

Summary
Vibrio cholerae uses a quorum-sensing (QS) system composed of the autoinducer 3,5-dimethylpyrazin-2-ol (DPO) and receptor VqmA (VqmAVc), which, together, repress genes for virulence and biofilm formation. vqmA genes exist in vibrios and in one vibriophage, VP882. Phage-encoded VqmA (VqmAPhage) binds to host-produced DPO, launching the phage lysis program via an antirepressor that inactivates the phage repressor by sequestration. The antirepressor interferes with repressors from related phages. Like phage VP882, these phages encode DNA-binding proteins and partner antirepressors, suggesting they too integrate host-derived information into their lysis-lysogeny decisions. VqmAPhage activates the host VqmAVc regulon, whereas VqmAVc cannot induce phage-mediated lysis, suggesting an asymmetry whereby the phage influences host QS while enacting its own lytic-lysogeny program without interference. We reprogram phages to activate lysis in response to user-defined cues. Our work shows that a phage, causing bacterial infections, and V. cholerae, causing human infections, rely on the same signal molecule for pathogenesis.
Keywords: phage, quorum sensing, autoinducer, lysis-lysogeny, VqmA, vibrio, antirepressor, kill switch
Graphical Abstract
eTOC Blurb:
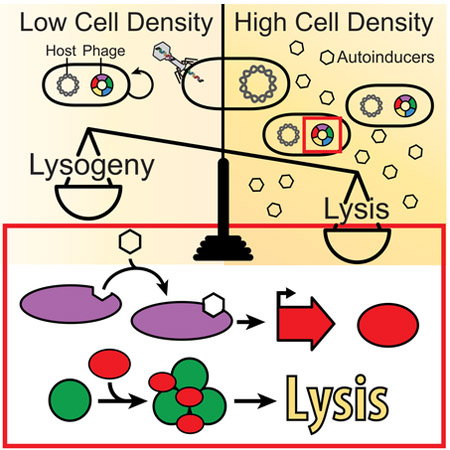
Bacteriophages can “listen in” on their host bacteria’s quorum sensing systems, shifting their lysis-lysogeny decisions based on host cell density
Introduction
Bacteria use the cell-cell communication process called quorum sensing (QS) to coordinate group behaviors. QS depends on the production, release, and group-wide detection of signal molecules called autoinducers (AI). QS is perhaps most well established in vibrios (Papenfort and Bassler, 2016). Within this genus, Vibrio cholerae is a particularly compelling model because QS regulates genes involved in the human disease cholera (Miller et al., 2002). We recently discovered a new QS circuit in V. cholerae, and other vibrios, consisting of a cytoplasmic receptor-transcription factor, VqmA and the AI, 3,5-dimethylpyrazin-2-ol (DPO) (Papenfort et al., 2017). Upon binding DPO, VqmA activates transcription of the vqmR gene encoding the small RNA (sRNA), VqmR. VqmR represses genes required for biofilm formation and virulence factor production.
In bacteria, virulence factors and the emergence of toxigenic subtypes from non-toxigenic strains can often be traced back to phages (Waldor and Friedman, 2005). Indeed, interaction with phages is thought to have significantly contributed to overall bacterial evolution (Faruque and Mekalanos, 2012). V. cholerae has provided insight into how phages have shaped their pathogenic hosts. The gene encoding the major V. cholerae virulence factor CTX, is located on the genome of the lysogenic CTXφ temperate phage (Waldor and Mekalanos, 1996). Only V. cholerae strains harboring CTXφ cause epidemic and pandemic cholera disease (Faruque and Mekalanos, 2012). Thus, while V. cholerae is considered the causative agent of cholera, the etiology must also include the CTXφ phage.
Much of our understanding of temperate phage biology stems from seminal work on phage lambda (Ptashne, 2004). Upon infection, temperate phages can lyse the host cell and propagate or they can lysogenize the host cell and remain dormant as prophages. In lambda, this fate determination switch is governed by the status of a single protein, the lambda cI repressor (Ptashne, 2004). If present in abundance, cI represses phage lytic genes and commits the phage to the lysogenic program. If cI is limiting or absent, for example, following inactivation due to the host stress response (SOS), repression is relieved, phage lytic genes are expressed, and the host cell is lysed. Evidence that phages exploit cues beyond host cell stress to drive the lysogeny-lysis switch has only recently been demonstrated in a phage-phage communication process, coined the arbitrium system (Erez et al., 2017). Briefly, SPbeta phage encode a peptide that they produce upon infection of Bacillus. The peptide is detected by prophages in neighboring cells via a phage-encoded receptor. The phage peptide-receptor pair repress lysis across the population.
The ability of host signaling molecules to double as cues that regulate phage fate decisions has never been demonstrated. Here, we discover that the vibriophage, VP882 has the capacity to respond to a host-produced QS AI. The VP882 phage-encoded protein, Gp56, which we rename VqmAPhage, is a viral DPO-binding QS receptor and transcription factor with homology to the vibrio QS receptor VqmA. DPO, via VqmAPhage, activates the phage lytic program via induction of expression of a phage gene that we name qtip (quorum triggered inactivator of cI protein). Qtip is a small (79 amino acid) protein that aggregates and inactivates the VP882 repressor of lysis. DPO-bound VqmAPhage recognizes the V. cholerae vqmR promoter whereas V. cholerae VqmA is unable to recognize the phage qtip promoter. This “one-sided conversation” could enable the VP882 prophage to influence host QS while executing its own lifestyle programs with immunity from host interference. To our knowledge, this is the first report of a phage-encoded receptor that senses a host-produced AI to mediate the lysis-lysogeny decision.
We report that phages related to VP882 encode DNA-binding transcription factors and small proteins in the identical genomic locations as VP882 vqmAPhage and qtip, respectively. Qtip aggregates the repressors from these phages but not the distant, prototypical lambda repressor. In the opposite vein, the protein encoded in the qtip location in one of these other phages, despite having no similarity to Qtip, can induce aggregation of the VP882 repressor. Our analysis suggests that phages, in addition to VP882, control their lysis-lysogeny decisions by tuning into host-produced signaling factors using a Qtip-like lysis de-repression mechanism. We reprogram the VP882 phage to be insensitive to native inputs but responsive to user-defined cues. These reprogrammable “kill-switches” could be useful for environmental, industrial, and medical applications.
Results
The phage VP882 genome encodes a VqmA quorum-sensing receptor.
V. cholerae VqmA (denoted VqmAVc) is a dual QS receptor-transcription factor (Liu et al., 2006; Papenfort et al., 2017). Predicted VqmA homologs (Figure S1 and S2A) consist of C-terminal DNA-binding domains and N-terminal PAS-domains, presumably for binding the ligand, DPO. To identify other DPO-binding proteins, we performed a bioinformatic search for proteins possessing the PAS fold-4 subtype domain (InterPro, IPR013656) present in V. cholerae VqmAVc. Of the 103,219 returned proteins, only one was in the virus category, Gp56 of the Myoviridae virus VP882, a non-integrating temperate phage from a pandemic V. parahaemolyticus O3:K6 strain (Lan et al., 2009). VP882 infects vibrios including V. parahaemolyticus and V. cholerae (Lan et al., 2009), however, the GC content of the VP882-encoded gp56 is markedly higher than host encoded vqmA genes (55.5% in phage VP882 versus 46.5% and 47.5% in V. parahaemolyticus and V. cholerae, respectively) suggesting the phage gene was not directly transferred from the vibrio host. Examination of the 38.2 kb VP882 genome (Figure 1A and Lan et al., 2009) shows that gp56 lies between repA and telN; two essential genes conserved across all known linear phage, which are required for replication and maintenance as linear plasmids, respectively (Ravin, 2015). The curious location of gp56, hereafter called VqmAPhage, within a critical region of the phage genome, suggested that it too might be essential for some crucial phage-related process.
Figure 1: Phage VP882 encodes a homolog of vibrio VqmA that binds DPO and promotes host cell lysis.

(A) Organization of a region of the phage VP882 genome. Colors denote genes characterized here. telN and repA are conserved across linear plasmid-like phage. gp56 (purple) encodes the VqmAPhage QS receptor, gp55 (red) encodes the Qtip antirepressor, gp59 (green) encodes the cI repressor, gp62 (blue) encodes the Q antiterminator, and gp69-71 (yellow) encode the lysis genes. This color coding is used throughout this work. (B) Growth curves of V. parahaemolyticus lysogenized with phage VP882 (diamonds) or cured of phage VP882 (circles) harboring inducible vqmAPhage. As indicated, vqmAPhage expression was induced with 0.2% ara. (C) Growth curves of V. parahaemolyticus lysogenized with phage VP882 harboring inducible vqmAPhage in minimal medium lacking threonine. As indicated, vqmAPhage expression was induced with 0.2% ara and DPO was present at 10 μM. (D) Quantitation of DPO from purified proteins in a DPO-dependent E. coli reporter assay. The left bar shows the 10 μM synthetic DPO standard. RLU denotes relative light units. (E) Quantitation of DPO from the extracts in panel D by LC-MS. The left bar shows the 100 μM synthetic DPO standard. AU denotes arbitrary units. Data are represented as mean ± std with n=3 biological replicates (B, D), n=3 technical replicates (E), and n=3 biological and n=3 technical replicates (C). See also Figures S1 and S2.
VqmAPhage induces host cell lysis.
To study the function of VqmAPhage, we cloned vqmAPhage onto a vector under an arabinose inducible promoter and introduced it into the V. parahaemolyticus clinical isolate in which the phage was originally discovered. Induction of VqmAPhage caused a precipitous decline in optical density (OD600) following initial growth suggesting that VqmAPhage promotes cell lysis (Figure 1B). A similar effect occurred when the original V. parahaemolyticus isolate carrying the VP882 phage was exposed to the DNA damaging agent mitomycin C (MMC), a known inducer of lytic development for many temperate phages including VP882 (Figure S2B and Lan et al., 2009). To determine if the effect of VqmAPhage was a phage-dependent process, we cured the V. parahaemolyticus host of the VP882 phage using plasmid incompatibility (Figure S2B and S2C). Arabinose-induction of vqmAPhage in the V. parahaemolyticus strain lacking the prophage did not cause lysis (Figures 1B and S2B). Consistent with this result, induction of vqmAPhage in bacterial hosts that do not harbor the VP882 prophage (Escherichia coli, V. cholerae, and a different isolate of V. parahaemolyticus) also did not cause lysis (Figure S2D). Thus, VqmAPhage promotes host cell lysis in a phage-dependent manner.
