

Summary
Loss of p53 function is invariably associated with cancer. Its role in tumor growth was recently linked to its effects on cancer stem cells (CSCs), although the underlying molecular mechanisms remain unknown. Here, we show that c-myc is a transcriptional target of p53 in mammary stem cells (MaSCs) and is activated in breast tumors as a consequence of p53 loss. Constitutive Myc expression in normal mammary cells leads to increased frequency of MaSC symmetric divisions, extended MaSC replicative-potential, and MaSC-reprogramming of progenitors, whereas Myc activation in breast cancer is necessary and sufficient to maintain the expanding pool of CSCs. Concomitant p53 loss and Myc activation trigger the expression of 189 mitotic genes, which identify patients at high risk of mortality and relapse, independently of other risk factors. Altogether, deregulation of the p53:Myc axis in mammary tumors increases CSC content and plasticity and is a critical determinant of tumor growth and clinical aggressiveness.
Keywords: p53, Myc, CSCs, mammary gland, SC self-renewal, cancer maintenance, reprogramming
Graphical Abstract
Highlights
-
•
Myc is overexpressed and deregulated in breast tumors because of p53 signaling attenuation
-
•
Myc activation favors SC symmetric divisions and SC reprogramming of progenitors
-
•
Myc activation is necessary and sufficient to sustain the cancer SC phenotype
-
•
Expression of 189 mitotic p53:Myc targets identifies high-risk breast cancer patients
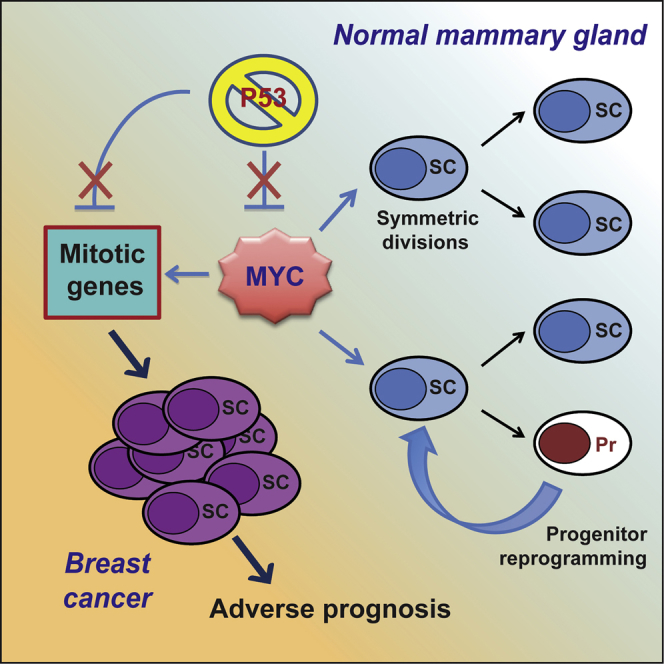
Santoro et al. demonstrate the existence of a regulatory axis instructed by the tumor-suppressor p53, executed by the Myc oncogene, and involving 189 p53:Myc targets. The authors show that activation of this axis maintains the expanding pool of cancer stem cells (SCs), thus promoting tumor growth, and predicts adverse prognosis.
Introduction
Loss of p53 function is considered a constant feature of cancer. p53 mutations and/or deletions are found in ∼50% of cases (Soussi and Wiman, 2007) and correlate with disease aggressiveness and worse prognosis (Miller et al., 2005). In the remaining tumors, p53 is wild-type (WT), but its function is frequently impaired because of alterations in the genes implicated in its regulation, such as the mouse double minute 2 (Mdm2) gene amplification or p14/ARF deletion, which enhance p53 degradation (Kruse and Gu, 2009).
Loss of p53 function is implicated in all phases of tumorigenesis: initiation, progression, metastasis, and tumor maintenance (Ventura et al., 2007, Xue et al., 2007, Martins et al., 2006). Traditionally, the role of p53 in cancer has been associated with its ability to activate DNA repair, apoptosis, senescence, or cell cycle arrest (Vousden and Prives, 2009). However, each of the above has been disputed as the biological mechanism underlying its tumor-suppressive function (Brady et al., 2011, Li et al., 2012b). Regulation of metabolism and autophagy have also been ascribed to p53, but their involvement is highly dependent on microenvironmental cues (Bieging et al., 2014, Vousden and Prives, 2009).
Recently, p53 has emerged as a negative regulator of adult stem cell (SC) self-renewal in the hematopoietic, neural, and mammary gland systems. Indeed, its loss leads to abnormal expansion of the SC compartment and increased repopulating ability in transplantation assays (Asai et al., 2011, Chen et al., 2008, Cicalese et al., 2009, Meletis et al., 2006). The underlying biological mechanisms are only partially characterized; p53 maintains the pool of quiescent hematopoietic SCs (Liu et al., 2009) and determines the balance between asymmetric and symmetric divisions of mammary SCs (MaSCs), with p53 loss favoring symmetric divisions (Cicalese et al., 2009).
Indirect evidence suggests that the effect of p53 on SCs is critical to its tumor-suppressive functions: (1) silencing of p53 in mouse brain and liver leads to the formation of undifferentiated tumors expressing SC markers (Friedmann-Morvinski et al., 2012, Tschaharganeh et al., 2014), (2) loss of p53 in breast cancer correlates with a SC-related transcriptional signature (Mizuno et al., 2010), and (3) restoration of p53 by the Mdm2-inhibitor Nutlin-3 (Nut3) (Vassilev et al., 2004) in ErbB2-overexpressing breast cancer decreases the number of cancer stem cells (CSCs) and significantly reduces tumor size, without any antiproliferative or apoptotic effect on bulk tumor cells (Cicalese et al., 2009).
Several bona fide p53 targets have been identified, possibly mediating the effects of p53 on SCs. For example, Mir34a is trans-activated by p53 and restricts the expansion of breast CSCs in mammosphere assays (Park et al., 2014). p53 also represses CD44 in breast cancer and Nestin in hepatocellular carcinoma, two CSC markers whose overexpression favors tumor growth (Godar et al., 2008, Tschaharganeh et al., 2014). p53 has also been reported as a transcriptional repressor of the c-myc proto-oncogene (Ho et al., 2005, Sachdeva et al., 2009, Li et al., 2012a), which regulates adult SCs particularly in the skin, hematopoietic, and neural compartments (Kerosuo et al., 2008, Laurenti et al., 2009, Watt et al., 2008) and induces a SC-like transcriptional pattern in immortalized mammary cells (Poli et al., 2018). However, whether any of these p53-regulated genes are critical effectors of p53 in SC homeostasis and tumor suppression remains unknown.
Here, we investigated the role of Myc as a p53 target in mammary CSCs, using transgenic mice overexpressing a mutated form of the breast cancer–associated ErbB2 oncogene (MMTV-ErbB2 mice) (Muller et al., 1988). In ErbB2 mammary tumors, higher numbers of CSCs follow abnormal self-renewing divisions characterized by increased frequency of symmetric divisions and extended replicative potential. Notably, these properties of CSCs are fully dependent on attenuated p53 signaling and contribute to tumor growth maintenance (Cicalese et al., 2009). We show that Myc is a direct target of p53, and its constitutive activation, as a consequence of p53 loss, is sufficient to maintain the transformed CSC phenotype in murine and human breast tumors.
Results
Loss of p53 Leads to Constitutive Myc Expression in ErbB2 Tumors
Western blot analyses showed marked Myc overexpression in unfractionated ErbB2 mammary tumors (Figure 1A). Myc overexpression was also observed in ErbB2-tumor mammosphere cultures, a cell population enriched in mammary SCs (MaSCs) and mammary progenitors (MaProgs) (Figures 1B and S1A). During the 5-day mammosphere growth (from single cells to formed mammospheres), Myc levels and the percentage of cycling cells decreased progressively in WT mammospheres, whereas they remained stable in the ErbB2-tumor mammospheres (Figures 1C and 1D).
Figure 1.

Myc is Overexpressed in Murine and Human Breast Tumors Because of Attenuation or Loss of p53 Signaling
(A and B) Western blot of Myc protein expression in (A) WT mammary gland and 5 independent ErbB2 tumors (T1–T5), and (B) two representative ErbB2 tumor mammosphere cultures (T19 and T20), untreated or treated with Nut3 (2.5 μM), as compared with one WT mammosphere culture. Mammospheres were analyzed at passage 3 (M3). Right: Relative Myc-protein expression for the untreated and Nut3-treated T19 and T20.
(C) Representative FACS-histograms of EdU (5-ethynyl-2′-deoxyuridine) incorporation in WT and ErbB2-tumor (T) cells at different times during mammosphere formation, as indicated. The percentage of EdU+ cells (cells in S phase) is shown for each time point.
(D and E) Western blot of Myc expression: (D) during WT and ErbB2 tumor mammosphere formation (24–120 h); and (E) in MCF10DCIS.com and primary cells from four PDX tumors (BC3, BC10, BC22, and BC26), untreated (UT) or treated with 2.5 or 10 μM Nut3 for 16 h in vitro. Right: Relative Myc-protein expression.
See also Figure S1.
