

SUMMARY
Although macrophages are armed with potent antibacterial functions, Mycobacterium tuberculosis (Mtb) replicates inside these innate immune cells. Determinants of macrophage intrinsic bacterial control, and the Mtb strategies to overcome them, are poorly understood. To further study these processes, we used an affinity tag purification mass spectrometry (AP-MS) approach to identify 187 Mtb-human protein-protein interactions (PPIs) involving 34 secreted Mtb proteins. This interaction map revealed two factors involved in Mtb pathogenesis - the secreted Mtb protein, LpqN, and its binding partner, the human ubiquitin ligase CBL. We discovered that an lpqN Mtb mutant is attenuated in macrophages, but growth is restored when CBL is removed. Conversely, Cbl−/− macrophages are resistant to viral infection, indicating that CBL regulates cell-intrinsic polarization between antibacterial and antiviral immunity. Collectively, these findings illustrate the utility of this Mtb-human PPI map for developing a deeper understanding of the intricate interactions between Mtb and its host.
Graphical Abstract
eTOC
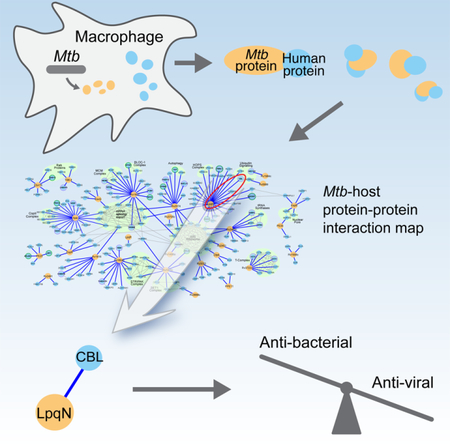
Penn et al. used an affinity tag purification mass spectrometry approach to generate an Mtb-human protein-protein interaction map, uncovering a connection between LpqN, a virulence factor in Mtb and CBL, a host ubiquitin ligase. CBL suppresses lpqN attenuation and acts as a switch for host anti-bacterial and anti-viral responses.
INTRODUCTION
Mycobacterium tuberculosis (Mtb) infection persists as a leading cause of death worldwide, with an estimated 2 billion people chronically infected and 1–2 million deaths annually (Zumla et al., 2015). The only vaccine, the live-attenuated Bacillus Calmette-Guerin strain developed nearly 100 years ago, offers very limited protection against Mtb (Mangtani et al., 2014). In addition, current treatment is cumbersome, requiring administration of multiple, potentially toxic antibiotics over a period of months (Gillespie et al., 2014). Thus, there remains a critical need to elucidate the mechanisms by which Mtb disrupts the host immune response in order to both optimize vaccine strategies as well as to explore therapies that promote sterilizing host immunity as an adjunct to antibiotics.
Mtb infection begins when airborne bacilli are inhaled and phagocytosed by alveolar macrophages (Torrelles and Schlesinger, 2017). This activates pattern-recognition receptors that bind bacterial constituents, leading to expression of proinflammatory cytokines such as interleukin 1 (IL-1) and tumor necrosis factor alpha (TNF-α) that are important for Mtb control (Mayer-Barber and Sher, 2015). Surprisingly, Mtb infection also activates secretion of Type-I interferons such as interferon beta (IFN-β) (Manzanillo et al., 2012), a hallmark cellular response to viral infection (McNab et al., 2015). IFN-β induction has been linked to Mtb’s ability to perforate the phagosome membrane through its Type VII/ESX-1 protein secretion system (Manzanillo et al., 2012; Novikov et al., 2011; Siméone et al., 2015; van der Wel et al., 2007), allowing communication between the bacterium and the host cytosol. This allows recognition of extracellular Mtb genomic DNA by cyclic GMP-AMP synthase (cGAS) (Collins et al., 2015; Wassermann et al., 2015; Watson et al., 2015; Wiens and Ernst, 2016) which activates interferon regulatory factor 3 (IRF3) to initiate IFN-β transcription. In vivo, IFN-β signaling counteracts anti-Mtb immunity (Manca et al., 2004; Manzanillo et al., 2012; Stanley et al., 2007), in-part by antagonizing the effects of interleukin-1 (IL-1) mediated resistance (Mayer-Barber et al., 2014). Moreover, elicitation of antiviral programs may serve to impede antibacterial responses of the host.
While cytokines such as IL-1, TNF-α, and interferon gamma (IFN-γ) are critical for activating macrophages to control Mtb growth (Mayer-Barber and Sher, 2015), the cell intrinsic mechanisms by which macrophages restrict Mtb are incompletely understood (Braverman and Stanley, 2017). For example, macrophages encode an array of activities capable of eliminating bacteria, including phagocytosis and subsequent delivery to toxic lysosomes (Alonso et al., 2007; Shiloh et al., 1999). Mtb has evolved the remarkable ability to replicate within this hostile environment as it is not only intrinsically resistant to low pH and oxidative damage (Darwin et al., 2003; Vandal et al., 2009), but can also inhibit fusion between phagosomes and lysosomes (Armstrong and Hart, 1971; Clemens, 1996; Rohde et al., 2007). Likewise, the mechanisms by which Mtb thwarts macrophage resistance mechanisms are unclear. Many bacterial pathogens inject secreted effector proteins into host cells, often to directly interact with host proteins and disrupt host cell function (Byndloss et al., 2017). The ability of Mtb to permeabilize its phagosome via ESX-1 and communicate with the host cell cytoplasm raises the possibility that it may similarly introduce secreted effectors to target host proteins. While elegant genetic studies have implicated dozens of secreted Mtb factors in virulence (Sassetti and Rubin, 2003; Zhang et al., 2013), whether these factors directly interact with host molecules to influence macrophage physiology is unknown.
Identifying the set of physical interactions between secreted Mtb and human proteins that occur in vivo will be crucial in understanding Mtb pathogenesis and may provide advanced approaches to combat infection. However, only a handful of Mtb-host protein-protein interactions (PPIs) have been characterized. For example, a previous yeast 2-hybrid screen targeting Mtb proteins identified an interaction between EsxH and the human ESCRT machinery (Mehra et al., 2013). Additional focused studies on individual Mtb factors have also identified interactions with the host proteins VPS33B and TLR2 (Bach et al., 2008; Pathak et al., 2007). However, the contribution of these interactions to Mtb virulence remains unclear either because of the known pleiotropic effects the bacterial factors have on the physiology of Mtb independent of the host (Siegrist et al., 2009), or because genetic disruption of the factors involved does not diminish Mtb virulence (Grundner et al., 2008).
Unbiased approaches for characterizing PPIs using mass spectrometry represent powerful ways to probe complex biological systems in an unbiased fashion (Beltrao et al., 2010; Collins et al., 2007; Krogan et al., 2006). Previously, using affinity tag purification combined with mass spectrometry (AP-MS), we have systematically identified host pathways required for virulence of a number of important human viruses (Shah et al., 2015), including human immunodeficiency virus (HIV) (Jäger et al., 2012), hepatitis C virus(HCV) (Ramage et al., 2015), and Kaposi’s sarcoma herpes virus (KSHV) (Davis et al., 2015). Likewise, by focusing on a set of Chlamydia trachomatis virulence factors secreted into host cells, we uncovered a unique pathway by which this difficult and persistent bacterial pathogen manipulates host cells (Elwell et al., 2017; Mirrashidi et al., 2015).
Here we report the results of an AP-MS study to identify the PPIs between Mtb secreted proteins and host factors. Using a combination of MS and bioinformatic analysis, we identified a set of high confidence physical interactions between Mtb and host proteins. Using this map, we have uncovered a previously undescribed secreted Mtb virulence factor essential for Mtb growth in vivo, LpqN, and have also identified the interacting host CBL ubiquitin ligase as a new restriction factor that regulates cell-intrinsic Mtb control. Surprisingly, while CBL-deficient macrophages are more permissive for Mtb growth, they are resistant to viral replication, indicating a role in modulating intrinsic control of antiviral versus antibacterial responses during Mtb infection. Collectively, these findings demonstrate the value of our Mtb-human PPI map as a resource to advance our understanding of the complex interaction between Mtb and its host.
RESULTS
Creation of an Mtb-host PPI network
To explore the mechanisms by which Mtb disrupts host immune function, we sought to identify physical interactions between secreted Mtb factors and host proteins using a systematic proteomic approach. We reasoned that since the Mtb-containing phagosome is permeabilized (Manzanillo et al., 2012; Novikov et al., 2011; Siméone et al., 2015; van der Wel et al., 2007), any protein secreted by the bacterium would have the potential opportunity to interact with host proteins in the cell. We began by analyzing highly controlled culture filtrates of Mtb, free of detectable cytoplasmic contamination. Using mass spectrometry (MS), we established a high-confidence set of bona-fide secreted proteins (Figure S1A; Table S1), and these data were further curated with additional published MS studies of Mtb protein localization (Målen et al., 2007). These conservative criteria, requiring direct biochemical evidence of secretion beyond the bacterial cell wall, were employed to minimize inclusion of non-secreted proteins, though it precludes analysis of secreted proteins present below the level of detection by MS. These analyses identified a high-confidence set of 105 secreted Mtb proteins, including 69 proteins with canonical Sec-transporter N-terminal signal sequences and 21 ESX-system substrates (Table S1).
