

Abstract
Humans are highly visual. Retinal ganglion cells (RGCs), the neurons that connect the eyes to the brain, fail to regenerate after damage, eventually leading to blindness. Here, we review research on regeneration and repair of the optic system. Intrinsic developmental growth programs can be reactivated in RGCs, neural activity can enhance RGC regeneration, and functional reformation of eye-to-brain connections is possible, even in the adult brain. Transplantation and gene therapy may serve to replace or resurrect dead or injured retinal neurons. Retinal prosthetics that can restore vision in animal models may too have practical power in the clinical setting. Functional restoration of sight in certain forms of blindness is likely to occur in human patients in the near future.
Sight is crucial for humans to navigate the world. Under normal, healthy conditions, our eyes and brain create sight so automatically that only when our visual pathways are damaged do we fully appreciate the extent to which eyesight defines our experience. For the many who suffer visual impairments, it is urgent that we discover strategies to regenerate retinal neurons and convert those strategies into clinically viable therapeutics.
Vision begins in the retina, the thin trilayered neural tissue at the back of the eye (Fig. 1); there, photoreceptors transform light information into electrical signals that the rest of the visual system can understand. The retinal interneurons—the horizontal, bipolar, and amacrine cells—then pass that information to the retinal ganglion cells (RGCs), the output neurons of the eye. There are ~30 different types of RGCs, each firing action potentials depending on the quality and location of visual stimuli in the environment (1). Those action potentials propagate down the optic nerves and into the brain, where they are translated into perceptions and light-mediated behaviors.
Fig. 1. Visual information is transmitted from the eye to the visual centers in the brain via the optic nerve.

(Top) Light reaching the retina is converted into electrical potentials that eventually cause action potentials in the ganglion cells (RGCs). (Middle) The myelinated optic nerve transmits action potentials (Bottom left) to the visual processing centers in the brain. (Bottom right) After damage, RGC axons degenerate. In the absence of therapeutic interventions, blindness ensues.
The importance of retinal ganglion cells
RGCs are a bottleneck for vision. Even when the rest of the visual system is healthy, if RGCs are dead or dysfunctional, vision is impossible. RGCs are protected by the sclera, the thick, durable tissue that encompasses the back of the eye. However, the path that RGC axons take to reach the brain renders them vulnerable to damage in response to impacts to the head or eye. Glaucoma, with its attendant elevated eye pressure, is the most common cause of irreversible blindness (2). Research on visual repair has therefore focused on sustaining RGCs after injury, encouraging axon regrowth down the optic nerve, and reestablishing their correct synaptic relationships. Unfortunately, mammalian RGC axons do not regenerate after damage and damaged RGCs eventually die, never to be replaced. So prevalent is this cause for blindness that the U.S. National Eye Institute has set forth as their “Audacious Goal” to discover means to repair RGC connections with the brain (https://nei.nih.gov/audacious).
The major questions that drive research on visual restoration and RGC repair are simple but challenging: What strategies support RGC axon regeneration after damage? Can regenerating RGC axons form functional synapses with their targets in the brain?
Extrinsic factors unique to the CNS limit optic nerve regeneration
Various environmental influences limit RGC regeneration. Although neurons with cell bodies in the peripheral nervous system (PNS) avidly regenerate, neurons such as RGCs, whose cell bodies reside in the central nervous system (CNS), fail to reextend after injury (3, 4). Even if the rodent optic nerve is completely transected, a peripheral nerve graft allows RGC axons to regenerate and form synapses with their targets in the brain (5, 6). Thus, the damage environment constrains the regeneration of mature rodent RGCs. Unfortunately for humans, the PNS nerve graft approach holds limited therapeutic potential because it involves massive neuro-surgeries. Nevertheless, these studies underscore the principle that RGCs can regenerate if given the appropriate milieu.