To assess whether Myc overexpression in ErbB2 tumor cells is a consequence of reduced p53 activity, we analyzed the effect of p53 restoration. Treatment with Nut3 restored p53 activity (Figure S1B) and reduced Myc expression in ErbB2-tumor (Figures 1B and S1A) and human p53-WT breast cancer samples (Figure 1E), including MCF10DCIS.com (Miller et al., 2000) and four distinct patient-derived xenografts (PDXs), whereas it had had no effect on p53−/− mammospheres (Figure S1C).
We then investigated the correlation between p53 activity and Myc expression in breast cancer samples from The Cancer Genome Atlas (TCGA). Analyses of Myc RNA levels in samples with WT (n = 673) or mutated (n = 294) p53 showed significantly elevated Myc in patients carrying the p53 mutations (Figures S1D and S1E), including those with either “missense” or “nonsense” mutations. Nonsense mutations usually generate truncated proteins and cause p53 loss of function, whereas missense mutations may account for either loss- or gain-of-function effects on p53. Consistently, loss of p53 function (as revealed by analyses of expression of the p53-target p21) and Myc overexpression were accentuated in the presence of nonsense mutations (Figures S1D–S1G), suggesting that tumor-associated p53 mutations are unable to downregulate Myc. Together, these results demonstrate constitutive and increased Myc expression in murine and human breast cancer because of loss or attenuation of p53 signaling.
Constitutive Myc Expression in the Normal Mammary Gland Leads to Expansion of MaSC Numbers
To analyze the effects of de-regulated Myc expression on the replicative potential of MaSCs, we performed mammosphere serial-replating assays with cells from a murine model harboring MycER (fusion of human Myc with a modified estrogen receptor) within the Rosa26 locus (Murphy et al., 2008). Notably, levels of MycER protein and the extent of its nuclear translocation in mammospheres (hereafter, R-MycER mammospheres) were entirely dependent on the concentration of 4-hydroxytamoxifen (4-OHT) (Figures S2A and S2B).
In the absence of 4-OHT, numbers of Rosa26-MycER control (R-Ctrl) mammospheres progressively decreased at each passage, reaching culture exhaustion after five to seven passages (Figure 2A), confirming that MaSC self-renewal is intrinsically limited (Cicalese et al., 2009). At low doses of 4-OHT (20 or 200 nM), we observed increased cumulative numbers of spheres (growth rate [GR], 121% and 161%, respectively; Figure 2A) and cells (GR, 132% and 162%, respectively; Figure 2B) and extended life span (mammosphere cultures could be passaged indefinitely; not shown). High doses of 4-OHT (500 or 1,000 nM), instead, induced accumulation of phosphorylated p53 and cleaved caspase 3 (Figure S2A), massive cell death (Figure S2C), and rapid exhaustion of the R-MycER mammosphere culture (Figure 2C). 4-OHT had no effect on the growth of WT mammospheres (Figure S2D). Together, these data demonstrate that the effects of Myc on the replicative potential of MaSCs depended strictly on its expression level: high levels induced p53 activation and apoptosis; low levels expanded the numbers of MaSCs and induced their immortalization.
Figure 2.

Constitutive Myc Expression Increases MaSC Replicative Potential
(A and B) Serial replating of Rosa26-MycER cells untreated (UT) or treated with low doses of 4-OHT (20 and 200 nM): graphs of cumulative (A) sphere and (B) cell number (n = 6). R2, coefficient of determination.
(C) Cumulative sphere graph of Rosa26-MycER cells UT, as in (A) or treated with high doses of 4-OHT (500 nM and 1 μM). Data from one representative experiment are shown (n = 2).
(D) Whole mount carmine staining of reconstituted glands after injection of 500 LTR-Ctrl and 100 LTR-MycER cells. Scale bar, 2 mm.
(A–C) Error bars represent SD. See also Figures S2 and S3.
We then investigated the effects of constitutive low MycER expression on mammary gland development in vivo. To avoid 4-OHT treatment, which severely perturbs mammary gland development (Figures S3A and S3B), we transduced mammospheres with a lentivirus constitutively expressing MycER (LTR-MycER) or the corresponding empty vector (LTR-Ctrl). LTR-MycER mammospheres express low levels of nuclear Myc, do not activate p53, and possess extended self-renewal (Pasi et al., 2011). Notably, the extent of Myc-target activation and the growth properties of these cells are nearly identical to R-MycER upon low concentrations of 4-OHT (Figures S3C and S3D).
Injection of 500 or 100 cells of either LTR-MycER or LTR-Ctrl mammospheres in the cleared fat pad of syngenic, 3-week-old, virgin recipients (6–7 mice per sample) gave rise to positive outgrowths with undistinguishable morphologies (Figure 2D). The calculated MaSC frequency was <1:1,839 (CI = 95%) in the LTR-Ctrl mammospheres, as reported (Cicalese et al., 2009), and significantly higher (p = 0.02) in the LTR-MycER mammospheres (∼1:368; CI = 95%), suggesting that low-constitutive Myc expression increases the numbers of MaSCs. Notably, we did not observe tumor formation in a cohort of 15 LTR-MycER-transplanted mice during a 1-year observation.
Constitutive Myc Expression Leads to Increased Symmetric Divisions of Normal MaSCs
To investigate the biological mechanisms underlying the effects of Myc on MaSC numbers, we analyzed the relative proportion of asymmetric versus symmetric self-renewing divisions in control (Ctrl) and MycER MaSCs (using either R-MycER mammospheres induced with 200 nM 4-OHT or WT mammospheres transduced with LTR-MycER).
Disaggregated primary mammospheres were plated as single cells in 20% methylcellulose for 24 h to allow formation of doublets and were analyzed for CD49f expression, a well-established MaSC marker (Shackleton et al., 2006, Stingl et al., 2006). Because cells expressing the highest level of CD49f are enriched for mammary repopulation capacity (Stingl et al., 2006), we analyzed only the doublets with the brightest fluorescence signal (CD49fhi). Divisions giving rise to two cells both expressing CD49f were scored as symmetric, whereas retention of the marker by only one of the daughter cells indicated an asymmetric division. The prevailing modality of division of CD49fhi MaSCs was asymmetric (54%) in Ctrl samples and symmetric (52%) in R-MycER cells (Figures 3A and 3B).
Figure 3.

Constitutive Myc Expression Increases the Rate of MaSC Symmetric Divisions
(A) Representative confocal images of asymmetric and symmetric divisions in, respectively, control (Ctrl) and 4-OHT-treated (MycER) Rosa26-MycER cells. Cells were stained with DAPI, αMyc, and αCD49f antibodies. Scale bar, 10 μm.
(B) Percentage of asymmetric and symmetric divisions in Ctrl and 4-OHT-treated (MycER) Rosa26-MycER cells based on the distribution of the CD49f marker. ∗chi-square p < 0.0001.
(C) Experimental scheme of the PKH26 label-retaining assay.
(D) Percentage of asymmetric and symmetric divisions in LTR-Ctrl and LTR-MycER PKHhigh cells (left) and R-Ctrl and R-MycER bulk mammospheres (right), based on Numb distribution. ∗chi square p < 0.0001.
(E) Immunofluorescence staining for DAPI, Myc, and Numb expression in 4-OHT-treated (200 nM) WT Ctrl (Ctrl) and Rosa26-MycER (MycER) doublets. Representative images depict an asymmetric (top) and a symmetric (bottom) division. Scale bar, 10 μm.
As a parallel approach, we evaluated the distribution of the cell fate determinant Numb. MaSCs were purified near homogeneity as PKHhigh cells, using the well-established PKH26 label-retaining assay (Figure 3C) (Cicalese et al., 2009, Pece et al., 2010), infected with LTR-Ctrl and LTR-MycER, and then treated with blebbistatin to block cytokinesis and to allow evaluation of Numb localization before cell division. As observed with CD49f, Numb distribution in the LTR-Ctrl binucleated MaSCs was asymmetric in ∼69% of the cases, whereas, in LTR-MycER MaSCs, ∼67% of the mitoses were symmetric (Figure 3D). These results were confirmed in R-Ctrl and R-MycER bulk dividing cells, where ∼36% and ∼66% of divisions scored, respectively, as symmetric (Figures 3D and 3E). We noticed that Numb and CD49f co-localized in ∼70% of all asymmetric divisions scored, suggesting that Numb segregates with MaSCs during asymmetric self-renewing mitosis, consistently with the role of Numb in the regulation of asymmetric divisions in MaSCs (Tosoni et al., 2015).
Together, these findings demonstrate that low levels of constitutive MycER expression increase the proportion of symmetric divisions in MaSCs and suggest that Myc controls MaSC numbers by regulating the modality of self-renewing divisions.
Low Levels of Constitutive Myc Expression Reprogram MaProg Cells into MaSCs
We next investigated whether low levels of constitutive Myc expression induce reprogramming of MaProgs into MaSCs, as a further mechanism of functional MaSC expansion by Myc. As MaProgs, we used PKH− cells purified from primary PKH-labeled mammospheres (Figure 4A). The PKH− subset consists of a heterogeneous population of actively cycling cells, which give rise to either epithelial or myoepithelial colonies in differentiation assays (Pece et al., 2010) but which are unable to form mammospheres upon serial replating or a mammary gland upon transplantation (Cicalese et al., 2009). Notably, PKH− cells downregulate MaSC-specific genes, including Krt5 and Krt14 (Table S1: RNA-seq of PKHhigh and PKH− cells).
Figure 4.

