

Horizontal gene transfer (HGT), natural selection, and gene duplication are three main engines that drive the adaptive evolution of microbial genomes. Previous studies indicated that HGT was a main adaptive mechanism in acidophiles to cope with heavy-metal-rich environments. However, evidences of HGT in Acidithiobacillus species in response to challenging metal-rich environments and the mechanisms addressing how metal resistance genes originated and evolved in Acidithiobacillus are still lacking. The findings of this study revealed a fascinating phenomenon of putative cross-phylum HGT, suggesting that Acidithiobacillus spp. recruited and consolidated additional novel functionalities during the adaption to challenging environments via HGT, gene duplication, and purifying selection. Altogether, the insights gained in this study have improved our understanding of the metal resistance strategies of Acidithiobacillus spp.
KEYWORDS: Acidithiobacillus spp., comparative genomics, evolution, horizontal gene transfer, metal resistance
ABSTRACT
Members of the genus Acidithiobacillus, which can adapt to extremely high concentrations of heavy metals, are universally found at acid mine drainage (AMD) sites. Here, we performed a comparative genomic analysis of 37 strains within the genus Acidithiobacillus to answer the untouched questions as to the mechanisms and the evolutionary history of metal resistance genes in Acidithiobacillus spp. The results showed that the evolutionary history of metal resistance genes in Acidithiobacillus spp. involved a combination of gene gains and losses, horizontal gene transfer (HGT), and gene duplication. Phylogenetic analyses revealed that metal resistance genes in Acidithiobacillus spp. were acquired by early HGT events from species that shared habitats with Acidithiobacillus spp., such as Acidihalobacter, Thiobacillus, Acidiferrobacter, and Thiomonas species. Multicopper oxidase genes involved in copper detoxification were lost in iron-oxidizing Acidithiobacillus ferridurans, Acidithiobacillus ferrivorans, and Acidithiobacillus ferrooxidans and were replaced by rusticyanin genes during evolution. In addition, widespread purifying selection and the predicted high expression levels emphasized the indispensable roles of metal resistance genes in the ability of Acidithiobacillus spp. to adapt to harsh environments. Altogether, the results suggested that Acidithiobacillus spp. recruited and consolidated additional novel functionalities during the adaption to challenging environments via HGT, gene duplication, and purifying selection. This study sheds light on the distribution, organization, functionality, and complex evolutionary history of metal resistance genes in Acidithiobacillus spp.
IMPORTANCE Horizontal gene transfer (HGT), natural selection, and gene duplication are three main engines that drive the adaptive evolution of microbial genomes. Previous studies indicated that HGT was a main adaptive mechanism in acidophiles to cope with heavy-metal-rich environments. However, evidences of HGT in Acidithiobacillus species in response to challenging metal-rich environments and the mechanisms addressing how metal resistance genes originated and evolved in Acidithiobacillus are still lacking. The findings of this study revealed a fascinating phenomenon of putative cross-phylum HGT, suggesting that Acidithiobacillus spp. recruited and consolidated additional novel functionalities during the adaption to challenging environments via HGT, gene duplication, and purifying selection. Altogether, the insights gained in this study have improved our understanding of the metal resistance strategies of Acidithiobacillus spp.
INTRODUCTION
The genus Acidithiobacillus is composed of a group of obligatory acidophilic chemolithotrophic Gram-negative bacteria that conduct energy conservation by oxidizing reduced inorganic sulfur compounds (RISCs), such as thiosulfate (S2O32−), hydrogen sulfide (H2S), and elemental sulfur (S0). This attribute has been exploited in the bioleaching of ores and has also been the main cause of the accelerating formation of polluting acid mine drainage (AMD) water, which brings disruptive consequences for terrestrial and aquatic ecosystems (1–5). Currently, 8 species have been reported in the genus Acidithiobacillus, including Acidithiobacillus albertensis, Acidithiobacillus caldus, Acidithiobacillus cuprithermicus, Acidithiobacillus ferridurans, Acidithiobacillus ferriphilus, Acidithiobacillus ferrivorans, Acidithiobacillus ferrooxidans, and Acidithiobacillus thiooxidans. Acidithiobacillus species are universally found at AMD sites where highly toxic inorganic forms of heavy metals and metalloids such as arsenic, mercury, copper, cadmium, and cobalt often dominate (1, 6–9). As revealed in previous studies, strains of A. ferrooxidans can adapt up to 800 mM Cu2+, 84 mM As3+, 1,071 mM Zn2+, 500 mM Cd2+, 1,000 mM Ni2+, and 50 mM Hg2+ (10, 11), while strains of A. thiooxidans and A. ferrooxidans are able to adapt up to 120 and 100 mM As5+ (12). It is obvious that Acidithiobacillus species surviving in acidic metal-rich leaching environments must possess certain highly efficient and advanced metal resistance mechanisms, rendering them ideal research models to improve our understanding of metal resistance. There are four main strategies that acidophiles generally utilize to increase resistance to metals, including the chelation and precipitation of toxic metals, the efflux of the toxic metals out of the cells by molecular pumps, the enzymatic conversion of metal ions to less toxic forms, and/or enhanced cell walls and membranes that are impermeable to toxic metal ions (13). These strategies are closely related to the genetic properties of the bacteria. The genes conferring toxic metal resistance are prone to clustering together as gene operons in Acidithiobacillus genomes, such as the mer operons that confer mercury resistance (14). However, the mechanisms driving the clustering of genes into operons are far from clear. For example, the mer operon generally contains merA, merD, merR, merP, merC, and merT, although their organization may be different. The reported genes related to mercury, arsenate, cadmium, zinc, cobalt, and copper resistance and their functions in bacteria are summarized in Table 1.
TABLE 1.
Genes involved in toxic metal resistance and transformation
| Category | Gene | Protein | Function | Reference(s) |
|---|---|---|---|---|
| Mercury detoxification | merA | Mercuric reductase | Reduce Hg(II) to gaseous mercury Hg(0) | 98 |
| merD | Regulatory protein | Downregulate mer operon expression | ||
| merR | Transcriptional activator/repressor | Regulate the overall expression of mer operon | ||
| merP | Periplasmic protein | Hg(II) scavenging | ||
| merC | Inner membrane spanning protein | Transport Hg(II) to the cytoplasm | ||
| merT | Inner membrane spanning protein | Transport Hg(II) to the cytoplasm | ||
| merB | Organomercury lyase | Break down organomercury | ||
| Arsenate/arsenite detoxification | arsR | Transcriptional regulator ArsR | Repress the basal expression of the ars operon | 99–101 |
| arsB | As(III) efflux pump protein | Pump As(III) out of the cell | ||
| arsC | As(V) reductase ArsC | Transform As(V) to As(III) | ||
| arsH | Organoarsenical oxidase | Impact the oxidation of arsenic | ||
| arsA | As(III) active ATPase | Convert membrane potential to ATP; provide from ATP hydrolysis to ArsB | ||
| arsD | As metallochaperone | Bind and transfer As(III) to ArsA; repress the upper expression levels of the ars operon | ||
| arrA | Large subunit of respiratory As(V) reductase | Provide energy by reducing As(V) to As(III) | ||
| arrB | Small subunit of respiratory As(V) reductase | |||
| aioA | Large subunit of As(III) oxidase | Provide energy by oxidizing As(III) to As(V) | ||
| aioB | Small subunit of As(III) oxidase | |||
| arsM | As(III) S-adenosylmethyltransferase | Catalyze the formation of volatile trimethylarsine from As(III) | ||
| Cadmium, zinc, cobalt, and copper detoxification | czc/cusA | Large inner membrane protein | Form a tripartite complex Czc/CusABC that pumps out cadmium, zinc, cobalt, and copper cations via the mechanism of cation-proton antiporter | 13, 26, 40–42 |
| czc/cusB | Periplasmic coupling protein | |||
| czc/cusC | Smaller outer membrane protein | |||
| czcD | Cation diffusion facilitator | Efflux of cadmium ions; regulation of the Czc system | 102 | |
| cop | Copper-translocating P-type ATPase | Pump Cu(I) ions from the cytosol to the periplasm | 103–105 | |
| mco | Multicopper oxidase | Oxidize Cu(I) to the less toxic form Cu(II) |
Horizontal gene transfer (HGT), natural selection, and gene duplication are three main engines that drive the adaptive evolution of microbial genomes, whereas their relative importance is still ambiguous (15, 16). HGT events generally occur via mobile genetic elements (MGEs) carrying functional genes that enhance the adaptability and versatility of the acceptors. Recent studies analyzing gene-transfer webs among a vast number of sequenced genomes revealed that more than 20% of the microbial genes may be acquired through HGT (17, 18). MGEs such as plasmids, phages, genomic islands (GIs), transposons, and insertion sequences are indicative of HGT events. GIs, which are large chromosomal regions usually found adjacent to or integrated within tRNA genes, represent a versatile gene pool (19). Deviant G+C contents, as well as incongruences between phylogenies of functional genes and species, can also be used as indicators of HGT events (20–23). Some previous studies have described the overall metal resistance mechanisms in acidophiles and indicated that HGT was a main adaptive mechanism by which the acidophiles cope with heavy-metal-rich environments (13, 24–27); for example, it was reported that A. ferrooxidans ATCC 53993 harbored GIs, inserted after a tRNA metabolism related gene (MiaB type), containing genes encoding mercury detoxification determinants (merA, merC, and merR) and copper translocating P-type ATPase (13).
However, direct evidence of HGT in other Acidithiobacillus species in response to challenging metal-rich environments and the mechanisms addressing how metal resistance genes originated and evolved in Acidithiobacillus are still lacking. The rapid development of next-generation sequencing technologies has enriched the public database with numerous sequenced genomes. Exploiting these resources, large-scale comparative genomic analyses are improving our understanding of extensive genetic diversity and evolutionary history. To date, 8 species have been reported in genus Acidithiobacillus, and strains of 6 species (A. thiooxidans, A. ferrooxidans, A. caldus, A. ferrivorans, A. albertensis, and A. ferridurans) have been sequenced. To reveal the genetic traits and phylogenetic history of metal resistance in genus Acidithiobacillus, a comparative genomic analysis of 37 strains within the genus Acidithiobacillus was carried out. In addition, molecular phylogenetic analysis, gene contents, molecular conservation, and linear analyses along with selective pressure and codon usage analyses were used to explore the evolution of toxic metal resistance genes in the genus Acidithiobacillus (28–31). Our results provide explicit evidence that HGT played a crucial role in driving the evolution of heavy metal resistance in Acidithiobacillus species surviving extreme environments. The insights gained in this study improve our understanding of the complex evolutionary history of toxic metal resistance genes in Acidithiobacillus spp. and its roles in the biogeochemical cycling of different metals.
RESULTS
Genomic features.
A summary of the features of 38 available sequenced genomes of Acidithiobacillus spp. are listed in Table 2. The G+C contents of the 38 genomes range from 52.6% to 61.5%. These genomes vary in size by approximately 2.48 Mb (ranging from 1.70 to 4.18 Mb) with coding sequence (CDS) numbers ranging from 1,617 to 4,359, suggesting substantial strain-to-strain variations.