MMC treatment, in addition to lysing the host, leads to production of VP882 phage particles and their release into culture fluids (Lan et al., 2009). Indeed, we could purify VP882 phage DNA from culture fluids of MMC-treated but not untreated V. parahaemolyticus harboring phage VP882 (Figure S2E). Induction of VqmAPhage in V. parahaemolyticus harboring phage VP882 also results in release of phage DNA (Figure S2E). Phage DNA could not be isolated if the phage was defective for the major capsid gene (gp07::Tn5) (Figure S2E). These results demonstrate that VqmAPhage, like MMC, launches the complete phage VP882 lytic cycle.
VqmAPhage is activated by the quorum-sensing autoinducer DPO.
VqmAVc and DPO were originally discovered as a QS receptor-AI pair in V. cholerae where they control biofilm formation and virulence factor production. Maximal transcriptional activation of the direct target of VqmAVc, vqmR, occurs only when VqmAVc is bound to DPO (Papenfort et al., 2017). Our finding that a phage genome encodes a VqmA homolog suggested that VqmAPhage activity could likewise be modulated by DPO. Vibrios produce DPO from threonine so growth in minimal medium lacking threonine eliminates DPO production (Papenfort et al., 2017), including in the lysogenized V. parahaemolyticus strain studied here. In the absence of DPO, induction of VqmAPhage in V. parahaemolyticus caused a low, basal level of cell lysis, whereas maximal host cell lysis occurred when DPO was supplied (Figure 1C). No lysis occurred when DPO was added in the absence of induction of VqmAPhage. Thus, DPO drives VqmAPhage activity.
To explore whether DPO acts directly on VqmAPhage, we overexpressed and purified the VqmAPhage protein from E. coli. Importantly, E. coli naturally produces DPO. We likewise overexpressed and purified VqmAVc. As a control, we overexpressed and purified V. cholerae LuxO, a DNA-binding QS transcription factor that does not bind DPO (Figure S2F and Papenfort et al., 2017). The three proteins were denatured, removed from suspension, and the remaining soluble fractions were extracted and assessed for DPO content by bioassay and liquid chromatography-mass spectrometry (LC-MS). Synthetic DPO was used as the standard. Figure 1D shows that the extracts made from the two VqmA proteins yielded activity in the DPO bioassay whereas the LuxO protein yielded none. The major component released from VqmAVc and VqmAPhage was DPO (Figure 1E). Indeed, one thousand-fold more DPO was present in each of the VqmA protein extracts than in the LuxO extract, which yielded the background level of DPO. Thus, VqmAPhage binds to and is activated by DPO which promotes the phage lytic lifecycle.
gp62 is the VqmAPhage-controlled gene required for lysis.
To identify phage genes required for lysis, we used in vitro Tn5 mutagenesis. Individual mutant phage were introduced into the phage-cured V. parahaemolyticus host harboring arabinose inducible vqmAPhage where they were assessed for the ability/inability to lyse the host when arabinose was added. Phage mutants with insertions in gp62 were defective for lysis (Figure 2A). By contrast, a phage carrying a Tn5 insertion immediately downstream of gp62 had the wild-type (WT) lysis phenotype. In trans expression of gp62 restored lysis to V. parahaemolyticus carrying the VP882 phage harboring gp62::Tn5 (Figure 2A). These results show that Gp62 is necessary for phage-mediated host lysis.
Figure 2: Gp62 acts downstream of VqmAPhage and is required for lysis.

(A) Lysis of V. parahaemolyticus lysogenized with VP882, VP882 gp62::Tn5, and VP882 with a Tn5 insertion downstream of gp62 (control Tn5). See key for introduced vectors/genes. (B) Lysis of V. parahaemolyticus lysogenized with VP882 vqmAPhage::Tn5 or VP882 gp62::Tn5. See key for introduced vectors/genes. Phages were transmitted to host strains by conjugation (see Methods). Where indicated, 50 ng mL−1 MMC was added. In both panels, all strains were grown in medium containing 0.2% arabinose. Data are represented as mean ± std with n=3 biological replicates.
We used epistasis analysis to define the relationship between VqmAPhage and Gp62. In trans expression of gp62 caused lysis in strains carrying either the wild-type VP882 phage or a VP882 phage with a Tn5 in vqmAPhage (Figure 2B). By contrast, in trans expression of vqmAPhage in a strain carrying the VP882 phage harboring gp62::Tn5 was lysis defective. Thus, Gp62 acts downstream of vqmAPhage. Consistent with this finding, strains carrying VP882 phage harboring vqmAPhage::Tn5 lyse upon MMC treatment, whereas strains carrying VP882 phage harboring gp62::Tn5 do not (Figure 2B). Our results suggest that DNA damage – mediated by Gp62 – and QS communication – mediated by DPO, VqmAPhage, and Gp62 – constitute two distinct triggers that promote phage VP882-induced host cell lysis.
Gp62 is a phage antiterminator that regulates an operon involved in lysis.
gp62 is predicted to encode a protein similar to Q from the lambdoid bacteriophage 82 (pairwise BLAST E-value, 5e-19; 30% identity), suggesting that Gp62 is likely a regulator of phage lytic genes. To determine what Gp62 controls, we focused on a region ~1 kb downstream of gp62 encoding a three-gene operon (gp69-71, Figure 1A). Four observations motivated us: First, the gp69-71 5’UTR harbors a rho-independent terminator, suggesting an antiterminator mechanism (Figure S3A). Second, gp70 encodes a protein with a lysozyme domain. Third, qRT-PCR showed that the gp69-71 operon is not expressed by the phage carrying the gp62::Tn5 mutation but is upregulated more than 500-fold prior to lysis when gp62 is present (Figure S3B). Fourth, introduction and induction of the gp69-71 operon was sufficient to cause lysis of E. coli lacking any other phage genes (Figure 3A). To explore the link between Gp62 and gp69-71 we engineered a Pgp69-lux transcriptional fusion that included the putative antiterminator sequence (Figure S3A). In recombinant E. coli, production of Gp62 protein was sufficient to activate the expression of the Pgp69-lux fusion (Figure 3B). Thus, the gp69-71 promoter/terminator is likely the direct target of Gp62. We hereafter refer to Gp62 as Q.
Figure 3: Gp62 is an antiterminator and Gp59 is a repressor of the gp69-71 operon encoding the phage lysin genes.

(A) Growth of E. coli carrying arabinose inducible gp69-71 on a plasmid in medium lacking (black) or containing (white) 0.4% arabinose. (B) E. coli carrying a plasmid with Pgp69-lux and a second empty plasmid or the plasmid with arabinose inducible gp62. Black; no arabinose, white 0.2% arabinose. (C) Pq-lux expression from V. parahaemolyticus harboring phage VP882 (denoted Lysogen), lacking VP882 (denoted Phage Cured), or lacking VP882 with gp59 expressed from a plasmid (denoted Phage Cured gp59). (D) Pq-lux expression from E. coli carrying an empty plasmid or the plasmid with arabinose inducible gp59. Black; no arabinose, white; 0.2% arabinose. (E) Western blot analysis of lysate from recA+ and ΔrecA E. coli carrying HIS-HALO-Gp59. Symbols: −; no MMC, +; 250 ng mL−1 MMC, M; Marker (PageRuler Plus; representative bands are labeled). (F) Pq-lux expression from the strains in panel E. Black; no MMC, white; 250 ng mL−1 MMC. (G) Time-course of Pq-lux (closed circles) and Pgp69-lux (open circles) production from V. parahaemolyticus lysogenized with phage VP882 following 50 ng mL−1 MMC treatment (top panel) and from V. parahaemolyticus lysogenized with phage VP882 and carrying arabinose inducible vqmAPhage following addition of 0.2% arabinose (bottom panel). Data are represented as mean ± std with n=3 biological replicates (A, B, C, D, F), and mean ± std with n=3 biological and n=3 technical replicates (G). See also Figure S3.
Gp59 is a phage lysis repressor.
Our results show that the Q antiterminator is required for phage-driven lysis. We wondered how Q is kept in check to enable the lysogenic state. To explore regulation, we made a Pq-lux transcriptional fusion. Twenty-five-fold more light was produced from this fusion in the V. parahaemolyticus strain that had been cured of the phage than in the strain carrying the phage (Figure 3C) suggesting that the prophage encodes a repressor of Pq that promotes lysogeny. An ORF, gp59, predicted to encode a DNA-binding repressor similar to cI (pairwise BLAST E-value, 5e-22; 37% identity) is located adjacent to but in the opposite orientation of the operon encoding the q gene. In trans expression of gp59 repressed Pq-lux in V. parahaemolyticus (Figure 3C). Moreover, Gp59 repression of Pq is likely direct: in recombinant E. coli, arabinose-induction of gp59 is sufficient to repress Pq-lux output 140-fold (Figure 3D). Thus, Gp59 is the phage VP882 repressor of lysis.
We suspected that, like lambda cI, Gp59 could be cleaved during the RecA-activated SOS response (Little, 1984), possibly explaining how MMC induces VP882 phage-mediated lysis (Figure S2B). To test this idea, we generated a functional HIS-HALO-Gp59 protein fusion (Figure S3C) and followed its fate in untreated and MMC-treated recA+ and ΔrecA E. coli cells. Western blot (Figure 3E) revealed a single band of the expected size (~60 kDa) in the untreated samples and lower-molecular-weight bands, presumably cleavage products, in the MMC-treated recA+ sample. The HIS-HALO-Gp59 protein in the ΔrecA sample was unaffected by MMC treatment, suggesting that RecA is required for MMC-mediated cleavage of Gp59. Consistent with this result, addition of MMC to recA+ E. coli carrying the Pq-lux reporter plasmid and HIS-HALO-Gp59 led to a significant increase in light production, whereas if the E. coli was ΔrecA, MMC had no effect (Figure 3F). These results suggest that Gp59 is subject to cleavage in a DNA-damage (MMC) and host-SOS response (recA) dependent manner, and moreover, that the activating effect of MMC on Pq coincides with this cleavage. We hereafter refer to Gp59 as cI.
DNA damage and quorum-sensing control phage lysis via activation of expression of the gene encoding the Q antiterminator.
