

Abstract
Microglia are engineers of the CNS both in health and disease. In addition to the canonical immunological roles of clearing damaging entities and limiting the spread of toxicity and death, microglia remodel the CNS throughout life. While they have been extensively studied in disease and injury, due to their highly variable functions, their precise role in these contexts still remains uncertain. Over the last decade we have greatly expanded our understanding of microglial function, including their essential homeostatic roles during development. Here, we review these developmental roles, identify parallels in disease, and speculate whether developmental mechanisms reemerge in disease and injury.
Introduction
Microglia, the parenchymal central nervous system (CNS) macrophage, are one of the most dynamic and paradoxical cell types in the brain. Along with other CNS macrophages, microglia derive from erythromyeloid progenitors in the yolk sac (Ginhoux et al., 2010) through a process that requires PU.1 and IRF8 (Kierdorf et al., 2013). They populate the developing CNS at early embryonic stages (Reemst et al., 2016) and expand by proliferation to eventually constitute 5–15% of total CNS cells in adulthood (Tay et al., 2016). Unlike other tissue macrophages, microglia persist for the life of the organism with slow turnover rates at steady state (Ajami et al., 2007; Askew et al., 2017; Réu et al., 2017; Tay et al., 2017).
Microglia have highly motile processes that contact neighboring CNS cells and are estimated to survey the entire brain every few hours (Nimmerjahn et al., 2005). With a vast sensome, microglia rapidly detect changes within their environment (Hickman et al., 2013) and respond primarily by the release of secreted factors or phagocytosis. Historically, microglia have been studied in contexts of disease and injury due to their contribution to the inflammatory milieu of the CNS. Yet, our understanding of their role in these contexts remains controversial since they can be neuroprotective but also exacerbate pathology through release of cytotoxic factors or phagocytosis of neuronal compartments (Salter and Stevens, 2017).
As dynamic cells, microglia make essential contributions to CNS development. They regulate neurogenesis, gliogenesis, and neuronal migration in addition to facilitating neural connectivity by regulating axon outgrowth, myelination, and remodeling synaptic connections (Sato, 2015; Frost and Schafer, 2016; Reemst et al., 2016; Li and Barres, 2017). As we learn more about the spectrum of microglia functions and how they are regulated, there is suggestion of intriguing interplay between their roles in development and disease. Here, we investigate whether developmental functions of microglia have parallels in pathological contexts. We discuss populations of microglia in development and disease that have gene expression profiles deviating from the microglial homeostatic signature. We review developmental roles of microglia and highlight related functions in disease and injury, including shared molecular pathways. We also draw attention to known developmental roles of disease-associated genes that are enriched in microglia. Further investigation of microglial function during normal CNS development is likely to yield significant insight into the roles of microglia in complex contexts of disease.
Microglial transcriptome in CNS development and disease
Adult microglia in the healthy CNS have a unique gene expression profile compared to other CNS cell types or myeloid cells (Gautier et al., 2012; Beutner et al., 2013; Chiu et al., 2013; Butovsky et al., 2014; Zhang et al., 2014) consistent with their specialized and essential functions. Although microglia gene expression overlaps with tissue macrophages and other immune cells, multiple transcriptional profiling studies have identified a distinct core signature that includes genes such as Tmem119, P2ry12, Hexb, Sall1 and Fcrls (Crotti and Ransohoff, 2016).
While all microglia express core signature genes, transcriptome analysis has also revealed remarkable heterogeneity in development (Hagemeyer et al., 2017; Wlodarczyk et al., 2017; De et al., 2018), across various brain regions (Butovsky et al., 2014; Grabert et al., 2016; De Biase et al., 2017; Stowell et al., 2017; Tay et al., 2017; Ayata et al., 2018), and in disease and injury (Hanisch, 2013; Wlodarczyk et al., 2014; Bachstetter et al., 2015; Kamphuis et al., 2016; Keren-Shaul et al., 2017; Friedman et al., 2018; Mrdjen et al., 2018), suggesting that microglial gene expression and phenotype is highly dynamic and context-dependent. As we discuss below, there are shifts in molecular phenotype during both CNS development and aging/disease. While the molecular changes occurring in these two contexts are distinct, overlap in sets of genes that are up and down-regulated imply that they are potentially related. Whether there are common mechanisms driving these gene expression changes under diverse conditions remains unclear.
Development of brain microglia
After populating the embryonic brain, microglia progress through a developmental program with stepwise changes in gene expression and chromatin accessibility that is linked to environmental changes (Gosselin et al., 2014; Lavin et al., 2014) as well as the functional demands of the developing CNS (Bennett et al., 2016; Matcovitch-Natan et al., 2016; Thion et al., 2018). Transcription factors PU.1 and IRF8 are required for the maturation of microglia precursors (Kierdorf et al., 2013) and key transcriptional regulators such as SALL1 and MAFB are essential for maintaining microglial homeostasis and identity (Buttgereit et al., 2016; Matcovitch-Natan et al., 2016; Holtman et al., 2017) (Figure 1). Through this developmental process there is gradual acquisition of expression of adult homeostatic genes, such as Tmem119, P2ry12, and Hexb (Butovsky et al., 2014; Bennett et al., 2016; Matcovitch-Natan et al., 2016). The proliferation, development, and survival of microglia are regulated by Interleukin 34 (IL-34) and Colony stimulating factor 1 (CSF1) signaling through the receptor CSF1R (Elmore et al., 2014; Chitu et al., 2016), while microglial specification and homeostatic gene expression is dependent upon Transforming growth factor β (TGFβ (Butovsky et al., 2014; Gosselin et al., 2014; Buttgereit et al., 2016) (Figure 1) as well as additional brain-derived signals (Bohlen et al., 2017). Although some intrinsic and extrinsic factors that regulate the differentiation and survival of microglia have been identified, the precise signals that govern transitions in microglial gene expression and phenotype across development remain to be defined.
Figure 1. Microglia have distinct functional states in development, health, and disease.
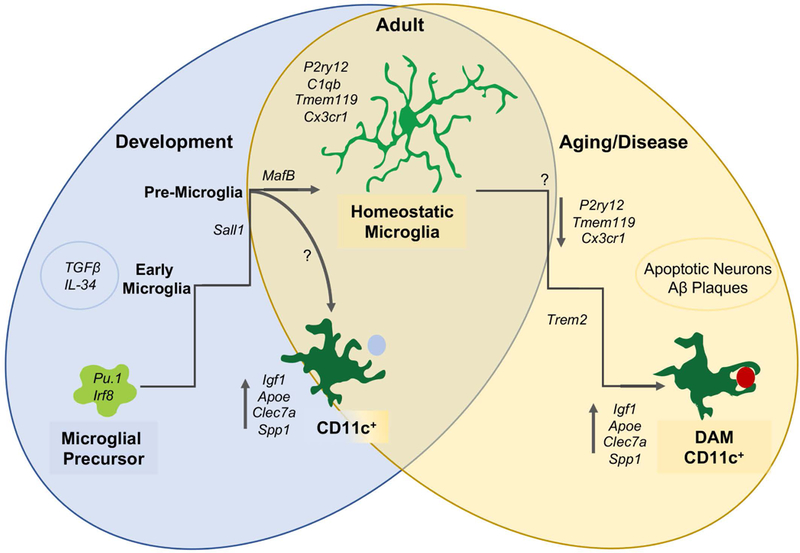
During early development, microglial precursors migrate to the brain and mature in a stepwise process into early, pre-, and adult microglia. During the early postnatal period, a subpopulation of CD11c-expressing microglia with a distinct profile localize to developing white matter tracts. During aging and disease, microglia downregulate homeostatic genes and upregulate disease-associated genes, a subset of which are common to developmental CD11c+ microglia. (DAM = disease-associated microglia).
Diversification of microglia in development
Interestingly, in early postnatal brain, there also exists a transcriptionally distinct subset of microglia marked by expression of CD11c (or Complement receptor 4) (Hagemeyer et al., 2017; Wlodarczyk et al., 2017). CD11c+ microglia transiently emerge in the developing white matter of the corpus callosum and cerebellum with peak density at postnatal day 3 (P3), representing approximately 15% of total brain microglia (Wlodarczyk et al., 2017). CD11c is not normally expressed by microglia (Wlodarczyk et al., 2015) but lineage analysis has confirmed that this developmental population derives from microglial precursors (Hagemeyer et al., 2017). Transcriptional analysis of CD11c+ microglia versus total brain microglia reveals enrichment of Insulin-like growth factor 1 (Igf1), as well as multiple other notable genes such as Spp1, Apoe, Gpnmb, and Clec7a, suggesting that this population represents a unique functional state of developing microglia (Figure 1). CD11c+ microglia are essential for the proper development of oligodendrocytes (Hagemeyer et al., 2017) and in regulating myelination via secretion of IGF1 (Wlodarczyk et al., 2017). Notably, clusters of repopulating microglia after ablation are CD11c+ and highly upregulate Igf1, Spp1 and Gpnmb (Wlodarczyk et al., 2017).
Disease-associated microglia
Recent population-wide transcriptome studies reveal microglia shift their molecular phenotype with age, injury, and disease (Holtman et al., 2015; Keren-Shaul et al., 2017; Krasemann et al., 2017; Friedman et al., 2018; Mrdjen et al., 2018). For example, microglia purified from mouse brain in aging and multiple models of neurodegeneration share a common disease-associated gene signature involving loss of homeostatic gene expression (P2ry12, Tmem119, Cx3cr1, etc) and induction of genes associated with phagocytosis (e.g., Axl, Clec7a) and lipid metabolism (e.g., Apoe, Lpl) (Holtman et al., 2015; Keren-Shaul et al., 2017; Krasemann et al., 2017) (Figure 1). These functional states diverge from resting/activated nomenclature and differ from the shifts in microglial gene expression in response to tumor, LPS stimulation, or viral infection although down-regulation of homeostatic genes is a common feature (Holtman et al., 2015; Friedman et al., 2018). Meta-analysis of microglial gene expression from diverse models of disease identified gene modules that are co-regulated in multiple contexts suggesting distinct activation states (Holtman et al., 2015; Friedman et al., 2018).
Interestingly, not all microglia are uniform in these disease states. Single cell analysis of microglia purified from brains of mouse models of Alzheimer’s disease (AD) identified microglia subsets dubbed “disease-associated microglia” (DAM) (Keren-Shaul et al., 2017). Acquisition of aspects of the DAM signature in neurodegeneration requires signaling through the triggering receptor expressed on myeloid cells 2 (TREM2) as well as apolipoprotein E (APOE) expression (Keren-Shaul et al., 2017; Krasemann et al., 2017) (Figure 1). Further meta-analysis has suggested that additional subsets of microglia expressing genes associated with proliferation and interferon-related response may be present in AD (Friedman et al., 2018). Thus there may be multiple subsets of microglia with distinct gene expression profiles, and potentially specialized functions, associated with a given disease state.
Notably, DAM cells were also enriched for Itgax, the gene encoding CD11c, and highly expressed Igf1 (Keren-Shaul et al., 2017). Itgax was also found to be upregulated in global molecular profiling of microglia in aging, AD, Multiple sclerosis (MS), and amyotrophic lateral sclerosis (ALS) (Holtman et al., 2015; Krasemann et al., 2017; Mrdjen et al., 2018). CD11c+ microglia are virtually absent in the healthy adult brain but increase with age (Wlodarczyk et al., 2015; Mrdjen et al., 2018), particularly in white matter (Raj et al., 2017). CD11c+ microglia associate with Aβ plaques in AD, and are also present in human AD patient brains (Kamphuis et al., 2016; Hopperton et al., 2017). CD11c+ microglia are largely considered to be neuroprotective in AD, as numbers are inversely associated with plaque load (Butovsky et al., 2006a). CD11c+ microglia also increase and are considered neuroprotective in experimental autoimmune encephalomyelitis (EAE), a mouse model of MS (Wlodarczyk et al., 2014; Wlodarczyk et al., 2015). While developmental CD11c+ microglia and EAE CD11c+ microglia share a unique set of genes, direct comparison indicates they are more molecularly distinct than similar (Wlodarczyk et al., 2017). Thus, CD11c+ microglia in development and in disease may represent diverse subclasses, although it remains possible that they have overlapping functions.
Summary
These studies hint that some aspects of developmental gene expression programs may reemerge in contexts of neurodegeneration, and that subsets of microglia associated with disease may share features with developmental microglia. In fact, genes that are expressed early in microglial development, such as Apoe, decline as microglia mature then are upregulated with disease (Krasemann et al., 2017). In addition, complement proteins such as C1q and C3 have high levels of expression during CNS development, aging, and neurodegeneration, but fairly low baseline expression throughout adulthood (Rajendran and Paolicelli, 2018) (Figure 3). There is also regional heterogeneity since microglia from adult cerebellum are enriched for genes characteristic of DAM as well as immature microglia, including genes associated with phagocytic and clearance functions (Ayata et al., 2018). Since the phenotype of microglia is highly context dependent, it is likely that additional populations of microglia representing specific functional states, and performing distinct tasks, will be identified. With increasing evidence that microglia can be both neuroprotective and neurotoxic, it is essential to understand how different functional states of microglia are acquired and modified. As we discuss below, overlapping molecular pathways may be involved in microglial function in development, injury, and disease, so more direct comparison of microglia in these different contexts is likely to provide deeper insight into their functional roles.
Figure 3. Genes implicated in disease are highly expressed in development and drive parallel functions.
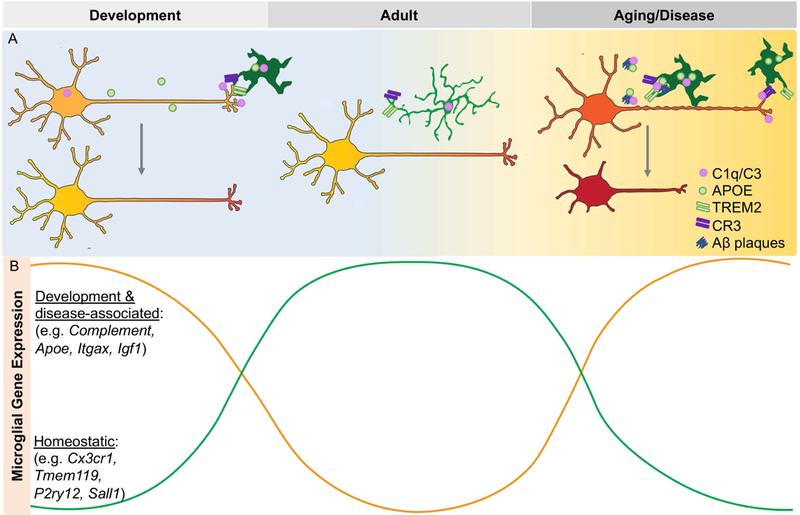
Microglia utilize the same signaling pathways to regulate synaptic pruning and phagocytosis in development and disease (A). Genes implicated in disease are developmentally regulated and have inverse expression with homeostatic genes (B).
Functional roles of microglia in CNS development, aging, and disease
The dynamic molecular phenotypes of microglia reflect their complex roles. They influence multiple aspects of neuronal health and function throughout life (Figure 2), with effects on proliferation, differentiation, survival, cell migration, and neural connectivity (Sato, 2015; Frost and Schafer, 2016; Reemst et al., 2016; Li and Barres, 2017).
Figure 2. Neurons are remodeled by microglia over their lifetime.
