

SUMMARY
Chemical modifications to DNA and histone proteins are involved in epigenetic programs underlying cellular differentiation and development. Regulatory networks involving molecular writers and readers of chromatin marks are thought to control these programs. Guided by this common principle, we established an orthogonal epigenetic regulatory system in mammalian cells using N6-methyladenine (m6A), a DNA modification not commonly found in metazoan epigenomes. Our system utilizes synthetic factors that write and read m6A, and consequently recruit transcriptional regulators to control reporter loci. Inspired by models of chromatin spreading and epigenetic inheritance, we used our system and mathematical models to construct regulatory circuits that induce m6A-dependent transcriptional states, promote their spatial propagation, and maintain epigenetic memory of the states. These minimal circuits were able to program epigenetic functions de novo, conceptually validating “read-write” architectures. This work provides a toolkit for investigating models of epigenetic regulation and encoding additional layers of epigenetic information in cells.
Graphical Abstract
eTOC BLURB
A synthetic, modular, and programmable read/write system allows isolated and orthogonal epigenetic control in mammalian cells
INTRODUCTION
Genetically identical cells can produce distinct gene expression and phenotypic states that persist through cell division, a capability that is fundamental to the processes of environmental adaptation, cellular differentiation and multicellular development. These heritable states, which do not involve changes in DNA sequence, are maintained and transmitted by self-propagating epigenetic mechanisms that persist in the absence of an initial stimulus. Chemical modifications to DNA and histone proteins have been implicated in these epigenetic programs (Berger, 2007; Bernstein et al., 2007; Feinberg, 2007; Kouzarides, 2007), and mechanisms for the propagation of certain modifications have been proposed (Bonasio et al., 2010; Moazed, 2011). These commonly invoke a core regulatory motif involving molecular species that perform basic operations on chromatin, namely “writers” that place marks and “readers” that interpret them. To investigate this core module and obtain an understanding of the basic principles of epigenetic control, it would be useful to develop a synthetic system that could establish and drive epigenetic states de novo.
Studies of natural chromatin systems have identified many molecular components that regulate the placement and recognition of DNA and histone modifications, and collectively these studies have proposed a set of minimal ingredients for a bona fide epigenetic system: (1) sequence-specific placement of a modification (establish); (2) recruitment of protein effectors to the modification to mediate transcriptional changes (read & regulate); and (3) a mechanism for self-propagation that persists in the absence of an inducing signal (propagate) (Gardner et al., 2011; Moazed, 2011) (Figure 1). Combined together, these modules are thought to regulate complex epigenetic phenomena, such as the formation of silent heterochromatic domains in a variety of organisms (Beisel and Paro, 2011; Grewal and Moazed, 2003; Moazed, 2011; Ratna et al., 2009). Here, a propagation mechanism is used to spread histone modifications along the chromatin template away from a nucleation site to create an altered domain. Once established, these domains and their transcriptional states can be maintained through cell division. While molecular details of these propagation mechanisms vary across chromatin systems and organisms, a common theme is a core “read-write” motif ( Figure 1). Exemplified by regulators such as wi6/Clr4 in S. pombe (Ragunathan et al., 2015) and HP1α/Suv39h in mammals (Lachner et al., 2001), these are believed to function as positive feedback loops by recognizing pre-existing marks and consequently mediating the placement of the same modification on a nearby or adjacent template (e.g. to enable re-establishment after cell division) (Al-Sady et al., 2013).
Figure 1: The basic functional modules of an epigenetic regulatory system.
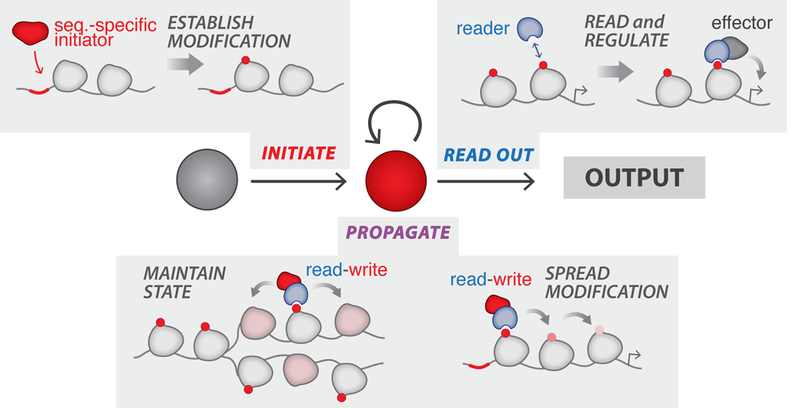
(1) Initiate: “initiators” establish chromatin modi fications at sequence-specific locations; (2) Read Out: “reader” proteins recognize modifications and mediate recruitment of regulators to establish transcriptional states; (3) Propagate: these states are propagated in the absence of the initial stimulus by read-write positive feedback mechanisms, whereby recognition of pre-existing marks is coupled to the placement of new modifications.
The complexity of natural chromatin networks can make it difficult to decipher the principles underlying epigenetic regulation. Our approach was to design a synthetic system by placing programmable control over the basic operations of writing and reading a chemical modification in cells. The functional modules of a minimum epigenetic system could be constructed with these operations, and subsequently used to engineer regulatory circuits in order to explore their capacity to generate higher-order behaviors, such as epigenetic memory. In principle, the synthetic approach has certain advantages because natural regulatory networks are extended with still many unclear links between chromatin modifications and regulators as well as pervasive cross-talk (Lee et al., 2010). Thus, a first design challenge to developing a synthetic system is establishing well-defined, orthogonal interactions. To address this, we exploited N6-methyladenine (m6A). In contrast to cytosine methylation, which is abundant in animals and typically acts to repress genes (Bernstein et al., 2007), m6A is rarely found in metazoan genomes, and its existence and potential function remain unclear in human cells (Heyn and Esteller, 2015; O’Brown and Greer, 2016). The orthogonal properties of DNA adenine methylation were previously harnessed to develop technology for mapping chromatin-associated proteins in eukaryotic genomes (Kind et al., 2013; van Steensel and Henikoff, 2000). By transplanting this modification into human cells, we hypothesized that we could establish defined interactions for reading and writing, minimize cross-interference with pre-existing chromatin systems, and enable rapid construction of regulatory circuits that encode new and desired functions. Analogously, in natural evolution, it has been proposed that the recent emergence of the phosphotyrosine modification presented similar opportunities for rapidly evolving signal transduction systems with new functions critical to metazoan biology (Lim and Pawson, 2010).
Here we have used m6A as the basis of encoding an additional, synthetic layer of epigenetic information in human cells. We developed synthetic factors that write and read m6A, and used them to build the functional modules required of an epigenetic system. By combining these modules and identifying relevant biochemical parameter spaces using a quantitative model, we created regulatory circuits with self-perpetuating properties that can drive epigenetic behaviors, such as tunable spatial propagation of m6A marks and epigenetic memory of m6A-dependent transcriptional states. Our synthetic system thus provides a platform for programming epigenetic functions in mammalian cells, and examining the core regulatory architectures underpinning epigenetic regulation.
RESULTS
Synthetic initiator enables targeted m6A enrichment at reporter loci
We first sought to develop a synthetic initiator module (synI) capable of establishing m6A marks in a sequence-specific manner at designer reporter loci integrated in the human genome. The general design of the module is a fusion of a Dam (E. coli DNA adenine methyltransferase) “writer” domain, which catalyzes methylation of adenines in GATC motifs, and an engineered zinc finger protein (ZF), which specifically binds a 20-bp synthetic binding sequence (BS) (Figures 2A and S1). We designed two classes of reporters for this study – the Clustered Reporter and Interspersed Reporter (Figures 2A and 3A) – and generated respective reporter cell lines by singly-integrating these constructs into the HEK293FT genome (see Methods, S1). The two reporters feature different arrangements of BS and GATC arrays placed upstream of a promoter driving expression of a destabilized EGFP (d2EGFP). The Clustered Reporter, which features BS motifs directly upstream of a long GATC array (spanning 1.5 kb), was designed to enable methylation measurements at single GATC resolution and accordingly to facilitate studies of spatial dynamics. The Interspersed Reporter, with intermixed BS and GATC motifs, was designed to couple m6A states with transcriptional reporter outputs and accordingly to facilitate temporal studies of transcriptional dynamics and memory.
Figure 2: Engineering a synthetic initiator to establish N6-methyladenine (m6A) DNA modifications at target reporter loci in human cells.
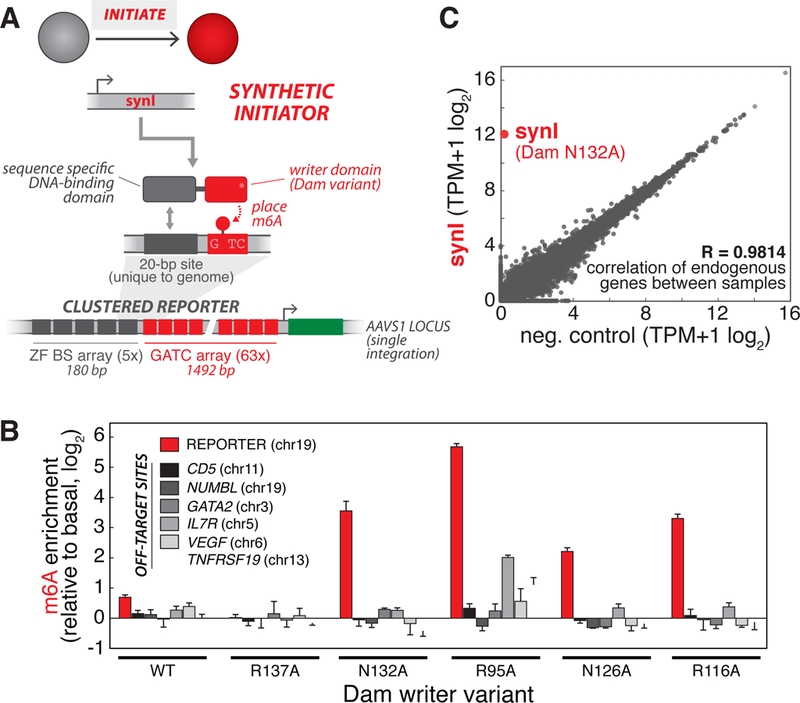
(A) Design of synthetic initiator module (synI). synI is a fusion of a Dam (DNA adenine methyltransferase) “writer” domain and an engineered zinc finger (ZF), which specifically binds a 20-bp synthetic binding sequence (BS). synI enables de novo placement of m6A marks at designer reporters integrated into 293FT cells. For these experiments, we used stable cell lines harboring a singly-integrated Clustered Reporter, with ZF BS and GATC arrays upstream of a pMinCMV driving expression of destabilized GFP (d2EGFP), as the background strain., (B) Screening Dam variants for synI factors that induce sequence-specific enrichment of m6A at target sites. Quantification of m6A enrichment at target reporter (red) and off-target, GATC-containing endogenous loci (grey shades) by transfected synI constructs composed of different Dam mutants. Off-target loci were chosen to represent different chromosomal locations. m6A enrichment is obtained by measuring fraction methylation at a single GATC probe site in the locus of interest using m6A-qPCR, and normalizing to basal methylation induced by the Dam variant not fused to ZF (see Methods, Figure S2). (n=3; error bars, SD)., (C) Expression of synI has minimal effect on the transcriptome. Correlation of transcriptome from RNA-seq measurements for reporter cells transfected with synI vs. empty plasmid. Correlation coefficient of endogenous genes between samples was calculated using log2 transformed expression values. mRNA corresponding to synI is labeled. The data are representative of two biological replicates., See also Figure S2.