Our results show that cI directly represses the gene encoding the antiterminator Q, and cI is cleaved upon DNA damage, which launches the phage lysis program. DPO-driven QS, mediated by VqmAPhage, also launches the phage lysis program. We sought to connect the occurrence of these two phage regulatory systems in the context of the natural V. parahaemolyticus host.
First DNA damage:
We added MMC to the V. parahaemolyticus lysogen harboring a plasmid carrying a reporter for the antiterminator Q (Pq-lux) or a reporter for its target operon (Pgp69-lux). Figure 3G shows that light production rapidly increased with a 10 min offset between activation of Pq-lux and activation of Pgp69-lux. The delay between the two reporters is consistent with a model in which the antiterminator Q is produced first and, only after, and as a consequence of Q production, are genes involved in lysis expressed.
Second QS:
We monitored light output from V. parahaemolyticus lysogens carrying either Pq-lux or Pgp69-lux and also the arabinose inducible VqmAPhage construct. Thirty min after arabinose addition, light production from the Pq-lux reporter occurred. Ten min later, light production commenced from the Pgp69-lux reporter (Figure 3G). Again, these results show that q expression occurs prior to genes for lysis. The time delay in the QS experiment roughly matches the delay in the MMC experiment. We conclude that DNA damage-induced lysis and QS-induced lysis converge at the point of activation of expression of q, after which, Q activates gp69 irrespective of which input launched the program.
VqmAPhage activates expression of a gene encoding an antirepressor.
We reasoned that the VqmAPhage transcription factor could act in one of three ways to initiate the Q-directed phage lysis program: by directly activating q expression, by directly repressing cI expression, thereby de-repressing q, or by indirectly repressing cI, again leading to derepression of q. To distinguish between these possibilities, we produced VqmAPhage from one plasmid and monitored Pq-lux expression from a second plasmid in E. coli. No change in light production occurred, excluding the first possibility that VqmAPhage directly activates q expression (Figure S4A, left two bars). To this system, we introduced a third plasmid carrying the phage cI gene. cI repressed Pq-lux expression, but again, no effect on reporter activity occurred when vqmAPhage was expressed, eliminating the second possibility that VqmAPhage directly represses cI (Figure S4A, middle two bars). To investigate the third possibility, that an additional, VqmAPhage-controlled intermediate component exists linking VqmAPhage to repression of cI and subsequent de-repression of q, we introduced the lysis-defective phage carrying Tn5 in q (see Figure 2) into E. coli harboring the plasmid with inducible vqmAPhage and the Pq-lux reporter plasmid. Addition of arabinose to this strain resulted in a twenty-fold increase in Pq-lux output (Figure S4A, right two bars), indicating that an element encoded on the phage is required to connect VqmAPhage to cI expression.
To identify the gene that VqmAPhage controls, we made a recombinant E. coli strain carrying arabinose inducible vqmAPhage, Pq-lux, and the cI repressor gene on a single plasmid. Into this strain, we introduced a library of phage genomic fragments on a vector. We screened the transformants for those that activated Pq-lux expression following induction of vqmAPhage. As shown in Figure 4A, we identified one ~600 bp phage genomic fragment that was sufficient (denoted Active Fragment). This fragment mapped to a region immediately upstream of the vqmAPhage locus and harbored only one complete ORF (gp55) of 240 bp (Figure 1A). We verified by electromobility shift assays (EMSA) that the VqmAPhage protein binds phage DNA immediately upstream of gp55 (Figure S4B).
Figure 4: VqmAPhage activates production of Qtip, a phage-encoded small protein that inactivates the cI repressor by sequestration.

(A) E. coli carrying a plasmid harboring Pq-lux, cI, and arabinose inducible vqmAPhage and a second empty vector, the vector containing a phage genomic fragment encoding gp55 (denoted Active Fragment), or the cloned gp55 gene under the tetA promoter. Where indicated, arabinose and aTc were provided at final concentrations of 0.2% and 100 ng mL−1, respectively. (B) Growth of V. parahaemolyticus harboring phage VP882 and a vector control, the vector with arabinose inducible vqmAPhage, or the vector with aTc inducible gp55. Arabinose, aTc, and MMC were added at 0.2%, 10 ng mL−1, and 50 ng mL−1 respectively. (C) SDS-PAGE analysis of proteins isolated from the following: Sample 1: cells producing HIS-Gp55 alone. Sample 2: cells producing HIS-Gp55 combined with cells producing the HALO-tag (not fused to cI). Sample 3: cells producing HIS-Gp55 combined with cells producing HALO-cI. Sample 4: cells producing HALO-cI in the absence of HIS-Gp55. Proteins were isolated by applying the indicated lysates to cobalt beads, which bind the HIS-tag fused to Gp55. Thus, proteins interacting with HIS-Gp55 are retained on the beads. Lysates (Input), wash (Wash), and protein remaining on cobalt beads (Beads). The gel was stained for HALO using HALO-TMR. Locations of HALO-cI and the HALO-tag are marked with black and white arrowheads, respectively. No other obvious protein bands were present in the bead sample containing HALO-cI and HIS-Gp55 suggesting that Gp55 is specific for cI. Marker (M, PageRuler Plus; representative bands are labeled). (D) Confocal microscopy of E. coli producing HIS-HALO-cI in the absence of Qtip (top left), HIS-HALO-cI combined with 100 ng mL−1 aTc induction of qtip (top right), HIS-HALO (not fused to cI) combined with 100 ng mL−1 induction of qtip (bottom left), HIS-HALO-cI combined with 250 ng mL−1 MMC (bottom right). (E) Confocal microscopy of E. coli producing HIS-HALO-cI and SNAP-Qtip. HALO-cI was visualized with HALO-TMR and SNAP with JF503. The medium contained 100 ng mL−1 aTc. (F) Confocal microscopy of E. coli producing HALO-tagged cIVP882, cIMJ1 or cIlambda (top row), co-producing Qtip (middle row) or co-producing ORF584 (bottom row). Qtip and ORF584 production were induced with 100 ng mL−1 aTc. (G) Growth of V. parahaemolyticus carrying phage VP882 and aTc inducible qtip or orf584. Black; no aTc, white; 10 ng mL−1 aTc. All scale bars are 3 pm. Data in A, B, and G are represented as mean ± std with n=3 biological replicates. See also Figures S4 and S5.
To show that Gp55 links VqmAPhage to cI repression and, in turn, to q de-repression, we cloned the gp55 ORF under a tetracycline inducible promoter on a plasmid (pTetA-gp55) and introduced it into the E. coli strain harboring the above combined reporter. Induction of gp55 was sufficient to generate Pq-lux activity (Figure 4A). We also introduced the pTetA-gp55 construct into the V. parahaemolyticus strain harboring the VP882 lysogen. Induction of gp55 expression caused host cell lysis comparable to when MMC was added or vqmAPhage was induced (Figure 4B). These results indicate that Gp55 is the effector connecting QS to derepression of q to the triggering of host cell lysis.
Gp55 is a 79 amino acid protein with no predicted domains and no significant homology to any protein in the NCBI database. The small size and lack of a DNA-binding domain led us to hypothesize that Gp55 acts post-translationally on cI. To test this notion, we individually produced HALO-cI and HIS-Gp55 from plasmids in recombinant E. coli, collected and combined the cell pellets from the two strains, lysed the cells, and purified the HIS-Gp55 protein. Figure 4C shows that cI binds to Gp55 during purification and Figure S4C shows that the effect of Gp55 on q promoter activity occurs only when cI is present. These results suggest that Gp55 is an antirepressor, which acts directly on the cI repressor and prevents cI from binding to q promoter DNA. We name Gp55 Qtip for Quorum triggered inactivator of cI protein.
To explore the Qtip mechanism of inactivation of cI in vivo, we monitored HALO-cI protein localization at the single-cell level in E. coli. In the absence of Qtip, the cI protein was uniformly dispersed in the cytoplasm (Figure 4D). Production of Qtip caused cI to form foci located primarily at the cell poles and Qtip colocalizes with cI (Figure 4D and 4E) suggesting that Qtip drives aggregation of the cI repressor. By contrast, in MMC-treated cells, the cI protein remained diffuse indicating that cleavage of cI does not cause foci formation (Figure 4D). Thus, both the QS and DNA damage inputs eliminate cI activity, derepress q, and cause lysis, however, their underlying mechanisms of action are different: DNA damage stimulates cI cleavage, whereas VqmAPhage-directed QS produces Qtip, which inactivates the cI repressor via aggregation.
Phage Qtip-type proteins and their host partner repressor proteins are conserved.
Qtip and its role in biology have not previously been reported raising questions about the generality of the sequestration mechanism we discovered. As mentioned, no obvious Qtip homologs exist in genome databases, therefore, we scanned databases for possible cI partners. We identified multiple putative repressors (Figure S4D), one within the Gram-negative marine bacterium, Marinobacterium jannaschii DSM 6295 (Bowditch et al., 1984), from which our own assembly of the raw data (DoE Joint Genome Portal, JGI Project ID: 1011030) showed the cI-like gene was located on a previously unreported ~35 kb contig with predicted repA and telN genes nearby. We confirmed that the repA gene from this element was functional by PCR amplification of the repA locus, ligation to an antibiotic resistance cassette, and demonstration that the resulting vector was maintained as a plasmid in E. coli (Figure S5A). This result suggested that M. jannaschii is lysogenized with a VP882-like phage that harbors the cI-like gene. We hereafter refer to this element as phage MJ1. Co-expression of phage VP882 Qtip with HALO-cI from MJ1 resulted in foci (Figure 4F). Aggregation also occurred when we co-expressed Qtip with a HALO-cI repressor from another vibriophage, VP58.5 (Figure S5B and Zabala et al., 2009). No aggregation occurred when phage VP882 Qtip was co-expressed with lambda HALO-cI (Figure 4F). Lambda cI has less similarity to the VP882, MJ1, and VP58.5 cI proteins than the three proteins have to one another (Figure S4D). Thus, Qtip sequesters closely-related but not distantly-related phage repressors.
