

SUMMARY
Natural killer (NK) cells develop from common progenitors but diverge into distinct subsets, which differ in cytokine production, cytotoxicity, homing and memory traits. Given their promise in adoptive cell therapies for cancer, a deeper understanding of regulatory modules controlling clinically beneficial NK phenotypes is of high priority. We report integrated “-omics” analysis of human NK subsets, which revealed super- enhancers associated with gene cohorts that may coordinate NK functions and localization. A transcription factor-based regulatory scheme also emerged, which is evolutionarily conserved and shared by innate and adaptive lymphocytes. For both NK and T lineages, a TCF1-LEF1-MYC axis dominated the regulatory landscape of long- lived, proliferative subsets that traffic to lymph nodes. In contrast, effector populations circulating between blood and peripheral tissues shared a PRDM1-dominant landscape. This resource defines transcriptional modules, regulated by feedback loops, which may be leveraged to enhance phenotypes for NK cell-based therapies.
Graphical Abstract
INTRODUCTION
Natural killer (NK) cells provide protection from viral infections or cancer via their cytolytic function and IFNγ production. A major clinical goal is to harness these NK cell functions for tumor immunotherapy (Baggio et al., 2017; Johnson and Miller, 2018; Romee et al., 2016). In addition, NK cells have been implicated in the control of HIV infection either directly or by antibody-mediated lysis of infected cells (Bradley et al., 2018; Ramsuran et al., 2018). Although NK cells lack antigen-specific receptors, recent studies indicate that some responses are characterized by expansion and memory, features originally thought to be restricted to adaptive immunity. NK cells with a memory or adaptive phenotype may be particularly effective in cellular therapies targeting tumors or chronic viral infections (Cooper et al., 2017; O’Sullivan et al., 2015).
In humans, circulating NK cells encompass two major subsets, known as CD56dim and CD56bright (Freud et al., 2017; Michel et al., 2016). While this distinction is based on relative expression of the cell surface molecule CD56, profound functional differences exist between human NK subsets. CD56dim cells predominate in blood, constituting ~90% of circulating NK populations. This subset has higher cytotoxic activity than CD56bright cells. Moreover, CD56dim NK cells preferentially express the activating Fc receptor CD16, endowing them with a capacity for antibody-dependent cellular cytotoxicity (ADCC) (Nagler et al., 1989). The CD56dim NK population also encompasses two subsets, CD57- and CD57+, the latter of which has adaptive features and expands in response to HCMV infection (O’Sullivan et al., 2015). The minor CD56bright population is more efficient than CD56dim NKs in the production of cytokines, including IFNγ, GM-CSF, and TNF-α (Freud et al., 2017; Michel et al., 2016).
Circulating NK subsets also show considerable differences in homing molecules. CD56bright cells express the chemokine receptor CCR7 and L-selectin, which drive their migration to secondary lymphoid organs (Allan et al., 2017; Fehniger et al., 2003). In contrast, CD56dim display a high density of CX3CR1 and CXCR1, which direct them into peripheral tissues. IL-2 and IL-15 promote activation and proliferation of all NK cells. However, CD56dim express the dimeric low affinity receptor for IL-2 (CD122/CD132), whereas CD56bright express the trimeric high affinity IL-2R (CD25/CD122/CD132) (Allan et al., 2017). In addition, the CD56bright population expresses IL-7R and c-Kit, which may contribute to homeostatic proliferation. NK cell activation is controlled by inhibitory receptors specific for MHC class I, with CD56dim selectively expressing the KIR and LILR families, whereas CD56bright display CD94/NKG2A. Phenotypic and functional differences between CD56dim and CD56bright subsets have been extended further by gene arrays and proteomics (Hanna et al., 2004; Wendt et al., 2006).
Developmental relationships between CD56bright and CD56dim NK cells remain unresolved; however, several studies indicate that the former is a precursor of the latter. An NK subset with intermediate features between CD56bright and CD56dim has been identified, corroborating this developmental trajectory (Freud et al., 2017; Yu et al., 2010). However, CD56dim also can convert into CD56bright cells, at least in vitro, in the presence of activating cytokines (Keskin et al., 2007). Several studies also have suggested that these subsets are terminally differentiated and arise from distinct precursors (Berrien-Elliott et al., 2015; Wu et al., 2014).
Several nuclear factors have been implicated in the development and function of CD56dim versus CD56bright cells. Patients with mutations in the GATA2 transcription factor (TF) lack CD56bright, but not CD56dim NK cells, supporting a model for their independent development (Mace et al., 2013). Mutations in the MCM4 gene, a DNA helicase associated with replication, specifically compromises the CD56dim population (Gineau et al., 2012). Despite these advances, little information exists on TF- orchestrated regulatory programs for functionally distinct human NK populations, information that will clearly be useful as NK-based cell therapies are optimized. We now report integrative analysis of enhancer and transcriptional landscapes for circulating human NK subsets compared to intra-epithelial innate lymphoid cells 1 (ieILC1), which reside in mucosal microenvironments and produce IFNγ (Fuchs et al., 2013; Simoni et al., 2017). Super-enhancer profiling identified novel genes that functionally specify the CD56dim and CD56bright subsets, including G-coupled protein receptors (G-PCRs), which may modulate human NK function in response to tissue-derived factors. Our analyses established parallels in function and homing potential between self-renewing T memory cells and the CD56bright population, while the molecular programming of CD56dim cells resembles that of effector T memory compartments. Importantly, key TFs governing these phenotypic modules comprise a regulatory scheme employed by both innate and adaptive lymphocytes for localization and effector function, which is evolutionarily conserved from humans to non-human primates and mice.
RESULTS
Transcriptional programs of human NK and ieILC1s from distinct microenvironments
To characterize the molecular programming for human NK and ieILC1 populations from distinct microenvironments, we initially compared transcriptomes of two circulating NK subsets, CD56bright or CD56dim, as well as tissue NK and ieILC1s derived from a mucosal-associated lymphoid tissue, the tonsil (Ts) (Table S1). Ts-NK and ieILC1 were defined as CD56+NKp44–CD94+ (Figure 1A), and CD56+NKp44+CD103+, respectively (Koues et al., 2016). Data for Ts-ILC3 were included for comparison with a distinct innate lymphoid lineage (Koues et al., 2016). Classification of the various subsets was confirmed by expression patterns for signature genes, including NCAM1 (CD56), ITGAM (CD11b) for cytolytic NK, CD27 for IFNγ-expressing NK (Fu et al., 2011), ZNF683 (HOBIT) and ITGAE (CD103) for ieILC1 (Cortez et al., 2017; Weizman et al., 2017) (Figure 1B). Principle component analysis (PCA) of transcriptomes revealed a striking segregation of CD56dim from the other subsets (Figure 1C). Transcriptional programs for the remaining three subsets formed a narrower spectrum of kinships, with Ts-NKs straddled by ieILC1 and CD56bright cells. Inspection of individual genes further validated these relationships. For example, when comparing CD56bright versus CD56dim NK cells, nearly 300 genes were differentially expressed (>2.5-fold) (Figure 1D), while only 42 genes attained the 2.5-fold threshold in CD56bright versus Ts-NK cells (Figure 1E). Numbers of significantly different genes in pair-wise comparisons between all four NK/ieILC1 subsets are presented in Figure 1F, revealing a gradient of transcriptome kinships ranging from near identity (Ts-NK vs CD56bright) to clear divergence (ieILC1 vs CD56dim).
Figure 1. Transcriptional landscape of NK/ieILC1s.

A) Strategy for CD56bright vs CD56dim (left) and tonsil NK (Ts-NK) sorting (right). B) Heat map highlighting differential expression of signature genes for NK/ieILC1 subsets. C) PCA of genes with variable expression among CD56bright, CD56dim, tonsil NK, ieILC1 and ILC3. D) Volcano plots showing significance and magnitude of expression changes between CD56bright (red) and CD56dim (blue) or E) between CD56bright (red) and Ts-NK (brown). Colored genes highlight > two-fold differences that are statistically significant (p < 0.01, T-test) F) Number of transcripts differentially regulated when comparing two cell types (≥ two-fold change and p < 0.01, T-test). G) Total number of transcripts unique to CD56bright, CD56dim and ieILC1 (> two-fold difference compared with other cell types). H) Fold change plot comparing gene expression in NK/ieILC21 populations. Colored circles represent transcripts expressed at least two-fold higher in one subset versus another. Triangles represent transcripts expressed at least two-fold higher in a single subset compared with all other three subsets.
We next established the core set of signature genes expressed specifically in each NK or ILC1 subset. Because transcriptomes of Ts- and CD56bright-NK cells were nearly identical, we limited core analyses to ieILC1, CD56bright, and CD56dim cells. As shown in Figure 1G, signature sets of genes ranged from ~20 (CD56bright) to ~160 for ieILC1. Relative expression of the signature genes are shown in Figures S1A-C. Ontology analysis of signature genes revealed that each NK/ieILC1 population was enriched for distinct functional categories, including cell migration, survival, IFNγ production, and degranulation (Figure S1D). In addition to cohorts of expected genes, each signature had tantalizing features that suggested subset-distinctive biology. For example, the ieILC1 program included several genes normally expressed in ILC3s (e.g., RORC and IL23R), perhaps reflecting functional plasticity in the latter cell type, which can convert to ILC1s under certain conditions (Cella et al., 2010).