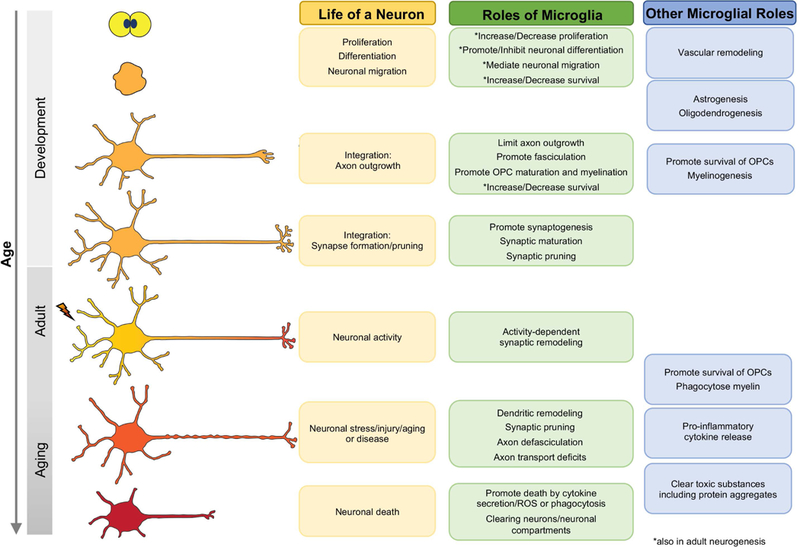
Microglia enter the neuroepithelium and influence progenitors, neuronal differentiation, migration, and survival. They then remodel neuronal structures throughout development, adulthood and in aging, injury, and disease. Microglia also regulate gliogenesis and vascular development, and clear damaging entities and release cytokines in aging and disease.
Neural progenitor proliferation and differentiation during development and adulthood
Microglia populate the CNS prior to the bulk of neurogenesis and interact with both neural progenitors and neurons, with influence on neurogenesis and gliogenesis in diverse contexts (Su et al., 2014; Sato, 2015). CNS progenitors undergo significant proliferative expansion in order to produce the requisite number of neurons, and microglia or microglial conditioned media (MCM) can enhance proliferation (Morgan et al., 2004; Antony et al., 2011), although the effects may be context dependent since there are examples where microglia have no effect (Walton et al., 2006) (Table 1). Interestingly, microglia can stimulate differentiation of different CNS populations depending on their activation state. They can promote and inhibit neurogenesis, stimulate oligodendrocyte development, and increase astrocyte differentiation (Aarum et al., 2003; Butovsky et al., 2006b; Walton et al., 2006; Nakanishi et al., 2007; Balasubramaniam et al., 2009; Antony et al., 2011; Shigemoto-Mogami et al., 2014) (Table 1). These varied effects of microglia on neural progenitor proliferation and differentiation are likely dependent upon their activation state and expression of specific regulatory factors, many of which remain undefined.
Table 1.
Examples of developmental functions of microglia
| Function | Promotes / Facilitates | Inhibits / Eliminates |
|---|---|---|
| Progenitor proliferation | •MCM increases cerebellar progenitor proliferation (Morgan et al., 2004). •PU.1−/− mice lack microglia.Neural precursor cell proliferation is reduced and rescued by adding microglia (Antony et al., 2011). •Csf1r knockouts have few microglia with cortical thinning, effects on proliferation and neuron production (Erblich et al., 2011; Nandi et al., 2012). But note direct effects of Csf1r in progenitors (Chitu et al., 2016). |
•Microglia have no effect on proliferation in cultures of postnatal subventricular neurospheres (Walton et al., 2006). |
| Neuronal and glial differentiation |
•Microglia promote neuronal differentiation of neural precursor cells (Aarum et al., 2003). •IL-4 treated microglia promote oligodendrogenesis while IFN-γ activated microglia enhance neurogenesis (Butovsky et al., 2006b). •Inhibiting microglia decreases neurogenesis and oligodendrogenesis in postnatal cortex (Shigemoto-Mogami et al., 2014). •IL-1β, IL-6, TNF-α, and IFN-γ enhance neurogenesis and oligodendrogenesis in vitro (Shigemoto-Mogami et al., 2014). •Microglia or MCM promote neurogenesis of postnatal subventricular neurospheres (Walton et al., 2006). •PU.1−/− cultures have reduced astrogenesis (Antony et al., 2011). •Microglial IL-6 and LIF promote the differentiation of astrocytes (Nakanishi et al., 2007). |
•Microglial Il-6 suppresses neurosphere generation from adult retinal cell suspensions (Balasubramaniam et al., 2009). |
| Adult neurogenesis | •Microglia in SVZ required for survival and migration of neuroblasts through the rostral migratory stream (Ribeiro Xavier et al., 2015) and in culture (Aarum et al., 2003). •Fractalkine signaling mediates exercise-induced increases in neurogenesis (Vukovic et al., 2012). •Exercise increased the proportion of microglia expressing Igf1 (Kohman et al., 2012). |
•Microglia engulf newly generated neuroblasts in the adult SGZ (Sierra et al., 2010). •Microglia in proliferative zones undergo age-related changes associated with reductions in neural stem cell proliferation and differentiation (Solano Fonseca et al., 2016). •Depleting microglia in aged animals increases neurogenesis (Vukovic et al., 2012). • Exogenous fractalkine reverses age-related deficits in neurogenesis (Bachstetter et al., 2011). |
| Neuronal and progenitor survival | •Microglia or conditioned medium increase survival of rat cerebellar granule neurons in culture (Morgan et al., 2004). •Microglia are required for the survival of developing layer V cortical neurons, in part by secretion of Igf1 (Ueno et al., 2013). |
•Microglia stimulate neuronal apoptosis through production of reactive oxygen species in cerebellum (Marín-Teva et al., 2004). •Microglial-mediated neuronal apoptosis requires Cd11b and Dap12 in developing hippocampus (Wakselman et al., 2008). •Microglial NGF stimulates RGC death via p75 (Frade and Barde, 1998). •Microglia engulf cortical neural precursor cells to limit neuron generation in embryo (Cunningham et al., 2013). |
| Neural circuit development | •Microglia required for laminar positioning of subsets of interneurons in somatosensory cortex (Squarzoni et al., 2014). •Microglia facilitate axon fasciculation in corpus callosum (Pont-Lezica et al., 2014a). •Microglia promote axon myelination (Hagemeyer et al., 2017; Wlodarczyk et al., 2017). •Microglial-derived BDNF is required for learning-dependent spine remodeling and formation. Disruption results in deficits in multiple learning tasks (Parkhurst et al., 2013). •Synapse formation in the somatosensory cortex (Miyamoto et al., 2016). |
•Microglia limit outgrowth of dopaminergic axons in the forebrain (Squarzoni et al., 2014). •Microglia eliminate excessive synapses from RGCs in the lateral geniculate nucleus (Stevens et al., 2007; Schafer et al., 2012). •In postnatal mouse hippocampus, microglial synaptic engulfment is required for normal synaptic maturation, functional connectivity, and behavior (Paolicelli et al., 2011; Zhan et al., 2014). |
The influence of microglia on neurogenesis is also evident in the adult brain. After CNS development is complete, neural stem cells and neuron production persist throughout life in discrete brain regions including the subgranular zone (SGZ) of the dentate gyrus and the subventricular zone (SVZ) of the lateral ventricle (Gage and Temple, 2013). Microglia influence survival and migration of neuroblasts as well as limit neuron number in adult proliferative zones (Aarum et al., 2003; Sierra et al., 2010; Ribeiro Xavier et al., 2015) (Table 1). In addition, microglia are important for environmental influences on adult neurogenesis (Gemma and Bachstetter, 2013; Valero et al., 2016). For example, microglia are responsible for aging-induced reductions in neurogenesis (Solano Fonseca et al., 2016), but also important for enhancement of neurogenesis in response to exercise. These changes appear to be in part mediated by variations in the level of fractalkine (Cx3cl1) signaling (Cx3cl1/Cx3cr1) (Bachstetter et al., 2011; Vukovic et al., 2012) as well as expression of neuroprotective factors such as IGF1 (Kohman et al., 2012) (Table 1). Thus, similar to developmental stages, microglia have dual impacts on adult neurogenesis. How microglia change phenotype in response to aging and environmental changes remains to be fully defined.
Neuronal and progenitor survival
In addition to regulating proliferation and neurogenesis, microglia can promote and limit survival. As professional phagocytes, their role in phagocytosis of dying or dead cells has long been appreciated (Bessis et al., 2007). However, recent evidence suggests microglia play a more active role in regulating survival. They can directly promote apoptosis using a variety of signaling mechanisms (Frade and Barde, 1998; Marín-Teva et al., 2004; Wakselman et al., 2008) (Table 1). Microglia also limit progenitor number in the developing brain through engulfment of non-apoptotic cells (Cunningham et al., 2013), a process termed phagoptosis (Brown and Neher, 2014). While some mechanisms mediating cell engulfment have been defined in vitro, the signals that drive phagoptosis in vivo are still largely undetermined. Microglia can also secrete factors that promote survival both in vitro and in vivo (Morgan et al., 2004; Ueno et al., 2013) (Table 1). These results suggest microglia have duties beyond a clean-up crew, although it is unclear how microglia decide who lives and who dies. In general, microglia can have diverse and sometimes opposing impacts on the CNS, likely dependent upon their functional state. One intriguing possibility is that subsets of microglia may be carrying out these diverse tasks in vivo.
Neural circuit formation and refinement
One of the most complex aspects of nervous system development is the establishment and refinement of neural circuits, and microglia are intimately involved at multiple steps (Paolicelli and Ferretti, 2017). For example, microglia impact laminar positioning of subsets of interneurons in the somatosensory cortex, which has lasting impacts on the balance of excitation and inhibition (Squarzoni et al., 2014). Microglia also associate with developing axons and promote fasciculation (Pont-Lezica et al., 2014b) and myelination (Hagemeyer et al., 2017; Wlodarczyk et al., 2017) but can also limit axon outgrowth (Squarzoni et al., 2014). In addition, microglia have very important roles at the synapse; in formation, maturation, and pruning (Schafer and Stevens, 2013; Wu et al., 2015; Frost and Schafer, 2016). During development, neurons make excessive synaptic connections which are subsequently removed by microglia during a period of activity dependent refinement (Stevens et al., 2007; Schafer et al., 2012). Diminished synaptic pruning has major impacts on functional connectivity as well as behavior (Zhan et al., 2014). In addition to phagocytic elimination of synapses, microglia also facilitate synaptic formation and maturation (Paolicelli et al., 2011; Miyamoto et al., 2016). Microglial brain derived neurotrophic factor (BDNF) is required for learning-dependent spine remodeling and loss of microglia or microglial BDNF results in deficits in a myriad of learning tasks in developing and adult mice (Parkhurst et al., 2013). These, and multiple other studies (Schafer and Stevens, 2013; Wu et al., 2015; Frost and Schafer, 2016), demonstrate that microglia remodel and refine circuits in a developmental and experience-dependent manner.
Neurodevelopmental and psychiatric diseases
Microglia play indispensable roles in the developing CNS, not only regulating neuronal number, position, and connectivity, but also impacting and supporting gliogenesis and myelination (Figure 2). Given these developmental roles, it is not surprising that microglia are linked to neurodevelopmental and psychiatric disorders. Autism spectrum disorder (ASD) is a group of neurological disorders marked by cognititve and social impairments. While the exact etiology of autism-related disorders is unknown, many ASD risk genes encode proteins of the immune system and maternal immune activation (MIA) is associated with ASD (Estes and McAllister, 2015). Additionally, immune dysregulation, gut microbiota, and microglial synaptic pruning are linked to ASD (Estes and McAllister, 2015; Edmonson et al., 2016). In mice, loss of microglial genes Cx3cr1 or Trem2 results in altered functional connectivity and behavioral phenotypes akin to those in ASD (Zhan et al., 2014; Filipello et al., 2018). In addition, microglia are implicated in Rett syndrome, an X-linked ASD that is primarily caused by mutations in the MECP2 gene (Amir et al., 1999). While the degree to which they drive pathology remains unclear (Derecki et al., 2012; Wang et al., 2015a), Rett syndrome microglia are toxic to neurons by elevated secretion of glutamate (Maezawa and Jin, 2010), and they excessively engulf synapses (Schafer et al., 2016).
Growing evidence suggests microglia are also important players in many psychiatric diseases such as depression, anxiety, and schizophrenia (Thion and Garel, 2017; Tay et al., 2018). These disorders have complex etiology where genetic and environmental factors are implicated, with increasing evidence for developmental origins. For example, MIA by either viral or bacterial infection during pregnancy has major impacts on microglial function and is a risk factor for neurodevelopmental disorders such as ASD and schizophrenia (Estes and McAllister, 2015). Furthermore, variants of complement component 4 (C4), which is involved in synaptic pruning by microglia, increases the risk of schizophrenia (Sekar et al., 2016). Interestingly, loss of Hoxb8 causes pathological grooming in mice similar to that of obsessive-compulsive disorder, trichotillomania and is associated with corticostriatal defects (Chen et al., 2010; Nagarajan et al., 2017). In the brain, the only cells of the Hoxb8 lineage are a subset of microglia (Chen et al., 2010; Nagarajan et al., 2017). We are just beginning to understand the extent to which microglia contribute to neurodevelopmental disorders and adult psychiatric disease, although it is clear that developmental roles of microglia have lasting impacts on CNS health and function.
Aging and neurodegeneration
With age, comes gradual changes to the CNS including increases in oxidative stress, cellular senescence, and cell death (Koellhoffer et al., 2017). As the brain’s surveyors, microglia are important sensors for brain aging, with important roles in remodeling and clearance of dying cells (Lucin and Wyss-Coray, 2009). However, there is evidence that microglia themselves drive age-related changes in cognition. Dysregulated inflammation and altered microglial function are thought to greatly contribute to aging and age-related dysfunction (Koellhoffer et al., 2017) as microglia undergo gene expression and functional changes both in mice and humans (Galatro et al., 2017; Olah et al., 2018). What drives microglial senescence is an important outstanding question. Interestingly, one study revealed that the kinetics of microglial aging is dependent on the brain region, showing that microglia in the cerebellum age faster than other CNS regions (Grabert et al., 2016). Since cerebellar microglia display specialized functions (Stowell et al., 2017) and have a unique transcriptional profile (Ayata et al., 2018), these data suggest that microglial functional states may impact microglial aging. Aged microglia have a DAM-like transcriptome signature (Holtman et al., 2015; Keren-Shaul et al., 2017; Krasemann et al., 2017) and show a predisposition for reactivity, dysregulated cytokine secretion, diminished energy production, increased oxidative stress, dystrophic morphology, and decreased phagocytic ability (von Bernhardi et al., 2010; Spittau, 2017). Aging likely impacts microglial function by both diminishing homeostatic functions, as well as biasing microglia towards a neurotoxic phenotype, both of which may increase the risk of neurodegeneration (Harry, 2013).