Figure 3: Programming m6A-dependent transcriptional states with engineered reader modules.
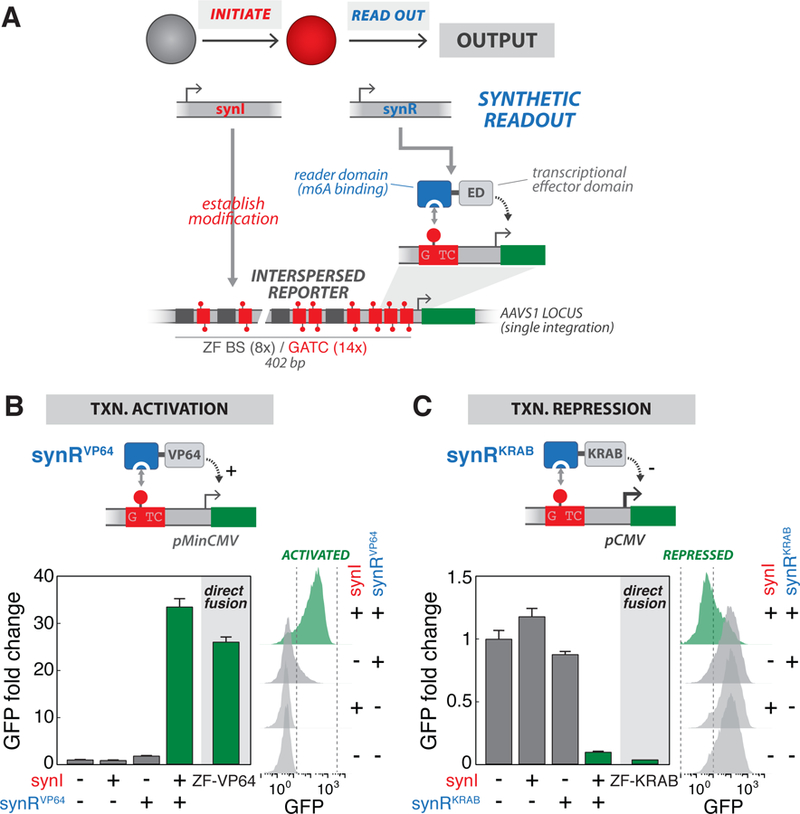
(A) Design of synthetic readout module (synR). synR is a fusion of an m6A “reader” domain (RD, binding domain of DpnI (aa146–254)) and a transcriptional effector domain (ED). m6A marks established by synI are specifically recognized by synR, which in turn regulates transcriptional activity of a reporter gene. For these experiments, we used stable cell lines harboring a singly-integrated Interspersed Reporter, with intermixed ZF BS and GATC sites upstream of a promoter (pMinCMV for activation or pCMV for repression), as the background strains., (B) Programming m6A-mediated transcriptional activation. Top: Schematic of the synRVP64 module, a fusion of DpnI m6A RD and VP64 transcriptional activation domain, which drives activation of a reporter gene via m6A recognition. Bottom: GFP fluorescence intensity, measured by flow cytometry, for cells transfected with indicated combinations of synI and synRVP64 expression constructs, or a direct ZF-VP64 fusion. Bottom left shows fold change of geometric mean GFP intensity normalized to the −/−condition (n=3; error bars, SD); bottom right shows raw flow cytometry distributions., (C) Programming m6A-mediated transcriptional repression. Top: Schematic of the synRKRAB module, a fusion of DpnI m6A RD and KRAB transcriptional repressive domain, which drives repression of a reporter gene via m6A recognition. Bottom: GFP fluorescence intensity, measured by flow cytometry, for cells transfected with indicated combinations of synI and synRKRAB expression constructs, or a direct ZF-KRAB fusion. See also Figure S3.
To identify a synI factor that can preferentially nucleate our reporter locus, we generated a library of Dam variants (DAM*, Figures 2A and S1). Because the wild-type Dam enzyme is known to be highly active, we chose to express the library at low levels (using a minimal CMV promoter, pMinCMV) to minimize global (non-specific) methylation (Figure S2G) (van Steensel and Henikoff, 2000). Additionally, we hypothesized that, by lowering intrinsic Dam activity and DNA affinity through mutations, we could identify a variant whose activity is more highly dependent on ZF binding (McNamara et al., 2002; Smith and Ford, 2007). We generated two expression constructs for each variant: fusion to ZF (synI, targeted) and mCherry (synINT, non-targeted) (Figures S2D and S2E). We then transfected each construct into the Clustered Reporter cell line, and used an adapted m6A-qPCR assay to measure adenine methylation frequency at the reporter (see Methods, Figures S2A-C). We found that single mutations to residues that mediate DNA phosphate group contact, which are known to affect the biochemical activity of Dam (Coffin and Reich, 2009; Horton et al., 2006), generally showed an enrichment in reporter m6A levels. Here, m6A enrichment is defined as targeted methylation (by synI) normalized to basal methylation, induced by the same Dam variant (synINT, non-targeted) (Figures S2D-G). In order to dentify a factor with minimal off-target activity, we screened these synI variants and compared m6A enrichment at the reporter (red) to ‘off-target’ endogenous loci (grey), chosen to represent different chromosomal locations and GATC frequencies (Figure 2B). We selected the ZF—Dam (N132A) fusion, which wil l henceforth be referred to as synI. SynI expression was found to have minimal effect on the 293FT transcriptome (Figures 2C, S2H and S2I), cell cycle and cell viability (Figures S2J and S2K). Together, these results establish a synthetic initiator module capable of sequence-specific placement of m6A marks at reporter loci in human cells.
Programming m6A-dependent transcriptional states with engineered reader modules
Chromatin modifications can modulate gene transcription through several mechanisms, including through reader proteins that recognize specific, or combinations of, marks and recruit transcriptional effector functions (Berger, 2007; Gardner et al., 2011; Kouzarides, 2007). Armed with the ability to nucleate m6A marks, we next sought to engineer reader modules (Haynes and Silver, 2011) that recognize and translate these modifications into defined transcriptional outputs. We designed a synthetic readout module (synR), composed of fusions of an m6A reader domain (RD, binding domain of S. pneumoniae DpnI), which selectively recognizes methylated GATC (Kind et al., 2013; Siwek et al., 2012), and modular transcriptional effector domains (EDs) (Figure 3A). We generated expression constructs for synR modules harboring different Ds: synRVP64 (VP64 activation domain), synRKRAB (KRAB repressive domain), and ynRHP1 (HP1α chromo shadow domain). We then transfected combinations of the constructs into Interspersed Reporter cell lines (harboring either pMinCMV for synR activators or full-length pCMV for synR repressors), and measured GFP reporter output (see Methods). The synR modules induced significant reporter activation or repression, only when expressed in combination with synI; moreover, these transcriptional changes were similar in levels to those induced by a direct transcriptional regulator (direct ZF-ED fusions) (Figures 3B-C and S3B-D). We further confirmed that reporter m6A levels ere enriched only in cells expressing synI (Figure S3A), and that the presence of GATC motifs was required for transcriptional regulation by synI and synR (Figure S3E). This two-module circuit (synI/synR) was found to function on both integrated and episomal reporters (Figures 3B-C and S3B-D) as well as to have minimal and rthogonal effects on the cellular transcriptome (Figure S3F). Taken together, we have developed a synthetic two-module regulatory system that utilizes engineered readers to establish m6A-dependent transcriptional states and logic.
To increase the versatility of this synthetic gene regulatory system, we next sought to develop a version in which synI activity could be readily directed to desired sequences without the need to redesign its DNA targeting domain. We created a CRISPR-guided version of the initiator by fusing the selected Dam (N132A) variant to the S. pyogenes dCas9 protein (synIdCas9) (Figures S1 and S3G). When expressed in combination with single gRNAs targeting various locations in the BS array of the Clustered Reporter, we found that synIdCas9 was capable of preferentially enriching m6A levels at our reporter for certain gRNAs (Figure S3G). Moreover, when combined with ynR, two-module circuits based on synIdCas9 (in place of ZF-based synI) were also able to drive transcriptional regulation of reporters (Figure S3H).
Finally, we wondered how our reporter constructs, which we have shown can be artificially modified and regulated by our synthetic m6A system, generally compare with naturally occurring GATC distributions in the human genome. This might inform future applications or improvements of our regulatory system for arbitrary genomic contexts, where GATC distributions cannot be precisely controlled. Based on a genome-wide bioinformatics analysis, we found that GATC sites are indeed naturally present in most human promoter regions, with a median of ~3–4 motifs (Figure S3I and S3J). Moreover, when we tested the performance of our synthetic system for a set of reporters harboring different numbers of ZF BS and GATC motifs, we found that it is possible to regulate reporters with GATC frequencies equivalent to those of natural promoter frequencies, albeit at more modest levels (Figure S3K).
Engineering spatial propagation with read-write regulatory circuits
Cells have mechanisms for propagating chromatin states in space and time, mechanisms that mediate the “spreading” of natural chromatin domains associated with silent transcription, and the faithful transmission of these altered domains across cell division. It is thought that a core regulatory feature driving these self-perpetuating mechanisms is the “read-write” system. (Al-Sady et al., 2013). Inspired by this, we focused first on developing a minimal read-write (RW) module to construct regulatory circuits that drive spatial propagation of m6A. To enable these studies, we developed a small molecule inducible initiator (synIIND), which uses abscisic acid (ABA)-induced dimerization to enable temporal control over initiation of m6A states (Liang et al., 2011) (Figure S4). We then designed a synthetic RW module (synRW), which is a fusion of m6A RD (binding domain of DpnI) and Dam writer domain (DAM*) (Figure 4A).
Figure 4: Engineering spatial propagation of m6A with three-module read-write circuits.