We next expressed each of these proteins in human cells as C-terminal Strep-tag fusions and identified co-purifying host proteins by MS. As expected, expression in 293T cells of the Sec-dependent substrates containing their natural signal sequences led to co-localization with the endoplasmic reticulum (Figure S1B). Thus, we expressed the protein predicted to correspond to the signal peptidase-processed form released by Mtb during natural infection and determined that these factors primarily localized to the cytoplasm (Figure S1B). We initially sought to express the entire set of Mtb secreted proteins directly in macrophages but, consistent with reports from other groups (Zhang et al., 2009), found this to be an inefficient process that yielded insufficient protein for MS analysis (Figure S1C). To circumvent this issue, yet still allow capture of macrophage-specific factors, we developed a two-step co-purification strategy (Figure 1A). After initially expressing and isolating the bacterial factors from 293T cells on Strep-tactin resin, the immobilized proteins were then incubated in an in vitro binding reaction with lysate from differentiated human U937 macrophages before subsequent AP-MS. This method was robust, as we readily expressed and purified 99 of the 105 bacterial proteins (Figure S1D). We analyzed a subset of bacterial factors both with and without addition of U937 lysate and found a substantial number of additional interactions specific to the macrophage lysate (Table S2).
Figure 1. AP-MS experimental design and analysis.

(A) Flow-chart of the AP-MS proteomics pipeline used for this study.
(B) Distribution of MiST scores for all of the interactions in the Mtb-host PPI network, as well as for a randomized control network.
(C) Number of high-confidence host interactors (MiST score >0.7, x-axis) for each of the 34 bacterial proteins (y-axis) that had at least one host interaction. Genes encoding bacterial proteins likely essential for Mtb growth are denoted by red font (Griffin et al., 2011). See also Figure S1.
To identify the set of interacting human proteins, we performed AP-MS in triplicate for each bacterial protein with the addition of GFP-Strep as a control (see Table S3 for raw MS data). To identify specific interactions, we utilized the MiST bioinformatic algorithm, which scores each interaction for specificity, reproducibility, and abundance using principal component analysis (Verschueren et al., 2015). Since no ‘gold-standard’ set of known host-Mtb protein interactions existed with which to optimize a score threshold, we used a score threshold of 0.7 to define high-confidence interactions. Using MiST (Figure 1B), the initial interaction dataset was distilled to a high-confidence network between 34 bacterial proteins and 187 specifically-interacting human proteins (Figure 1C; Figure 3; Table S4). As a control, when the AP-MS data were randomized and then analyzed by MiST, only three host proteins exceeded the MiST 0.7 threshold (Figure 1B), providing further support that we defined a high-quality set of PPIs. To independently validate these interactions, we defined a high-priority set of 34 host interactors with annotated immune-related functions and expressed them in 293T cells as 3xFLAG-tagged proteins with their Strep-tagged bacterial partner. Reciprocal co-immunoprecipitation of the tagged human proteins with their putative bacterial interaction partner verified 25 of 34 (74%) of the interactions, underscoring the reliability of the AP-MS coupled with bioinformatic analysis (Figure S1E).
Figure 3. Mtb-host protein-protein interaction network.

A network representation of the 34 Mtb proteins (yellow circles) and 187 human proteins (light blue circles), with blue edges representing the interactions identified in this study. Human-human interactions (thin grey lines) were defined by the CORUM and STRING databases, with known protein complexes highlighted in green. Proteins differentially phosphorylated and/or ubiquitylated upon Mtb infection are indicated by concentric circles.
Features of the Mtb-Human PPI Map
Bioinformatic analyses of the interactome revealed several striking features of the Mtb-host interaction. First, we found that only 34% of the Mtb proteins we targeted provided high-scoring PPIs with host proteins, which is in contrast to our previous findings with viral proteomes – HIV (89%), HCV (100%), and KSHV (75%) (Davis et al., 2015; Jäger et al., 2012; Ramage et al., 2015). This suggests that, unlike viruses that are completely dependent on host functions for replication, many of the Mtb-secreted proteins are involved in bacterial cell-autonomous processes. While we had previously found C. trachomatis also had a high percentage of proteins with host interactions (66%), this could reflect the fact that the subset of virulence factors investigated was enriched for mediating contacts with host cells (Mirrashidi et al., 2015). Second, comparison of the host-pathogen interactomes of Mtb, HIV, KSHV and C. trachomatis revealed that while there was some overlap among the host proteins bound by the different pathogens, the majority of proteins in each of the datasets were pathogen-specific. In particular, 138 of 187 host proteins in the Mtb–host interaction map are Mtb-specific (Figure 2A, Table S5). However, comparison of the functional pathways represented by these interactions revealed several commonalities between the pathogens (Figure 2B, Table S6). In particular, both Mtb and C. trachomatis datasets were enriched for proteins involved in vesicular transport, consistent with their common intracellular lifestyles. Moreover, despite some commonalities between the datasets, the interactomes reflected distinct host pathways, suggesting that these pathogens have evolved unique strategies to establish a replicative niche.
Figure 2. Secreted Mtb proteins interact with a distinct, rapidly-evolving set of human proteins.

(A) Venn diagram representing the overlap of human proteins identified in four pathogen-host PPI maps. The significance of the overlap in the PPI networks between pairwise comparisons of Mtb with HIV, C. trachomatis, and KSHV is p=1×10−9, p=2.1×10−5 and p=0.008 respectively, using Fisher’s exact test. This data is also presented in Table S7.
(B) Host biological processes enriched in each PPI map for Mtb, C. trachomatis, HIV, and KSHV. Processes enriched >2-fold with p-value <0.05 after Benjamini-Hochberg multiple comparisons correction, are displayed.
(C) Evolutionary rates of Mtb-interacting proteins within the chimpanzee lineage using SniPRE (left) and within the human lineage using iHS analysis (right). The distributions of evolutionary scores across host proteins partitioned into two groups: Mtb-interacting proteins shown in blue and non-interacting proteins shown in white, with higher values indicating diversifying selection. Analysis of HIV, Chlamydia and KSHV interactomes are shown for comparison. Solid line denotes median, dashed lines denote upper/lower quartile. *p=0.02, **p<0.01.
Evolutionary Analysis of the Mtb-host interactome
Our data also offer an opportunity to empirically test evolutionary hypotheses regarding Mtb and its host. There are numerous examples of host antiviral restriction factors that are inhibited by direct binding of viral effector proteins. As a consequence, many of these restriction factors experience selective pressure to diversify rapidly (positive selection) and escape the effects of the viral protein - an example of the “Red Queen Hypothesis” of coevolution (Duggal and Emerman, 2012). We hypothesized that if secreted Mtb effectors physically interact with host proteins, then these host proteins might show similar evidence of positive selection. We began by using SnIPRE (Eilertson et al., 2012), which compares the relative rate of non-synonymous substitutions between human and chimpanzee orthologs and detects positive selection over a 5–7 million-year span, but identified no evidence of selective pressure over this timeframe (Figure 2C, left panel). However, examination over more recent evolution within the human lineage itself (10,000–30,000-year span), revealed a significant amount of recent diversification (Figure 2C, right panel). In the human population, positively selected alleles quickly sweep to high frequency before recombination separates these variants from the surrounding genome. As a result, loci that have experienced recent positive selection will exhibit longer than expected haplotypes in the human genome. The integrated Haplotype Score (iHS) measures haplotype block lengths to identify variant alleles with abnormally long haplotypes, which are likely undergoing positive selection (Szpiech and Hernandez, 2014; Voight et al., 2006). In contrast to our findings over longer evolutionary timeframes, iHS detected a shift in the distribution of evolutionary rates, with increased diversification for the set of Mtb-interacting host proteins (Figure 2C, right panel, Table S7). To control for detection bias in our MS methods, we analyzed the set of non-interacting proteins identified in our affinity purifications and found no such shift. We also analyzed the interactomes of other pathogens and found rapid diversification in HIV-interacting host proteins, but not in the interactomes of pathogens associated with lower mortality – KSHV and C. trachomatis. Thus, our findings are unlikely to be the result of a bias introduced by either MS detection or bioinformatic filtering. Rather, our data suggest that the set of Mtb-interacting host proteins are rapidly diversifying, potentially as a result of their interactions with Mtb proteins.
Topology of the Mtb-host interactome
Individual proteins often associate into larger protein complexes to carry out cellular processes (Alberts, 1998). To help interpret the Mtb-host PPI map in this light, we used previously published host PPI data to arrange the identified host proteins into complexes. To this end, we used data from the CORUM and STRING databases to overlay known human PPIs within the Mtb interactome (Figure 3). This analysis revealed a number of host complexes targeted by Mtb proteins, including a connection between bacterial Apa and five subunits of the COP II vesicular trafficking complex as well as an association with the uncharacterized Mtb protein, Rv3722c and seven components of the CCT chaperone complex. Furthermore, we uncovered a connection between Rv1804c and Rv1075c and the host STRIPAK signal transduction complex, which has been recently linked to innate immunity and autophagy targeting (Liu et al., 2016; Neisch et al., 2017). Interestingly, Rv1075c also interacts with components of the SET1 histone methyltranferase complex, COMPASS, which regulates transcriptional elongation by RNAPII (Herz et al., 2013; Krogan et al., 2003). This connection suggests that Mtb may regulate host transcriptional regulation by hijacking this complex using one of its secreted proteins.