Aging is the biggest risk factor for neurodegenerative diseases such as AD, which are associated with decline in cognitive function, neuronal loss, aggregated and misfolded proteins as well as dysregulated neuroinflammation (Doty et al., 2015). Recent genome-wide association studies show the most common risk alleles for developing AD are expressed in myeloid cells and are microglia specific or highly expressed in microglia in the CNS, including: TREM2, CD33, CR1, ABCA7, SHIP1, and APOE (Hansen et al., 2018; Tansey et al., 2018). Interestingly, many of these genes are involved in important microglia processes such as phagocytosis, lipid metabolism, and cytokine secretion (Mhatre et al., 2015) (Table 1). The ability of microglia to clear Aβ plaques in AD brains is considered neuroprotective, but microglia may also drive pathology by the secretion of pro-inflammatory cytokines (Gonzalez et al., 2014), engulfment of neuronal synapses (Hong et al., 2016a), and the spread of pathological tau (Asai et al., 2015). While increasing evidence suggests that microglia are central to the development and progression of AD, their exact roles remain elusive.
Notably, mutations in microglial genes can directly promote neurodegeneration (Bianchin et al., 2010). Mutations in TREM2 or TYROBP cause Nasu-Hakola disease (NHD), or polycyctic lipomembranous osteodysplasia and sclerosing leukoencephaolopathy (PLOSL) which is an early onset neurodegenerative disease affecting the frontal lobe (Dardiotis et al., 2017). Mutations or deletions in TREM2 are also linked to variants of frontotemporal dementia (FTD), Parkinson’s disease (PD), and ALS (Yeh et al., 2017). Furthermore, mutations in CSF1R, which is essential for microglial development and survival, cause Hereditary diffuse leukoencephalopathy with spheroids (HDLS), a debilitating disease characterized by behavior, cognitive, and motor changes (Rademakers et al., 2012). Thus, microglial dysfunction can directly cause neurodegeneration.
In spinal cord there is evidence that microglia contribute to ALS, which is characterized by progressive degeneration of motor neurons, in some cases linked to mutations in Cu, Zn superoxide dismutase 1 (SOD1) (Renton et al., 2013; Brites and Vaz, 2014). Mutations in microglial SOD1 lead to disease (Clement et al., 2003) while replacement with wild type microglia slows disease progression and prolongs the lifespan of mutant animals (Lee et al., 2012). Co-cultures of microglia and motoneurons suggest microglia may be more neuroprotective early in disease and highly express BDNF, but at later stages microglia increase production of pro-inflammatory factors, which may promote motor neuron death (Liao et al., 2012), although it is possible that infiltrating cells contributed to this shift in phenotype.
Microglia have also been implicated in retinal neurodegenerative diseases including age-related macular degeneration (AMD) and glaucoma, which are the leading causes of blindness (Silverman and Wong, 2018). AMD primarily affects photoreceptors along with associated retinal pigmented epithelium (Al-Zamil and Yassin, 2017). During AMD, there is recruitment of microglia into the degenerating photoreceptor layer, where they can secrete pro-inflammatory cytokines and contribute to loss by engulfing non apoptotic rods (Zhao et al., 2015). Glaucoma is the progressive decline and loss of retinal ganglion cells (RGCs), the main projection neuron of the retina (Chang and Goldberg, 2012) and neuroinflammation and immune cells have been implicated in contributing to pathology (Soto and Howell, 2014; Williams et al., 2017). Microglia become activated in human glaucoma (Neufeld, 1999; Yuan and Neufeld, 2001), in various animal models of RGC pathology, such as axotomy, acute ocular hypertension (eg. (Johnson et al., 2007; Ebneter et al., 2010; Guo et al., 2011; Roh et al., 2012)), and in the DBA/2J mouse model of chronic glaucoma (Bosco et al., 2008; Bosco et al., 2011). Gene profiling studies of retina in multiple animal models of glaucoma show early upregulation of genes linked to microglia activation, including multiple complement components (Miyahara et al., 2003; Ahmed et al., 2004; Steele et al., 2006; Johnson et al., 2007; Guo et al., 2011; Howell et al., 2011a), and retinal microglia become activated in acute ocular hypertension and in the DBA/2J mouse well before RGC structural degeneration (Lam et al., 2003; Ebneter et al., 2010; Bosco et al., 2011; Roh et al., 2012). Manipulations that alter the functional state of microglia can alter the course of RGC degeneration, including inhibition by minocycline, high dose irradiation, or loss of fractalkine signaling (Anderson et al., 2005; Nakazawa et al., 2006; Bosco et al., 2008; Bosco et al., 2012; Breen et al., 2016), although their precise role remains to be defined. Thus, microglia are active participants in neurodegeneration thoughout the CNS.
Summary
With the advancement of methodologies and tools to probe microglial function, we have greatly enhanced our understanding of the lifelong impacts microglia have on CNS development, homeostasis, and function (Figure 2). As CNS remodelers, microglia are also essential players in injury such as stroke (Benakis et al., 2015; Guruswamy and ElAli, 2017) and traumatic brain injury (TBI) (Donat et al., 2017), as well as regeneration (Carpentier and Palmer, 2009; Martino et al., 2011; Kokaia et al., 2012). Following injury, microglia can also be both helpful and harmful, in part dependent on their phenotype (Xu et al., 2017). Therefore in development, following injury, and in disease, microglial influence on CNS integrity is complex, likely contingent upon timing, situational factors, and their functional state. In addition, microglia perceive and disseminate environmental signals, further complicating the effects that microglia have on CNS homeostasis. Therefore, it is essential to understand how these functional states are driven and maintained, as similar states may have conflicting outcomes on the CNS in different contexts. Altogether, these data demonstrate that microglial function greatly impacts CNS health throughout life.
Shared pathways of microglia in developmental and disease states
It is likely that there are core functions and key regulatory pathways that are active across the lifespan. It is therefore instructive to consider examples where molecular pathways involved in developmental functions also serve as critical regulators of microglia in disease. Here, we highlight a few signaling pathways that mediate developmental microglial roles that reemerge in disease states.
Secreted Factors
Secreted proteins, including cytokines, are essential for host response during immune processes but also mediate normal signaling between cells of the CNS (Deverman and Patterson, 2009). Microglia utilize secreted proteins and factors to communicate with stem cells, neurons, glia as well as other immune cells including those in the circulating blood. A whole myriad of cytokines including interleukins, interferons, tumor necrosis factors (TNFs), and members of the TGF-β family can modulate cell proliferation, neurogenesis, gliogenesis, cell migration, and apoptosis during neural development (Borsini et al., 2015). In addition, with increased feasibility of stem cell therapies, communication between microglia and engrafted neural stem or progenitor cells via secreted factors is an important area of research (Kokaia et al., 2012). Factors secreted by microglia also influence the course of disease. For example, cytokine dysregulation has been implicated in psychiatric disorders such as depression (Zunszain et al., 2013), as well as in AD and PD (Nuzzo et al., 2014). Neurodegenerative diseases, are often accompanied by neuroinflammation, which is defined as microglial activation and pro-inflammatory cytokine production (Carpentier and Palmer, 2009). Interestingly, microglial derived factors also affect neuron number and function indirectly. Microglial IL-1β TNFα, and C1q induce a neurotoxic subtype of reactive astrocyte termed A1 that is considered harmful and can be found in AD, Huntington’s disease, PD, ALS, and MS (Liddelow et al., 2017). In this section, we discuss the role of two secreted factors that play a central role in both development and disease.
Igf1
One example of a signaling pathway utilized in neuron-microglia crosstalk in development and disease is IGF1. It is expressed by many CNS cell types with peak expression during development, and low levels throughout life except in neurogenic zones (Dyer et al., 2016). IGF1 promotes proliferation, neuronal survival, neuronal differentiation, and maturation during development and in adulthood (Nieto-Estévez et al., 2016). IGF1 is also important for vascularization (Bach, 2015) and gliogenesis (Dyer et al., 2016). Interestingly, overexpression of Igf1 increases hippocampal neurogenesis and synaptogenesis during postnatal development (Kusky et al., 2000) and IGF1 can restore synaptic deficits in iPSC-derived neurons from individuals with a neurodevelopmental disorder Phelan-McDermid syndrome (Shcheglovitov et al., 2013).
Although many cell types can express IGF1, microglia are a major source in the brain (Suh et al., 2013; Labandeira-Garcia et al., 2017) with high expression embryonically (Thion et al., 2018) and a reemergence with age, injury, or disease (O’Kusky and Ye, 2012) (Figure 3). Microglial IGF1 is required for survival of Layer V cortical neurons postnatally (Ueno et al., 2013). Also as discussed earlier, a subset of microglia that are CD11c+ in developing white matter highly express IGF1 (Hagemeyer et al., 2017; Wlodarczyk et al., 2017). Either microglial depletion, or specific loss of IGF1 in CD11c+ microglia, results in reduced expression of mature myelination genes, defects in myelination (Hagemeyer et al., 2017; Wlodarczyk et al., 2017) and diminished development of oligendrocytes (Hagemeyer et al., 2017) suggesting that microglial IGF1 is important for oligodendrocyte development and survival consistent with culture experiments (Butovsky et al., 2006b). IGF1 is linked to exercise-induced increases in adult neurogenesis (Llorens-Martín et al., 2009) and microglial IGF1 increases coincident with increased neurogenesis (Kohman et al., 2012). Environmental enrichment also increases microglial IGF1 expression concurrent with increased adult neurogenesis (Ziv et al., 2006).
Microglial IGF1 expression may also be an essential regulator in injury and disease. Microglia upregulate IGF1 expression following various brain injuries (O’Kusky and Ye, 2012). For example, microglial IGF1 expression is increased in damaged brain regions following ischemia (Beilharz et al., 1998; O’Donnell et al., 2002) and ablation of microglia, which are the primary source of IGF1 in these lesions, increases infarction size and the number of apoptotic cells (Lalancette-Hébert et al., 2007). SGZ proliferation following status epilepticus requires IGF1 derived from microglia (Choi et al., 2008). Interestingly, microglia also upregulate IGF1 following cuprizone-induced demyelination (Gudi et al., 2011).
IGF1 is considered a hub gene in microglia sorted from aged mice (Holtman et al., 2015) and is upregulated in disease associated microglia as mentioned above (Chiu et al., 2013; Keren-Shaul et al., 2017; Krasemann et al., 2017). Increased microglial IGF1 stimulates adult neurogenesis, and IGF1 and IGF2 can promote dendritic spine formation and restore hippocampal excitatory synaptic transmission (Pascual-Lucas et al., 2014), which could alleviate behavioral and cognitive deficits associated with aging and disease (Morel et al., 2017) as adult hippocampal neurogenesis decreases in neurodegenerative diseases such as AD and PD (Shohayeb et al., 2018). Plaque-associated microglia strongly express IGF1 (Kamphuis et al., 2016), and an increase in CD11c+ IGF1+ microglia is associated with decreased plaque formation, increased neurogenesis, and reduced cognitive decline in a mouse model of AD following T-cell based vaccination with glatiramer acetate (Butovsky et al., 2006a). IGF1 is thought to control Aβ clearance by modulation of carrier proteins, Aβ degradation via insulin degrading enzyme, and Aβ uptake and lysosomal degradation in addition to reducing the phosphorylation of tau (Gasparini and Xu, 2003; Werner and LeRoith, 2014). In other disease models, microglial IGF1 reduces photoreceptor degeneration in a model of retinitis pigmentosa (Arroba et al., 2011). Microglia are important souces of IGF1 in MS (Wlodarczyk et al., 2015) or following spinal cord injury, which is important for oligodendrocyte precursor survival, differentiation, and remyelination (Alizadeh and Karimi‐Abdolrezaee, 2016). Microglia progressively upregulate IGF1 in a mouse model of ALS, and IGF1 promotes motor neuron survival (Chiu et al., 2013). Because microglia can secrete neurotrophic factors such as IGF1 as well as neurotoxic ones (Chiu et al., 2013), it will be important to determine whether the discrete subsets of microglia identified by recent profiling studies have contrasting influences on the CNS in disease states.
Apoe
Another important secreted factor that is expressed by microglia is Apolipoprotein E (APOE), a lipoprotein that is important for the transport of cholesterol in the brain (Puglielli et al., 2003). APOE is predominately expressed by astrocytes and microglia in the CNS but is vital for neuronal access to cholesterol, which is important for axonal growth, synaptic formation and remodeling (Zhang and Liu, 2015). Brain imaging studies have revealed AD risk alleles of APOE affect brain development in human patients. Infant ε4 carriers have lower white matter myelin content, measured by myelin water fraction, and reduced gray matter volume in several regions which will be later affected by AD (Dean et al., 2014). ε4 and ε3 carriers under 21 years of age have reduced cortical thickness of the left entorhinal regions, which is the first affected region in AD, compared to aged matched ε2 carriers, a phenomenon which did not progress over time (Shaw et al., 2007). In addition, mice expressing the human ε4 allele have significant synaptic deficits, displaying reduced excitatory synaptic activity at 1 month of age that progressively worsens by 7 months of age (Klein et al., 2010). This could be mediated in part through cholesterol availability regulated by APOE, which is required for synapse development (Mauch et al., 2001). Furthermore, there is evidence that APOE is important for synaptic plasticity as mutant mice display deficits (Pfrieger, 2003; Kim et al., 2014). Additionally, loss of any one of the APOE receptors results in abnormal brain development, including LRP, Megalin, and Very low density lipoprotein receptor, further indicating the importance for APOE signaling on brain development (Herz and Beffert, 2000).
APOE polymorphic alleles are the main genetic determinants of developing AD, and the APOE ε4 risk allele is the strongest common genetic risk factor (Bertram et al., 2010). Importantly, APOE associates with Aβ and regulates plaque metabolism, aggregation, and deposition (Kanekiyo et al., 2014). In addition, certain APOE isoforms can have pro-inflammatory effects and can reduce adult neurogenesis (Liu et al., 2013). While the exact mechanism in which APOE drives pathology remains unknown, several studies have shown that APOE is required for Aβ deposition. Apoe is highly upregulated in DAM and its signaling through TREM2 is thought to be a primary driver of this microglial functional state (Holtman et al., 2015; Keren-Shaul et al., 2017; Krasemann et al., 2017). Whether similar pathways are involved in development is unknown.
Phagocytosis/phagoptosis
One of the most specialized functions of microglia is phagocytosis, which is utilized during development and disease to maintain homeostasis, remodel, and refine. Microglia eliminate debris, dead, or dying cells (Bilimoria and Stevens, 2015). However, there is increasing evidence microglial phagocytosis is a more complex and active process, above and beyond the clearance of harmful entities. In addition to phagocytosis of synapses (discussed below), microglia can clear dendrites and axons (Vilalta and Brown, 2018). Interestingly, there is increasing evidence for phagoptosis, or the elimination of stressed but viable cells (Brown and Neher, 2014). Phagoptosis has been observed extensively in culture, and several molecular pathways have been identified including phosphatidylserine and complement (Neher et al., 2011; Neher et al., 2012). As previously mentioned, microglia engulf non-apoptotic, mitotic neural precursor cells in the developing brain (Cunningham et al., 2013) and newborn neuroblasts during adult neurogenesis (Sierra et al., 2010) to limit neuron production.