MaProgs Are Reprogrammed into Normal MaSCs by Constitutive Myc Expression
(A) Experimental scheme for the isolation of PKH− cells from WT or Rosa26-MycER mice and the generation of control (Ctrl) and MycER expressing (MycER) progenitor cultures.
(B and C) Cumulative sphere (B) and cell (C) number graphs of Rosa26-MycER PKH− cells cultured in the absence (UT) or presence of low doses of 4-OHT (20 and 200 nM) during serial replating (n = 3). Error bars represent SD.
(D) Limiting dilution transplantation of LTR-Ctrl or LTR-MycER PKH− cells in the cleared fat pad of 3-week-old, female mice. Overall test for differences p = 0.000116.
(E) Recipients were transplanted with PKH−-LTR-MycER cells obtained from GFP-transgenic mice; GFP+ cells from a pooled group of 8 glands were FACS-sorted and re-transplanted into WT FVB mice. The presence of positive outgrowths was scored by whole mount carmine staining (WM, left) and analysis of GFP expression (right). Scale bars: 2 mm (left) and 100 μm (right).
(F) Representative images of organoids formed by purified Basal (Lin−CD24+CD49fhi) and LUM cells (Lin−CD24hiCD49f+) infected with LTR-Ctrl or LTR-MycER, respectively (LUM Ctrl and LUM MycER cells). Scale bar, 400 μm.
(G) Absolute number of compact, acinar, and budding organoids counted for three consecutive passages (P1, P2, and P3) in 5% matrigel cultures originating from basal (top) and control (Ctrl) or MycER luminal cells (LUM; bottom). Data from one representative experiment are shown (n = 3).
(H) Hierarchical clustering of 93 MaSC and MaProg genes; mean expression levels of PKHhigh cells (n = 2), PKH−-LTR-MycER (n = 3) and bulk LTR-MycER (n = 3) mammospheres, relative to the corresponding values of PKH− cells (n = 3) are shown as a continuous variable from blue to red. Columns correspond to genes and rows correspond to experimental samples.
We first evaluated whether low Myc expression confers mammosphere formation capacity on PKH− cells, by culturing PKH− cells obtained from Rosa26-MycER mammospheres in nonadherent conditions. As expected, in the absence of 4-OHT treatment, PKH−-R-Ctrl cells did not form mammospheres (Figure S4A) and were rapidly exhausted after few serial passages (GR, −74%; Figure 4B). Conversely, treatment with low-dose 4-OHT (20 or 200 nM) induced formation of mammospheres (Figure S4A), which expanded progressively after serial replating and acquired a near-immortal phenotype (Figures 4B and 4C). Notably, the growth rate of PKH−-R-MycER mammospheres treated with 200 nM 4-OHT (167%; Figure 4B) was similar in equally treated bulk R-MycER (161%; Figure 2A) and untreated PKH−-LTR-MycER mammospheres (Pasi et al., 2011).
We then examined whether low Myc expression confers a MaSC phenotype on PKH− cells by transplanting WT PKH− cells infected with LTR-Ctrl or LTR-MycER (Figure 4A) into the cleared fat pad of prepubertal mice, under limiting dilution conditions. As expected, PKH−-LTR-Ctrl did not reconstitute the cleared fat pad at any of the injected cell concentrations, thus ruling out the possibility of MaSC contamination in our progenitor preparations. In contrast, whole mount staining of the transplanted glands showed reconstitution of the fat pad by PKH−-LTR-MycER cells, with a calculated SC frequency of ∼1:100,000 (Figure 4D), suggesting that low levels of Myc can reprogram MaProgs into bona fide MaSCs, albeit at relatively low frequency.
In vivo self-renewal potential of the reprogrammed PKH−-LTR-MycER progenitors was evaluated by serial transplantation, using PKH− cells from GFP-transgenic mice (Hadjantonakis et al., 1998). GFP+ PKH− cells were transduced with LTR-MycER, transplanted into the cleared fat pad of WT mice and then retransplanted as GFP+ cells. As shown in Figure 4E, we obtained GFP+ outgrowths upon serial transplantation, proving the existence of long-living MaSCs and suggesting extended self-renewal of Myc-reprogrammed PKH− cells in vivo.
In vivo differentiation potential was evaluated by morphological, lineage marker, and functional analyses of transplanted glands. Hematoxylin-eosin staining and digital image analysis of tissue sections showed that the percentage of area occupied by epithelial structures was ∼2% in each acquired field of both PKH−-LTR-MycER and WT control outgrowths (WT Ctrl; Figure S4B). Staining for Ki67 confirmed that proliferation rates were comparable between the two groups (Figure S4B), as well as the expression of markers associated with myoepithelial (cytokeratin 14 [K14]) or luminal (cytokeratin 8 [K8]) differentiation (Figure S4C). Remarkably, anti-beta-casein staining of whole mounts showed milk presence in all mammary glands of recipients mated 10 weeks after the injection of PKH−-LTR-MycER cells and analyzed on day 18.5 of the pregnancy (Figure S4C). Finally, we observed no tumor formation in six additional mice/group kept under observation for 1 year (not shown), although some degree of hyperplasia and/or dysplasia was noted in all mammary glands obtained from both PKH−-LTR-MycER and WT Ctrl cells, likely because of active regeneration after transplantation (Figure S4B).
Together, these data demonstrate that low levels of Myc expression bestow MaProgs with mammosphere-initiating potential and the ability to form a fully functional mammary gland upon transplantation, suggesting that Myc is able to reprogram a heterogeneous population of committed progenitors into bona fide MaSCs, without inducing transformation.
To investigate reprogramming of lineage-restricted progenitor cells, we tested the effect of MycER expression on the differentiation and self-renewal potential of luminal (LUM; Lin-CD24hiCD49f+) and double-negative stromal (DN; Lin-CD24–CD49f–) cells (Figure S4D), which are devoid of any repopulating activity (Shackleton et al., 2006, Stingl et al., 2006), using an in vitro organoid formation assay (Panciera et al., 2016). For the positive control, we used basal cells (Lin-CD24+CD49fhi), which are enriched in mammary-repopulating units. As expected, basal cells formed compact and acinar organoids (Figure 4F) that could be serially passaged, albeit decreasing in number (Figure 4G). Up to passage 2, organoids were prevalently compact (Figures 4F and 4G). Strikingly, we could confirm the presence of functional myoepithelial cells by the contractile movements of compact organoids (Video S1). LUM Ctrl cells instead formed mainly acinar organoids or irregular (“budding”) structures (Figures 4F and 4G). However, in line with previous reports that used immortalized human mammary cells (Poli et al., 2018), Myc overexpression bestowed LUM cells with enhanced self-renewal (Figure 4G) and differentiation capacity, indicated by the formation of predominantly compact (Figures 4F and 4G) and contracting organoids (Video S2). Finally, MycER expression had no effect on the morphology of DN stromal cells (Figure S4E). Thus, constitutive Myc expression reprograms primary murine lineage-restricted luminal progenitors.
Finally, we analyzed the transcriptional effects of low Myc expression on PKH− cells, by RNA sequencing (RNA-seq). To that end, genes up- or downregulated in Myc-overexpressing PKH− cells (obtained by comparing PKH−-LTR-MycER mammospheres versus PKH− cells: PKH−MycER-DEGs; Table S1) were crossed with data sets of WT PKHhigh versus PKH− cells (Table S1), and with published MaSC data sets (Kendrick et al., 2008, Lim et al., 2010, Soady et al., 2015). We identified a core of 1,100 differentially expressed genes (DEGs; 369 UP and 731 DOWN), which were coherently regulated in Myc-reprogrammed mammospheres and PKHhigh MaSCs (Tables S1 and S2), including upregulated MaSC-specific (e.g., Ppp1r14a, Stk39, Nt5e, Fam101b, Ctnnal1, and Ldb2) and downregulated LUM-specific (e.g., Tbx3 and Wnt5a) genes (Table S2). Notably, the expression profile of Myc-reprogrammed mammospheres was almost identical to WT bulk mammospheres infected with LTR-MycER (Table S1: Myc-DEGs; Figures 4H and S4F). These data support the conclusion that PKH− cells acquire a multipotent MaSC-like transcriptional profile upon constitutive Myc-expression.
Constitutive Myc Expression Is Necessary and Sufficient to Maintain CSC Numbers in ErbB2 Mammary Tumors
Downregulation of Myc expression by RNA interference (Rubinson et al., 2003) resulted in rapid and complete growth arrest in both WT (Figures S5A and S5B) and ErbB2-tumor mammospheres (not shown). As an alternative approach to investigate whether Myc expression is critical to maintaining the pool of ErbB2-tumor CSCs, we used a doxycycline-inducible, dominant-negative mutant of Myc (Omomyc) (Soucek et al., 2013). Doxycycline treatment of Omomyc-transduced mammospheres (0.5 μM; Figure S5C) induced a significant drop in ErbB2-tumor sphere count, whereas it had no effect on WT (Figure 5A) or control empty-vector ErbB2-tumor (Figure S5D) spheres. Thus, impairing constitutive Myc expression in ErbB2-tumors leads to depletion of the CSC pool.
Figure 5.