Inhibitory effects of myelin proteins
Normally, myelin insulates axons, increasing conduction velocity of electrical signals (Fig. 1). In the PNS, where regeneration is inherent to the system, Schwann cells provide myelination. In the CNS, Oligodendrocytes are the myelinating glial cells and have an inhibitory effect on axon regeneration. Oligodendrocytes present a variety of proteins inhibitory to axon re-growth, including myelin-associated glycoprotein, the neurite-outgrowth inhibitor “Nogo,” oligodendrocyte-myelin glycoprotein, and semaphorins (7, 8). Neutralization of these proteins has been shown to enhance RGC axon regeneration in vitro (7). However, experiments assessing the consequences of removing these proteins in vivo reveal little or no regeneration (9), challenging whether these proteins actually constitute major brakes on regeneration. Neutralizing Nogo can enhance regeneration if RGCs are shifted into a growth state (10), but overall, the effects of reducing myelin-associated proteins on RGC regeneration are subtle. Thus, attention has expanded to consider other extrinsic influences that might underlie RGC regenerative failure and that might constitute targets for enhancing regeneration in the clinic.
Reactive scarring and inflammation
As with any injury, damage to the optic pathway recruits cellular and molecular processes to buffer the injury response, some of which affect the regenerative potential of RGCs (11). Astrocytes—the glial cells that support synapse development, transmission, and plasticity (12)—create physical and molecular barriers after injury that can prevent RGC axons from regrowing. Some of these include chondroitin sulfate proteoglycans (CSPGs) and tenasins (11, 13). Lesions of the rodent spinal cord induced in a context of minimal astrocyte reactivity allow for robust axon regeneration, even through myelin (14), underscoring the extent to which astroglial scarring might limit RGC regeneration. Other work shows, however, that glial scars can actually promote regeneration in the rodent spinal cord (15). The field of visual repair awaits studies that evaluate the role of scar-related factors in the optic nerve in vivo. This needs to be addressed directly in the visual system because the wiring architecture of the eye-to-brain pathway differs vastly from that of the spinal column. Spinal lesions typically injure axons at locations that allow for collateral (side-branch) sprouting around the lesion, something not possible for RGC lesions located near the optic nerve head (Fig. 1).
Lesions can cause local production of inflammation-related cytokines such as interleukin-6, leukemia inhibitory factor, and ciliary neurotrophic factor (CNTF) from glia and other non-neural cells (16, 17). In the adult, CNTF up-regulates a transcriptional pathway involving suppression of cytokine signaling factor 3 (SOCS3) in RGCs, thus limiting axon regeneration (18). In the absence of SOCS3, CNTF can, however, enhance regeneration by activating gp130-dependent kinase signaling (19). Thus, the pathways that affect RGC regeneration depend on the signaling context, which imposes complexity on potential therapeutic strategies.
Zinc released from amacrine interneurons after injury is internalized by RGCs and limits their regeneration (20). Other extrinsic factors, however, can promote regeneration: Lens injury causes macrophages to release oncomodulin, which supports RGC axon extension via a Ca++/calmodulin pathway (21). Lesion-reactive cells and proteins in both the eye and in the optic nerve can either help or hurt regeneration. The key is to discover when and why.
Intrinsic factors that limit RGC regeneration
RGC axons down-regulate their axon growth speed more than 1000-fold as they transition from embryonic to postnatal ages, likely because of molecular programs intrinsic to RGCs (22). As RGCs mature, they down-regulate expression of phosphorylated mammalian target of rapamycin (phosphor-mTOR), a growth-promoting molecule. Deletion of an mTOR inhibitor, phosphatase and tensin homolog (PTEN) in RGCs, greatly enhances their axonal regeneration capacity after injury (23). Some regenerated axons even extend from the lesion site located just behind the eye all the way to the optic chiasm, a distance of many millimeters. This degree of regeneration represents a triumph for the field, and when combined with knockdown of SOCS3, mTOR enhancement causes even more RGCs to regenerate (24).
Combining mTOR activation and SOCS3 inhibition is promising, but caveats pertain when considering their clinical applications. First, the increase in phosphor-mTOR has to be in place before axon injury in order for regeneration to occur (25). Second, mTOR broadly affects cell growth (26) and thus may cause retinal tumor formation (27). Any therapeutic approach that relies on enhancing mTOR signaling thus would have to include safeguards. Third, mTOR enhancement alone (or mTOR plus SOCS3 deletion) triggers regeneration of RGC axons only as far as the chiasm (23–25). This suggests that there are inhibitory cues at the optic chiasm and that more potent stimulators of RGC regeneration may be needed to inspire RGC axon growth into the brain. In some mice, axons regenerate to the optic chiasm but then turn away from the brain and grow into the other optic nerve, toward the contralateral eye (28). Thus, not all regeneration is productive. Other manipulations such as knockdown of the growth-inhibiting transcriptional repressor KLF4 can also encourage RGCs regeneration (29), but again, not the full distance back into the brain.