The region intervening the phage MJ1 telN and repA genes lacks a predicted vqmAPhage gene. Rather, there is an ORF encoding a different, putative transcription factor. Adjacent to that ORF, encoded in the opposite orientation, is a small gene (orf584). This arrangement exactly parallels the genomic organization of phage VP882 qtip-vqmAPhage. Despite orf584 and qtip having no detectable sequence similarity (Figure S5C), we hypothesized that phage MJ1 orf584 encodes a protein with a Qtip-like function. We cloned and induced phage MJ1 orf584, and it caused aggregation of phage MJ1 cI and phage VP882 cI but not lambda cI (Figure 4F). ORF584 leads to a stripe of cI localization along the length of the cell, whereas Qtip leads to foci at the poles (Figure 4F). Like Qtip, ORF584 is sufficient to disrupt cI repressor activity, as its induction leads to de-repression of phage VP882 Pq expression (Figure S5D) and lysis of V. parahaemolyticus lysogenized with phage VP882 (Figure 4G). These results suggest that ORF584 and Qtip are functionally analogous: they induce aggregation of their native and related phage repressors, launching the phage lysis programs.
Inspired by our above findings, we inspected genomes of nine linear plasmid-like phages (Figures S5E and S6A). In 5 of the 9 cases, in the qtip-vqmAPhage location, we could identify small genes and oppositely-oriented ORFs with strongly predicted DNA-binding domains (Figure S6A). Only phage VP882 harbors an obvious VqmAPhage homolog; we speculate that these additional phage-encoded DNA binding proteins respond to other host-produced cues to activate transcription of their partner genes encoding Qtip-like antirepressors.
VqmAPhage can substitute for VqmAVc but not the reverse.
The conservation between VqmAPhage and vibrio VqmA made us wonder if there was any connection between host QS and phage biology. VqmA-directed QS has only been studied in V. cholerae, where VqmAVc, DPO, VqmR, and the downstream targets were discovered (Liu et al., 2006; Papenfort et al., 2015, 2017). Therefore, we transferred the VP882 prophage to V. cholerae for exploration of this question.
We first tested whether VqmAPhage could replace VqmAVc. As a control we show that, as expected, a ΔvqmAVc V. cholerae strain carrying a PvqmR-mKate2 reporter produces no fluorescence because VqmA is required for activation of vqmR expression (Figure 5A and Papenfort et al., 2015). Reintroduction of vqmAVc or introduction of vqmAPhage on a plasmid restored expression of PvqmR-mKate2 showing that vqmAPhage complements the V. cholerae ΔvqmAVc mutation (Figure 5A). Using EMSAs, we found that both VqmAVc and VqmAPhage bind the V. cholerae vqmR promoter (Figures 5B and S6B). By contrast, while VqmAPhage activates a Pqtip-lux reporter and VqmAPhage binds qtip promoter DNA, V. cholerae VqmA does neither (Figure 5B and 5C and Figure S6B). Consistent with this finding, arabinose induction of vqmAVc does not trigger lysis of V. cholerae carrying the VP882 prophage while vqmAPhage does (Figure 5D). Thus, at least with respect to the tests we have performed, VqmAPhage can substitute for V. cholerae VqmAVc, but the reverse is not the case; V. cholerae VqmAVc cannot carry out VqmAPhage functions. Figure 6A summarizes our results: Regulation of the phage VP882 lysis-lysogeny switch relies on the presence of one of two inputs, DNA damage or host DPO. The latter functions through VqmAPhage. The consequences of DPO regulation of VqmAPhage is activation of phage lysis genes via Qtip, cI, and Q as well as possible modulation of the host QS output via activation of PvqmR.
Figure 5: Asymmetric gene regulation by VqmAPhage and VqmAVc.

(A) Fluorescence from PvqmR-mKate2 in ΔvqmA V. cholerae carrying a vector control, vqmAVc under its native promoter, or arabinose inducible vqmAPhage. The medium contained 0.2% arabinose. RFU denotes relative mKate2 fluoresence units. (B) Electrophoretic mobility shift assay with PvqmR DNA (left, white arrowheads) and Pqtip DNA (right, black arrowheads) together with VqmAVc, VqmAPhage, or no protein (denoted -). Proteins used at 130 nM and 16.25 nM (denoted 8x and 1x, respectively). DNA probes = 540 pM. (C) E. coli carrying a plasmid with Pqtip-lux (black) or PvqmR-lux (white) and a second plasmid with either arabinose inducible vqmAPhage or vqmAVc. Medium contained 0.2% arabinose. We estimate that VqmAVc binds to its own (PvqmR) promoter 456-fold better than the non-cognate promoter (Pqtip), while VqmAPhage binds its target promoter (Pqtip) 4.5-fold better than the non-cognate promoter (PvqmR). PvqmR is activated 7-fold more strongly by the cognate VqmA (VqmAVc) than by the non-cognate VqmAPhage. In contrast, Pqtip is activated 278-fold more strongly by the cognate VqmA (VqmAPhage) than by the non-cognate VqmAVc. See Methods for details on quantitative comparisons. (D) Growth of ΔvqmA V. cholerae lysogenized with phage VP882 and either a vector carrying vqmAVc (left set of bars) or vqmAPhage (right set of bars). Black; no arabinose, white; 0.2% arabinose. In A, C, and D, data are represented as mean ± std with n=3 biological replicates. See also Figure S6.
Figure 6: Quorum-sensing controls the lysis-lysogeny fate decision in phage VP882.

(A) Phage VP882 (multi-colored ring) can lyse or lysogenize its vibrio host. In the lysogenic state, Q (blue) production is repressed by cI (green). Lysis depends on inactivation of cI activity, and that is mediated by two independent inputs, host DNA damage or QS. Host DNA damage (lightning bolt) leads to RecA-assisted proteolysis (scissors) of the cI repressor. The QS input is mediated by VqmAPhage (purple) binding to the host-produced DPO AI, which is derived from threonine via the Tdh enzyme. VqmAPhage bound to DPO activates expression of qtip (red). Qtip aggreagates the cI protein. Irrespective of the input, reduced cI activity leads to derepression of q and subsequent expression of genes involved in lysis (yellow). VqmAPhage, when bound to host DPO, also activates transcription of the host VqmA QS target, vqmR, leading to production of the sRNA VqmR. The VqmR regulon includes genes required for biofilm formation. (B) Growth of WT and Δtdh V. cholerae lysogens. Left; carrying inducible vqmAPhage on a plasmid. Right; carrying inducible qtip on a plasmid. Top; no addition. Bottom; with 100 μM DPO. All strains carry phage VP882 vqmAPhage::Tn5. When present, as indicated in the associated keys, the inducers of vqmAPhage and qtip (arabinose and aTc, respectively), were present at 0.035% and 10 ng mL−1, respectively. (C) qPCR of phage DNA prepared from the samples in (B). Black; Δtdh, white; WT V. cholerae. Viral Load is the amount of VP882-specific DNA in the induced samples relative to the uninduced samples, in all cases relative to DNA of a non-phage plasmid (see Methods). Data are represented as mean ± std with n=3 biological replicates (B) and as mean ± sem with n=3 biological replicates and n=4 technical replicates (C). See also Figures S6 and S7.
Host QS controls the phage VP882 lytic cycle
Our results show that DPO, in conjunction with VqmAphage, drives the phage VP882 lysis-lysogeny decision. If so, host QS should influence the phage lifecylce. To verify this idea, we introduced a mutant VP882 phage carrying vqmAPhage::Tn5 into wild-type and Δtdh V. cholerae, each carrying the vector encoding arabinose-inducible vqmAPhage. This strategy restricted VqmAPhage production to that from the inducible promoter on the plasmid. We measured V. cholerae growth over time by OD600 and we measured phage production (viral load) by quantitative PCR of viral preparations made from identical cultures that were grown in parallel to those from which we obtained the growth data (Figure 6B and 6C). The Δtdh V. cholerae strain underwent minimal lysis and there was less than a 3-fold-change in viral load following induction of vqmAPhage (Figure 6B and 6C, respectively). By contrast, induction of vqmAphage caused WT V. cholerae to lyse and viral load to increase 15-fold relative to when vqmAPhage was not induced (Figure 6B and 6C, respectively). Thus, the VP882 phage fate switch can be driven by endogenously produced host DPO. We also supplied synthetic DPO to the WT and Δtdh strains. In this case, lysis occurred and viral load increased in both strains to similar extents (Figure 6B and 6C, respectively). Morevoer, in the WT strain, 25% more lysis occurred and viral load increased an additional 35% when exogenous DPO was provided compared to when only endogenously-produced DPO was present (Figure 6B and 6C). Thus, given a fixed amount of VqmAPhage, DPO becomes the determining factor controlling lysis by the phage.
To decouple QS regulation from Qtip-directed inactivation of cI, we produced qtip from an inducible promoter, thus bypassing the need for VqmAPhage. Near-complete lysis and increased viral production occured in both the WT and Δtdh strains in response to qtip expression and both processes were insensitive to DPO (Figure 6B and 6C). Similar results were obtained when MMC was added instead of induction of qtip (Figures S6C and 6C), suggesting that, at least under our test conditions, inactivation of cI by Qtip or by RecA-assisted cleavage promotes the same outcomes to the host and the phage populations. However, input from Qtip enables the phage lysis-lysogeny decision to be connected to the cell density of the host.
Phage VP882 as a QS-activated kill switch.
The plasmid-like nature of phage VP882 allows it to be maintained in non-native hosts via its endogenously-encoded replication machinery. Among the limited number of plasmid-like prophages that have been discovered, phage VP882 is unique in that it also encodes a receptor for a host-produced QS AI, which we have shown here activates the phage lytic cycle. This singular arrangement inspired us to explore possibilities for synthetic control of VP882 with the goal of generating a recombinant phage with highly specific, quorum-controlled lytic triggers.
In contrast to the widespread production of DPO by diverse bacteria, the response to DPO, consisting of DPO binding to VqmA and activation of the vqmR promoter is, as far as is known, limited to the vibrio genus. We exploited this asymmetry to design a V. cholerae-specific phage kill switch. To do this, we cloned the VP882 phage q gene under the V. cholerae vqmR promoter (called PvqmR-q) on a plasmid. We introduced this plasmid into E. coli and V. cholerae each lysogenized with the identical lysis-defective phage carrying a Tn5 in the q gene (see Figure 2 and Figure 7A). Introduction of the plasmid carrying PvqmR-q did not alter growth of E. coli (Figure S6D). However, no colonies could be recovered from V. cholerae suggesting that Q protein, made as a consequence of DPO-VqmAVc-directed activation of the vqmR promoter, killed the V. cholerae recipient (Figure 7A and 7B). In contrast to the WT parent, no growth defect/no lysis occurred if the host was ΔvqmA V. cholerae. Indeed, ~5 orders of magnitude more colonies were recovered from the ΔvqmA V. cholerae strain verifying that VqmAVc is required to transduce the DPO information to the vqmR-q promoter on the phage (Figure 7A and 7B).