Myc Is the Downstream Effector of p53 Loss in Breast CSCs
(A) Relative sphere number of WT (n = 2) and ErbB2-tumor (ErbB2; n = 3) cells transduced with the TET-inducible Omomyc vector. Spheres were counted at the end of the second passage in the absence (UT) or presence of 0.5 μM Doxycycline (+Dox). ∗paired t test p < 0.001.
(B and C) Cumulative sphere (B) and cell (C) number graphs of LTR-Ctrl and LTR-MycER ErbB2-tumor mammospheres (n = 4), in the absence or presence of 2.5 μM Nut3. ∗paired t test p < 0.05.
(D) Top: schematic representation of the experimental outline. Mice transplanted with LTR-Ctrl (n = 13) or LTR-MycER (n = 12) ErbB2-tumor cells were intraperitoneally [i.p.] injected with vehicle (DMSO, n = 7 for LTR-Ctrl and n = 6 for LTR-MycER) or with Nut3 (n = 6 for both LTR-Ctrl and LTR-MycER). Bottom: box plot of tumor volumes for each experimental group at the end of treatment. ∗t test p < 0.05.
(E) Tumor-free survival curve for mice injected with LTR-Ctrl and LTR-MycER DCIS cells untreated or Nut3-treated as mammospheres in vitro. Numbers of mice per group as indicated. ∗Mantel-Cox test between LTR-Ctrl treated versus untreated, p < 0.01.
(A–C) Error bars represent SD. See also Figure S5.
To establish whether constitutive Myc expression is sufficient for CSC maintenance in ErbB2-tumors, we experimentally uncoupled p53 and Myc and studied the effects of constitutive Myc expression on CSCs in the presence of functional p53. To this end, we expressed LTR-MycER in ErbB2-tumor mammospheres and treated cells with Nut3. Notably, in the lentiviral vector, MycER is under the control of the heterologous EF1α promoter, thus allowing assessment of the effect of constitutive Myc expression on CSCs independently of Nut3 and any transcriptional effect of functional p53 (Figure S5E).
The growth rate of LTR-Ctrl ErbB2-tumor mammospheres was reduced by treatment with 2.5 μM Nut3 (sphere-GR from 194% to 115%; p = 0.019; cell-GR from 171% to 58%; p = 0.004; Figures 5B and 5C), as reported by Cicalese et al. (2009), whereas it was not modified by expression of LTR-MycER (sphere-GR from 194% to 210%; p = 1; cell-GR from 171% to 226%; p = 0.1; Figures 5B and 5C). Nut3 treatment had no effect on the growth properties of LTR-MycER ErbB2-tumor (cumulative sphere and cell numbers), which showed the typical exponential growth of control ErbB2-tumor mammospheres (sphere-GR: 194% for both; cell-GR: 168% versus 171%; Figures 5B and 5C). Thus, in the context of enforced Myc expression, restoration of p53 function in ErbB2-tumor mammospheres does not prevent CSC expansion, suggesting that constitutive Myc expression is sufficient to maintain CSC growth properties.
To validate these findings in vivo, we transplanted LTR-Ctrl or LTR-MycER ErbB2-tumor cells and treated the recipients with either Nut3 or vehicle only. As expected, Nut3 significantly reduced tumor volume in control mice, with no effect in the LTR-MycER-transplanted cohort (Figure 5D). These observations were confirmed in MCF10DCIS.com cells (ductal carcinoma in situ [DCIS]), a human cell line with tumor-initiating cell frequency of ∼1:50 cells (Figure S5F). LTR-Ctrl and LTR-MycER DCIS mammospheres were treated in vitro with Nut3 (5 μM) and injected into non-obese diabetic-severe combined immunodeficiency (NOD/SCID) mice. Although Nut3 treatment significantly depleted the CSC content of LTR-Ctrl DCIS cells, diminishing their ability to seed a tumor upon transplantation, it had no effect on LTR-MycER DCIS, which formed tumors with the same frequency and latency of untreated control (Figure 5E).
Together, these results indicate that constitutive Myc expression is sufficient to maintain CSC properties in murine and human mammary cancer, independent of p53 function.
P53 and Myc Deregulate a Set of Mitotic Genes in ErbB2-Tumors
We first investigated whether p53 and Myc are epistatically linked as well in normal mammary cells. In normal mammospheres, levels of endogenous Myc transcripts increased progressively in the presence of heterozygous or homozygous deletions of p53 (Figure S6A) and gradually decreased upon acute p53 overexpression (Figure S6B) or following genotoxic treatment (Figure S6C). Chromatin immunoprecipitation sequencing (ChIP-seq) experiments in NMuMG mammary epithelial cells (Termén et al., 2013) showed p53 enrichment at the 3′-UTR of the c-myc gene, under unperturbed growth conditions. Stabilization of p53 following Adriamycin treatment resulted in two additional binding sites, including the transcription start site (TSS; Figure 6A), and marked downregulation of Myc levels (Figure S6D), suggesting that Myc is directly repressed by p53.
Figure 6.

p53 and Myc Co-regulate Mitotic Genes in CSCs
(A) Genome browser visualization of p53 binding on the c-myc gene.
(B) GSEA pre-ranked analysis showing enrichment of the Myc signature in the p53−/−. Left: upregulated genes; Right: downregulated genes. NES, normalized enrichment scored.
(C) Venn diagram of p53-Myc DEGs in non-transformed mammospheres: intercross between p53-DEGs and Myc-DEGs. Numbers represent genes coherently up- and downregulated and relative percentages.
(D) GSEA pre-ranked analysis, as in (B) showing enrichment of the Myc signature in the ErbB2-DEGs (top) and the Nut3-DEGs (bottom).
(E) Left, Venn diagram of p53 and/or Myc DEGs in ErbB2 tumors: intercross between the union of p53- and Myc-DEGs (red circle) and ErbB2-DEGs (blue circle). Right: pie chart, characterization of the p53 and/or Myc DEGs in only p53-dependent (blue), only Myc-dependent (red), and common p53-Myc DEGs (green).
(F) Pathway analysis of the ErbB2 p53-Myc common DEGs using MSigDB (http://software.broadinstitute.org/gsea/msigdb/index.jsp). The top 15 up- and downregulated pathways are shown ranked by q-value.
To identify p53:Myc common targets in normal mammospheres, we generated RNA-seq data sets of p53-responsive (p53−/− versus WT mammospheres: p53-DEGs) and Myc-responsive (LTR-MycER-expressing versus WT mammospheres: Myc-DEGs) genes (Table S1). Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) of Myc-DEGs in p53-DEGs showed a strong correlation between upregulated and downregulated genes, respectively (Figure 6B). The intercross between p53-DEGs and Myc-DEGs showed that a large fraction of the ∼14,000 genes expressed in normal mammospheres are regulated by both p53 and Myc (WT p53-Myc common DEGs: n = 3,292; Figure 6C). Notably, the WT p53-Myc common DEGs represent ∼50% of their respective regulatory potential (Figure 6C). Together, these data show that Myc is a direct target of p53 in normal MaSCs and progenitors, and that the p53:Myc epistatic axis regulates a large fraction of the expressed genes in normal MaSCs and progenitors.
We then searched for genes deregulated in ErbB2 tumor cells through the p53:Myc axis (p53 loss and Myc activation). A list of all tumor-associated gene deregulations was generated by RNA-seq analyses of ErbB2 tumor versus WT mammospheres (ErbB2-DEGs; Table S1).
First, we investigated the presence and p53 dependency of the identified Myc transcriptional program in ErbB2-tumors. GSEA of Myc-DEGs in ErbB2-DEGs showed a strong correlation, including both up- and downregulation (Figure 6D, top). Strikingly, the enrichment-curve profile of Myc-DEGs was inversed in the set of genes modulated by Nut3 (ErbB2-tumor + Nut3 versus ErbB2 tumor mammospheres; Nut3-DEGs; Table S1; Figure 6D, bottom). In conclusion, the Myc transcriptional program is activated in ErbB2 tumor cells, mirroring Myc overexpression in normal mammary cells and is highly dependent on reduced p53 activity.
Second, we intercrossed the ErbB2-DEGs with the union of p53- and Myc-DEGs and found that ∼80% of them (n = 2,843) are p53 and/or Myc responsive genes (Figure 6E, left). Of those, ∼53% are regulated by both p53 and Myc (“Common”), ∼30% are regulated by p53 (“p53-dependent”) in a Myc-independent manner, and ∼17% are regulated by Myc (“Myc-dependent”) independently of p53 (Figure 6E, right). Notably, ChIP-seq analyses of NMuMG epithelial cells revealed that ∼40% of the ErbB2-p53-Myc common DEGs were directly bound by Myc; among which, 64% were also bound by p53 (Table S3). In conclusion, ∼40% of DEGs identified in ErbB2-tumor cells respond to both p53 loss and Myc activation, either as a result of the p53:Myc epistasis or through direct co-binding.