Notably, many SNPs associated with increased risk for AD are in genes encoding proteins involved in microglial phagocytosis, such as APOE, ApoJ, ABCA7, and CR1 (Neher et al., 2012), suggesting that alterations in phagocytosis play a significant role in AD. Diminished clearance of apoptotic cells may contribute to a neuroinflammatory and damaging environment as neuronal apoptosis is increased in AD (Broe et al., 2001) and genes associated with phagocytosis such as Milk fat globe EGF-like factor 8 (Mfge8) are reduced (Boddaert et al., 2007). Morever, there is evidence microglia actively drive pathology by phagoptosis (Brown and Neher, 2014). In AD, Aβ may stimulate phagoptosis and contribute to neuronal loss as Aβ can stimulate the release of microglial MFGE8, which binds to and opsonizes viable neurons and stimulates their phagocytosis by microglia in culture (Neniskyte and Brown, 2013). In a genetic model of retinal photoreceptor degeneration, microglia engulf non-apoptotic rod photoreceptors, and ablation of microglia or inhibition of microglial phagocytosis results in neuroprotection and increased visual function (Zhao et al., 2015). Furthermore, following brain ischemia, inhibition of phagocytosis by loss of Mer receptor tyrosine kinase (MerTK) and MFGE8 results in diminished neuronal loss and brain atrophy and long term improvement of motor function (Neher et al., 2013). Altogether, these results suggest that abberant microglial phagocytosis contributes to and exacerbates brain and retina pathology. There is evidence that phagoptosis may also be beneficial. Following status epilepticus, microglia target and engulf viable newborn neurons in the SGZ, removing the excess newborn cells generated following a seizure to restore homeostasis (Luo et al., 2016). Therefore, microglial phagocytosis is a highly complex process with essential roles during development. Increasing evidence suggests that dysregulation of phagocytosis may significantly contribute to disease.
Complement
While multiple signaling pathways are involved in phagocytosis, here we discuss two important pathways with important roles in development and disease. The first is complement signaling, which has been implicated in many aspects of microglial function including phagocytosis (Stephan et al., 2012; Zabel and Kirsch, 2013). The complement system consists of 30 proteins classically regarded as the immune surveillance system, detecting foreign invaders, orchestrating immune responses, and promoting attack of membranes or phagocytosis (Ricklin et al., 2010). Complement has important and unexpected roles, from fertilization to organogenesis (Coulthard et al., 2018; Hawksworth et al., 2018). Complement has widely been implicated in aging, neuronal injury, and neurodegeneration in brain, retina, and spinal cord (McGeer et al., 2005; Bonifati and Kishore, 2007; McGeer et al., 2017). Remarkably in the CNS, components of the complement cascade are used to maintain homeostasis in a distinct, but parallel manner, where microglia are effectors for complement activity.
While complement can have diverse functions, perhaps the most well-studied and exquisitely characterized role that is mediated by microglia is the developmental regulation of synaptic refinement [reviewed in detail in (Ji et al., 2013; Miyamoto et al., 2013; Wu et al., 2015; Paolicelli and Ferretti, 2017)]. For example, during refinement of visual pathways RGC axons from both eyes form overlapping inputs onto neurons of the lateral geniculate nucleus (LGN). Complement component 1q (C1q) and complement component 3 (C3) tag a subset of synapses for removal by microglia, which exclusively express the receptor complement receptor 3 (CR3, Cd11b), (Stevens et al., 2007; Schafer et al., 2012).
Alterations in complement signaling contribute to neurodevelopmental diseases. Knockouts of C1q, an initiator of the complement pathway, have increased predisposition to seizures and increased density of excitatory synapses, which is likely due to a lack of synaptic pruning by microglia (Chu et al., 2010). Aberrant complement signaling can lead to neurodevelopment disorders. C1q, C3, and C4 mRNA levels are decreased in ASD patients and knockdown of C3 in mice causes social interaction deficits (Fagan et al., 2017). Interestingly, genes encoding immune proteins including C1Q, C3, and C3R were found to be hypomethylated and overexpressed in other cortical regions of patients with ASD (Nardone et al., 2014). In addition, SNPs in C4, as previously mentioned, are associated with schizophrenia (Sekar et al., 2016). These data suggest that dysregulation of complement during development is detrimental.
There is particularly compelling evidence for the role of complement in the pathogenesis of AD. Complement proteins such as C1q and C3 increase substantially in models of neurodegeneration (Reichwald et al., 2009) and in human AD brains compared to controls (Walker and McGeer, 1992). One central function for microglia and complement in disease is the pathological elimination of synapses in a similar manner to that seen in development (Stephan et al., 2012; Siskova and Tremblay, 2013; Hong et al., 2016b; Rajendran and Paolicelli, 2018). Early loss of synapses preceding neuronal death is a hallmark of AD (West et al., 1994). In humans, synapse loss highly correlates with cognitive impairment, more than the presence of Aβ plaques or neurofibrillary tangles (Terry et al., 1991). C1q deposits on synapses prior to Aβ deposition and inhibition of C1q, C3, or receptor CR3 attenuates early synaptic loss. Furthermore, Aβ exacerbates synaptic pruning by increasing C1q localization with synapses and promoting microglial engulfment of synapses via CR3 (Hong et al., 2016a). Knockout of C3 in another mouse model of AD also resulted in increased synapse and neuron density, and the authors found this rescued performance on learning and memory tasks despite an overall increase in Aβ plaques (Shi et al., 2017). This data is consistent with recent work (Hong et al., 2016a) and suggests that C3 also drives synapse elimination. These studies suggest that synaptic elimination by microglia is a major driver of cognitive decline, but also highlight the duality of microglia function. One important question that remains is whether microglia passively remove inactive or defective synaptic inputs or whether they play a more active role in synapse removal due to their own intrinsic dysfunction. Recent work illustrating that loss of TDP-43, a DNA-RNA binding protein that regulates microglial phagocytosis, was found to exacerbate synaptic loss (Paolicelli et al., 2017), arguing that defective microglia are sufficient to eliminate synapses. In addition, because the preservation of synapses correlates with cognitive improvements, this argues that removed synapses are part of active circuits.
Complement signaling is involved in other aspects of pathology including Aβ plaque clearance, increased Aβ plaque aggregation, a potentiation of plaque neurotoxicity, and increased neuroinflammation (Bonifati and Kishore, 2007; Orsini et al., 2014; Rajendran and Paolicelli, 2018). Interestingly, mutations in the gene encoding CR1, the receptor for proteolytic fragments, C3b and C4b, is a major AD risk allele (Crehan et al., 2012). CR1 binding with C3b, which deposits on plaques promotes engulfment (Bradt et al., 1998; Rogers et al., 2006; Crehan et al., 2013). Similarly to children carrying APOE mutations, CR1 risk allele carriers have reduced gray matter volume in the entorhinal cortex as young adults suggesting potential roles in brain development (Bralten et al., 2011).
In retinal neurodegeneration, complement also plays a role in glaucoma (Mirzaei et al., 2017). C1q is localized to RGC compartments (Stevens et al., 2007) and knockout in the DBA/2J mouse is neuroprotective (Howell et al., 2011b), while paradoxically knockout of C3 worsens pathology (Harder et al., 2017), suggesting that the roles of complement are complex. Targeted inhibition of the C3 activation step by retinal gene therapy restricts the progression of neurodegeneration (Bosco et al. 2018). AMD is associated with mutations in several complement proteins and the pathogenic role of complement system has received great attention (Anderson et al., 2010; Warwick et al., 2014; Bora et al., 2015; McHarg et al., 2015). Complement-based therapies are under active development for retinal diseases such as AMD (Xu and Chen, 2016). The role of microglia as effectors for changes in complement activity in retinal disease remains to be fully defined and future work will determine whether complement mediates synaptic elimination or phagoptosis in retinal neurodegeneration.
Trem2/Tyrobp
TREM2 is a receptor of the immunoglobulin superfamily primarily expressed by microglia in the CNS (Kiialainen et al., 2005). Less is known regarding TREM2 function during development. TREM2 is highly expressed by microglia in the postnatal brain, with regional variations and age-related decline in expression (Chertoff et al., 2013). Interestingly, microglia in white matter tracts of corpus callosum and the neurogenic subventricular zone highly express TREM2 in addition to proteins involved with antigen presentation as well as phagocytosis (Chertoff et al., 2013). Recent RNA-sequencing of 4-month-old Trem2 KO mouse hippocampus and cortex suggests TREM2 may be required for normal brain development. Trem2 KO mice have altered expression of Amyloid precursor protein (APP), the main component of Aβ plaques, and endothelial cell genes, suggesting that TREM2 may also be important for vascular changes in AD (Carbajosa et al., 2018). Importantly, this work shows that these changes are early and unrelated to AD pathology. Recently, another study uncovered a novel function for microglial TREM2 during development. The authors found that Trem2 KO animals had altered microglial density and increased morphological complexity, accompanied with molecular changes such as increased P2RY12 and reduced CD68 in the postnatal mouse brain. Furthermore, 2–3 week-old Trem2 KOs had increased neuronal activity and increased synapse density which was due to reduced synaptic engulfment. These synaptic changes led to decreased functional connectivity and impairments in social and repetitive behaviors akin to ASD symptoms (Filipello et al., 2018). Interestingly, the authors also found that TREM2 protein levels negatively correlate with severity of ASD symptoms in human patients. These results issue a new role for TREM2 in mediating synaptic pruning and refinement. Consistent with this, animals with mutations in TREM2 adaptor protein, TYRO protein tyrosine kinase-binding protein (Tyrobp) also have defects in synaptic function (Roumier et al., 2004). It is interesting to note that patients with TREM2 null mutations have symptoms very early in life, during adolescence when developmental processes are not complete (Giraldo et al., 2013).
Several mutations in TREM2 and TYROBP are linked to increased risk for AD including rare TREM2 variant R47H which increases the probability of developing AD by 3–4 fold (Guerreiro et al., 2012; Jonsson et al., 2012). TREM2 function is also linked to several other neurodegenerative disorders, suggesting a shared pathway and/or microglial function drives pathology in these different contexts (Yeh et al., 2017). Disease-associated mutations in TREM2 are mainly found in the extracellular Ig-like V type domain (Yeh et al., 2017) where it binds many different ligands including phospholipids such as exposed phosphatidylserine (PS) on apoptotic neurons, anionic molecules on bacteria, glycolipids from cells and myelin, and lipoproteins containing APOE (Takahashi et al., 2005; Diaye et al., 2009; Kleinberger et al., 2014; Atagi et al., 2015; Bailey et al., 2015; Yeh et al., 2016). How do disease-linked mutations affect TREM2 function? Several studies illustrate that mutant TREM2 reduces binding and association with ligands including APOE (Kleinberger et al., 2014; Atagi et al., 2015; Bailey et al., 2015; Yeh et al., 2016), and are considered loss of function. These results suggest that reduced uptake of Aβ may contribute to increased risk of AD. However, loss of TREM2 in different AD mouse models has variable outcomes on plaque deposition, neuronal survival, and microglial activation (Yeh et al., 2017).
In addition to phagocytosis of Aβ plaques via APOE, TREM2 and TYROBP signal microglial activation and survival (Yeh et al., 2017). TREM2 and TYROBP are thought to promote an anti-inflammatory microglial state, and loss of either exacerbates secretion of pro-inflammatory cytokines in response to TLR stimulation (Hamerman et al., 2005; Hamerman et al., 2006; Turnbull et al., 2006). Increasing evidence suggests microglia also stably envelop plaques with their processes creating a barrier and limiting nearby axonal dystrophy, a process that decreases with age (Condello et al., 2015). Loss of TREM2 reduces the prevalence of microglial association with plaques and increases axonal dystrophy in a mouse model of AD and human patients (Yuan et al., 2016). A recent study also found that TREM2 may be important for microglial energy demands and metabolism as loss of Trem2 in a mouse model of AD and patients carrying Trem2 risk variants displayed increased autophagy and metabolic deficiencies (Ulland et al., 2017).
Furthermore, profiling studies have found that TREM2 is a major driver in shifting the gene expression of microglia into the DAM signature, which are adjacent to Aβ plaques (Wang et al., 2015b; Keren-Shaul et al., 2017; Krasemann et al., 2017). Whether that is due to a reduced association of plaques or direct downstream signaling pathways mediated by TREM2 remains unclear. In addition, an important outstanding question is whether or not this signature confers neuroprotective or neurotoxic features. Given the role of TREM2 during development, it is intriguing to speculate that TREM2 could also drive aberrant synapse elimination by microglia in AD. This phenomenon would help explain the multiplicity of outcomes that loss of TREM2 exerts on disease severity.
Summary
Overall, these studies underscore that many disease risk alleles or microglial genes associated with disease have important developmental functions. This suggests that in some cases patients harboring disease associated mutations may have neurodevelopmental abnormalities. It is intriguing to speculate that altered developmental processes may confer predisposition to later neurodegenerative disease. In general, it is clear that microglia function both early in life and later in disease to influence normal and pathological aging.
Perspectives and future research directions
Many proteins classically associated with immune system function are also expressed in the CNS (Boulanger, 2009). Therefore, CNS cell types and microglia speak the same language, allowing crosstalk that has implications throughout the life of an organism. These interactions are essential for building the CNS, for experience and activity-dependent adaptation, as well as during injury, age, and disease. While microglia are crucial players in these processes, studies in all of these contexts has revealed complex and often opposing roles. During development they can promote and limit neurogenesis. They similarly mediate exercise induced increases in adult neurogenesis as well as age-related decline. During disease, they can limit the spread of toxic protein aggregates but not without also pruning synaptic connections driving cognitive impairments.
Why do microglial functions have opposing consequences on CNS development? And how do microglia promote proliferation versus differentiation or survival versus death? In development, the CNS is a rapidly evolving environment, where multiple cell types must communicate and integrate in a coordinated manner. The responsive and adaptive nature of microglia make them the perfect collaborators. However, their versatility in an ever-changing environment can bring about opposing roles. While some of the conflicting evidence for microglial function may be explained by contextual differences (embryonic vs postnatal, culture conditions, etc.), these studies accentuate the complexity of microglia interactions and their dichotomous nature. In contexts of injury, disease, and aging, there is added complexity as peripheral cells recruited to the CNS also have important contributions, and only recently have the tools become available to clearly distinguish these from microglia (Prinz and Priller, 2017). Future work will determine the influential factors driving these functions at specific stages of development.
If microglial function is so context dependent, how can they perform similar functions in development and disease? Despite being distinct contexts, development, injury, and disease of the CNS have one major thing in common: there is remodeling on the cellular and circuit level. And while microglia are extremely versatile cells, they do have a limited toolkit. Different functional states likely play a major part in whether microglia are harmful or helpful in various different contexts, and subsets of microglia may be carrying out these various tasks in vivo. Profiling of CD11c+ microglia both in development and disease reveal a distinct overlap in expression of a set of genes including Igf1, Clec7a, and Spp1 (Keren-Shaul et al., 2017; Wlodarczyk et al., 2017). While these cells are more different from each other than alike, the common gene set gives us mechanistic insight. For example, what do microglia surrounding Aβ plaques and microglia in developing white matter have in common? In both contexts, lipid metabolism and phagocytosis are essential. Future work will elucidate the intrinsic and extrinsic factors driving these diverse functional states.
The functional state of microglia is also influenced by other factors including biological sex, diet, stress, gut microbiota, and perinatal inflammation, and may be an important link between environmental insults and brain development and disease (Thion and Garel, 2017; Tay et al., 2018). Recent evidence suggests that microglia may have an immune “memory” mediated by epigenetic changes, whereby past systemic experiences can alter response to AD pathology (Hoeijmakers et al., 2017; Wendeln et al., 2018). We are just beginning to understand the epigenetic landscape of microglia and how certain regulators of chromatin accessibility impact microglial development and disease. HDAC1 and HDAC2, for example, are important for proper microglial development and function in disease, with little effect on microglia during adult steady state (Datta et al., 2018). In addition, polycomb repressive complex 2 (PRC2) restricts expression of genes associated with phagocytosis in a brain region-dependent manner (Ayata et al., 2018). DAM genes such as Apoe, Clec7a, and Axl are among the genes that are regionally modulated, so it is interesting to speculate that PRC2 may help regulate shifts in gene expression in disease as well. Understanding how early life events, including genetic and environmental factors, may increase the propensity to develop neurodegenerative diseases or affect outcomes after brain injury is of utmost importance.
