

Abstract
Mitochondria are increasingly recognized as key mediators of acute cellular stress responses in asthma. However, the distinct roles of regulators of mitochondrial physiology on allergic asthma phenotypes are currently unknown. The mitochondrial Ca2+ uniporter (MCU) resides in the inner mitochondrial membrane and controls mitochondrial Ca2+ uptake into the mitochondrial matrix. To understand the function of MCU in models of allergic asthma, in vitro and in vivo studies were performed using models of functional deficiency or knockout of MCU. In primary human respiratory epithelial cells, MCU inhibition abrogated mitochondrial Ca2+ uptake and reactive oxygen species (ROS) production, preserved the mitochondrial membrane potential and protected from apoptosis in response to the pleiotropic Th2 cytokine IL-13. Consequently, epithelial barrier function was maintained with MCU inhibition. Similarly, the endothelial barrier was preserved in respiratory epithelium isolated from MCU−/− mice after exposure to IL-13. In the ovalbumin-model of allergic airway disease, MCU deficiency resulted in decreased apoptosis within the large airway epithelial cells. Concordantly, expression of the tight junction protein ZO-1 was preserved, indicative of maintenance of epithelial barrier function. These data implicate mitochondrial Ca2+ uptake through MCU as a key controller of epithelial cell viability in acute allergic asthma.
Keywords: mitochondrial Ca2+ uniporter, allergic asthma, respiratory epithelium, oxidative stress, epithelial barrier function
Graphical Abstract
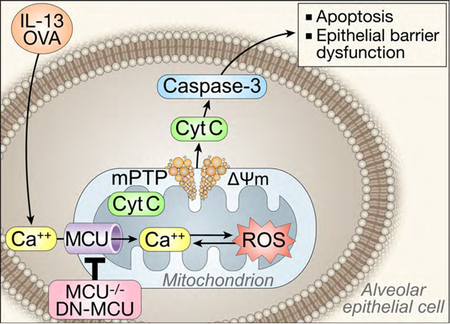
Introduction
The airway epithelium is an essential controller of numerous responses to allergens, including inflammatory, immune and regenerative processes [1]. The epithelium forms a physiologic barrier to extraneous allergens and pollutants and is comprised of mucus, apical junction complexes and airway surface liquids [2, 3]. Of note, inhaled toxins and certain inflammatory cytokines can disrupt barrier integrity, which might represent a risk factor for allergen sensitization [1]. In addition, airway barrier dysfunction likely initiates signal transduction cascades that alter epithelial cell fate [1]. Recently, dysfunctional apical junctional complexes have been documented in the respiratory epithelium of asthmatic patients [4], although the precise mechanisms involved and consequences for this phenomenon are not clear. Therefore, understanding the regulation of airway epithelial barrier function is important in asthma immunology and pathophysiology.
Excessive reactive oxygen species (ROS) have a significant effect on the development of tissue injury and epithelial barrier dysfunction observed in asthma [5, 6]. Mitochondria are key players in the development of oxidant stress-related illnesses by serving as the primary source of cellular ROS [7–10]. Mitochondria serve to buffer intracellular Ca2+ levels, which has been proposed to drive mitochondrial ROS production. Indeed, the “love-hate” triangle between mitochondrial Ca2+, ROS, and ATP is well-established, in which mitochondrial Ca2+ uptake increases the activity of Ca2+-dependent dehydrogenases that in turn increase ATP production and mitochondrial ROS generation [7, 11]. In the case of mitochondrial Ca2+ overload, in conjunction with pathophysiological accumulation of ROS, cells respond by sustained opening of the high conductance cyclosporin A-sensitive permeability transition pore [12]. This in turn leads to the rapid collapse of the mitochondrial membrane potential (ΔΨm) and the swelling of mitochondria. Subsequent loss of cytochrome c leads to cellular apoptosis [13, 14]. These data led us to hypothesize that there is a considerable conceptual overlap between asthma pathophysiology and mitochondrial biology in the aspects of oxidative stress, apoptosis, and Ca2+ homeostasis [15–18].
Mechanistically, mitochondrial Ca2+ uptake was discovered to be mediated by the mitochondrial calcium uniporter (MCU) located in the organelle’s inner membrane [19]. In models of cardiac disease, overexpression of MCU in cardiomyocytes increases cell death in response to challenge by proapoptotic stimuli [20]. By contrast, suppressing MCU with Ru360, a pharmacological antagonist related to ruthenium red, protects against ischemia-reperfusion injury [21]. Despite the clear evidence for the role in mitochondrial Ca2+ uptake and excessive mitochondrial ROS generation in asthma, no studies to date have explored whether the mechanisms of the “love-hate” triangle exist in airway epithelia or whether MCU inhibition can prevent this process. Utilizing primary human epithelial cells as well as an in vivo murine model of experimental allergic asthma, we investigated the role of MCU as a key regulator of mitochondrial ROS-dependent effects, in particular airway epithelial cell death and loss of barrier function.
Materials and Methods
Epithelial cell culture and DN-MCU adenoviral expression
Primary human airway epithelial cells (HAEC) were obtained from the Cells and Tissue Core, Division of Pulmonary Medicine, University of Iowa [22]. Cells were grown in keratinocyte serum-free media (KSFM, Gibco, Waltham, MA) and routinely tested for mycoplasma contamination. Cells were grown until confluent and then infected with either adenovirus containing the cDNA for mitochondrial-targeted GFP (Mt-GFP, empty vector) or Mt-DNMCU (both at 10 MOI) for 48 hrs prior to treatment with recombinant human IL-13 (10 ng/ml, cat. 213-ILB/CF, R&D Systems, Minneapolis, MN) for 48 or 72 hrs. Adenoviral vectors were generated by the University of Iowa Gene Vector Core Facility.
Primary murine tracheal epithelial cells (MTBEC) were isolated from CD1 mice or MCU knockout mice (KO, CD1 strain) as previously described [23]. Cells were plated onto collagen (BD Biosciences)– coated transwell plates and maintained in MTEC Plus culture medium as described previously [23]. Cells were grown until confluent and then challenged with recombinant murine IL-13 (10 ng/ml, cat. 413-ML-005/CF, R&D Systems) for 4 days.
Isolation of mitochondrial fractions
Freshly collected HAEC were suspended in ice-cold mannitol, sucrose, EGTA (MSE) buffer (5 mM 3-(N-morpholino) propanesulfonic acid, 70 mM sucrose, 2 mM ethyleneglycol-bis-(b-aminoethyl ether)-N, N9-tetraacetic acid, 220 mM mannitol, pH 7.2, with KOH), then homogenized. Nuclei and unbroken cells were pelleted by centrifugation twice at 600Xg for 5 min. The crude mitochondrial and cytosolic fraction was obtained from the supernatant by centrifugation at 8,500Xg for 10 min. The pellet was then resuspended in 100 μl MSE with protease and phosphatase inhibitors. Finally, samples were centrifuged at 8,500Xg for 10 min and the supernatant (cytoplasmic fraction) was isolated. The pellet (mitochondrial fraction) was resuspended in 50μl MSE with protease and phosphatase inhibitors for subsequent immunoblotting.
Immunoblotting
Immunoblotting was performed on whole cell lysates, cytoplasmic (for cytochrome c) or mitochondrial fractions (see above). To obtain whole cell lysates, HAEC or lung homogenates were lysed in RIPA buffer and centrifuged at 6000 × g for 10 min at 4°C to separate insoluble components. Protein concentration was measured via DC Assay according to manufacturer protocol (Bio-Rad, Hercules, CA). Lysates were mixed with 5X sample buffer, resolved by SDS-PAGE electrophoresis and transferred to PVDF membranes. After blocking in 5% dry milk in TBS with 0.05% Tween 20 for 60 min, the membranes were incubated in primary antibody against MCU (1:500, HPA016480, Sigma), COXIV (1:1000, cat. 4850, Cell Signaling), cytochrome c (cytoplasmic fraction, 1:500, cat. 4280, Cell Signaling), ZO-1 (1:500, cat. AB2272, Millipore), Bax (1:500, cat. Sc-70407, Santa Cruz), Bcl-2 (1:500, cat. sc492, Santa Cruz) or GAPDH (1:1000, cat. 2118, Cell Signaling) overnight, followed by anti-rabbit (1:2000, cat. sc-2004, Bio-Rad) or anti-mouse (1:2000, cat. 7076, Cell Signaling) IgG secondary antibody for 60 min at room temperature. Proteins were visualized with the enhanced chemiluminescence detection method (Licor, Lincoln, NE). Densitometric analysis was assessed by ImageStudioLite software (Licor). After normalization, data were calculated as fold change.