Recently, our groups carried out a global analysis of changes in the host proteome with respect to post-translational modifications, including phosphorylation and ubiquitination, in the presence of Mtb infection (Table S8). Overlaying this dataset with our PPI map revealed that we uncovered 22 and 12 host proteins with altered phosphorylation and ubiquitylation, respectively (Figure 3). These data suggest that these proteins might both be regulated by the host in response to Mtb and targeted by Mtb effectors making them high-priority targets for future study.
LpqN is an Mtb virulence factor
To begin to identify the functionally relevant interactions between Mtb secreted proteins and macrophages, we used a genetic approach to disrupt bacterial and host components of the PPI map and determined the effects during infection. Initially, we selected a set of 10 bacterial factors whose host interactors had known immune-related functions or were post-translationally modified in response to Mtb infection, and we generated or obtained previously-created Mtb mutants with these genes disrupted. These mutants were evaluated for growth in primary macrophages, and four strains with impaired growth relative to wild-type Mtb were carried forward for further analysis (Figure 4A, data not shown). For each mutant, we created two isogenic strains carrying either a wild-type copy of the disrupted gene under the control of its native promoter on an integrated plasmid, or an empty control plasmid. Each plasmid also contained a unique DNA barcode that allowed us to determine the relative abundance of each of the eight strains within pooled infections. We infected mice with this mixture of strains via the aerosol route, recovered bacilli from lung homogenates at multiple time-points, and quantified the relative proportion of each strain by qPCR (Figure 4A). Two of the mutants competed equally with their cognate “complementation” strains, and the Rv2469c mutant appeared to slightly out-compete the complemented strain but the lpqN::Tnhimar1 mutant (lpqN mutant) was rapidly depleted relative to lpqN::Tnhimar1::plpqN (lpqN complemented). Importantly, the lpqN mutant competed equally with the complemented strain when the pool was grown in culture (Figure S2), and the two strains grew with indistinguishable kinetics in axenic culture (Figure 4B), demonstrating that the lpqN mutant was specifically attenuated in the host.
Figure 4. The LpqN-Interacting protein CBL is a host restriction factor for Mtb.

(A) In vivo competition assay. Bacterial mutant strains (lpqN/Rv0583c, Rv2469c, Rv1804c, Rv0999) and cognate complemented strains were pooled and used to infect mice via the respiratory route. At the indicated times, bacteria were recovered from lung homogenates and the relative proportion of each strain was quantified by qPCR using unique sequence tags present in each strain. The Rv2469c mutant appeared to slightly out-compete the complemented strain, although this difference was not significant.
(B) Growth curve of the indicated strains in standard 7H9 mycobacterial media. Representative data from two independent experiments is shown.
(C) Luminescent bacterial growth assay. BMMs were infected with the indicated strains carrying the LuxBCADE reporter operon at an effective MOI=1. Relative luminescent units (RLU) were quantified at the indicated times and mean RLU relative to t=0 is plotted. Mean ± SEM are displayed from four replicate samples. Representative data of three independent experiments are shown. * p<0.05 by t-test.
(D) Phosphoproteomic analysis. RAW264.7 cells were isotopically labeled and infected with the indicated bacteria. Lysates were analyzed by quantitative LC-MS-MS and the fold-increase for the CBL S450 phosphosite is shown. Mean ± SEM are displayed for two biological replicates, each with two technical replicates.
(E) Luminescent bacterial growth assay in a RAW264.7 cell clone with CRISPR-induced homozygous frameshift mutations in the Cbl locus. Representative data of 3 independent experiments. Similar results were observed with three independent Cbl−/− clones and two independent control clones. Scramble indicates a non-targeting gRNA. *p=0.004 by t-test.
(F) LpqN-Strep or GFP-Strep was expressed in 293T cells and purified with Strep-tactin resin under native conditions, followed by SDS-PAGE and western blotting using antibodies that recognize CBL.
(G) Luminescent bacterial growth assay in Cbl−/− and Cbl+/+ BMMs infected with the lpqN mutant. *p<0.0001 by t-test.
(H) Direct CFU enumeration of intracellular bacteria from experiment shown in (G).
(I,J) Luminescent bacterial growth assays in Cbl−/− and Cbl+/+ BMMs infected with the lpqN complemented strain (I) (*p=0.003 by t-test), and WT Mtb CDC1551 strain (J) (*p<0.001 by t-test). Mean ± SEM of four replicate samples is displayed. Representative data from two independent experiments is shown.
(K) Restriction by CBL was derived by determining the ratio of bacterial growth in Cbl−/− and Cbl+/+ BMMs at the final timepoint using data from (G,I,J). Error bars denote SEM.
See also Figures S2, S3 and S4. (L) Luminescent growth assay of ESX-1-deficient strain (∆eccC-LuxBCADE) in BMMs. Growth of wild-type Mtb from Figure 4J plotted for comparison.
To test whether the lpqN mutant was also attenuated during infection of macrophages, we used a bioluminescent growth assay in which the isogenic lpqN mutant and complemented strains were transformed with the luxBCADE operon from Vibrio harveyi, and their growth in macrophages was monitored by quantifying luminescence over time. As with our observations in infected mice, we found that the lpqN mutant was also significantly attenuated in primary macrophages (Figure 4C), suggesting that LpqN functions during the initial bacterial encounter with the innate immune system. Collectively, these results demonstrate that LpqN is critical for bacterial growth in both ex vivo macrophages and in mice, thus establishing LpqN as a Mtb virulence determinant for the first time.
LpqN and the ubiquitin ligase CBL interact physically and genetically
We hypothesized that LpqN functions to promote bacterial growth through its interaction with host proteins, either by blocking their function or by ‘hijacking’ them for pathogenic purposes. To test this idea, we used CRISPR/Cas9-based mutagenesis in RAW 264.7 cells to determine whether disruption of LpqN-interacting host factors was able to rescue the growth defect of the lpqN mutant. As noted above, our related studies using MS to examine host protein post-translational modifications during Mtb infection demonstrated increased phosphorylation of the ubiquitin ligase CBL, a LpqN-interacting protein, and suggested a possible role for CBL in antibacterial immunity (Figure 4D; Table S8). Indeed, while mutagenesis of several other LpqN interactors failed to rescue the lpqN mutant phenotype, we observed that disruption of Cbl rescued the growth of the lpqN mutant in RAW264.7 cells (Figures 4E and S3A).
We verified the physical interaction between LpqN and CBL by expressing LpqN-Strep in 293T cells. Endogenous CBL protein that copurified was detected by western blotting, verifying the interaction detected by MS (Figure 4F). Additional in vitro pull-down experiments using LpqN and CBL proteins produced in E. coli also revealed direct interaction between these two factors (Figure S4). We further explored the genetic interaction between lpqN and Cbl using primary bone marrow-derived macrophages (BMMs) deficient in CBL. To avoid the potential confounding effects of the two related CBL family ubiquitin ligases, we analyzed BMMs lacking the CblB and CblC genes, and carrying either a wild-type or floxed Cbl locus that could be deleted with ~90% efficiency by addition of 4-hydroxy-tamoxifen to the culture media - hereafter designated Cbl+/+ and Cbl−/− respectively (Mukhopadhyay et al., 2016). We infected Cbl+/+ and Cbl −/− primary macrophages with the lpqN mutant strain and monitored bacterial growth over time. Consistent with our observations in Cbl−/− RAW264.7 cells (Figure 4E), the lpqN mutant was markedly attenuated in Cbl+/+ BMMs (Figure 4G). Importantly, in Cbl−/− BMMs, we observed significant rescue of the lpqN mutant intracellular growth phenotype, with a 15-fold increase in bacterial growth (Figure 4G). This result was further confirmed by plating macrophage lysates and directly enumerating bacterial colony-forming units (CFU) (Figure 4H).
We also noted that both the lpqN complemented strain and the parental WT CDC1551 Mtb strain grew ~2-fold faster in Cbl−/− macrophages, indicating that CBL imposes some restriction on wild-type M. tuberculosis (Figures 4I, 4J), however, the effect of CBL activity was much more pronounced with the lpqN mutant (Figure 4K). Importantly, the growth of an attenuated ESX-1 mutant of Mtb, which is unable to permeabilize its phagosome, was unaffected in Cbl−/− macrophages (Figure 4L), as were the growth of other intracellular bacteria including Listeria monocytogenes and Salmonella enterica serovar Typhimurium (S. Typhimurium, Figures S3B and S3C). Thus, there is a genetic interaction between bacterial lpqN and host Cbl, whereby growth of the lpqN mutant strain is selectively enhanced upon loss of host Cbl, but strains expressing LpqN are relatively insensitive to CBL activity. This genetic interaction strongly suggests that LpqN and CBL function in a common pathway. Collectively, the combined biochemical and genetic data suggest a model whereby CBL acts as a host restriction factor that limits Mtb growth, and LpqN acts as a virulence factor to block the normal functions of CBL and promote bacterial replication.