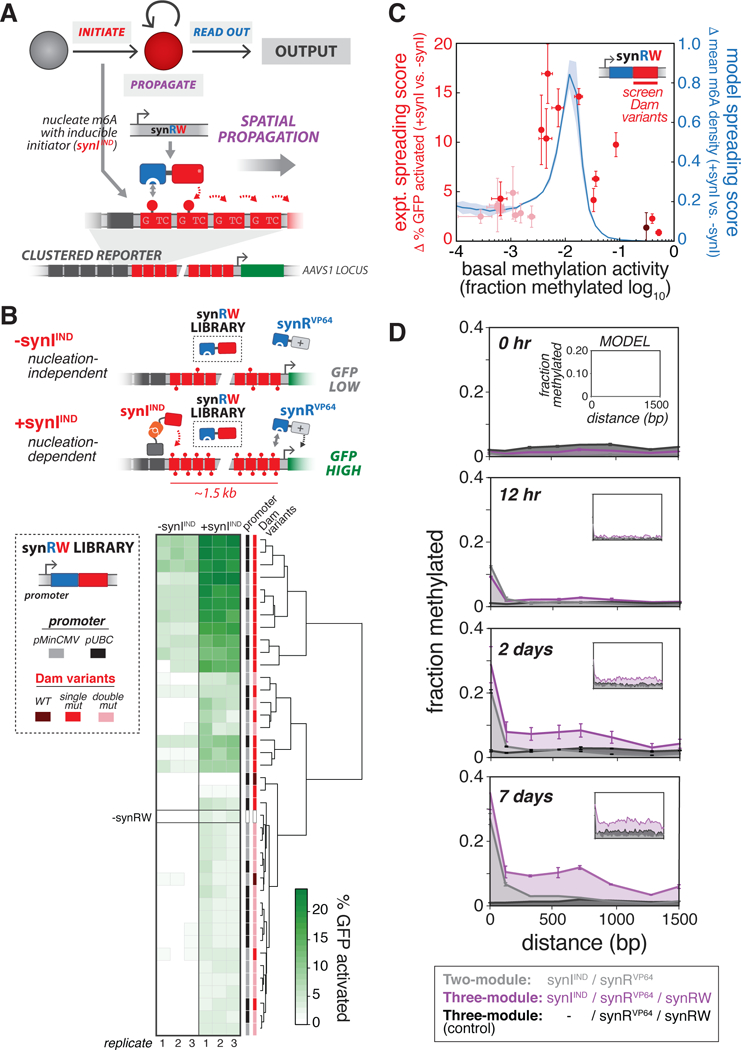
(A) Design of synthetic read-write module (synRW) for propagating m6A modifications over a domain. synRW is a fusion of DpnI m6A RD and a Dam writer domain (Dam variant). synRW recognizes pre-existing m6A marks and catalyzes methylation of nearby GATC motifs, creating local reinforcement and spreading of m6A to larger domains. For these experiments, we used Clustered Reporter cell lines, with pMinCMV driving expression of d2EGFP, as the background strain., (B) Screening synRW variants for nucleation-dependent spreading behavior. Top: Schematic of the “spatial propagation screen” design. A library of synRW variants was screened for the ability to activate a distal reporter gene in cells with synthetic initiator (+synIIND, GFP High), but not in cells lacking initiator (-synIIND, GFP Low). Bottom left: The synRW library featuring different promoter strengths and Dam mutant writers. Each member of the library was individually transfected into Clustered Reporter lines stably expressing synRVP64, either with (+synIIND) or without (-synIIND) stable expression of the inducible initiator. Bottom right: Screen results. Heat map of % GFP activated cells, quantified by flow cytometry 4 days following transfection of synRW (with continuous 200 μM ABA induction of synIIND). Hierarchical clustering analysis based on similarity in % GFP activated cells is shown. (C)Summary and modeling of spatial propagation screen. We defined quantitative metrics to score propagation propensity for synRW library members: “expt. spreading score” (red; n=3, error bars, SD) is the difference between % GFP activated cells with and without synIIND measured from the experimental screen; “model spreading score” (blue; mean ± SD) is the difference between model-computed m6A density at promoter-proximal sites with and without synI (see Methods, Figure S5). Shades of red correspond to Dam mutant writers: WT, single mutants, double mutants (dark to light, respectively)., (D) m6A profiles measured across the GATC array and over time for cells stably expressing the three-module “propagation circuit” (purple) or circuits lacking either synRW (grey) or synIIND (black). Cells were continuously induced with 200 μM ABA. Model simulations are shown in insets (with bZF =10, bDpnI =100, see Methods, Figure S5). (n=3; error bars, SD)., See also Figures S4-S6.
Our objective was to identify RW circuit designs capable of driving spatial propagation. Specifically, we sought to identify synRW variants that, when combined with synIIND and synR modules, could spread m6A from a nucleation site. To do this, we generated a synRW variant library (varying synRW expression levels and Dam mutants), and devised a simple, phenotypic screen for spatial propagation behavior (Figure 4B). For a full description of the library, screen, and analysis, see Methods. Briefly, the screen leverages the Clustered Reporter’s long GATC domain (with 20 bp inter-GATC spacing) separating the nucleation site from the reporter gene. Devoid of a propagation mechanism, reporter cells stably expressing the two-module circuit (synIIND/synRVP64) re not activated (Figure S6A). As a result, the synRW library can be screened in these cells for candidates that produce reporter activation (+synIIND, Figure 4B), as well as in cells lacking initiator to screen out spurious cases for which downstream reporter activation is induced independent of m6A nucleation (-synIIND, Figure 4B). Variants emerging from this screen would, in principle, represent potential candidates for the RW module of three-module regulatory circuits (synIIND/synRVP64/synRW) that drive inducible spatial propagation leading to reporter activation.
Clustering analysis of our screen results based on GFP expression revealed a strong clustering by synRW Dam mutants (Figure 4B). Specifically, we observed a relationship between methylation activity of synRW and propagation, quantified by a “spreading score” metric we developed to score the phenotypic outcomes of the screen (Figure 4C, Methods). This suggested to us that intermediate writer activity (i.e. intermediate levels of Dam methylation activity) may be an important design criterion for the synRW module to produce these propagation phenotypes. To explore the generality of this result, we turned to quantitative modeling, adapting a previously described model of chromatin spreading dynamics (Hodges and Crabtree, 2012) to capture the essential features of our system (see Methods, Figure S5). We used our model to simulate and interrogate how synRW properties, such as methylation writer activity, affect spatial m6A profiles; this revealed a similar relationship to that observed from our experimental screen (Figure 4C, blue line).
Supported by our screen and simulation results, we selected a high-scoring, intermediate-activity synRW (pUBC: DpnI—Dam (R95A)), and integrated this construct to generate cells stably expressing a full read-write “propagation circuit” synIIND/synRVP64/synRW). We then tested the circuit by triggering m6A nucleation and easuring m6A profiles across the domain over time. Cells expressing our circuit exhibited a growing m6A domain over time, in contrast to a control circuit lacking synRW (Figures 4D and S6C). Moreover, and as desired, robust propagation was dependent on m6A nucleation, as cells lacking initiator (-synIIND) did not exhibit propagating m6A domains (Figure S6C). Interestingly, and consistent with recent studies on the dynamics of heterochromatin formation in mammalian cells (Hathaway et al., 2012), establishment of this m6A domain occurred over relatively slow time-scales (~days). Consistent with our previous observations for synI, expression of synRW has no adverse effects on the cell cycle and viability (Figures S2J and S2K), and led to only modest increases in methylation at chosen off-target, endogenous loci (Figure S2L). These results demonstrate the development of a three-module regulatory circuit that can drive spatial m6A propagation in order to synthetically modify a domain and regionally control the expression of genes.
We next explored how different compositions and expression levels of the modular components making up the propagation circuit affect spreading dynamics. A synI “trigger” and constitutive synRW were both required for m6A propagation (Figures 4D and S6C). However, the synR module, while necessary for screening and coupling m6A activity to transcriptional output states, is presumably not essential to the circuit’s ability to drive m6A spreading (acting only to compete with synRW for binding sites). To test this, we generated stable cell lines with a propagation circuit lacking synR and measured m6A profiles after triggering nucleation. Indeed, these cells also exhibited propagating m6A domains with similar profiles to those of the full three-module circuit, albeit with slightly elevated overall m6A levels across the domain by day 7; these elevated m6A profiles were also observed for control circuits lacking synI or synRW (Figure S6D). These results suggest that using competitors for methylated GATC substrates – i.e. “futile” reader modules like synR – may provide strategies for tuning methylation levels and suppressing basal writing at desired time points, presumably through altering the kinetics of propagation.
Clustering analysis of our screen results revealed no obvious correlation or sensitivity in spreading behavior for the two different promoters used to drive synRW expression (pMinCMV and pUBC) (Figure 4B). We reasoned this was likely because the transfection conditions used in screening produced saturating concentrations of synRW for reactions on a limited number of available methylated GATC sites. To further investigate how synRW levels may affect propagation dynamics, we used our screen-selected synRW variant (pUBC: DpnI—Dam (R95A)) to perform a dosing experiment, in which we varied the concentration of synRW plasmid transfected into Clustered Reporter cells (with vs. without synIIND) and quantified difference in methylation at the promoter proximal end of the GATC array (Figure S6E). By varying plasmid concentrations, we observed a dose-dependent relationship in propagation outcomes as a function of synRW levels. The dose response curve varied in time, shifting to lower synRW threshold concentrations by day 4 (the saturation peak was well below the concentration used in the screen (100 ng), suggesting that the conditions used in the original screen were likely producing saturating synRW for both promoters).
Next we investigated the behavior of the propagation circuit for different configurations of the reporter. First we tested the effect of varying GATC density, which may have implications for potential applications of our synthetic epigenetic regulatory system in natural genomic contexts where GATC spatial frequencies cannot be controlled. We generated three Clustered Reporter variant lines with inter-GATC spacers of increasing length (50 bp, 212 bp, 800 bp) (Figure S6F); our canonical reporter uses 20 bp spacers, the shortest possible while still allowing individual GATC measurements with m6A-qPCR. m6A profiles induced by our propagation circuit (with transfected synRW) showed a monotonic decrease in the capacity for spreading from 50 bp to 800 bp spacers, where we observed complete extinction.
Finally, inspired by recent observations that H3K9me3 profiles grow symmetrically from a nucleation site (Hathaway et al., 2012), we wondered whether our synthetic read-write circuit could also drive symmetric propagation patterns. To test this, we exploited our CRISPR-guided initiator (synIdCas9), which could be directed to the center of the reporter GATC array with a single gRNA to induce m6A enrichment (Figures 5A and 5B). Upon triggering a central nucleation site, cells expressing the synRW module exhibited bi-directional enrichment in m6A, while control circuits did not (Figure 5C).
Figure 5: Synthetic propagation circuits induce bi-directional spreading.
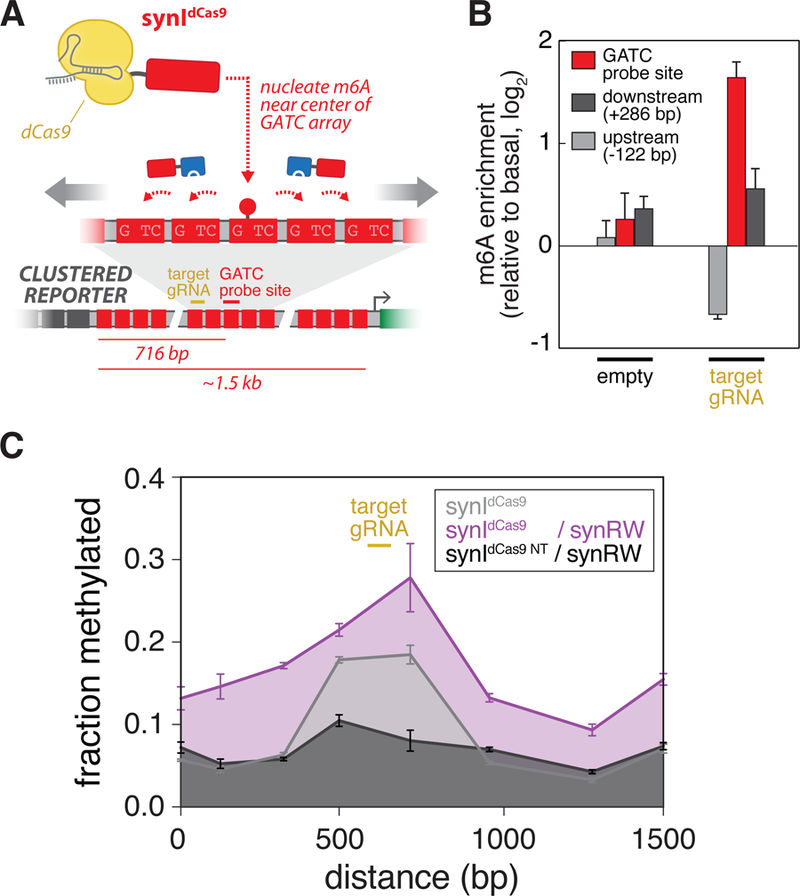
(A) Using a CRISPR-guided synthetic initiator (synIdCas9) to investigate m6A propagation symmetry. synIdCas9 is recruited to the center of the Clustered Reporter GATC array to establish a nucleation site and to observe synRW-induced m6A propagation away from the site (see Figures S3G, S3H).(B) synIdCas9 enables targeted m6A enrichment near the center of the GATC reporter array. Quantification of m6A enrichment at a central GATC probe site (red; 716-bp downstream from ZF array) and sites located downstream (dark grey; +286-bp) and upstream (light grey; −122-bp) of probe site following transfection of Clustered Reporter lines with synIdCas9 and single gRNA (or empty) cassette. m6A enrichment is obtained as previously described, normalizing to basal methylation induced by synIdCas9 alone.(n=3; error bars, SD). (C) m6A profiles measured across the GATC array for Clustered Reporter cell lines transfected with: synIdCas9 and target gRNA (grey); synIdCas9, target gRNA, and synRW (purple); synIdCas9, empty gRNA, and synRW (black). (n=3; error bars, SD). See also Figure S6.