O-Cresolphthalein Ca2+ assay
For measurement of mitochondrial Ca2+ content, HAEC were suspended in ice-cold mannitol, sucrose (MS) buffer that was free of EGTA to avoid Ca2+ chelation. Mitochondria were pelleted and diluted in calcium assay buffer (Cat. 700551, Cayman Chemical, Ann Arbor, MI) homogenized and sonicated (2 × 10 s at 40% of maximal power output). Ca2+ content in the supernatant was determined spectrophotometrically using the O-Cresolphthalein Complex one calcium assay kit (Cat. 701220, Cayman Chemical). Values were normalized to total protein concentration measured via DC Assay according to manufacturer protocol (Bio-Rad).
Calcium retention capacity (CRC) assay
Calcium Green-5N was used to monitor extramitochondrial Ca2 in HAEC assay as previously described [24, 25]. HAEC were infected with empty vector or DN-MCU virus. Cells were grown for 48 hrs, harvested using trypsin and washed in PBS. The CRC assay was performed in 106 HAEC in 100 µl respiration buffer containing 120 mM KCl, 10 mM Tris-HCl pH 7.4, 5 mM MOPS, 5 mM Na2HPO4, 10 mM glutamate, 2 mM malate, 0.002% digitonin, 0.5 mM thapsigargin to inhibit ER Ca2+ uptake and 1 µM Calcium Green-5N (cat. C3739, Molecular Probes, Waltham, MA). Calcium Green-5N fluorescence was monitored at 485 nm excitation; 535 nm emission, after adding CaCl2 (100 µM free Ca2+ at 3-min intervals at 30 °C). Values were normalized to baseline.
Mitochondrial Ca2+assay
To study changes in mitochondrial Ca2+, cells were loaded with the AM ester of Rhod-2 (5 μm; Kd = 570 nm, cat. R1245MP, Molecular Probes) at 36.5°C for 15 min in culture medium, followed by a subsequent 30 min period for hydrolysis of the dye. In AM-ester form, the dye carries a positive charge, which allows accumulation in negatively charged mitochondria, in which the ester bond is hydrolyzed. Because the hydrolysis might also occur in the cytosol, cells were also incubated with the mitochondria-specific fluorescent marker (mitoTracker, cat. M22426, Molecular Probes) to ascertain mitochondrial localization. After washout, plates were transferred to the recording chamber mounted on a Leica P8 STED (Buffalo Grove, IL) confocal laser-scanning system. MitoTracker (5 μm) was loaded simultaneously with the Ca2+ ion-sensitive probes. After 1 min of baseline recording, agonist histamine (100 μM, cat. HY125, Sigma, St. Louis, MO) or IL-13 (10ng/ml) was added and confocal images were recorded every 5 sec up to 6 min. All fluorescent probes were purchased from Molecular Probes. The fluorescent signal over time was analyzed with Fiji software from NIH ImageJ. All images were taken at the same time and using the same imaging settings. Data were reported as difference in signal over baseline.
In additional experiments, cells loaded with Rhod-2 were imaged at 2 min after addition of IL-13 using a LSM 510 confocal microscope (Carl Zeiss, San Diego, CA), and analyzed with NIH ImageJ. All images were taken at the same time and using the same imaging settings. Data are presented as difference in signal over baseline.
Confocal imaging of mitochondrial matrix membrane potential
The mitochondrial membrane potential was measured in live cultured HAEC using tetramethylrhodamine methyl ester (TMRM, 100 nM, cat. T668, Molecular Probes). Cells were imaged using a LSM 510 confocal microscope (Carl Zeiss, San Diego, CA), and analyzed with NIH ImageJ. All images were taken at the same time and using the same imaging settings. Data are presented as fold change over control.
For determination of the mitochondrial matrix membrane potential by fluorescence-activated cell sorting (FACS), HAEC were washed and resuspended in PBS with 2.5 mM CaCl2, 1 mM MgCl2, 5 mM pyruvate, and 1% BSA. Cells were labeled with 2 µM TMRM for 20 min at 37°C. Depolarization of HAEC was induced with 10 µM Antimycin A (Sigma) as a positive control. After labeling cells samples were analyzed with Muse Cell Analyzer (Millipore Sigma, Burlington, MA). Two cell populations are distinguished by cytofluorimetric separation: depolarized “positive” and “negative” cells with preserved membrane potential. Data were calculated as fold change from control.
Mitochondrial superoxide detection with mitoSOX
ROS were measured in live cultured HAEC using the dihydroethidium derivative mitoSOX red (5 μM, cat. D1168, Invitrogen, Waltham, MA). Cells were imaged using a LSM 510 confocal microscope (Carl Zeiss), and analyzed with NIH ImageJ. All images were taken at the same time and using the same imaging settings. Data are presented as fold change over control.
For MitoSOX determination via FACS, HAEC or MTBEC were washed and resuspended in PBS with 2.5 mM CaCl2, 1 mM MgCl2, 5 mM pyruvate, and 1% BSA. Cells were labeled with 0.5 µM MitoSOX (cat. D1168, Invitrogen) for 20 min at 37°C. Maximal ROS production was induced with 10 µM Antimycin A (Sigma) as a positive control. After labeling cells samples were analyzed with a Muse Cell Analyzer (Millipore Sigma, Burlington, MA). Based on mitochondrial superoxide production induced by Antimycin, this signal was gated to distinguish two populations: ROS “negative” and ROS “positive” cells. Data were expressed as fold change from control.
Transepithelial electrical resistance and conductance
HAEC or MTBEC were seeded on collagen-coated cell culture inserts (Transwell-clear, diameter 12 mm, 0.4 μm pores; Corning, Acton, MA) in multi-well plates and cultured under submerged conditions in DMEM-F-12 (Gibco) or MTBEC media [23] in the presence of 10% fetal bovine serum (FBS) and penicillin/streptomycin (1%) until confluent. Then, the medium was removed from the upper compartment to allow polarization at the air–liquid interface (ALI). Transepithelial electrical resistance (TEER) of HAEC monolayers was measured with an epithelial voltmeter (EVOM2, World Precision Instruments, Sarasota, FL). The resistance of the epithelial monolayer was calculated by subtracting the baseline electrical resistance of the supporting filter and buffer medium without cells from the total electrical resistance determined with the monolayer. The conductance was calculated as the reciprocal of TEER (1/Ω × cm2).
Endothelial barrier permeability
TRITC-Dextran (50μl, 4.4 kDa, 1mg/ml, cat. T1037, Sigma) was added to the apical chamber (total volume 1mg/ml) of HAEC or MTBEC cultured at the ALI. After 20 min, 100μl of media were collected from the lower chamber. TRITC fluorescence was measured on a SpectroMax ELISA plate reader. Data were calculated as the percent of control (set as 100%).
Cell viability analysis
Following adenoviral expression of DN-MCU or Mt-GFP in HAEC for 72 hrs (10 MOI), cell viability was assessed by trypan blue staining min (0.4% (w/v) trypan blue solution, cat. 15250061, ThermoFisher, Waltham, MA). Cells were counted using a hemocytometer. Viable and nonviable cells were recorded separately. Data were calculated as the means of independent experiments.
Annexin V Assay
HAEC were infected with adenovirus expressing DN-MCU or Mt-GFP (48 hrs, 10 MOI), and exposed to IL-13 or untreated media (72 hrs), and then trypsinized, resuspended in media, and labeled for Annexin V using an Annexin V/Dead Cell kit (cat #MCH100105, Merck Millipore, Billerica, MA, USA) as recommended by the manufacturer. As a positive control, samples were treated for 24 hrs with hydrogen peroxide (100μM, Sigma-Aldrich). The samples were analyzed with a Muse automated cell analyzer (Merck Millipore). Based on labeling for Annexin V in apoptotic cells and for the nuclear dye 7-aminoactinomycin D (7-AAD) in dead cells, this assay distinguished four populations by cytofluorimetric separation: live (7-AAD negative, Annexin V negative), non-apoptotic dead (7-AAD positive, Annexin V negative), apoptotic live (7-AAD negative, Annexin V positive), and apoptotic dead (7-AAD positive, Annexin V positive) cells. Data were calculated as fold change from control. Scatterplots were generated using software from the FlowJo program (FlowJo LLC).
Caspase-3 Activity Assay
HAEC were infected with adenovirus expressing DN-MCU or Mt-GFP (48 hrs, 10 MOI), and exposed to IL-13 (72 hrs) prior to lysis in RIPA buffer and centrifugation at 6000 × g for 10 min at 4°C to separate insoluble components. Protein concentration was measured via DC Assay according to manufacturer protocol (Bio-Rad). Cell lysates were then assessed for caspase-3 activity according to manufacturer’s instructions (cat. KH01091, Invitrogen). Values were normalized to total protein concentration.