Third, we investigated intracellular pathways regulated by the p53:Myc axis in ErbB2 tumor cells. To that end, the 1,494 ErbB2-p53-Myc common DEGs (801 upregulated and 693 downregulated) were submitted to the molecular signature database (MSigDB) available on the GSEA website (https://www.broadinstitute.org/gsea). Strikingly, the most significantly enriched pathways were in the 801 p53-Myc upregulated DEGs (q < 0.05), and were all cell-cycle related, with particular emphasis on pathways controlling mitosis (Figure 6F). Manual curation of the corresponding gene lists revealed a total of 189 “mitotic” genes, including genes involved in anaphase-promoting complex or cyclosome (APC/C)-mediated mitotic spindle checkpoint, G2/M checkpoint, mitosis entry or progression, spindle assembly at metaphase, association to kinetochore complexes, M-phase-promoting factor, assembly and motility of the spindle microtubules (Table S3). Intersection of the 189 mitotic genes with Nut3-DEGs showed that 181 of them are repressed by Nut3 treatment in the ErbB2-tumor mammospheres, thus showing a correlation between the 189 mitotic DEGs and the ErbB2-tumor CSC phenotype (e.g., expansion and immortality).
Together, our data demonstrate the existence of 189 p53 and Myc common targets implicated in the regulation of mitosis (mitotic signature [MitSig]) and upregulated in the ErbB2-tumor mammospheres, suggesting that these genes are crucial p53:Myc targets for the regulation of CSC numbers.
Expression of the MitSig Predicts Patient Clinical Outcome Independently of Other Known Risk Factors
Finally, we investigated the relevance of p53:Myc MitSig in the clinical setting. We interrogated TCGA and classified 1,032 cases of primary breast cancer into two clusters, according to MitSig gene expression: 384 cases were assigned as “UP” and 648 as “DOWN” (Figure S7A). MitSig upregulation (UP) correlated with increased cell cycle, proliferation, and apoptosis (Figure S7B), as defined by the pathway activation score based on reverse-phase protein array (Akbani et al., 2014).
To investigate the prognostic value of MitSig, we performed survival analyses in four independent breast-cancer data sets with a follow-up of >100 months (Desmedt et al., 2007, Ivshina et al., 2006, Pawitan et al., 2005, Wang et al., 2005). Expression data from those four studies contained information on 161 MitSig genes. Hierarchical clustering of all 892 cases segregated patients into signature UP (n = 366) and DOWN (n = 526) cohorts (Figure 7A). Disease-free survival (DFS) of the UP group was significantly shorter in the overall population (Figure 7B), as well as within each individual study (Figure S7C). We then evaluated the effect of MitSig on survival, as compared to well-established prognostic factors. Cox multiple-regression analysis (including age, tumor grade and size, lymph node status, estrogen receptor [ER] status, and p53 status as possible covariates; Table S4) showed that MitSig is an independent predictor of DFS (Figure 7C). Of note, the ER status, which has strong prognostic power (Cianfrocca and Goldstein, 2004), was independent from the signature (Figure S7D).
Figure 7.

MitSig Expression Correlates with Breast Cancer Aggressiveness and Poor Prognosis (GEO: GSE1456, GSE2034, GSE4922, GSE7390)
(A) Hierarchical clustering of 892 breast cancer patients according to the log2-normalized probe intensity of 161 of the 189 genes of the MitSig (values are expressed as Z score). Color code of ER status, PAM50 classification, and probe-intensity scale defined in the legend on the right.
(B and E) Disease-free survival (DFS) curve of all patients (B) and luminal B patients (E) grouped in DOWN and UP cohorts based on the expression of 161 genes of the MitSig. Log-rank test, p = 3.34e−08 and Hazard ratio (HR) = 1.93, 95% CI, 1.54–2.42 (B). Log-rank test p = 9.88e−4 and HR = 1.78, 95% CI, 1.26–2.52 (E).
(C) Forest plot for Cox multiple regression analysis of MitSig (in red) versus individual covariates (in blue). LN_status, lymph node status.
(D) Histogram showing the distribution of breast cancer patients with MitSig classification UP and DOWN within each molecular subtype (basal; HER2+; LumB, luminal B; LumA, luminal A; and normal, normal-like).
Finally, we compared directly the performance of MitSig against the PAM50 classifier. PAM50 is a multigene expression-based system that assigns patients to five “intrinsic subtypes” (luminal A, luminal B, basal, HER2+, and normal-like) with high prognostic significance (Parker et al., 2009). Proliferation-associated genes have a major role in the PAM50 signature, in particular for the distinction between luminal A and B groups, which can be approximated by immunohistochemistry for Ki67 (Cheang et al., 2009). We reclassified patients into PAM50 subtypes (Figure 7A) by established computational algorithms (Gendoo et al., 2016). MitSig upregulation was significantly associated with the PAM50 classification (chi-square p = 3.93e−93). In particular, it strongly correlated with the basal (92% overlap) and HER2+ (75%) groups and was anti-correlated with the luminal A (4%) and normal-like (3%). The luminal B group, instead, was almost equally dichotomized in two populations (Figure 7D) with significantly different DFS (78 versus 54 months, respectively; Figure 7E), suggesting that MitSig has distinct features with respect to the PAM50 proliferative signature. Notably, not all MitSig genes were predicted to favor proliferation upon overexpression because many encode proteins that negatively regulate cell-cycle progression (e.g., Chek2, Atm, Ercc6l, Rad1, Rbl1, Brca1, and Brca2), whereas others control the proper execution of mitosis (e.g., Alms1 and Asp, which regulate planar polarization and symmetric divisions, respectively).
Altogether, these analyses show that MitSig expression is, by itself, predictive of clinical outcome, independently of other known risk factors, suggesting that it represents the link between p53 loss of function and CSC-specific biological functions.
Discussion
We reported here that Myc is activated in mammary tumors as a consequence of attenuated p53 signaling and that activated Myc is necessary and sufficient to sustain the pool of expanding mammary CSCs. In the ErbB2-driven model of mammary gland carcinogenesis, p53 is WT but hypo-functional because of enhanced Mdm2 activity (Zhou et al., 2001). We found that, in ErbB2 tumor mammospheres, Myc levels, and transcriptional activity are up- and deregulated and that p53 restoration results in the downregulation of Myc protein and transcriptional activity. These observations hold true as well for normal mammospheres, in which Myc transcription is repressed by p53 and inversely correlates with p53 gene dosage and activation. Altogether, these data demonstrate the existence of a regulatory p53:Myc axis in normal and cancer MaSCs and progenitors. Notably, this axis regulates a large fraction of the expressed genes in normal and tumor mammary cells.
Perturbation of the p53:Myc axis leads to critical biological changes in normal MaSC homeostasis. Indeed, we showed that constitutive Myc expression in mammary epithelial cells increases the rate of MaSC symmetric divisions, extends MaSC self-renewal, and induces MaProg reprogramming into MaSCs. The first two phenotypes have also been described as consequences of p53 loss of function (Cicalese et al., 2009). Unpublished observations from our group demonstrated that p53 silencing facilitates reprogramming of MaProgs in vitro and in vivo (unpublished data). The effects of p53:Myc on progenitor reprogramming are coherent with the known role of p53 and Myc in somatic cell reprogramming, where they exert inhibitory and promoting functions, respectively. In the case of induced pluripotent SCs (iPSCs), p53 loss and Myc expression favor reprogramming, yet they remain dispensable in the presence of the other iPSC-inducing factors (Oct4, Sox2, and Klf4) (Takahashi and Yamanaka, 2006). In the mammary system, instead, they are sufficient to endow MaProgs with MaSC characteristics.
In the tumor setting, the three phenotypes described here are the biological outputs of a program instructed in mammary tumors by loss of p53 and mediated by Myc activation, which leads to the continuous expansion of the CSC pool and the maintenance of tumor growth. In this respect, it is hard to dissect the relative contribution of each mechanism to CSC maintenance. However, if one considers the large amount of bulk tumor cells at various differentiation stages, reprogramming, at any rate, is likely to have dramatic effects in vivo because of the potentially unlimited supply of new CSCs.
Analyses of the p53:Myc downstream targets unraveled mechanistic insights into the above-mentioned phenotypes. We showed that the activity of the p53:Myc epistasis converges toward the regulation of 189 mitotic genes (MitSig), which are downregulated by p53 in normal mammospheres and upregulated by Myc in the ErbB2 tumors. Most important, the expression of a large part of these genes (181 out of 189) is strictly connected to the maintenance of the CSC phenotype because they are downregulated in the Nut3-treated tumors. Nut3-treated CSCs, indeed, regain functional p53, lose Myc expression, and, as a result, lose their unlimited self-renewal capacity.
Regardless of the underlying mechanisms, MitSig represents the molecular fingerprint of CSC-specific functions and, not surprisingly, its expression in breast cancer patients segregates with adverse prognosis. Several p53-dependent and Myc-dependent gene expression signatures have been linked to breast tumor aggressiveness, either directly or indirectly (Ben-Porath et al., 2008, Brasó-Maristany et al., 2016, Kim et al., 2010, Miller et al., 2005, Mizuno et al., 2010). However, none of them points to CSC-specific cellular mechanisms, and their overlap with our MitSig is limited. MitSig differs because of the combination of (1) its derivation from a MaSC-enriched population obtained from primary tissues, and (2) its definition within a precise genetic context (p53 alterations) commonly encountered in the clinic.
The potential of MitSig as a stratification biomarker might be clinically relevant in breast cancer. In particular, the finding that it segregates two almost equally sized populations with clearly different outcomes within the luminal B subtype deserves further evaluation. This PAM50 subtype comprises patients with heterogeneous clinical-biological characteristics and outcomes, for which prognostic and/or predictive markers are highly sought after (Ades et al., 2014). Notably, several gene-expression signatures have been tested against the intrinsic molecular classification, and all defined these cases as uniformly high risk (Sotiriou and Pusztai, 2009). In a direct comparison, 93% and 83% of luminal B patients were defined as high risk using the Oncotype DX and Mammaprint stratification platforms, respectively (Fan et al., 2006). Thus, integrating MitSig into gene expression based prognostic and/or predictive scores, such as PAM50, may help identify patients at high risk of relapse who may benefit from treatment intensification.