Reconnection to targets in the brain
A few studies show regeneration of RGC axons beyond the optic chiasm into the brain. Enhancement of mTOR combined with augmentation of adenosine 3′,5′-monophosphate (cAMP) and injections of oncomodulin promoted long-range regeneration of RGC axons (30) to brain structures, including the dorsal lateral geniculate nucleus (dLGN), which relays visual information to the cortex. Mice that received the combined treatment of PTEN-knockdown/cAMP-increase/oncomodulin also recovered some visual function, although the connections eventually regressed.
Therefore, strategies must support both RGC regeneration and long-term survival of circuits. Some of the same factors that can promote regeneration also can promote degeneration of RGCs after injury. Removal of dual leucine zipper kinase (DLK) from RGCs enhances their survival after optic nerve injury by altering apoptotic pathways but also limits PTEN-knockdown–based enhancement of axon regeneration (31).
Neural activity facilitates RGC axon regeneration
During development, spontaneous and visually driven electrical activity refine RGC connections (32) and enhance RGC axon outgrowth by increasing responsiveness to trophic factors (22). In the adult animal, increasing RGC firing permits their axons to regenerate through optic nerve lesions (Fig. 2) (25, 33) and boosts the impact of molecular stimulants of axon growth such as mTOR (25). Conversely, reducing activity levels of RGCs inhibits their survival after damage (25). Increasing RGC firing alone was not sufficient to support RGC axon regeneration into the brain unless phosphor–mTOR was increased (25).
Fig. 2. Electrical activity can promote RGC axon regeneration.

Increasing the activity of RGCs after optic nerve damage can facilitate the repair of degenerating axons in the optic nerve (25, 33), with many extending past the site of damage into the brain and partially restoring sight in animal models (25).
RGC axons that regenerate back to their targets apparently fail to undergo myelination and therefore suffer slower conduction of electrical potentials (34). RGC firing induces myelinating oligodendrocytes during development (35) but apparently not during adulthood. This informs us that only some of the mechanisms that initially establish visual circuitry are available for reactivation in adulthood; others may need to be replaced.
Target and cell-type specificity of regenerated connections
Can regenerating RGC axons rewire appropriately in the brain? One idea is that adult mammalian CNS circuits avoid regeneration because it is better to have no regeneration than incorrect regeneration. Evidence, however, indicates that regenerated RGCs form correct connections and avoid targets incorrect for their function (25). Similarly, in the spinal cord the central branch of dorsal root neurons (DRGs) regenerates in a topographic- and laminar-specific manner, unless myelin-associated factors such as Nogo are perturbed (36). Axon-guidance molecules that serve in development to direct RGC axons to their targets may thus get up-regulated after injury, a feature known to occur in cold-blooded vertebrates that naturally add new RGCs across their life span (37).
Thresholds for reversing blindness
Regeneration of axons is only part of the story; restoration of functional visual capacity is required. The mere presence of regenerated RGC axons at a target in the brain does not predict visual function (25, 34). For example, combining RGC activity with mTOR enhancement led to the recovery of the mice’s visual reflex to avoid overhead looming stimuli (25), but even when RGC axons regenerated to the dLGN, those connections failed to lead to enhanced depth perception—a well-established property of the retino-dLGN-cortical pathway (25). The fact that some visual functions are restored whereas others are not, even in the presence of structural regeneration, may reflect the lack of axon myelination or perhaps failure of correct proteins to reaccumulate at retino-dLGN synapses (38). The good news is that with modern approaches such as single-cell RNA-sequencing that enable the genetic makeup of specific sets of neurons to be evaluated, one can now compare the molecular attributes of normal and regenerated CNS neurons and synapses.
Transplantation of RGCs
Therapeutic intervention with regeneration-inducing stimuli should be successful only during a limited period after injury, when RGCs are still alive. Although the mammalian retina may harbor a stem cell population in the ciliary margin (39), there is no evidence they replace RGCs damaged by injury or disease. The lack of endogenous RGC replacement in mammals is in stark contrast to the scenario in fish and amphibians, which add new RGCs throughout their life span, a feature thought to arise at least in part from the presence of a specific proneural transcriptional factor, Ascl1, made by retinal Müller glia in cold blooded vertebrates but not by mammalian Müller glia (40).