Induction of OVA-mediated allergic asthma in mice
MCU−/− mice in CD1 background were a kind gift from Dr. Toren Finkel. Additional CD1 WT were obtained through Charles Rivers Labs. Equal numbers of 8–10 week old male and female mice were used. Allergic asthma was modeled in vivo by challenge OVA as previously described [24, 25]. Mice were sensitized by i.p. injection of 10 µg of OVA (cat. A7641, Sigma) mixed with 1 mg of alum (or saline alone, for control mice) on days 0 and 7. Mice were subsequently challenged with inhaled OVA (cat. A5503, Sigma, 1% solution in 0.9% saline, 40-min challenge) or saline on days 14–17. Mice were then euthanized on day 18.
Immunofluorescence and immunohistochemistry
For immunofluorescence imaging, HAEC were fixed with 4% PFA for 20 min at room temperature followed by 0.1% Triton X-100 permeabilization for 10 min. The cells were then incubated with ZO-1 (1:100, rabbit anti-human or mouse, 1:500, cat. AB2272, Millipore, Bilerica, MA), followed by Alexa Fluor 488-labeled goat anti-rabbit IgG secondary antibody (1:2500, cat. R37116, Thermo Scientific). Cells were imaged at 63x or 100x magnification using a LSM 510 confocal microscope (Carl Zeiss). Brightness and contrast were adjusted with ImageJ software. As control, cells were visualized and imaged using epifluorescence microscopy without primary to image GFP staining. The methods used to quantify the staining intensity involved two steps: 1) count the mean density, 2) divide the mean area [26]. Data were calculated as fold change from control.
For immunohistochemistry, lungs were fixed with 4% paraformaldehyde, paraffin-embedded, and 5μm-tissue sections cut. After antigens retrieval by boiling the tissue sections in 0.01 M citrate buffer, pH 6.0, for 15 min, the sections were washed in dH2O for 5 min, followed by quenching of endogenous peroxidases with 3% hydrogen peroxide containing 1% sodium azide for 10 min. After permeabilization in 0.1% Triton-X-100, the sections were blocked in goat anti-rabbit serum, incubated anti-ZO-1 (rabbit anti-human or mouse, 1:100, cat. AB2272, Millipore) for 24 hrs, goat-anti-biotin (1:2000, DAB, Sigma) for 1 hr at room temperature, and horseradish peroxidase-conjugated secondary antibody for 30 min at room temperature. Staining was developed with diaminobenzidine (DAB, Sigma-Aldrich) substrate for 3 min at room temperature. Sections then were counter-stained with hematoxylin and mounted with Perma-Mount medium (Thermo Scientific). Images were taken on an Olympus BX-61 light microscope (Olympus, Center Valley, PA) at 20-x objective. Sections were evaluated in a blinded fashion and staining intensity was scored using NIH Image J software (ImageJ64, version 1.48, National Institutes of Health). Specifically, the intensity of the nuclear DAB staining was considered as background and values were divided by the mean area.
TUNEL staining of mouse lung tissue
Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) staining was performed with the InSitu Cell Death Detection Kit, TMR Red Kit as recommended by the manufacturer (12156792910; Roche, Indianapolis, IN). Nuclei were counterstained with TO-PRO-3 (Invitrogen). Sections were mounted with VectaShield mounting media (VectaShield, Burlingame, CA). Images (3–4/lung) were acquired with a Zeiss 710 Confocal Microscope at 20X or 63X magnification. All images were acquired with the same imaging settings. TUNEL positive cells were imaged using NIH ImageJ software and calculated as the percentage of TUNEL-positive cells relative to the total number of cells in large airways.
Statistical analysis
All data are presented as mathematical mean values ± standard error of the mean. Student’s two-tailed t-test, one-way ANOVA or two-way ANOVA followed by Tukey’s test was used to assess statistical differences. p-values ≤ 0.05 were considered statistically significant.
Results
Expression of dominant negative MCU in airway epithelial cells blunts IL-13-mediated mitochondrial Ca2+ uptake
Our first goal was to develop a model of MCU inhibition in airway epithelial cells. Here we applied a recently reported approach to inhibit MCU activity through expression of a dominant-negative MCU construct with point mutations of Asp260 and Glu263 to Gln [27] (D260Q, E263Q, denoted “DN-MCU”). This approach has been validated to block Ca2+ conductance by MCU and thereby abolish IMCU. [27]. To validate whether DN-MCU inhibits endogenous MCU function, primary human airway epithelial cells (HAEC) were infected with DN-MCU adenovirus or a control vector (Mt-GFP). Isolation of mitochondrial fractions of HAEC after infection showed a significant increase in total MCU expression compared to GFP controls, demonstrating overexpression in mitochondria of HAEC (Figure 1A). Next, we determined the baseline Ca2+ concentrations in mitochondria of HAEC expressing Mt-GFP or DN-MCU. Overexpression of DN-MCU diminished the Ca2+ concentration by about 50% (Figure 1B). In addition, experiments using Calcium Green-5N to measure mitochondrial Ca2+ uptake showed a rapid increase in Calcium Green-5N fluorescence followed by a decline in the fluorescence intensity of the calcium sensor indicating increased mitochondrial uptake (Figure 1C). Over the same range of calcium concentration, HAEC expressing DN-MCU did not demonstrate evidence for mitochondrial uptake (Figure 1C). We next confirmed that DN-MCU efficiently blocks mitochondrial matrix Ca2+ uptake after Ca2+ mobilization by Histamine [28, 29]. For this purpose, we loaded HAEC infected with adenovirus expressing Mt-GFP or DN-MCU with the mitochondrial Ca2+ indicator, Rhod-2. As expected, DN-MCU abolished the histamine-induced increase in matrix Ca2+ uptake (Figure 1D). These data validate that our model of DN-MCU overexpression in airway epithelial cells inhibits MCU activity.
Figure 1. MCU inhibition blocks mitochondrial matrix Ca2+ uptake in human airway epithelial cells.
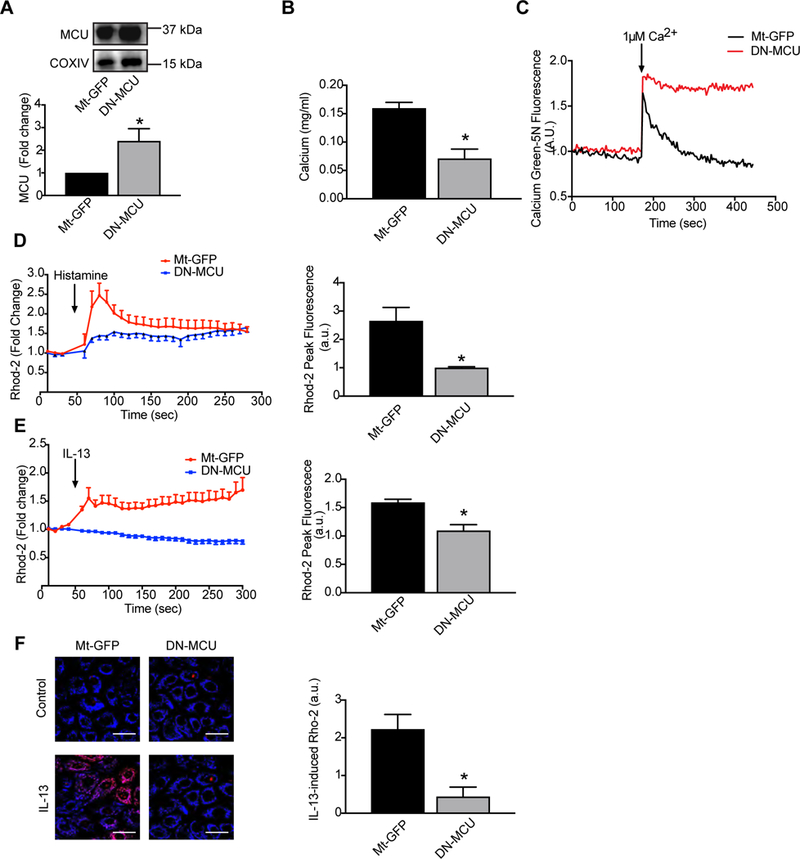
HAEC were infected with empty virus (Mt-GFP) or DN-MCU (both at 10 MOI) for 48 hrs. (A) Immunoblots for MCU in isolated mitochondrial fractions from Mt-GFP or DN-MCU-infected HAEC. COXIV: loading control for mitochondria. (B) Ca2+ concentration by o-cresolphthalein assay in mitochondria. Data were normalized to total mitochondrial protein measured by BCA assay (n = 3 independent experiments). (C) Comparison of mitochondrial calcium levels in HAEC infected with control or DN-MCU virus. Arrow indicates calcium addition. Increasing mitochondrial calcium results in a rapid increase in the fluorescent signal but the subsequent decline in the fluorescent signal, representing mitochondrial calcium uptake, is only observed in control-infected cells. D) Mitochondrial matrix Ca2+ uptake by Rhod-2 fluorescence. Arrow denotes when histamine was added. Peak mitochondrial matrix Ca2+ concentration represented as Rhod-2 fluorescence intensity. (n = 4 independent experiments). (E) Mitochondrial matrix Ca2+ uptake by Rhod-2 fluorescence. Arrow denotes when IL-13 was added. Peak mitochondrial matrix Ca2+ uptake represented as Rhod-2 fluorescence intensity (n = 3 independent experiments). (F) Representative images and quantification of Rhod-2 fluorescence two minutes after addition of IL-13 (n = 3 independent experiments). All data are the means ± SEM. Student’s two-tailed t-test was used. * p < 0.05 vs. Mt-GFP.