TABLE 2.
General features of bacterial genomes used in this study
| Species | Strain | GenBank accession no. | Level | Size (Mb) | GC% | No. of genes | No. of proteins |
|---|---|---|---|---|---|---|---|
| A. thiooxidans | A01 | GCA_000559045.1 | Contig | 3.8 | 53.1 | 4,131 | 3,719 |
| A. thiooxidans | Licanantay | GCA_000709715.1 | Contig | 3.9 | 52.8 | 4,359 | 3,774 |
| A. thiooxidans | DMC | GCA_001705625.1 | Contig | 3.9 | 53.1 | 4,196 | 3,772 |
| A. thiooxidans | A02 | GCA_001705645.1 | Contig | 3.7 | 53.0 | 4,034 | 3,639 |
| A. thiooxidans | GD1-3 | GCA_001705695.1 | Contig | 3.9 | 52.9 | 4,225 | 3,824 |
| A. thiooxidans | BY-02 | GCA_001705725.1 | Contig | 3.8 | 53.1 | 4,196 | 3,683 |
| A. thiooxidans | JYC-17 | GCA_001705755.1 | Contig | 3.8 | 53.1 | 4,176 | 3,737 |
| A. thiooxidans | DXS-W | GCA_001705805.1 | Contig | 3.9 | 52.9 | 4,229 | 3,833 |
| A. thiooxidans | ZBY | GCA_001756595.1 | Contig | 3.8 | 53.2 | 4,106 | 3,714 |
| A. thiooxidans | CLST | GCA_002079865.1 | Scaffold | 4.0 | 52.4 | 4,045 | 3,619 |
| A. thiooxidans | ATCC 19377 | GCA_000227215.2 | Contig | 3.0 | 53.2 | 3,106 | 2,847 |
| A. ferrooxidans | BY0502 | GCA_001652185.1 | Contig | 3.0 | 56.8 | 3,186 | 2,821 |
| A. ferrooxidans | CCM 4253 | GCA_003233765.1 | Contig | 3.2 | 58.6 | 3,278 | 3,049 |
| A. ferrooxidans | IO-2C | GCA_003309025.1 | Contig | 2.7 | 58.7 | 2,822 | 2,620 |
| A. ferrooxidans | YQH-1 | GCA_001418795.1 | Scaffold | 3.1 | 58.6 | 3,189 | 2,956 |
| A. ferrooxidans | DLC-5 | GCA_000732185.1 | Contig | 4.2 | 57.6 | 4,289 | 4,006 |
| A. ferrooxidans | ATCC 23270 | CP001219.1 | Complete | 3.0 | 58.8 | 3,087 | 2,909 |
| A. ferrooxidans | ATCC 53993 | CP001132.1 | Complete | 2.9 | 58.9 | 2,926 | 2,779 |
| A. ferrooxidans | Hel18 | GCA_001559335.1 | Contig | 3.1 | 58.6 | 3,179 | 2,955 |
| Acidithiobacillus sp. | SH | GCA_002847505.1 | Contig | 2.9 | 54.3 | 2,987 | 2,799 |
| Acidithiobacillus sp. | NORP59 | GCA_002733395.1 | Contig | 3.6 | 52.6 | 3,525 | 3,260 |
| Acidithiobacillus sp. | GGI-221 | GCA_000179815.2 | Contig | 3.2 | 58.6 | 4,079 | 4,007 |
| Acidithiobacillus sp. | UBA2486 | GCA_002341825.1 | Contig | 1.7 | 58.5 | 1,617 | 1,596 |
| A. albertensis | DSM 14366 | GCA_001931655.1 | Contig | 3.5 | 52.5 | 3,622 | 3,354 |
| A. ferrivorans | SS3 | CP002985.1 | Complete | 3.2 | 56.6 | 3,206 | 3,016 |
| A. ferrivorans | PRJEB5721 | GCA_900174455.1 | Scaffold | 3.5 | 56.5 | 3,555 | 3,296 |
| A. ferrivorans | CF27 | GCA_000750615.1 | Contig | 3.4 | 56.4 | 3,557 | 3,236 |
| A. ferrivorans | YL15 | GCA_001685225.1 | Contig | 3.0 | 56.6 | 3,087 | 2,778 |
| A. ferrivorans | PQ33 | GCA_001857665.2 | Contig | 3.3 | 56.6 | 3,382 | 3,077 |
| A. ferrivorans | 21-59-9 | GCA_002255305.1 | Scaffold | 2.7 | 59.0 | 3,208 | 2,909 |
| A. caldus | ATCC 51756 | GCA_000175575.2 | Complete | 3.0 | 61.4 | 3,005 | 2,849 |
| A. caldus | SM-1 | GCA_000221025.1 | Complete | 3.2 | 60.9 | 3,254 | 3,106 |
| A. caldus | MTH-04 | GCA_001650235.2 | Complete | 3.0 | 61.5 | 3,102 | 2,713 |
| A. caldus | DX | GCA_001756675.1 | Contig | 3.1 | 61.0 | 3,179 | 2,861 |
| A. caldus | ZBY | GCA_001756725.1 | Contig | 3.2 | 61.0 | 3,210 | 2,902 |
| A. caldus | ZJ | GCA_001756745.1 | Contig | 3.1 | 61.0 | 3,223 | 2,884 |
| A. caldus | S1 | GCA_001756775.1 | Contig | 2.8 | 60.9 | 2,906 | 2,396 |
| A. ferridurans | JCM 18981 | AP018795.1 | Complete | 2.9 | 58.4 | 3,043 | 2,802 |
Phylogenic analyses and pangenome analyses.
To associate the distributions of metal resistance genes in Acidithiobacillus spp. with their phylogenetic affiliations, we constructed a phylogenetic tree of the Acidithiobacillus spp. and closely related species on the basis of 16S rRNA gene sequences using neighbor-joining (NJ) methods (see Fig. S1 in the supplemental material). The results showed that Acidithiobacillus species are phylogenetically close to Thermithiobacillus spp. Since no 16S rRNA gene sequence could be retrieved from the genome of Acidithiobacillus sp. strain NORP59, we constructed a phylogenetic tree of the 38 Acidithiobacillus genomes with their phylogenetic affiliations on the basis of whole-genome sequences (see Fig. S2). The results showed that Acidithiobacillus sp. NORP59 forms a clade with Thioglobus sp. strain MED-G25, distant from the standard strains of Acidithiobacillus spp. In addition, Acidithiobacillus sp. NORP59 showed low values of BLASTN-based average nucleotide identity (ANI; average 75%) and tetranucleotide composition regression (Tetra; average 0.758) in comparison with those of other available Acidithiobacillus strains (see Tables S1 and S2), indicating that strain NORP59 was not a member of genus Acidithiobacillus; thus, it was excluded from any further analyses.
To reveal the genomic features specific to each strain, we identified orthologous groups among the 37 Acidithiobacillus genomes using the Bacterial Pan Genome Analysis (BPGA) pipeline (32). Our analysis of the 37 genomes revealed a pangenome containing 141,937 putative protein-coding genes in the genus Acidithiobacillus. Of these 141,937 genes, 16,317 (11.5%) were clustered into the core genome of Acidithiobacillus spp., 95,704 (67.4%) were represented in the accessory genome, and 5,332 (3.8%) were identified as strain-specific genes. The numbers of specific genes ranged from 0 to 811 (Fig. 1A). The results of BacMet database annotation revealed that the accessory genome had a higher proportion (8.2% [7,846 genes]) of genes involved in heavy metal resistance activities (Fig. 1C) than the core genome (5.2% [851 genes]) and a higher proportion of unique genes (0.3% [16 genes]). We constructed a robust phylogenetic tree of the 37 genomes on the basis of the concatenation of the 110 single-copy core genes in each genome using the NJ method (see Fig. S3). The phylogenetic trees, inferred using ML and UPGMA methods (see Fig. S4 and S5), were congruent with the NJ phylogenetic tree, showing that the phylogenetic tree was highly reliable. A chronogram for NJ phylogenetic tree was constructed with a calibration point to explore the divergence times of Acidithiobacillus (Fig. 2). The most recent common ancestor (MRCA) of Acidithiobacillus (i.e., the emergence time of A. caldus) was estimated to have emerged around 800 million years ago (MYA) (Fig. 2), whereas the MRCA of A. thiooxidans, A. ferrooxidans, A. ferrivorans, and A. ferridurans was estimated to have emerged around 439 MYA (Fig. 2). These well-supported core gene trees showed that some strains in different reported species were grouped together, such as A. ferridurans JCM 18981 and A. ferrooxidans IO-2C, Acidithiobacillus sp. strain GGI-221 and strains of A. ferrooxidans, and A. albertensis DSM 14366 and strains of A. thiooxidans. These results suggested that there were mistakes in the classification of Acidithiobacillus spp. The ANI approach was further applied to evaluate the genomic similarity of microorganisms (33). The ANI results (Table S1) justified the conclusion from the phylogenetic analysis. All 37 strains could be classified into 9 species based on an ANI of ≥96%. A comparison of strains JCM 18981 and IO-2C resulted in a high ANI (98.9%), suggesting that they belong to the same species (A. ferridurans). A comparison between strains A. albertensis DSM 14366 and A. thiooxidans spp. resulted in a high ANI (97.3%), suggesting that they belong to the same species (A. thiooxidans). A comparison between strains GGI-221 and A. ferrooxidans spp. resulted in a high ANI (99.5%), suggesting that they belong to the same species (A. ferrooxidans). Thus, in this article, strain A. ferrooxidans IO-2C was renamed A. ferridurans IO-2C, strain A. albertensis DSM 14366 was renamed A. thiooxidans DSM 14366, and Acidithiobacillus sp. strain GGI-221 was renamed A. ferrooxidans GGI-221.
FIG 1.

Pangenome analysis of strains in the genus Acidithiobacillus. (A) Petal diagram of the pangenome. Each strain is represented by a colored oval. The center is the number of orthologous coding sequences shared by all strains (i.e., the core genome). Numbers in nonoverlapping portions of each oval show the numbers of CDSs unique to each strain. The total numbers of protein-coding genes within each genome are listed below the strain names. (B) Mathematical modeling of the pangenome and core genome of Acidithiobacillus. (C) Proportions of genes involved in heavy metal resistance in unique genes, accessory genome, core genome, and pangenome according BacMet database annotation.
FIG 2.

Chronogram of the 37 Acidithiobacillus strains. The value near each internal branch is the estimated emerge time for that branch. Nodes with fossil record corrections are indicated with a red star.
Model extrapolation of the pangenome of Acidithiobacillus.
Vernikos et al. (34) stated that a mathematical extrapolation of the pangenome would be highly reliable provided that sufficient genomes (more than five) are involved. As shown by the deduced power law regression function (Ps [n] = 4451.42n0.36465) (Fig. 1B), the pangenome of Acidithiobacillus had a parameter (γ) of 0.365, falling into the range of 0 < γ < 1, suggesting that the pangenome is open (35). The deduced exponential regression (Fc [n] =2052.65e−0.0714494n) revealed that the extrapolated curve of the core genome followed a steep slope, reaching a minimum of 441 gene families after the 37th genome was added (Fig. 1B). The numbers of core genes were relatively constant, such that an extra genome added would not significantly affect the size of core genome, which was consistent with the notion that the core genes were conserved genes universally present in all strains (36).