Three general approaches to replace RGCs include (i) syngeneic transplantation of adult induced pluripotent stem cells (iPSs) that have been programmed to assume RGC-phenotypes, (ii) allogeneic transplantation of RGCs from healthy eyes into host eyes, and (iii) possible reprogramming of endogenous Müller glia into RGCs (40). iPS cells with RGC-like characteristics have been created in vitro (41) but not yet used to rebuild functional eye-to-brain circuitry.
Allogeneic transplantation of RGCs led to substantial integration of RGCs into existing retinal circuitry in rats (42). The transplanted RGCs adopted on-type or off-type or on-off-type photic responses to light signals and extended axon projections from the eye and to central visual targets in the brain (Fig. 3) (42). Thus, the isolation of RGCs from the retinas of recently deceased humans for transplantation into recipient humans may actually represent a clinically viable strategy for curing otherwise irreversible forms of blindness.
Fig. 3. Transplantation of RGCs to restore vision.

(Left and top) Injected donor RGCs (red) differentiate and integrate into the retina after nerve damage, whereas endogenous RGCs (blue) degenerate. (Bottom right) A subset of the transplanted RGCs extend axons (red cables) down the optic nerve and ultimately reach the brain (41).
Synthetic materials for replacing light-driven electrical responses
When blindness results from disabled retinal light sensing, two approaches are likely to prove helpful: (i) prosthetic devices that directly stimulate the RGCs or (ii) gene therapy to activate quiescent remaining photoreceptors. Flexible microarrays could be implanted into the human eye to convert light to electrical signals and then passed to RGCs (Fig. 4, A and B). The efficiency of some of the modern implantable electrode arrays is comparable with light stimulation in terms of generation of action potentials in the inner retina of animal models (43). The precision with which these devices can stimulate the visual pathways is impressive; some are starting to move them from the laboratory to the clinic.
Fig. 4. Retinal prostheses and virally introduced light-sensitive ion channels can potentially restore sight.
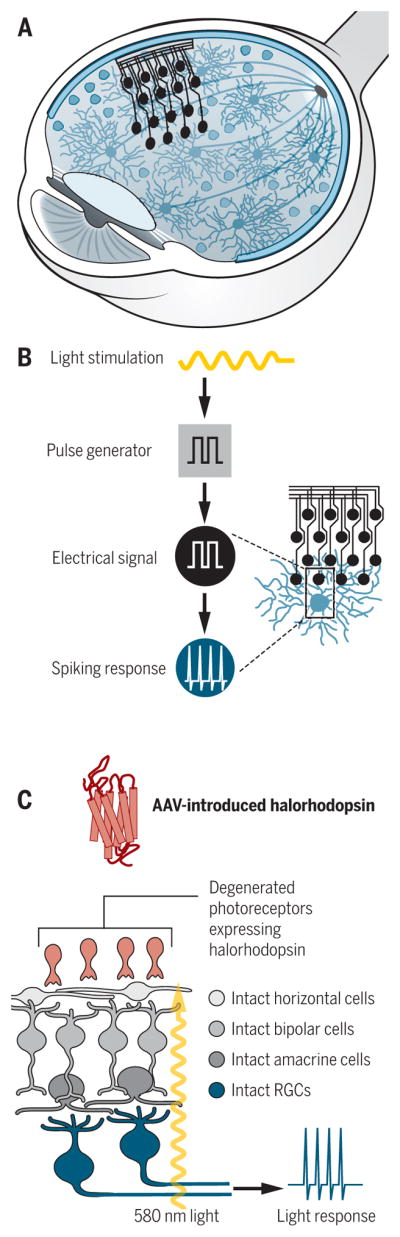
(A) Electrode arrays can be surgical implanted in the retina. (B) Incoming light is converted into electrical signals by electrodes, which generate spiking in RGCs, restoring eye-to-brain communication and, potentially, sight. (C) Adeno-associated viruses (AAVs) can be used to deliver light-sensitive ion channels (halorhodopsin) to degenerated photoreceptors, allowing light stimulation at the appropriate wavelength to generate spiking in the RGCs.