In addition to overall differences between transcriptomes, two-by-two-fold comparisons of all cell populations identified a limited cohort of genes for blood versus tonsil subsets, likely reflecting their distinct microenvironments (Figure 1H). Notably, ts-NK and ieILC1 expressed CRTAM, encoding an adhesion molecule that facilitates tissue retention (Cortez et al., 2014). Blood NKs expressed the gene for TGFBR3, which modulates TGFβ signaling and is distinctive from the classical TGFBR2 receptor that mediates TGFβ imprinting in ILC1s (Cortez et al., 2016). Despite their divergence from other cell subsets, CD56dim NKs shared a small set of genes that was also expressed by ieILC1s. This gene cohort includes those encoding a pair of NK cell receptors (TIGIT and CD160), suggesting that subpopulations of the two cell types can respond to similar cues. Together, transcriptome analyses established a spectrum of functional relationships between NK cells and ieILC1s in humans, as well as defined cohorts of genes that endow each with their specialized roles in innate immunity.
Expression programs reveal a developmental trajectory for circulating NK cells
Prior studies have shown that CD56dim cells include two subsets, CD57– and CD57+, the latter of which expand in HCMV seropositive individuals and encompass ‘adaptive’ NK cells (Lee et al., 2015; O’Sullivan et al., 2015; Schlums et al., 2015). To probe developmental relationships between circulating NK subsets, we examined gene and protein expression profiles. Mass cytometry corroborated segregation of blood NK cells into three major subsets (Figure 2A), CD56brightCD16–CD57–, CD56dimCD16+CD57– (referred to as CD57–) and CD56dimCD16+CD57+ (CD57+). Expression levels of proteins detected by a panel of 41 antibodies was used to probe developmental trajectories. Pseudotime analysis of CyTOF data predicted a developmental trajectory with two clear end-points: CD57+ and CD56bright. The CD57– population emerged as a transitional stage, with a mostly linear trajectory from the CD57+ population. However, a subset of CD57– cells projected off this main pathway due to differential expression of IL2-R and/or NKG2A (Figures 2A and S2A).
Figure 2. Characterization of blood NK populations.

A) Analysis of peripheral NK cells by a 41 antibody CyTOF panel. Relative levels of CD56, CD16 and CD57 are indicated (grey: min, orange: max). B) Pseudotime projection (Monocle) of CyTOF analysis. Cells were defined as CD56bright: Red, CD56dimCD57-: magenta, CD56dimCD57int: purple, or CD56dimCD57+: blue. C) PCA of genes with variable expression among CD56bright, CD57−, CD57+, ieILC1 and ILC3. D) Bulk RNA-seq expression values (RPKM) of XCL1 and PRF1. r2 regression values are indicated above. Boxes represent interquartile ranges (IQR) and whiskers represent values within 1.5xIQR. E) Volcano plots showing significance and magnitude of expression changes between CD56bright (red), CD57+ (blue) or CD57− (magenta) subsets. Colored genes highlight > two-fold differences that are statistically significant (p < 0.01, T-test). G) Number of transcripts differentially regulated when comparing two cell types (≥ two-fold change and p < 0.01, T-test).
The protein-based developmental trajectory was further supported by gene expression. PCA of RNA-seq data placed the transcriptome of CD57– cells squarely between the CD56bright and CD57+ populations (Figure 2C). Indeed, analysis of signature CD56bright versus CD56dim genes revealed a linear relationship, with CD57– cells having intermediate expression at the RNA and protein levels (Figure 2D, S2B-C). Pairwise comparison of gene expression data showed remarkable similarity between the CD57– cells and CD56bright populations, with more of a distinction between the CD57– and CD57+ subsets (Figure 2E, F). Together, single-cell and bulk expression analyses uncovered a distant relationship between CD56bright and CD57+ NK cells, which was largely bridged by the CD57– subset.
Human NK Cell Regulomes
As an initial step in understanding mechanisms that drive distinctive aspects of NK cell expression programs, we subjected circulating NK subsets to epigenome analyses, which were compared with published data for tonsillar ieILC1 and ILC3 (Koues et al., 2016). We identified locations of all active or poised regulatory elements using Assay for Transposase-Accessible Chromatin coupled with high throughput sequencing (ATAC- seq). We assessed relative enhancer activities using a Chromatin Immunoprecipitation (ChIP) sequencing assay for the histone modification H3K27ac, performed on bulk CD56dim or CD56bright populations. Initially, we focused on enhancers, classified as ATAC+ regions, but excluding promoters, CTCF sites or regions accessible in only a single replicate (Figure S3A). Clustering analysis of either ATAC or H3K27ac data revealed consistent segregation of each cell population into unique branches, indicating negligible impact of donor diversity (Figures 3A and S3B). Similar to gene expression, the accessible regulome of CD56bright NK cells was closely related to the CD57– subset (Figure 3A, p < 0.05, bootstrapping analysis).
Figure 3. Chromatin landscape of ieILC1, ILC3 and blood NK cells.

A) Clustering analysis of ATAC values for enhancers in CD56bright, CD57−, CD57+, ieILC1, and ILC3 subsets. Colors represent unique clusters (K-means). B) PCA of ATAC enhancer values. Red: CD56bright, Blue: CD57+, Magenta: CD57−, Green: ieILC1, Purple: ILC3. C-D) UCSC screenshots showing C) KLF43 and D) PRF1 loci. Green tracks represent H3K27ac (0–35 RPKM) and brown tracks show ATAC-seq (0–100 RPKM). Gene location, coding regions and directionality are indicated on top. Red boxes indicate selected enhancers. Heatmaps on the right represent relative gene expression (Txn: blue: gene minimum to red: gene max RPKM). E) SNP enrichment for enhancers differentially active in the given cell types. Shown are the degrees of significance (- log10P values, binomial test) for SNPs classified as immune disorders or cancer (left), or individual GWAS traits (right). F) UCSC screenshot of TNFSF13B (top) or IKZF3 (bottom) loci. Dashes under the screenshots represent GWAS SNPs, and red boxes highlight selected SNPs.
We created a master list of “hyper-accessible enhancers” that reproducibly surpass a stringent cutoff for ATAC-seq signals (see Methods) and identified elements that were differentially regulated in ILC populations (Figure S3C and Table S2). We reduced ATAC data for the hyper-accessible enhancers into two components by PCA (Figure 3B). The greatest contributor of variation, PC1, separated regulomes of blood NK versus tissue-resident ILCs. This segregation of regulomes is illustrated by blood NK- specific activity of an enhancer near the KLF3 transcriptional repressor (Figure 3C). PC2 separated cellular subsets primarily based on their cytotoxicity (CD57+>CD57–, ieILC1>CD56bright, ILC3). As an example, an enhancer neighboring the signature cytotoxicity gene, PRF1, exhibited a similar gradient of accessibility (Figure 3D). Other PC2-dominant loci associated with enhancers having the converse gradient of activities, including one situated proximal to an ILC3-enriched gene encoding leukemia inhibitory factor (Figure S3D) (Cella et al., 2010). In all examples, gene expression tracked closely with the accessibility of highlighted enhancers (Figures 3C-D), as well as H3K27ac loads for relevant cell populations. The transcriptional patterns were retained on a global level for “hyper-accessible enhancers” (Figure S3E). Thus, in addition to cell type-selective elements, we identified two modules of chromatin activity and enhancers: one that distinguished human NK versus ILC subsets and a second that orchestrated expression programs for cytotoxicity.
We also used chromatin-based analyses to explore whether NK regulomes contribute to the etiology of human diseases, including autoimmunity. Indeed, the collection of active enhancers in all NK/ieILC1s was enriched for single nucleotide polymorphisms (SNPs) associated with immune system diseases but not with cancer or other non-immune traits (Figure 3E). Most of these SNPs were localized to enhancers active in all cell populations examined. However, SNPs associated with a susceptibility to tonsillar inflammation (Pickrell et al., 2016) were enriched only in ieILC1-active enhancers, consistent with their tissue origin (Figure 3E, Table S3). This finding indicates that the tonsillar microenvironment impacts a distinct set of enhancers in ieILC1s regulating genes involved in the magnitude of acute inflammatory responses. The major SNPs driving this enrichment in ieILC1-enhancers are associated with loci encoding IKZF1 (encoding IKAROS), NFKB1 and TNFSF13B (encoding BAFF). The disease-associated SNP in TNFSF13B (Figure 3F) is predicted to disrupt an enhancer binding site for the GATA family of TFs, which may reduce BAFF expression.
A second category of differential SNP enrichment is associated with inflammatory bowel diseases (IBD), and localize to enhancers with preferential activity in ieILC1 and CD56dim NK cells (Table S3). A major driver of this enrichment was a set of SNPs in IKZF3 (Aiolos), whose expression pattern parallels the aforementioned enhancer activity (Figure 3F). Thus, the IKZF3-associated SNP may alter expression of this repressive TF in ieILC1 and CD56dim NK cells, contributing to initiation or progression of IBD. Our analyses suggest that enhancer-associated DNA variants may contribute to inflammatory autoimmune diseases via changes in NK gene expression.
Super-Enhancers Identify Novel NK Signaling Pathways
Highly transcribed genes that endow cell type functions are often associated with dense enhancer regions, termed super-enhancers (SEs) (Hnisz et al., 2013). These broad, hyperactive regulatory regions are assigned an SE status based on their extreme breadth and density of H3K27ac (Koues et al., 2016). In addition to transcriptional regulation, SEs can serve as discovery tools for genes with critical functions in a given cell type. We applied an SE detection algorithm to our ChIP data as a means of identifying core genes for CD56bright and CD56dim NK populations, which were compared with previously published ieILC1 SEs (Koues et al., 2016). Several SE-associated genes are highlighted in Figures 4A and S4, including TFs (e.g., PRDM1, RUNX2, TCF7, BACH2, PLZF), cell surface receptors (e.g., TGBR2), and signaling molecules (e.g., PRKCH, CD247). As expected, genes associated with SEs called specifically in one of the three cell types correlated with their higher levels of expression (Figure 4B). Furthermore, a substantial fraction of SEs and associated gene expression were conserved between human and mouse NK subsets (Figures 4C and S4B-E).