CBL regulates the balance between cell-intrinsic antibacterial and antiviral responses.
CBL has been well-characterized as a ubiquitin ligase responsible for the ubiquitylation and degradation of activated receptor protein tyrosine-kinases (Levkowitz et al., 1999). Since ubiquitylated proteins localize to the Mtb phagosome and are proposed to function as a targeting mechanism for autophagy-mediated host defense (Ponpuak et al., 2010; Watson et al., 2012), we initially hypothesized that CBL might directly contribute to this process. However, the frequency of ubiquitin colocalization with either wild-type or lpqN mutant bacteria after 4h of infection was unaffected in Cbl−/− BMMs (Figure 5A), demonstrating that this ligase is not required for ubiquitin deposition around invading Mtb.
Figure 5. CBL represses antiviral responses during Mtb infection.

(A) Cbl−/− and Cbl+/+ BMMs infected with wild-type and lpqN mutant Mtb for 4h and immunostained for polyubiquitin. Percent colocaliztion of >500 phagosomes counted per condition, scored blinded to sample identity. Mean ± SEM displayed. Scale bars = 10 microns.
(B) BMMs infected with either wild-type or lpqN mutant Mtb for 6 hours, and analyzed by RT-qPCR for the proinflammatory cytokine TNF-α or the antiviral response genes IFN-β and IFIT1. t-tests were used for statistical analysis.
(C) Luminescent growth assay. BMMs were treated with DNA (Lipo+DNA), transfection reagent alone (Lipo), or IFN-β (250U) as indicated. Mean ± SEM of four replicate samples are shown. IFN-β was added 4h pre-infection and DNA was delivered 1h post-infection. *p=0.003 by t-test.
Given the role of CBL as a regulator of signaling, we investigated whether the ligase modulates macrophage inflammatory responses to bacterial infection by monitoring expression of key cytokines activated in response to Mtb. While we observed a modest decrease in TNF-α mRNA levels between Cbl+/+ and Cbl−/− cells, we noted a dramatic increase in the expression of the antiviral cytokine IFN-β (Figure 5B). Likewise, IFIT1, another target of antiviral signaling, was also increased in Cbl−/− cells (Figure 5B). Thus, the ability of Mtb to activate antiviral responses, likely through the exposure of bacterial DNA to the cytoplasmic sensor cGAS (Manzanillo et al., 2012), is amplified in the absence of CBL, indicating that this ligase functions to negatively regulate these responses.
These data support a model in which CBL-deficient macrophages are skewed towards more robust antiviral responses, but which come at the cost of decreased antibacterial resistance. In this way, CBL would promote cellular resistance by counteracting the antiviral response induced by Mtb via the cGAS/STING pathway. Indeed, our finding that ESX-1 mutants, which fail to activate antiviral signaling during infection (Stanley and Cox, 2013; Watson et al., 2015), are attenuated in Cbl−/− macrophages is consistent with the idea that CBL is unnecessary to control bacterial growth in the absence of an antiviral signal (Figure 4L). To further investigate the idea that a CBL-regulated antiviral program promotes Mtb growth, we tested whether ectopic activation of antiviral responses by experimentally delivering DNA to the host cell cytosol would rescue the growth defects of ESX-1 mutant bacteria. In this way, activation of the antiviral pathway negatively regulated by CBL would occur in the absence of phagosomal permeabilization, effectively bypassing the ESX-1 requirement. In macrophages transfected with DNA we observed a significant rescue of the ESX-1 mutant with impaired bacterial killing over the first 24h, an effect that is enhanced in Cbl−/− macrophages (Figure 5C). This suggests that antiviral signaling is sufficient to blunt the antibacterial activity of macrophages and that CBL functions to counteract this effect. Importantly, addition of IFN-β did not rescue growth of the ESX-1 mutant, consistent with the idea that CBL regulates an antiviral program that is independent of Type I IFN (Figure 5C).
Given the reciprocal effects of antiviral and antibacterial responses mediated by CBL, a simple prediction is that Cbl−/− cells may be resistant to viral infection. To test this, we infected Cbl−/− and Cbl+/+ BMMs with Sendai Virus (SeV) and Herpes Simplex Virus 1 (HSV-1) and evaluated the release of viral particles 24h after infection. Deletion of Cbl conferred resistance to both viruses, with fewer viral particles released, and a reduction in viral cytopathic effects (Figure 6A, 6B). Thus, while loss of CBL results in increased sensitivity to Mtb infection, it also creates a more restrictive environment for viral replication. This is consistent with recent findings that siRNA-mediated depletion of CBL results in stabilization of IRF3 and increased antiviral signaling (Zhao et al., 2016), though when we evaluated whether CBL similarly regulated IRF3 during Mtb infection, we detected no change in protein levels (Figure 6C).
Figure 6. CBL represses antiviral responses during viral infection.

(A) BMMs infected with Sendai virus (SeV) for 24h, and the relative number of virions in the supernatant were quantified by RT-qPCR; Mean ± SEM displayed. Phase-contrast image (10x) at 24h post-infection. Scale bars = 100 microns.
(B) Infection with HSV-1 for 24h, analyzed as above. t-tests were used for statistical analysis.
(C) BMMs were infected with lpqN mutant Mtb for 6h and analyzed by Western blot.
(D) Model of balance between antiviral and antibacterial cell-intrinsic immune response pathways mediated by CBL. Host macrophages tailor responses to distinct kinds of pathogens at the earliest stages of infection by activating cell-intrinsic immune pathways tailored to the threat, e.g. virus or bacterium. These programs appear to be mutually antagonistic, as activation of antiviral pathways comes at the cost of antibacterial immunity during Mtb infection. Our work indicates that CBL functions to influence this balance by inhibiting viral responses and promoting antibacterial immunity.
Although the exact mechanism by which CBL influences the balance between intrinsic antibacterial and antiviral defense, permissiveness of Cbl−/− macrophages is unlikely to result from increased Type I interferon itself as Mtb replicates normally in macrophages deficient for Type I IFN production (Irf3−/−) or signaling (Ifnar−/−) (Manzanillo et al., 2012; Stanley et al., 2007). Thus, it appears that CBL may control a broader cell-intrinsic antibacterial program by inhibiting antiviral responses, with one component being Type I IFN activation (Figure 6D).
DISCUSSION
Despite the impact of TB on mankind, surprisingly little is known about the physical interactions between Mtb proteins and its human host proteome. Our understanding of how many enteric pathogens interact with host cells via secretion of virulence factors that target host pathways has shed light on the evolution of intricate interactions that underlie the host-pathogen interface. Because Mtb is a remarkably successful bacterial pathogen, capable of persisting for the lifetime of the host, it seems likely that Mtb would similarly introduce secreted effectors to manipulate host pathways and forestall host immunity. However, the evidence that Mtb actually employs this strategy is scant. There are only a handful of characterized interactions between Mtb and host proteins, and it remains unclear how they impact Mtb virulence. We have addressed this question using our AP-MS pipeline to globally identify 187 high confidence, physical interactions between Mtb and host proteins. It is likely that not all of these will contribute significantly to bacterial virulence during the course of a natural Mtb infection. However, this proteomic approach, when combined with genetic analysis, represents a powerful way to uncover biologically relevant interactions between pathogens and their host cells. Indeed, with this approach we have identified an Mtb virulence factor, LpqN, and additionally have identified the CBL ubiquitin ligase as a protein that interacts both physically and genetically with LpqN and functions as a host restriction factor for Mtb, limiting bacterial growth in macrophages. Interestingly, LpqN is also physically associated with the ribosome and the HOPS complex, which is involved in vesicle fusion. Further work will be required to determine the functional relevance of these and the many other connections we uncovered in the our protein-protein interaction map.
CBL had not been implicated in innate responses to bacteria, and the mechanisms by which it contributes to host immunity await discovery. Ubiquitin ligases play key roles in targeting of intracellular pathogens to autophagy, but our data indicates that CBL plays a regulatory role modulating cell-intrinsic responses to infection. Indeed, we noted a surprising increase in the cellular antiviral response in the absence of CBL, with a concomitant decrease in the efficiency of antibacterial processes. Importantly, CBL had no effect on the growth of an ESX-1 mutant strain of Mtb that does not trigger antiviral responses. This finding that the antibacterial properties of CBL are only manifest in the setting of an antiviral host response is consistent with the hypothesis that CBL is acting as a regulator of the antiviral response to control bacterial infection (Figure 6D).