Engineering epigenetic memory with read-write regulatory circuits
In principle, RW positive feedback loops could also provide a mechanism to establish epigenetic memory. Thus, we wondered whether our propagation circuit could mediate the maintenance and transmission of an induced transcriptional state through cell division, as compared with the transient state induced by a direct transcription factor (TF). To test this, we first followed the response of Interspersed Reporter cells stably expressing either an inducible ZF-VP64IND or the m6A two-module circuit (synIIND/synRVP64) to a transient pulse of ABA (Figure 6A). Both exhibited rapid reporter activation followed by deactivation upon removal of ABA; however, the m6A system exhibited a delay in the time-scale of deactivation (Figure 6B). In contrast to direct TF recruitment, the decay dynamics of the two-module circuit depends on the lifetime of the modified m6A state, which in the absence of an active removal mechanism is largely governed by passive dilution through semiconservative replication. We tested this by blocking DNA replication using Aphidicolin (APC). As expected, without APC, m6A levels roughly halved every two days with a concomitant loss of GFP activated cells, whereas blocking DNA replication led to persistence of m6A and corresponding GFP states (Figures 6C and S7A).
Figure 6: Engineering epigenetic memory with three-module read-write circuits.
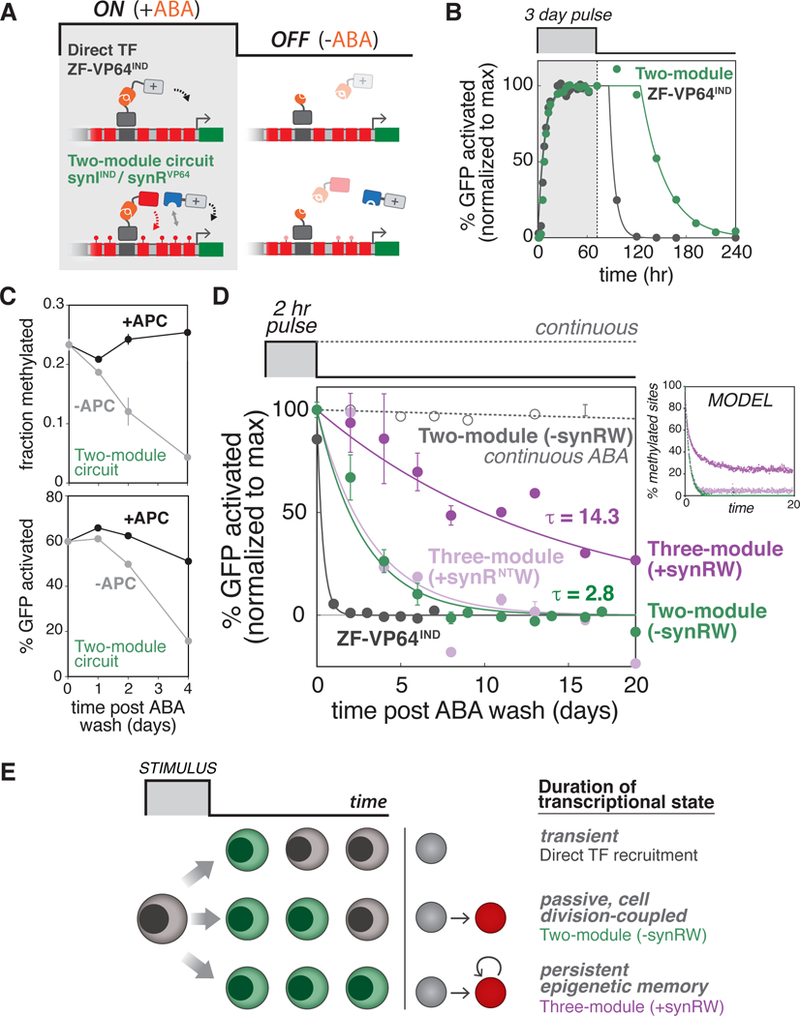
(A) Schematic depicting ON (+ABA) and OFF (-ABA) states with direct TF recruitment (ZF-VP64IND, top) and m6A two-module regulatory circuit (synIIND/synRVP64, bottom). Addition of ABA mediates dimerization to form an active ZF-VP64IND (top) or synIIND initiator (bottom). Removal of ABA allows analysis of deactivation dynamics and epigenetic memory. For these experiments, we used Interspersed Reporter cell lines, with pMinCMV driving expression of d2EGFP, as the background strain., (B) Activation/deactivation dynamics for ZF-VP64IND (grey) vs. two-module m6A circuit (green) in response to a transient (3 day) ABA pulse. The percentage of GFP activated cells was quantified by flow cytometry (see Figure S4E). Dots are data points (n=3; error bars, SD); lines represent sigmoidal curve fits to ON phase and exponential fits with time delay to OFF phase., (C) Maintenance of m6A (top) and transcriptional state (bottom) by inhibiting DNA replication. Cells stably expressing m6A two-module circuit were induced with a 3 day ABA pulse, then ABA was washed out and cells were maintained in media with or without 5 μg/mL APC. Fraction methylation and percentage of GFP activated cells were quantified at the indicated time points following ABA washout. (n = 3; error bars, SD)., (D) Three-module propagation circuit induces epigenetic memory. Deactivation dynamics following transient (2 hr) ABA pulse for cells stably expressing: three-module propagation circuit (purple), the circuit lacking synRW (green), and the circuit with a reader-defective synRNTW (light purple), or cells stably expressing ZF-VP64IND (grey). Also plotted are cells stably expressing the two-module circuit continuously induced with 200 μM ABA (grey, dotted line). The percentage of GFP activated cells was quantified by flow cytometry. Dots are data points (n=3; error bars, SD); lines represent exponential fits to OFF phase. To the right are model simulation results of m6A maintenance for the different m6A circuit components (same color code). The percentage of methylated GATC sites (out of 14 available in the Interspersed Reporter) are plotted as a function of time (see Methods)., (E) Proposed model for engineering transcriptional states with varying durations and epigenetic memory using synthetic m6A operations. Direct TF recruitment induces transient transcriptional states (top). Depositing m6A marks facilitates passive, cell-division coupled persistence of the induced transcriptional state (middle). Regulatory circuits utilizing read-write positive feedback can induce persistent epigenetic memory (bottom)., See also Figures S4 and S7.
We next tested whether our three-module propagation circuit could transform this ‘passive’, cell division-coupled state into a more durable memory. The time-scale of deactivation of the two-module circuit (without RW) is approximately the same for all induction pulse lengths tested (Figure S4F), which is once again consistent with a passive dilution model governed by cell division. Thus, for our inducing signal, we selected a short (2 hr) ABA pulse because it is sufficient to induce the activated state in cells (longer pulses led to only slight increases in fraction of activated cells), and tested whether our circuit could create a memory of this transient signal. We subjected cells to this short pulse of ABA and tracked the percentage of activated cells following removal of ABA for a total of 20 days (Figure 6D). Our propagation circuit led to higher levels of maintenance of GFP activated cells (dark purple), extending the decay time significantly beyond ZF-VP64IND (grey) and the two-module circuit lacking synRW (green), which as fully lost by 8 days. Notably, with the three-module RW circuit, approximately half of the cellular population remained GFP-positive for ~10 generations. To ensure this was not simply the result of a Dam dosage effect (by addition of synRW module), but indeed dependent on the RW mechanism, we also tested a control circuit in which the synRW RD was replaced with mCherry (synRNTW). This abolished memory, returning the deactivation phase to two-module circuit levels (-synRW, light purple). Model simulations of this experiment, using the same model parameter set from the spatial dynamics simulations, effectively reproduced these dynamics (Figures 6D and S7E, Methods). The memory induced by our circuit resembled GFP maintenance profiles resulting from blocking DNA replication with APC, and as before was dependent on m6A initiation by synIIND (Figures S7B and S7C). Additionally, cells stably expressing the propagation circuit were actively dividing and transmitting the epigenetic state to progeny, not simply slowing/halting cell growth (Figure S7D).
Taken together, we have demonstrated that minimal synthetic circuits utilizing RW positive feedback can establish epigenetic memory of transient signals. Additionally, by combining different modular operations, we found that the duration of a transcriptional state can be tuned from: (1) a highly transient state regulated by direct TF recruitment, to (2) a passive, cell division-coupled persistent state regulated by circuits that deposit and recognize m6A, and finally to (3) longer lasting epigenetic memory regulated by circuits that additionally utilize RW modules (Figure 6E).
DISCUSSION
Chromatin is a substrate for a complex assortment of chemical modifications made to DNA and histone proteins. These dynamic modifications can influence genome structure and orchestrate the recruitment of effector protein complexes, thereby playing essential roles in regulating gene transcription and other biological processes (Berger, 2007; Bernstein et al., 2007; Feinberg, 2007; Kouzarides, 2007). Additionally, certain modifications have been implicated as carriers of epigenetic information, and significant efforts have been focused on understanding how these marks contribute to mechanisms for transmission of heritable transcriptional states independent of DNA sequence (Bonasio et al., 2010; Moazed, 2011). Here we used a synthetic biology approach to construct an epigenetic regulatory system in mammalian cells from first principles. Specifically, we developed a suite of synthetic factors that execute reading and writing operations with the orthogonal DNA chemical modification m6A, and assembled them into regulatory architectures believed to be core engines for driving epigenetic functions. Importantly, by creating variants of these factors that capture a range of biochemical properties and then combining them into circuits both experimentally and computationally, we were able to identify operating regimes for an array of epigenetic behaviors.
The first component of our system (synI) is a factor that enables sequence-specific establishment of m6A. We found that to achieve targeted m6A enrichment at artificial reporter loci, it was necessary to reduce the intrinsic activity of WT Dam by mutating residues that interact with the DNA phosphate group; similar variants (e.g. K9A) were also found to be effective in the DamID/DamIP system (Xiao and Moore, 2011). These results suggest that DNA specificity of WT Dam, which likely has strong evolutionary preference for GATC to support critical functions such as DNA repair and immunity, can be reprogrammed by protein engineering efforts aimed at first reducing binding energy. Our selected Dam variant was shown to function with two different DNA-targeting technologies (ZFs and CRISPR/dCas9), and the CRISPR-guided version of synI in particular may open up future possibilities for artificially modifying and regulating endogenous loci. However, this will require assessing the genome-wide specificities (and off-target profiles) of synI, a critical and yet unresolved issue, especially for the CRISPR-guided version which showed higher propensity for m6A enrichment at our limited panel of off-target loci (Figure S3G). More broadly, the efficacy of our system at endogenous loci would need to be established before it can be translated into an epigenome and cellular engineering platform, including characterizing its performance at more complex genomic contexts (nucleosome positioning, barrier elements, etc.).