IL-13 induces apoptosis and barrier dysfunction in colonic epithelial cells [30] and diminishes the capacity of respiratory epithelial cells to maintain barrier function [31]. Since mitochondria play key roles in activating apoptosis and MCU deletion protects from cell death [32], we asked whether IL-13 induces mitochondrial Ca2+ uptake via MCU. Indeed, we observed an increase in mitochondrial Ca2+ uptake by IL-13 that was abolished with overexpression of DN-MCU (Figure 1E, F).
MCU inhibition abolishes mitochondrial ROS production and preserves mitochondrial membrane potential
Mitochondrial matrix Ca2+ uptake is well known to enhance mitochondrial ROS (Mt-ROS) production We therefore examined whether MCU inhibition blocks Mt-ROS production in airway epithelia. To determine if inhibition of MCU function modulates IL-13-induced changes, the same time point was assessed, HAEC were treated with IL-13 for 48 hrs as we previously reported [24, 33]. As expected, controls exposed to IL-13 showed an increase in mitochondrial superoxide production. In contrast, DN-MCU significantly blunted IL-13-induced Mt-ROS production compared to control (Figure 2A–C).
Figure 2. MCU inhibition abolishes ROS production and preserves the membrane potential.
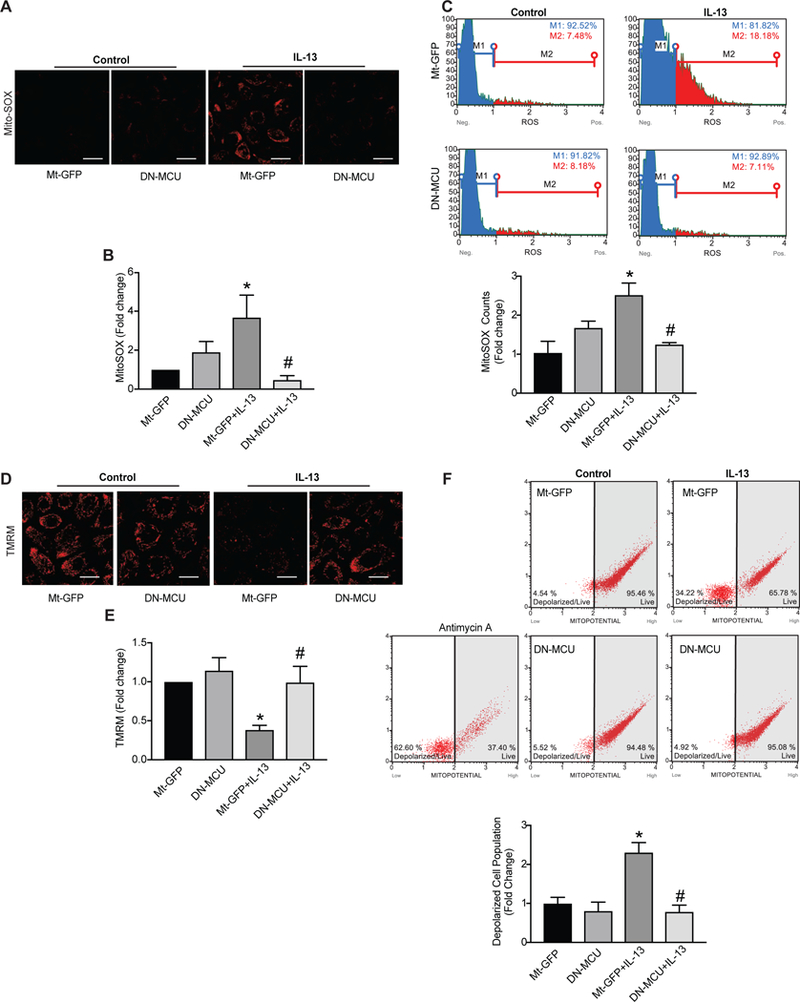
(A) Representative images and (B) quantification of mitochondrial ROS production in HAEC infected with Mt-GFP (empty) or DN-MCU adenovirus (10 MOI) prior to exposure to IL-13 (10 ng/ml) for 2 days (n = 5 independent experiments; data were quantified for 5–6 images per treatment at 63x magnification and represented as percent of control). Scale bars = 100μm. (C) Scatter plots and quantification of Mt-ROS of MitoSOX fluoresence by flow cytometry (Representative plots are shown, n = 4 independent experiments, represented as fold change). Representative images of mitochondrial membrane potential staining by TMRM fluorescence. Quantification of TMRM (n=5 independent experiments; data were quantified for 5–6 images per treatment at 63x magnification, represented as percent of control). Scale bars= 100μm. (F) Scatter plots and quantification of TMRM staining by flow cytometry (Representative plots are shown, n = 4 independent experiments, represented as fold change). All data are the means ± SEM. One-way ANOVA with Tukey post hoc test used. * p < 0.05 vs. control, # p < 0.05 versus Mt-GFP/IL-13.
Increased Ca2+ uptake triggers Mt-ROS production, which in turn promotes opening of the mitochondrial transition pore (MPTP) and depolarization of the mitochondrial membrane potential (ΔΨm). Here, we hypothesized that inhibition of MCU would preserve the ΔΨm. Thus, we measured ΔΨm in HAEC following stimulation with IL-13 for 72 hrs by two complementary methods. As expected, IL-13 treatment in control cells dissipated ΔΨm, whereas inhibition of MCU preserved it (Figure 2D–F).
DN-MCU expression protects against cytokine-induced cell death
Prolonged opening of MPTP, such as in the setting of excessive Mt-ROS, results in leakage of cytochrome c into the cytoplasm and subsequent activation of apoptotic pathways [14, 34]. Thus, we examined whether DN-MCU prevented epithelial cell apoptosis after IL-13 exposure. As compared to control-treated cells, expression of DN-MCU in HAEC prevented the marked decrease in cell number after exposure to IL-13 (Fig. 3A). Moreover, significantly fewer Annexin V-positive cells were seen with MCU inhibition. These findings support that the decrease in cell number after IL-13 exposure is at least in part through activation of apoptosis that can be prevented by DN-MCU (Fig. 3B).
Figure 3. Apoptotic cell death and mitochondrial cytochrome c release by IL-13 are reduced by MCU inhibition.
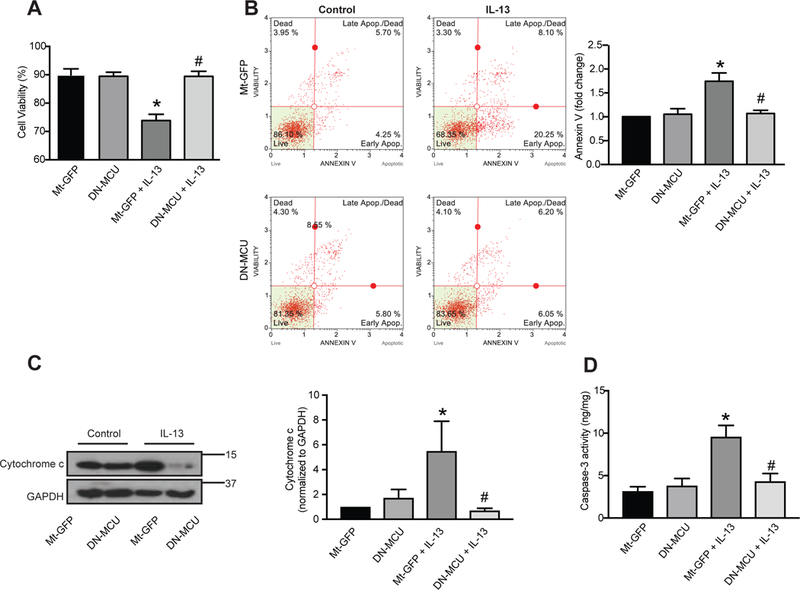
HAEC were infected with Mt-GFP or DN-MCU (10 MOI) for 48 hrs and exposed to IL-13 (10 ng/ml) for 72 hrs. (A) Cell viability by trypan blue staining (n = 5 independent experiments, measured as the ratio of viable cells to total cells). (B) Scatter plots and quantitation of apoptosis based on FITC-Annexin-V/7-AAD staining (n = 5 independent experiments). (C) Representative immunoblots and densitometric analysis for cytochrome C isolated from cytoplasmic fractions of cells (n = 6 independent experiments, results represented as percent of control). (D) Caspase-3 activity was measured via caspase-3 activity assay (n = 4 independent experiments, results are normalized to total protein concentration). All data are the means ± SEM. One-way ANOVA with Tukey post hoc test used. * p < 0.05 vs. control; # p < 0.05 vs. Mt-GFP/IL-13.