Introduction of light-sensitive ion-gated channels such as channelopsins to restore light sensitivity to sick photoreceptors (Fig. 4C) is being validated in mouse models of retinitis pigmentosa and in ex vivo human retinas. This approach has been shown to be capable of driving RGC firing in response to light and can activate visual circuits sufficiently well to drive visually guided behaviors in mice (44). Work in humans and nonhuman primates also suggest that such approaches can lead to recovery of the ability to detect motion, read words, and recognize high-contrast objects (45, 46). Although diseases that mainly affect the photoreceptors may leave RGCs intact, they may also indirectly alter RGC wiring (47). Therefore, therapeutic restoration of light sensitivity to degenerated photoreceptors may also require steps to enhance RGC regeneration and central plasticity in order to restore accurate vision.
Paths forward
The potential to stave off and reverse certain forms of blindness is starting to emerge as a realistic goal for the next 5 to 10 years, and perhaps even sooner. The phase in which replacement of damaged eye-to-brain circuits has proven possible has arrived, albeit in animal models. The three categories of approaches used to produce these effects—gene therapy for intrinsic growth-enhancing pathways, increasing RGC electrical activity, and cell transplantation—in theory are all clinically feasible. The goal now is to determine which specific molecular pathways are safe to trigger in humans and how to combine those with protocols that support RGC survival and regrowth. Research will determine whether these methods enhance visual function in humans, and whether that function derives from RGC axon regeneration, from central plasticity or both (48). Meanwhile, prosthetic implants or gene therapy with light-activated channels are both now ready for testing in humans. It seems likely that a combination of therapies may be needed to get full recovery of visual function. Regardless, the idea of regenerating eye-to-brain connections in humans is becoming an exciting and realistic possibility.
Acknowledgments
Work in the laboratory of these authors was supported by the National Eye Institute, the Glaucoma Research Foundation, the McKnight Foundation, the Pew Charitable Trusts, and the E. Matilda Ziegler Foundation for the Blind.
REFERENCES AND NOTES
- 1.Baden T, et al. Nature. 2016;529:345–350. doi: 10.1038/nature16468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quigley HA, Broman AT. Br J Ophthalmol. 2006;90:262–267. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldberg JL, Barres BA. Annu Rev Neurosci. 2000;23:579–612. doi: 10.1146/annurev.neuro.23.1.579. [DOI] [PubMed] [Google Scholar]
- 4.Case LC, Tessier-Lavigne M. Curr Biol. 2005;15:R749–R753. doi: 10.1016/j.cub.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Richardson PM, McGuinness UM, Aguayo AJ. Nature. 1980;284:264–265. doi: 10.1038/284264a0. [DOI] [PubMed] [Google Scholar]
- 6.Aguayo AJ, Vidal-Sanz M, Villegas-Pérez MP, Bray GM. Ann N Y Acad Sci. 1987;495:1–9. doi: 10.1111/j.1749-6632.1987.tb23661.x. [DOI] [PubMed] [Google Scholar]
- 7.Schwab ME. Curr Opin Neurobiol. 2004;14:118–124. doi: 10.1016/j.conb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Goldberg JL, et al. J Neurosci. 2004;24:4989–4999. doi: 10.1523/JNEUROSCI.4390-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng B, et al. Proc Natl Acad Sci USA. 2005;102:1205–1210. doi: 10.1073/pnas.0409026102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer D, He Z, Benowitz LI. J Neurosci. 2004;24:1646–1651. doi: 10.1523/JNEUROSCI.5119-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fitch MT, Silver J. Exp Neurol. 2008;209:294–301. doi: 10.1016/j.expneurol.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuchero JB, Barres BA. Development. 2015;142:3805–3809. doi: 10.1242/dev.129304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yiu G, He Z. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies SJ, et al. Nature. 1997;390:680–683. doi: 10.1038/37776. [DOI] [PubMed] [Google Scholar]