The specific antiviral pathways regulated by CBL, and exactly how this impairs antibacterial functions in the context of Mtb infection remain unknown. IFN-β has been suggested to impair clearance of the related pathogen Mycobacterium leprae (Teles et al., 2013), and disruption of either IRF3 or IFNAR have previously been shown to modestly impact Mtb growth in mice (Manzanillo et al., 2012; Stanley et al., 2007). However, IFN-β signaling itself does not alter Mtb growth cell-intrinsically in isolated macrophages, as we observe with CBL. This suggests additional, uncharacterized antiviral pathways exist, and are activated by Mtb, in addition to the known IRF3-IFN-β-IFNAR pathway. The existence of additional antiviral pathways is further supported by our findings in a related study using proteomics to examine host protein post-translational modifications during Mtb infection (Parry et. al. in preparation). In that work, we identified the IRF7-regulated antiviral response as a potent facilitator of Mtb replication. The effects mediated by IRF7 and CBL share several similarities. Unlike the IRF3-IFNAR pathway, they both alter Mtb replication cell-intrinsically in isolated macrophages. The effects of perturbing IRF7 and CBL are also both specific to Mtb, as the replication of neither S. Typhimurium nor L. monocytogenes are altered. It remains to be determined whether CBL and IRF7 are actually functioning in the same pathway, or whether they modulate parallel antiviral pathways. The finding that two different regulators of the macrophage antiviral response both impact the ability of Mtb to replicate inside macrophages suggests that this is an essential element in TB pathogenesis. We postulate that Mtb might have evolved the ability to release bacterial DNA into the host in order to trigger the macrophage antiviral response, and that this antiviral response antagonizes antibacterial activity. Effectors such as LpqN could then potentially function to prolong or amplify such a signal and create a more permissive intracellular environment for bacterial growth.
From within its replicative niche in host macrophages, Mtb is able to undermine the host immune response and establish a chronic, often lifelong infection – but how it actually does so remains mysterious. The interaction map reported here represents a unique resource to generate testable hypotheses regarding the Mtb-host interface and to identify the host pathways that dictate TB pathogenesis.
STAR METHODS
CONTACT AND REAGENT RESOURCE SHARING
Please contact the Lead Contact Nevan Krogan (nevan.krogan@ucsf.edu) for further information, reagents, and tools.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Macrophages
Bone marrow-derived macrophages (BMMs) were isolated by flushing the femurs from 8–10-week-old C57BL/6 female mice. They were cultured in high-glucose DMEM supplemented with 20% heat-inactivated FCS and 10% conditioned media from 3T3-MCSF cells. All cells were cultured at 37°C with 5% CO 2. Cbl+/+ and Cbl−/− cells were generated by isolating BMMs from Cbl+/+ [B6.Cg-Cblbtm1Hua, B6.Cg-Cblctm1Pngr, Gt(ROSA)26Sortm1(cre/ERT)Nat/J, Gt(ROSA)26Sortm1.1(CAG-EGFP)Fsh/Mmjax] and Cbl−/− [B6.Cg-Cbltm2Hua, B6.Cg-Cblbtm1Hua, B6.Cg-Cblctm1Pngr, Gt(ROSA)26Sortm1(cre/ERT)Nat/J, Gt(ROSA)26Sortm1.1(CAG-EGFP)Fsh/Mmjax] (Mukhopadhyay et al., 2016) in the presence 1uM 4-hydroxytamoxifen (Sigma).
Cell Lines
RAW 264.7 mouse macrophages (ATCC TIB-71; male) and 293T cells (ATCC® CRL-3216; female) were purchased from ATCC and cultured according to ATCC recommendations with addition of 20mM HEPES pH7.4.
Bacterial strains
All Mtb strains were cultured in 7H9 liquid media (BD) supplemented with 10% Middlebrook OADC (Sigma), 0.5% glycerol, 0.05% Tween80 in roller bottles at 37°C. Transposon mutants were obtained from ATCC and carry Himar1 transposon insertions with the Kanamycin resistance gene in the indicated locus. Transposon insertion sites were validated by PCR and Sanger sequencing (Lamichhane et al., 2003). For genetic complementation studies, the predicted promoter region and open reading frame were cloned into the SacI site of the integrating vector pBU (Vultos et al., 2006) which had been modified to confer Zeocin resistance, and strains were selected with 25μg/ml Zeocin. Control strains carried pBU with a ~100 nucleotide fragment of GFP as unique sequence tag (see Key Resources Table). For luminescent growth assays, strains carried the containing a codon-optimized luxBCADE operon expressed from a MOPS promoter carried on the pmv306-Hyg integrating vector (Craney et al., 2007). The ΔeccC strain was made by homologous recombination using the pMSG361 vector (Rosenberg et al., 2015).
METHOD DETAILS
Mtb secreted protein analysis
Mtb Erdman strain was grown in 7H9+OADC (BD Biosciences), +0.05% Tween-80 (Sigma) until mid-log, then transferred to Sauton’s minimal media + 0.05% Tween-80 for 5d, and then finally to Sauton’s media with 0.005% Tween for 5d. Bacteria were pelleted and supernatants concentrated with a 3K MWCO filter (Millipore). Bacteria were lysed by boiling, followed by 10 minutes of sonication in 1% SDS. 20μg each of cell lysate and culture filtrate were separated by SDS-PAGE and western blot performed against GroEL to verify that there was no detectable contamination of the culture filtrate by cytoplasmic proteins. For MS analysis, protein was resolved on SDS-PAGE, visualized by Coomassie, and gel slices subjected to trypsin digestion as described below.
In vivo competition assay
12-week-old female C57BL/6 mice were inoculated by aerosol with a pool of four mutant strains and cognate complemented strains delivered at ~75 CFU of each strain using a Madison chamber device. At the indicated times, lungs of 4–5 mice were homogenized and plated on 7H10 plates. ~10,000 individual colonies were scraped into Trizol (Thermo Fisher Scientific), samples were lysed via bead-beating, RNA was removed per manufacturer instructions, and genomic DNA was isolated via extraction with 4 M guanidine thiocyanate/50mM Sodium Citrate/1M Tris base. 0.2 volumes sodium acetate and 0.4 volumes ethanol was added, and DNA was purified using RNeasy columns (Qiagen). qPCR primers were designed to detect unique sequence tags inserted in each strain’s genome using either a fragment of the GFP gene (mutant strains) or the unique junction between the pBU vector & complementation cassette (complemented strains). Each primer set was verified to detect only its specific strain. qPCR was performed with Taq polymerase (NEB) and SYBR green I (Sigma) detection.
Ex vivo luminescent bacterial growth assay
Mtb strains carrying the luxBCADE operon were prepared for inoculation by washing twice in PBS, removing aggregates with a 200 RCF spin, followed by gentle sonication to generate a fine bacterial suspension. Bacteria were opsonized in 10% heat-inactivated horse serum and macrophages infected at an MOI=2 by spinning inoculated plates for 10 min at 400 RCF. Monolayers were washed, and bacterial luminescence measured at the indicated times using a Spectromax L (Molecular Dynamics). Macrophage growth media was changed daily for cell lines and every 48h for BMM cultures.
CRISPR mutagenesis
CRISPR guides were designed to target regions in the first exon of target genes using an online bioinformatic tool (http://crispr.mit.edu), and cloned into the BsmB1 site of the pXPR_001 vector (a gift from Feng Zhang, Addgene plasmid # 52961). Viral particles were produced in Lenti-X cells (Clontech) per manufacturer’s instructions and used to transduce RAW264.7 cells. These were selected in 5ug/ml Puromycin (InVivoGen), and single-cell clones isolated. Mutations were identified by using PCR to amplify the first exon by PCR followed by Sanger sequencing. CRISPR CBL mutants were independently confirmed by western blot with a mouse anti-c-CBL antibody (Santa Cruz Biotechnology cat# sc-1651).
Western blots
Protein in lysates was quantified by BCA (Pierce Fisher Scientific). 20μg of cell lysate was separated by SDS-PAGE (Bio-Rad TGX), and transferred onto nitrocellulose membranes. After probing with the indicated antibodies, membranes were then imaged on an Odyssey scanner (Li-cor).
Affinity purification
Mtb genes were amplified by PCR from Erdman strain genomic DNA. For genes with a predicted signal peptide using SignalP 3.0 (Bendtsen et al., 2004), the portion of the open reading frame corresponding to the mature protein was amplified. Genes were cloned into PCDNA4 with a C-terminal 2x-Strep tag and expressed in 293T cells using calcium phosphate transfection. Cells were lysed 36h later in IP buffer (50mM Tris 7.4, 150 mM NaCl, 1mM EDTA, 0.05% NP-40) with phosphatase and protease inhibitors (Roche) and tagged proteins immobilized on Strep-Tactin resin (IBA). Macrophage lysate was generated from U937 cells differentiated with 10nM PMA for 72h and then similarly lysed in IP buffer. 10mg of macrophage lysate was added to each immobilized bacterial factor and binding allowed to proceed overnight. The resin was then washed 4x in IP buffer, and twice in IP buffer without NP-40 before elution in 10mM biotin. For reciprocal immunoprecipitation using FLAG-tagged human proteins the human factors were cloned as 3xFLAG-tag fusion proteins in pCDNA4. Combinations of host and bacterial factors were co-transfected into 293T cells using calcium phosphate and immunoprecipitation with anti-FLAG M2 antibody was performed using the same lysis and wash conditions as above.