Identification of putative mobile genetic elements.
Mobile genetic elements (MGEs) are specific genome segments capable of intra- and extracellular movements, carrying gene(s) encoding putative functions, which are considered indicators of HGT events (37). In this study, MGEs including transposases, integrases, genomic islands (GIs), and phage-associated genes were identified and compared in all genomes. Each Acidithiobacillus genome in this study contained a number of transposons (see Table S3). The number of transposon copies per genome ranged from 143 (A. thiooxidans strain CLST) to 334 (A. caldus strain SM-1). Members of the IS3, IS5, IS21, and IS110 families were the most common. The numbers of GI copies per genome ranged from 25 (A. ferrivorans strain SS3) to 66 (A. thiooxidans CLST) (see Table S4). The numbers of prophages and/or prophage remnants ranged from 0 to 6 (see Table S5). The BacMet annotation results revealed that the genomes of Acidithiobacillus spp. contained a vast repertoire of metal resistance genes, with an average 160 BacMet hits in each genome. A visualization of the chromosomes of representative strains of different species of Acidithiobacillus (see Fig. S6) revealed that many of these genes were distributed within genomic island regions or flanking other mobile genetic elements such as transposons, indicating that putative HGT events contributed greatly to the adaptive survival of Acidithiobacillus spp. in toxic metal-rich niches.
Estimation of gene family gains and losses.
We further investigated how the metal-resistance-related gene families might have been gained and lost during the evolution of A. thiooxidans, A. ferrooxidans, A. ferrivorans, A. ferridurans, and A. caldus (see Fig. S7). Gene family gains were observed on the branches from A. ferridurans, A. caldus, and the most recent common ancestor (MRCA) of A. ferridurans and A. ferrooxidans. In all, new gene families arose over time probably via HGT, with only a few gene families being lost.
Case study of Hg-related genes.
The overall distributions and organization of Hg resistance genes in 37 Acidithiobacillus strains are summarized in Fig. S8. Hg resistance genes merA, merC, and merR were detected in most Acidithiobacillus genomes, while merD, merP, and merT were only detected in A. ferrooxidans, A. thiooxidans, and A. ferrivorans genomes. The gene merB that encodes organomercury lyase was missing in Acidithiobacillus genomes. The mer genes were prone to group together as mer clusters in Acidithiobacillus genomes, which could be categorized into six subgroups (merCAD, merRTPA, merTAR, merACR, merAR, and merA). Some strains in this study possessed two different mer clusters in their genomes (Fig. S8). However, the mer clusters were not detected in Acidithiobacillus sp. strain SH, possibly resulting from incomplete sequencing.
(i) Origin and evolution of mer clusters. The divergent distribution and organization of mer genes in Acidithiobacillus raised a question as to their origin and evolution. The deviant G+C contents can be used to detect HGT (20, 21). Herein, we detected the G+C contents of mer clusters and their corresponding genomes. The results showed that the G+C contents of the merCAD clusters were higher than those of the genomes in A. ferrooxidans strains (62.8% versus 57.6% to 61.0%, respectively); the G+C contents of respective merAPTR clusters were higher than those of the genomes in A. thiooxidans strains (60.5% to 61.1% versus 52.4% to 53.2%), A. ferrooxidans strains (62.6% to 62% versus 58.6%), and A. ferridurans strains (63.7% to 64.1% versus 58.4% to 61.3%). The merTAR and merACR gene clusters also showed variations of G+C contents between mer gene clusters and their corresponding genomes (see Fig. S9). To further elucidate the evolution of the mer gene clusters, we compared the chromosomal regions flanking the mer gene clusters among the 37 Acidithiobacillus strains and found that the genes in the upstream and downstream regions were conserved among strains of the same species except for merACR clusters (see Fig. S10). Several indicators of HGT, such as flanking tRNA genes and genes encoding mobile element proteins, were found near the merRTPA and merACR gene clusters. The results showed that strains of the same species generally shared identical insertion sites, whereas strains from different species had different insertion sites (Fig. S10), suggesting that mer clusters may be acquired more than once in Acidithiobacillus species. merACR gene clusters were found in different insertion sites regardless of whether the species were different or the same, suggesting that merACR clusters may be acquired via different HGT events (Fig. S10).
To elucidate the origin of mer genes clusters in Acidithiobacillus, a NJ phylogenetic tree was constructed on the basis of the MerA protein sequences. As shown in Fig. 3, the strains were separated into three group: group I included merCAD and merRTPA clusters, group II represented merACR clusters, and group III contained merRTAC, merTA, and merAR clusters and stand-alone merA genes. The phylogenetic tree revealed that the group I and mer genes of Burkholderiales members were sister groups, while those in group II were more similar to mer genes from Acidihalobacter spp., and those in group III were more similar to mer genes of Thiobacillus spp. and Acidiferrobacter thiooxydans, implying that the group I mer cluster may have been acquired via HGT from members of Burkholderiales, the group II mer cluster from Acidihalobacter spp., and the group III mer cluster from Thiobacillus spp. or Acidiferrobacter spp. in early evolutionary history. In addition, A. thiooxidans Licanantay containing cluster merRTAC branches near the base of the merTA and merAR clade. We inferred that among group III, gene clusters merRTAC, merTA, and merAR likely originated from A. thiooxidans, followed by gene loss in A. thiooxidans and HGT to A. ferrivorans. Besides, only stand-alone merA genes were detected in strains of A. caldus. The phylogenetic tree revealed that merA1 and merA2 genes of A. caldus form separate groups, in which merA2 genes of A. caldus were more similar to those from A. ferrivorans. The genes upstream and downstream were conserved for merA1 and merA2 genes, respectively (Fig. S10). Several genes encoding mobile element proteins which are indicators of HGT were found near the merA2 genes. All these indicated that merA2 genes of A. caldus arose from early HGT events, most likely from members within genus Acidithiobacillus.
FIG 3.
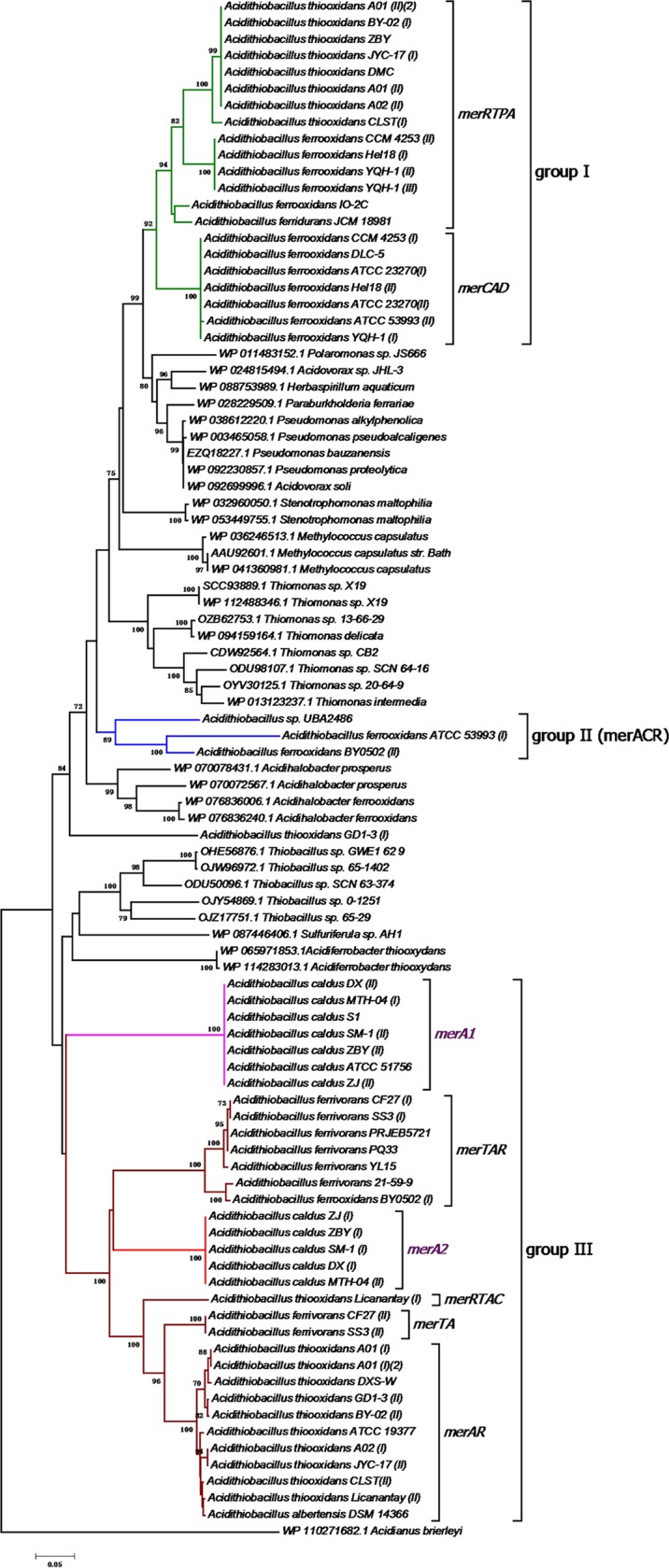
Neighbor-joining (NJ) phylogenetic tree of the MerA protein sequences derived from Acidithiobacillus species strains and other representative species. Bootstrap values indicated at each node are based on a total of 1,000 bootstrap replicates. Branches representing group I mer clusters are in green, those representing group II mer clusters are in blue, and those representing group III mer clusters are in red and pink.
Case study of As-related genes.
The overall distribution and organization of As resistance genes in Acidithiobacillus spp. are summarized in Fig. S11. As resistance genes (ars genes), including arsR, arsB, and arsC, were detected in all Acidithiobacillus genomes, while arsA and arsD were only detected in A. ferrivorans genomes and some strains from other species, including A. ferrooxidans strain DLC-5 and A. ferridurans IO-2C (A. ferrooxidans IO-2C), and arsH genes were only detected in genomes of A. ferrooxidans (within arsCRBH cluster) and A. thiooxidans (stand alone) (Fig. S11). Stand-alone arsM genes were also detected in all species. However, aio and arr, involved in arsenite oxidation and respiratory reduction of arsenate, were not found in Acidithiobacillus genomes, suggesting that cytoplasmic As(V) reduction and As(III) extrusion are the main As resistance strategies used in genus Acidithiobacillus spp. The ars genes in Acidithiobacillus genomes grouped together as ars clusters, including four subgroups, arsADCR, arsRB, arsBRC, and arsBRCH. The arsB gene of A. caldus strain MTH-04 was intercepted by an exogenous insertion sequence, which may result in malfunction of this arsB gene, rendering A. caldus MTH-04 vulnerable to toxic arsenic ions (Fig. S11).