Biological sex is another important determinant in both development and disease. For example, more men develop ASD, while significantly more women suffer from MS. There is also a strong link between disease-risk and sexually dimorphic inflammatory states (Hanamsagar and Bilbo, 2016). There is growing appreciation for sex-related differences of microglia in their development (Thion et al., 2018), ability to phagocytose (Nelson et al., 2017; Yanguas-Casás et al., 2017) and microglial activation states (Dorfman et al., 2017; Turano et al., 2017) which can lead to changes in disease outcomes (Villa et al., 2018). Interestingly, in the hippocampus, complement signaling is sexually divergent (Mangold et al., 2017). Elucidating how biological sex modulates microglial function will shed light on mechanisms involved in sexually dimorphic outcomes in disease and injury.
If similar proteins are involved in developmental and disease processes, are microglial functions really the same? While the context may change, microglia utilize the same sensors, signaling pathways, and cellular machinery to impact their environment. It is interesting to note that expression of certain proteins, including APOE, IGF1, Complement, and CD11c are high in development and in disease, which low baseline levels throughout adulthood (Figure 3). The finding that microglia utilize the complement system to signal for the removal of synaptic compartments in development and in AD is arguably the most compelling evidence that developmental mechanisms can be redeployed in disease. These data further highlight the remodeling nature of microglia, and how the same function can be beneficial in development, but detrimental in disease. Future therapeautic interventions will have to avoid disrupting important developmental or homeostatic functions.
Many AD risk variants are found in genes that are essential for aspects of neural development which prompts the question, how early are precipitating events that initiate or catalyze degenerative processes? Human brain imaging studies revealing AD risk variants associate with altered brain structure at early developmental stages argues that neurodegeneration and neurodevelopment are inextricably linked. Recent work assessing TREM2 function during postnatal ages further emphasizes this (Filipello et al., 2018). Whether neurodegenerative diseases have developmental origins, or whether dysfunction in developmental processes increase the risk of neurodegeneration, remain very important questions. One thing remains certain, understanding microglial function and phenotype in development will help unravel their complex roles in disease.
Table 2.
Developmental functions of select microglial genes associated with disease
| Gene/Pathway | Development | Disease |
|---|---|---|
| Apoe | •Mutations linked to reduced white matter and gray matter volume in infants (Dean et al., 2014) and reduced volume of the entorhinal cortex in children and adolescents (Shaw et al., 2007). •Human mutations cause synaptic deficits and reduced excitatory activity in mice (Klein et al., 2010). •Important for synaptic plasticity (Kim et al., 2014). |
•AD risk allele (Bertram et al., 2010). •Regulates plaque metabolism, aggregation, and deposition (Kanekiyo et al., 2014). •Disease-associated gene (Keren-Shaul et al., 2017) and may drive functional shift (Krasemann et al., 2017). |
| Trem2/Tyrobp | •Potential endothelial cell regulation and required for normal levels of developmental APP (Carbajosa et al., 2018). •Synaptic pruning and refinement, proper functional connectivity and behavior (Filipello et al., 2018). •Long term potentiation, synaptic glutamate receptor and TrkB expression (Roumier et al., 2004). |
•AD risk allele (Guerreiro et al., 2012; Jonsson et al., 2012). •Mutations linked to NHD, FTD, PD, and ALS (Dardiotis et al., 2017; Yeh et al., 2017). •Phagocytosis receptor for Aβ plaques via APOE (Atagi et al., 2015; Bailey et al., 2015; Yeh et al., 2017). •Microglial association with plaques (Yuan et al., 2016). •Microglial energy homeostasis (Ulland et al., 2017). •Promotes anti-inflammatory state (Hamerman et al., 2005; Hamerman et al., 2006; Turnbull et al., 2006). •Disease-associated gene and required for functional shift (Keren-Shaul et al., 2017; Krasemann et al., 2017). |
| Complement | Microglial roles: • Synaptic pruning and refinement (Ji et al., 2013; Miyamoto et al., 2013; Wu et al., 2015; Paolicelli and Ferretti, 2017). • C1q KO mice have increased seizures (Chu et al., 2010). • Variants of C4 increase risk for schizophrenia; deficits in synaptic pruning in mice (Sekar et al., 2016). |
•CR1 is AD risk allele (Crehan et al., 2012). •Synaptic pruning (CR3/C1q/C3) (Hong et al., 2016a). •Plaque phagocytosis (CR1 and C3) and plaque aggregation (Bradt et al., 1998; Rogers et al., 2006; Bonifati and Kishore, 2007; Crehan et al., 2013). •Increases inflammatory state (Bonifati and Kishore, 2007; Orsini et al., 2014). |
| Igf1 | Microglial roles: • Survival of Layer V cortical neurons during postnatal development (Ueno et al., 2013). • Survival and maturation of oligodendrocytes (Butovsky et al., 2006b). • Proper myelinogenesis in corpus callosum and cerebellum (Wlodarczyk et al., 2017). • Linked to exercise or enriched environment induced increases in adult neurogenesis (Ziv et al., 2006; Kohman et al., 2012). |
•Promotes cell survival following ischemia (Lalancette-Hébert et al., 2007). •Required for SGZ proliferation following seizure (Choi et al., 2008). •Increases survival of photoreceptors in retinitis pigmentosa (Arroba et al., 2011). •Promotes motor neuron survival in ALS* (Chiu et al., 2013). •Correlated with decreased Aβ formation and increased neurogenesis in AD model (Butovsky et al., 2006a). • Aβ degradation and uptake* (Gasparini and Xu, 2003; Werner and LeRoith, 2014). •Hub gene in aged microglia and a disease associated gene (Holtman et al., 2015; Keren-Shaul et al., 2017; Krasemann et al., 2017). |
didn’t directly test whether microglia-derived
Acknowledgments
This work was supported by the U.S. Department of Health and Human Services, National Institutes of Health, National Eye Institute: EY012274 and EY025967.
References
- Aarum J, Sandberg K, Haeberlein SL, Persson MA. 2003. Migration and differentiation of neural precursor cells can be directed by microglia. Proc Natl Acad Sci U S A 100:15983–15988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed F, Brown KM, Stephan DA, Morrison JC, Johnson EC, Tomarev SI. 2004. Microarray analysis of changes in mRNA levels in the rat retina after experimental elevation of intraocular pressure. Invest Ophthalmol Vis Sci 45:1247–1258. [DOI] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. 2007. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10:1538–1543. [DOI] [PubMed] [Google Scholar]
- Al-Zamil WM, Yassin SA. 2017. Recent developments in age-related macular degeneration: a review. Clinical Interventions in Aging 12:1313–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh A, Karimi‐Abdolrezaee S. 2016. Microenvironmental regulation of oligodendrocyte replacement and remyelination in spinal cord injury. The Journal of Physiology 594:3539–3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genetics 23:185. [DOI] [PubMed] [Google Scholar]
- Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Rickman CB, Bok D, Hageman GS, Johnson LV. 2010. The Pivotal Role of the Complement System in Aging and Age-related Macular Degeneration: Hypothesis Re-visited. Progress in retinal and eye research 29:95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MG, Libby RT, Gould DB, Smith RS, John SW. 2005. High-dose radiation with bone marrow transfer prevents neurodegeneration in an inherited glaucoma. Proc Natl Acad Sci U S A 102:4566–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony JM, Paquin A, Nutt SL, Kaplan DR, Miller FD. 2011. Endogenous microglia regulate development of embryonic cortical precursor cells. J Neurosci Res 89:286–298. [DOI] [PubMed] [Google Scholar]
- Arroba AI, Álvarez-Lindo N, van Rooijen N, de la Rosa EJ. 2011. Microglia-Mediated IGF-I Neuroprotection in the rd10 Mouse Model of Retinitis Pigmentosa. Investigative Ophthalmology & Visual Science 52:9124–9130. [DOI] [PubMed] [Google Scholar]
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kügler S, Ikezu T. 2015. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nature Neuroscience 18:1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, Tipton T, Chapman MA, Riecken K, Beccari S, Sierra A, Molnár Z, Cragg MS, Garaschuk O, Perry VH, Gomez-Nicola D. 2017. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Reports 18:391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atagi Y, Liu C-C, Painter MM, Chen X-F, Verbeeck C, Zheng H, Li X, Rademakers R, Kang SS, Xu H, Younkin S, Das P, Fryer JD, Bu G. 2015. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). Journal of Biological Chemistry 290:26043–26050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata P, Badimon A, Strasburger HJ, Duff MK, Montgomery SE, Loh Y-HE, Ebert A, Pimenova AA, Ramirez BR, Chan AT, Sullivan JM, Purushothaman I, Scarpa JR, Goate AM, Busslinger M, Shen L, Losic B, Schaefer A. 2018. Epigenetic regulation of brain region-specific microglia clearance activity. Nature Neuroscience 21:1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach LA. 2015. Endothelial cells and the IGF system. Journal of Molecular Endocrinology 54:R1–R13. [DOI] [PubMed] [Google Scholar]
- Bachstetter AD, Morganti JM, Jernberg J, Schlunk A, Mitchell SH, Brewster KW, Hudson CE, Cole MJ, Harrison JK, Bickford PC, Gemma C. 2011. Fractalkine and CX 3 CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol Aging 32:2030–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Van Eldik LJ, Schmitt FA, Neltner JH, Ighodaro ET, Webster SJ, Patel E, Abner EL, Kryscio RJ, Nelson PT. 2015. Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathologica Communications 3:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CC, DeVaux LB, Farzan M. 2015. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. Journal of Biological Chemistry 290:26033–26042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniam B, Carter DA, Mayer EJ, Dick AD. 2009. Microglia derived IL-6 suppresses neurosphere generation from adult human retinal cell suspensions. Exp Eye Res 89:757–766. [DOI] [PubMed] [Google Scholar]
- Beilharz EJ, Russo VC, Butler G, Baker NL, Connor B, Sirimanne ES, Dragunow M, Werther GA, Gluckman PD, Williams CE, Scheepens A. 1998. Co-ordinated and cellular specific induction of the components of the IGF/IGFBP axis in the rat brain following hypoxic–ischemic injury. Molecular Brain Research 59:119–134. [DOI] [PubMed] [Google Scholar]
- Benakis C, Garcia-Bonilla L, Iadecola C, Anrather J. 2015. The role of microglia and myeloid immune cells in acute cerebral ischemia. Frontiers in Cellular Neuroscience 8:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, Weissman IL, Chang EF, Li G, Grant GA, Hayden Gephart MG, Barres BA. 2016. New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America 113:E1738–E1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Lill CM, Tanzi RE. 2010. The Genetics of Alzheimer Disease: Back to the Future. Neuron 68:270–281. [DOI] [PubMed] [Google Scholar]
- Bessis A, Bechade C, Bernard D, Roumier A. 2007. Microglial control of neuronal death and synaptic properties. Glia 55:233–238. [DOI] [PubMed] [Google Scholar]
- Beutner C, Linnartz-Gerlach B, Schmidt SV, Beyer M, Mallmann MR, Staratschek-Jox A, Schultze JL, Neumann H. 2013. Unique transcriptome signature of mouse microglia. Glia 61:1429–1442. [DOI] [PubMed] [Google Scholar]
- Bianchin MM, Martin KC, de Souza AC, de Oliveira MA, Rieder CR. 2010. Nasu-Hakola disease and primary microglial dysfunction. Nat Rev Neurol 6:2 p following 523. [DOI] [PubMed] [Google Scholar]
- Bilimoria PM, Stevens B. 2015. Microglia function during brain development: New insights from animal models. Brain Res 1617:7–17. [DOI] [PubMed] [Google Scholar]
- Boddaert J, Kinugawa K, Lambert J-C, Boukhtouche F, Zoll J, Merval R, Blanc-Brude O, Mann D, Berr C, Vilar J, Garabedian B, Journiac N, Charue D, Silvestre S, Duyckaerts C, Amouyel J-P, Mariani J, Tedgui A, Mallat Z. 2007. Evidence of a Role for Lactadherin in Alzheimer’s Disease. The American Journal of Pathology 170:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, Barres BA. 2017. Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron 94:759–773.e758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati DM, Kishore U. 2007. Role of complement in neurodegeneration and neuroinflammation. Molecular Immunology 44:999–1010. [DOI] [PubMed] [Google Scholar]
- Bora NS, Matta B, Lyzogubov VV, Bora PS. 2015. Relationship between the complement system, risk factors and prediction models in age-related macular degeneration. Molecular Immunology 63:176–183. [DOI] [PubMed] [Google Scholar]
- Borsini A, Zunszain PA, Thuret S, Pariante CM. 2015. The role of inflammatory cytokines as key modulators of neurogenesis. Trends in Neurosciences 38:145–157. [DOI] [PubMed] [Google Scholar]
- Bosco A, Anderson SR, Breen KT, Romero CO, Steele MR, Chiodo VA, Boye SL, Hauswirth WW, Tomlinson S, Vetter ML. 2018. Complement C3-Targeted Gene Therapy Restricts Onset and Progression of Neurodegeneration in Chronic Mouse Glaucoma. Molecular Therapy 26:2379–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco A, Crish SD, Steele MR, Romero CO, Inman DM, Horner PJ, Calkins DJ, Vetter ML. 2012. Early reduction of microglia activation by irradiation in a model of chronic glaucoma. PLoS One 7:e43602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco A, Inman DM, Steele MR, Wu G, Soto I, Marsh-Armstrong N, Hubbard WC, Calkins DJ, Horner PJ, Vetter ML. 2008. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci 49:1437–1446. [DOI] [PubMed] [Google Scholar]
- Bosco A, Steele MR, Vetter ML. 2011. Early microglia activation in a mouse model of chronic glaucoma. J Comp Neurol 519:599–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger LM. 2009. Immune proteins in brain development and synaptic plasticity. Neuron 64:93–109. [DOI] [PubMed] [Google Scholar]
- Bradt BM, Kolb WP, Cooper NR. 1998. Complement-dependent Proinflammatory Properties of the Alzheimer’s Disease β-Peptide. The Journal of Experimental Medicine 188:431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bralten J, Franke B, Arias-Vásquez A, Heister A, Brunner HG, Fernández G, Rijpkema M. 2011. CR1 genotype is associated with entorhinal cortex volume in young healthy adults. Neurobiology of Aging 32:2106.e2107–2106.e2111. [DOI] [PubMed] [Google Scholar]
- Breen KT, Anderson SR, Steele MR, Calkins DJ, Bosco A, Vetter ML. 2016. Loss of Fractalkine Signaling Exacerbates Axon Transport Dysfunction in a Chronic Model of Glaucoma. Frontiers in Neuroscience 10:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brites D, Vaz AR. 2014. Microglia centered pathogenesis in ALS: insights in cell interconnectivity. Frontiers in Cellular Neuroscience 8:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broe M, Shepherd CE, Milward EA, Halliday GM. 2001. Relationship between DNA fragmentation, morphological changes and neuronal loss in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathologica 101:616–624. [DOI] [PubMed] [Google Scholar]
- Brown GC, Neher JJ. 2014. Microglial phagocytosis of live neurons. Nat Rev Neurosci 15:209–216. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. 2014. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, Schwartz M. 2006a. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proceedings of the National Academy of Sciences 103:11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, Martino G, Schwartz M. 2006b. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci 31:149–160. [DOI] [PubMed] [Google Scholar]
- Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, Nishinakamura R, Becher B, Greter M. 2016. Sall1 is a transcriptional regulator defining microglia identity and function. Nature Immunology 17:1397. [DOI] [PubMed] [Google Scholar]