Epigenome editing tools that allow modifications to be induced at specific genomic sites have recently emerged to help understand causal links between chromatin modifications and gene regulation, and to explore therapeutic strategies for a number of diseases (Braun et al., 2017; de Groote et al., 2012; Hilton et al., 2015; Maeder et al., 2013; Mendenhall et al., 2013; Park et al., 2016; Thakore et al., 2016). Among these, there has been significant previous effort dedicated to developing site-specific DNA methylation tools, typically utilizing bacterial cytosine methyltransferases to enable CpG methylation and heritable gene silencing (Nomura and Barbas, 2007; Smith and Ford, 2007; Xiong et al., 2017). Instead, our approach was to leverage N6-methyladenine (m6A). While m6A is a well-established and abundant DNA modification in bacteria, much less is known about its existence and role in eukaryotic chromosomes. With the advent of sensitive detection techniques, studies have recently emerged reporting the discovery of DNA m6A in the genomes of certain eukaryotes, including C. reinhardtii, D. melanogaster, and C. elegans, and putative roles in transcriptional regulation, nucleosome positioning, embryogenesis, and epigenetic inheritance have been suggested (Fu et al., 2015; Greer et al., 2015; Zhang et al., 2015). Its presence in mammalian genomes has been more elusive and potentially controversial. Though recent reports claimed to observe trace amounts of DNA m6A in mouse embryonic stem cells and human cells (Wu et al., 2016; Xiao et al., 2018), validation studies using ultrasensitive UHPLC-MS methods find no evidence for it, potentially attributing its observation to bacterial DNA or mammalian mRNA sources (Schiffers et al., 2017).
We next combined m6A deposition using synI with recruitment of transcriptional regulators using engineered synR readers. This minimal two-module circuit was sufficient to establish m6A-dependent transcriptional states. Moreover, the modular architecture of synR provided flexibility to encode different logical outputs depending on the fusion domain. In principle, other types of regulatory and effector domains could be recruited to m6A-modified domains using this mechanism, providing a platform for combinatorial or enhanced recruitment of regulatory function through both genomic and epigenomic signatures.
An epigenetic mark in the strict sense is one that can persist after removal of an initial stimulus. The “read-write” motif is believed to be a core regulatory mechanism driving this self-amplification and contributing to the epigenetic inheritance of diverse chromatin modifications, but direct investigations of the RW principle have been difficult (Moazed, 2011). Using this as a blueprint, we engineered a synthetic “read-write” module, and tested whether it provides a positive feedback mechanism capable of propagating and facilitating heritable maintenance of m6A states. Critically, our synthetic RW circuits displayed self-perpetuating behaviors and were able to program epigenetic functions de novo that do not rely on potentially confounding, endogenous chromatin mechanisms. Moreover, the behaviors we programmed exhibited hallmarks of natural epigenetic phenomena. For example, m6A distributions in our spatial propagation studies resembled those of propagating H3K9 methylation in HP1-induced heterochromatin, which underlies gene silencing and position-effect variegation; and both were shown to propagate symmetrically away from a nucleation site (Hathaway et al., 2012). Additionally, our synthetic circuits were capable of inducing epigenetic memory, enabling transcriptional states to be heritably maintained for >10 days in the majority of cells upon removal of an inducing signal.
Our synthetic approach enables investigation into how epigenetic behaviors are regulated by different configurations of the underlying molecular modules. For example, spatial propagation and epigenetic memory were both strictly dependent on the initiation module (synI). Furthermore, we showed how spatial propagation could, in principle, be tuned by adjusting synRW levels through a pseudo dose-response curve (within a range where available methylated GATC sites are presumably not limiting). Finally, though propagation did not strictly depend on the synR module, this additional factor could be used to help set/lower methylation levels, including basal levels. It is interesting to speculate that “futile” reader domains, such as synR, could be operating to compete with and accordingly adjust the effective concentration of synRW via its dose-response curve to shape evolving m6A profiles.
By recombining variants of a reader and writer domain, we were able to generate synthetic modules that reconstitute the functional requirements of an epigenetic regulatory system. The modular simplicity of this design may highlight why this core regulatory solution seems to appear in different cellular contexts and for diverse chemical modifications, ranging from DNA methylation to histone post-translational modifications (PTMs) and protein phosphorylation. The modularity of the system may also provide a facile means for building additional layers of regulation atop this core circuitry. Indeed, it has recently been suggested that positive feedback based exclusively on RW mechanisms may constitute weaker forms of epigenetic inheritance (Audergon et al., 2015; Ragunathan et al., 2015), and that additional heritable feedback loops may be necessary to increase stability of epigenetic states. In support of this, the epigenetic memory we engineered based on minimal synthetic RW circuits was not as stable or long-lived as some of the memory observed in a recent study in which silencers were dynamically recruited to reporters (Bintu et al., 2016). Additional mechanisms for enhancing epigenetic memory/inheritance have been proposed –including sequence-specific elements (Laprell et al., 2017; Wang and Moazed, 2017), chromatin remodeling (Kundu et al., 2007), cooperativity between modifications (Rudner et al., 2005; Rusche et al., 2002) – and a recent intriguing example showed that positive feedback involving siRNA amplification can directly couple to and strengthen (RW) histone PTM feedback loops (Yu et al., 2018). In principle, additional regulatory elements, connections, and feedback loops could be constructed within our synthetic system to explore their functional roles.
Early pioneering work in synthetic biology illustrated the power of using minimal synthetic genetic circuits to explore cellular regulation and engineer emergent properties in bacteria, such as cellular memory and oscillations (Elowitz and Leibler, 2000; Gardner et al., 2000). As interest in eukaryotic and especially mammalian systems has grown recently, it is becoming increasingly important to develop foundational tools and frameworks for manipulating the different organizational layers of mammalian gene regulation. Our work provides an initial set of molecular building blocks and a circuit engineering framework to aid in connecting the individual regulatory components making up complex systems like chromatin to its salient, higher-order properties like epigenetic memory and spreading. Much like synthetic circuits in bacteria provided strong evidence for quantitative control mechanisms underlying switch-like and oscillatory behaviors in natural systems, our work offers insights into the long-standing “read-write” propagation hypothesis in chromatin biology. In the future, our synthetic platform may also be adapted to provide new tools/systems for cell and epigenome engineering, such as for manipulating genome architecture, molecular recording through epigenomic changes, and new forms of dynamic gene expression control. Finally, we envision our system could be expanded to incorporate other molecular operations, such as erasing, and chemical modifications with which to synthetically encode and regulate additional layers of epigenetic information in cells.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Abscisic acid | Sigma Aldrich | Cat# A1049–250MG |
| Aphidicolin from Nigrospora sphaerica | Sigma Aldrich | Cat# A0781–5MG |
| Polyethylenimine (PEI) | Polysciences | Cat# 23966–1 |
| FxCycle Far Red | Thermo Fisher Scientific | Cat# F10348 |
| RNaseA | Qiagen | Cat# 19101 |
| Hygromycin B | Gibco | Cat# 10–687-010 |
| Puromycin Dihydrochloride | Gibco | Cat# A1113803 |
| Blasticidin S HCl | Gibco | Cat# A1113903 |
| LightCycler® 480 SYBR Green I Master | Roche | Cat# 4887352001 |
| DpnII (1,000 units; 10,000 units/ml) | NEB | Cat# R0543S |
| Critical Commercial Assays | ||
| Genomic DNA extraction: DNeasy Tissue kit | Qiagen | Cat# 69506 |
| Click-iT Plus EdU Pacific Blue Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat# C10636 |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74134 |
| QIAshredder | Qiagen | Cat# 79656 |
| CellTrace Far Red Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34564 |
| Deposited Data | ||
| Raw RNA sequencing data | This study | SRA: PRJNA488081 |
| Human genome sequence | UCSC Genome Browser | Version hg38; https://genome.ucsc.edu / |
| Experimental Models: Cell Lines | ||
| HEK293FT | Thermo Fisher Scientific | Cat# R70007 |
| Human: Interspersed Reporter (pMinCMV) | This study | MP153 |
| Human: Clustered Reporter. (pMinCMV) | This study | MP175 |
| Human: Intrs. pMinCMV Reporter + synRVP64 | This study | MP243 |
| Human: Intrs. pMinCMV Reporter + synIIND + synRVP64 | This study | MP244 |
| Human: Clust. pMinCMV Reporter + synRVP64 | This study | MP252 |
| Human: Clust. pMinCMV Reporter + synIIND + synRVP64 | This study | MP253 |
| Human: Intrs. pMinCMV Reporter + synRVP64 + synRW(pUBC-DpnI-Dam(R95A)) |
This study | MP422 |
| Human: Intrs. pMinCMV Reporter + synIIND + synRVP64 + synRW(pUBC-DpnI-Dam(R95A)) |
This study | MP423 |
| Human: Clust. pMinCMV Reporter + synRVP64 + synRW(pUBC-DpnI-Dam(R95A)) |
This study | MP427 |
| Human: Clust. pMinCMV Reporter + synIIND + synRVP64 + synRW(pUBC-DpnI-Dam(R95A)) |
This study | MP428 |
| Additional derived cell lines and further information | This study | Table S2 |
| Oligonucleotides | ||
| no GATC reference (GFP) forward qPCR primer GTGAACCGCATCGAGCTGAAG |
This study | N/A |
| no GATC reference (GFP) reverse qPCR primer TGTTGCCGTCCTCCTTGAAGTC |
This study | N/A |
| GATC (20bp from ZF BS) forward qPCR primer TAAAGGCTTACTGAGCACTA |
This study | N/A |
| GATC (20bp from ZF BS) reverse qPCR primer TGTGATTCAGAGACAACTTC |
This study | N/A |
| GATC (140bp from ZF BS) forward qPCR primer AATCGTTGCGTAATCTACAA |
This study | N/A |
| GATC (140bp from ZF BS) reverse qPCR primer TTGCGAAAGTTGGAGAAATA |
This study | N/A |
| Additional oligonucleotide pairs and qPCR conditions | This study | Table S3 |
| Recombinant DNA | ||
| pUBC ZF-VP64 | This study | pMP258 |
| Inters. (8XZFBS 14XGATC)-pMinCMV-GFPd2-RbGpA pGK-PuroR-T2A-mCh-BGHpA AAVS1 donor |
This study | pMP472 |
| Clust. (5XZFBS 63XGATC)-pMinCMV-GFPd2-RbGpA pGK-PuroR-T2A-mCh-BGHpA AAVS1 donor |
This study | pMP498 |
| pMinCMV-NLS-ABI1cs-ZF-NLS-P2A-Dam(N132A)- PYL1cs-HA pGK-BlastR |
This study | pMP597 |
| pUBC-DpnI(aa146–254)-VP64-V5 pGK-ZeoR | This study | pMP650 |
| pUBC-DpnI(aa146–254)-3XFLAG-Dam(R95A) pGK-HygroR | This study | pMP926 |
| pUBC-mCh-3XFLAG-Dam(R95A) pGK-HygroR | This study | pMP967 |
| gRNA_AAVS1-T2 plasmid | (Mali et al., 2013) | Addgene: #41820 |
| VP12 humanSpCas9-Hf1 plasmid | (Kleinstiver et al.,2016) | Addgene: #72247 |
| Additional recombinant DNA constructs and further information |
This study | Table S1 |
| Software and Algorithms | ||
| FlowJo v8 | FlowJo, LLC. | https://www.flowjo.com/solutions/flowjo/downloads |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| TopHat | (Trapnell et al., 2009) | http://ccb.jhu.edu/software/tophat/index.shtml |
| featureCounts | (Liao et al., 2014) | http://bioinf.wehi.edu.au/featureCounts/ |
| EMBOSS-dreg | (Rice et al., 2000) | http://www.bioinformatics.nl/cgibin/emboss/dreg |
| Other | ||
| Attune NxT Flow Cytomete | Thermo Fisher Scientific |
Attune NxT |
| LightCycler 480 Instrument II | Roche | Cat# 05015243001 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ahmad Khalil (khalil@bu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
The background cell line for all experiments in this study was 293FT cell line (Thermo Fisher Scientific). Cells were cultured in Dulbecco’s modified Eagle’s medium with L-Glutamine, 4.5 g/L Glucose and Sodium Pyruvate (DMEM, Thermo Fisher Scientific) supplemented with 10% Tet-system approved fetal bovine serum (FBS, Clontech), 1% GlutaMAX supplement (Thermo Fisher Scientific), 1% MEM Non-Essential Amino Acids (NEAA, Thermo Fisher Scientific) solution and 1% penicillin-streptomycin (Thermo Fisher Scientific). Cells were split every 3 days, and maintained at 37°C and 5% CO2 in a humidified incubator.
