) to afford 6 (3.3 mg). Fraction A4c4 (30.2 mg) was purified by HPLC (MeOH/H2O, 70:30) to yield 13 (14.0 mg) and 14 (2.2 mg). Fraction A4c5 (70.3 mg) was purified by HPLC (MeOH/H2O, 70:30) to obtain (Z)-N-(3-hydroxy-4-methoxyphenethyl)-3-methyldodec-2-enamide (5.0 mg), 3 (6.3 mg), and 7 (2.6 mg). Fraction A4c6 (90.2 mg) was purified by HPLC (MeOH/H2O, 80:20) to obtain 17 (5.2 mg), 12 (11.3 mg), and (Z)-N-(4-hydroxyphenethyl)-3-methyldodec-2-enamide (23.5 mg). Fraction A6 (1.5 g) was subjected to a Sephadex LH-20 column, using CH2Cl2/MeOH (1:1) as eluent, to obtain three fractions (A6a–A6c). Fraction A6c (580.1 mg) was fractionated on an ODS column, eluting with MeOH/H2O (65:35, 70:30, 75:25, and 80:20), to afford five fractions (A6c1–A6c5). Fraction A6c2 (19.3 mg) was purified by HPLC (MeOH/H2O, 70:30) to yield 16 (2.6 mg) and 2 (2.0 mg). Following the same protocol as for fraction A6c2, 4 (1.2 mg) and 10 (1.3 mg) were separated from fraction A6c4 (10.5 mg) using MeOH/H2O (80:20) as a mobile phase.
Verrupyrroloindoline (1): colorless oil; (c 0.06, CHCl3); UV (MeOH) λmax (log ε) 205 (3.96), 242 (3.55), 288 (3.76); IR (KBr) vmax 3437, 2932, 2873, 1677, 1607, 1466, 1383, 1237, 1148, 1105, 1048, 756 cm−1; 1H and 13C NMR data, Tables 1 and 2; HRESIMS m/z 323.1369 [M + Na]+ (calcd for C17H20N2O3Na, 323.1372).
Verrubenzospirolactone (2): colorless oil; (c 0.10, CHCl3); UV (MeOH) λmax (log ε) 203 (4.68), 296 (3.37); IR (KBr) νmax 3444, 2929, 2862, 1743, 1647, 1610, 1512, 1450, 1413, 1381, 1186, 1049 cm−1; 1H and 13C NMR data, Tables 1 and 2; HRESIMS m/z 375.1567 [M + Na]+ (calcd for C22H24O4Na, 375.1572).
10-Deoxocapillosanane D (3): colorless oil; (c 0.04, CHCl3); UV (MeOH) λmax (log ε) 240 (3.73); IR (KBr) νmax 3432, 2925, 1645, 1459, 1385, 1051, 997 cm−1; 1H and 13C NMR data, Tables 1 and 2; HRESIMS m/z 203.1791 [M + H − H2O]+ (calcd for C15H23, 203.1800).
(4S*,5S*)-4-Hydroxy-5-(hydroxymethyl)-2,3-dimethyl-4-pentylcy-clopent-2-en-1-one (4): colorless oil; (c 0.05, MeOH); UV (MeOH) λmax (log ε) 232 (3.68); IR (KBr) νmax 3393, 2923, 2855, 1693, 1647, 1382, 1048 cm−1; 1H and 13C NMR data, Tables 1 and 2; HRESIMS m/z 249.1465 [M + Na]+ (calcd for C13H22O3Na, 249.1467).
(S)-4-Hydroxy-5-methylene-2,3-dimethyl-4-pentylcyclopent-2-en-1-one (5): colorless oil; (c 0.10, MeOH); UV (MeOH) λmax (log ε) 259 (3.72); ECD (MeOH) 257 (Δε +2.53) nm; IR (KBr) νmax 3454, 2927, 2861, 1694, 1631, 1386, 1033 cm−1; 1H and 13C NMR data, Tables 1 and 2; HRESIMS m/z 231.1359 [M + Na]+ (calcd for C13H20O2Na, 231.1361).