Figure 4. Super-enhancers in NK and ieILC1.

A) Rank order of increasing H3K27ac enrichment at enhancer loci for each NK/ieILC1 subset. Gene names are indicated next to SE rankings. B) Average expression for genes that are most proximal (< 30 kb) to cell type-specific SEs: ieILC1 (green), CD56dim (blue), and CD56bright (red). Boxes represent interquartile ranges (IQR) and whiskers represent values within 1.5xIQR (* P < 0.05, Student’s T test). C) Percent of human NK SEs that overlap conserved and active mouse NK SEs. Human NK enhancers are classified as shared NK, CD56bright- or CD56dim-specific. D) Venn diagram showing unique or shared SEs in human NK and ieILC1s. A subset of genes for different SE activity profiles is highlighted. E) UCSC screenshot showing GPR183 and GPR18 locus (see Figure change and p < legend). Red boxes specify SEs. F) FACS sorted CD56bright NK cells were cultured for 72 hours in IL-12 and IL-18 in presence or absence of 7α,25-dihydroxycholesterol. IFNγ levels from culture supernatants were determined by CBA. Data are represented as mean + SD. * p < 0.05, Student’s T test.
Most of the SEs were specific to only one or two NK/ieILC1s (Figure 4D). For example, an NK-specific SE spanned NMUR1, encoding a G-PCR for the neuropeptide neuromedin U, which is expressed in NK cells but not ieILC1s (Figure S4C). Given the increasing appreciation of crosstalk between neural and immune systems (Veiga- Fernandes and Artis, 2018), NMUR1 is an attractive target for future functional studies in NK subsets. More globally, SE analysis identified a collection of G-PCR genes with a wide variety of expression profiles (Table S4). The G-PCR encoded by CMKLR1 is covered by an SE only in CD56dim NK cells (Figure S4D), correlating with its cell type- restricted expression. This chemokine receptor promotes egress of CD56dim NK cells to sites of tumor-associated inflammation in response to chemerin (Pachynski et al., 2012; Parolini et al., 2007). A second CD56dim-specific SE spans GPR141, which encodes a conserved G-PCR with unknown ligands or function, but with a potentially important role in NK biology (Figure S4E). As shown in Figure 4E, the locus encompassing GPR18 and GPR183 was covered by an SE in CD56bright and ieILC1s but not in CD56dim NK cells, again correlating with gene expression. GPR183, also known as EBI2, is a receptor for oxysterols with roles in lymphocyte trafficking and inflammatory cytokine expression (Emgård et al., 2018). To test whether CD56bright cells were responsive to the GPR183 ligand, 7α,25-dihydroxycholesterol (7α,25-dhc), we cultured this NK subset in the activating cytokines IL-12+IL-18, either with or without 7α,25-dhc. As shown in Figure 4F, the GPR183 ligand attenuated IFN-γ production by CD56bright NK cells. Thus, SE analysis provides a discovery tool to identify novel genes with functions in NK cells.
Transcription factors that control NK/ieILC1 regulomes
To understand how ILC-NK subsets coordinate the activation and repression of distinct enhancer and SE collections, we analyzed differentially regulated cis-elements for enrichment of TF motifs. This agnostic approach yielded a similar set of enriched motifs whether accessibility (ATAC-seq) or activation (H3K27ac-seq) data were fed into the HOMER algorithm (see Methods). As shown in Figures 5A and S5A, differentially active enhancers appear to be targeted by TF families that distinguish each NK/ieILC1 subset or are shared specifically by all NK populations. When paired with gene expression (Figure S5B), single members of a TF family usually correlated with motif utilization (Figure 5A). Comparisons between ieILC1 and NK cells identified enhancers with preferential ieILC1 activity that were significantly enriched for motifs predicted to bind SMAD transcription factors, which mediate TGFβ imprinting. When compared with ieILC1s, active enhancers in circulating NK subsets were enriched for T-box motifs, which are targets of the type 1 “master regulators” T-BET and EOMES (Gordon et al., 2012). Moreover, NK subsets were enriched for co-occurring T-box and RUNX motifs within individual enhancers (Figure S5C); a finding supported further by publicly available ChIP-seq data (Table S5). As one example, an evolutionarily conserved RUNX+T-box enhancer was located near the SELL gene, which encodes CD62L, and was more accessible in CD56bright NK. The enhancer was bound by both RUNX3 and T- BET in a human B cell line, and by T-BET in primary mouse NK cells (Figure 5B).
Figure 5. Enriched TF motifs in NK and ieILC1 regulomes.

A) Motif utilization for differentially regulated enhancers based on ATAC data. Motif utilization is represented as relative levels ranging from low (blue) to high (red) and is pre-filtered for significance (p < 0.001, binomial test). Motifs were assigned to a TF family using HOMER and hocomoco databases. The right panel shows expression of selected genes in the corresponding TF family (blue: 23 intensity, red: 28 RPKM). B) UCSC screenshots of the human (top) and mouse SELL locus (bottom) showing ATAC data from indicated cell types. The bottom tracks in each panel show ChIP-seq data for TBX21 or RUNX3 in either a human B cell line (GM12878) or mouse splenic NK cells. The middle panel shows a conserved RUNX-TBOX composite motif for one of the enhancers. C) UCSC screenshot of the MYC locus. The black track (bottom) represents TCF7 ChIP-seq from K562 cells (ENCODE). D) Total RNA per cell for each NK subset. E) GSEA enrichment of ZEB2 repressed genes from Claudia X. et al, 2015, showing relative levels in CD56bright and CD56dim NK transcriptomes. E) UCSC screenshot of the CCR7 locus with the bottom track showing ChIP-seq data for eGFP-ZEB2 from the K562 cell line (ENCODE). F) A representation of CD56bright and CD56dim NK transcription factor regulatory circuits, as predicted from combined expression and motif analysis. Also included are key effector genes of both subsets.
The collection of enhancers that display preferential activity in CD56bright NK cells were enriched for RUNT, HMG, PRDM1, E-box, and NFKB motifs (Figure 5A). RUNT motifs are targeted by RUNX family members, which are important players in lymphocyte biology (Collins et al., 2009). Although RUNX1 and RUNX3 were expressed in all blood NK subsets, RUNX2 was highly restricted to CD56bright cells at the RNA and protein levels (Figures 5A and S2B). These data suggest that a distinctive set of enhancers were activated in the CD56bright compartment either because they were unique targets of RUNX2 or required levels of total RUNX family proteins that were surpassed when this additional member was expressed. Consistent with the latter possibility, there were no identifiable differences in RUNX motifs for CD56bright-specific enhancers versus those shared with CD56dim cells (Figure S5C).
Preferential use of HMG motifs in CD56bright cells likely corresponds to their enhanced expression of TCF7 and LEF1 (Figure 5A), two homologous TFs with some functional overlap (Held et al., 2003). Both of these HMG family proteins have been implicated in self-renewal programs of memory T and hematopoietic stem cells, and therefore may control homeostatic proliferation of CD56bright NK cells (Wu et al., 2012). Importantly, NK-specific deletion of Tcf7 in mice leads to defects in NK cell survival (Jeevan-Raj et al., 2017). In this context, CD56bright accessibility was enriched at TCF1 ChIP-seq peaks from K562 cells (Figure S5D). This included two regulatory elements near MYC, which is expressed at higher levels in the CD56bright subset, and is known to coordinate programs for cell proliferation, survival and biomass production (Figure 5C). Indeed, CD56bright NK had higher levels of total RNA per cell than the MYC-CD57+ NK population (Figure 5D). Consistent with their transitional status and intermediate levels of MYC expression, CD57- cells had intermediate levels of total RNA. These findings were confirmed by RNA-seq using external ERCC spike-ins for normalization (Chou et al., 2014).
Motif analysis also revealed that the CD56bright molecular program is suppressed in CD57+ NK cells via expression of several transcriptional repressors (Figure 5A). Specifically, the PRDM1 motif was enriched in active enhancers of CD56bright NK cells. This finding is supported by differential expression of PRDM1, which encodes the BLIMP1 repressor -- high in CD56dim (enhancer repression) and low in CD56bright (not repressed). Importantly, BLIMP1 silences genes associated with lymphoid self-renewal and survival, including MYC and IL7R (Küçük et al., 2011), and is required for mouse effector NK development (Kallies et al., 2011). The other TF that binds PRDM1 motifs is ZNF683 (HOBIT). However, ZNF683 expression is restricted to ieILC1, reflecting its known function in tissue residency (Mackay et al., 2016). Another repressor, BACH2, enforces a self-renewal program in T memory cells via repression of BLIMP1 and downstream effector pathways (Herndler-Brandstetter et al., 2018). Indeed, BACH2, which binds a PRDM1 enhancer, is preferentially expressed in CD56bright cells, where BLIMP1 is extinguished (Figures 5A and S5E).
Transcripts for the ZEB2 repressor were elevated in both CD56dim subsets. Gene Set Enrichment Analysis (GSEA) confirmed preferential expression of ZEB2 (Figure 5E) and PRDM1 (Figure S5F) targets in CD56bright NK cells (Dominguez et al., 2015), in which these repressors were not expressed. As an example, the gene encoding CCR7, a chemokine receptor that promotes homing of CD56bright into lymph nodes, is flanked by an enhancer that was attenuated in CD56dim cells, corresponding to their expression of the ZEB2 repressor. The same regulatory element is bound by ZEB2 in K562 cells, in which this enhancer is silent (Figure 5E). In addition to repressor binding, the collection of enhancers specifically augmented in CD56dim cells were enriched for bZIP motifs, which can facilitate either transcriptional activation or repression. Two members of the bZIP family, MAF and NFIL3, were expressed at higher levels in CD56dim versus CD56bright NK cells (Figure 5A). These TFs have been implicated in helper T cell differentiation (Gabryšová et al., 2018) and NK cell responsiveness to IL-15 (Firth et al., 2013), respectively. Importantly, CD56dim populations require expression of MCM complex genes, some of which are under MAF control (Figure S5F) (Chihara et al., 2018).