Cell line generation
Cell lines used in this study are listed in Table S2, and were generated by genome-integrating constructs into the 293FT cell line, a fast-growing variant of the human female embryonic kidney 293 cell line stably expressing the SV40 large T antigen (Thermo Fisher Cat# R70007). The 293FT cell line was authenticated by morphology check with microscope.
Reporter lines were generated by site-specific integration of reporter constructs (Figure S1) using CRISPR/Cas9-mediated homologous recombination into the AAVS1 (PPP1R2C) locus as follows: 60,000 cells were plated in a 48-well plate and co-transfected the following day with 70 ng of gRNA_AAVS1-T2 plasmid (Addgene 41820), 70 ng of VP12 humanSpCas9-Hf1 plasmid (Addgene 72247), and 175 ng of donor reporter plasmid using PEI. Donor reporter plasmids contain flanking arms homologous to the AAVS1 locus, a puromycin resistance cassette, and constitutive mCherry expression (Figure S1). After transfection, cells were cultured in 2 µg/mL puromycin selection for at least 2 weeks with splitting 1:10 every 3 days, then monoclonal populations for each reporter cell line were isolated by limiting dilution in 96-well plates.
All other stable lines were generated by lentiviral integration of indicated constructs (encoding synI, synR, synRW modules and/or respective controls) into specific reporter lines. Lentivirus was produced by PEI co-transfection of 293FT cells with the donor plasmid, along with packaging vectors pCMVR8.74 (Addgene 22036), pAdVAntage (Promega), and pMD2.G (Addgene 12259). Virus was harvested with centrifugation (300g, 5 min) and was added/incubated into specific reporter lines for three days, followed by selection in appropriate selection media: blasticidin (10 µg/ml), zeocin (100 µg/ml), and/or hygromycin (200 µg/ml).
METHOD DETAILS
Cloning and plasmid construction
Plasmid constructs used in this study are listed in Table S1 and their designs described in Figure S1. All constructs were constructed using standard molecular biology techniques and Gibson isothermal assembly. Donor plasmids for CRISPR/Cas9-induced reporter knock-in were constructed by PCR and subsequent Gibson assembly of components into the pCAGEN mammalian expression vector (Addgene 1160), digested with SalI/HindIII. Donor plasmids for lentiviral integration were constructed by PCR and subsequent Gibson assembly of components into pFUGW (Addgene 14883), digested with PacI/XhoI. During cloning, plasmids were transformed and prepped in E. coli TOP10 (Thermo Fisher Scientific). After sequence-verification, final reporter vectors were transformed and propagated in the dam-/dcm-strain, E. coli K12 ER2925 (NEB).
Transfection
For all transient transfection experiments, plasmid constructs (Figure S1) were transfected into indicated stable cell lines using polyethylenimine (PEI, 7.5 mM linear PEI stock, nitrogen/phosophorus ratio of 20, Polysciences). 60,000 cells were plated in 48-well plates and transfected the following day with a total of 100–300 ng DNA, including a pCAG-iRFP720 (Table S1) transfection control plasmid. Cells were collected and prepared for either flow cytometry or qPCR analysis 3 days after transfection, unless otherwise noted.
m6A-qPCR assay for measuring adenine methylation
We adapted a previously described qPCR-based assay to quantitatively measure adenine methylation at specific genomic sequences/loci (van Steensel and Henikoff, 2000). To obtain fraction methylated values reported throughout this paper, we used the assay to calculate the ratio of amplified DNA, protected from DpnII digestion, for a GATC site(s) of interest relative to a non-GATC reference site, which serves as an internal control to account for variation in DNA amount in each sample (Figures S2A-C). First, total genomic DNA (gDNA) was isolated using a DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s instructions (with addition of 4 µL of 100 mg/mL RNase A), eluting in 300 µL elution buffer. A 35 µL aliquot of the resulting gDNA was incubated for 16 hr at 37°C with or without 2 u nits of DpnII (NEB), followed by a 20 min heat inactivation at 65oC. Next, four qPCR reactions were prepared to amplify: (1) DpnII-digested GATC site(s), (2) undigested GATC site(s), (3) DpnII-digested reference site, (4) undigested reference site (Figure S2B). Specifically, qPCR reactions using a 1:10 dilution of the digested gDNA samples were prepared using LightCycler 480 SYBR Green I Master Kit (Roche) according to the manufacturer’s instruction. qPCR reactions were performed on a LightCycler 480 Instrument II (Roche) with a total reaction volume of 20 µL (5 µL of DNA, 0.5 µM of forward primer, 0.5 µM of reverse primer, 10 µL of 2X SYBR Green Master Mix), using the following cycle conditions: (i) pre-incubation: 95°C for 10 min; (ii) amplification (45 cycles): 95°C fo r 10 s, [annealing temperature] for 20 s, 72°C for [extension time]; (iii) melting curve: 95° C for 5 s, 65°C for 1 min, 97°C at ramp rate 0.11°C/s; (iv) cooling: 40°C for 10 s. PCR pri mer sequences (listed in Table S3) were designed to flank the GATC site of interest or reference site. Annealing temperatures and extension times for specific primer sets are also listed in Table S3. Fraction methylated is then computed from the resulting qPCR Ct values using the ΔΔCt method/equation shown in Figure S2B.
To obtain “m6A enrichment” (e.g. for a synI factor), fraction methylation at a GATC probe site in the locus of interest was measured using m6A-qPCR and normalized to basal methylation induced by the Dam variant not fused to ZF (synINT, mCherry-Dam fusion) (Figures S2D and S2E).
Flow cytometry
Flow cytometry measurements were performed using an Attune NxT Flow Cytometer (Thermo Fisher Scientific) equipped with a high-throughput auto-sampler. Typically, 50,000 or 70,000 events were acquired for transient transfection or stable cell line experiments, respectively. Cells were gated by forward (FSC) and side scatter (SSC) distributions, and either iRFP or mCherry expression for transfection- or integration-positive populations, respectively. For experiments with transient transfection of synI/synR modules (Figure 3), geometric means of the GFP fluorescence distributions were calculated using FlowJo (Treestar Software). GFP fold change was then calculated by normalizing mean GFP intensity to reporter-only controls, unless otherwise noted. For spreading and memory experiments (with integrated constructs), the percentage of GFP activated cells was quantified using a GFP+ gate that contains ~0.5–1% of negative control cells (Figure S4E). Flow cytometer laser/filter configurations used in this study were: Click-iT Plus Edu Pacific Blue (405 nm laser, 440/50 emission filter), EGFP (488 nm, 510/10), mCherry (561 nm, 615/25), FxCycle Far Red or CellTrace Far Red (638 nm, 670/14), iRFP-720 (638 nm, 720/30).
Cell cycle assay
Cells cultured for 3 days with or without 200 µM abscisic acid (ABA, Sigma Aldrich) were labeled with Click-iT EdU Pacific Blue (Click-iT Plus EdU Pacific Blue Flow Cytometry Assay Kit, Thermo Fisher Scientific) to monitor DNA replication and FxCycle Far Red Stain (Thermo Fisher Scientific) to measure DNA content. Labeling was performed according to the manufacturers’ instructions, with a 1.5 hr incubation in 10 µM EdU, and 30 min additional incubation with 200 nM FxCycle Far Red Stain and 1 µL of RNaseA (100 mg/mL). Cells were analyzed using flow cytometry.
RNA Sequencing
RNA-seq measurements were performed on two biological replicates per experimental condition. Total RNA was purified from ~1 million cells using the RNeasy Plus Mini Kit (Qiagen) and QIAshredder (Qiagen), according to the manufacturer’s instructions, three days following transfection. Sequencing libraries were prepared at the Tufts University Core Facility (TUCF Genomics) using the TruSeq Stranded mRNA Library Prep Kit (Illumina). 50-bp single-end reads were sequenced on an Illumina HiSeq 2500.
DNA sequences for our synthetic constructs (reporter, synI, synR, ZF-VP64) were appended to the human UCSC genome (version hg19), and genome indices were built using the Bowtie 2 software (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml). Sequencing reads were aligned to this indexed genome using the TopHat software (http://ccb.jhu.edu/software/tophat/index.shtml), and the mapped reads were counted for genomic features using featureCounts (http://bioinf.wehi.edu.au/featureCounts/). Differential expression analysis was performed in R using the DESeq2 analysis package. Multiple hypothesis correction was performed using the Benjamini-Hochberg procedure with a FDR of < 1%.
Screen for spatial propagation
Our objective was to devise a phenotypic screen that would allow for identification of three-module, read-write (RW) circuit designs that drive spatial propagation behaviors. Specifically, we sought to identify synRW variants that, when coupled with synIIND and synR modules, could propagate m6A across a domain in a manner that is dependent on m6A nucleation. Below we provide a detailed description of how we generated a library of synRW variants, developed a phenotypic screen to identify synRW candidates, and finally analyzed the screen results.
Generation of synRW module library
We created a synRW library by varying two biochemical properties: expression level and Dam writer activity. To vary expression level, we placed synRW expression under the control of two promoters of different strength: pMinCMV (weak) and pUBC (strong) (Figure S6B). To vary writer activity, we used a series of Dam mutants that have a range of methylation activities (Figures 4B and S1). Most of these mutations target residues responsible for mediating DNA phosphate group contact, either within or flanking the GATC sequence, which are known to affect the biochemical activity of the molecule (Coffin and Reich, 2009; Horton et al., 2006). In total, this collection represented 11 single residue mutants and 9 double residue mutants, along with WT Dam. The affinity of the DpnI reader domain represents another potential, tunable biochemical property of the synRW module; however, we chose to keep the reader domain fixed across the library because (1) less has been done to identify mutants and characterize their biochemical properties (relative to Dam) and (2) we wanted to maintain a manageable library size to transform, culture, and assay in arrayed format.
We used Gibson isothermal assembly to construct the library, and then cloned the collection into a lentiviral vector (Figure S1).
Screen design
To identify three-module RW circuits that can drive spatial propagation, we developed a phenotypic screen in Clustered Reporter cell lines. The screen leverages the long GATC domain (~1.5 kb) that separates a nucleation site (ZF BS array) from the reporter gene (Figure 4B). Devoid of a mechanism for propagation across this domain, reporter cells stably expressing the two-module circuit (synIIND/synRVP64) do not activate the reporter (Figure S6A), since marks established at the distal nucleation site cannot effectively mediate promoter regulation by synR. We therefore screened the synRW library in these cells for candidates that lead to reporter activation (+synIIND, Figure 4B), as well as in cells lacking synIIND to screen out spurious cases for which reporter activation is independent of m6A nucleation (-synIIND, Figure 4B). Variants emerging from this screen would represent potential candidates for the RW module of three-module regulatory circuits (synIIND/synRVP64/synRW) that drive inducible spatial propagation leading to reporter activation.