(i) Origin and evolution of ars clusters. The distribution and organization of ars genes in Acidithiobacillus raise a question as to their evolution. The results showed that the G+C contents of the arsADCR clusters were higher than those of the genomes in A. ferrivorans strains (60.0% to 63.3% versus 56.4% to 58.6%, respectively); the G+C contents of respective arsBRC clusters were slightly higher than those of the genomes in A. caldus strains (62.3% to 63.1% versus 60.9% to 61.5%) and A. thiooxidans strains (53.1% to 55.3% versus 52.4% to 54.3%), showing the variation of G+C contents between clusters and the corresponding genomes (see Fig. S12). A comparison of the chromosomal regions flanking the ars gene clusters among Acidithiobacillus strains revealed that the genes in the upstream and downstream regions were conserved among strains of the same species (see Fig. S13). There were several genes encoding mobile element proteins (transposases or recombinases) located near the arsADCR and arsBRC clusters, and there were tRNA genes flanking the arsBRC clusters of A. caldus, indicating putative HGT events (Fig. S13). Strains of the same species share the same insertion sites, whereas strains of different species had different insertion sites, suggesting that ars clusters may have been acquired more than once via different HGT events. The flanking regions of the arsBRC gene clusters in strain A. thiooxidans were homologous to the corresponding regions of strains A. ferrivorans SS3 and A. ferrooxidans ATCC 23270 that have no arsBRC; the same phenomenon was found in arsBR gene clusters and arsH genes. These results suggested that ars genes may have been acquired by different HGT events followed by gene loss in some strains (Fig. S13).
To gain insights into the origin of ars genes clusters, NJ phylogenetic trees were constructed on the basis of on the concatenated ArsBRC protein sequences and ArsAD protein sequences. As shown in Fig. 4, the phylogenetic analysis revealed that the strains possessing arsBRC clusters formed a monophyletic clade. Acidithiobacillus spp. likely obtained the arsBRC cluster via HGT from Acidihalobacter, followed by rearrangement and gene loss, since ars gene clusters of Acidihalobacter prosperus branch near the base of the clade of Acidithiobacillus. NJ phylogenetic trees based on concatenated ArsAD protein sequences showed that the ars clusters of Acidithiobacillus and ars clusters of Alcaligenes faecalis, Pseudomonas hunanensis, and Sulfuriferula spp. were sister groups (Fig. 5), implying that Alcaligenes faecalis, Pseudomonas hunanensis, and Sulfuriferula spp. are likely donors of the arsADCR cluster.
FIG 4.

Neighbor-joining phylogenetic tree of concatenated ArsBRC protein sequences derived from Acidithiobacillus species strains and other representative species. Bootstrap values indicated at each node are based on a total of 1,000 bootstrap replicates. Branches representing ars clusters of Acidithiobacillus spp. are in blue.
FIG 5.

Neighbor-joining phylogenetic tree of concatenated ArsAD protein sequences derived from Acidithiobacillus species strains and other representative species. Bootstrap values indicated at each node are based on a total of 1,000 bootstrap replicates. Branches representing ars clusters of Acidithiobacillus spp. are marked in red.
Stand-alone arsM genes encoding As(III) S-adenosylmethyltransferase were found located distant from canonical ars gene clusters. NJ phylogenetic trees constructed on the basis of the ArsM protein sequences (see Fig. S14) revealed that the Acidithiobacillus strains formed three separate subgroups, of which subgroup I comprises ArsMs from A. thiooxidans, A. ferrooxidans, A. ferridurans, A. ferrivorans, and Acidithiobacillus sp. strain SH, grouping with ArsM from Thiomonas. Subgroup II comprises two deviant ArsMs from A. ferrooxidans and A. thiooxidans (GenBank accession no. WP_113525444.1 and WP_080707752.1, respectively), grouping with ArsMs from Melaminivora and Pseudomonas. Subgroup III comprises ArsMs from A. caldus, grouping with ArsMs from members of Rhodospirillales. Incongruences between the phylogenies of ArsMs and species imply that the subgroup I arsM genes may have been acquired via HGT from members of Thiomonas, subgroup II arsM genes from members of the genera Melaminivora and Pseudomonas, and subgroup III arsM genes from members of Rhodospirillales in early evolutionary history.
Case study of Cd-, Zn-, Co-, and Cu-related genes.
Gene clusters encoding Czc/CusABC homologs, which are involved in the detoxification of divalent cations (cadmium and zinc and cobalt) (38, 39) and monovalent cations (copper and silver) (13, 40, 41), were detected in all Acidithiobacillus genomes (see Fig. S15). In addition, genes encoding CzcD and genes encoding copper-translocating P-type ATPase (Cop) were detected in all Acidithiobacillus genomes, most of which located next to czcABC gene clusters. However, genes that encode multicopper oxidase were only detected in strains of A. thiooxidans and A. caldus. The above-mentioned genes represent the dominant genetic traits that contribute resistance to Cd, Zn, Co, and Cu in Acidithiobacillus genomes. Considerable numbers of additional copies of czc-like and cop-like genes were also present. The czc clusters were divided into two subgroups, czcABC and czcCAB (Fig. S15). The gene clusters in the organization of czcABC were observed in strains across all species of Acidithiobacillus, of which strains of A. ferrooxidans and A. ferridurans possess other copies of czcABC located on different sites, suggesting that czc clusters may be acquired more than once. There are also tRNA genes and genes encoding mobile element proteins flanking czcABC clusters (Fig. S15). Each A. caldus strain possesses a czcCAB cluster in addition to an existing czcABC cluster (Fig. S15). The czc gene clusters exhibited more than 90% identity within each subgroup and over 80% identity between two subgroups. Actually, the czc genes were detected in all strains of Acidithiobacillus. The genes in the upstream and downstream regions of a czcABC cluster were conserved among strains of all species of Acidithiobacillus (Fig. S15A), which indicated that this czcABC gene cluster was acquired before the divergence of genus Acidithiobacillus. Notably, cop genes encoding copper-translocating P-type ATPase located upstream of the czcABC clusters are conserved across all species of Acidithiobacillus (Fig. S15A).
(i) Origin and evolution of czc clusters. The G+C contents of the czcABC clusters are higher than those of the genomes in Acidithiobacillus spp. (62.2% to 64.2% versus 52.9% to 61.5%, respectively) except in A. caldus, in which the G+C contents of the clusters are lower than of the A. caldus genomes (59.4% to 59.5% versus 60.9% to 61.4%, respectively); the G+C contents of respective czcBAC clusters are higher than those of the genomes in A. caldus strains (64.9% to 66.0% versus 60.9% to 61.5%, respectively), presenting variations of G+C contents between clusters and the corresponding genomes (see Fig. S16). To gain insights into the origin of czc genes clusters in Acidithiobacillus, a NJ phylogenetic tree was constructed on the basis of concatenated CzcABC protein sequences. As shown in Fig. 6, the strains possessing czcABC and czcBAC clusters formed separate groups. Notably, the phylogeny reveals that the czcABC of Acidithiobacillus and czc clusters of Thiomonas spp. and Acidihalobacter spp. are sister groups, whereas czcBAC of Acidithiobacillus and czc clusters of another clade of Thiomonas spp. are sister groups. These results implied that the czcABC cluster may have been acquired via HGT from members of Acidihalobacter and Thiomonas, while czcBAC may have been acquired from members of Thiomonas.
FIG 6.

Neighbor-joining phylogenetic tree of concatenated CzcA, CzcB, and CzcC protein sequences derived from Acidithiobacillus species strains and other representative species. Bootstrap values indicated at each node are based on a total of 1,000 bootstrap replicates. Branches representing czcABC clusters are in green; czcBAC clusters formed in another group are in blue.
(ii) mco genes in Acidithiobacillus spp. Stand-alone mco genes that encode multicopper oxidase, which oxidizes Cu+ to the less toxic Cu2+ form, were found in all strains of A. thiooxidans and A. caldus in this study. An NJ phylogenetic tree based on the multicopper oxidase protein sequences revealed that the multicopper oxidases of Acidithiobacillus were adjacent to those of Acidihalobacter spp., implying that Acidithiobacillus and Acidihalobacter have a common multicopper oxidase ancestor (see Fig. S17). Synteny of the chromosomal regions flanking the mco genes showed that multicopper oxidase was probably lost in strains of A. ferrivorans, A. ferridurans, and A. ferrooxidans (see Fig. S18).
(iii) czc-like and cop-like genes in Acidithiobacillus. Our studies revealed that there are multiple czc-like genes and cop-like genes present in the 37 genomes, including 1 to 5 pairs of czcA-like genes, 1 to 5 pairs of czcB-like genes, 1 to 4 pairs of czcC-like genes, 1 to 4 pairs of czcD-like genes, and 1 to 3 pairs of cop-like genes (see Fig. S20, S21, S22, S23, and S24). An alignment of residues of CzcA and CzcA-like proteins revealed that the “DDE” motifs essential for CzcA function (42) are highly conserved (see Fig. S25). Alignments of residues of Cop and Cop-like proteins revealed that the metal binding domains (CASC …. CASC), translocation domains (CPCAMGLA), and phosphorylation domains (FDKTGTLT) (43) are conserved, indicating that these genes may be functional (see Fig. S26). A phylogenetic analysis revealed that czc-like and cop-like genes from the same species clustered together and that there were incongruences between the phylogenies of functional genes and species, which suggested that multiple czc-like and cop-like genes may have arisen from gene duplication and HGT (Fig. S20, S21, S22, S23, and S24).
Assessment of functionality of metal resistance genes.
We also assessed indirectly the functionality of above-mentioned metal-resistance-related genes by assessing the strength of natural selection acting on these genes, as well as the codon adaption indexes (CAIs) of these genes. The results showed that 83.3% of the above-mentioned metal resistance genes had a rate of nonsynonymous substitutions that was lower than the rate of synonymous substitutions (dN/dS < 1), suggesting purifying selection on these genes (Fig. 7 and Table S6). Only a merA gene from Acidithiobacillus thiooxidans strain A02 (dN/dS = 1.51), a cop gene from Acidithiobacillus ferrooxidans strain CCM 4253 (dN/dS = 1.39), and an arsH gene from Acidithiobacillus ferridurans JCM 18981 (dN/dS = 1.82) showed dN/dS ratios of >1, indicating that they might be under positive selection. The lowest dN/dS ratio was observed for arsB genes (average dN/dS = 0.06) and arsM genes (average dN/dS = 0.04), showing strong purifying selection. All these metal resistance genes with dN/dS ratios of <1 were supposed to be most essential for the growth of Acidithiobacillus spp. However, merA genes from A. ferrooxidans, arsR genes from A. thiooxidans, merC genes from A. caldus, merP genes from A. thiooxidans and A. ferridurans, merC genes from A. caldus, mco genes from A. thiooxidans, and some cop-like and czc-like genes showed dN/dS ratios of ≈1, indicating that the selection force had relaxed on them (Fig. 7 and Table S6). Highly expressed genes in bacteria often have a stronger codon bias than genes expressed at lower levels, due to translational selection (44–46). In this study, the expression levels of metal-resistance-related genes in the Acidithiobacillus spp. were predicted using the CAI as a numerical estimator (47, 48). Approximately 53.2% of the metal resistance genes were predicted to be highly expressed genes using CAI cutoff values of greater than 0.73 in A. thiooxidans, greater than 0.75 in A. caldus, and greater than 0.76 in A. ferrooxidans, A. ferrivorans, and A. ferridurans (Fig. 8 and see Table S7). The cutoff values were indicated with average CAI values of Tu genes in each species.