To corroborate that MCU inhibition blocks apoptosis in HAEC, the IL-13-mediated cytochrome c release was measured using Western blot analysis. After 72 hrs exposure to cytokine, IL-13 induced a significant increase of cytochrome c in cytoplasmic fractions of cell lysates (Fig. 3C). Compared to control, IL-13-treated cells in the presence of DN-MCU had significantly reduced levels of cytochrome c (Fig. 3C).
Finally, we measured caspase-3 activation since different upstream pathways converge on caspase-3 induction for final apoptotic execution. HAEC exposed to IL-13 demonstrated a significant increase in caspase-3 activity compared to untreated control cells. In contrast, no change in caspase-3 activity occurred in HAEC expressing DN-MCU after IL-13 exposure (Fig. 3D).
DN-MCU decreases IL-13-induced impairment of barrier function
Epithelial cell apoptosis in acute asthma can result in increased permeability of the epithelial layer [35]. To directly test the role of MCU in epithelial barrier function, we measured HAEC transepithelial electric resistance (TEER) (and its inverse, or conductance). In HAEC infected with control virus, incubation with IL-13 for 3 days significantly lowered TEER and increased conductance compared to no cytokine treatment. By contrast, conductance/TEER did not change in cells infected with the DN-MCU after exposure to IL-13 (Figure 4A, B). We also assessed epithelial barrier permeability in additional assays in which TRITC-labeled dextran was applied to the apical surface and recovered from the basolateral chamber. While IL-13 treatment strongly enhanced barrier permeability in monolayers of HAEC infected with control adenovirus, barrier function was significantly attenuated in DN-MCU-infected cells (Figure 4C).
Figure 4. MCU inhibition in HAEC decreases IL-13-mediated conductance and epithelial monolayer permeability.
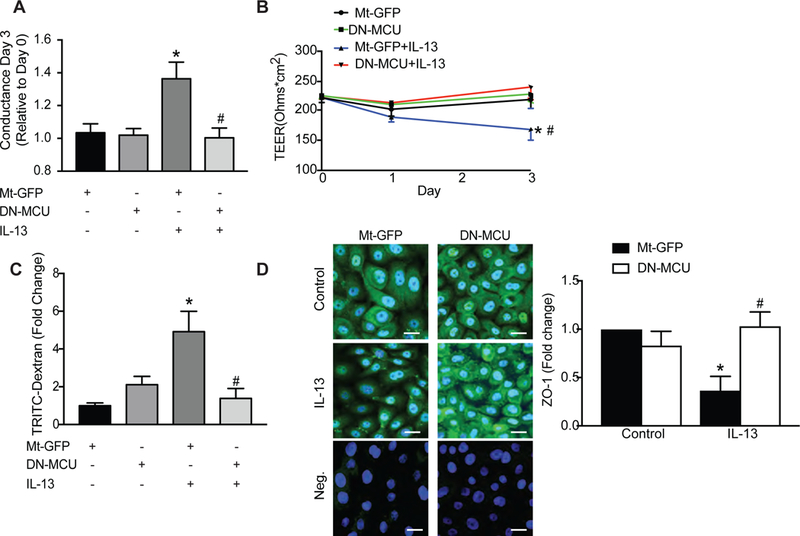
(A, B) HAEC in Transwell inserts were grown to confluence. Fresh medium was added every 2 days until cells excluded basolateral media. Cells were then infected with Mt-GFP or DN-MCU (10 MOI) for 48 hrs and TEER (ohm × cm2) was monitored after IL-13 treatment (72 hr, 10 ng/ml). Conductance was calculated based on TEER values. (C) Epithelial permeability was assessed by the amount of TRITC-dextran (1 mg/ml) detected in the media of the basolateral compartment of the Transwell and represented as fold-change over GFP-control cells. (D) Representative immunofluorescent images for ZO-1 (green) in HAEC expressing Mt-GFP or DN-MCU after 72 hr treatment with IL-13 or control (63x). Nuclei TO-PRO (blue), negative staining control without primary antibody for Mt-GFP or DN-MCU. Scale bar 100μm. All data are means ± SEM for 3 independent experiments (n = 3 samples/treatment group). Analysis used one-way ANOVA with Tukey post hoc test. * p < 0.05 vs. control; # p < 0.05 vs. Mt-GFP/IL-13.
To confirm that MCU inhibition preserves barrier function, we also investigated the expression of the tight junction protein zona occludens-1 (ZO-1), a well-established marker of junctional integrity [36]. Mt-GFP or DN-MCU infected cells had formed cell-cell contacts with a similar pattern of staining in all cells (Figure 4D). In contrast, after exposure of control cells to IL-13, ZO-1 staining was less intense (Figure 4D). These IL-13-mediated effects were absent when DN-MCU was overexpressed (Figure 4D). These data are in line with previous studies that patients with asthma have decreased ZO-1 expression in the pulmonary epithelium [31, 36]. Together, these findings support a role for MCU in airway epithelial cell barrier dysfunction.
Reduced IL-13-mediated barrier dysfunction and mitochondrial ROS production in MCU−/− primary airway epithelial cells
To further confirm our findings in HAEC, murine tracheal bronchial airway epithelial cells were isolated from WT or MCU−/− mice in CD1 background [37]. Assessment of barrier function by analysis of TEER and TRITC-dextran movement in primary MTBEC from MCU−/− mice had a significantly higher resistance compared to WT cells (Figure 5A). After treatment with IL-13 for 4 days, the movement of TRITC-dextran over the MTBEC monolayer was significantly greater in WT controls compared to in MCU−/− MTBEC (Figure 5B).
Figure 5. Reduced IL-13-mediated mitochondrial ROS generation and permeability in MCU knockout tracheal epithelial cells.
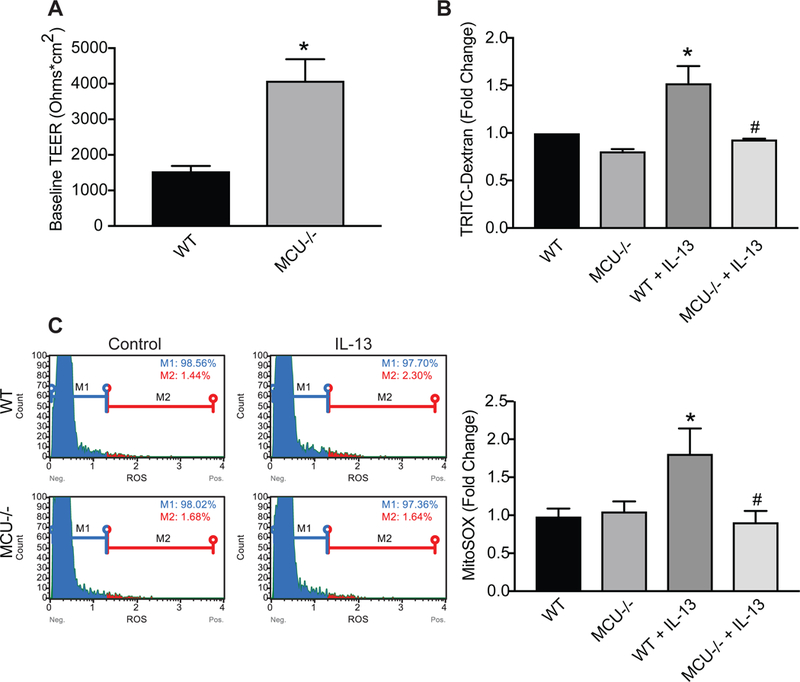
(A) WT or MCU−/− MTBEC in Transwell inserts were grown to confluence and TEER (ohm × cm2) was measured after cells excluded media from the apical side indicating monolayer establishment. (B) MTBEC were exposed to 10 ng/ml IL-13 for 4 days and epithelial permeability was assessed by the amount of TRITC-dextran (1 mg/ml) detected in the media of the basolateral compartment of the Transwell. Data are represented as fold-change over WT. (C) Quantitation of Mt-ROS with MitoSOX using flow cytometry. Representative plots are shown. All data are the means ± SEM for 3 independent experiments (n = 3 samples/treatment group). Student’s two-tailed t-test was used for (A), one-way ANOVA with Tukey post hoc test for (B, C). * p < 0.05 vs. control; # p < 0.05 vs. CD1/IL-13.