Figure 7: Reprogrammed phage as kill switches.

(A) A V. cholerae-specific phage kill switch. Phage VP882 q::Tn5 (depicted as the multi-colored ring lacking the blue q gene) exists as a lysogen that is unable to lyse host cells. A functional copy of q, under a V. cholerae-specific promoter (PvqmR-q, depicted as the plasmid carrying the blue gene) is not activated in E. coli (left) because E. coli lacks VqmA, but is activated in V. cholerae that possesses VqmA (middle, blue waves showing Q production and yellow waves showing lysis,). ΔvqmA V. cholerae, by contrast does not activate q (right). We note that all three strains produce DPO. The key for this kill switch is the selective presence of VqmAVc only in the WT V. cholerae host. (B) Killing of WT V. cholerae and growth of ΔvqmA V. cholerae, WT V. parahaemolyticus, and WT V. vulnificus using the V. cholerae-specific targeting strategy from panel A. Relative colonies recovered is the number of exconjugant colonies obtained after the plasmid carrying PvqmR-q was introduced into the indicated strains compared to when a control plasmid was introduced. All strains carry phage VP882 q::Tn5 as a lysogen. (C) S. typhimurium-specific kill switch. Growth of S. typhimurium carrying a plasmid with aTc inducible hilD, the Pinvf-q kill switch, and phage VP882 harboring q::Tn5 (left two bars), inducible hilD and the Pinvf-q kill switch without phage VP882 q::Tn5 (middle two bars), inducible hilD and phage VP882 q::Tn5 without the PinvF-q kill switch (right two bars). Black; no aTc, white; 2 ng mL−1 aTc. In B and C, data are represented as mean ± std with n=3 biological replicates. See also Figure S6.
To test for cross-activation of the V. cholerae vqmR-based kill switch in other vibrios, we introduced the plasmid with PvqmR-q into V. parahaemolyticus and V. vulnificus strains lysogenized with the lysis-defective phage VP882 harboring q::Tn5. Figure 7B shows that the same number of colonies were recovered following introduction of the PvqmR-q kill switch or a control plasmid, despite the fact that both V. parahaemolyticus and V. vulnificus naturally encode vqmA homologs and they make DPO. Moreover, the number of colonies recovered in both cases was 5 orders of magnitude higher than when the killing module was introduced into V. cholerae (Figure 7B). We interpret this result to mean that V. parahaemolyticus and V. vulnificus VqmA cannot recognize the V. cholerae vqmR promoter, and, therefore, they do not activate PvqmR-q expression and, in turn, the downstream lytic functions. Taken together, the PvqmR-q construct is recognized in a V. cholerae VqmAVc-specific manner and thus, this construct only kills V. cholerae, not closely related vibrios.
Phage VP882 kill switches specific for other pathogens.
To demonstrate the modularity of the species-specific kill switch, we engineered it to target a human pathogen unrelated to vibrios, Salmonella typhimurium. InvF is a transcriptional activator of genes encoding proteins essential for S. typhimurium pathogenicity. invF is indirectly activated by the HilD transcription factor. Specifically, HilD activates expression of hilA, encoding a second transcription factor, HilA, that directly activates expression of invF (Lucas and Lee, 2001). We fused the invF promoter to the phage q gene (PinvF-q) and introduced the construct on a plasmid into S. typhimurium lysogenized with phage VP882 harboring q::Tn5, and also carrying a plasmid with tetracycline inducible hilD (pTetA-hilD). This S. thypimurium strain grew without defect (Figure 7C), showing that q is not expressed. Addition of low level aTc (2 ng mL−1) to induce hilD, caused a dramatic decline in growth (Figure 7C). Isogenic S. typhimurium strains containing the PinvF-q and pTetA-hilD plasmids but lacking the VP882 phage or with pTetA-hilD and the phage, but lacking the PinvF-q plasmid were unaffected by aTc (Figure 7C). These results show that S. typhimurium killing is phage- and kill switch-dependent.
Our results demonstrate that we can exploit phage VP882 q::Tn5 as a template, and by exchanging the promoter driving q expression, we can engineer phage kill switch systems that are specific for different bacterial species. In our examples, we used plasmids to carry the PvqmR and PinvF to facilitate testing in different hosts lysogenized with the q::Tn5 phage mutant. However, these fusions can be integrated onto the phage VP882 genome, making the recombinant phage the only required element for the kill switch. Finally, as we have uncovered several points of regulation within the phage lytic program, including the repressor cI and three activators, VqmAPhage, Qtip, and Q that function upstream and downstream of the cI repressor, we can imagine programming phages to contain different logic circuits and multiple input dependencies.
Discussion
We discovered that a phage encodes a QS receptor, VqmAPhage, which binds the host-produced AI DPO, and in turn, activates the phage lytic pathway. Lytic development in phage VP882 is sensitive to at least two inputs, DNA damage and QS (Figure 6A). The first input, DNA damage, leads to proteolysis of the phage VP882 cI repressor, which occurs in a host RecA-dependent fashion and requires no other phage components. QS constitutes the second input, and involves the production of the bacterial host cell density-dependent factor, DPO, to activate a phage-dependent process consisting of VqmAPhage-induced expression of qtip; Qtip sequesters the phage cI repressor and leads to lysis. To our knowledge, phage VP882 represents the first example of a phage lysis-lysogeny decision mediated by a phage-encoded receptor monitoring a host-produced QS AI.
One caveat of our studies is that we induced vqmAPhage expression in all of our analyses. Analogous to other temperate phages such as lambda, the only phage gene strongly expressed during lysogeny is that encoding the cI repressor (Ptashne, 2004). We do not know the cue that naturally activates vqmAPhage expression, however, we have identified two feedback mechanisms that could amplify VqmAPhage production, once initiated. First, VqmAPhage activates its own expression (Figure S7A). Second, phage plasmid copy number increases in the absence of cI (Figure S7B) similar to the plasmid-like phage N15 (Lobocka et al., 1996). Qtip-directed inactivation of cI thus elevates phage copy number, increasing the pool of vqmAPhage and qtip DNA that can be transcribed. Aided by these two mechanisms, we suspect that low-level production of VqmAPhage, triggered by some environmental stimulus or unstable state, is sufficient to drive the phage lysis program. Identifying natural induction conditions is the subject of our future work.
The Qtip mechanism is reminiscent of small phage antirepressors such as those from coliphage P1 (Coi, 7.7 kDa; (Heinzel et al., 1992)), 186 (Tum, 16 kDa; (Shearwin et al., 1998)), and N15 (AntC, 8.2 kDa; (Mardanov and Ravin, 2007)) which engage in non-covalent interactions with partner repressor proteins, thereby, inhibiting repressor activity. Unlike in phage VP882, the promoters driving these antirepressors are LexA-controlled, indicating they are induced exclusively by DNA-damage, not by QS.
While VqmAPhage and Qtip appear unique to phage VP882, their locations in the VP882 genome, between two essential genes present in all known plasmid-like phages, allowed us to identify ORFs with putative DNA binding domains and Qtip-like proteins in the identical locations in other plasmid-like phage genomes. Our examination of one such phage, MJ1, shows that its Qtip-like protein, ORF584, can substitute for Qtip, sequester a non-cognate repressor protein, albeit with a different sub-cellular localization, and launch the phage VP882 lysis program. By contrast, Qtip and ORF584 do not function with the distantly-related lambda repressor.
With respect to vibriophage, the antirepressor, RstC, encoded by the CTX satellite phage RS1, which is integrated into some V. cholerae genomes induces aggregation of multiple RstR repressor variants that share little identity (Davis et al., 2002). Davis hypothesized that RstC recognizes a common structural motif in the RstR proteins. Similarly, Qtip can aggregate multiple repressor proteins, and moreover, different Qtip-like antirepressors with little amino acid identity can aggregate the same phage repressor protein. To our knowledge, this latter feature is new.
We uncovered an asymmetric binding pattern: VqmAPhage binds its own (qtip) and host (vqmR) target promoters, whereas VqmAVc only binds its own (vqmR) target promoter (Figure 5). The lack of sequence similarity between PvqmR and Pqtip (Figure S6B and Papenfort et al., 2015), coupled with the ability of VqmAPhage to bind both, could represent a strategy by which the VP882 phage can tune-in and respond to host QS, while maintaining the ability to execute its own Qtip-mediated lysis-lysogeny pathway without host interference. We wonder how this asymmetry plays out in natural settings. As one clue, database analyses show that V. cholerae strain FORC 055, has two CRISPRs matching regions of the VP882 genome (Figure S7C) suggesting that V. cholerae is vulnerable to phage VP882 and needs to defend itself.
We also consider how phage-vibrio interactions could occur during human infection. V. cholerae encounters the host and associated microbiota upon infection. DPO is produced by the host microbiota from the threonine-rich resource, mucin, and DPO represses V. cholerae biofilm formation and virulence. Thus, the human host and its microbiota “team up” to defend against V. cholerae using DPO. DPO is also produced and detected by V. cholerae leading to dispersal, a crucial step in the V. cholerae lifecycle because it maximizes dissemination to new hosts. We now propose that a phage also uses DPO to trigger dissemination, in this case, via host V. cholerae cell lysis at high host-cell population density. This strategy likely maximizes phage infection of the next V. cholerae cell. Thus, interactions across the eukaryotic, bacterial, and viral kingdoms all rely on one QS AI, DPO. Presumably, each entity in these combined beneficial and parasitic partnerships is garnering the information encoded in the DPO molecule to optimize its survival and reproduction.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Bonnie L. Bassler (bbassler@princeton.edu).