FIG 7.

Distributions and ranges of selection pressures on different metal resistance genes of Acidithiobacillus spp.
FIG 8.

Ranges of CAI values of different metal resistance genes of Acidithiobacillus spp. with Tu gene as a reference.
DISCUSSION
In the present work, we characterized the genomic features of Acidithiobacillus spp., focusing on the distribution, organization, complex evolutionary history, and functionality of metal resistance genes. With the rapid advances in genomic analysis technology, new measurements such as ANI are being developed to evaluate the genomic similarity between bacteria (33). In this study, we redetermined the phylogenetic status of 38 Acidithiobacillus spp. by using ANI and phylogenetic trees. Our results showed that strains DSM 14366, GGI-221, and IO-2C were misnamed. We therefore reclassified strain A. ferrooxidans IO-2C as A. ferridurans IO-2C, A. albertensis DSM 14366 as A. thiooxidans DSM 14366, and Acidithiobacillus sp. strain GGI-221 as A. ferrooxidans GGI-221. Low ANI values for Acidithiobacillus sp. strain NORP59 in comparison to those of other Acidithiobacillus strains and a distant phylogenetic relation indicated that strain NORP59 is not a member of genus Acidithiobacillus. Our study revealed the complex taxonomy of genus Acidithiobacillus and demonstrated the usefulness and accuracy of advanced bioinformatics measurements such as ANI in rectifying inaccurate identifications from previous studies.
There is generally a positive correlation between the abundance of mobile genetic elements and the frequency of HGT (49). We found that Acidithiobacillus spp. contain 0 to 6 prophages and/or prophage remnants, 143 to 334 insertion elements (IS), and 25 to 66 GIs per genome, suggesting that Acidithiobacillus spp. are rich in mobile genetic elements. The existence of transposable elements and prophages near the heavy metal resistance genes and gene clusters suggested that they may be involved in HGT of heavy metal resistance genes, which is consistent with previous studies illustrating that HGT was a major mechanism that confers upon bacteria crucial traits such as metal resistance for adaptation to toxic metal-containing habitats (15, 50–52). In addition, BacMet database annotation revealed that the accessory genome had a higher proportion (8.2% [7,846 genes]) of genes related to heavy metal resistance activity (Fig. 1C), and models of extrapolation revealed that the pangenome of 37 Acidithiobacillus spp. is “open,” which is in line with the results showing that metal-resistance-associated gene family gain events overcame loss events. We inferred that during evolutionary niche adaption, Acidithiobacillus spp. accessorized their genome armaments with genetic elements transferred from members outside their species or genus, with the help of MGEs. The “accessory genome” was therefore considered a massive resource that provided Acidithiobacillus spp. with unprecedented elasticity to improve their fitness while adapting to harsh conditions (53–55). Our results also showed that the transformation of heavy metals to less toxic forms followed by their efflux out of cells was an often-used metal resistance strategy of Acidithiobacillus spp., which is also a process contributing to the biogeochemical cycling of metals.
We implemented a combination of several approaches to discover putative HGT events, including deviant G+C content detection, phylogenetic analysis, mobile genetic element detection, and chromosomal synteny analysis, since it was difficult to identify HGT events via deviant G+C contents alone if HGT happened to occur between organisms with similar G+C contents (56). The different gene contents and organization of metal resistance genes suggested a complicated evolutionary history of these genes and also suggested different genetic requirements for effective cellular metal detoxification and regulation of metal resistance operons. We supposed Thiomonas spp. to be the donors of the arsM gene and czcABC and czcBAC gene clusters in Acidithiobacillus spp., which was reasonable since members of Thiomonas spp. are ubiquitous in heavy-metal-contaminated environments, especially in AMD sites, the same habitats as Acidithiobacillus species, and usually contained a vast flexible gene pool. For instance, the genome of Thiomonas arsenitoxydans contained a circular chromosome and a plasmid (pTHI) where more than 20 GIs (ThGEI-A to ThGEI-S) resided, which were associated with heavy metal resistance and conjugation (57–59). In addition, the median G+C content of Thiomonas species genomes was 64.0%, which was close to those of czcABC (64.2%) and czcBAC (64.9%) gene clusters in Acidithiobacillus spp. All these indicate that Thiomonas spp. are the most possible sources of multiple heavy metal resistance operons that were acquired by Acidithiobacillus spp. via cross-phylum HGT. Similar traits were also shown in case of Acidihalobacter spp., the putative cross-phylum donors of merACR, arsBRC, and the multicopper oxidase-encoding gene, which are a group of halophilic, mesophilic, heavy-metal-tolerant extreme acidophiles that share the same habitats with Acidithiobacillus spp., with an average genome G+C content (61.9%) similar to those of merACR (60.0%) and arsBRC (61.7%) gene clusters in Acidithiobacillus spp. (60, 61). This could also be applied to Acidiferrobacter spp. that inhabit AMD sites, which were predicted to be the cross-phylum donors of the merAPTR gene cluster and rusticyanin-encoding gene (62). Multiple czc-like genes and cop-like genes were present in the genomes of Acidithiobacillus spp., which may arise from gene duplication. Metal resistance proteins encoded by genes such as czc and cop are in high demand for Acidithiobacillus spp. for coping with harsh metal-rich environments. The presence of duplicate metal resistance genes may be beneficial by enhancing the regulatory elasticity of Acidithiobacillus spp., simply because extra amounts of protein or RNA products are generated (63).
The gene contexts of metal resistance genes in different Acidithiobacillus species were not conserved, which indicated that these genes may be acquired more than once via different HGT events. Synteny of the chromosomal regions flanking the mco genes showed that multicopper oxidases were possibly lost in strains of A. ferridurans, A. ferrivorans, and A. ferrooxidans during evolution, since mco genes in A. thiooxidans experienced relaxed selection pressure (dN/dS ≈ 1) and A. thiooxidans arose before the speciation of A. ferridurans, A. ferrivorans, and A. ferrooxidans in evolutionary history (see Fig. S7 in the supplemental material). We supposed that the function of this enzyme was replaced in A. ferridurans, A. ferrivorans, and A. ferrooxidans by rusticyanin, a periplasmic protein with an affinity for Cu+, forming part of an iron-oxidizing supercomplex and supposed to confer a higher level of copper resistance (64, 65). Rusticyanin was absent in species incapable of oxidizing iron, such as A. thiooxidans and A. caldus, and was acquired via HGT from Acidiferrobacter spp. to A. ferridurans, A. ferrivorans, and A. ferrooxidans, as revealed by a phylogenetic analysis (see Fig. S19). Most nonsynonymous substitutions are deleterious, especially for functionally important genes, since the functionality of important proteins might be substantially affected even if small changes happens in its sequences, resulting in a dN/dS if <1 for these genes considering that mutants of these genes are prone to be eliminated by purifying (negative) selection. On the contrary, when new mutations are beneficial, conferring adaptive advantages, these genes might be under diversifying (directional or positive) selection, reflected by a dN/dS of >1, namely, there are more nonsynonymous than synonymous nucleotide substitutions. Nonsynonymous nucleotide substitutions and synonymous substitutions will accumulate at similar rates (dN/dS ≈ 1) if genes become nonfunctional in the population, since there is no selection pressure acting to maintain the adaptive variant (neutral evolution), and these genes are more prone to loss during evolution (66). The results showed that 83.3% of metal resistance genes in Acidithiobacillus species had a lower rate of nonsynonymous substitution than of synonymous substitution (dN/dS < 1), which is in consistent with the assumption of predominant purifying selection on functionally important metal resistance genes. Besides, approximately 53.2% of the metal resistance genes were predicted to be highly expressed via the CAI (codon adaption index). There is a close correlation between codon usage bias and gene expressivity, which results from the selection for efficient and accurate translation of highly expressed genes (67, 68). The CAI was used to measure the synonymous codon usage bias for a given DNA sequence with an ever-improving algorithm by comparing the similarity between the synonymous codon usage of a gene and the synonymous codon frequency of a reference data set of more than 30 highly expressed genes derived from prokaryotes and lower eukaryotes, which can serve as a reliable indicator of codon usage bias as well as a numerical estimator of gene expressivity for genes of prokaryotes and lower eukaryotes (65, 69). Widespread low dN/dS ratios and relative high CAI values across metal-resistance-related genes emphasized their indispensable functions in supporting the growth of these extremophiles in the face of harsh environments. Altogether, our research revealed that Acidithiobacillus spp. recruited and consolidated additional novel resistant functionalities during adaption to challenging metal-rich environments via HGT, gene duplication, and purifying selection. However, further experimental confirmations of the functionalities and expression levels of metal-resistance-related genes in Acidithiobacillus spp. are still necessary for an advanced understanding of these genes.
Concluding remarks.
We performed a comparative genomic analysis of 37 strains within the genus Acidithiobacillus to explore the distribution, organization, functionality, and complex evolutionary history of metal resistance genes in Acidithiobacillus spp. The results indicated that the evolutionary history of toxic metal resistance genes in Acidithiobacillus spp. involved a combination of gene gain and loss, HGT, gene duplication, and gene family expansion. Gene clusters involved in toxic metal resistance, such as mer, ars, and czc-cus, were found to be acquired by early horizontal gene transfer (HGT) events from sources related to Burkholderiales, Rhodospirillales, Acidihalobacter, Thiobacillus, Acidiferrobacter, Alcaligenes, Pseudomonas, Sulfuriferula, Melaminivora, and Thiomonas, many of which share habitats with Acidithiobacillus spp., whereas the multicopper oxidase genes of iron-oxidizing A. ferridurans, A. ferrivorans, and A. ferrooxidans involved in copper detoxification were lost and replaced by rusticyanin genes during evolution. An assessment of the functionality of metal resistance genes showed that 83.3% of metal resistance genes in Acidithiobacillus spp. were under purifying selection (dN/dS < 1) and 53.2% of them were highly expressed. The insights gained in this study have greatly improved our understanding of the metal resistance strategies of Acidithiobacillus spp.
MATERIALS AND METHODS
A total of 38 sequenced strains belonging to genus Acidithiobacillus were collected from the NCBI database, containing one to three sequenced standard strains in each species and four unidentified strains. The general features of the bacterial genomes used in this study are summarized in Table 2.
Average nucleotide identity.
Comparisons of average nucleotide identities (ANIs) based on a BLAST algorithm (70) and tetranucleotide frequency correlation coefficient (Tetra) (71) were conducted using the web server JSpeciesWS (33, 72) with default parameters.
Phylogenic analyses.