- 15.Anderson MA, et al. Nature. 2016;532:195–200. doi: 10.1038/nature17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cafferty WB, et al. J Neurosci. 2001;21:7161–7170. doi: 10.1523/JNEUROSCI.21-18-07161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Müller A, Hauk TG, Fischer D. Brain. 2007;130:3308–3320. doi: 10.1093/brain/awm257. [DOI] [PubMed] [Google Scholar]
- 18.Park KK, et al. Mol Cell Neurosci. 2009;41:313–324. doi: 10.1016/j.mcn.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Leibinger M, et al. J Neurosci. 2009;29:14334–14341. doi: 10.1523/JNEUROSCI.2770-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, et al. Proc Natl Acad Sci USA. 2017;114:E209–E218. doi: 10.1073/pnas.1616811114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin Y, et al. Nat Neurosci. 2006;9:843–852. doi: 10.1038/nn1701. [DOI] [PubMed] [Google Scholar]
- 22.Goldberg JL, et al. Neuron. 2002;33:689–702. doi: 10.1016/s0896-6273(02)00602-5. [DOI] [PubMed] [Google Scholar]
- 23.Park KK, et al. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun F, et al. Nature. 2011;480:372–375. doi: 10.1038/nature10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim JHA, et al. Nat Neurosci. 2016;19:1073–1084. doi: 10.1038/nn.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cantrup R, et al. PLOS ONE. 2012;7:e32795. doi: 10.1371/journal.pone.0032795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Magri L, Galli R. Cell Mol Life Sci. 2013;70:2887–2898. doi: 10.1007/s00018-012-1196-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo X, et al. Exp Neurol. 2013;247:653–662. doi: 10.1016/j.expneurol.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore DL, et al. Science. 2009;326:298–301. doi: 10.1126/science.1175737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Lima S, et al. Proc Natl Acad Sci USA. 2012;109:9149–9154. doi: 10.1073/pnas.1119449109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watkins TA, et al. Proc Natl Acad Sci USA. 2013;110:4039–4044. doi: 10.1073/pnas.1211074110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huberman AD, Feller MB, Chapman B. Annu Rev Neurosci. 2008;31:479–509. doi: 10.1146/annurev.neuro.31.060407.125533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S, et al. Proc Natl Acad Sci USA. 2016;113:1937–1942. doi: 10.1073/pnas.1523645113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bei F, et al. Cell. 2016;164:219–232. doi: 10.1016/j.cell.2015.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barres BA, Raff MC. Nature. 1993;361:258–260. doi: 10.1038/361258a0. [DOI] [PubMed] [Google Scholar]
- 36.Harvey P, Gong B, Rossomando AJ, Frank E. Proc Natl Acad Sci USA. 2010;107:11585–11590. doi: 10.1073/pnas.1003287107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.King CE, et al. Exp Neurol. 2003;183:593–599. doi: 10.1016/s0014-4886(03)00211-5. [DOI] [PubMed] [Google Scholar]
- 38.Narushima M, et al. Neuron. 2016;91:1097–1109. doi: 10.1016/j.neuron.2016.07.035. [DOI] [PubMed] [Google Scholar]
- 39.Tropepe V, et al. Science. 2000;287:2032–2036. doi: 10.1126/science.287.5460.2032. [DOI] [PubMed] [Google Scholar]
- 40.Ueki Y, et al. Proc Natl Acad Sci USA. 2015;112:13717–13722. doi: 10.1073/pnas.1510595112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka T, et al. Sci Rep. 2015;5:8344. doi: 10.1038/srep08344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Venugopalan P, et al. Nat Commun. 2016;7:10472. doi: 10.1038/ncomms10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goetz GA, Palanker DV. Rep Prog Phys. 2016;79:096701. doi: 10.1088/0034-4885/79/9/096701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Busskamp V, et al. Science. 2010;329:413–417. doi: 10.1126/science.1190897. [DOI] [PubMed] [Google Scholar]
- 45.Weiland JD, Liu W, Humayun MS. Annu Rev Biomed Eng. 2005;7:361–401. doi: 10.1146/annurev.bioeng.7.060804.100435. [DOI] [PubMed] [Google Scholar]
- 46.Picaud S, Sahel JA. C R Biol. 2014;337:214–222. doi: 10.1016/j.crvi.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 47.Jones BW, et al. J Comp Neurol. 2003;464:1–16. doi: 10.1002/cne.10703. [DOI] [PubMed] [Google Scholar]
- 48.Gall C, et al. PLOS ONE. 2016;11:e0156134. doi: 10.1371/journal.pone.0156134. [DOI] [PMC free article] [PubMed] [Google Scholar]