Experimental Model and Subject Details
Unless otherwise indicated, E. coli, V. cholerae, V. parahaemolyticus, and V. vulnificus were grown with aeration in Luria-Bertani (LB-Miller, BD-Difco) broth at 37°C. Low salt LB (Lennox) broth w as used for S. typhimurium, and M. jannaschii was grown in Marine Broth 2216 (BD-Difco) at room temperature. M9 minimal medium supplemented with 200 mM NaCl (for V. parahaemolyticus and V. cholerae) was used where indicated. Strains used in this study are listed in Table S1. Unless otherwise noted, antibiotics, were used at: 50 U mL−1 polymyxin B (Pb, Sigma), 100 μg mL−1 ampicillin (Amp, Sigma), 100 μg mL−1 kanamycin (Kan, GoldBio), 80 μg mL−1 Zeocin (Zeo, Thermo), and 5 μg mL−1 or 10 μg mL−1 chloramphenicol (Cm, Sigma). Cm concentration was 10 μg mL−1 when it was the only antibiotic present and 5 μg mL−1 when used in conjunction with other antibiotics. When multiple plasmids were simultaneously present within a single strain, care was exercised to limit the number of passages. Inducers were used as follows: E. coli: 250 ng mL−1 mitomycin C (MMC, Sigma), 100 ng mL−1 anhydrotetracycline (aTc, Clontech), 0.2% L-arabinose (ara, Sigma), 0.5 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG, GoldBio). Vibrios: 50 ng mL−1 MMC, 10 ng mL−1 aTc, 0.2% ara. S. typhimurium: 2 ng mL−1 aTc.
Method Details
DPO Isolation from Protein Extracts and DPO Bioassay
The HIS-VqmAPhage, HIS-VqmAVc, and HIS-LuxO proteins were produced and purified as described (Papenfort et al., 2017), with the exception that HIS-VqmAVc was treated with thrombin CleanCleave (Sigma) post-purification. Briefly, preparations containing DPO were obtained from E. coli BL21(DE3) producing HIS-VqmAPhage (pJES-095), HIS-VqmAVc (pKP-445), and HIS-LuxO (pWN-001) based on a previously reported method (Papenfort et al., 2017). Briefly, the three strains were grown and protein production was induced by adding 0.5 mM IPTG at OD600 = 0.5-0.7. Following an additional 4 h of growth, cell pellets were collected from 1 L cultures. The pellets were resuspended in 10 mL of lysis buffer (20 mM sodium phosphate pH 7.4, 500 mM NaCl, and 30 mM imidazole) and the cells were lysed by sonication (Branson). The lysates were subjected to centrifugation at 13,000 g × 30 min at 4°C, and each clarified supernatant was applied to 3 mL Ni-NTA Superflow (Qiagen) resin. The resins were washed with two rounds of lysis buffer, two rounds of lysis buffer lacking imidazole, and, finally, two rounds of water (10 mL for each wash step). Next, the resins were resuspended in an equivalent volume of 40% ethanol and heated at 70°C for 10-20 min. De natured protein and Ni-NTA resin were removed by centrifugation (13,000 g × 10 min) leaving released DPO in the supernatants. Supernatants were dried and resuspended in 100 μL water and then assessed for DPO by \ bioassay or LC-MS (LC-MS is described below). The bioassay was performed as in (Papenfort et al., 2017) with some modifications, and is shown in Figure 1D. Overnight cultures of E. coli BW25113 tdh::kanR harboring pKP-367 and pJES-045 were back diluted 1:100 (from LB) into M9 minimal medium containing 0.4 mM leucine, 0.01% arabinose, and appropriate antibiotics. The cultures were grown to OD600 ~0.2, and 198 μL aliquots dispensed into wells of a 96 well plate. Aliquots of the small molecule preparations from above were added to each well (2 μL per well). Control wells received 2 μL of water or 2 μL of 1 mM synthetic DPO (10 μM final concentration). OD600 and bioluminescence were measured in a platereader, as described below (see Growth, Lysis, and Reporter Assays and Table S4 for timing).
Liquid Chromatography-Mass Spectrometry for DPO Detection
Standards were prepared in 50% MeOH. Samples and standards were loaded onto a 1 mm × 75 mm C12 column (ACE 3 C18 PFP, Mac-Mod) using a Shimadzu HPLC system and PAL auto-sampler (20 μL per injection) at a flow rate of 70 μL min−1. The column was maintained at 45°3 using a column oven. The column was connected inline to an electrospray source coupled to an LTQ-Orbitrap XL mass spectrometer (Thermo). Caffeine (2 pM μL−1 in 50% acetonitrile with 0.1% formic acid) was injected as a lock mass through a tee at the column outlet using a syringe pump at 45 μL min−1 (Harvard PHD 2000). Chromatographic separation was achieved with a linear gradient from 1% to 45% B in 6 min (A: 0.1% formic acid, B: 0.1% formic acid in acetonitrile) with an initial 1 min hold at 1% B and followed by 4 min wash at 100% B and equilibration for 8 min with 1% B (total 20 min program). Electrospray ionization was achieved using a spray voltage of 4.50 kV aided by sheath gas (Nitrogen) at a flow rate of 12 (arbitrary units) and auxiliary gas (Nitrogen) flow rate of 1 (arbitrary units). Full scan MS data were acquired in the Orbitrap at a resolution of 60,000 in profile mode from the m/z range of 110-220. Raw files were imported into Skyline v4.1 (MacCoss Lab) and peak areas for DPO were extracted using the small molecule workflow.
Cloning Techniques
Primers and dsDNA (gene blocks) used for plasmid construction are listed in Tables S2 and S6, respectively, both obtained from Integrated DNA Technologies. Plasmids are listed in Table S3. Plasmid combinations are additionally organized according to the figures and strains in which they appear in Table S4. The publicly available VP882 annotations associated with the available sequence (NC_009016.1; (Lan et al., 2009)) were used for reference and cloning with the exception of cI and q (gp59 and gp62, respectively), which, based on our own experimental characterization and sequence alignments, we deemed to start 27 codons downstream from the currently annotated start site of cl (gp59) and 39 codons downstream from the currently annotated start site of q (gp62). Gibson assembly, intramolecular reclosure, and traditional cloning methods, including blunt and restriction enzyme-based cloning, were employed for all cloning (see Table S5). PCR with Q5 High Fidelity Polymerase was used to generate insert and backbone DNA. Primer pairs and templates are described in Table S5. Gibson assembly relied on the HiFi DNA assembly mix. Intramolecular reclosure used the KLD enzyme mix. In traditional cloning, inserts were treated with DpnI and backbones with DpnI and CIP. Ligations were performed with T4 DNA ligase. All enzymes used in cloning were obtained from NEB. Constructs were initially transformed into TOP10 E. coli (Invitrogen). DNA was introduced by electroporation using 0.1 cm gap cuvettes (USA Scientific) with a Bio-Rad MicroPulser.
Phage Curing and Phage/Plasmid DNA Transfer
Plasmids and VP882 phage were introduced into vibrios by triparental matings using an E. coli donor, E. coli helper, and the vibrio recipient based on previously described protocols (Bassler et al., 1993). Briefly, matings were conducted by spotting a 3:3:1 mixture of donor, helper, and recipient cells, respectively, onto LB plates and incubating at 37°3 for 6-12 h, afterwhich the mixtures were streaked onto medium containing Pb and the appropriate antibiotic to select for vibrios carrying the transferred element. V. parahaemolyticus was cured of phage VP882 via introduction of a plasmid (pJES-174) containing a fragment harboring the native VP882 origin of replication and plating with selection for the plasmid. The strain was subsequently cured of pJES-174 by growth in the absence of antibiotic selection. In S. typhimurium (Figure 7C), plasmids and phages were introduced using electroporation and conjugation, respectively, as follows: The non-conjugatable pTetA-hilD plasmid (pJES-171) was first introduced into WT S. typhimurium by electroporation, conferring Amp resistance. The q::Tn5 phage or the Pinvf-q kill switch (pJES-167) was next transferred into this strain by conjugation, followed by selection with Amp and Cm or Amp and Kan, respectively (Figure 7C, left and center sets of bars, respectively). Finally, when pJES-171, pJES-167, and q::Tn5 phage were required simultaneously (Figure 7C, right set of bars), pJES-167 was conjugated into the Amp- and Cm-resistant S. typhimurium strain harboring pJES-167 and the q::Tn5 phage with plating on Amp, Cm, and Kan. The efficiency of the V. cholerae-specific kill switch was quantified by calculating the difference in CFUs obtained from the indicated vibrio strains when mated with E. coli carrying a control plasmid (pJY-014) compared to when mated with E. coli carrying the PvqmR-q kill switch plasmid (pJES-117).
Growth, Lysis, and Reporter Assays
Unless otherwise noted, overnight cultures were back diluted 1:1000 into fresh medium with appropriate antibiotics and strains were grown to OD600 0.5-0.6. Cultures were back diluted 1:20, and grown to OD600 0.1, before being dispensed (200 μL) into 96 well plates (Corning Costar 3904). MMC, ara, or aTc was added as specified. Wells that did not receive treatment received an equivalent volume of water. Plates were shaken at 37°C and a BioTek Syne rgy Neo2 Multi-Mode reader was used to measure OD600, OD600 and bioluminescence, or OD600 and fluorescence. Measurement times for single time point assays are provided in Table S4. In Figure S2D, V. parahaemolyticus was monitored at 6 h, V. cholerae at 8 h, and E. coli at 12 h. Relative light units (RLU) and relative fluorescence units (RFU) were calculated by dividing the bioluminescence and fluorescence readings, respectively, by the OD600 at that time. In the case of fluorescence assays, M9 supplemented with 0.4% casamino acids (BD-Difco) was used and readings were made by excitation at 588 nm and emission at 633 nm. For data in Figure 3G, the RLU of each sample that received MMC or ara was subtracted from the value of the control sample that received water. In Figures 6B and S6C, cultures were back-diluted 1:167 (6 μL into 1 mL) directly from glycerol stocks into M9 supplemented with 0.4% casamino acids and appropriate antibiotics before being dispensed into wells containing the indicated inducers, DPO, or water (control). The glycerol stocks were prepared from three independent colonies of each strain at a single OD600. Care was taken to ensure that the OD600 between these strains was identical prior to storage. Parallel cultures of the same strains were used for viral preparations (Figure 6C). Additional details on procedures and growth for viral preparations are noted in the appropriate section below.