To associate the distribution of metal resistance genes in Acidithiobacillus spp. with their phylogenetic affiliations, we constructed the phylogenetic tree of the Acidithiobacillus spp. with closely related species on the basis of 16S rRNA gene sequences using the neighbor-joining method in MEGA v7.0 (73) and with rRNA genes predicted using the RNAmmer v1.2 (74). Since no 16S rRNA gene sequence could be retrieved from the genome of Acidithiobacillus sp. strain NORP59, we also constructed a phylogenetic tree of the 38 Acidithiobacillus species genomes with their phylogenetic affiliations on the basis of whole-genome sequences with CVTree3 (75). A phylogenetic tree of the 37 Acidithiobacillus species genomes on the basis of the concatenation of the 110 single-copy core genes in a genome was constructed with the maximum likelihood (ML) method using MEGA v7.0 (73) with Jones-Taylor-Thornton (JTT) model (76) and 1,000 bootstraps, rooted by Escherichia coli and Salmonella enterica. The ambiguous areas of alignment were removed by Gblocks 0.91 b (77) before constructing the tree. A neighbor-joining tree and UPGMA tree were also constructed with MEGA v7.0 (73), both under a p-distance model with 1,000 bootstraps of the above-mentioned core genes. A chronogram for the NJ phylogenetic tree of the 37 Acidithiobacillus species with branch lengths reflecting divergence times was inferred using the divergence time between Escherichia coli and Salmonella enterica (140 MYA [millions of years ago]) (78) as calibration points with Timetree Wizard, a built-in program of MEGA v7.0 (73) with the JTT matrix-based model (76) and the real-time method (79).
Genes for toxic metal resistance.
Genes involved in heavy metal resistance in Acidithiobacillus genomes were identified by performing BLASTP searches against the BacMet database (80), which contains genes with experimentally confirmed metal resistance functions. Genes meeting the following cutoffs were considered reliable hits: E value, <1e−50; sequence similarity, >35%. The evolutionary history of genes for toxic metal resistance was inferred using MEGA v7.0 (73) with the neighbor-joining method and 1,000 bootstraps.
Pangenome analysis and model extrapolations of Acidithiobacillus.
The Bacterial Pan Genome Analysis (BPGA) pipeline (32) was used to identify orthologous groups among Acidithiobacillus testing genomes and to extrapolate the pangenome models of Acidithiobacillus spp. by applying default parameters. The set of genes shared among all testing strains was defined as their core genome, the set of genes shared with more than two but not all testing strains was defined as their accessory genome, the set of genes not shared with other strains in each testing strain was defined as the unique genes, and the set of nonhomologous genes with all testing genomes was defined as the pangenome. The details of all testing strains used are listed in Table 2. Genes involved in heavy metal resistance in the pangenome, core genome, and accessory genome and unique genes were identified by performing BLASTP searches against the BacMet database (80) with cutoffs of an E value of <1e−50 and a sequence similarity of >35%.
In this study, the size of the Acidithiobacillus pangenome was extrapolated by implementing an power law regression function, Ps = κnγ, using a built-in program of the BPGA pipeline (32), in which Ps represents the total number of nonorthologous gene families within its pangenome, n represents the number of tested strains, and both κ and γ are free parameters (35). An exponent γ of <0 suggests the pangenome is “closed,” where the size of the pangenome reaches a constant value as extra genomes are added. Conversely, the species is predicted to harbor an open pangenome for γ values between 0 and 1. In addition, the size of the core genome was extrapolated by fitting into an exponential decay function, Fc = κcexp(-n/τc) + Ω, with a built-in program of the BPGA pipeline (32), where Fc is the number of core gene families, and κc, τc, and Ω are free parameters (81).
Estimation of gene family gains and losses.
BadiRate (82) was used with the “BDI-FR-CWP” method to estimate the number of metal-resistance-related gene family gains and losses during the evolution of Acidithiobacillus. The analysis was conducted only on 5 representative Acidithiobacillus strains, including A. thiooxidans CLST, A. ferrooxidans ATCC 53993, A. ferrivorans PQ33, A. ferridurans JCM 18981, and A. caldus SM-1, because the estimation required complete sets of testing gene families, which are only available in species with considerable phylogenetic distance and have high-quality (complete or nearly complete) genome databases. The orthologous groups of metal resistance genes based on reciprocal best hits within each Acidithiobacillus species was inferred using the CD-HIT software (global sequence identity cutoff was 0.9 and bandwidth of alignment cutoff was 20 amino acids [aa]) (83). These data were used to construct a gene family size file (a matrix [column, species; row, number of orthologous groups]). The topology of the core-gene tree was used as the reference tree. The number of gene families related to metal resistance in each internal node was inferred using those numbers in branch species and the phylogenetic branch lengths. Gene family gains and losses in each phylogenetic branch were then estimated using the Wagner parsimony algorithm under the BDI stochastic model and free rates (FR) model, assuming that each branch has its own turnover rates.
Prediction of mobile genetic elements.
The ISFinder platform (84) was used to predict and classify insertion sequences (IS) and transposases distributed over Acidithiobacillus genomes, with default criteria. The IslandViewer 4 platform (85), which integrates two prediction methods that make use of sequence composition, i.e., IslandPath-DIMOB (86) and SIGI-HMM (87), and a comparative genomic islands prediction method IslandPick (88), were used to identify putative genomic islands (GIs). PHASTER (Phage Search Tool Enhanced Release) (89) was used for the identification and annotation of prophage sequences within Acidithiobacillus genomes. In addition, tRNA genes were predicted using the tRNAscan-SE v2.0 (90). The genes involved in heavy metal resistance were visualized, along with the tRNAs, insertion sequences, prophage sequences, and putative genomic islands, as well as the core genes, accessory genes, and unique genes, using BRIG-0.95 (91).
Selective pressure analysis and expressivity prediction.
The strength of natural selection was assessed by estimating the relative rates of nonsynonymous (dN) to synonymous (dS) nucleotide substitutions of orthologous metal resistance genes by using the HyPhy package (92) with the adaptive branch-site random effects likelihood (aBSREL) method (93) via the datamonkey web server (94). A gene in the node or tip of a given tree was considered under positive (diversifying) selection (dN/dS > 1) or negative (purifying) selection (dN/dS < 1) if the P value was <0.05 using the likelihood ratio test after correcting for multiple testing. The CAI (codon adaption index) was used as a numerical estimator of gene expression level (47, 48), a method previously described by Wu et al. (95). CAIcal (96) was used to calculate the CAI values of the above-mentioned metal resistance genes. CAI values range from 0 to 1.0, with a higher CAI value indicating a higher expression level. Putative highly expressed (PHX) genes related to toxic metal resistance in the Acidithiobacillus spp. were inferred, with Tu genes as the references, which encode elongation factors that are supposed to be highly expressed across most organisms (97).
Supplementary Material
ACKNOWLEDGMENTS
We thank the NCBI for providing the genome sequences of Acidithiobacillus species strains.
This work was funded by the National Natural Science Foundation of China (NSFC; grants 31570113, 41807332, 41877345, 91851206 [Major Research Plan of "Mechanism Underlying Elemental Cycling on the Earth by Microorganisms in Hydrosphere" launched by NSFC], and 41573072) and open funding of the State Key Laboratory of Comprehensive Utilization of Low-Grade Refractory Gold Ores (Zijin Mining Group Co., Ltd.; no. 738010212).
We also thank the Hunan International Scientific and Technological Cooperation Base of Environmental Microbiology and Application, China. We declare no competing interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02153-18.
REFERENCES
- 1.Chen LX, Hu M, Huang LN, Hua ZS, Kuang JL, Li SJ, Shu WS. 2015. Comparative metagenomic and metatranscriptomic analyses of microbial communities in acid mine drainage. ISME J 9:1579. doi: 10.1038/ismej.2014.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marrero J, Coto O, Goldmann S, Graupner T, Schippers A. 2015. Recovery of nickel and cobalt from laterite tailings by reductive dissolution under aerobic conditions using Acidithiobacillus species. Environ Sci Technol 49:6674–6682. doi: 10.1021/acs.est.5b00944. [DOI] [PubMed] [Google Scholar]
- 3.Xia L, Liu X, Zeng J, Yin C, Gao J, Liu J, Qiu G. 2008. Mechanism of enhanced bioleaching efficiency of Acidithiobacillus ferrooxidans after adaptation with chalcopyrite. Hydrometallurgy 92:95–101. doi: 10.1016/j.hydromet.2008.01.002. [DOI] [Google Scholar]
- 4.Valdés J, Pedroso I, Quatrini R, Dodson RJ, Tettelin H, Blake R, Eisen JA, Holmes DS. 2008. Acidithiobacillus ferrooxidans metabolism: from genome sequence to industrial applications. BMC Genomics 9:597. doi: 10.1186/1471-2164-9-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao Y, Liu X, Fang J, Liang Y, Zhang X, Meng D, Yin H. 2017. Responses of zinc recovery to temperature and mineral composition during sphalerite bioleaching process. AMB Express 7:190. doi: 10.1186/s13568-017-0491-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farasin J, Andres J, Casiot C, Barbe V, Faerber J, Halter D, Heintz D, Koechler S, Lièvremont D, Lugan R, Marchal M, Plewniak F, Seby Y, Bertin PN, Arsène-Ploetze F. 2015. Thiomonas sp. CB2 is able to degrade urea and promote toxic metal precipitation in acid mine drainage waters supplemented with urea. Front Microbiol 6:993. doi: 10.3389/fmicb.2015.00993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lièvremont D, Bertin PN, Lett MC. 2009. Arsenic in contaminated waters: biogeochemical cycle, microbial metabolism and biotreatment processes. Biochimie 91:1229–1237. doi: 10.1016/j.biochi.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 8.Oremland RS, Stolz JF. 2005. Arsenic, microbes and contaminated aquifers. Trends Microbiol 13:45–49. doi: 10.1016/j.tim.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Cheng H, Hu Y, Luo J, Xu B, Zhao J. 2009. Geochemical processes controlling fate and transport of arsenic in acid mine drainage (AMD) and natural systems. J Hazard Mater 165:13–26. doi: 10.1016/j.jhazmat.2008.10.070. [DOI] [PubMed] [Google Scholar]
- 10.Sugio T, Komoda T, Okazaki Y, Takeda Y, Nakamura S, Takeuchi F. 2010. Volatilization of metal mercury from organomercurials by highly mercury-resistant Acidithiobacillus ferrooxidans MON-1. Biosci Biotechnol Biochem 74:1007–1012. doi: 10.1271/bbb.90888. [DOI] [PubMed] [Google Scholar]