Mitochondrial superoxide production by mitoSox was increased in WT, but not in MCU−/− MTBEC exposed to IL-13 after 4 days (Figure 5C). These results are consistent with our findings with overexpression of the DN-MCU mutant and demonstrate that loss of expression or functional knockout of MCU activity reduces cytokine-mediated airway epithelial cell Mt-ROS and barrier dysfunction.
Decreased apoptosis and preserved ZO-1 expression in respiratory epithelium in MCU−/− mice after OVA challenge
To investigate the effects of MCU deficiency in a murine model of allergic asthma in vivo, we performed studies using MCU−/− or WT mice following sensitization and challenge with ovalbumin (OVA) [25]. Based on the in vitro findings of preserved barrier function under MCU inhibition, we tested whether MCU deletion in vivo protects from apoptosis in acute allergic asthma. As expected, TUNEL-positive cells were readily detected in large airway epithelium from WT mice as compared to saline controls (Figure 6A, B). By contrast, OVA-challenged MCU−/− mice had significantly fewer TUNEL-positive cells.
Figure 6. MCU knockout protects from respiratory epithelial cell death and barrier breakdown after OVA exposure in vivo.
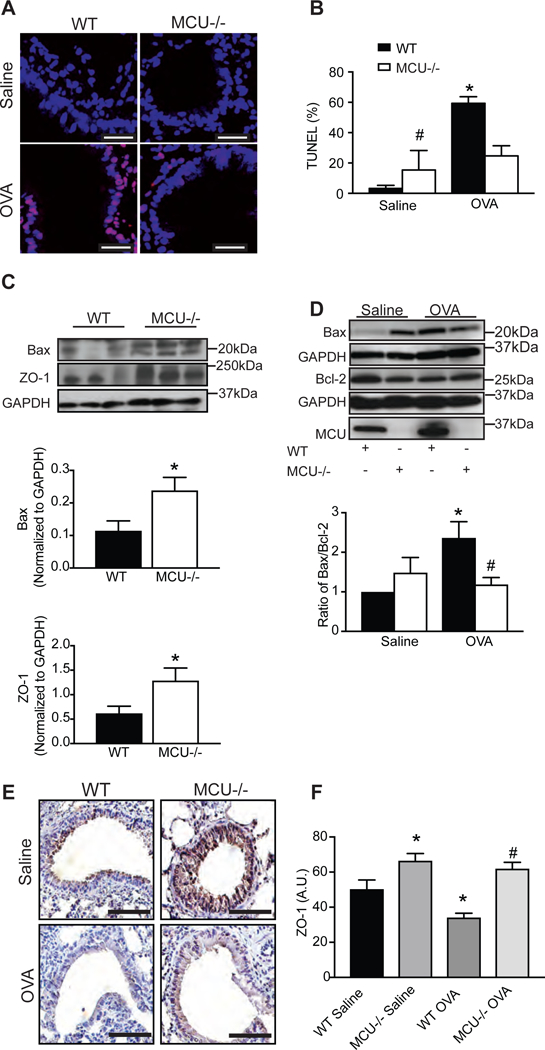
(A) Representative staining (TUNEL red; nuclei TO-PRO blue) and (B) quantification of apoptotic cells. Cells were counted in 4–5 visual fields per condition (63x magnification). Scale bar 100μm. (C) Representative immunoblot and quantification of Bax or ZO-1 in lung homogenates (n = 8 WT saline mice and 7 MCU−/− saline mice). (D) Representative immunoblot for Bax, Bcl-2 and MCU. Quantification of Bax/Bcl-2 ratio in WT and MCU KO mice (n = 8 WT saline mice, 6 WT OVA-challenged mice, 7 MCU−/− saline and 7 MCU−/−OVA-challenged mice). Data are represented as fold change over WT saline control. (E, F) Representative immunohistochemistry and scoring for ZO-1 in lung sections of OVA- or saline-challenged WT or MCU−/− mice. Scale bar 50μm. Student’s two-tailed t-test was used for (C); one-way ANOVA with Tukey post hoc test used for all other measurements. * p < 0.05 vs. saline; # p < 0.05 vs. WT mice treated with OVA.
MCU deletion selectively in cardiac myocytes prevented cell death and alleviated ischemia-reperfusion injury [38]. In a previous study utilizing cardiac myocyte-specific DN-MCU mice, mRNA expression of Bcl-2 and protein levels of Bax were increased [27]. Similar to these findings, compared to WT saline controls, MCU−/− mice had increased Bax protein expression (Figure 6C). Further analysis of the expression of the two proapoptotic and antiapoptotic proteins, Bax and Bcl-2, respectively, showed an increase in the ratio of Bax/Bcl-2 in the lungs of WT mice exposed to OVA compared to controls (Figure 6D). However, analysis of the ratio of Bax/Bcl-2 showed no difference in expression between MCU−/− controls compared to saline-treated WT mice (Figure 6D). In addition, MCU−/− mice treated with OVA did not show increased expression compared to saline controls or OVA-treated WT mice (Figure 6D). Moreover, Bcl-2 protein levels were decreased in WT, but not in MCU−/− mice after OVA treatment, probably as a result of altered Bax expression. These data support the hypothesis that MCU is involved in allergen-mediated apoptosis of airway epithelium.
To test a marker of barrier function integrity, we examined ZO-1 protein levels in lung homogenates from MCU−/− and WT mice. In lung lysates from MCU−/− mice, ZO-1 expression was increased compared to WT mice (Figure 6C). Further analysis of ZO-1 protein by immunostaining demonstrated increased ZO-1 expression in MCU−/− mice as compared to WT controls (Figure 6E, F). OVA-treated WT mice had significantly reduced ZO-1 staining within the airway epithelia. In contrast, large airway epithelial cell ZO-1 from MCU−/− did not significantly change after OVA treatment (Figure 6E, F). These data are consistent with our in vitro data and strongly suggest that MCU has a key role in barrier integrity in respiratory epithelium in allergic asthma in vivo.
Discussion
The objective of this study was to define the role of MCU in the relationship between mitochondrial Ca2+ uptake and ROS production in airway epithelium and its implication in Th2-type cytokine- and allergen-mediated responses. Here we report that in the presence of allergic stimuli, MCU modulates Mt-ROS generation, epithelial barrier integrity and apoptosis.
MCU, the channel responsible for mitochondrial matrix Ca2+ uptake, has been recently identified and is now being extensively studied through the use of genetic and pharmacologic agents to inhibit MCU activity [20, 39]. Somewhat surprisingly, mice with global MCU knockout are viable, with no obvious baseline abnormalities [37]. However, the most striking phenotypes associated with MCU deficiency were observed in ROS-dependent models [32, 40, 41]. While asthma is also a ROS-driven disease, little work has been performed to understand the role of MCU in the lung [42, 43] Thus, our data add to the existing literature and position MCU as a regulator of mitochondrial dysfunction in asthma.
Mitochondrial Ca2+ overload is thought to dissipate the inner ΔΨm via MPTP opening and enhance formation of ROS [44]. Dissipation of ΔΨm is associated with release of cytochrome c and induction of apoptosis [11]. Indeed, apoptotic activity, indicated by increased cytochrome c release, occurs in OVA-induced experimental allergic asthma [45]. However, conclusive evidence for this proposed mechanism, and in particular the precise role of MCU, is lacking. Our data are in agreement with previous reports in other cell or tissue types demonstrating a role for MCU in cell death [18, 32, 38, 46–48]. For example, knockdown of MCU in HeLa cells and primary cerebellar granule neurons decreases mitochondrial Ca2+ uptake following histamine stimulation and attenuates cell death induced by oxidative stress [32]. In mice, deletion of MCU protects against cell death in an in vivo ischemia-reperfusion (IR) injury model by preventing the activation of the mitochondrial permeability transition pore (MPTP) [38, 48]. Finally, knockdown of the TRV1 Ca2+ respiratory epithelium abolishes mitochondrial Ca2+ overload and decreases apoptosis in response to IL-13 [47], further suggestive of a link between mitochondrial Ca2+ and apoptosis specifically in airway epithelial cells. Here, we demonstrate that in addition to histamine [48], IL-13 promotes mitochondrial matrix Ca2+ uptake in respiratory epithelial cells, likely as a result of IP3-dependent ER-Ca2+ release [49]. Indeed, IL-13 and other allergens induce sustained Ca2+ elevations in monocytes and alveolar epithelial cells, followed by activation of Ca2+ release-activated Ca2+ (CRAC) channels [49–51].