To perform the screen, we transfected 100 ng of each synRW construct (and 50 ng of pCAG-iRFP720 transfection marker) into two cell lines (60,000 cells, in triplicates): (1) Clustered Reporter cells stably expressing the two-module circuit (synIIND/synRVP64), and (2) Clustered Reporter cells stably expressing only synRVP64. Cells triggered at the same time (6 hr after transfection) and induced continuously thereafter with 200 μM of ABA were harvested 4 days after transfection, whereupon half of the cells were assayed for GFP activation by flow cytometry (Figure 4B) and the remaining half collected for m6A-qPCR analysis of methylation profiles.
Screen analysis
To examine our screen results, define thresholds, and guide circuit designs, we performed hierarchical cluster analysis on the GFP expression patterns (similarity in % GFP activated data, treating each replicate individually; heatmap.2 function in R). Satisfyingly, the unbiased analysis distinguished -synIIND from +synIIND cells (vertical dendrogram not shown in Figure 4B). The analysis also revealed a number of interesting features. Library members divided into two parental clusters: one with strong GFP activation in the +synIIND case (top) and the other with weak or no GFP activation (bottom) (Figure 4B). We used these clusters to define a threshold of circuits exhibiting functional (top) vs. non-functional (bottom) propagation phenotypes, with the -synRW control circuit occupying the latter.
Examining molecular components within these clusters, we find that the non-functional cluster possessed all the Dam double mutants (as well as WT Dam), whereas the functional cluster was composed entirely of Dam single mutants. These results suggest that methylation activity of the synRW module could be a key factor in the design of synthetic propagation circuits. On the other hand, the two promoters we tested were scattered across parental and sub clusters, meaning there was no obvious preference in phenotypic outcomes for either promoter. This suggests that the transfection conditions used for screening likely produced saturating concentrations of synRW for reactions on a limited number of available methylated GATC sites. We tested whether this was the case by performing an analogous experiment with the other engineered reader module: synR. Indeed, when we transfected Interspersed Reporter cells with a two-module circuit, placing expression of synRVP64 under the control of either weak pMinCMV or strong pUBC, we observed a similar insensitivity in reporter output (Figure S6B).
The unbiased analysis of our propagation screen revealed a strong clustering of circuits based on synRW Dam mutants (Figure 4B). To further examine the relationship between writer methylation activity and the results of the screen, we defined a quantitative metric to score propagation propensity: “expt. spreading score” is computed as the difference (Δ) in % GFP activated for cells with and without synIIND (Figures 4B,C). Higher spreading scores correspond to circuits that drive high levels of reporter activation preferentially in cells with m6A nucleation (+synIIND). In Figure 4C, the spreading score for each synRW library member is plotted as a function of its basal Dam methylation activity, as previously measured (Figures S2D and S2F). (Plotted is the average value for both promoters since screen results for each synRW were similar across both promoters – a plot of the full library can be found in Figure S5D). Based on this analysis, we find that the highest scoring circuits contain synRW variants with intermediate methylation activity, potentially an important design feature of this module for producing the propagation phenotype.
We selected three synRW variants representing low, intermediate and high Dam writer activity to further investigate (Figures S5D-G). We measured corresponding m6A profiles across the Clustered Reporter GATC array in cells that were transfected with or without the synRW module (see Methods). As expected, a low-activity synRW module produced no enrichment in m6A across the array over control cells lacking synRW (Figure S5G, left). A high-activity synRW module, on the other hand, led to significantly higher levels of m6A over cells lacking synRW; however, this came at a cost, as m6A levels were enriched in cells with or without synthetic initiator (Figure S5G, right). We speculate that the corresponding low reporter/spreading score (Figure S5F) is likely a result of global off-target m6A enrichment induced by the high-activity synRW, similar to what we observed with high-activity synI factors, which could serve to titrate synR factors away from the reporter. Finally, profiles measured for the intermediate-activity synRW showed evidence for an enlarged m6A domain in cells expressing both synIIND and synRW (Figure S5G, middle). These results further validate the “spreading score” as a useful phenotypic metric for identifying circuits capable of driving spatial propagation behaviors.
Model of m6A spatial dynamics
Model description
Despite the molecular complexities inherent to chromatin regulatory systems, previous studies have shown that behaviors like nucleation and propagation of histone modifications along a chromosome can be captured by simple general models (Hathaway et al., 2012; Hodges and Crabtree, 2012). We therefore hypothesized that such models could also be used to capture spatial propagation of m6A by synthetic RW circuits, providing a general guide for their design and construction.
We adapted a previously described chromatin spreading model (Hodges and Crabtree, 2012) in order to explore how the properties of synI and synRW affect m6A spatial dynamics. We modeled the array of GATC sites in the Clustered Reporter as a discrete, one-dimensional lattice with 63 sites (Figure S5). Each position (j) on the lattice (Ij) corresponds to a GATC site in the reporter, where methylated and unmethylated states are denoted by values of Ij = 1 and Ij = 0, respectively (Figure S5A). Four reactions govern this model: basal (non-targeted) methylation, sequence-specific nucleation, reader-mediated propagation, and mark turnover. We implemented these reactions with the following rules:
(1) Basal methylation by synI or synRW occurs at a rate ksynI_act or ksynRW_act, respectively, to convert any unmethylated GATC site (Ij = 0) to methylated (Ij = 1).
(2) Sequence-specific nucleation by synI occurs at a rate bZF • ksynI_act to convert the first lattice site from unmethylated (I1 = 0) to methylated (I1 = 1). ksynI_act is the basal methylation rate of the synI Dam writer domain, while bZF serves as a specificity multiplier to increase the methylation frequency by synI at the nucleation site.
(3) Reader-mediated propagation by synRW occurs at a rate bDpn1•ksynRW_act to convert unmarked sites (Ij = 0) to methylated, if any neighboring GATC sites are marked (Ij-1 = 1 or Ij+1 = 1). ksynRW_act is the basal methylation rate of the synRW Dam writer domain, while bDpn1 serves as a specificity multiplier to increase the methylation frequency of unmarked sites adjacent to marked ones.
(4) Mark turnover occurs at rate kturn during which any methylated GATC site (Ij =1) can be converted to unmethylated (Ij =0).
Model parameterization
For all simulations, we set kturn = 0.05 hr−1 to approximate dilution of m6A modifications by cell division (~20 hr doubling time). In the model, synI activity is described by the non-specific methylation (ksynI_act) of each Dam mutant as well as the specificity (bZF) conferred by the ZF domain at the nucleation site (I1). To approximate the range of non-specific methylation activity (ksynI_act) values across the Dam library, we used the following relationship:
where fmeth_basal is the experimentally obtained values of basal methylation for each Dam mutant (Figure S2D) and kturn = 0.05 hr-1. We found that non-specific activity values ranged from ksynI_act = 10−6 to 101 hr-1. Subsequently, to parameterize the ZF specificity multiplier (bZF) for each synI in the library, we calculated steady-state methylation (fmeth) by synI across a range of ksynI_act and bZF values with the following relationship:
where fmeth_target is the experimentally obtained values of targeted methylation for each Dam mutant (Figure S2E) and kturn = 0.05 hr-1. We found that our synI module library is captured by specificity multiplier values, ranging between bZF = 1 – 100 (Figure S5B). We chose values of ksynI_act = 5 •10−4 hr−1 and bZF = 10, which closely approximated the behavior of the selected synI (Dam N132A) featured in our three-module propagation circuit.
Stochastic simulations of spatial propagation
To model the synRW library screen in Figure 4B, we ran stochastic simulations of synRW activity for a range of ksynRW_act (10−6 to 101 hr−1 as obtained above for ksynI_act) and bDpn1 values, with and without synI. We defined a “model spreading score” to assess m6A propagation from the nucleation site ( j =1) for each synRW library member. This was defined as the difference in mean m6A density at the 15 most downstream GATC sites (j = 49 to j = 63), with and without synI (Figure S5C). All stochastic simulations were implemented using the Gillespie algorithm and were run to steady-state (t = 5000 hr).
We found that the model spreading score distribution closely resembled the experimental spreading score distribution for bDpn1 values between 50 and 200 (Figures S5D and S5E, see also Figure 4C with bDpn1 = 100). Moreover, the model results showed a similar relationship between methylation activity of synRW and propagation propensity, where intermediate methylation levels were predicted to yield the highest scoring circuits. Interestingly, the model also predicted that, if reader specificity could be increased (higher bDpn1), then one could generate synRW factors that drive high levels of propagation with lower writer activity (Figure S5E).
In general, model-generated m6A spatial profiles agreed well with experimentally measured profiles for the range of synRW variants, representing low, intermediate and high Dam writer activity (Figure S5G). For example, high-activity synRW was predicted to produce nucleation-independent methylation, while low-activity synRW was predicted to yield weak spreading from the nucleation site. Furthermore, our model captured the dynamics of the growing m6A domain induced by our three-module “propagation circuit” (with bDpn1 = 100, ksynRW _act = 3.4•10−4 hr−1) (Figure 4D). Taken together, this simple, four-parameter model can effectively capture the essential features of our m6A synthetic propagation system.
In order to increase the predictive power of this model, future iterations could include off-target methylation by synI and synRW (at other genomic loci), as well as incorporate transcriptional regulation by synR. Describing these properties may be necessary for predicting targeted reporter activity from the distribution of methylation across the genome.
Epigenetic memory experiments
~120,000 cells were initially plated in multiple wells of a 6-well plate, and incubated either with or without 200 µM ABA. ABA was washed out at indicated times by aspirating out ABA-containing media and adding back fresh media. At indicated time points following ABA washout, approximately half of the cells were re-plated and continued in culture, while the rest were harvested for downstream analysis. For Aphidicolin (APC) experiments, re-plated cells were continued in culture with or without 5 µg/mL Aphidicolin (APC).
Model of epigenetic memory
Stochastic simulations of epigenetic memory
We applied our model framework to investigate epigenetic memory induced by synthetic RW circuits. Specifically, we modeled the effect of different synRW variants on the methylation decay dynamics of an initially fully methylated Interspersed Reporter. The Interspersed Reporter was described as a one-dimensional lattice with 14 methylated GATC sites (Ij=1– 14 = 1). We tracked methylation across the lattice in the presence and absence of synRW (bDpn1 = 100, ksynRW_act = 3.4•10−4 hr−1), as well as for a reader-defective synRNTW (bDpn1 = 0, ksynRW_act = 3.4•10−4 hr−1). Simulated methylation dynamics showed close agreement with experiments, wherein methylation profiles decay rapidly due to passive dilution in simulations without synRW or with a control synRNTW, and show maintenance of methylation over 20 days in simulations with a functional synRW (Figure 6D). Finally, we simulated decay profiles for synRW variants across a range of bDpn1 values (Figure S7E). We found that higher bDpn1 values corresponded to a larger percentage of the lattice retaining its methylation state over time. These results suggest that epigenetic memory could be strengthened by improving the binding activity of the synRW reader domain in future designs.
Cell proliferation assay
~120,000 cells were initially plated in multiple wells of a 6-well plate, and incubated either with or without 200 µM ABA. ABA was washed out after 3 days by aspirating out ABA-containing media and adding back fresh media. Cells were harvested with trypsin and brought to suspension. Cells were then stained with 5 µM CellTrace Far Red, according to manufacturer’s instructions for labeling cells in suspension (CellTrace Far Red Cell Proliferation Kit, Thermo Fisher Scientific). About half of the stained cells were analyzed with flow cytometry (Day 0), while the rest were re-plated for continued culture. Thereafter at indicated time points following ABA washout, approximately half of the cells were re-plated and continued in culture, while the rest were resuspended in media for flow cytometry analysis.