Mass Spectrometry
Purified proteins eluates were digested with trypsin for LC-MS/MS analysis. Samples were denatured and reduced in 2M urea, 10 mM NH4HCO3, 2 mM DTT for 30 min at 60C, then alkylated with 2 mM iodoacetamide for 45 min at room temperature. Trypsin (Promega) was added at a 1:100 enzyme:substrate ratio and digested overnight at 37C. Following digestion, samples were concentrated using C18 ZipTips (Millipore) according to the manufacturer’s specifications. Desalted samples were evaporated to dryness and resuspended in 0.1% formic acid for mass spectrometry analysis.
For the MS study to establish a high-confidence set of Mtb secreted proteins culture filtrates were resolved by SDS-PAGE. The gel was partitioned into sequential slices and each of these was individually subjected to in-gel trypsin digestion followed by desalting and LC-MS/MS analysis on a Thermo Scientific LTQ XL linear ion trap mass spectrometer. The LTQ XL system was equipped with a LC Packings Ultimate HPLC with an analytical column (10 cm × 75 um I.D. packed with ReproSil Pur C18 AQ 5 um particles). The system delivered a gradient from 5% to 30% ACN in 0.1% formic acid over one hour, and collected data in a data-dependent fashion. The LTQ XL collected one full scan followed by 10 collision-induced dissociation MS/MS scans of the 10 most intense peaks from the full scan. Dynamic exclusion was enabled for 30 seconds with a repeat count of 1.
For the primary AP-MS study used to establish the interactome, digested peptide mixtures were analyzed by LC-MS/MS on a Thermo Scientific LTQ XL linear ion trap mass spectrometer as above.
For the secondary analysis, where AP-MS was performed without inclusion of U937 macrophage lysate, the digested peptide mixtures were analyzed by LC-MS/MS on a Thermo Scientific Velos Pro dual linear ion trap 238 mass spectrometer equipped with an Easy-nLC II HPLC with a pre-column (2 cm × 100 um I.D. packed with 239 ReproSil Pur C18 AQ 5 um particles) and an analytical column (10 cm × 75 um I.D. packed with ReproSil Pur 240C18 AQ 3 um particles). A gradient was delivered from 5% to 30% ACN in 0.1% formic acid over one hour. The mass spectrometer collected data in a data-dependent fashion with one full scan followed by 20 collision-242 induced dissociation MS/MS scans of the 20 most intense peaks from the full scan. Dynamic exclusion was 243 enabled for 30 seconds with a repeat count of 1.
The raw data was matched to protein sequences by the Protein Prospector algorithm (Clauser et al., 1999). Data were searched against a database containing Swiss Prot Human protein sequences (downloaded March 6, 2012) and concatenated to a decoy database where each sequence was randomized in order to estimate the false positive rate. The searches considered a precursor mass tolerance of 1 Da and fragment ion tolerances of 0.8 Da and considered variable modifications for protein N-terminal acetylation, protein N-terminal acetylation and oxidation, glutamine to pyroglutamate conversion for peptide N-terminal glutamine residues, protein N-terminal methionine loss, protein N-terminal acetylation and methionine loss, and methionine oxidation, and constant modification for carbamidomethyl cysteine. Prospector data was filtered using a maximum protein expectation value of 0.01 and a maximum peptide expectation value of 0.05.
Our global identification of phosphorylation and ubiquitylation during Mtb infection of RAW264.7 macrophages is described in a separate manuscript (Johnson et. al., in preparation) and will be submitted to ProteomeXchange via PRIDE.
MiST
AP-MS samples were scored with Mass spectrometry Interaction STatistics (MiST) algorithm, using the MiST reproducibility (0.45), specificity (0.50) and abundance (0.05) weights previously published (Davis et al., 2015). All bait-prey pairs with a MIST score ≥ 0.70 were considered confident interactions and were combined with human protein interactions from the CORUM and STRING databases. The resulting network diagram was plotted using Cytoscape, v.3.1.2 411 (Smoot et al., 2011). For the subset re-analysis without U937 lysate, MiST with identical settings was used but additional control Strep-tag purifications added to the analysis matrix in MiST to compensate for the smaller number of experimental samples and establish robust specificity scoring.
Evolutionary Analysis
For analysis over a 5–7 million-year timescale, we employed SnIPRE (Eilertson et al., 2012). We focused our analysis on the human lineage by removing sites that are inferred to have received mutations along the chimpanzee lineage based on an alignment of closely related primates. For analysis of more recent evolution in the primate lineage, we used a modified version of integrated Haplotype Score (iHS) (Voight et al., 2006) as implemented in (Szpiech and Hernandez, 2014).
In Vitro Interaction Studies
Proteins were cloned into the pH3C vector as either a Strep-Tag fusion (LpqN) or FLAG-Tag fusions (CBL, GFP). Factors were individually expressed in BL21 cells by overnight induction with IPTG at room-temperature. Cells were lysed in PBS + protease inhibitors using lysozyme and sonication. Total cell lysates containing ~10μg of each tagged protein was combined, subjected to Strep-Tactin purification, and interacting proteins were detected by Western blotting as above.
Gene Expression Analysis
Infected macrophages were lysed in Trizol (Thermo Fisher Scientific) and then purified with silica spin-columns (Purelink, Ambion) per manufacturer instructions. cDNA was generated using 500ng total RNA with the Superscript III First Strand Synthesis Kit (Invitrogen) and subjected to qPCR as described above with normalization to the beta-actin transcript.
Immunofluorescence microscopy
Coverslips were fixed for 20min in 4% PFA, permeabilized in 0.05% Saponin (Sigma) and incubated with the indicated antibodies for 3h at RT. Mycobacteria were visualized either by expression of mCherry or by labeling with SytoBC (Fisher). All samples were mounted and images acquired on a Keyence BZ-X700 microscope. Z-stacks at 0.5μM were acquired and maximum intensity projections generated. To quantify colocalization positive phagosomes were scored for each marker by a microscopist blinded to sample identity.
Viral Infections
Primary BMDMs were inoculated at 1.5E5 cells in each well of a 24-well plate and had either Sendai virus or human Herpes Simplex virus-1 added and were spun at 1500RMP for 1 hour to inoculate virus at MOI=1. 24 hours after infection cell supernatants were removed and nucleic acid isolated using a Quiagen QIAamp MinElute Virus Spin Kit. For Sendai virus RT-qPCR was performed and for Herpes Simplex virus-1 qPCR was then performed.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
For RT-qPCR, each sample was amplified in triplicate and transcript levels normalized to the beta-actin gene and the standard error of the mean was calculated. Each experiment was repeated at least two times from separate biological samples with representative data from one biological replicate shown. For luminescent growth assays, each sample was quantified in quadruplicate wells and standard-error of the mean was calculated. Each experiment was repeated at least three times from separate biological samples with representative data from one biological replicate shown. For in vivo competition assays, the sequence tags were amplified in triplicate qPCR reactions for each mouse, and the ratio between mutant and complement calculated. For the set of mice at each time point, the mean mutant:complement ratio and standard-error of mean were calculated. A Kruskal-Wallis test was used to calculate p-values for comparisons among sets of mice.
Bioinformatic Analysis of Host Proteins
The set of high-confidence host interacting proteins was analyzed using the DAVID Bioinformatics Resources 6.8 functional annotation tool. Uniprot accession numbers were matched with Uniprot keyword categorization to map protein function and determine enrichment relative to the human proteome. Thresholds for inclusion were set to two-fold enrichment with a Benjamini–Hochberg corrected p-value of <0.05 to adjust for multiple hypotheses testing. Categories were manually curated to remove redundant and nonspecific categorizations. For overlap analysis between datasets, a Fisher’s exact test was used with the background proteome defined empirically as the set of all proteins detectable by our Velos Pro dual linear ion trap instrument in 293T cell-related experiments (10,782 proteins).
DATA AND SOFTWARE AVAILABILITY
The MiST algorithm is available freely on github (https://github.com/kroganlab/mist). All raw microscopy images and blots have been deposited to Mendeley Data (http://dx.doi.org/10.17632/x3v7rb83bg.1).