In addition to the proposed apoptosis induction via Ca2+ influx, Mt-ROS production and release of cytochrome C release, our data also imply that the loss of MCU has additional effects on apoptosis by regulating Bax protein levels. Of note, a previous study using conditionally expressing DN-MCU in hearts of adult mice showed increased Bax protein expression [27]. While predominantly cytosolic at baseline, upon activation Bax becomes a mitochondrial outer membrane component of the mitochondrial permeability pore. Thus, Bax induces mitochondrial outer membrane permeabilization, leading to the release of proteins normally found in the space between the inner and outer mitochondrial membranes and cell death [52–54]. Of note, Bax expression is regulated by cytosolic Ca2+ as was evidenced by a study showing enhanced mRNA levels of Bax in pancreatic acinar cells after increased intraceullar Ca2+ [55]. Since apoptosis was inhibited with ablation of MCU in our study, we speculate that Bax activation is inhibited by MCU deficiency. Although the mechanism is unclear, we speculate that similar to other published evidence [29, 50], exposure of mice to OVA increases ER-localized Ca2+ release which is buffered by MCU in WT mice. However, the reduction in mitochondrial Ca2+ uptake in MCU−/− mice may lead to increased intracellular Ca2+ concentrations that alter localization, expression and activation of Bax [52].
The bronchial epithelium provides a physical barrier to protect the lung parenchyma from external stimuli that induce the production of inflammatory cytokines. Consistent with this mechanism, human asthmatic patients have strong evidence of epithelial desquamation [56], which likely leads to airway remodeling and a more chronic and persistent inflammatory phenotype [4]. Primary airway epithelial cells obtained from patients with asthma have diminished epithelial barrier function and decreased expression of ZO-1 [35, 57], an intracellular membrane protein which clusters and stabilizes adhesive components of the apical junction complex. In our studies, we found that MCU deficiency preserves ZO-1 expression following allergen challenge. We also detected increased protein levels of ZO-1 in MCU−/− mice as compared to littermates. There is some evidence for ROS-mediated changes in ZO-1 expression [58], which requires further investigation in the context of allergic asthma.
Conclusions
These data implicate that mitochondrial Ca2+ uptake through MCU activity in the presence of allergen is a mechanism by which epithelial cell integrity and survival is compromised. Mitochondrial matrix Ca2+ uptake induces Mt-ROS production and opening of MPTP. As a result, the mitochondrial membrane potential dissipates and cytochrome C is released and activates apoptosis. In turn, respiratory epithelial cell death results in loss of epithelial barrier function. Thus, maintaining epithelial layer integrity via MCU inhibition may be a novel approach for the treatment of allergic asthma. Given that the respiratory epithelium can be easily targeted with inhaled agents, such a formulation would have a direct effect on the impacted cell type with fewer undesired effects on other tissues. For example, proof-of-concept studies from our group established the utility of nanoparticle-based treatments, delivered by inhalation, as a novel therapeutic modality for allergic asthma [59].
Highlights.
IL-13 promotes mitochondrial matrix Ca2+ uptake, ROS production and apoptosis in respiratory epithelium
MCU inhibition blocks mitochondrial ROS production and membrane depolarization
MCU inhibition prevents apoptosis and epithelial barrier dysfunction
Inhibition of MCU reduces allergen-mediated epithelial cell death in vivo
Acknowledgements
This work was supported by a University of Iowa Carver College of Medicine Innovative Science grant (to IMG) and the Iowa Center for Research by Undergraduates (to CJW). We are indebted to Dr. Steven Pennington and Kayla Ladd for technical assistance and Dr. Kristina W. Thiel for assistance in the preparation of the manuscript.
Footnotes
Conflict of interest
All authors declare that no conflict of interest exists.
References
- [1].Ganesan S, Comstock AT, Sajjan US, Barrier function of airway tract epithelium, Tissue Barriers 1 (2013) e24997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Martin LD, Krunkosky TM, Voynow JA, Adler KB, The role of reactive oxygen and nitrogen species in airway epithelial gene expression, Environ Health Perspect 106 Suppl 5 (1998) 1197–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rezaee F, Georas SN, Breaking barriers. New insights into airway epithelial barrier function in health and disease, Am J Respir Cell Mol Biol 50 (2014) 857–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Holgate ST, Epithelium dysfunction in asthma, J Allergy Clin Immunol 120 (2007) 1233–1244; quiz 1245–1236. [DOI] [PubMed] [Google Scholar]
- [5].Kim SR, Kim DI, Kim SH, Lee H, Lee KS, Cho SH, Lee YC, NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation, Cell Death Dis 5 (2014) e1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rao R, Oxidative stress-induced disruption of epithelial and endothelial tight junctions, Front Biosci 13 (2008) 7210–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Adam-Vizi V, Starkov AA, Calcium and mitochondrial reactive oxygen species generation: how to read the facts, J Alzheimers Dis 20 Suppl 2 (2010) S413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ahmad T, Aggarwal K, Pattnaik B, Mukherjee S, Sethi T, Tiwari BK, Kumar M, Micheal A, Mabalirajan U, Ghosh B, Sinha Roy S, Agrawal A, Computational classification of mitochondrial shapes reflects stress and redox state, Cell Death Dis 4 (2013) e461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Reddy PH, Mitochondrial Dysfunction and Oxidative Stress in Asthma: Implications for Mitochondria-Targeted Antioxidant Therapeutics, Pharmaceuticals (Basel) 4 (2011) 429–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rizzuto R, De Stefani D, Raffaello A, Mammucari C, Mitochondria as sensors and regulators of calcium signalling, Nat Rev Mol Cell Biol 13 (2012) 566–578. [DOI] [PubMed] [Google Scholar]
- [11].Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS, Calcium, ATP, and ROS: a mitochondrial love-hate triangle, Am J Physiol Cell Physiol 287 (2004) C817–833. [DOI] [PubMed] [Google Scholar]
- [12].Gogvadze V, Orrenius S, Zhivotovsky B, Multiple pathways of cytochrome c release from mitochondria in apoptosis, Biochim Biophys Acta 1757 (2006) 639–647. [DOI] [PubMed] [Google Scholar]
- [13].Patron M, Raffaello A, Granatiero V, Tosatto A, Merli G, De Stefani D, Wright L, Pallafacchina G, Terrin A, Mammucari C, Rizzuto R, The mitochondrial calcium uniporter (MCU): molecular identity and physiological roles, J Biol Chem 288 (2013) 10750–10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cai J, Yang J, Jones DP, Mitochondrial control of apoptosis: the role of cytochrome c, Biochim Biophys Acta 1366 (1998) 139–149. [DOI] [PubMed] [Google Scholar]
- [15].Mabalirajan U, Ghosh B, Mitochondrial dysfunction in metabolic syndrome and asthma, J Allergy (Cairo) 2013 (2013) 340476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gutterman DD, Mitochondria and reactive oxygen species: an evolution in function, Circ Res 97 (2005) 302–304. [DOI] [PubMed] [Google Scholar]
- [17].La Rovere RM, Roest G, Bultynck G, Parys JB, Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy, Cell Calcium 60 (2016) 74–87. [DOI] [PubMed] [Google Scholar]
- [18].Orrenius S, Zhivotovsky B, Nicotera P, Regulation of cell death: the calcium-apoptosis link, Nat Rev Mol Cell Biol 4 (2003) 552–565. [DOI] [PubMed] [Google Scholar]
- [19].Kirichok Y, Krapivinsky G, Clapham DE, The mitochondrial calcium uniporter is a highly selective ion channel, Nature 427 (2004) 360–364. [DOI] [PubMed] [Google Scholar]
- [20].De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R, A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter, Nature 476 (2011) 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Garcia-Rivas Gde J, Carvajal K, Correa F, Zazueta C, Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo, Br J Pharmacol 149 (2006) 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Karp PH, Moninger TO, Weber SP, Nesselhauf TS, Launspach JL, Zabner J, Welsh MJ, An in vitro model of differentiated human airway epithelia. Methods for establishing primary cultures, Methods Mol Biol 188 (2002) 115–137. [DOI] [PubMed] [Google Scholar]