(4S,5S)-4-Hydroxy-4-(ethylpropanoate)-2,3-dimethyl-5-butylcy-clopent-2-en-1-one (6): colorless oil; (c 0.08, MeOH); UV (MeOH) λmax (log ε) 229 (3.77); IR (KBr) νmax 3446, 2930, 2866, 1776, 1707, 1650, 1461, 1384, 1165, 1018 cm−1; 1H and 13C NMR data, Tables 1 and 2; HRESIMS m/z 305.1726 [M + Na]+ (calcd for C16H26O4Na, 305.1729).
Sinularone D (8): colorless oil; (c 0.10, MeOH); lit. (c 0.09, MeOH).16
(4S*,5S*)-4-Hydroxy-5-(ethoxymethyl)-2,3-dimethyl-4-pentylcy-clopent-2-en-1-one (16): colorless oil; (c 0.10, MeOH); UV (MeOH) λmax (log ε) 233 (3.63); IR (KBr) νmax 3440, 2928, 2865, 1700, 1651, 1458, 1382, 1110, 1042 cm−1; 1H NMR (600 MHz, CDCl3) δ 3.96 (1H, dd, J = 9.6, 4.0 Hz, H-6a), 3.54 (2H, q, J = 6.6 Hz), 3.52 (1H, dd, J = 10.2, 9.6 Hz, H-6b), 2.84 (1H, dd, J = 10.2, 4.0 Hz, H-5), 1.98 (3H, s, H3-13), 1.86 (1H, ddd, J = 13.2, 12.0, 4.2 Hz, H-7a), 1.78 (1H, ddd, J = 13.2, 12.6, 5.4 Hz, H-7b), 1.71 (3H, s, H3-12), 1.22 (3H, t, J = 6.6 Hz), 1.23 (2H, m, H-9), 1.23 (2H, m, H-10), 0.85 (1H, m, H-8a), 0.85 (3H, t, J = 6.6 Hz, H3-11), 0.80 (1H, m, H-8b); 13C NMR (150 MHz, CDCl3) δ 202.7 (C, C-1), 168.2 (C, C-3), 136.1 (C, C-2), 81.3 (C, C-4), 66.8 (CH2, C-6), 66.8 (CH2), 59.4 (CH, C-5), 36.6 (CH2, C-7), 32.1 (CH2, C-9), 25.2 (CH2, C-8), 22.4 (CH2, C-10), 15.2 (CH3), 14.0 (CH3, C-11), 11.3 (CH3, C-13), 7.8 (CH3, C-12); HRESIMS m/z 277.1776 [M + Na]+ (calcd for C15H26O3Na, 277.1780).
Isosinularone I (17): colorless oil; (c 0.08, MeOH); UV (MeOH) λmax (log ε) 204 (4.24), 228 (sh); IR (KBr) νmal 3448, 2926, 2861, 1727, 1636, 1383, 1277, 1120, 1074 cm−1; 1H NMR (600 MHz, CDCl3) δ 3.40 (1H, dq, J = 12.0, 6.6 Hz), 3.14 (1H, dq, J = 12.0, 6.6 Hz), 2.34 (2H, m, H2-16), 1.98 (1H, ddd, J = 13.8, 12.0, 3.6 Hz, H-5a), 1.86 (3H, s, H3-19), 1.84 (3H, s, H3-18), 1.67 (1H, m, H-5b), 1.62 (2H, m, H2-15), 1.30 (2H, m, H-6), 1.25 (16H, H2-7-H2-14), 1.17 (3H, t, J = 6.6 Hz); 13C NMR (150 MHz, CDCl3) δ 178.4 (C, C-17), 172.0 (C, C-1), 156.6 (C, C-3), 126.9 (C, C-2), 110.1 (C, C-4), 35.8 (CH2, C-5), 34.1 (CH2, C-16), 29.5 (CH2, C-7), 29.5 (CH2, C-8), 29.5 (CH2, C-9), 29.5 (CH2, C-10), 29.4 (CH2, C-11), 29.4 (CH2, C-12), 29.2 (CH2, C-13), 29.1 (CH2, C-14), 24.9 (CH2, C-15), 22.6 (CH2, C-6), 10.9 (CH3, C-19), 8.4 (CH3, C-18); HRESIMS m/z 367.2495 [M − H]− (calcd for C21H35O5, 367.2484).