Supplementary Material
KEY RESOURCE TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-CBL | Santa Cruz Bio. | RRID:AB_2244054 |
| Rabbit monoclonal anti-IRF-3 | Cell Signaling | RRID:AB_1904036 |
| Rabbit monoclonal anti-phospho-IRF-3 (S396) | Cell Signaling | RRID:AB_823547 |
| Rabbit monoclonal anti-β-Actin | Cell Signaling | Cat# 8457T |
| Mouse monoclonal anti-Ubiquitin | Millipore | RRID:AB_2043482 |
| Rabbit polyclonal anti-V-ATPase A1 | Santa Cruz Bio. | RRID:AB_2258865 |
| Mouse monoclonal anti-FLAG tag | Sigma | Cat #F1804 |
| Mouse monoclonal anti-Strep tag | IBA Life Sciences | Cat # 2–1509-001 |
| Bacterial and Virus Strains | ||
| Mtb: CDC1551 Strain | BEI Resources | Cat # CNR-13649 |
| Mtb: CDC1551, lpqN::Tnhimar1 | BEI Resources | Cat # NR-18454 |
|
Mtb: CDC1551, lpqN::Tnhimar1, attBMS6 ::pBUGFP, attBTn5 ::pmv306Lux |
This paper | N/A |
|
Mtb: CDC1551, lpqN::Tnhimar1, attBMS6::pBUlpqN, attBTn5 ::pmv306Lux |
This paper | N/A |
| Mtb: CDC1551, Rv0999::Tnhimar1 | TARGET Consortium | Cat# JHU0999–616 |
|
Mtb: CDC1551, Rv0999::Tnhimar1, attBMS6 ::pBUGFP, attBTn5 ::pmv306Lux |
This paper | N/A |
|
Mtb: CDC1551, Rv0999::Tnhimar1, attBMS6::pBURv0999, attBTn5 ::pmv306Lux |
This paper | N/A |
| Mtb: CDC1551, Rv1804c::Tnhimar1 | BEI Resources | Cat# NR-14706 |
|
Mtb: CDC1551, Rv1804c::Tnhimar1, attBMS6 ::pBUGFP, attBTn5 ::pmv306Lux |
This paper | N/A |
|
Mtb: CDC1551, Rv1804c::Tnhimar1, attBMS6::pBURv1084c, attBTn5 ::pmv306Lux |
This paper | N/A |
| Mtb: CDC1551, Rv2469c::Tnhimar1 | BEI Resources | Cat # NR-18289 |
|
Mtb: CDC1551, Rv2469c::Tnhimar1, attBMS6 ::pBUGFP, attBTn5 ::pmv306Lux |
This paper | N/A |
|
Mtb: CDC1551, Rv2469c::Tnhimar1, attBMS6 ::pBURv2469, attBTn5 ::pmv306Lux |
This paper | N/A |
| Mtb: Erdman Strain | W.R. Jacobs, Jr. | BEI Cat # NR-15404 |
| Mtb: Erdman, ΔeccCa1-ΔeccCb1, attBTn5 ::pmv306Lux | (Rosenberg et al., 2015) | N/A |
| Mtb: Erdman::pmv261Zeo_MCherry | This paper | N/A |
| Sendai virus Cantell Strain | ATCC | Cat# VR-907 |
| Human herpesvirus 1 KOS, Strain 17 | ATCC | Cat# VR-1493 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Trypsin | Promega | V5280 |
| Strep-Tactin Resin | IBA Biosciences | 2–1201-010 |
| FLAG M2 Resin | Sigma | M8823–1ML |
| Deposited Data | ||
| All raw microscopy images and blots have been deposited to Mendeley Data (http://dx.doi.org/10.17632/x3v7rb83bg.1) |
||
| Experimental Models: Cell Lines | ||
| Mouse: RAW267.4 | ATCC | TIB-71 |
| Human: U937 | ATCC | CRL-1593.2 |
| Human: Lenti-X 293T | Clontech | Cat# 632180 |
| Human: 293T | ATCC | Cat # CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Jackson Laboratory | Cat#: 000664 |
| Mouse: B6.Cg-Cblbtm1Hua, B6.Cg-Cblctm1Pngr, Gt(ROSA)26Sortm1(cre/ERT)Nat/J, Gt(ROSA)26Sortm1.1(CAG-EGFP)Fsh/Mmjax |
(Mukhopadhyay et al., 2016) | N/A |
| Mouse: B6.Cg-Cbltm2Hua, B6.Cg-Cblbtm1Hua, B6.Cg- Cblctm1Pngr, Gt(ROSA)26Sortm1(cre/ERT)Nat/J, Gt(ROSA)26Sortm1.1(CAG-EGFP)Fsh/Mmjax |
(Mukhopadhyay et al., 2016) | N/A |
| Oligonucleotides | ||
| See Table S9 for oligonucleotides, genomic barcodes, and complementation sequences |
||
| Recombinant DNA | ||
| pXPR_001 (lentiCRISPR V2) | (Sanjana et al., 2014) | Addgene plasmid # 52961 |
| pCDNA4/TO | Thermo | Cat# V102020 |
| pH3c-LIC | DNASU | Cat# EvNO00292960 |
| pBU | Vultos et al., 2006 | N/A |
| pmv261 | (Stover et al., 1991) | N/A |
| pmv306 | (Stover et al., 1991) | N/A |
| Software and Algorithms | ||
| MiST | (Verschueren et al., 2015) | https://github.com/kroganlab/mist |
Highlights.
Creation of a Mtb-host protein-protein interaction map using mass spectrometry
LpqN is a novel Mtb virulence factor that associates with CBL, a host ubiquitin ligase
Removal of CBL rescues the attenuated lpqN Mtb mutant
CBL acts as a switch between anti-viral and anti-bacterial responses in host
ACKNOWLEDGMENTS
We thank members of the Cox, Krogan and Stanley (UCB) labs for comments on the manuscript and for invaluable discussions. We thank Mayumi Naramura for the gift of Cbl−/−macrophages. This work was supported by NIH grants P01 AI063302 (N.J.K., J.S.C., D.A.P.), P50 GM082250 (N.J.K.), U19 AI106754 (N.J.K.), DP1 AI124619 (J.S.C.), and R01 AI120694 (N.J.K. and J.S.C.). B.P.H. was supported by an A.P. Giannini award and NIH K08 (K08AI104766).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTEREST
Daniel A. Portnoy has a financial interest in Aduro Biotech, and both he and the company stand to benefit from commercialization of this research.
REFERENCES
- Alberts B (1998). The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell 92, 291–294. [DOI] [PubMed] [Google Scholar]
- Alonso S, Pethe K, Russell DG, and Purdy GE (2007). Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc Natl Acad Sci USA 104, 6031–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong JA, and Hart PD (1971). Response of cultured macrophages to mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J Exp Med 134, 713–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach H, Papavinasasundaram KG, Wong D, Hmama Z, and Av-Gay Y (2008). Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host & Microbe 3, 316–322. [DOI] [PubMed] [Google Scholar]
- Beltrao P, Cagney G, and Krogan NJ (2010). Quantitative genetic interactions reveal biological modularity. Cell 141, 739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, Heijne, von G, and Brunak S (2004). Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340, 783–795. [DOI] [PubMed] [Google Scholar]
- Braverman J, and Stanley SA (2017). Nitric Oxide Modulates Macrophage Responses to Mycobacterium tuberculosis Infection through Activation of HIF-1α and Repression of NF-κB. J Immunol 199, ji1700515–ji1701816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byndloss MX, Rivera-Chávez F, Tsolis RM, and Bäumler AJ (2017). How bacterial pathogens use type III and type IV secretion systems to facilitate their transmission. Curr Opin Microbiol 35, 1–7. [DOI] [PubMed] [Google Scholar]
- Clauser KR, Baker P, and Burlingame AL (1999). Role of accurate mass measurement (+/−10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Analytical Chemistry 71, 2871–2882. [DOI] [PubMed] [Google Scholar]
- Clemens DL (1996). Characterization of the Mycobacterium tuberculosis phagosome. Trends Microbiol 4, 113–118. [DOI] [PubMed] [Google Scholar]
- Collins AC, Cai H, Li T, Franco LH, Li X-D, Nair VR, Scharn CR, Stamm CE, Levine B, Chen ZJ, et al. (2015). Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host & Microbe 17, 820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, Miller KM, Maas NL, Roguev A, Fillingham J, Chu CS, Schuldiner M, Gebbia M, Recht J, Shales M, et al. (2007). Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446, 806–810. [DOI] [PubMed] [Google Scholar]
- Craney A, Hohenauer T, Xu Y, Navani NK, Li Y, and Nodwell J (2007). A synthetic luxCDABE gene cluster optimized for expression in high-GC bacteria. Nucleic Acids Res 35, e46–e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin KH, Ehrt S, Gutierrez-Ramos J-C, Weich N, and Nathan CF (2003). The Proteasome of Mycobacterium tuberculosis Is Required for Resistance to Nitric Oxide. Science 302, 1963–1966. [DOI] [PubMed] [Google Scholar]
- Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Dollen, Von J, Maher MC, Johnson T, Newton W, et al. (2015). Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol Cell 57, 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggal NK, and Emerman M (2012). Evolutionary conflicts between viruses and restriction factors shape immunity. Nat Rev Immunol 12, 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilertson KE, Booth JG, and Bustamante CD (2012). SnIPRE: selection inference using a Poisson random effects model. PLoS Comput Biol 8, e1002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell CA, Czudnochowski N, Dollen, Von J, Johnson JR, Nakagawa R, Mirrashidi K, Krogan NJ, Engel JN, and Rosenberg OS (2017). Chlamydia interfere with an interaction between the mannose-6-phosphate receptor and sorting nexins to counteract host restriction. Elife 6, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie SH, Crook AM, McHugh TD, Mendel CM, Meredith SK, Murray SR, Pappas F, Phillips PPJ, Nunn AJ, REMoxTB Consortium (2014). Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N Engl J Med 371, 1577–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, and Sassetti CM (2011). High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7, e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundner C, Cox JS, and Alber T (2008). Protein tyrosine phosphatase PtpA is not required for Mycobacterium tuberculosis growth in mice. FEMS Microbiol Lett 287, 181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, et al. (2012). Global landscape of HIV-human protein complexes. Nature 481, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, et al. (2006). Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643. [DOI] [PubMed] [Google Scholar]
- Lamichhane G, Zignol M, Blades NJ, Geiman DE, Dougherty A, Grosset J, Broman KW, and Bishai WR (2003). A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: application to Mycobacterium tuberculosis. Proc Natl Acad Sci USA 100, 7213–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, Lavi S, Iwai K, Reiss Y, Ciechanover A, et al. (1999). Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell 4, 1029–1040. [DOI] [PubMed] [Google Scholar]