Based on the reporter results in Figure 5C, we made several quantitative comparisons to assess binding of the different VqmA proteins to their cognate and non-cognate promoters. Those values are presented in the legend of Figure 5C and were calculated as follows:
We estimated VqmAVc binding at the cognate vqmR promoter compared to the non-cognate qtip promoter by dividing the average RLU value obtained for the VqmAVc-to-PvqmR interaction by that of the VqmAVc-to-Pqtip interaction. We estimated binding of VqmAPhage to its cognate qtip promoter compared to the non-cognate vqmR promoter by dividing the average RLU value obtained for the VqmAPhage-to-Pqtip interaction by that of the VqmAPhage-to-PvqmR interaction. We estimated the extent to which PvqmR is activated by its cognate VqmAVc compared the non-cognate VqmAPhage by dividing the average RLU value obtained for the VqmAVc-to-PvqmR interaction by that of the VqmAPhage-to-PvqmR interaction. We estimate the extent to which Pqtip is activated by its cognate VqmAPhage compared to the non-cognate VqmAVc by dividing the average RLU value obtained for the VqmAPhage-to-Pqtip interaction by that of the VqmAVc-to-Pqtip interaction.
Phage DNA Isolation
Phage DNA was purified from phage particles present in the supernatants of lysed cells (virion) and from host cell pellets lysogenized with VP882 (prophage).
Prophage DNA was purified using the FosmidMAX kit (Lucigen) according to the manufacturer’s protocol for 1.5 mL cultures.
Virion DNA was isolated using a Phage DNA isolation kit (Norgen Biotek) including the DNase I and Proteinase K treatment steps indicated in the manufacturer’s protocol. In Figure S2E, for each sample (each lane in Figure S2E), elutions from no less than 10 purifications were pooled, subjected to electrophoresis on a 0.5% agarose gel, and the contents from the location on the gel corresponding to the VP882 genome size was extracted. PCR and sanger sequencing were used to verify that the band consisted of VP882 DNA. DNA purified from the gel-extracted bands was visualized on an agarose gel, presented in Figure S2E.
In vitro Tn5 (IVT) Mutagenesis
VP882 phage DNA was used as the target for IVT. VP882 prophage DNA was isolated as described above, with the addition of a final digestion step to remove residual genomic DNA. Removal was accomplished by treatment with EcoRI, a restriction site that is absent in the VP882 genome, followed by Plasmid-Safe DNase (Lucigen). The transposon was constructed by PCR amplification of pRE112 (Edwards et al., 1998) with primers JSO-929 × 930 and JSO-931 × 932 (see Table S2) to make recombinant VP882 phage carrying an oriR6ky-oriT-cmR Tn5 transposon. To engineer the recombinant VP882 phage carrying the araC-pBAD-vqmAPhage-oriT-kanR Tn5 transposon, PCR amplification of pJES-052 with JSO-1029 × 1031 was used (Table S2). All IVT reactions were carried out in a PCR thermocycler with EZ-Tn5 transposase (Lucigen) as described by the manufacturer for in vitro insertion reactions.
RT-qPCR
Overnight cultures of V. parahaemolyticus carrying pJES-052 and either WT phage VP882 or phage VP882 q::Tn5 were back diluted 1:1000 and grown with shaking at 37°C. Upon reaching OD600 0.2-0.4, cultures were divided in half, ara (0.2% final) was added to one aliquot and an equal volume of water to the other. These preparations were allowed to continue to grow at 37°C for 60 min. A total of 1.5-2 OD600 worth of cells was treated with RNAProtect Bacteria Reagent (Qiagen) according to the supplier’s protocol. The cells were pelleted at 4,000 RPM for 10 min at 4°C. Pellets were stored at −80°C prior t o processing. Total RNA was isolated from three independent cultures per condition using the RNeasy Mini Kit (Qiagen). The samples were treated with DNAse using a TURBO DNA-free Kit (Thermo). cDNA was prepared from 1.5 μg RNA as described (Tu and Bassler, 2007) using SuperscriptIII reverse transcriptase (Thermo). SYBR Green mix (Quanta) and Applied Biosystems QuantStudio 6 Flex Real-Time PCR detection system (Thermo) were used for real-time PCR. Each cDNA sample was amplified in technical quadruplicate. Data were analyzed by a comparative CT method, in which the indicated target gene (vqmAPhage or gp69-71) was normalized to an internal bacterial control gene (hfq). The reference sample for all comparisons was the WT V. parahaemolyticus strain lysogenized with phage VP882 q::Tn5 to which no arabinose was added.
qPCR and Viral Preparations
Viral preparations consisted of purified non-chromosomal DNA (ZR BAC kit, Zymo Research) prepared from 2 mL of cells of the strains in Figures 6B and S6C. The viral preparations used for Figure 6C were made identically to those in Figures 6B and S6C except that the supplemented compounds or water control were withheld until the samples had reached an OD600 of 0.2. Following addition of the indicated compounds or water control, the cultures were returned to growth for 3 h prior to harvesting. This alteration in procedure was necessary to enable growth of sufficient cells for use with the viral preparation kit. 0.5 ng of purified non-chromosomal DNA was used for each qPCR reaction. qPCR reactions were performed as described above for qRT-PCR reactions. Data were analyzed by a comparative CT method in which the VP882 phage-specific primer set (JSO-1399 × 1400) was normalized to the non-phage plasmid-specific primer set (JSO-1401 × 1402). The reference sample for each comparison was the isogenic strain (Δtdh or WT) that was not induced.
Western Blot to Assess HALO-cI.
The pJES-134 plasmid carrying the HIS-HALO-cI protein construct was transformed into recA+ E. coli T7Express (NEB) and ΔrecA E. coli BLR(DE3) (Novagen). Overnight cultures were back-diluted 1:100 into fresh medium and grown to OD600 ~ 0.4-0.6. The cultures were divided in half. To one aliquot, MMC (250 ng mL−1 final conc.) was added, and to the other aliquot, an equivalent volume of water was added. Samples were incubated for an additional 2.5 h. 1 mL of culture was collected from each sample, the cells were pelleted (13,000 g × 1 min), and resuspended in HALO western lysis buffer (30 μL B-PER complete (Thermo), 1x Halt protease inhibitor cocktail (Thermo), 0.5 mM EDTA, 5 μM HALO-TMR ligand (Promega), 1 μL benzonase (Millipore)). The samples were incubated in the dark at 37°C for 15-20 min before addition of 10 μL Laemmli sample buffer (Bio-Rad), followed by incubation at 70°C for 15-20 min. 10 μL of each sample was separated by SDS-PAGE in 4–20% Mini-Protein TGX gels (Bio-Rad) and imaged using an ImageQuant LAS 4000 (GE) imager under the SYBR-Green setting.
Co-Immunoprecipitation
Cultures of E. coli BL21(DE3) producing HIS-Qtip, HALO-cI, and HALO were grown overnight, back-diluted 1:100, and grown to OD600 ~ 0.5-0.8. 0.5 mM IPTG was added, followed by 4 h continued incubation. The cultures were moved to ice and 75 mL of cells containing HIS-Qtip were divided into three equal aliquots of 25 mL and placed in 50 mL conical tubes. To one tube, 25 mL of cells that had produced HIS-Qtip were added. To a second tube, 25 mL of cells that had produced HALO-cI were added. To the third tube, 25 mL of cells that had produced HALO were added. Each mixture was made in triplicate and immediately pelleted at 4,000 RPM for 10 min at 4°C . The cell pellets were stored at −80°C.
Cell lysis was carried out by resuspending each pellet in 1 mL of lysis buffer (20 mM Tris-HCl pH 8, 150 mM NaCl, 1x protease inhibitor cocktail, and benzonase) followed by sonication. 50 μL of magnetic cobalt based beads (Thermo, Dynabeads His-Tag Isolation and Pulldown) were added to each lysate and the samples incubated at RT for 10 – 20 min with gentle agitation. The samples were placed in a magnetic stand separator (Thermo) for 2 min. Proteins that remained associated with the magnetic particles were retained on the tube-wall facing the magnet while unbound and non-specifically bound proteins were removed via three washes with wash buffer (lysis buffer lacking protease inhibitor and benzonase). The washed magnetic beads were resuspended in 100 μL of wash buffer. 30 μL aliquots were taken as specified (input, first wash, cobalt beads). HALO-TMR ligand (5 μM) was added to each aliquot and the samples incubated for 10 – 20 min at RT, protected from light. 10 μL of 4x Laemmli buffer was added to each sample followed by incubation in a PCR machine at 70°C for 15 min, after which the samples were separated by SDS-PAGE and imaged using an ImageQuant LAS 4000 imager under the SYBR-Green setting. Samples were diluted prior to loading onto a gel, as follows: Input, 1:5; Wash 1:2; Beads 1:4.
Electrophoretic Mobility Shift Assays (EMSA)
5’ biotinylated forward primers and unmodified reverse primers (Table S2) were used to make probes. Purified VP882 DNA was used as the template to make all the probes except for the one for PvqmR. In that case, V. cholerae genomic DNA was used. EMSAs were performed using the LightShift Chemiluminescent EMSA Kit (Thermo) according to the manufacterer’s general protocol. Briefly, the indicated quantities of proteins and probes noted in the figures/legends were mixed in binding buffer (10 mM Tris-HCl pH 8, 1 mM EDTA, 1 mM DTT, 50 mM KCl, 1.5 μg/mL poly(dI-dC), 50 μg/mL BSA, and 2.5% glycerol), and incubated at RT for 15-20 min. The samples were subsequently loaded onto a Novex 6% DNA Retardation Gel (Thermo) at 4°C and electrop horesed in 1x TBE at 100 V for 1.5-2.5 h. The samples were transferred from the gel to a charged nylon membrane (Amersham Hybond-XL) in 0.5x TBE at 300 mA for 30-40 min at 4°C. The membrane was cross-linked in a Stratagene Stratalinker 2400 UV crosslinker using the auto-crosslink setting. The membrane was subsequently treated with the LightShift kit buffers as follows: Incubation in Blocking Buffer for 15 min, then Blocking Buffer with Stabilized Streptavidin-Horseradish Peroxidase Conjugate for 15 min, washed with four successive rounds of Wash Buffer (5 min each), incubated in Substrate Equilibration Buffer for 5 min, and lastly, incubated in a 1:1 mixture of Luminol and Enhancer solution. After 2 min of incubation in the dark, the membrane was imaged using a ImageQuant LAS 4000 imager using the chemiluminescence setting. Exposure times never exceeded 1 min.