- Carbajosa G, Malki K, Lawless N, Wang H, Ryder JW, Wozniak E, Wood K, Mein CA, Dobson RJB, Collier DA, Neill MJ, Hodges AK, Newhouse SJ. 2018. Loss of Trem2 in microglia leads to widespread disruption of cell co-expression networks in mouse brain. Neurobiology of Aging 69:151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier PA, Palmer TD. 2009. Immune influence on adult neural stem cell regulation and function. Neuron 64:79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EE, Goldberg JL. 2012. Glaucoma 2.0: Neuroprotection, Neuroregeneration, Neuroenhancement. Ophthalmology 119:979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S-K, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G, Capecchi MR. 2010. Hematopoietic Origin of Pathological Grooming in Hoxb8 Mutant Mice. Cell 141:775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chertoff M, Shrivastava K, Gonzalez B, Acarin L, Giménez-Llort L. 2013. Differential Modulation of TREM2 Protein during Postnatal Brain Development in Mice. PLOS ONE 8:e72083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Gokhan Ş, Nandi S, Mehler MF, Stanley ER. 2016. Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends in Neurosciences 39:378–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S, Myers RM, Maniatis T. 2013. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep 4:385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y-S, Cho H-Y, Hoyt KR, Naegele JR, Obrietan K. 2008. IGF-1 Receptor-Mediated ERK/MAPK Signaling Couples Status Epilepticus to Progenitor Cell Proliferation in the Subgranular Layer of the Dentate Gyrus. Glia 56:791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, Prince DA. 2010. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proceedings of the National Academy of Sciences of the United States of America 107:7975–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boill, xe e S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH, Julien JP, Goldstein LSB, Cleveland DW. 2003. Wild-Type Nonneuronal Cells Extend Survival of SOD1 Mutant Motor Neurons in ALS Mice. Science 302:113–117. [DOI] [PubMed] [Google Scholar]
- Condello C, Yuan P, Schain A, Grutzendler J. 2015. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nature Communications 6:6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulthard LG, Hawksworth OA, Woodruff TM. 2018. Complement: The Emerging Architect of the Developing Brain. Trends in Neurosciences 41:373–384. [DOI] [PubMed] [Google Scholar]
- Crehan H, Hardy J, Pocock J. 2013. Blockage of CR1 prevents activation of rodent microglia. Neurobiology of Disease 54:139–149. [DOI] [PubMed] [Google Scholar]
- Crehan H, Holton P, Wray S, Pocock J, Guerreiro R, Hardy J. 2012. Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology 217:244–250. [DOI] [PubMed] [Google Scholar]
- Crotti A, Ransohoff RM. 2016. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity 44:505–515. [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Martinez-Cerdeno V, Noctor SC. 2013. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci 33:4216–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardiotis E, Siokas V, Pantazi E, Dardioti M, Rikos D, Xiromerisiou G, Markou A, Papadimitriou D, Speletas M, Hadjigeorgiou GM. 2017. A novel mutation in TREM2 gene causing Nasu-Hakola disease and review of the literature. Neurobiology of Aging 53:194.e113–194.e122. [DOI] [PubMed] [Google Scholar]
- Datta M, Staszewski O, Raschi E, Frosch M, Hagemeyer N, Tay TL, Blank T, Kreutzfeldt M, Merkler D, Ziegler-Waldkirch S, Matthias P, Meyer-Luehmann M, Prinz M. 2018. Histone Deacetylases 1 and 2 Regulate Microglia Function during Development, Homeostasis, and Neurodegeneration in a Context-Dependent Manner. Immunity 48:514–529.e516. [DOI] [PubMed] [Google Scholar]
- De Biase LM, Schuebel KE, Fusfeld ZH, Jair K, Hawes IA, Cimbro R, Zhang HY, Liu QR, Shen H, Xi ZX, Goldman D, Bonci A. 2017. Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron 95:341–356.e346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Van Deren D, Peden E, Hockin M, Boulet A, Titen S, Capecchi MR. 2018. Two distinct ontogenies confer heterogeneity to mouse brain microglia. Development 145:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean DC Iii, , Jerskey BA, Chen K, et al. 2014. Brain differences in infants at differential genetic risk for late-onset alzheimer disease: A cross-sectional imaging study. JAMA Neurology 71:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, Kipnis J. 2012. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverman BE, Patterson PH. 2009. Cytokines and CNS development. Neuron 64:61–78. [DOI] [PubMed] [Google Scholar]
- Diaye E-N, Branda CS, Branda SS, Nevarez L, Colonna M, Lowell C, Hamerman JA, Seaman WE. 2009. TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. The Journal of Cell Biology 184:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donat CK, Scott G, Gentleman SM, Sastre M. 2017. Microglial Activation in Traumatic Brain Injury. Frontiers in Aging Neuroscience 9:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfman MD, Krull JE, Douglass JD, Fasnacht R, Lara-Lince F, Meek TH, Shi X, Damian V, Nguyen HT, Matsen ME, Morton GJ, Thaler JP. 2017. Sex differences in microglial CX3CR1 signalling determine obesity susceptibility in mice. Nature Communications 8:14556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doty KR, Guillot-Sestier M-V, Town T. 2015. The role of the immune system in neurodegenerative disorders: Adaptive or maladaptive? Brain Research 1617:155–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer AH, Vahdatpour C, Sanfeliu A, Tropea D. 2016. The role of Insulin-Like Growth Factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Neuroscience 325:89–99. [DOI] [PubMed] [Google Scholar]
- Ebneter A, Casson RJ, Wood JP, Chidlow G. 2010. Microglial activation in the visual pathway in experimental glaucoma: spatiotemporal characterization and correlation with axonal injury. Investigative ophthalmology & visual science 51:6448–6460. [DOI] [PubMed] [Google Scholar]
- Edmonson CA, Ziats MN, Rennert OM. 2016. A Non-inflammatory Role for Microglia in Autism Spectrum Disorders. Frontiers in Neurology 7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN. 2014. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82:380–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW. 2011. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One 6:e26317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes ML, McAllister AK. 2015. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nature reviews. Neuroscience 16:469–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan K, Crider A, Ahmed AO, Pillai A. 2017. Complement C3 Expression Is Decreased in Autism Spectrum Disorder Subjects and Contributes to Behavioral Deficits in Rodents. Molecular Neuropsychiatry 3:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, Erreni M, Markicevic M, Starvaggi-Cucuzza C, Otero K, Piccio L, Cignarella F, Perrucci F, Tamborini M, Genua M, Rajendran L, Menna E, Vetrano S, Fahnestock M, Paolicelli RC, Matteoli M. 2018. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 48:979–991.e978. [DOI] [PubMed] [Google Scholar]
- Frade JM, Barde Y-A. 1998. Microglia-Derived Nerve Growth Factor Causes Cell Death in the Developing Retina. Neuron 20:35–41. [DOI] [PubMed] [Google Scholar]
- Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee S-H, Haddick PCG, Ngu H, Modrusan Z, Larson JL, Kaminker JS, van der Brug MP, Hansen DV. 2018. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Reports 22:832–847. [DOI] [PubMed] [Google Scholar]
- Frost JL, Schafer DP. 2016. Microglia: Architects of the Developing Nervous System. Trends in Cell Biology 26:587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH, Temple S. 2013. Neural stem cells: generating and regenerating the brain. Neuron 80:588–601. [DOI] [PubMed] [Google Scholar]
- Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, Veras MM, Pereira TF, Leite REP, Möller T, Wes PD, Sogayar MC, Laman JD, den Dunnen W, Pasqualucci CA, Oba-Shinjo SM, Boddeke EWGM, Marie SKN, Eggen BJL. 2017. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nature Neuroscience 20:1162. [DOI] [PubMed] [Google Scholar]
- Gasparini L, Xu H. 2003. Potential roles of insulin and IGF-1 in Alzheimer’s disease. Trends in Neurosciences 26:404–406. [DOI] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua W-J, Hansen TH, Turley SJ, Merad M, Randolph GJ, the Immunological Genome C, Gautier EL, Jakubzick C, Randolph GJ, Best AJ, Knell J, Goldrath A, Miller J, Brown B, Merad M, Jojic V, Koller D, Cohen N, Brennan P, Brenner M, Shay T, Regev A, Fletcher A, Elpek K, Bellemare-Pelletier A, Malhotra D, Turley S, Jianu R, Laidlaw D, Collins J, Narayan K, Sylvia K, Kang J, Gazit R, Garrison BS, Rossi DJ, Kim F, Rao TN, Wagers A, Shinton SA, Hardy RR, Monach P, Bezman NA, Sun JC, Kim CC, Lanier LL, Heng T, Kreslavsky T, Painter M, Ericson J, Davis S, Mathis D, Benoist C. 2012. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nature Immunology 13:1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemma C, Bachstetter AD. 2013. The role of microglia in adult hippocampal neurogenesis. Front Cell Neurosci 7:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. 2010. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science (New York, N.Y.) 330:841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldo M, Lopera F, Siniard A, Corneveaux JJ, Schrauwen I, Carvajal J, Muñoz C, Ramirez-Restrepo M, Myers AJ, Caselli RJ, Kosik KS, Reiman EM, Huentelman MJ. 2013. Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and Alzheimer’s disease. Neurobiology of aging 34: 10.1016/j.neurobiolaging.2013.1002.1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez H, Elgueta D, Montoya A, Pacheco R. 2014. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J Neuroimmunol 274:1–13. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Link V, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, Glass CK. 2014. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159:1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, McColl BW. 2016. Microglial brain region-dependent diversity and selective regional sensitivities to ageing. Nature neuroscience 19:504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudi V, Škuljec J, Yildiz Ö, Frichert K, Skripuletz T, Moharregh-Khiabani D, Voß E, Wissel K, Wolter S, Stangel M. 2011. Spatial and Temporal Profiles of Growth Factor Expression during CNS Demyelination Reveal the Dynamics of Repair Priming. PLoS ONE 6:e22623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JSK, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert J-C, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J St., George-Hyslop P, Singleton A, Hardy J. 2012. TREM2 Variants in Alzheimer’s Disease. New England Journal of Medicine 368:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Johnson EC, Cepurna WO, Dyck JA, Doser T, Morrison JC. 2011. Early gene expression changes in the retinal ganglion cell layer of a rat glaucoma model. Investigative ophthalmology & visual science 52:1460–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruswamy R, ElAli A. 2017. Complex Roles of Microglial Cells in Ischemic Stroke Pathobiology: New Insights and Future Directions. International Journal of Molecular Sciences 18:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemeyer N, Hanft K-M, Akriditou M-A, Unger N, Park ES, Stanley ER, Staszewski O, Dimou L, Prinz M. 2017. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta neuropathologica 134:441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL. 2006. Cutting Edge: Inhibition of TLR and FcR Responses in Macrophages by Triggering Receptor Expressed on Myeloid Cells (TREM)-2 and DAP12. The Journal of Immunology 177:2051. [DOI] [PubMed] [Google Scholar]
- Hamerman JA, Tchao NK, Lowell CA, Lanier LL. 2005. Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nature Immunology 6:579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanamsagar R, Bilbo SD. 2016. Sex differences in neurodevelopmental and neurodegenerative disorders: Focus on microglial function and neuroinflammation during development. The Journal of steroid biochemistry and molecular biology 160:127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch U-K. 2013. Functional diversity of microglia – how heterogeneous are they to begin with? Frontiers in Cellular Neuroscience 7:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Hanson JE, Sheng M. 2018. Microglia in Alzheimer’s disease. The Journal of Cell Biology 217:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder JM, Braine CE, Williams PA, Zhu X, MacNicoll KH, Sousa GL, Buchanan RA, Smith RS, Libby RT, Howell GR, John SWM. 2017. Early immune responses are independent of RGC dysfunction in glaucoma with complement component C3 being protective. Proceedings of the National Academy of Sciences of the United States of America 114:E3839–E3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harry GJ. 2013. Microglia During Development and Aging. Pharmacology & therapeutics 139:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawksworth OA, Coulthard LG, Mantovani S, Woodruff TM. 2018. Complement in stem cells and development. Seminars in Immunology 37:74–84. [DOI] [PubMed] [Google Scholar]
- Herz J, Beffert U. 2000. Apolipoprotein E receptors: linking brain development and Alzheimer’s disease. Nature Reviews Neuroscience 1:51–58. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. 2013. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16:1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers L, Ruigrok SR, Amelianchik A, Ivan D, van Dam A-M, Lucassen PJ, Korosi A. 2017. Early-life stress lastingly alters the neuroinflammatory response to amyloid pathology in an Alzheimer’s disease mouse model. Brain, Behavior, and Immunity 63:160–175. [DOI] [PubMed] [Google Scholar]
- Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, Wes PD, Moller T, Orre M, Kamphuis W, Hol EM, Boddeke EWGM, Eggen BJL. 2015. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathologica Communications 3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtman IR, Skola D, Glass CK. 2017. Transcriptional control of microglia phenotypes in health and disease. The Journal of Clinical Investigation 127:3220–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B. 2016a. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Dissing-Olesen L, Stevens B. 2016b. New insights on the role of microglia in synaptic pruning in health and disease. Current Opinion in Neurobiology 36:128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopperton KE, Mohammad D, Trépanier MO, Giuliano V, Bazinet RP. 2017. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: a systematic review. Molecular Psychiatry 23:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, Barbay JM, King BL, Marchant JK, Hibbs M, Stevens B, Barres BA, Clark AF, Libby RT, John SW. 2011a. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. The Journal of clinical investigation 121:1429–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, Barbay JM, King BL, Marchant JK, Hibbs M, Stevens B, Barres BA, Clark AF, Libby RT, John SW. 2011b. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Invest 121:1429–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji K, Akgul G, Wollmuth LP, Tsirka SE. 2013. Microglia Actively Regulate the Number of Functional Synapses. PLoS ONE 8:e56293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EC, Jia L, Cepurna WO, Doser TA, Morrison JC. 2007. Global changes in optic nerve head gene expression after exposure to elevated intraocular pressure in a rat glaucoma model. Invest Ophthalmol Vis Sci 48:3161–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K. 2012. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. New England Journal of Medicine 368:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, Kooijman L, Schetters S, Orre M, Hol EM. 2016. Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1862:1847–1860. [DOI] [PubMed] [Google Scholar]