The identified p53:Myc:mitosis axis could also provide important opportunities in terms of targeted therapies. Interestingly, 8 of 189 MitSig genes, when carrying a gain-of-function mutation, predict sensitivity to specific drugs in breast cancer (Brca1, Brca2, Aurkb, AurkA, Check2, Cdk6, Ccnd1, and Atm). This opens the possibility of selectively hitting the effectors of the p53:Myc axis in tumors, instead of taking the challenge of drugging p53 or Myc directly. As a therapeutic option, this approach could have an enormous effect on patients who have no alternative to chemotherapy, especially those in the triple-negative breast cancer subgroup that is characterized by high MitSig expression.
p53 and Myc have been implicated in the pathogenesis of different tumors, and their role in sustaining CSCs might represent a critical and general mechanism of tumorigenesis. Somatic mutations or deletion of the p53 gene are found in almost every type of cancer and, even in the presence of WT alleles, functional inactivation is a highly common event. Genetic alterations of Myc, instead, are much less frequent. Myc expression, however, is elevated or deregulated in a much higher fraction of tumors (Vita and Henriksson, 2006), possibly because of perturbations in the p53:Myc axis. Notably, p53 and Myc were recently identified as the central hubs in key protein networks specifically deregulated in the CD34+ compartment of chronic myeloid leukemia patients (Abraham et al., 2016). Thus, p53 loss and Myc activation might be integral to the oncogenic process itself, through the maintenance of the CSC pool in most tumors. The continuous generation of cells with CSC properties might represent an invariable trait of many tumors, likely the most aggressive ones, and the manipulation of the p53:Myc axis seems to constitute a potential means to modulate tumor development. In this context, MitSig, as the key downstream molecular player, could serve as a powerful diagnostic tool for efficient patient stratification and a source of potentially druggable hits for targeted therapies.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE-Cy7 rat monoclonal anti-mouse CD45 (clone 30-F11) | Thermo Fisher | Cat#25-0451-82; RRID:AB_469625 |
| Pe-Cy7 rat monoclonal anti-mouse TER-119 (clone TER-119) | Thermo Fisher | Cat#25-5921-82; RRID:AB_469661 |
| PE-Cy7 rat monoclonal anti-mouse CD31 (PECAM) (clone 390) | Thermo Fisher | Cat#25-0311-82; RRID:AB_469615 |
| APC rat monoclonal anti-mouse CD326 (EpCAM) (clone G8.8) | Thermo Fisher | Cat#17-5791-82; RRID:AB_1659714 |
| PE rat monoclonal anti-mouse CD24 (clone M1/69) | Thermo Fisher | Cat#12-0242-82; RRID:AB_465602 |
| APC rat monoclonal anti-human/mouse CD49f (clone GoH3) | Thermo Fisher | Cat#17-0495-82; RRID:AB_2016694 |
| eFluor®450 rat monoclonal anti-human/mouse CD49f (clone GoH3) | Thermo Fisher | Cat#48-0495-82; RRID:AB_11042564 |
| PE armenian hamster monoclonal anti-mouse CD61 (clone 2C9.G3) | Thermo Fisher | Cat#12-0611-82; RRID:AB_465718 |
| Rabbit monoclonal anti-human/mouse c-Myc (clone Y69) | Abcam | Cat# ab32072; RRID:AB_731658 |
| Rat monoclonal anti-human/mouse CD49f (clone GoH3) | BD Biosciences | Cat# 555734; RRID:AB_2296273 |
| Mouse monoclonal anti-Numb (clone AB21) | Colaluca et al., 2008 | N/A |
| Rabbit monoclonal anti-Ki67 (clone SP6) | Thermo Fisher | Cat#RM-9106-S; RRID:AB_149707 |
| Mouse polyclonal anti-Cytokeratin 14 | Covance | Cat#PRB-155P; RRID:AB_292096 |
| Mouse monoclonal anti-Cytokeratin 18 (clone Ks18.04) | Progen | Cat#61028; RRID:AB_1541026 |
| Rabbit polyclonal anti-βcasein | Santa Cruz | Cat#sc30042; RRID:AB_2084343 |
| Rabbit polyclonal anti-GFP | Abcam | Cat#ab6556; RRID:AB_305564 |
| Mouse monoclonal anti-Vinculin (clone hVIN-1) | Sigma-Aldrich | Cat# V9131; RRID:AB_477629 |
| Rabbit polyclonal anti-human/ mouse c-Myc | Cell Signaling | Cat#9402; RRID:AB_2151827 |
| Rabbit polyclonal anti-human/mouse phosphoS15 p53 | Cell Signaling | Cat#9284; RRID:AB_331464 |
| Mouse monoclonal anti-human/mouse p21 (clone F-5) | Santa Cruz | Cat#sc-6246; RRID:AB_628073 |
| Rabbit polyclonal anti-human/mouse cleaved Casp3 | Cell Signaling | Cat#9661; RRID:AB_2341188 |
| Rabbit polyclonal anti-mouse p53 | Novocastra | Cat#NCL-L-p53-CM5p; RRID:AB_563933 |
| Rabbit polyclonal anti-human/mouse c-Myc (N-262) | Santa Cruz | Cat#sc-764; RRID:AB_631276 |
| Biological Samples | ||
| Patient derived xenograft (PDX) samples | Luisa Lanfrancone, Target Identification and Validation (TIV) unit, IEO | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4-hydroxytamoxifen (4-OHT) | Sigma | Cat#H7904; CAS#68047-06-3 |
| Nutlin-3 (Nut3) | Sigma | Cat#N6287; CAS#548472-68-0 |
| Adriamycin (Doxorubicin hydrochloride) | Sigma | Cat#D1515; CAS#25316-40-9 |
| Poly-HEMA | Sigma | Cat#P3932; CAS#25249-16-5 |
| Critical Commercial Assays | ||
| Click-iT® EdU Alexa Fluor® 647 Flow Cytometry Assay Kit | Thermo Fisher | Cat#C10424 Lot#1293070 |
| PKH26 red fluorescent cell linker kit for general membrane labeling (PKH26) | Sigma | Cat#PKH26GL |
| Deposited Data | ||
| TCGA breast cancer RNA-seq and clinical data | TCGA Research Network | https://cancergenome.nih.gov/ |
| Raw and analyzed ChIP-seq and RNA-seq data: Regulation of p53- and Myc-targets in normal and tumoral murine mammary epithelial cells | This paper | GEO: GSE87004 |
| Affymetrix Array U133A data: Gene expression of breast cancer tissue in a large population-based cohort of Swedish patients | Pawitan et al., 2005 | GEO: GSE1456 |
| Affymetrix Array U133A data: Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer | Wang et al., 2005 | GEO: GSE2034 |
| Affymetrix Array U133A data: Genetic Reclassification of Histologic Grade Delineates New Clinical Subtypes of Breast Cancer | Ivshina et al., 2006 | GEO: GSE4922 |
| Affymetrix Array U133A data: Dataset of microarray experiments from primary breast tumors used to validate the 76-gene signature (VERIDEX) | Desmedt et al., 2007 | GEO: GSE7390 |
| Experimental Models: Cell Lines | ||
| MCF10DCIS.com | Miller et al., 2000 | RRID:CVCL_5552 |
| NMuMG | ATCC; Termén et al., 2013 | RRID:CVCL_0075 |
| Experimental Models: Organisms/Strains | ||
| Mouse: GFP/FVB: FVB.Cg-Tg(ACTB-EGFP)B5Nagy/J | Jackson lab | Stock#003516 |
| Mouse: MMTV-ErbB2: FVB-Tg(MMTV-Erbb2)NK1Mul/J | Jackson lab | Stock#005038 |
| Mouse: p53−/−: B6.129S2-Trp53tm1Tyj/J | Jackson lab | Stock#002101 |
| Mouse: Rosa26-MycER (R26-MER) | Murphy et al., 2008 | N/A |
| Mouse: NOD/SCID: NOD.CB17-Prkdcscid/NCrHsd | Envigo | N/A |
| Oligonucleotides | ||
| Primer: GusB Forward GTGGGCATTGTGCTACCTC | This paper | N/A |
| Primer: GusB Reverse ATTTTTGTCCCGGCGAAC | This paper | N/A |
| Primer: c-Myc Forward TTTGTCTATTTGGGGACAGTGTT | This paper | N/A |
| Primer: c-Myc Reverse CATCGTCGTGGCTGTCTG | This paper | N/A |
| Primer: Ncl Forward CATGGTGAAGCTCGCAAAG | This paper | N/A |
| Primer: Ncl Reverse TCACTATCCTCTTCCACCTCCTT | This paper | N/A |
| Primer: Cad Forward GATCATCATGGGGGAGAAAG | This paper | N/A |
| Primer: Cad Reverse CCAAGCGTGAGAAGGAGAAC | This paper | N/A |
| Primer: Odc1 Forward GCTAAGTCGACCTTGTGAGGA | This paper | N/A |
| Primer: Odc1 Reverse AGCTGCTCATGGTTCTCGAT | This paper | N/A |
| Recombinant DNA | ||
| pWPI | Didier Trono | Addgene #12254 |
| pWPI-MycER | Pasi et al., 2011 | N/A |
| pLL3.7 | Rubinson et al., 2003 | Addgene #11795 |
| pLL3.7-mMyc (AP4619/20) - puro | Bruno Amati | N/A |
| insert: mMyc hairpin TGAATTTCTATCACCAGCAATTCAAGAGAT TGCTGGTGATAGAAATTCTTTTTTC | ||
| pLL3.7-mMyc (AP4621/22) – puro insert: mMyc hairpin TGGAGAT GATGACCGAGTTATTCAAGAGATAACTCGGTCATCAT CTCCTTTTTTC |
Bruno Amati | N/A |
| pTRIPZ | Open Biosystems | Cat#RHS4750 |
| pTRIPZ-RFP-Omomyc | Annibali et al., 2014 | N/A |