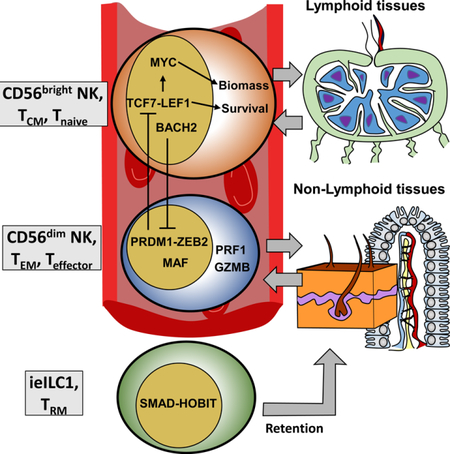
Together, integrative -omics profiling of human blood NK populations defined a TF- based regulatory scheme for maintaining transcriptional identities of CD56bright versus effector CD56dim cells (Figure 5F). In brief, CD56bright phenotypes are positively coordinated by TCF1/LEF1 and MYC, while BACH2 represses effector phenotypes associated with CD56dim NK cells. Conversely, BLIMP1 and ZEB2 suppress the CD56bright features at the CD56dim stage, while MAF, and likely other TFs, coordinate expression of major effector genes.
A conserved regulatory network of TFs for lymphoid cells
The counterbalance of positive and negative transcriptional regulation orchestrating NK phenotypes, as well as the identity of responsible TFs, bears a striking resemblance to regulatory strategies that compartmentalize self-renewal, localization, and effector functions in other lymphoid lineages. A striking example of this potentially conserved regulatory paradigm is in the CD8 T cell memory compartment. Similar to CD56bright NK cells, self-renewing CD8 memory cells (T stem cell memory, TSCM and T central memory, TCM) transit between blood and lymph node, and co-express TCF1 and BACH2 (Herndler-Brandstetter et al., 2018). By analogy to CD56dim NK cells, effector and effector memory (TEM) CD8 subsets are highly cytotoxic and co-express BLIMP1 and MAF (Im et al., 2016).
To explore more comprehensively the parallel between human subsets, we compared our NK and publicly available CD8 transcriptome data (Gattinoni et al., 2011). Indeed, CD56bright NK, TSCM, and TCM had strikingly similar expression profiles, which were also related to the naïve CD8 T cell transcriptome (Figures 6A-B). In contrast, gene expression programs of CD57+ NK cells most significantly overlap with the TEM subset. These parallels also were observed at the chromatin level. Promoters that were differentially accessible in CD56bright NK cells had higher levels of the activating H3K4me3 modification in naïve CD8 T cells, but were hypomethylated in TEM (Figure 6C). The converse was true for accessible CD57+ promoters. These integrative analyses revealed a unifying regulatory strategy deployed by both innate and adaptive lymphocytes in humans.
Figure 6. Evolutionarily conserved signatures of NK and CD8 cells.

A) Transcripts differentially regulated in peripheral human NK and CD8 subsets (left), and expression of orthologous genes from mouse cells (right). Heatmaps show relative probe intensity (human CD8 data) or RPKM values of transcripts across replicates. B) Percentage of CD56bright or CD57+ specific genes overlapping CD8 Tnaive, TSCM, TCM and TEM gene sets. * p < 0.01 Binomial enrichment. C) Average Tnaive and TEM H3K4me3 density across promoters specifically accessible in CD56bright or CD57+ cells. D) GSEA enrichment of genes selectively expressed in CD27+CD11b- (left) or CD27-CD11b+ (right) cells from Robinette M. L. et al, 2015 showing relative levels in CD56bright and CD56dim NK transcriptomes. E) Overlap between conserved CD56bright and CD56dim cell- type selective enhancers with either CD27+CD11b- or CD27-CD11b+ ATAC peaks. F) UCSC screenshots of the human (top) and mouse (bottom) TCF7 locus showing ATAC or H3K4me3 data from indicated cell type. The bottom track for mouse data shows ChIP-seq data for BLIMP1 from mouse CD8 cells.
To determine whether this regulatory scheme is evolutionally conserved, we examined mouse NK and CD8 T cell subsets. NK cell development in mice proceeds from a poorly cytotoxic precursor (CD11b-, CD27+), through a double positive intermediate (CD11b+CD27+), terminating in a poorly proliferative, cytotoxic NK cell (CD11b+CD27-) (Figure S6A) (Chiossone et al., 2009). Initially, we compared transcriptomes of human blood and mouse splenic NK subsets, as well as mouse CD8 memory T cells (Best et al., 2013; Robinette et al., 2015). As shown in Figure 6A, expression of key genes corroborated parallels between the precursor-like populations in human and mouse (CD56bright, CD11b–CD27+ NK, TCM), including the master regulatory TFs, Tcf7-Lef1 and Myc. Human effector NK cells (CD57+) were most similar to the mouse CD11b+ subset and TEM. These overall patterns were confirmed by GSEA of differentially expressed genes in mouse and human (Figure 6D). In addition, core genes in the regulatory scheme were differentially expressed in sorted NK subsets from macaques (Hong et al., 2013), further underscoring the evolutionarily conservation of this paradigm (Figure S6B).
We also found similarities between human blood and mouse splenic NK subsets at the regulome level. Globally, the set of evolutionarily conserved enhancers specific for CD56bright NK cells was preferentially active in the mouse CD27+CD11b– subset (Figure 6E), while the converse was true for enhancers active in circulating CD56dim cells. Indeed, enhancers distinguishing human CD56bright versus CD56dim subsets that were conserved in their mouse counterparts exhibited an enrichment for the expected TF motifs (Figure S6C). Many of the conserved enhancers with differential activity were proximal to genes identified in the core set of signature transcripts shown in Figure 5A. For example, the TCF7 locus contained three CD56bright-specific enhancers that were conserved evolutionarily and were accessible in the CD27+CD11b– subset (Figure 6F). Strikingly, one enhancer bound the repressor BLIMP1 in splenic CD8 T cells (Mackay et al., 2016), likely attenuating its activity in mature subsets from both mouse and human (Figure 6D). Together, comparative transcriptome and epigenome analyses revealed an evolutionarily conserved regulatory scheme that spans at least two lymphoid lineages and coordinates expression of genes responsible for precursor-like or effector phenotypes.
DISCUSSION
The collection of IFNγ-producing innate lymphocytes, which are important for controlling microbial infections and transformed cells, display a wide range of functional phenotypes in both humans and mice (Cortez and Colonna, 2016). In mouse, these cells can be grouped phenotypically as helper ILC1s versus cytotoxic NK, which diverge as separate lineages (Constantinides et al., 2015; Klose et al., 2014). In humans, however, functional and lineage relationships between ILC1s and subsets of NK cells, including those found in blood, are not as well defined (Freud et al., 2017; Michel et al., 2016). Given the high priority status of NKs in cell-based therapies for cancer, we require a deeper understanding of the regulatory modules controlling phenotypes for clinical efficacy, such as cytotoxicity, IFNγ production, homing, proliferation, self- renewal, and memory.
Our studies approached this problem using integrative –omics analysis of human NK/ieILC1s from distinct microenvironments and functional subsets, including helper and adaptive phenotypes. At both the transcriptional and epigenetic levels, we find that the adaptive subset, CD57+ cytotoxic NK cells, segregated away from a spectrum of more closely related subsets (CD57–, CD56bright, tonsillar NK, and ieILC1s). Beyond this, a small set of signature genes distinguished the five subsets from one another, with a substantial mixing and matching of transcriptional modules to create patchwork expression programs. For example, RUNX2, an osteoblast-restricted gene, was selectively expressed in CD56bright NK cells (Komori, 2010). KLF3, originally thought to be an erythrocyte TF (Crossley et al., 1996; Vu et al., 2011), was selectively expressed in circulating type 1 cells, but not those found in mucosal tissue. Unexpectedly, circulating CD56dim NK cells shared a small set of genes with ieILC1s residing in a mucosal microenvironment, further underscoring the patchwork nature of NK/ieILC1 expression programs. Although additional studies are required, the CD56dim-ieILC1 shared module may derive from a unique combination of TF expression that includes ZNF683 (HOBIT, ieILC1), PRDM1 (BLIMP1, CD56dim), and IKZF3 (AIOLOS, both).
A major finding to emerge from this study is a conserved regulatory scheme deployed by both innate and adaptive lymphocytes, which is coordinated by a cohort of TFs. The scheme functionally compartmentalizes effector cells that circulate between blood and peripheral tissues from self-renewing precursors that traffic to lymph nodes. The latter regulatory scheme utilizes the transcriptional activators TCF1-LEF1 and MYC in NK (CD56bright), T memory and T naïve compartments. Moreover, human B, T, and NK cells suppress effector pathways via expression of BACH2, which may be under the control of AHR signaling (Richer et al., 2016). The BACH2-mediated brake appears to be released in all of these cells upon expression of BLIMP1, which feeds back to repress the TCF1-LEF1 and MYC module, while effector programs are activated by MAF. In support of this unified model, TCF1 promotes renewal and maintenance of hematopoietic stem cells (Wu et al., 2012), TSCM, which exhibit a heightened capacity to undergo IL-7– and IL-15–driven homeostatic proliferation (Gattinoni et al., 2011), CD4+ T follicular helper cells (Choi et al., 2015), and Ly49H+ MCMV-specific naïve and memory NK cells (Lau et al., 2018). With regard to translational implications, TCF1 also is expressed in a subset of CXCR5+CD8+ T cells that proliferate in response to anti-PD1 treatments for chronic infection and cancer (Im et al., 2016).