Conformational Modeling.
The electrostatic potential of molecule 4b was calculated at the Hartree–Fock level with the 6–31G* basis set using Gaussian09. The general AMBER force field (gaff) and the ff99SB force field were used for 4b and CHCl3 molecules, respectively. Molecule 4b was solvated in a box of CHCl3 molecules with a shell of 10 Å. Energy minimization and molecular dynamic (MD) simulation were performed with the AMBER 11 packages. First, in order to remove bad contacts, the whole system was subjected to 500 steps of minimization without any restriction. Then, the system was heated gradually from 0 to 300 K in the NVT ensemble for 100 ps and equilibrated at 300 K for 100 ps prior to MD simulation. The 500 ps of MD simulation was accomplished at 300 K under a constant pressure of 1 bar. The particle mesh Ewald method was employed to deal with the long-range electrostatic interactions under periodic boundary conditions. The SHAKE method was used to constrain hydrogen atoms, and the time step was set to 2 fs. Finally, Ptraj, a module of AMBER 11 packages, was used to monitor the distance between H2-6 and H2-7 in 4b.
ECD Calculation.
The MD simulations were accomplished for a pair of enantiomers of 5 following the same protocol as depicted for 4b except that methanol was used as solvent for 5. The lowest energy conformers were optimized by CAM-B3LYP/6–311++G** in methanol. The solvent effects were included using the polarizable continuum model. The theoretical ECD spectra were obtained according to Boltzmann weighting of each conformer.
Anti-HIV-1 Activity Assay.
The XTT-tetrazolium primary NCI screen for anti-HIV activity was performed as described previously.21–24 The CEM-SS human lymphocytic target cell line was maintained in RPMI 1640 medium (Gibco) without phenol red and supplemented with 5% fetal bovine serum, 50 μg/mL gentamicin, and 2 mM l-glutamine (complete medium). Exponentially growing cells were pelleted and resuspended in complete medium at a concentration of 2 × 105 cells/mL. Frozen virus stock solution (Haitian variant HIV-1RF, 3.54 × 106 SFU/mL) was thawed immediately prior to use and resuspended in complete medium to yield 1.2 × 105 SFU/mL. Each test compound was dissolved in 100% DMSO and then diluted to the desired initial concentrations in complete medium.
Uninfected CEM-SS cells were plated in 50 μL of complete medium at a density of 1 × 104 cells. Diluted HIV-1 virus was then added in a volume of 50 μL to yield a multiplicity of infection of 0.6. Appropriate cell, virus, and positive control (AZT) were incorporated in each experiment, and the final volume in each microtiter well was 200 μL. Quadruplicate and duplicate wells were used for virus-infected and uninfected cells, respectively. Plates were incubated for 6 days at 37 °C in an atmosphere containing 5% CO2. Subsequently, aliquots of cell-free supernatant were removed from each well. Cellular growth then was estimated on the remaining contents of each well using the XTT assay as previously described.23,24
Assay for Inhibition of Nitric Oxide Production.
A previously established protocol25 was followed except that 30 mM dexamethasone in DMSO was used as the positive control and each test compound (30 mM in DMSO) was diluted to 1–30 μM before the experiment.
Supplementary Material
Chart 1.

ACKNOWLEDGMENTS
This work was supported by grants from NSFC (No. 21202123), ZJNSF (No. LQ12B02002), CSC (No. 201408330121), and Start-Up Funding from Wenzhou Medical University (No. QTJ10018). This research was also supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnat-prod.6b00031.
1D and 2D NMR and HRESIMS spectra of the new compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).MarinLit database, Department of Chemistry; University of Canterbury: http://www.chem.canterbury.ac.nz/marinlit/marinlit.shtml.