Confocal Microscopy
Overnight cultures, started from single colonies of E. coli (T7Express) carrying HALO and/or SNAP fusions (Table S4) were back-diluted 1:1000, and grown at 37°C to OD 600 ~ 0.2. Either MMC, aTc, or ethanol in water was added. The so-treated samples were returned to shaking at 37°C for an additional 60 min prior to live-cell staining and imaging. Live-cell staining was performed using a protocol adapted from (Ke et al., 2016). Briefly, HALO-TMR was added to each sample at 10 μM. The samples were incubated in the dark for 15-20 min at 37°C. The cells were pelleted at 13,000 g × 1 min, washed twice with M9 medium, and resuspended in 50-150 μL of fresh M9 medium. 5-10 μL of each sample was spotted onto a glass coverslip and overlaid with a small amount of M9-agar. The samples were imaged on a Leica SP8 Confocal microscope. HALO-TMR was excited with 561 nm light and detected within a range of 579-622 nm. For SNAP-Qtip co-localization experiments, the SNAP-specific dye JF503 (Grimm et al., 2017) was added at 10 μM at the same time as the HALO-TMR reagent was added. SNAP was visualized by excitation at 504 nm and detection within 516-540 nm. All images were acquired under the same conditions (laser power and gain) in LASX (Leica Microsystems). Images were imported into Fiji for processing, which consisted solely of adjusting brightness and contrast (min-max).
Quantification and Statistical Analysis
Data are presented as the mean ± std unless otherwise indicated in the figure legends. The number of technical and independent biological replicates for each experiment is indicated in the figure legends or methods. No blinding or randomization was used in these experiments.
All data, excluding those generated from microscopy, were recorded, analyzed, and plotted using Microsoft Excel and GraphPad Prism 6. Sequencing results, plasmid maps, and genomes were assembled using SnapGene (GSL Biotech), UGENE (Unipro), Geneious (Biomatters Limited), and ApE (M. Wayne Davis). Raw M. jannaschii DSM 6295 sequencing data were accessed from the Joint Genome Portal (DoE) at: https://genome.jgi.doe.gov/portal/MarianDSM6295/MarjanDSM6295.info.html. Phage MJ1 was identified from assembly of raw data in Geneious. Imaging data were collected, processed, and prepared using LASX and Fiji, as described.
Data and Software Availability
All experimental data that support the findings of this study, including the re-assembly of the publicly available M. jannaschii raw data mentioned here, are available from the corresponding author upon request.
Supplementary Material
Highlights:
A phage encodes a quorum-sensing receptor that detects a host produced autoinducer
Phage detection of the autoinducer controls the phage lytic-lysogeny fate switch
A new phage protein, Qtip, sequesters and inactivates the phage cI repressor
Recombinant phages are engineered to execute lysis in response to user-defined cues
Acknowledgments
This work was supported by the Howard Hughes Medical Institute, NIH Grant 2R37GM065859, National Science Foundation Grant MCB-1713731 and the Max Planck Society-Alexander von Humboldt Foundation (to B.L.B.). J.E.S. was supported by the Department of Defense (DoD) through the National Defense Science & Engineering Graduate Fellowship (NDSEG) Program. We thank Jian-Ping Cong, Jen Munko, and Kai Papenfort for assistance in locating VP882 and the Food Industry Research and Development Institute for providing the VP882 lysogen. We thank Nina Molin Høyland-Kroghsbo for thoughtful discussions and Tharan Srikumar and Saw Kyin for help in the Princeton Proteomics & Mass Spectrometry Core.
Footnotes
Supplemental Information
Supplemental information includes six tables and seven figures and can be found with this article online.
Declaration of Interests
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, and Mori H (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol 2, 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler BL, Wright M, Showalter RE, and Silverman MR (1993). Intercellular signalling in Vibrio harveyi: sequence and function of genes regulating expression of luminescence. Mol. Microbiol 9, 773–786. [DOI] [PubMed] [Google Scholar]
- Bina XR, Wong EA, Bina TF, and Bina JE (2014). Construction of a tetracycline inducible expression vector and characterization of its use in Vibrio cholerae. Plasmid 76, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose JL, Rosenberg CS, and Stabb EV (2008). Effects of luxCDABEG induction in Vibrio fischeri: enhancement of symbiotic colonization and conditional attenuation of growth in culture. Arch. Microbiol 190, 169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowditch RD, Baumann L, and Baumann P (1984). Description of Oceanospirillum kriegii sp. nov. andO. jannaschii sp. nov. and assignment of two species ofAlteromonas to this genus as O. commune comb. nov. and O. vagum comb. nov. Curr. Microbiol 10, 221–229. [Google Scholar]
- Davis BM, Kimsey HH, Kane AV, and Waldor MK (2002). A satellite phage-encoded antirepressor induces repressor aggregation and cholera toxin gene transfer. EMBO J. 21, 4240–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RA, Keller LH, and Schifferli DM (1998). Improved allelic exchange vectors and their use to analyze 987P fimbria gene expression. Gene 207, 149–157. [DOI] [PubMed] [Google Scholar]
- Erez Z, Steinberger-Levy I, Shamir M, Doron S, Stokar-Avihail A, Peleg Y, Melamed S, Leavitt A, Savidor A, Albeck S, et al. (2017). Communication between viruses guides lysis-lysogeny decisions. Nature 541, 488–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruque SM, and Mekalanos JJ (2012). Phage-bacterial interactions in the evolution of toxigenic Vibrio cholerae. Virulence 3, 556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, Muthusamy AK, Liang Y, Brown TA, Lemon WC, Patel R, Lu R, Macklin JJ, Keller PJ, Ji N, et al. (2017). A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 14, 987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel T, Velleman M, and Schuster H (1992). C1 repressor of phage P1 is inactivated by noncovalent binding of P1 Coi protein. J. Biol. Chem 267, 4183–4188. [PubMed] [Google Scholar]
- Ke N, Landgraf D, Paulsson J, and Berkmen M (2016). Visualization of Periplasmic and Cytoplasmic Proteins with a Self-Labeling Protein Tag. J. Bacteriol 198, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan S-F, Huang C-H, Chang C-H, Liao W-C, Lin I-H, Jian W-N, Wu Y-G, Chen S-Y, and Wong H (2009). Characterization of a New Plasmid-Like Prophage in a Pandemic Vibrio parahaemolyticus O3:K6 Strain. Appl. Environ. Microbiol 75, 2659–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little JW (1984). Autodigestion of lexA and phage lambda repressors. Proc. Natl. Acad. Sci 81, 1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Hsiao A, Joelsson A, and Zhu J (2006). The Transcriptional Regulator VqmA Increases Expression of the Quorum-Sensing Activator HapR in Vibrio cholerae. J. Bacteriol 188, 2446–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobocka MB, Svarchevsky AN, Rybchin VN, and Yarmolinsky MB (1996). Characterization of the primary immunity region of the Escherichia coli linear plasmid prophage N15. J. Bacteriol 178, 2902–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas RL, and Lee CA (2001). Roles of hilC and hilD in Regulation of hilA Expression in Salmonella enterica Serovar Typhimurium. J. Bacteriol 183, 2733–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardanov AV, and Ravin NV (2007). The Antirepressor Needed for Induction of Linear Plasmid-Prophage N15 Belongs to the SOS Regulon. J. Bacteriol 189, 6333–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarter LL (1998). OpaR, a Homolog of Vibrio harveyi LuxR, Controls Opacity of Vibrio parahaemolyticus. J. Bacteriol 180, 3166–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MB, Skorupski K, Lenz DH, Taylor RK, and Bassler BL (2002). Parallel Quorum Sensing Systems Converge to Regulate Virulence in Vibrio cholerae. Cell 110, 303–314. [DOI] [PubMed] [Google Scholar]
- Papenfort K, and Bassler BL (2016). Quorum sensing signal–response systems in Gram-negative bacteria. Nat. Rev. Microbiol 14, 576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfort K, Förstner KU, Cong J-P, Sharma CM, and Bassler BL (2015). Differential RNA-seq of Vibrio cholerae identifies the VqmR small RNA as a regulator of biofilm formation. Proc. Natl. Acad. Sci 112, E766–E775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfort K, Silpe JE, Schramma KR, Cong J-P, Seyedsayamdost MR, and Bassler BL (2017). A Vibrio cholerae autoinducer–receptor pair that controls biofilm formation. Nat. Chem. Biol 13, 551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M (2004). A Genetic Switch, Third Edition: Phage Lambda Revisited (Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- Ravin NV (2015). Replication and Maintenance of Linear Phage-Plasmid N15. Microbiol. Spectr 3. [DOI] [PubMed] [Google Scholar]
- Shearwin KE, Brumby AM, and Egan JB (1998). The Tum Protein of Coliphage 186 Is an Antirepressor. J. Biol. Chem 273, 5708–5715. [DOI] [PubMed] [Google Scholar]
- Thelin KH, and Taylor RK (1996). Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect. Immun 64, 2853–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu KC, and Bassler BL (2007). Multiple small RNAs act additively to integrate sensory information and control quorum sensing in Vibrio harveyi. Genes Dev. 21, 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldor MK, and Friedman DI (2005). Phage regulatory circuits and virulence gene expression. Curr. Opin. Microbiol 8, 459–465. [DOI] [PubMed] [Google Scholar]
- Waldor MK, and Mekalanos JJ (1996). Lysogenic Conversion by a Filamentous Phage Encoding Cholera Toxin. Science 272, 1910–1914. [DOI] [PubMed] [Google Scholar]
- Wei Y, Ng W-L, Cong J, and Bassler BL (2012). Ligand and antagonist driven regulation of the Vibrio cholerae quorum-sensing receptor CqsS. Mol. Microbiol 83, 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Sharo AG, Stone HA, Wingreen NS, and Bassler BL (2016). Vibrio cholerae biofilm growth program and architecture revealed by single-cell live imaging. Proc. Natl. Acad. Sci 113, E5337–E5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabala B, Hammerl JA, Espejo RT, and Hertwig S (2009). The Linear Plasmid Prophage Vp58.5 of Vibrio parahaemolyticus Is Closely Related to the Integrating Phage VHML and Constitutes a New Incompatibility Group of Telomere Phages. J. Virol 83, 9313–9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental data that support the findings of this study, including the re-assembly of the publicly available M. jannaschii raw data mentioned here, are available from the corresponding author upon request.