| pBABE-p53ER | Vater et al., 1996 | N/A |
| Software and Algorithms | ||
| ELDA | Hu and Smyth 2009 | http://bioinf.wehi.edu.au/software/elda/ |
| FlowJo 9.3-2 (or later versions) | FowJo, LLC | https://www.flowjo.com/ |
| ImageJ | Schneider et al., 2012 | https://imagej.net/Welcome |
| bwa (version 0.6.2-r126) | Li and Durbin 2010 | http://bio-bwa.sourceforge.net |
| samtools rmdup tools (version 0.1.18) | Li et al., 2009 | https://github.com/samtools/samtools |
| MACS (version 1.4) | Zhang et al., 2008 | https://github.com/taoliu/MACS/ |
| GIN tool | Cesaroni et al., 2008 | N/A |
| Tophat2 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| HTSeq (v0.5.3p9) | Anders et al., 2015 | https://github.com/simon-anders/htseq |
| DESeq2 | Anders and Huber 2010 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSEA v2.2.0 | Subramanian et al., 2005 | https://www.broadinstitute.org/gsea/ |
| ClustVis | Metsalu and Vilo 2015 | https://biit.cs.ut.ee/clustvis/ |
| cBioportal | Cerami et al., 2012, Gao et al., 2013 | http://www.cbioportal.org/ |
| Mouse Gene Database (MGI) | Blake et al., 2017 | http://www.informatics.jax.org/homology.shtml |
| Broad GDAC Firehose [https://doi.org/10.7908/C11G0KM9] | Broad Institute | https://gdac.broadinstitute.org/ |
| Aroma R package | Bengtsson et al., 2008 | https://cran.rstudio.com/web/packages/aroma.affymetrix/index.html |
| Other | ||
| Tamoxifen diet for rodents | Envigo | TD.130859 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pier Giuseppe Pelicci (piergiuseppe.pelicci@ieo.it).
Experimental Model and Subject Details
Cell lines and PDX samples
MCF10DCIS.com (Miller et al., 2000) and patient derived xenografts were kindly provided by Dr. Luisa Lanfrancone, and cultured as previously described (D’Alesio et al., 2016). Nut3 (Sigma, N6287) was used in vitro at a final concentration of 2.5 or 10 μM as mentioned. Prior to transplantation in NOD/SCID mice (see Transplantation Experiments), MCF10DCIS.com were cultured as mammospheres (see Method Details) and treated with 5 μM Nut3. NMuMG cells were obtained from ATCC and cultured in adhesion at 37°C, 5% CO2, in DMEM media (Lonza) supplemented with 10% fetal bovine serum (FBS; Microgem), 2 mM L-Glutamine (EuroClone) and 10 μg/mL insulin (Roche). p53 activation was mediated by the addition of Adriamycin (Doxorubicin hydrochloride, Sigma, D1515) at a final concentration of 5 μM.
Mouse models
MMTV-ErbB2 (Muller et al., 1988) and GFP transgenic mice (Hadjantonakis et al., 1998) were in the FVB background; p53−/−, p53+/− and Rosa26-MycER (Murphy et al., 2008) in C57/BL6J. In vivo studies were performed after approval from our fully authorized animal facility and our institutional welfare committee (OPBA) and notification of the experiments to the Ministry of Health (as required by the Italian Law; IACUCs Numbers: 19/2013; 757/2015; 537/2017), in accordance with EU directive 2010/63.
4-OHT administration in vivo
Female WT C57/BL6J mice were tested for tolerability of long-term administration of 4-OHT and its effects on the mammary gland tissue. 3- and 8-week-old mice were fed with 4-OHT-containing or standard diet for 14 days, then sacrificed and their mammary glands digested and analyzed by FACS for the expression of epithelial SC markers. 4-OHT-containing food was purchased from Harlan (TD. 130859) and, according to manufacturer’s instructions, ∼40 mg 4-OHT/kg of body weight were provided per day, assuming 20-25 g body weight and 3-4 g intake.
Transplantation Experiments
Transplantation of WT mammary cells was performed in cleared fat pads of 3-week-old female FVB mice anaesthetized with 2.5% Avertin in PBS (100% avertin: 10 g of tribromoethanol in 10 mL of tertamyl alcohol, both Sigma). Transplanted mammary glands were collected 12 weeks later, fixed in 4% formaldehyde and colored with carmine alum for whole mount staining (Deome et al., 1959). Fat pads were scored as positive when the ductal branching originated from a central region of the cleared fad pad and its directionality was variable in different parts of the fat pad.
For in vivo tumor growth assays, 200,000 ErbB2-tumor or 10,000 MCF10DCIS.com cells were injected orthotopically in one inguinal mammary gland per mouse in 8-week-old female FVB or NOD/SCID virgin recipients, respectively, without prior clearing of the fat pad. Secondary ErbB2-tumors developed with a latency of 40-50 days and full penetrance.
Nut3 treatment in vivo
Female FVB mice transplanted with LTR-Ctrl or LTR-MycER ErbB2-tumor cells were treated with Nut3 (Sigma, 20 mg/kg body weight) or DMSO (both diluted 1:1 with PBS) by i.p. injections once every 2 days for 2 weeks, upon tumor appearance (tumor volume < 650 mm3). Mice were sacrificed at the end of the treatment, and tumor volumes calculated [V = (length × Widthˆ2)/2] to evaluate Nut3 effect on tumor growth.
Primary mouse mammary epithelial cell culture
Mammary glands were collected, cells isolated, and mammosphere cultures established as described previously (Cicalese et al., 2009). For organoid self-renewal assays, isolated MaProgs were cultured for 7 days in adhesion on collagen-coated plates (Corning) and, then, seeded at a density of 10,000 cells/well in 6-well ultralow attachment plates (Corning) in 5% matrigel (Corning) mammary colony medium (Panciera et al., 2016). Colonies were counted at the end of each passage (14 days), recovered in ice-cold HBSS, incubated with 400 μL Accutase (Sigma) for 5 minutes at 37°C and mechanically dissociated to obtain single-cell suspensions for serial replating.
Primary ErbB2-tumor cells were occasionally kept in short-term cultures in adhesion using DMEM/F12 (1:1, Lonza/GIBCO) supplemented with 10% FBS (HyClone, GE Healthcare Life Science), 10 mM HEPES (Sigma), 5 μg/mL insulin (Roche), 0.5 μg/mL hydrocortisone (Sigma), 10 ng/mL epidermal growth factor (EGF, Tebu-Bio), and 10 ng/mL cholera toxin (Sigma). In vitro treatments with Nut3 (Sigma, N6287), Adriamycin (Sigma, D1515) and 4-OHT (Sigma, H7904) were performed as indicated in the corresponding Results sections and Figures.
Method Details
Mammosphere growth curves
For the modeling of mammosphere growth curves, primary mammospheres were dissociated mechanically and re-plated (at 20,000 cells/mL) to obtain secondary mammospheres in 6-well low-adhesion plates coated with poly-HEMA (Sigma, P3932). After 7 days, the newly formed mammospheres were counted, collected, and manually dissociated by pipetting. At each passage, the number of retrieved mammospheres reflects the number of mammosphere-initiating cells present in the original culture, and the number of cells counted after dissociation allows the evaluation of the number of cells per sphere formed. Cumulative sphere and cell curves were calculated based on the ratio between plated spheres and obtained spheres and cells, respectively. The number of plated spheres was derived from the total number of cells divided by the size of the mammospheres (number of cells/number of spheres) over the passages, assuming no changes in the average mammosphere size in a culture (Cicalese et al., 2009).
Cell Cycle Analysis
Clik-iT Plus EdU kit for flow cytometry assay was purchased from Life Technologies (Thermo Fisher, C10424). WT FVB and ErbB2-tumor mammospheres were labeled each day of culture (from day 1 to day 5) for 1.5 h and stained according to the manufacturer’s protocol. Cells were then fixed in ethanol 100% for 1 h on ice and stained with propidum iodide (PI) solution (final 2.5 μg/mL). Cells were then incubated with RNaseA (final concentration 0.25 mg/mL) at 4°C for a minimum of 3 h; the fluorescence signal was acquired on a FACS Canto II (BD Bioscience) and files analyzed with the FlowJo 9.3-2 analysis software.