- 11.Dopson M, Bakeraustin C, Koppineedi PR, Bond PL. 2003. Growth in sulfidic mineral environments: metal resistance mechanisms in acidophilic micro-organisms. Microbiology 149:1959–1970. doi: 10.1099/mic.0.26296-0. [DOI] [PubMed] [Google Scholar]
- 12.Leng F, Li K, Zhang X, Li Y, Zhu Y, Lu J, Li H. 2009. Comparative study of inorganic arsenic resistance of several strains of Acidithiobacillus thiooxidans and Acidithiobacillus ferrooxidans. Hydrometallurgy 98:235–240. doi: 10.1016/j.hydromet.2009.05.004. [DOI] [Google Scholar]
- 13.Navarro CA, Diego VB, Jerez CA. 2013. Heavy metal resistance strategies of acidophilic bacteria and their acquisition: importance for biomining and bioremediation. Biol Res 46:363–371. doi: 10.4067/S0716-97602013000400008. [DOI] [PubMed] [Google Scholar]
- 14.Zhang X, She S, Dong W, Niu J, Xiao Y, Liang Y, Liu X, Zhang X, Fan F, Yin H. 2016. Comparative genomics unravels metabolic differences at the species and/or strain level and extremely acidic environmental adaptation of ten bacteria belonging to the genus Acidithiobacillus. Syst Appl Microbiol 39:493–502. doi: 10.1016/j.syapm.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 15.Hemme CL, Green SJ, Rishishwar L, Prakesh O, Pattenato A, Chakraborty R, Deutschbauer AM, Van Nostrand JD, Wu L, He Z, Jordan IK, Hazen TC, Arkin AP, Kostka JE, Zhou J. 2016. Lateral gene transfer in a heavy metal-contaminated-groundwater microbial community. mBio 7:e02234-15. doi: 10.1128/mBio.02234-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Liu X, Liang Y, Guo X, Xiao Y, Ma L, Miao B, Liu H, Peng D, Huang W, Zhang Y, Yin H. 2017. Adaptive evolution of extreme acidophile Sulfobacillus thermosulfidooxidans potentially driven by horizontal gene transfer and gene loss. Appl Environ Microbiol 83:e03098-16. doi: 10.1128/AEM.03098-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Popa O, Hazkani-Covo E, Landan G, Martin W, Dagan T. 2011. Directed networks reveal genomic barriers and DNA repair bypasses to lateral gene transfer among prokaryotes. Genome Res 21:599–609. doi: 10.1101/gr.115592.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Polz MF, Alm EJ, Hanage WP. 2013. Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends Genet 29:170–175. doi: 10.1016/j.tig.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sullivan JT, Ronson CW. 1998. Evolution of rhizobia by acquisition of a 500-kb symbiosis island that integrates into a phe-tRNA gene. Proc Natl Acad Sci U S A 95:5145–5149. doi: 10.1073/pnas.95.9.5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ochman H, Lawrence JG, Groisman EA. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 21.Xie J-B, Du Z, Bai L, Tian C, Zhang Y, Xie J-Y, Wang T, Liu X, Chen X, Cheng Q, Chen S, Li J. 2014. Comparative genomic analysis of N2-fixing and non-N2-fixing Paenibacillus spp.: organization, evolution and expression of the nitrogen fixation genes. PLoS Genet 10:e1004231. doi: 10.1371/journal.pgen.1004231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolhuis H, Severin I, Confurius-Guns V, Wollenzien UI, Stal LJ. 2010. Horizontal transfer of the nitrogen fixation gene cluster in the cyanobacterium Microcoleus chthonoplastes. ISME J 4:121–130. doi: 10.1038/ismej.2009.99. [DOI] [PubMed] [Google Scholar]
- 23.Khaldi N, Collemare J, Lebrun MH, Wolfe KH. 2008. Evidence for horizontal transfer of a secondary metabolite gene cluster between fungi. Genome Biol 9:R18. doi: 10.1186/gb-2008-9-1-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Niu J, Liang Y, Liu X, Yin H. 2016. Metagenome-scale analysis yields insights into the structure and function of microbial communities in a copper bioleaching heap. BMC Genet 17:21. doi: 10.1186/s12863-016-0330-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dopson M, Holmes DS. 2014. Metal resistance in acidophilic microorganisms and its significance for biotechnologies. Appl Microbiol Biotechnol 98:8133–8144. doi: 10.1007/s00253-014-5982-2. [DOI] [PubMed] [Google Scholar]
- 26.Nies DH. 2003. Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol Rev 27:313–339. doi: 10.1016/S0168-6445(03)00048-2. [DOI] [PubMed] [Google Scholar]
- 27.Hemme CL, Deng Y, Gentry TJ, Fields MW, Wu L, Barua S, Barry K, Tringe SG, Watson DB, He Z, Hazen TC, Tiedje JM, Rubin EM, Zhou J. 2010. Metagenomic insights into evolution of a heavy metal-contaminated groundwater microbial community. ISME J 4:660–672. doi: 10.1038/ismej.2009.154. [DOI] [PubMed] [Google Scholar]
- 28.Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA. 2015. Evidence for lateral gene transfer between archaea and bacteria from genome sequence of Thermotoga maritima. Nature 399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- 29.Dagan T, Artzy-Randrup Y, Martin W. 2008. Modular networks and cumulative impact of lateral transfer in prokaryote genome evolution. Proc Natl Acad Sci U S A 105:10039–10044. doi: 10.1073/pnas.0800679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brochier-Armanet C, Forterre P. 2007. Widespread distribution of archaeal reverse gyrase in thermophilic bacteria suggests a complex history of vertical inheritance and lateral gene transfers. Archaea-an International Microbiological J 2:83–93. doi: 10.1155/2006/582916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rice SA, Lampson BC. 1995. Phylogenetic comparison of retron elements among the myxobacteria: evidence for vertical inheritance. J Bacteriol 177:37–45. doi: 10.1128/jb.177.1.37-45.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaudhari NM, Gupta VK, Dutta C. 2016. BPGA- an ultra-fast pan-genome analysis pipeline. Sci Rep 6:24373. doi: 10.1038/srep24373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richter M, Rosselló-Móra R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vernikos G, Medini D, Riley DR, Tettelin H. 2015. Ten years of pan-genome analyses. Curr Opin Microbiol 23:148–154. doi: 10.1016/j.mib.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 35.Tettelin H, Riley D, Cattuto C, Medini D. 2008. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol 11:472–477. doi: 10.1016/j.mib.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Sievert SM. 2014. Pan-genome analyses identify lineage- and niche-specific markers of evolution and adaptation in Epsilonproteobacteria. Front Microbiol 5:110. doi: 10.3389/fmicb.2014.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ullrich SR, González C, Poehlein A, Tischler JS, Daniel R, Schlömann M, Holmes DS, Mühling M. 2016. Gene loss and horizontal gene transfer contributed to the genome evolution of the extreme acidophile “Ferrovum”. Front Microbiol 7:797. doi: 10.3389/fmicb.2016.00797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chakraborty J, Das S. 2014. Characterization and cadmium-resistant gene expression of biofilm-forming marine bacterium Pseudomonas aeruginosa JP-11. Environ Sci Pollut Res 21:14188–14201. doi: 10.1007/s11356-014-3308-7. [DOI] [PubMed] [Google Scholar]
- 39.Nies DH. 1995. The cobalt, zinc, and cadmium efflux system CzcABC from Alcaligenes eutrophus functions as a cation-proton antiporter in Escherichia coli. J Bacteriol 177:2707–2712. doi: 10.1128/jb.177.10.2707-2712.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colombi E, Straub C, Künzel S, Templeton MD, Mccann HC, Rainey PB. 2016. Evolution of copper resistance in the kiwifruit pathogen Pseudomonas syringae pv. actinidiae through acquisition of integrative conjugative elements and plasmids. Environ Microbiol 19:819–832. doi: 10.1111/1462-2920.13662. [DOI] [PubMed] [Google Scholar]
- 41.Masami N, Masao G, Katsumi A, Tadaaki H. 2004. Nucleotide sequence and organization of copper resistance genes from Pseudomonas syringae pv. actinidiae. Eur J Plant Pathol 110:223–226. doi: 10.1023/B:EJPP.0000015360.88352.a9. [DOI] [Google Scholar]
- 42.Goldberg M, Pribyl T, Juhnke S, Nies DH. 1999. Energetics and topology of CzcA, a cation/proton antiporter of the resistance-nodulation-cell division protein family. J Biol Chem 274:26065–26070. doi: 10.1074/jbc.274.37.26065. [DOI] [PubMed] [Google Scholar]
- 43.Orell A, Navarro CA, Arancibia R, Mobarec JC, Jerez CA. 2010. Life in blue: copper resistance mechanisms of bacteria and Archaea used in industrial biomining of minerals. Biotechnol Adv 28:839. doi: 10.1016/j.biotechadv.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 44.Zhao F, Yu C, Liu Y. 2017. Codon usage regulates protein structure and function by affecting translation elongation speed in Drosophila cells. Nucleic Acids Res 45:8484–8492. doi: 10.1093/nar/gkx501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brandis G, Hughes D. 2016. The selective advantage of synonymous codon usage bias in Salmonella. PLoS Genet 12:e1005926. doi: 10.1371/journal.pgen.1005926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu CH, Dang Y, Zhou Z, Wu C, Zhao F, Sachs MS, Liu Y. 2015. Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol Cell 59:744–754. doi: 10.1016/j.molcel.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carbone A, Zinovyev A, Képès F. 2003. Codon adaptation index as a measure of dominating codon bias. Bioinformatics 19:2005–2015. doi: 10.1093/bioinformatics/btg272. [DOI] [PubMed] [Google Scholar]
- 48.Sharp PM, Li WH. 1987. The codon adaptation index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Springael D, Top EM. 2004. Horizontal gene transfer and microbial adaptation to xenobiotics: new types of mobile genetic elements and lessons from ecological studies. Trends Microbiol 12:53–58. doi: 10.1016/j.tim.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 50.Li LG, Xia Y, Zhang T. 2017. Co-occurrence of antibiotic and metal resistance genes revealed in complete genome collection. ISME J 11:651–662. doi: 10.1038/ismej.2016.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Villegas-Torres MF, Bedoya-Reina OC, Salazar C, Vives-Florez MJ, Dussan J. 2011. Horizontal gene transfer among microorganisms isolated from arsenic polluted soil. Int Biodeterior Biodegradation 65:147–152. doi: 10.1016/j.ibiod.2010.10.007. [DOI] [Google Scholar]
- 52.Cai L, Liu G, Rensing C, Wang G. 2009. Genes involved in arsenic transformation and resistance associated with different levels of arsenic-contaminated soils. BMC Microbiol 9:4. doi: 10.1186/1471-2180-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sugawara M, Epstein B, Badgley BD, Unno T, Xu L, Reese J, Gyaneshwar P, Denny R, Mudge J, Bharti AK, Farmer AD, May GD, Woodward JE, Médigue C, Vallenet D, Lajus A, Rouy Z, Martinez-Vaz B, Tiffin P, Young ND, Sadowsky MJ. 2013. Comparative genomics of the core and accessory genomes of 48 Sinorhizobium strains comprising five genospecies. Genome Biol 14:R17. doi: 10.1186/gb-2013-14-2-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Croll D, McDonald BA. 2012. The accessory genome as a cradle for adaptive evolution in pathogens. PLoS Pathog 8:e1002608. doi: 10.1371/journal.ppat.1002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kung VL, Ozer EA, Hauser AR. 2010. The accessory genome of Pseudomonas aeruginosa. Microbiol Mol Biol Rev 74:621–641. doi: 10.1128/MMBR.00027-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hirsch AM, Mckhann HI, Reddy A, Liao J, Fang Y, Marshall CR. 1995. Assessing horizontal transfer of nifHDK genes in eubacteria: nucleotide sequence of nifK from Frankia strain HFPCcI3. Mol Biol Evol 12:16–27. doi: 10.1093/oxfordjournals.molbev.a040184. [DOI] [PubMed] [Google Scholar]