In contrast, TCF7-LEF1 were both poorly expressed in the adaptive CD56dimCD57+ NK subset, which exhibits a more terminally differentiated state reminiscent of CD8+ TEM and CD4+TH1 cells (Choi et al., 2015; Herndler-Brandstetter et al., 2018). Our analyses identified conserved enhancers in TCF7 and LEF1 loci that are targeted by BLIMP1 in differentiated mouse cells, likely repressing their expression. A similar switch is governed by these TFs in mouse counterparts of human NK subsets, as well as in macaques, a non-human primate. The parallels between regulatory programs in NK and T cells may extend to additional TFs. Specifically, we observed a reciprocity between ZEB1 and ZEB2 expression in proliferative precursors (CD56bright, TSCM, TCM) and effector cells (CD56dim, TEM), respectively (Guan et al., 2018).
Our integrative and single cell analyses revealed potential developmental relationships between circulating human NK cells. Transcriptome, epigenome and protein expression data displayed gradients of activity from CD56bright to CD57- transitional cells to the CD57+ NK subset. This trajectory is consistent with some reports indicating that CD56bright cells can serve as precursors to the CD56dim subset (Freud et al., 2017; Yu et al., 2010). Thus, the TCF7-LEF1-MYC module in CD56bright NKs may function to preserve progenitor populations, which can continually replenish CD57+ adaptive cells following induction of BLIMP1. However, we do not exclude alternative cause-effect relationships between transcriptional modules, especially since fate mapping studies of NK cells are not possible in humans. Distinct expression programs may reflect homing potential or localization of NK subsets, rather than a discrete developmental pathway. In support of this scenario, all cells sharing the TCF1-LEF1-MYC module, including naïve CD8 T cells, traffic between the blood and secondary lymphoid organs, as indicated by expression of CD62L and CCR7. Indeed, we found that expression programs of tonsillar NK cells were nearly indistinguishable from the CD56bright subset. In contrast, CD56dim NK and TEM cells, which share the BLIMP1-MAF module, traffic between blood and non- lymphoid organs. Consistent with recent studies, a third regulatory module driven by the TF HOBIT (ZNF683) appears to drive retention of ieILC1 and resident memory T cells (TRM) in a variety of tissues (Jameson and Masopust, 2018).
Our approach, including the characterization of histone modifications in scarce NK subsets, allowed us to identify super-enhancers, which associate with genes endowing a given cell type with its unique identity (Hnisz et al., 2013). Especially intriguing were SEs with distinct activities in NK subsets, such as those proximal to a substantial set of G-PCRs. Given the role of G-PCRs in transmitting microenvironmental cues, the distinctive set of receptors may govern migration of circulating NK cells to different tissues, expression of cytokines, cytotoxic responses, or CD56bright to CD56dim differentiation. Indeed, the cholesterol metabolite 7α,25-dhc, which is produced by virally infected cells (Blanc et al., 2013; Cyster et al., 2014), attenuated IFN-γ expression by CD56bright NK cells, presumably through their high levels of GPR183 expression. Consistent with IFN-γ inhibition, GPR183 signals through Gαi to inhibit cAMP production (Rosenkilde et al., 2006). Additionally, an NK-restricted SE coats the NMUR1 gene, suggesting that its continued expression may be important for CD56bright and CD56dim NK functions, perhaps by enabling communication with the peripheral nervous system (Veiga-Fernandes and Artis, 2018). A number of G-PCRs (e.g., GPR141) and other SE- associated genes emerging from our data remain uncharacterized, especially with regard to their functions in NK cell biology, thus providing a valuable resource for future studies.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD56 PE (RRID: AB_10598372) | eBioscience | Cat#12–0567 |
| Anti-NKp44 AlexaFluor647 (RRID: AB_647153) | BDPharmingen | Cat#558564 |
| Anti-CD103 BV605 (RRID: AB_2564283) | Biolegend | Cat#350218 |
| Anti-CD94 PE-Cy7 (RRID: AB_2632752) | Biolegend | Cat#305515 |
| Anti-H3K27ac (RRID: AB_2118291) | Abcam | Ab4729 |
| Anti-CD3 FITC (RRID: AB_314060) | Biolegend | Cat#300406 |
| Anti-CCR6 BV421 (RRID: AB_2561356) | Biolegend | Cat#353408 |
| Anti-CD3 PerCP-Cy5.5 (RRID: AB_1518743) | eBioscience | Cat#45–0036 |
| Anti-CD19 PerCP-Cy5.5 (RRID: AB_2043821) | eBioscience | Cat#45–0199 |
| Anti-CD94 FITC (RRID: AB_314534) | Biolegend | Cat#305504 |
| Anti-NKp44 PE (RRID: AB_647239) | BDPharmingen | Cat#558563 |
| CD56 Microbeads | Miltenyi Biotec | Cat#130–050-401 |
| Anti-NKp44 PE-Cy7 | Biolegend | Cat#325116 |
| Anti-CD127 BV421 | Biolegend | Cat#351309 |
| Anti-CD117 BV510 | Biolegend | Cat#313219 |
| Anti-CCR7 PE | Biolegend | Cat#353204 |
| Anti-S100A4 PE | Biolegend | Cat#370003 |
| Anti-CXCR2 PE | Biolegend | Cat#320705 |
| Anti-AHR unconjugated | Santa Cruz Biotechnology |
Cat#sc-133088 |
| Anti-mouse IgG2b PE | SouthernBiotech | Cat#109209 |
| Anti-TIGIT BV605 | Biolegend | Cat#372711 |
| Anti-CD27 PE | eBioscience | Cat#12–0279-42 |
| Anti-CD6 FITC | BDPharmingen | Cat#559491 |
| Anti-CD57 FITC | eBioscience | Cat#11–0577-41 |
| Anti-TCF1 PE | Biolegend | Cat#655207 |
| Anti-Ksp37 PE | Biolegend | Cat#346606 |
| Anti-CD57 biotin | Biolegend | Cat#346606 |
| Anti-IL18R biotin | Biolegend | Cat#313806 |
| Anti-CX3CR1 PE | Biolegend | Cat#341604 |
| Anti-CD62L BV605 | Biolegend | Cat#304833 |
| Anti-Gata3 PE | eBioscience | Cat#12–9966-41 |
| Anti-CD56 APC | eBioscience | Cat#17–056-42 |
| Anti-RUNX2 | Cell Signaling Technology |
Cat#12556S |
| Anti-Rabbit PE | SouthernBiotech | Cat#4050–09 |
| Streptavidin BV421 | BDPharmingen | Cat#563259 |
| DX5 microbeads | Miltenyi | Cat#130–052-501 |
| Anti- mouse NK1.1 BV421 | Biolegend | Cat#108731 |
| Anti-mouse CD3 APC | eBioscience | Cat#17–0031-83 |
| Anti-mouse CD19 APC | BDPharmingen | Cat#550992 |
| Anti-mouse CD11b PE | eBioscience | Cat#12–0112-85 |
| Anti-mouse CD27 FITC | eBioscience | Cat#11–0271-81 |
| Cytof Panel | ||
| NKp44–167Er | Ebioscience | Cat#16–3369-85 |
| CD45–089y | Fluidigm | Cat#3089003B |
| CD196–141Pr | Fluidigm | Cat#3141003A |
| CD19–142Nd | Fluidigm | Cat#3142001B |
| CD117–143Nd | Fluidigm | Cat#3143001B |
| CD4–145Nd | Fluidigm | Cat#3145001B |
| CD8a-146ND | Fluidigm | Cat#3146001B |
| CD25–149Sm | Fluidigm | Cat#3149010B |
| FceR-150Nd | Fluidigm | Cat#315001B |
| CD138–150Nd | Fluidigm | Cat#3150012B |
| CD103–151Eu | Fluidigm | Cat#3151011B |
| TCRgd-152S | Fluidigm | Cat#3152008B |
| TIGIT-153Eu | Fluidigm | Cat#3153019B |
| CD3–154Sm | Fluidigm | Cat#3154003B |
| CD85j-156Gd | Fluidigm | Cat#3156020B |
| CD194–158Gd | Fluidigm | Cat#3158006A |
| CD161–159Tb | Fluidigm | Cat#3159004B |
| CD39–160Gd | Fluidigm | Cat#3160004B |
| CD27–162Dy | Fluidigm | Cat#3162009B |
| CD45RO-165Ho | Fluidigm | Cat#3165011B |
| CD34–166Er | Fluidigm | Cat#3166012B |
| CD127–168Er | Fluidigm | Cat#3168017B |
| CD159–169Tm | Fluidigm | Cat#3169013B |
| CD45RA-170Er | Fluidigm | Cat#3170010B |
| CD226–171Yb | Fluidigm | Cat#3171013B |
| CD354–172Yb | Fluidigm | Cat#3172022B |
| CD94–174Yb | Fluidigm | Cat#3174015B |
| CD56–176Yb | Fluidigm | Cat#3176003B |
| CD16–299Bi | Fluidigm | Cat#3209002B |
| CD7–147Sm | Fluidigm | Cat#3147009B |
| PD1–155Gd | Fluidigm | Cat#3155007B |
| CD26–161Dy | Fluidigm | Cat#3161003B |
| CD57–163Dy | Fluidigm | Cat#3163003B |
| CD158–173Yb | Fluidigm | Cat#3173002B |
| ICOS-148Nd | Fluidigm | Cat#3148019B |
| Cisplatin | Enzo life Sciences | Cat#NC0503617 |
| CyPBS | Rockland | Cat#MB-008 |
| BSA | Sigma | Cat#A3059 |
| Sodium Azide | Sigma | Cat#71289 |
| EDTA | Hoefer | Cat#GR123–100 |
| PFA 16% | Electtron Microscopy
Sciences |
Cat#201192A |
| Intercalator-Ir125 | Fluidigm | Cat#201192A |
| Biological Samples | ||
| Adult Blood | Missisipi Valley Regional Blood Center, MO |
|
| Human tonsils, as surgical waste | Children Hospital St Louis, MO |
|
| Chemicals, Peptides, and Recombinant Proteins | ||
| 7α,25-dihydroxycholesterol | Avanti Polar | Cat#700080 |