Human genome sequence analysis of GATC motifs
Promoter sequences, defined as the region 1500 bp upstream of each TSS, for all human transcripts by chromosome were retrieved from the UCSC genome browser (version hg38) using ‘GENCODE v24’ track. The startcoordinates of the GATC motif within the retrieved promoter sequences were mapped out using the EMBOSS-dreg bioinformatics tool (http://www.bioinformatics.nl/cgi-bin/emboss/dreg). The retrieved coordinates were further analyzed and plotted for the number of GATC motif occurrences and inter-GATC distances using custom R scripts.
Supplementary Material
HIGHLIGHTS.
A synthetic epigenetic regulatory system in human cells using m6A DNA modification
Engineered writers and readers of m6A enable construction of regulatory circuits
Read-write circuits drive spatial propagation and hallmarks of chromatin spreading
Read-write circuits enable epigenetic memory of transcriptional states
ACKNOWLEDGEMENTS
We thank M. Maeder, J. Sander and J.K. Joung for generously providing the zinc finger (ZF) construct, H. VanBenschoten for help with cloning, C.-H. Wang and P. Mehta for modeling discussions, and members of the Khalil laboratory for insightful comments on the manuscript. We thank the Tufts University Core Facility (TUFC) for their Next-Gen sequencing services. This work was supported by a NIH-NIGMS Ruth L. Kirschstein Postdoctoral Fellowship (1F32GM105272–01A1 to A.J.K.), a NSF Expeditions in Computing grant (CCF-1522074 to A.S.K.), a NSF grant (MCB-1713855 to A.S.K.), and a DARPA grant (W911NF-11–2-0056 to A.S.K.). A.S.K. also acknowledges funding from the NIH Director’s New Innovator Award (1DP2AI131083–01), NSF CAREER Award (MCB-1350949), and DARPA Young Faculty Award (D16AP00142).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
A provisional US patent has been filed based on this work.
DATA AND SOFTWARE AVAILABILITY
Raw RNA-seq data for 293FT transcriptome analysis upon expression of synI and/or synR newly reported in this paper is available at SRA: PRJNA488081.
QUANTIFICATION AND STATISTICAL ANALYSIS
FlowJo was used to extract geometric mean fluorescence values or the percentage of GFP activated cells from flow cytometry measurements. Microsoft Excel and GraphPad Prism software were used to process data. Statistical details such as N and error calculations are provided in figure legends.
REFERENCES
- Al-Sady B, Madhani HD, and Narlikar GJ (2013). Division of labor between the chromodomains of HP1 and Suv39 methylase enables coordination of heterochromatin spread. Mol Cell 51, 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audergon PN, Catania S, Kagansky A, Tong P, Shukla M, Pidoux AL, and Allshire RC (2015). Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science 348, 132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisel C, and Paro R (2011). Silencing chromatin: comparing modes and mechanisms. Nat Rev Genet 12, 123–135. [DOI] [PubMed] [Google Scholar]
- Berger SL (2007). The complex language of chromatin regulation during transcription. Nature 447, 407–412. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Meissner A, and Lander ES (2007). The mammalian epigenome. Cell 128, 669–681. [DOI] [PubMed] [Google Scholar]
- Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, Uno N, Oshimura M, and Elowitz MB (2016). Dynamics of epigenetic regulation at the single-cell level. Science 351, 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, Tu S, and Reinberg D (2010). Molecular signals of epigenetic states. Science 330, 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun SMG, Kirkland JG, Chory EJ, Husmann D, Calarco JP, and Crabtree GR (2017). Rapid and reversible epigenome editing by endogenous chromatin regulators. Nat Commun 8, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin SR, and Reich NO (2009). Escherichia coli DNA adenine methyltransferase: the structural basis of processive catalysis and indirect read-out. J Biol Chem 284, 18390–18400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groote ML, Verschure PJ, and Rots MG (2012). Epigenetic Editing: targeted rewriting of epigenetic marks to modulate expression of selected target genes. Nucleic Acids Res 40, 10596–10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, and Leibler S (2000). A synthetic oscillatory network of transcriptional regulators. Nature 403, 335–338. [DOI] [PubMed] [Google Scholar]
- Feinberg AP (2007). Phenotypic plasticity and the epigenetics of human disease. Nature 447, 433–440. [DOI] [PubMed] [Google Scholar]
- Fu Y, Luo GZ, Chen K, Deng X, Yu M, Han D, Hao Z, Liu J, Lu X, Dore LC, et al. (2015). N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell 161, 879–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner KE, Allis CD, and Strahl BD (2011). Operating on chromatin, a colorful language where context matters. J Mol Biol 409, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner TS, Cantor CR, and Collins JJ (2000). Construction of a genetic toggle switch in Escherichia coli. Nature 403, 339–342. [DOI] [PubMed] [Google Scholar]
- Greer EL, Blanco MA, Gu L, Sendinc E, Liu J, Aristizabal-Corrales D, Hsu CH, Aravind L, He C, and Shi Y (2015). DNA Methylation on N6-Adenine in C. elegans. Cell 161, 868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SI, and Moazed D (2003). Heterochromatin and epigenetic control of gene expression. Science 301, 798–802. [DOI] [PubMed] [Google Scholar]
- Hathaway NA, Bell O, Hodges C, Miller EL, Neel DS, and Crabtree GR (2012). Dynamics and memory of heterochromatin in living cells. Cell 149, 1447–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes KA, and Silver PA (2011). Synthetic reversal of epigenetic silencing. J Biol Chem 286, 27176–27182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, and Esteller M (2015). An Adenine Code for DNA: A Second Life for N6-Methyladenine. Cell 161, 710–713. [DOI] [PubMed] [Google Scholar]
- Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, and Gersbach CA (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges C, and Crabtree GR (2012). Dynamics of inherently bounded histone modification domains. Proc Natl Acad Sci U S A 109, 13296–13301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JR, Liebert K, Bekes M, Jeltsch A, and Cheng X (2006). Structure and substrate recognition of the Escherichia coli DNA adenine methyltransferase. J Mol Biol 358, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, and van Steensel B (2013). Single-cell dynamics of genome-nuclear lamina interactions. Cell 153, 178–192. [DOI] [PubMed] [Google Scholar]
- Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, and Joung JK (2016). High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T (2007). Chromatin modifications and their function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Kundu S, Horn PJ, and Peterson CL (2007). SWI/SNF is required for transcriptional memory at the yeast GAL gene cluster. Genes Dev 21, 997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachner M, O’Carroll D, Rea S, Mechtler K, and Jenuwein T (2001). Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116–120. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprell F, Finkl K, and Muller J (2017). Propagation of Polycomb-repressed chromatin requires sequence-specific recruitment to DNA. Science 356, 85–88. [DOI] [PubMed] [Google Scholar]
- Lee JS, Smith E, and Shilatifard A (2010). The language of histone crosstalk. Cell 142, 682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang FS, Ho WQ, and Crabtree GR (2011). Engineering the ABA plant stress pathway for regulation of induced proximity. Sci Signal 4, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lim WA, and Pawson T (2010). Phosphotyrosine signaling: evolving a new cellular communication system. Cell 142, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, Ho QH, Sander JD, Reyon D, Bernstein BE, et al. (2013). Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol 31, 1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara AR, Hurd PJ, Smith AE, and Ford KG (2002). Characterisation of site-biased DNA methyltransferases: specificity, affinity and subsite relationships. Nucleic Acids Res 30, 3818–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK, and Bernstein BE (2013). Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol 31, 1133–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D (2011). Mechanisms for the inheritance of chromatin states. Cell 146, 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura W, and Barbas CF 3rd (2007). In vivo site-specific DNA methylation with a designed sequence-enabled DNA methylase. J Am Chem Soc 129, 8676–8677. [DOI] [PubMed] [Google Scholar]
- O’Brown ZK, and Greer EL (2016). N6-Methyladenine: A Conserved and Dynamic DNA Mark. Advances in experimental medicine and biology 945, 213–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Keung AJ, and Khalil AS (2016). The epigenome: the next substrate for engineering. Genome Biol 17, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragunathan K, Jih G, and Moazed D (2015). Epigenetics. Epigenetic inheritance uncoupled from sequence-specific recruitment. Science 348, 1258699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratna P, Scherrer S, Fleischli C, and Becskei A (2009). Synergy of repression and silencing gradients along the chromosome. J Mol Biol 387, 826–839. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, and Bleasby A (2000). EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16, 276–277. [DOI] [PubMed] [Google Scholar]
- Rudner AD, Hall BE, Ellenberger T, and Moazed D (2005). A nonhistone protein-protein interaction required for assembly of the SIR complex and silent chromatin. Molecular and cellular biology 25, 4514–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusche LN, Kirchmaier AL, and Rine J (2002). Ordered nucleation and spreading of silenced chromatin in Saccharomyces cerevisiae. Molecular biology of the cell 13, 2207–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffers S, Ebert C, Rahimoff R, Kosmatchev O, Steinbacher J, Bohne AV, Spada F, Michalakis S, Nickelsen J, Muller M, et al. (2017). Quantitative LC-MS Provides No Evidence for m(6) dA or m(4) dC in the Genome of Mouse Embryonic Stem Cells and Tissues. Angewandte Chemie 56, 11268–11271. [DOI] [PubMed] [Google Scholar]
- Siwek W, Czapinska H, Bochtler M, Bujnicki JM, and Skowronek K (2012). Crystal structure and mechanism of action of the N6-methyladenine-dependent type IIM restriction endonuclease R.DpnI. Nucleic Acids Res 40, 7563–7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AE, and Ford KG (2007). Specific targeting of cytosine methylation to DNA sequences in vivo. Nucleic Acids Res 35, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakore PI, Black JB, Hilton IB, and Gersbach CA (2016). Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat Methods 13, 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Henikoff S (2000). Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat Biotechnol 18, 424–428. [DOI] [PubMed] [Google Scholar]
- Wang X, and Moazed D (2017). DNA sequence-dependent epigenetic inheritance of gene silencing and histone H3K9 methylation. Science 356, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TP, Wang T, Seetin MG, Lai Y, Zhu S, Lin K, Liu Y, Byrum SD, Mackintosh SG, Zhong M, et al. (2016). DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature 532, 329–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao CL, Zhu S, He M, Chen, Zhang Q, Chen Y, Yu G, Liu J, Xie SQ, Luo F, et al. (2018). N(6)-Methyladenine DNA Modification in the Human Genome. Mol Cell 71, 306–318 e307. [DOI] [PubMed] [Google Scholar]
- Xiao R, and Moore DD (2011). DamIP: using mutant DNA adenine methyltransferase to study DNA-protein interactions in vivo. Curr Protoc Mol Biol Chapter 21, Unit21 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong T, Meister GE, Workman RE, Kato NC, Spellberg MJ, Turker F, Timp W, Ostermeier M, and Novina CD (2017). Targeted DNA methylation in human cells using engineered dCas9-methyltransferases. Scientific reports 7, 6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Wang X, and Moazed D (2018). Epigenetic inheritance mediated by coupling of RNAi and histone H3K9 methylation. Nature 558, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, Yin R, Zhang D, Zhang P, Liu J, et al. (2015). N6-methyladenine DNA modification in Drosophila. Cell 161, 893–906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.