- Liu B, Zheng Y, Yin F, Yu J, Silverman N, and Pan D (2016). Toll Receptor-Mediated Hippo Signaling Controls Innate Immunity in Drosophila. Cell 164, 406–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca C, Reed M, Sherry Freeman, Mathema B, Kreiswirth B, Barry C, and Kaplan G (2004). Differential Monocyte Activation Underlies Strain-Specific Mycobacterium tuberculosis Pathogenesis. Infect Immun 72, 5511–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangtani P, Abubakar I, Ariti C, Beynon R, Pimpin L, Fine PEM, Rodrigues LC, Smith PG, Lipman M, Whiting PF, et al. (2014). Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clinical Infectious Diseases : an Official Publication of the Infectious Diseases Society of America 58, 470–480. [DOI] [PubMed] [Google Scholar]
- Manzanillo PS, Shiloh MU, Portnoy DA, and Cox JS (2012). Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host & Microbe 11, 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer-Barber KD, and Sher A (2015). Cytokine and lipid mediator networks in tuberculosis. Immunol Rev 264, 264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, et al. (2014). Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 511, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Målen H, Berven FS, Fladmark KE, and Wiker HG (2007). Comprehensive analysis of exported proteins from Mycobacterium tuberculosis H37Rv. Proteomics 7, 1702–1718. [DOI] [PubMed] [Google Scholar]
- McNab F, Mayer-Barber K, Sher A, Wack A, and O’garra A (2015). Type I interferons in infectious disease. Nat Rev Immunol 15, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra A, Zahra A, Thompson V, Sirisaengtaksin N, Wells A, Porto M, Koster S, Penberthy K, Kubota Y, Dricot A, et al. (2013). Mycobacterium tuberculosis type VII secreted effector EsxH targets host ESCRT to impair trafficking. PLoS Pathog 9, e1003734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirrashidi KM, Elwell CA, Verschueren E, Johnson JR, Frando A, Dollen, Von J, Rosenberg O, Gulbahce N, Jang G, Johnson T, et al. (2015). Global Mapping of the Inc-Human Interactome Reveals that Retromer Restricts Chlamydia Infection. Cell Host & Microbe 18, 109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay C, Triplett A, Bargar T, Heckman C, Wagner K-U, and Naramura M (2016). Casitas B-cell lymphoma (Cbl) proteins protect mammary epithelial cells from proteotoxicity of active c-Src accumulation. Proc Natl Acad Sci USA 113, E8228–E8237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neisch AL, Neufeld TP, and Hays TS (2017). A STRIPAK complex mediates axonal transport of autophagosomes and dense core vesicles through PP2A regulation. J Cell Biol 216, 441–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, Myers TG, Rabin RL, Trinchieri G, Sher A, et al. (2011). Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1β production in human macrophages. J Immunol 187, 2540–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak SK, Basu S, Basu KK, Banerjee A, Pathak S, Bhattacharyya A, Kaisho T, Kundu M, and Basu J (2007). Direct extracellular interaction between the early secreted antigen ESAT-6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nat Immunol 8, 610–618. [DOI] [PubMed] [Google Scholar]
- Ponpuak M, Davis AS, Roberts EA, Delgado MA, Dinkins C, Zhao Z, Virgin HW, Kyei GB, Johansen T, Vergne I, et al. (2010). Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 32, 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramage HR, Kumar GR, Verschueren E, Johnson JR, Dollen, Von J, Johnson T, Newton B, Shah P, Horner J, Krogan NJ, et al. (2015). A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol Cell 57, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde K, Yates RM, Purdy GE, and Russell DG (2007). Mycobacterium tuberculosis and the environment within the phagosome. Immunol Rev 219, 37–54. [DOI] [PubMed] [Google Scholar]
- Rosenberg OS, Dovala D, Li X, Connolly L, Bendebury A, Finer-Moore J, Holton J, Cheng Y, Stroud RM, and Cox JS (2015). Substrates Control Multimerization and Activation of the Multi-Domain ATPase Motor of Type VII Secretion. Cell 161, 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Meth 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti CM, and Rubin EJ (2003). Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA 100, 12989–12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PS, Wojcechowskyj JA, Eckhardt M, and Krogan NJ (2015). Comparative mapping of host-pathogen protein-protein interactions. Curr Opin Microbiol 27, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh MU, Macmicking JD, Nicholson S, Brause JE, Potter S, Marino M, Fang F, Dinauer M, and Nathan C (1999). Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity 10, 29–38. [DOI] [PubMed] [Google Scholar]
- Siegrist MS, Unnikrishnan M, McConnell MJ, Borowsky M, Cheng T-Y, Siddiqi N, Fortune SM, Moody DB, and Rubin EJ (2009). Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc Natl Acad Sci USA 106, 18792–18797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siméone R, Sayes F, Song O, Gröschel MI, Brodin P, Brosch R, and Majlessi L (2015). Cytosolic access of Mycobacterium tuberculosis: critical impact of phagosomal acidification control and demonstration of occurrence in vivo. PLoS Pathog 11, e1004650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot ME, Ono K, Ruscheinski J, Wang P-L, and Ideker T (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley SA, and Cox JS (2013). Host-pathogen interactions during Mycobacterium tuberculosis infections. Curr Top Microbiol Immunol 374, 211–241. [DOI] [PubMed] [Google Scholar]
- Stanley SA, Johndrow JE, Manzanillo P, and Cox JS (2007). The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol 178, 3143–3152. [DOI] [PubMed] [Google Scholar]
- Stover CK, la Cruz de, V.F., Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, et al. (1991). New use of BCG for recombinant vaccines. Nature 351, 456–460. [DOI] [PubMed] [Google Scholar]
- Szpiech ZA, and Hernandez RD (2014). selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol. Biol. Evol 31, 2824–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teles RMB, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, Komisopoulou E, Kelly-Scumpia K, Chun R, Iyer SS, et al. (2013). Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science 339, 1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrelles JB, and Schlesinger LS (2017). Integrating Lung Physiology, Immunology, and Tuberculosis. Trends Microbiol 25, 688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, and Peters PJ (2007). M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129, 1287–1298. [DOI] [PubMed] [Google Scholar]
- Vandal OH, Nathan CF, and Ehrt S (2009). Acid resistance in Mycobacterium tuberculosis. J Bacteriol 191, 4714–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschueren E, Dollen J Von, Cimermancic P, Gulbahce N, Sali A, and Krogan NJ (2015). Scoring Large-Scale affinity purification mass spectrometry datasets with MiST. Curr Protoc Bioinformatics 2015, 8.19.1–8.19.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, and Pritchard JK (2006). A map of recent positive selection in the human genome. PLoS Biol 4, e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vultos TD, Méderlé I, Abadie V, Pimentel M, Moniz-Pereira J, Gicquel B, Reyrat J-M, and Winter N (2006). Modification of the mycobacteriophage Ms6 attP core allows the integration of multiple vectors into different tRNAala T-loops in slow- and fast-growing mycobacteria. BMC Mol. Biol 7, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, Schmid-Burgk JL, Schmidt T, Hornung V, Cole ST, et al. (2015). Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host & Microbe 17, 799–810. [DOI] [PubMed] [Google Scholar]
- Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, and Cox JS (2015). The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host & Microbe 17, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RO, Manzanillo PS, and Cox JS (2012). Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiens KE, and Ernst JD (2016). The Mechanism for Type I Interferon Induction by Mycobacterium tuberculosis is Bacterial Strain-Dependent. PLoS Pathog 12, e1005809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Edwards JP, and Mosser DM (2009). The expression of exogenous genes in macrophages: obstacles and opportunities. In Macrophages and Dendritic Cells, (Totowa, NJ: Humana Press; ), pp. 123–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Reddy MC, Ioerger TR, Rothchild AC, Dartois V, Schuster BM, Trauner A, Wallis D, Galaviz S, Huttenhower C, et al. (2013). Tryptophan biosynthesis protects mycobacteria from CD4 T-cell-mediated killing. Cell 155, 1296–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Zhu H, Yu J, Li H, Ge J, and Chen W (2016). c-Cbl-mediated ubiquitination of IRF3 negatively regulates IFN-β production and cellular antiviral response 28, 1683–1693. [DOI] [PubMed] [Google Scholar]
- Zumla A, George A, Sharma V, Herbert RHN, Baroness Masham of Ilton, Oxley A, and Oliver M (2015). The WHO 2014 global tuberculosis report--further to go. Lancet Glob Health 3, e10–e12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MiST algorithm is available freely on github (https://github.com/kroganlab/mist). All raw microscopy images and blots have been deposited to Mendeley Data (http://dx.doi.org/10.17632/x3v7rb83bg.1).