| TRizol | Invitrogen | 15596026 |
| T4 DNA Polymerase | New England Biolabs | M0203L |
| Klenow DNA Polymerase | New England Biolabs | M0210L |
| T4 PNK | New England Biolabs | M0201L |
| Klenow (3’−5’ Exo-) | New England Biolabs | M0212L |
| Quick DNA Ligase | New England Biolabs | M2200L |
| Phusion HF Master Mix | New England Biolabs | M0531L |
| Recombinant Human IL-12p80 | Peprotech | Cat#20012p80H |
| Recombinant Human IL-18 | Peprotech | Cat#200–18 |
| Critical Commercial Assays | ||
| Nextera DNA Library Preparation Kit | Illumina | FC-121–1030 |
| Nextera Index Kit | Illumina | FC-121–1011 |
| NEBNext ChIP-Seq Library Prep Master
Mix Set for Illumina |
NEB | E6240L |
| EZ nuclei isolation kit | Sigma | NUC101–1KT |
| Low Bind DNA Tubes | Ambion | AM12450 |
| MaxTract High Density Tubes | Qiagen | 129046 |
| Agencourt AMPure XP | Beckman Coulter | A63880 |
| RNeasy Plus Micro kit | Qiagen | Cat#74034 |
| Human Th1/Th2/Th17 Cytokine Kit BD
Cytometric Bead Array |
BD Biosciences | Cat#560484 |
| Deposited Data | ||
| Raw and Analyzed Data | This paper | GSE112813 |
| ATAC- and H3K27ac-seq of ieILC1 and ILC3 | Koues O.I. and Collins P.L. et al 2016 |
GSE77299 |
| Microarray of Zeb2 Knockout CD8 cells | Dominguez et al. 2015 | GSE72408 |
| Mouse CD11b+/− and CD27+/− NK RNA-seq | Immgen consortium | GSE109125 |
| Mouse CD11b+/− and CD27+/− NK ATAC-seq | Immgen consortium | GSE100738 |
| Mouse NK BLIMP1 ChIP-seq | Mackay L.K. et al, 2016. | GSE79339 |
| Human GM12878 TBX21, and K562
eGFP-ZEB2 or TCF7 ChIP-seq |
ENCODE Consortium | https://www.encodeproject.org/ |
| Mouse NK TBX21 ChIP-seq | Shish, HY et al, 2017 | GSE77695 |
| Mouse CD8 PRDM1 and MAF knockout expression data | Chihara et al., 2018 | GSE113968 |
| Macaque expression data | Hong et al 2013 | Private Communication |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 |
http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Novoalign | Hercus et al, 2012 | www.novocraft.com |
| R | R open license | https://www.r-project.org/ |
| pvclust | Suzuki et al, 2006 | https://cran.r-project.org |
| ggplot2 | Wickham H., 2016 | http://ggplot2.tidyverse.org/ |
| OrthoRetriever | Kasprzyk A., 2011 | lighthouse.ucsf.edu/orthoretriever/ |
| Picard | MIT open license | https://broadinstitute.github.io/picard/ |
| MACs2 | Zhang Y et al, 2008 | https://github.com/taoliu/MACS |
| ChIPSeeker | Yu G. Et all, 2015 | http://bioconductor.org |
| HOMER | Heinz S. et al, 2010 | http://homer.ucsd.edu/homer/ |
| CrossMap | Zhao, H. et al, 2013 | crossmap.sourceforge.net/ |
| BEDTools | Quinlan A.R. and Hall I.M, 2010 |
bedtools.readthedocs.io |
| UCSC Genome Browser | Karolchik et al, 2003 | https://genome.ucsc.edu/ |
| Monocle | Qiu et al, 2017 | https://bioconductor.org/ |
| Gene Ontology | Ashburner et al, 2000 | http://www.geneontology.org/ |
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marco Colonna (mcolonna@wustl.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell Purification and Stimulation of Human Tonsil and Blood Cells.
All human tissues and cells used in this study were obtained under the approval of the institutional review boards at Washington University in Saint Louis. Tonsils were obtained from elective tonsillectomies (Children’s Hospital in Saint Louis) and provided as surgical waste, with no identifiers attached. Human blood cells and tonsil cell suspensions were enriched by CD56 Microbeads MACS sorting. iILC1s and ILC3s were then FACS sorted based on expression of NKp44, CD103 and CCR6, as previously described (Cella 2010 PNAS). Tonsil NK cells were defined as CD56+NKp44-CD103-CD94+ cells. Peripheral Blood NK cells were sorted based on expression of CD56 and CD57 among CD3 negative cells. Cell purity was normally > 98%. For stimulation experiments, after sorting CD56high cells were plated in 200µl of complete RPMI medium (1X104 cells/well) and stimulated with rIL-12 (10ng/ml) + rIL-18 (50ng/ml) in the presence or absence of 7α,25- dihydroxycholesterol (1 µM). Cytokines were quantified in supernatants after 72 hs with the human Th1/Th2/Th17 CBA kit.
Animal Studies.
Spleens from six week old sex-matched CD57BL/6 litter-mates were dissected and strained through mesh, and purified using FACS (BD, Aria II). Mouse NK were sorted as CD3-, CD19-, NK1.1+, and on basis of CD27 and CD11b staining. Cell purity was normally > 98%. Mice were maintained in specific pathogen-free facilities at Washington University in Saint Louis. All Studies were conducted in accordance with the Washington University Animal Studies Committee.
METHODS DETAILS
Cytof and Flow Cytometric Analysis.
CD56-enriched cells were washed with Cy-FACS buffer (CyPBS; 0.1% BSA, 0.02% Sodium Azide, 2mM EDTA) stained on ice for an hour. After two washes cells were stained with cisplatin for 1 min, washed again twice, fixed in 4% PFA for 15 min, spun down and re-suspended in Intercalator-Ir125 overnight. Cells were washed and counted and analyzed on a Cytof 2 mass cytometer (Fluidigm). Data were processed using the R package monocle (Bioconductor).
Gene Expression Analysis.
RNA was isolated with the RNeasy Plus Micro kit according to procedures recommended by the manufacturer. RNA was amplified and hybridized to the Affymetrix Human Gene 1.0 ST array platform (Figure 1) or processed for RNA-seq. Raw array expression data were analyzed using GenePattern and normalized by RMA. For RNA-seq, RNA was prepared using SMARTer Universal Low Input RNA Kit (Clontech). ERCC-spike in analysis and normalization was performed per manufactures protocol (Thermo Fisher). Sequences were aligned, and RPKM values were determined using STAR aligner. For conservation analysis, gene IDs were converted between species using OrthoRetriever. Normalized macaque expression data was retrieved from Hong et al 2013, and expression data was presented as heat maps.
Ultra-low Input ChIP and ATAC-seq.
Aliquots of 100,000 cells were suspended in 20 ul EZ nuclei isolation buffer (Sigma-Aldrich). Samples were then digested using 20 ul MNase buffer (NEB 10X MNase buffer, 1 ul MNase, 3mM DTT) for 5 minutes at 37’C. Reactions were halted by addition of EDTA, Trition and DOC. Digested nucleosomes were diluted in complete ChIP immunoprecipitation buffer (20mM Tris-HCL pH8.0, 2mM EDTA, 150 mM NaCl, 0.1% Triton X-100, 5mM sodium butyrate, EDTA and protease inhibitors (Roche)), precleared using Dynabeads (Invitrogen) for one hour at 4’C and subjected to IP overnight at 4’C (H3K27ac, Abcam). Bead chromatin complexes were washed using low-salt buffer (0.1%SDS, 1%Trition X-100, 2mM EDTA, 20mM Tris pH8.0, 150 mM NaCl), high-salt buffer (0.1%SDS, 1% Trition X-100, 2 mM EDTA, 20 mM Tris pH8.0, 500 mM NaCl) and eluted with an SDS buffer (1% SDS, 100 mM NAHCO3) for 1 hour at 65’C. DNA was purified using Maxtract tubes (Qiagen), then precipitated overnight. For ATAC-seq, aliquots of 50,000 cells were processed as per manufacturer’s instructions (Nextera DNA Library Preparation Kit, Illuminia).
ChIP- and ATAC-seq data processing.
For quality assessment, FASTQC, Samtools and Picard were used to generate sequencing quality, chromosome coverage and read duplication statistics. Samples that failed FastQC, had >50% mitochondrial reads or >50% read duplication were excluded for further processing. Data sets passing initial QC were pre-processed to remove duplicate reads using Picard’s markDuplicates. The collection of statistically significant peaks (MACs2) was curated to remove promoters (UCSC knowntranscripts), CTCF sites (encode GM12878 peaks), gene deserts (regions >100 kb from any transcript), and non-autosomal regions using BEDTools intersect with settings -v and -wa. Super enhancer ranking of H3K27ac ChIP-seq data sets were identified using the HOMER findPeaks algorithm (program options findPeaks.pl <Tag directory> -superslope 1000 –k 0) and plotted using R. Gene(s) were assigned to peaks or super enhancers using ChIPSeeker (Bioconductor), and analyzed for differential enhancer activities. For conservation analysis, conserved regions were identified using CrossMap and BEDTools.
SNP Analyses.