- Kanekiyo T, Xu H, Bu G. 2014. ApoE and Aβ in Alzheimer’s Disease: Accidental Encounters or Partners? Neuron 81:740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I. 2017. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 1691276–1290.e1217. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Holscher C, Muller DN, Luckow B, Brocker T, Debowski K, Fritz G, Opdenakker G, Diefenbach A, Biber K, Heikenwalder M, Geissmann F, Rosenbauer F, Prinz M. 2013. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci 16:273–280. [DOI] [PubMed] [Google Scholar]
- Kiialainen A, Hovanes K, Paloneva J, Kopra O, Peltonen L. 2005. Dap12 and Trem2, molecules involved in innate immunity and neurodegeneration, are co-expressed in the CNS. Neurobiology of Disease 18:314–322. [DOI] [PubMed] [Google Scholar]
- Kim J, Yoon H, Basak J, Kim J. 2014. Apolipoprotein E in Synaptic Plasticity and Alzheimer’s Disease: Potential Cellular and Molecular Mechanisms. Molecules and Cells 37:767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RC, Mace BE, Moore SD, Sullivan PM. 2010. Progressive loss of synaptic integrity in human apoE4 targeted replacement mice and attenuation by apoE2. Neuroscience 171:1265–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin J-J, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C. 2014. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Science Translational Medicine 6:243ra286. [DOI] [PubMed] [Google Scholar]
- Koellhoffer EC, McCullough LD, Ritzel RM. 2017. Old Maids: Aging and Its Impact on Microglia Function. International Journal of Molecular Sciences 18:769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohman RA, DeYoung EK, Bhattacharya TK, Peterson LN, Rhodes JS. 2012. Wheel running attenuates microglia proliferation and increases expression of a proneurogenic phenotype in the hippocampus of aged mice. Brain, Behavior, and Immunity 26:803–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia Z, Martino G, Schwartz M, Lindvall O. 2012. Cross-talk between neural stem cells and immune cells: the key to better brain repair? Nat Neurosci 15:1078–1087. [DOI] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, Mazaheri F, Hartmann K, Madi A, Ulrich JD, Glatzel M, Worthmann A, Heeren J, Budnik B, Lemere C, Ikezu T, Heppner FL, Litvak V, Holtzman DM, Lassmann H, Weiner HL, Ochando J, Haass C, Butovsky O. 2017. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47:566–581.e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusky JR, Ye P, Ercole AJ. 2000. Insulin-Like Growth Factor-I Promotes Neurogenesis and Synaptogenesis in the Hippocampal Dentate Gyrus during Postnatal Development. The Journal of Neuroscience 20:8435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labandeira-Garcia JL, Costa-Besada MA, Labandeira CM, Villar-Cheda B, Rodríguez-Perez AI. 2017. Insulin-Like Growth Factor-1 and Neuroinflammation. Frontiers in Aging Neuroscience 9:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Gowing G, Simard A, Weng YC, Kriz J. 2007. Selective Ablation of Proliferating Microglial Cells Exacerbates Ischemic Injury in the Brain. The Journal of Neuroscience 27:2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam TT, Kwong JM, Tso MO. 2003. Early glial responses after acute elevated intraocular pressure in rats. Invest Ophthalmol Vis Sci 44:638–645. [DOI] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. 2014. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell 159:1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Seong J, Kim SH, Lee SJ, Cho YJ, An J, Nam D-H, Joo KM, Cha CI. 2012. Replacement of microglial cells using Clodronate liposome and bone marrow transplantation in the central nervous system of SOD1G93A transgenic mice as an in vivo model of amyotrophic lateral sclerosis. Biochemical and Biophysical Research Communications 418:359–365. [DOI] [PubMed] [Google Scholar]
- Li Q, Barres BA. 2017. Microglia and macrophages in brain homeostasis and disease. Nature Reviews Immunology 18:225. [DOI] [PubMed] [Google Scholar]
- Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. 2012. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Experimental Neurology 237:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung W-S, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA. 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C-C, Kanekiyo T, Xu H, Bu G. 2013. Apolipoprotein E and Alzheimer disease: risk, mechanisms, and therapy. Nature reviews. Neurology 9:106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens-Martín M, Torres-Alemán I, Trejo JL. 2009. Reviews: Mechanisms Mediating Brain Plasticity: IGF1 and Adult Hippocampal Neurogenesis. The Neuroscientist 15:134–148. [DOI] [PubMed] [Google Scholar]
- Lucin KM, Wyss-Coray T. 2009. Immune Activation in Brain Aging and Neurodegeneration: Too Much or Too Little? Neuron 64:110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Koyama R, Ikegaya Y. 2016. Microglia engulf viable newborn cells in the epileptic dentate gyrus. Glia 64:1508–1517. [DOI] [PubMed] [Google Scholar]
- Maezawa I, Jin L-W. 2010. Rett Syndrome Microglia Damage Dendrites and Synapses by the Elevated Release of Glutamate. The Journal of neuroscience : the official journal of the Society for Neuroscience 30:5346–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangold CA, Wronowski B, Du M, Masser DR, Hadad N, Bixler GV, Brucklacher RM, Ford MM, Sonntag WE, Freeman WM. 2017. Sexually divergent induction of microglial-associated neuroinflammation with hippocampal aging. Journal of Neuroinflammation 14:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. 2004. Microglia Promote the Death of Developing Purkinje Cells. Neuron 41:535–547. [DOI] [PubMed] [Google Scholar]
- Martino G, Pluchino S, Bonfanti L, Schwartz M. 2011. Brain regeneration in physiology and pathology: the immune signature driving therapeutic plasticity of neural stem cells. Physiol Rev 91:1281–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, Ben-Yehuda H, David E, Zelada González F, Perrin P, Keren-Shaul H, Gury M, Lara-Astaiso D, Thaiss CA, Cohen M, Bahar Halpern K, Baruch K, Deczkowska A, Lorenzo-Vivas E, Itzkovitz S, Elinav E, Sieweke MH, Schwartz M, Amit I. 2016. Microglia development follows a stepwise program to regulate brain homeostasis. Science 353:6301. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller E-C, Otto A, Pfrieger FW. 2001. CNS Synaptogenesis Promoted by Glia-Derived Cholesterol. Science 294:1354. [DOI] [PubMed] [Google Scholar]
- McGeer EG, Klegeris A, McGeer PL. 2005. Inflammation, the complement system and the diseases of aging. Neurobiology of Aging 26:94–97. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Lee M, McGeer EG. 2017. A review of human diseases caused or exacerbated by aberrant complement activation. Neurobiology of Aging 52:12–22. [DOI] [PubMed] [Google Scholar]
- McHarg S, Clark SJ, Day AJ, Bishop PN. 2015. Age-related macular degeneration and the role of the complement system. Molecular Immunology 67:43–50. [DOI] [PubMed] [Google Scholar]
- Mhatre SD, Tsai CA, Rubin AJ, James ML, Andreasson KI. 2015. Microglial Malfunction: The Third Rail in the Development of Alzheimer’s Disease. Trends in Neurosciences 38:621–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaei M, Gupta VB, Chick JM, Greco TM, Wu Y, Chitranshi N, Wall RV, Hone E, Deng L, Dheer Y, Abbasi M, Rezaeian M, Braidy N, You Y, Salekdeh GH, Haynes PA, Molloy MP, Martins R, Cristea IM, Gygi SP, Graham SL, Gupta VK. 2017. Age-related neurodegenerative disease associated pathways identified in retinal and vitreous proteome from human glaucoma eyes. Scientific Reports 7:12685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyahara T, Kikuchi T, Akimoto M, Kurokawa T, Shibuki H, Yoshimura N. 2003. Gene microarray analysis of experimental glaucomatous retina from cynomologous monkey. Invest Ophthalmol Vis Sci 44:4347–4356. [DOI] [PubMed] [Google Scholar]
- Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, Koizumi S, Moorhouse AJ, Yoshimura Y, Nabekura J. 2016. Microglia contact induces synapse formation in developing somatosensory cortex. Nature Communications 7:12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto A, Wake H, Moorhouse AJ, Nabekura J. 2013. Microglia and synapse interactions: fine tuning neural circuits and candidate molecules. Front Cell Neurosci 7:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel GR, León ML, Uriarte M, Reggiani PC, Goya RG. 2017. Therapeutic potential of IGF-I on hippocampal neurogenesis and function during aging. Neurogenesis 4:e1259709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan SC, Taylor DL, Pocock JM. 2004. Microglia release activators of neuronal proliferation mediated by activation of mitogen-activated protein kinase, phosphatidylinositol-3-kinase/Akt and delta–Notch signalling cascades. Journal of Neurochemistry 90:89–101. [DOI] [PubMed] [Google Scholar]
- Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, Lelios I, Heppner FL, Kipnis J, Merkler D, Greter M, Becher B. 2018. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 48:380–395.e386. [DOI] [PubMed] [Google Scholar]
- Nagarajan N, Jones BW, West PJ, Marc RE, Capecchi MR. 2017. Corticostriatal circuit defects in Hoxb8 mutant mice. Mol Psychiatry 00:1–10. [DOI] [PubMed] [Google Scholar]
- Nakanishi M, Niidome T, Matsuda S, Akaike A, Kihara T, Sugimoto H. 2007. Microglia-derived interleukin-6 and leukaemia inhibitory factor promote astrocytic differentiation of neural stem/progenitor cells. European Journal of Neuroscience 25:649–658. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Nakazawa C, Matsubara A, Noda K, Hisatomi T, She H, Michaud N, Hafezi-Moghadam A, Miller JW, Benowitz LI. 2006. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci 26:12633–12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi S, Gokhan S, Dai X-M, Wei S, Enikolopov G, Lin H, Mehler MF, Stanley ER. 2012. The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Developmental Biology 367:100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardone S, Sharan Sams D, Reuveni E, Getselter D, Oron O, Karpuj M, Elliott E. 2014. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Translational Psychiatry 4:e433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher JJ, Emmrich JV, Fricker M, Mander PK, Théry C, Brown GC. 2013. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proceedings of the National Academy of Sciences of the United States of America 110:E4098–E4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher JJ, Neniskyte U, Brown GC. 2012. Primary Phagocytosis of Neurons by Inflamed Microglia: Potential Roles in Neurodegeneration. Frontiers in Pharmacology 3:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher JJ, Neniskyte U, Zhao J-W, Bal-Price A, Tolkovsky AM, Brown GC. 2011. Inhibition of Microglial Phagocytosis Is Sufficient To Prevent Inflammatory Neuronal Death. The Journal of Immunology 186:4973. [DOI] [PubMed] [Google Scholar]
- Nelson LH, Warden S, Lenz KM. 2017. Sex differences in microglial phagocytosis in the neonatal hippocampus. Brain, Behavior, and Immunity 64:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neniskyte U, Brown GC. 2013. Lactadherin/MFG-E8 is essential for microglia-mediated neuronal loss and phagoptosis induced by amyloid β. Journal of Neurochemistry 126:312–317. [DOI] [PubMed] [Google Scholar]
- Neufeld AH. 1999. Microglia in the optic nerve head and the region of parapapillary chorioretinal atrophy in glaucoma. Archives of Ophthalmology 117:1050–1056. [DOI] [PubMed] [Google Scholar]
- Nieto-Estévez V, Defterali Ç, Vicario-Abejón C. 2016. IGF-I: A Key Growth Factor that Regulates Neurogenesis and Synaptogenesis from Embryonic to Adult Stages of the Brain. Frontiers in Neuroscience 10:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. 2005. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 308:1314. [DOI] [PubMed] [Google Scholar]
- Nuzzo D, Picone P, Caruana L, Vasto S, Barera A, Caruso C, Di Carlo M. 2014. Inflammatory Mediators as Biomarkers in Brain Disorders. Inflammation 37:639–648. [DOI] [PubMed] [Google Scholar]
- O’Donnell SL, Frederick TJ, Krady JK, Vannucci SJ, Wood TL. 2002. IGF-I and microglia/macrophage proliferation in the ischemic mouse brain. Glia 39:85–97. [DOI] [PubMed] [Google Scholar]
- O’Kusky J, Ye P. 2012. Neurodevelopmental effects of insulin-like growth factor signaling. Frontiers in neuroendocrinology 33:230–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah M, Patrick E, Villani A-C, Xu J, White CC, Ryan KJ, Piehowski P, Kapasi A, Nejad P, Cimpean M, Connor S, Yung CJ, Frangieh M, McHenry A, Elyaman W, Petyuk V, Schneider JA, Bennett DA, De Jager PL, Bradshaw EM. 2018. A transcriptomic atlas of aged human microglia. Nature Communications 9:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini F, De Blasio D, Zangari R, Zanier ER, De Simoni M-G. 2014. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Frontiers in Cellular Neuroscience 8:380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT. 2011. Synaptic pruning by microglia is necessary for normal brain development. Science 333:1456–1458. [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, Ferretti MT. 2017. Function and Dysfunction of Microglia during Brain Development: Consequences for Synapses and Neural Circuits. Frontiers in Synaptic Neuroscience 9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Jawaid A, Henstridge CM, Valeri A, Merlini M, Robinson JL, Lee EB, Rose J, Appel S, Lee VMY, Trojanowski JQ, Spires-Jones T, Schulz PE, Rajendran L. 2017. TDP-43 Depletion in Microglia Promotes Amyloid Clearance but Also Induces Synapse Loss. Neuron 95:297–308.e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, , Lafaille JJ, Hempstead BL, Littman DR, Gan WB. 2013. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155:1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Lucas M, Viana da Silva S, Di Scala M, Garcia-Barroso C, González-Aseguinolaza G, Mulle C, Alberini CM, Cuadrado-Tejedor M, Garcia-Osta A. 2014. Insulin-like growth factor 2 reverses memory and synaptic deficits in APP transgenic mice. EMBO Molecular Medicine 6:1246–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW. 2003. Cholesterol homeostasis and function in neurons of the central nervous system. Cellular And Molecular Life Sciences: CMLS 60:1158–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pont-Lezica L, Beumer W, Colasse S, Drexhage H, Versnel M, Bessis A. 2014a. Microglia shape corpus callosum axon tract fasciculation: functional impact of prenatal inflammation. Eur J Neurosci 39:1551–1557. [DOI] [PubMed] [Google Scholar]
- Pont-Lezica L, Beumer W, Colasse S, Drexhage H, Versnel M, Bessis A. 2014b. Microglia shape corpus callosum axon tract fasciculation: functional impact of prenatal inflammation. European Journal of Neuroscience 39:1551–1557. [DOI] [PubMed] [Google Scholar]
- Prinz M, Priller J. 2017. The role of peripheral immune cells in the CNS in steady state and disease. Nature Neuroscience 20:136. [DOI] [PubMed] [Google Scholar]
- Puglielli L, Tanzi RE, Kovacs DM. 2003. Alzheimer’s disease: the cholesterol connection. Nature Neuroscience 6:345. [DOI] [PubMed] [Google Scholar]
- Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, Lash J, Wider C, Wojtas A, DeJesus-Hernandez M, Adamson J, Kouri N, Sundal C, Shuster EA, Aasly J, MacKenzie J, Roeber S, Kretzschmar HA, Boeve BF, Knopman DS, Petersen RC, Cairns NJ, Ghetti B, Spina S, Garbern J, Tselis AC, Uitti R, Das P, Van Gerpen JA, Meschia JF, Levy S, Broderick DF, Graff-Radford N, Ross OA, Miller BB, Swerdlow RH, Dickson DW, Wszolek ZK. 2012. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 44:200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj D, Yin Z, Breur M, Doorduin J, Holtman IR, Olah M, Mantingh-Otter IJ, Van Dam D, De Deyn PP, den Dunnen W, Eggen BJL, Amor S, Boddeke E. 2017. Increased White Matter Inflammation in Aging- and Alzheimer’s Disease Brain. Frontiers in Molecular Neuroscience 10:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Paolicelli RC. 2018. Microglia-Mediated Synapse Loss in Alzheimer’s Disease. The Journal of Neuroscience 38:2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reemst K, Noctor SC, Lucassen PJ, Hol EM. 2016. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Frontiers in Human Neuroscience 10:566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichwald J, Danner S, Wiederhold K-H, Staufenbiel M. 2009. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. Journal of Neuroinflammation 6:35–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Chiò A, Traynor BJ. 2013. State of play in amyotrophic lateral sclerosis genetics. Nature Neuroscience 17:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Réu P, Khosravi A, Bernard S, Mold JE, Salehpour M, Alkass K, Perl S, Tisdale J, Possnert G, Druid H, Frisén J. 2017. The Lifespan and Turnover of Microglia in the Human Brain. Cell Reports 20:779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro Xavier AL, Kress BT, Goldman SA, Lacerda de Menezes JR, Nedergaard M. 2015. A Distinct Population of Microglia Supports Adult Neurogenesis in the Subventricular Zone. The Journal of Neuroscience 35:11848–11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D, Hajishengallis G, Yang K, Lambris JD. 2010. Complement: a key system for immune surveillance and homeostasis. Nature Immunology 11:785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Li R, Mastroeni D, Grover A, Leonard B, Ahern G, Cao P, Kolody H, Vedders L, Kolb WP, Sabbagh M. 2006. Peripheral clearance of amyloid β peptide by complement C3-dependent adherence to erythrocytes. Neurobiology of Aging 27:1733–1739. [DOI] [PubMed] [Google Scholar]