- (2).Chen BW; Chao CH; Su JH; Huang CY; Dai CF; Wen ZH; Sheu JH Tetrahedron Lett. 2010, 51, 5764–5766. [Google Scholar]
- (3).Cheng SY; Huang KJ; Wang SK; Duh CY Mar. Drugs 2011, 9, 1469–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Radhika P; Rao PVS; Anjaneyulu V; Asolkar RN; Laatsch HJ Nat. Prod 2002, 65, 737–739. [DOI] [PubMed] [Google Scholar]
- (5).Shen S; Zhu H; Chen D; Liu D; van Ofwegen L; Proksch P; Lin W Tetrahedron Lett. 2012, 53, 5759–5762. [Google Scholar]
- (6).Pawlik JR Chem. Rev 1993, 93, 1911–1922. [Google Scholar]
- (7).Lai D; Li Y; Xu M; Deng Z; van Ofwegen L; Qian P; Proksch P; Lin W Tetrahedron 2011, 67, 6018–6029. [Google Scholar]
- (8).Kamel HN; Slattery M Pharm. Biol 2005, 43, 253–269. [Google Scholar]
- (9).Zhao M; Yin J; Jiang W; Ma M; Lei X; Xiang Z; Dong J; Huang K; Yan P Mar. Drugs 2013, 11, 1162–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yin J; Zhao M; Ma M; Xu Y; Xiang Z; Cai Y; Dong J; Lei X; Huang K; Yan P Mar. Drugs 2013, 11, 455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zhao M; Li X; Zhao F; Cheng S; Xiang Z; Dong J; Huang K; Yan P Chem. Pharm. Bull 2013, 61, 1323–1328. [DOI] [PubMed] [Google Scholar]
- (12).Jackson AH; Shannon PVR; Wilkins DJJ Chem. Soc., Chem. Commun 1987, 9, 653–654. [Google Scholar]
- (13).Cheng SY; Huang KJ; Wang SK; Wen ZH; Chen PW; Duh CY J. Nat. Prod 2010, 73, 771–775. [DOI] [PubMed] [Google Scholar]
- (14).Chen D; Chen W; Liu D; van Ofwegen L; Proksch P; Lin WJ Nat. Prod 2013, 76, 1753–1763. [DOI] [PubMed] [Google Scholar]
- (15).Zhang NX; Tang XL; van Ofwegen L; Xue L; Song WJ; Li PL; Li GQ Chem. Biodiversity 2015, 12, 273–283. [DOI] [PubMed] [Google Scholar]
- (16).Shi H; Yu S; Liu D; van Ofwegen L; Proksch P; Lin W Mar. Drugs 2012, 10, 1331–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wu J; Tsujimori M; Hirai H; Kawagishi H Biosci., Biotechnol., Biochem 2011, 75, 783–785. [DOI] [PubMed] [Google Scholar]
- (18).Lin YF; Kuo CY; Wen ZH; Lin YY; Wang WH; Su JH; Shen JH; Sung PJ Molecules 2013, 18, 8160–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Pope SAS; Burtin GE; Clayton PT; Madge DJ; Muller DP R. Bioorg. Med. Chem 2001, 9, 1337–1343. [DOI] [PubMed] [Google Scholar]
- (20).Kazlauskas R; Marwood JF; Wells RJ Aust. J. Chem 1980, 33, 1799–1803. [Google Scholar]
- (21).Weislow OS; Kiser R; Fine DL; Bader J; Shoemaker RH; Boyd MR J. Natl. Cancer Inst 1989, 81, 577–586. [DOI] [PubMed] [Google Scholar]
- (22).Gulakowski RJ; McMahon JB; Stately PG; Moran RA; Boyd MR J. Virol. Methods 1991, 33, 87–100. [DOI] [PubMed] [Google Scholar]
- (23).Gustafson KR; Munro MHG; Blunt JW; Cardellina JH II; McMahon JB; Gulakowski RJ; Cragg GM; Cox PA; Brinen LS; Clardy J; Boyd MR Tetrahedron 1991, 47, 4547–4554. [Google Scholar]
- (24).Gustafson KR; Cardellina JH II; McMahon JB; Gulakowski RJ; Ishitoya J; Szallasi Z; Lewin NE; Blumberg PM; Weislow OS; Beutler JA; Buckheit RW Jr.; Cragg GM; Cox PA; Bader JP; Boyd MR J. Med. Chem 1992, 35, 1978–1986. [DOI] [PubMed] [Google Scholar]
- (25).Yan P; Deng Z; van Ofwegen L; Proksch P; Lin W Mar. Drugs 2010, 8, 2837–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.