SNP annotation of enhancer regions was accomplished using TraseR (Bioconductor) and plotted with Prism software (GraphPad). Predictions for TF motif disruption were generated by HaploReg (Broad Institute).
Figure Compilation.
Figures were compiled using Microsoft PowerPoint. Low resolution text or UCSC snapshot gene representations were cropped and replaced using PowerPoint. Heatmaps for either motif enrichment or the expression data appended to the right of UCSC snapshots were first generated using Morpheus (Broad Institute), then copied into PowerPoint boxes using color matching tools.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of primary data were performed with R or Prism software (GraphPad). For statistical comparisons between two groups (data from cytokine bead arrays, volcano plots, pairwise ATAC-, H3K27ac- and RNA-seq comparisons), we used Student’s 2-tailed T-test. Data are expressed as mean ± SD and significance levels are indicated in the figure legends. For comparisons between multiple groups (expression, ATAC- and H3K27ac-seq), data were first filtered to include only genes or enhancers with signals above a minimum threshold in multiple replicates using KOverA() from the R package GeneFilter (Bioconductor). Significance at the 0.001 threshold was then determined by ANOVA using row.oneway.anova() from the R package HybridMTests (Bioconductor). Significantly enriched genes or enhancer values (probe intensities or normalized reads) were then subjected to hierarchical clustering and PCA using R libraries. PCA was plotted using the R package, ggplot2. For hierarchical clustering, data were first converted into distances using as.dist(1-cor()), then clustered using hclust( ,method=“ward.D”)”. Colors based upon K-means clustering were assigned using the R package, dendextend.
Gene set enrichment in various NK subsets and significance values were determined using GSEA (Broad Institute). Overlaps between enriched gene sets for CD8 and NK subsets was determined by a binomial distribution, using the R function pbinom().
For TF motif analysis, enhancers were assigned as differentially active if quantile normalized RPKMs for either ATAC or H3K27ac data were significantly enriched in one cell type by more than two-fold, when compared with another cellular subset (Student’s T test, p < 0.01). Centers of ATAC peaks (200 bp) were extracted from differentially active enhancers and analyzed by HOMER using findMotifsGenome.pl with default settings. Significance was determined using Homer’s binomial distribution reporting and only significant motifs are presented (p < 0.001).
DATA AND SOFTWARE AVAILABILITY
The accession number for the raw data files for microarray, RNA-, ChIP, and ATAC-seq analysis reported in this paper is the NCBI GEO repository. Accession number GSE112813.
Supplementary Material
Highlights:
Regulome analysis reveals a spectrum of molecular programs for human ILC1-NK cells
Conserved regulatory schemes for localization–function shared by NK and T cells
TCF1-MYC enforce progenitor-like, lymph node localization modules
PRDM1-ZEB2-MAF dominate effector and non-lymphoid localization programs
A combination of single cell and systems biology approaches determines the molecular programs defining the identity and function of human tissue resident type I innate lymphoid cells and natural killer cells.
ACKNOWLEDGEMENTS
This work was supported by NIH grants AI120606, AI 134035 (E.M.O., Colonna); AI11852, CA188286 (E.M.O.); AI095542, DE021255 (Colonna); CA176695 (Cella); AI 090735 (RPJ); OD 011132 (Yerkes P51 Base Grant); TR000448 (WU-ICTS); and CA91842 (Siteman Cancer Center). We thank Maxim Artymov for helpful suggestions, Takeshi Egawa for reagents, J. Michael Billingsley and Premeela Rajakumar for assistance with transcriptional profiling and data analysis, and the Genome Technology Access Center for help with –omics analyses.
DECLARATION OF INTERESTS
Henoch Hong is a Merck employee. The other authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Allan DSJ, Cerdeira AS, Ranjan A, Kirkham CL, Aguilar OA, Tanaka M, Childs RW, Dunbar CE, Strominger JL, Kopcow HD, et al. (2017). Transcriptome analysis reveals similarities between human blood CD3− CD56bright cells and mouse CD127+ innate lymphoid cells. Sci. Rep 7, 3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggio L, Laureano ÁM, Silla LM da R., and Lee DA (2017). Natural killer cell adoptive immunotherapy: Coming of age. Clin. Immunol 177, 3–11. [DOI] [PubMed] [Google Scholar]
- Berrien-Elliott MM, Wagner JA, and Fehniger TA (2015). Human Cytokine-Induced Memory-Like Natural Killer Cells. J. Innate Immun 7, 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best JA, Blair DA, Knell J, Yang E, Mayya V, Doedens A, Dustin ML, Goldrath AW, Consortium TIGP, Monach P, et al. (2013). Transcriptional insights into the CD8+ T cell response to infection and memory T cell formation. Nat. Immunol 14, 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ, et al. (2013). The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley T, Peppa D, Pedroza-Pacheco I, Li D, Cain DW, Henao R, Venkat V, Hora B, Yue Chen, Vandergrift Nathan A., et al. (2018). RAB11FIP5 expression and altered natural killer cell function are associated with induction of HIV broadly neutralizing antibody responses. Cell. In Press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Otero K, and Colonna M (2010). Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc. Natl. Acad. Sci. U. S. A 107, 10961–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, Nyman J, Marjanovic ND, Kowalczyk MS, Wang C, et al. (2018). Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 558, 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, and Walzer T (2009). Maturation of mouse NK cells is a 4-stage developmental program. Blood 113, 5488– 5496. [DOI] [PubMed] [Google Scholar]
- Choi YS, Gullicksrud JA, Xing S, Zeng Z, Shan Q, Li F, Love PE, Peng W, Xue H-H, and Crotty S (2015). LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat. Immunol 16, 980–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C, Pinto AK, Curtis JD, Persaud SP, Cella M, Lin C-C, Edelson BT, Allen PM, Colonna M, Pearce EL, et al. (2014). c-Myc-induced transcription factor AP4 is required for host protection mediated by CD8+ T cells. Nat. Immunol 15, 884– 893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins A, Littman DR, and Taniuchi I (2009). RUNX proteins in transcription factor networks that regulate T-cell lineage choice. Nat. Rev. Immunol 9, 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinides MG, Gudjonson H, McDonald BD, Ishizuka IE, Verhoef PA, Dinner AR, and Bendelac A (2015). PLZF expression maps the early stages of ILC1 lineage development. Proc. Natl. Acad. Sci. U. S. A 112, 5123–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MA, Fehniger TA, and Colonna M (2017). Is There Natural Killer Cell Memory and Can It Be Harnessed by Vaccination? Vaccination Strategies Based on NK Cell and ILC Memory. Cold Spring Harb. Perspect. Biol a029512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez VS, and Colonna M (2016). Diversity and function of group 1 innate lymphoid cells. Immunol. Lett 179, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez VS, Cervantes-Barragan L, Song C, Gilfillan S, McDonald KG, Tussiwand R, Edelson BT, Murakami Y, Murphy KM, Newberry RD, et al. (2014). CRTAM controls residency of gut CD4+CD8+ T cells in the steady state and maintenance of gut CD4+ Th17 during parasitic infection. J. Exp. Med 211, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez VS, Cervantes-Barragan L, Robinette ML, Bando JK, Wang Y, Geiger TL, Gilfillan S, Fuchs A, Vivier E, Sun JC, et al. (2016). Transforming Growth Factor-β Signaling Guides the Differentiation of Innate Lymphoid Cells in Salivary Glands. Immunity 44, 1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, White AJ, Gilfillan S, Cella M, and Colonna M (2017). SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling. Nat. Immunol 18, 995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley M, Whitelaw E, Perkins A, Williams G, Fujiwara Y, and Orkin SH (1996). Isolation and characterization of the cDNA encoding BKLF/TEF-2, a major CACCC-box-binding protein in erythroid cells and selected other cells. Mol. Cell. Biol 16, 1695–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster JG, Dang EV, Reboldi A, and Yi T (2014). 25-Hydroxycholesterols in innate and adaptive immunity. Nat. Rev. Immunol 14, 731–743. [DOI] [PubMed] [Google Scholar]
- Dominguez CX, Amezquita RA, Guan T, Marshall HD, Joshi NS, Kleinstein SH, and Kaech SM (2015). The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J. Exp. Med 212, 2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emgård J, Kammoun H, García-Cassani B, Chesné J, Parigi SM, Jacob J-M, Cheng H-W, Evren E, Das S, Czarnewski P, et al. (2018). Oxysterol Sensing through the Receptor GPR183 Promotes the Lymphoid-Tissue-Inducing Function of Innate Lymphoid Cells and Colonic Inflammation. Immunity 48, 120–132.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehniger TA, Cooper MA, Nuovo GJ, Cella M, Facchetti F, Colonna M, and Caligiuri MA (2003). CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: a potential new link between adaptive and innate immunity. Blood 101, 3052–3057. [DOI] [PubMed] [Google Scholar]