- Roh M, Zhang Y, Murakami Y, Thanos A, Lee SC, Vavvas DG, Benowitz LI, Miller JW. 2012. Etanercept, a widely used inhibitor of tumor necrosis factor-alpha (TNF-alpha), prevents retinal ganglion cell loss in a rat model of glaucoma. PloS one 7:e40065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumier A, Bechade C, Poncer JC, Smalla KH, Tomasello E, Vivier E, Gundelfinger ED, Triller A, Bessis A. 2004. Impaired synaptic function in the microglial KARAP/DAP12-deficient mouse. J Neurosci 24:11421–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, Stevens B. 2017. Microglia emerge as central players in brain disease. Nature Medicine 23:1018. [DOI] [PubMed] [Google Scholar]
- Sato K 2015. Effects of Microglia on Neurogenesis. Glia 63:1394–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Heller CT, Gunner G, Heller M, Gordon C, Hammond T, Wolf Y, Jung S, Stevens B. 2016. Microglia contribute to circuit defects in Mecp2 null mice independent of microglia-specific loss of Mecp2 expression. eLife 5:e15224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. 2012. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74:691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Stevens B. 2013. Phagocytic glial cells: sculpting synaptic circuits in the developing nervous system. Curr Opin Neurobiol 23:1034–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, Genovese G, Rose SA, Handsaker RE, Schizophrenia Working Group of the Psychiatric Genomics C, Daly MJ, Carroll MC, Stevens B, McCarroll SA. 2016. Schizophrenia risk from complex variation of complement component 4. Nature 530:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P, Lerch JP, Pruessner JC, Taylor KN, Rose AB, Greenstein D, Clasen L, Evans A, Rapoport JL, Giedd JN. 2007. Cortical morphology in children and adolescents with different apolipoprotein E gene polymorphisms: an observational study. The Lancet Neurology 6:494–500. [DOI] [PubMed] [Google Scholar]
- Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF, Dolmetsch RE. 2013. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, Lemere CA. 2017. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Science Translational Medicine 9:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto-Mogami Y, Hoshikawa K, Goldman JE, Sekino Y, Sato K. 2014. Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. J Neurosci 34:2231–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohayeb B, Diab M, Ahmed M, Ng DCH. 2018. Factors that influence adult neurogenesis as potential therapy. Translational Neurodegeneration 7:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M. 2010. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7:483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman SM, Wong WT. 2018. Microglia in the Retina: Roles in Development, Maturity, and Disease. Annual Review of Vision Science 4:45–77. [DOI] [PubMed] [Google Scholar]
- Siskova Z, Tremblay ME. 2013. Microglia and synapse: interactions in health and neurodegeneration. Neural Plast 2013:425845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solano Fonseca R, Mahesula S, Apple DM, Raghunathan R, Dugan A, Cardona A, O’Connor J, Kokovay E. 2016. Neurogenic Niche Microglia Undergo Positional Remodeling and Progressive Activation Contributing to Age-Associated Reductions in Neurogenesis. Stem Cells and Development 25:542–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto I, Howell GR. 2014. The Complex Role of Neuroinflammation in Glaucoma. Cold Spring Harbor Perspectives in Medicine 4:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spittau B 2017. Aging Microglia—Phenotypes, Functions and Implications for Age-Related Neurodegenerative Diseases. Frontiers in Aging Neuroscience 9:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squarzoni P, Oller G, Hoeffel G, Pont-Lezica L, Rostaing P, Low D, Bessis A, Ginhoux F, Garel S. 2014. Microglia modulate wiring of the embryonic forebrain. Cell Rep 8:1271–1279. [DOI] [PubMed] [Google Scholar]
- Steele MR, Inman DM, Calkins DJ, Horner PJ, Vetter ML. 2006. Microarray analysis of retinal gene expression in the DBA/2J model of glaucoma. Invest Ophthalmol Vis Sci 47:977–985. [DOI] [PubMed] [Google Scholar]
- Stephan AH, Barres BA, Stevens B. 2012. The Complement System: An Unexpected Role in Synaptic Pruning During Development and Disease. Annual Review of Neuroscience 35:369–389. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. 2007. The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178. [DOI] [PubMed] [Google Scholar]
- Stowell RD, Wong EL, Batchelor HN, Mendes MS, Lamantia CE, Whitelaw BS, Majewska AK. 2017. Cerebellar microglia are dynamically unique and survey Purkinje neurons in vivo. Developmental Neurobiology 78:627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su P, Zhang J, Zhao F, Aschner M, Chen J, Luo W. 2014. The interaction between microglia and neural stem/precursor cells. Brain Res Bull 109:32–38. [DOI] [PubMed] [Google Scholar]
- Suh H-S, Zhao M-L, Derico L, Choi N, Lee SC. 2013. Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: differential regulation by inflammatory mediators. Journal of Neuroinflammation 10:37–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Rochford CDP, Neumann H. 2005. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. The Journal of Experimental Medicine 201:647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey KE, Cameron D, Hill MJ. 2018. Genetic risk for Alzheimer’s disease is concentrated in specific macrophage and microglial transcriptional networks. Genome Medicine 10:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay TL, Bachade C, apos Andrea I, St-Pierre M-K, Henry MS, Roumier A, Tremblay M-E. 2018. Microglia Gone Rogue: Impacts on Psychiatric Disorders across the Lifespan. Frontiers in Molecular Neuroscience 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay TL, Hagemeyer N, Prinz M. 2016. The force awakens: insights into the origin and formation of microglia. Current Opinion in Neurobiology 39:30–37. [DOI] [PubMed] [Google Scholar]
- Tay TL, Mai D, Dautzenberg J, Fernández-Klett F, Lin G, Sagar Datta M, Drougard A, Stempfl T, Ardura-Fabregat A, Staszewski O, Margineanu A, Sporbert A, Steinmetz LM, Pospisilik JA, Jung S, Priller J, Grün D, Ronneberger O, Prinz M. 2017. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nature Neuroscience 20:793. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. 1991. Physical basis of cognitive alterations in alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Annals of Neurology 30:572–580. [DOI] [PubMed] [Google Scholar]
- Thion MS, Garel S. 2017. On place and time: microglia in embryonic and perinatal brain development. Current Opinion in Neurobiology 47:121–130. [DOI] [PubMed] [Google Scholar]
- Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, Blecher R, Ulas T, Squarzoni P, Hoeffel G, Coulpier F, Siopi E, David FS, Scholz C, Shihui F, Lum J, Amoyo AA, Larbi A, Poidinger M, Buttgereit A, Lledo P-M, Greter M, Chan JKY, Amit I, Beyer M, Schultze JL, Schlitzer A, Pettersson S, Ginhoux F, Garel S. 2018. Microbiome Influences Prenatal and Adult Microglia in a Sex-Specific Manner. Cell 172:500–516.e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turano A, Lawrence JH, Schwarz JM. 2017. Sex differences in microglial activation in the developing rat brain. Brain, Behavior, and Immunity 66:e5. [Google Scholar]
- Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, Colonna M. 2006. Cutting Edge: TREM-2 Attenuates Macrophage Activation. The Journal of Immunology 177:3520. [DOI] [PubMed] [Google Scholar]
- Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, Yamashita T. 2013. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci 16:543–551. [DOI] [PubMed] [Google Scholar]
- Ulland TK, Song WM, Huang SC-C, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E, Gilfillan S, Cella M, Virgin HW, Unanue ER, Wang Y, Artyomov MN, Holtzman DM, Colonna M. 2017. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 170:649–663.e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero J, Paris I, Sierra A. 2016. Lifestyle Shapes the Dialogue between Environment, Microglia, and Adult Neurogenesis. ACS Chemical Neuroscience 7:442–453. [DOI] [PubMed] [Google Scholar]
- Vilalta A, Brown GC. 2018. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. The FEBS Journal 285:3566–3575. [DOI] [PubMed] [Google Scholar]
- Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G, Lolli F, Marcello E, Sironi L, Vegeto E, Maggi A. 2018. Sex-Specific Features of Microglia from Adult Mice. Cell Reports 23:3501–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bernhardi R, Tichauer JE, Eugenin J. 2010. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J Neurochem 112:1099–1114. [DOI] [PubMed] [Google Scholar]
- Vukovic J, Colditz MJ, Blackmore DG, Ruitenberg MJ, Bartlett PF. 2012. Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J Neurosci 32:6435–6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakselman S, Bechade C, Roumier A, Bernard D, Triller A, Bessis A. 2008. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci 28:8138–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DG, McGeer PL. 1992. Complement gene expression in human brain: comparison between normal and Alzheimer disease cases. Molecular Brain Research 14:109–116. [DOI] [PubMed] [Google Scholar]
- Walton NM, Sutter BM, Laywell ED, Levkoff LH, Kearns SM, Marshall GP 2nd, Scheffler B, Steindler DA. 2006. Microglia instruct subventricular zone neurogenesis. Glia 54:815–825. [DOI] [PubMed] [Google Scholar]
- Wang J, Wegener JE, Huang T-W, Sripathy S, De Jesus-Cortes H, Xu P, Tran S, Knobbe W, Leko V, Britt J, Starwalt R, McDaniel L, Ward C, Parra D, Newcomb B, Lao U, Flowers DA, Cullen S, Jorstad NL, Yang Y, Glaskova L, Vigneau S, Kozlitina J, Reichardt SD, Reichardt HM, Gärtner J, Bartolomei MS, Fang M, Loeb K, Keene CD, Bernstein I, Goodell M, Brat DJ, Huppke P, Neul J, Bedalov A, Pieper AA. 2015a. Wild type microglia do not arrest pathology in mouse models of Rett syndrome. Nature 521:E1–E4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich Jason D, Young Katherine L, Robinette Michelle L, Gilfillan S, Krishnan Gokul M, Sudhakar S, Zinselmeyer Bernd H, Holtzman David M, Cirrito John R, Colonna M. 2015b. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 160:1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warwick A, Khandhadia S, Ennis S, Lotery A. 2014. Age-Related Macular Degeneration: A Disease of Systemic or Local Complement Dysregulation? Journal of Clinical Medicine 3:1234–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendeln A-C, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, Wagner J, Häsler LM, Wild K, Skodras A, Blank T, Staszewski O, Datta M, Centeno TP, Capece V, Islam MR, Kerimoglu C, Staufenbiel M, Schultze JL, Beyer M, Prinz M, Jucker M, Fischer A, Neher JJ. 2018. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556:332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner H, LeRoith D. 2014. Insulin and insulin-like growth factor receptors in the brain: Physiological and pathological aspects. European Neuropsychopharmacology 24:1947–1953. [DOI] [PubMed] [Google Scholar]
- West MJ, Coleman PD, Flood DG, Troncoso JC. 1994. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. The Lancet 344:769–772. [DOI] [PubMed] [Google Scholar]
- Williams PA, Marsh-Armstrong N, Howell GR. 2017. Neuroinflammation in glaucoma: A new opportunity. Experimental Eye Research 157:20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarczyk A, Cedile O, Jensen KN, Jasson A, Mony JT, Khorooshi R, Owens T. 2015. Pathologic and Protective Roles for Microglial Subsets and Bone Marrow- and Blood-Derived Myeloid Cells in Central Nervous System Inflammation. Frontiers in Immunology 6:463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarczyk A, Holtman IR, Krueger M, Yogev N, Bruttger J, Khorooshi R, Benmamar‐Badel A, de Boer‐Bergsma JJ, Martin NA, Karram K, Kramer I, Boddeke EWGM, Waisman A, Eggen BJL, Owens T. 2017. A novel microglial subset plays a key role in myelinogenesis in developing brain. The EMBO Journal 36:3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarczyk A, Løbner M, Cédile O, Owens T. 2014. Comparison of microglia and infiltrating CD11c(+) cells as antigen presenting cells for T cell proliferation and cytokine response. Journal of Neuroinflammation 11:57–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dissing-Olesen L, MacVicar BA, Stevens B. 2015. Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends in Immunology 36:605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Wang Z, Li J, Wu H, Peng Y, Fan L, Chen J, Gu C, Yan F, Wang L, Chen G. 2017. The Polarization States of Microglia in TBI: A New Paradigm for Pharmacological Intervention. Neural Plasticity 2017:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanguas-Casás N, Crespo-Castrillo A, de Ceballos ML, Chowen JA, Azcoitia I, Arevalo MA, Garcia-Segura LM. 2017. Sex differences in the phagocytic and migratory activity of microglia and their impairment by palmitic acid. Glia 66:522–537. [DOI] [PubMed] [Google Scholar]
- Yeh FL, Hansen DV, Sheng M. 2017. TREM2, Microglia, and Neurodegenerative Diseases. Trends in Molecular Medicine 23:512–533. [DOI] [PubMed] [Google Scholar]
- Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. 2016. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 91:328–340. [DOI] [PubMed] [Google Scholar]
- Yuan L, Neufeld AH. 2001. Activated microglia in the human glaucomatous optic nerve head. Journal of Neuroscience Research 64:523–532. [DOI] [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird Thomas D, Paul Steven M, Luo W, Colonna M, Baddeley D, Grutzendler J. 2016. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 90:724–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel MK, Kirsch WM. 2013. From development to dysfunction: Microglia and the complement cascade in CNS homeostasis. Ageing Research Reviews 12:749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, Gross CT. 2014. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nature Neuroscience 17:400. [DOI] [PubMed] [Google Scholar]
- Zhang J, Liu Q. 2015. Cholesterol metabolism and homeostasis in the brain. Protein & Cell 6:254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. 2014. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. The Journal of Neuroscience 34:11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Zabel MK, Wang X, Ma W, Shah P, Fariss RN, Qian H, Parkhurst CN, Gan WB, Wong WT. 2015. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Molecular Medicine 7:1179–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M. 2006. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nature Neuroscience 9:268. [DOI] [PubMed] [Google Scholar]
- Zunszain PA, Hepgul N, Pariante CM. 2013. Inflammation and Depression In: Cowen PJ, Sharp T, Lau JYF, editors. Behavioral Neurobiology of Depression and Its Treatment. Berlin, Heidelberg: Springer Berlin Heidelberg; pp 135–151. [Google Scholar]