- 57.Slyemi D, Moinier D, Brochier-Armanet C, Bonnefoy V, Johnson DB. 2011. Characteristics of a phylogenetically ambiguous, arsenic-oxidizing Thiomonas sp., Thiomonas arsenitoxydans strain 3AsT sp. nov. Arch Microbiol 193:439–449. doi: 10.1007/s00203-011-0684-y. [DOI] [PubMed] [Google Scholar]
- 58.Arsène-Ploetze F, Koechler S, Marchal M, Coppée JY, Chandler M, Bonnefoy V, Brochier-Armanet C, Barakat M, Barbe V, Battaglia-Brunet F, Bruneel O, Bryan CG, Cleiss-Arnold J, Cruveiller S, Erhardt M, Heinrich-Salmeron A, Hommais F, Joulian C, Krin E, Lieutaud A, Lièvremont D, Michel C, Muller D, Ortet P, Proux C, Siguier P, Roche D, Rouy Z, Salvignol G, Slyemi D, Talla E, Weiss S, Weissenbach J, Médigue C, Bertin PN. 2010. Structure, function, and evolution of the Thiomonas spp. genome. PLoS Genet 6:e1000859. doi: 10.1371/journal.pgen.1000859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bryan CG, Marchal M, Battaglia-Brunet F, Kugler V, Lemaitre-Guillier C, Lièvremont D, Bertin PN, Arsène-Ploetze F. 2009. Carbon and arsenic metabolism in Thiomonas strains: differences revealed diverse adaptation processes. BMC Microbiol 9:127. doi: 10.1186/1471-2180-9-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khaleque HN, Corbett MK, Ramsay JP, Kaksonen AH, Boxall NJ, Watkin ELJ. 2017. Complete genome sequence of Acidihalobacter prosperus strain F5, an extremely acidophilic, iron- and sulfur-oxidizing halophile with potential industrial applicability in saline water bioleaching of chalcopyrite. J Biotechnol 262:56–59. doi: 10.1016/j.jbiotec.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 61.Dopson M, Holmes DS, Lazcano M, McCredden TJ, Bryan CG, Mulroney KT, Steuart R, Jackaman C, Watkin ELJ. 2016. Multiple osmotic stress responses in Acidihalobacter prosperus result in tolerance to chloride ions. Front Microbiol 7:2132. doi: 10.3389/fmicb.2016.02132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hallberg KB, Hedrich S, Johnson DB. 2011. Acidiferrobacter thiooxydans, gen. nov. sp. nov.; an acidophilic, thermo-tolerant, facultatively anaerobic iron- and sulfur-oxidizer of the family Ectothiorhodospiraceae. Extremophiles 15:271–279. doi: 10.1007/s00792-011-0359-2. [DOI] [PubMed] [Google Scholar]
- 63.Zhang J. 2003. Evolution by gene duplication: an update. Trends Ecol Evol 18:292–298. doi: 10.1016/S0169-5347(03)00033-8. [DOI] [Google Scholar]
- 64.Navarro CA, von Bernath D, Martínez-Bussenius C, Castillo RA, Jerez CA. 2016. Cytoplasmic CopZ-like, periplasmic rusticyanin and AcoP proteins are possible new copper resistance determinants in Acidithiobacillus ferrooxidans ATCC 23270. Appl Environ Microbiol 82:1015–1022. doi: 10.1128/AEM.02810-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walter RL, Ealick SE, Friedman AM, Blake Ii RC, Proctor P, Shoham M. 1996. Multiple wavelength anomalous diffraction (MAD) crystal structure of rusticyanin: a highly oxidizing cupredoxin with extreme acid stability. J Mol Biol 263:730. doi: 10.1006/jmbi.1996.0612. [DOI] [PubMed] [Google Scholar]
- 66.Nielsen R. 2005. Molecular signatures of natural selection. Annu Rev Genet 39:197–218. doi: 10.1146/annurev.genet.39.073003.112420. [DOI] [PubMed] [Google Scholar]
- 67.Zhou M, Guo J, Cha J, Chae M, Chen S, Barral JM, Sachs MS, Liu Y. 2013. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 495:111–115. doi: 10.1038/nature11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hiraoka Y, Kawamata K, Haraguchi T, Chikashige Y. 2010. Codon usage bias is correlated with gene expression levels in the fission yeast Schizosaccharomyces pombe. Genes Cells 14:499–509. doi: 10.1111/j.1365-2443.2009.01284.x. [DOI] [PubMed] [Google Scholar]
- 69.Kalhoefer D, Thole S, Voget S, Lehmann R, Liesegang H, Wollher A, Daniel R, Simon M, Brinkhoff T. 2011. Comparative genome analysis and genome-guided physiological analysis of Roseobacter litoralis. BMC Genomics 12:324–324. doi: 10.1186/1471-2164-12-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. 2007. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 71.Teeling H, Meyerdierks A, Bauer M, Amann R, Glöckner FO. 2004. Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ Microbiol 6:938–947. doi: 10.1111/j.1462-2920.2004.00624.x. [DOI] [PubMed] [Google Scholar]
- 72.Richter M, Rosselló-Móra R, Oliver Glöckner F, Peplies J. 2015. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32:929. doi: 10.1093/bioinformatics/btv681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zuo G, Hao B. 2015. CVTree3 web server for whole-genome-based and alignment-free prokaryotic phylogeny and taxonomy. Genomics Proteomics Bioinformatics 13:321–331. doi: 10.1016/j.gpb.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jones DT, Taylor WR, Thornton JM. 1992. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 77.Castresana J. 2000. GBLOCLKS: selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 78.Bergthorsson U, Ochman H. 1998. Distribution of chromosome length variation in natural isolates of Escherichia coli. Mol Biol Evol 15:6. doi: 10.1093/oxfordjournals.molbev.a025847. [DOI] [PubMed] [Google Scholar]
- 79.Tamura K, Battistuzzi FU, Billing-Ross P, Murillo O, Filipski A, Kumar S. 2012. Estimating divergence times in large molecular phylogenies. Proc Natl Acad Sci U S A 109:19333–19338. doi: 10.1073/pnas.1213199109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pal C, Bengtsson-Palme J, Rensing C, Kristiansson E, Larsson DGJ. 2014. BacMet: antibacterial biocide and metal resistance genes database. Nucleic Acids Res 42:D737–D743. doi: 10.1093/nar/gkt1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit y Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, Khouri H, Radune D, Dimitrov G, Watkins K, O'Connor KJ, Smith S, Utterback TR, White O, Rubens CE, Grandi G, Madoff LC, Kasper DL, Telford JL, Wessels MR, Rappuoli R, Fraser CM. 2005. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A 102:13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Librado P, Vieira FG, Rozas J. 2012. BadiRate: estimating family turnover rates by likelihood-based methods. Bioinformatics 28:279–281. doi: 10.1093/bioinformatics/btr623. [DOI] [PubMed] [Google Scholar]
- 83.Li W, Godzik A. 2006. Cd-Hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 84.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. 2006. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bertelli C, Laird MR, Williams KP, Lau BY, Hoad G, Winsor GL, Brinkman FS. 2017. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res 45:W30–W35. doi: 10.1093/nar/gkx343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hsiao W, Wan I, Jones SJ, Brinkman FS. 2003. IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19:418–420. doi: 10.1093/bioinformatics/btg004. [DOI] [PubMed] [Google Scholar]
- 87.Waack S, Keller O, Asper R, Brodag T, Damm C, Fricke W, Surovcik K, Meinicke P, Merkl R. 2006. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7:142. doi: 10.1186/1471-2105-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Langille MG, Hsiao WW, Brinkman FS. 2008. Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinformatics 9:329. doi: 10.1186/1471-2105-9-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS. 2016. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44:W16–W21. doi: 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Alikhan NF, Petty NK, Zakour NLB, Beatson SA. 2011. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pond SL, Frost SD, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 93.Smith MD, Wertheim JO, Weaver S, Murrell B, Scheffler K, Kosakovsky Pond SL. 2015. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol Biol Evol 32:1342–1353. doi: 10.1093/molbev/msv022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Weaver S, Shank SD, Spielman SJ, Li M, Muse SV, Kosakovsky Pond SL. 2 January 2018. Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Molecular Biology & Evolution doi: 10.1093/molbev/msx335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu G, Culley DE, Zhang W. 2005. Predicted highly expressed genes in the genomes of Streptomyces coelicolor and Streptomyces avermitilis and the implications for their metabolism. Microbiology 151:2175–2187. doi: 10.1099/mic.0.27833-0. [DOI] [PubMed] [Google Scholar]
- 96.Puigbò P, Bravo IG, Garcia-Vallve S. 2008. CAIcal: a combined set of tools to assess codon usage adaptation. Biol Direct 3:38. doi: 10.1186/1745-6150-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Puigbo P, Romeu A, Garcia-Vallve S. 2008. HEG-DB: a database of predicted highly expressed genes in prokaryotic complete genomes under translational selection. Nucleic Acids Res 36:D524–D527. doi: 10.1093/nar/gkm831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Boyd ES, Barkay T. 2012. The mercury resistance operon: from an origin in a geothermal environment to an efficient detoxification machine. Front Microbiol 3:349. doi: 10.3389/fmicb.2012.00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yuan C, Lu X, Qin J, Rosen BP, Le XC. 2008. Volatile arsenic species released from Escherichia coli expressing the AsIII S-adenosylmethionine methyltransferase gene. Environ Sci Technol 42:3201–3206. doi: 10.1021/es702910g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Qin J, Rosen BP, Zhang Y, Wang G, Franke S, Rensing C. 2006. Arsenic detoxification and evolution of trimethylarsine gas by a microbial arsenite S-adenosylmethionine methyltransferase. Proc Natl Acad Sci U S A 103:2075–2080. doi: 10.1073/pnas.0506836103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chang JS, Yoon IH, Kim KW. 2018. Arsenic biotransformation potential of microbial arsH responses in the biogeochemical cycling of arsenic-contaminated groundwater. Chemosphere 191:729–737. doi: 10.1016/j.chemosphere.2017.10.044. [DOI] [PubMed] [Google Scholar]
- 102.Anton A, Große C, Reißmann J, Pribyl T, Nies DH. 1999. CzcD Is a heavy metal ion transporter involved in regulation of heavy metal resistance in Ralstonia sp. strain CH34. J Bacteriol 181:6876–6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Williams CL, Neu HM, Gilbreath JJ, Michel SLJ, Zurawski DV, Merrell DS. 2016. Copper resistance of the emerging pathogen Acinetobacter baumannii. Appl Environ Microbiol 82:6174–6188. doi: 10.1128/AEM.01813-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Grass G, Rensing C. 2001. CueO is a multi-copper oxidase that confers copper tolerance in Escherichia coli. Biochem Biophys Res Commun 286:902–908. doi: 10.1006/bbrc.2001.5474. [DOI] [PubMed] [Google Scholar]
- 105.Rensing C, Fan B, Sharma R, Mitra B, Rosen BP. 2000. CopA: an Escherichia coli Cu(I)-translocating P-type ATPase. Proc Natl Acad Sci U S A 97:652–656. doi: 10.1073/pnas.97.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.