- Firth MA, Madera S, Beaulieu AM, Gasteiger G, Castillo EF, Schluns KS, Kubo M, Rothman PB, Vivier E, and Sun JC (2013). Nfil3-independent lineage maintenance and antiviral response of natural killer cells. J. Exp. Med 210, 2981–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freud AG, Mundy-Bosse BL, Yu J, and Caligiuri MA (2017). The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity 47, 820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu B, Wang F, Sun R, Ling B, Tian Z, and Wei H (2011). CD11b and CD27 reflect distinct population and functional specialization in human natural killer cells. Immunology 133, 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, and Colonna M (2013). Intraepithelial Type 1 Innate Lymphoid Cells Are a Unique Subset of IL-12- and IL-15-Responsive IFN-γ-Producing Cells. Immunity 38, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabryšová L, Alvarez-Martinez M, Luisier R, Cox LS, Sodenkamp J, Hosking C, Pérez-Mazliah D, Whicher C, Kannan Y, Potempa K, et al. (2018). c-Maf controls immune responses by regulating disease-specific gene networks and repressing IL-2 in CD4+ T cells. Nat. Immunol 19, 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. (2011). A human memory T cell subset with stem cell–like properties. Nat. Med 17, 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gineau L, Cognet C, Kara N, Lach FP, Dunne J, Veturi U, Picard C, Trouillet C, Eidenschenk C, Aoufouchi S, et al. (2012). Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J. Clin. Invest 122, 821–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, Lindsten T, and Reiner SL (2012). The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 36, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan T, Dominguez CX, Amezquita RA, Laidlaw BJ, Cheng J, Henao-Mejia J, Williams A, Flavell RA, Lu J, and Kaech SM (2018). ZEB1, ZEB2, and the miR- 200 family form a counterregulatory network to regulate CD8 + T cell fates. J. Exp. Med 215, 1153–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Bechtel P, Zhai Y, Youssef F, McLachlan K, and Mandelboim O (2004). Novel insights on human NK cells’ immunological modalities revealed by gene expression profiling. J. Immunol 173, 6547–6563. [DOI] [PubMed] [Google Scholar]
- Held W, Clevers H, and Grosschedl R (2003). Redundant functions of TCF-1 and LEF-1 during T and NK cell development, but unique role of TCF-1 for Ly49 NK cell receptor acquisition. Eur. J. Immunol 33, 1393–1398. [DOI] [PubMed] [Google Scholar]
- Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, Lietzenmayer M, Kroehling L, Takumi A, Kometani K, et al. (2018). KLRG1+ Effector CD8+ T Cells Lose KLRG1, Differentiate into All Memory T Cell Lineages, and Convey Enhanced Protective Immunity. Immunity 48, 716–729.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong HS, Rajakumar PA, Billingsley JM, Reeves RK, and Johnson RP (2013). No monkey business: why studying NK cells in non-human primates pays off. Front. Immunol 4, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson SC, and Masopust D (2018). Understanding Subset Diversity in T Cell Memory. Immunity 48, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeevan-Raj B, Gehrig J, Charmoy M, Chennupati V, Grandclément C, Angelino P, Delorenzi M, and Held W (2017). The Transcription Factor Tcf1 Contributes to Normal NK Cell Development and Function by Limiting the Expression of Granzymes. Cell Rep 20, 613–626. [DOI] [PubMed] [Google Scholar]
- Johnson JK, and Miller JS (2018). Current strategies exploiting NK-cell therapy to treat haematologic malignancies. Int. J. Immunogenet 45, 237–246. [DOI] [PubMed] [Google Scholar]
- Kallies A, Carotta S, Huntington ND, Bernard NJ, Tarlinton DM, Smyth MJ, and Nutt SL (2011). A role for Blimp1 in the transcriptional network controlling natural killer cell maturation. Blood 117, 1869–1879. [DOI] [PubMed] [Google Scholar]
- Keskin DB, Allan DSJ, Rybalov B, Andzelm MM, Stern JNH, Kopcow HD, Koopman LA, and Strominger JL (2007). TGFbeta promotes conversion of CD16+ peripheral blood NK cells into CD16- NK cells with similarities to decidual NK cells. Proc. Natl. Acad. Sci. U. S. A 104, 3378–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, et al. (2014). Differentiation of Type 1 ILCs from a Common Progenitor to All Helper-like Innate Lymphoid Cell Lineages. Cell 157, 340–356. [DOI] [PubMed] [Google Scholar]
- Komori T (2010). Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res 339, 189–195. [DOI] [PubMed] [Google Scholar]
- Koues OI, Collins PL, Cella M, Robinette ML, Porter SI, Pyfrom SC, Payton JE, Colonna M, and Oltz EM (2016). Distinct Gene Regulatory Pathways for Human Innate versus Adaptive Lymphoid Cells. Cell 165, 1134–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küçük C, Iqbal J, Hu X, Gaulard P, De Leval L, Srivastava G, Au WY, McKeithan TW, and Chan WC (2011). PRDM1 is a tumor suppressor gene in natural killer cell malignancies. Proc. Natl. Acad. Sci. U. S. A 108, 20119–20124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CM, Adams NM, Geary CD, Weizman O-E, Rapp M, Pritykin Y, Leslie CS, and Sun JC (2018). Epigenetic control of innate and adaptive immune memory. Nat. Immunol 19, 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Zhang T, Hwang I, Kim A, Nitschke L, Kim M, Scott JM, Kamimura Y, Lanier LL, and Kim S (2015). Epigenetic Modification and Antibody-Dependent Expansion of Memory-like NK Cells in Human Cytomegalovirus-Infected Individuals. Immunity 42, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace EM, Hsu AP, Monaco-Shawver L, Makedonas G, Rosen JB, Dropulic L, Cohen JI, Frenkel EP, Bagwell JC, Sullivan JL, et al. (2013). Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset. Blood 121, 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay LK, Minnich M, Kragten NAM, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Freestone D, et al. (2016). Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463. [DOI] [PubMed] [Google Scholar]
- Michel T, Poli A, Cuapio A, Briquemont B, Iserentant G, Ollert M, and Zimmer J (2016). Human CD56bright NK Cells: An Update. J. Immunol 196, 2923–2931. [DOI] [PubMed] [Google Scholar]
- Nagler A, Lanier LL, Cwirla S, and Phillips JH (1989). Comparative studies of human FcRIII-positive and negative natural killer cells. J. Immunol 143, 3183–3191. [PubMed] [Google Scholar]
- O’Sullivan TE, Sun JC, and Lanier LL (2015). Natural Killer Cell Memory. Immunity 43, 634–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachynski RK, Zabel BA, Kohrt HE, Tejeda NM, Monnier J, Swanson CD, Holzer AK, Gentles AJ, Sperinde GV, Edalati A, et al. (2012). The chemoattractant chemerin suppresses melanoma by recruiting natural killer cell antitumor defenses. J. Exp. Med 209, 1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parolini S, Santoro A, Marcenaro E, Luini W, Massardi L, Facchetti F, Communi D, Parmentier M, Majorana A, Sironi M, et al. (2007). The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood 109, 3625– 3632. [DOI] [PubMed] [Google Scholar]
- Pickrell JK, Berisa T, Liu JZ, Ségurel L, Tung JY, and Hinds DA (2016). Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet 48, 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsuran V, Naranbhai V, Horowitz A, Qi Y, Martin MP, Yuki Y, Gao X, Walker-Sperling V, Del Prete GQ, Schneider DK, et al. (2018). Elevated HLA-A expression impairs HIV control through inhibition of NKG2A-expressing cells. Science 359, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richer MJ, Lang ML, and Butler NS (2016). T Cell Fates Zipped Up: How the Bach2 Basic Leucine Zipper Transcriptional Repressor Directs T Cell Differentiation and Function. J. Immunol 197, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, Gilfillan S, Colonna M, Shaw L, Yu B, et al. (2015). Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat. Immunol 16, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, Leong JW, Abdel-Latif S, Schneider SE, Willey S, et al. (2016). Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med 8, 357ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkilde MM, Benned-Jensen T, Andersen H, Holst PJ, Kledal TN, Lüttichau HR, Larsen JK, Christensen JP, and Schwartz TW (2006). Molecular pharmacological phenotyping of EBI2. An orphan seven-transmembrane receptor with constitutive activity. J. Biol. Chem 281, 13199–13208. [DOI] [PubMed] [Google Scholar]
- Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, Han H, Chiang SCC, Foley B, Mattsson K, et al. (2015). Cytomegalovirus Infection Drives Adaptive Epigenetic Diversification of NK Cells with Altered Signaling and Effector Function. Immunity 42, 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoni Y, Fehlings M, Kløverpris HN, McGovern N, Koo S-L, Loh CY, Lim S, Kurioka A, Fergusson JR, Tang C-L, et al. (2017). Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity 46, 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Fernandes H, and Artis D (2018). Neuronal-immune system cross-talk in homeostasis. Science 359, 1465–1466. [DOI] [PubMed] [Google Scholar]
- Vu TT, Gatto D, Turner V, Funnell APW, Mak KS, Norton LJ, Kaplan W, Cowley MJ, Agenès F, Kirberg J, et al. (2011). Impaired B Cell Development in the Absence of Krüppel-like Factor 3. J. Immunol 187, 5032–5042. [DOI] [PubMed] [Google Scholar]
- Weizman O-E, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, Degli- Esposti MA, Leslie CS, Sun JC, and O’Sullivan TE (2017). ILC1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell 171, 795–808.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt K, Wilk E, Buyny S, Buer J, Schmidt RE, and Jacobs R (2006). Gene and protein characteristics reflect functional diversity of CD56dim and CD56bright NK cells. J. Leukoc. Biol 80, 1529–1541. [DOI] [PubMed] [Google Scholar]
- Wu C, Li B, Lu R, Koelle SJ, Yang Y, Jares A, Krouse AE, Metzger M, Liang F, Loré K, et al. (2014). Clonal tracking of rhesus macaque hematopoiesis highlights a distinct lineage origin for natural killer cells. Cell Stem Cell 14, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Seay M, Schulz VP, Hariharan M, Tuck D, Lian J, Du J, Shi M, Ye Z, Gerstein M, et al. (2012). Tcf7 Is an Important Regulator of the Switch of Self- Renewal and Differentiation in a Multipotential Hematopoietic Cell Line. PLoS Genet 8, e1002565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Mao HC, Wei M, Hughes T, Zhang J, Park I, Liu S, McClory S, Marcucci G, Trotta R, et al. (2010). CD94 surface density identifies a functional intermediary between the CD56bright and CD56dim human NK-cell subsets. Blood 115, 274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the raw data files for microarray, RNA-, ChIP, and ATAC-seq analysis reported in this paper is the NCBI GEO repository. Accession number GSE112813.