- [23].You Y, Richer EJ, Huang T, Brody SL, Growth and differentiation of mouse tracheal epithelial cells: selection of a proliferative population, Am J Physiol Lung Cell Mol Physiol 283 (2002) L1315–1321. [DOI] [PubMed] [Google Scholar]
- [24].Jaffer OA, Carter AB, Sanders PN, Dibbern ME, Winters CJ, Murthy S, Ryan AJ, Rokita AG, Prasad AM, Zabner J, Kline JN, Grumbach IM, Anderson ME, Mitochondrial-targeted antioxidant therapy decreases transforming growth factor-beta-mediated collagen production in a murine asthma model, Am. J. Respir. Cell Mol. Biol 52 (2015) 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sanders PN, Koval OM, Jaffer OA, Prasad AM, Businga TR, Scott JA, Hayden PJ, Luczak ED, Dickey DD, Allamargot C, Olivier AK, Meyerholz DK, Robison AJ, Winder DG, Blackwell TS, Dworski R, Sammut D, Wagner BA, Buettner GR, Pope RM, Miller FJ Jr., Dibbern ME, Haitchi HM, Mohler PJ, Howarth PH, Zabner J, Kline JN, Grumbach IM, Anderson ME, CaMKII is essential for the proasthmatic effects of oxidation, Sci Transl Med 5 (2013) 195ra197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen YC, Statt S, Wu R, Chang HT, Liao JW, Wang CN, Shyu WC, Lee CC, High mobility group box 1-induced epithelial mesenchymal transition in human airway epithelial cells, Sci Rep 6 (2016) 18815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, Wagner BA, Soto J, McCormick ML, Kutschke W, Weiss RM, Yu L, Boudreau RL, Abel ED, Zhan F, Spitz DR, Buettner GR, Song LS, Zingman LV, Anderson ME, Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart, Proc Natl Acad Sci U S A 112 (2015) 9129–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ying X, Minamiya Y, Fu C, Bhattacharya J, Ca2+ waves in lung capillary endothelium, Circ Res 79 (1996) 898–908. [DOI] [PubMed] [Google Scholar]
- [29].Delmotte P, Yang B, Thompson MA, Pabelick CM, Prakash YS, Sieck GC, Inflammation alters regional mitochondrial Ca(2)+ in human airway smooth muscle cells, Am J Physiol Cell Physiol 303 (2012) C244–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Heller F, Fromm A, Gitter AH, Mankertz J, Schulzke JD, Epithelial apoptosis is a prominent feature of the epithelial barrier disturbance in intestinal inflammation: effect of pro-inflammatory interleukin-13 on epithelial cell function, Mucosal Immunol 1 Suppl 1 (2008) S58–61. [DOI] [PubMed] [Google Scholar]
- [31].Ahdieh M, Vandenbos T, Youakim A, Lung epithelial barrier function and wound healing are decreased by IL-4 and IL-13 and enhanced by IFN-gamma, Am J Physiol Cell Physiol 281 (2001) C2029–2038. [DOI] [PubMed] [Google Scholar]
- [32].Liao Y, Hao Y, Chen H, He Q, Yuan Z, Cheng J, Mitochondrial calcium uniporter protein MCU is involved in oxidative stress-induced cell death, Protein Cell 6 (2015) 434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sebag SC, Koval OM, Paschke JD, Winters CJ, Jaffer OA, Dworski R, Sutterwala FS, Anderson ME, Grumbach IM, Mitochondrial CaMKII inhibition in airway epithelium protects against allergic asthma, JCI Insight 2 (2017) e88297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Galindo MF, Jordan J, Gonzalez-Garcia C, Cena V, Chromaffin cell death induced by 6-hydroxydopamine is independent of mitochondrial swelling and caspase activation, J Neurochem 84 (2003) 1066–1073. [DOI] [PubMed] [Google Scholar]
- [35].de Boer WI, Sharma HS, Baelemans SM, Hoogsteden HC, Lambrecht BN, Braunstahl GJ, Altered expression of epithelial junctional proteins in atopic asthma: possible role in inflammation, Can J Physiol Pharmacol 86 (2008) 105–112. [DOI] [PubMed] [Google Scholar]
- [36].Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, Randi AM, Schwartz MA, Matter K, Balda MS, ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation, J Cell Biol 208 (2015) 821–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T, The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter, Nat Cell Biol 15 (2013) 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD, The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart, Cell Rep 12 (2015) 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK, Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter, Nature 476 (2011) 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, Bao W, Pavlidis S, Barnes PJ, Kanerva J, Bittner A, Rao N, Murphy MP, Kirkham PA, Chung KF, Adcock IM, Copdmap, Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease, J Allergy Clin Immunol 136 (2015) 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].De Stefani D, Rizzuto R, Pozzan T, Enjoy the Trip: Calcium in Mitochondria Back and Forth, Annu Rev Biochem 85 (2016) 161–192. [DOI] [PubMed] [Google Scholar]
- [42].Gu L, Larson-Casey JL, Carter AB, Macrophages utilize the mitochondrial calcium uniporter for profibrotic polarization, FASEB J 31 (2017) 3072–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rimessi A, Bezzerri V, Patergnani S, Marchi S, Cabrini G, Pinton P, Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis, Nat Commun 6 (2015) 6201. [DOI] [PubMed] [Google Scholar]
- [44].Peng TI, Jou MJ, Oxidative stress caused by mitochondrial calcium overload, Ann N Y Acad Sci 1201 (2010) 183–188. [DOI] [PubMed] [Google Scholar]
- [45].Mabalirajan U, Dinda AK, Kumar S, Roshan R, Gupta P, Sharma SK, Ghosh B, Mitochondrial structural changes and dysfunction are associated with experimental allergic asthma, J. Immunol 181 (2008) 3540–3548. [DOI] [PubMed] [Google Scholar]
- [46].Pan L, Huang BJ, Ma XE, Wang SY, Feng J, Lv F, Liu Y, Liu Y, Li CM, Liang DD, Li J, Xu L, Chen YH, MiR-25 protects cardiomyocytes against oxidative damage by targeting the mitochondrial calcium uniporter, Int J Mol Sci 16 (2015) 5420–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rehman R, Bhat YA, Panda L, Mabalirajan U, TRPV1 inhibition attenuates IL-13 mediated asthma features in mice by reducing airway epithelial injury, Int Immunopharmacol 15 (2013) 597–605. [DOI] [PubMed] [Google Scholar]
- [48].Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M, Elrod JW, The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition, Cell Rep 12 (2015) 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sozzani P, Cambon C, Vita N, Seguelas MH, Caput D, Ferrara P, Pipy B, Interleukin-13 inhibits protein kinase C-triggered respiratory burst in human monocytes. Role of calcium and cyclic AMP, J Biol Chem 270 (1995) 5084–5088. [DOI] [PubMed] [Google Scholar]
- [50].Jairaman A, Maguire CH, Schleimer RP, Prakriya M, Allergens stimulate store-operated calcium entry and cytokine production in airway epithelial cells, Sci Rep 6 (2016) 32311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Matsuwaki Y, Wada K, White T, Moriyama H, Kita H, Alternaria fungus induces the production of GM-CSF, interleukin-6 and interleukin-8 and calcium signaling in human airway epithelium through protease-activated receptor 2, Int Arch Allergy Immunol 158 Suppl 1 (2012) 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ, BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis, Science 300 (2003) 135–139. [DOI] [PubMed] [Google Scholar]
- [53].Lalier L, Cartron PF, Juin P, Nedelkina S, Manon S, Bechinger B, Vallette FM, Bax activation and mitochondrial insertion during apoptosis, Apoptosis 12 (2007) 887–896. [DOI] [PubMed] [Google Scholar]
- [54].Green DR, Kroemer G, The pathophysiology of mitochondrial cell death, Science 305 (2004) 626–629. [DOI] [PubMed] [Google Scholar]
- [55].Yu JH, Kim H, Kim KH, Calcium-dependent apoptotic gene expression in cerulein-treated AR42J cells, Ann N Y Acad Sci 1010 (2003) 66–69. [DOI] [PubMed] [Google Scholar]
- [56].Truong-Tran AQ, Carter J, Ruffin R, Zalewski PD, New insights into the role of zinc in the respiratory epithelium, Immunol Cell Biol 79 (2001) 170–177. [DOI] [PubMed] [Google Scholar]
- [57].Wawrzyniak P, Wawrzyniak M, Wanke K, Sokolowska M, Bendelja K, Ruckert B, Globinska A, Jakiela B, Kast JI, Idzko M, Akdis M, Sanak M, Akdis CA, Regulation of bronchial epithelial barrier integrity by type 2 cytokines and histone deacetylases in asthmatic patients, J Allergy Clin Immunol 139 (2017) 93–103. [DOI] [PubMed] [Google Scholar]
- [58].Fukui A, Naito Y, Handa O, Kugai M, Tsuji T, Yoriki H, Qin Y, Adachi S, Higashimura Y, Mizushima K, Kamada K, Katada K, Uchiyama K, Ishikawa T, Takagi T, Yagi N, Kokura S, Yoshikawa T, Acetyl salicylic acid induces damage to intestinal epithelial cells by oxidation-related modifications of ZO-1, Am J Physiol Gastrointest Liver Physiol 303 (2012) G927–936. [DOI] [PubMed] [Google Scholar]
- [59].Morris AS, Sebag SC, Paschke JD, Wongrakpanich A, Ebeid K, Anderson ME, Grumbach IM, Salem AK, Cationic CaMKII Inhibiting Nanoparticles Prevent Allergic Asthma, Mol Pharm 14 (2017) 2166–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]