PKH26 based label retaining assay
Primary heterozygous Rosa26-MycER, WT and GFP transgenic FVB mammary cells were resuspended at the concentration of 10 million cells/mL and stained for 5 minutes at room temperature by adding an equal volume of a PKH26 mix (1:2500 PKH26 in PBS) (Sigma, PKH26GL), light protected. The cells were then washed twice with culture medium and plated to obtain primary mammospheres. PKH-labeled mammospheres were collected after 7 days, and mechanically dissociated to obtain single cell suspensions. After a filtering step with a 40 μm cell strainer, cells were subjected to FACS sorting (Influx cell sorter equipped with a 488 nm laser and with a band pass 575/26 nm optical filter for PKH26 fluorescence detection, BD). PKHhigh cells were sorted as the brightest 1.5% population. The gate for the PKH− population was selected according to the basal fluorescence of unstained cells and usually included the 25% of live cells. The obtained PKH− cells were cultured as mammospheres (plated for growth curves in the presence or absence of 4-OHT or infected with lentiviral vectors) and used in transplantation assays.
FACS of epithelial cell sub-populations
Single cells isolated from the mammary tissue were mixed with digested organoids. Organoids were derived from the collection of aggregates that did not pass through each of the cell strainers used. This material was further digested with trypsin/EDTA (Lonza), dispase (5 U/mL, Stem Cell Technologies) and DNase (1 mg/mL, Stem Cell Technologies). Inactivation of the enzymes was performed with ice-cold PBS supplemented with 2% FBS. The cell suspension of single cells and digested organoids was blocked in BSA 10% and then stained for mammary stem and progenitor cell markers as listed below:
-
•
Lineage cocktail (Lin-): anti-CD45 (eBioscience, clone 30-F11); anti-Ter119 (eBioscience, clone Ter119); anti-CD31 (eBioscience, clone 390); all PE-Cy7 conjugated (1:300)
-
•
Anti-CD24 (eBioscience, clone M1/69) PE conjugated (1:200)
-
•
Anti-CD49f (eBioscience, clone GoH3) APC conjugated (1:100)
-
•
Anti-CD49f (eBioscience, clone GoH3) eFluor®450 conjugated (1:100)
-
•
Anti-CD61 (eBioscience, clone 2C9.C3) PE conjugated (1:40)
-
•
Anti-EpCAM (eBioscience, clone G8.8) APC conjugated (1:200).
In selected experiments, Lin- cells were isolated by column-based negative selection that was performed through the EasySep Mouse Epithelial Cell Enrichment Kit (Stem Cell Technologies).
Samples were acquired on a FACS Canto II (BD Biosciences) and analyzed with FlowJo 9.3-2 (or later versions). Cell sorting was performed on MoFlo Astrios (Beckman Coulter) or FACSAria Fusion (BD Biosciences).
Viral Infections
293-T and Phoenix-ECO packaging cells were cultured in DMEM supplemented with 10% FBS, 2 mM glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin. For lentiviral production, 293-T cells were transfected following the calcium-phosphate procedure with a mixture of: 2.5 μg of pRSV (Rev), 5 μg of pMDL/pRRE (Gag and Pol), 3 μg of pENV (VSV-G), and 10 μg of the lentiviral vector per plate. The same procedure was applied for retroviral production in Phoenix-ECO cells, which were transfected with: 5 μg of PKAT2, 10 μg of retroviral vector. 62.5 μL of 2M CaCl2 were added to the DNA mix and brought to a total volume of 500 μL with water. The mix was added drop-wise to 500 μL of bubbling 2X HBS (HEPES buffered saline: 250 mM HEPES pH 7.0, 250 mM NaCl and 150 mM Na2HPO4). After 15 minutes of incubation, the precipitate was distributed on exponentially growing (∼70% confluent) cells. The medium was replaced 12-16 h later with mammosphere medium or mammary gland (MG) medium (Panciera et al., 2016) deprived of EGF and FGF. Viral supernatant was collected 24 and 48 h later and filtered through a 0.45 μm syringe-filter.
Cells from dissociated primary mammospheres were resuspended in the corresponding viral supernatants supplemented with growth factors and Polybrene (4 μg/mL; Sigma, H9268) to a final concentration of 50,000 cell/mL and subjected to 2 cycles of infection in suspension (overnight and 6 h). Then, the cells were plated in fresh mammosphere medium to obtain secondary mammospheres. Isolated MaProgs, instead, were infected in adhesion on collagen-coated 6-well plates (LUM: 100,000 cells/well; DN: 50,000 cells/well), using MG viral supernatants supplemented with growth factors and Polybrene (5 μg/mL). At the end of a single round of overnight incubation, viral supernatants were replaced by fresh MG media and the infected cells cultured for 7 days prior to serial replating in 5% matrigel mammary colony medium for organoid formation and self-renewal assays.
In selected experiments, the viral supernatant was concentrated by ultra-centrifugation at 20,000 rpm for 2 h at 4°C, and the viral pellet obtained was resuspended in PBS at 1000X concentration. The viral stock was frozen (−80°C) and subsequently used to infect target cells in order to achieve high multiplicity of infection (MOI). The concentrated virus was employed for the infection of ErbB2 primary tumor cells in adhesion, prior to the injection in syngenic recipient mice. This alternative protocol allows shorter culture periods than the mammosphere assay.
The infection efficiency with LTR-Ctrl and LTR-MycER lentiviruses was generally high (∼80%) and MycER expressing cells exhibited a selective advantage upon passaging in vitro. For the in vivo experiment with ErbB2 cells shown in Figure 5D, the infection efficiency was lower (∼60% for LTR-Ctrl and ∼30% for LTR-MycER). Infected cells, however, were sorted (as GFPpos) prior to transplantation. Finally, for the experiments with Omomyc and corresponding control vector (Figures 5A and S5C-D) (Annibali et al., 2014) or pBABE-p53ER (Figure S6B) (Vater et al., 1996), puromycin selection was applied for one passage prior to the induction with doxycycline (0.5 μΜ; Sigma, D9891) or 4-OHT (200 nM; Sigma, H7904), accordingly.
Modality of SC mitotic division
Doublets in 20% methylcellulose were fixed in 2% formaldehyde and transferred onto poly-D-lysine coated glass slides (Corning). To visualize dividing cells prior to cytokinesis we treated PKHhigh cells from LTR-Ctrl and LTR-MycER with 25 μM blebbistatin for 36 h (Straight et al., 2003). Immunofluorescence was performed as described below. Confocal microscopy was performed on a Leica TCS SP2 microscope equipped with a 63X oil-immersion objective lens (HCX Plan-Apochromat 63X NA 1.4 Lbd Bl; Leica).
Immunofluorescence
Fixed single cells from dissociated mammospheres were permeabilized for 10 minutes with 0.1% Triton X-100 in PBS at room temperature, blocked with donkey serum and stained with specific primary and secondary antibodies. Samples were analyzed under an UpRight BX61 (Olympus) fluorescence microscope with a 60X/1.35 oil objective (Olympus) and images were acquired through MetaMorph® Microscopy Automation & Image Analysis Software (Molecular Devices).
Primary antibodies used were: a rabbit monoclonal c-Myc antibody (1:250 in blocking solution, clone Y69, ab32072 Abcam); a rat monoclonal anti-CD49f (1:250 in blocking solution, clone GoH3, #555734, BD); a monoclonal mouse anti-Numb antibody (clone AB21) (Colaluca et al., 2008). Fluorochrome-conjugated secondary antibodies were obtained from Jackson ImmunoResearch Laboratories.
Immunohistochemistry
For the preparation of paraffin-embedded sections: hydrated whole-mounted tissues were sequentially treated for 1 h at room temperature with 70%, 80%, 95% ethanol, 3 times with 100% ethanol, twice with xylene and twice for 2 h at 58°C with paraffin. The specimens were then embedded in paraffin and sectioned with a microtome at 5 μm thickness. Slides were stained with hematoxylin-eosin for histological analysis or stained by immunohistochemistry. Paraffin sections were deparaffinized with histolemon (Carlo Erba) for 10 minutes (twice) and hydrated through graded alcohol series (100%, 95%, 70% ethanol and water) for 5 minutes each. Antigen unmasking was performed in boiling citrate buffer (10 mM sodium citrate, 0.05% Tween20, pH 6.0) for 30 to 50 minutes, followed by incubation with 3% hydrogen peroxide in distilled water for 10 minutes at room temperature. Slides were subsequently pre-incubated with an antibody buffer (2% BSA, 5% FBS, 0.02% Tween20 in TBS) for 20 minutes at room temperature and then stained with primary antibody overnight at 4°C. After 2 washes with TBS, the slides were incubated with a secondary antibody (DAKO EnVision system HRP, rabbit or mouse) for 30 minutes at room temperature and washed twice again in TBS. The sections were subsequently incubated in a peroxidase substrate solution (DAB DAKO) for 2 to 10 minutes, rinsed in water, counterstained with hematoxylin for 30 s, dehydrated through graded alcohol series (water and 70%, 95%, 100% ethanol) for 5 minutes each, and ultimately mounted with Eukitt (Kindler GmbH). Primary antibodies used: rabbit anti-Ki67 (Thermo Scientific; RM-9106-S; 1:200), rabbit anti-Cytokeratin 14 (Covance; PRB-155P; 1:500), mouse anti-Cytokeratin 18 (Progen; 61028; 1:20), rabbit anti-β-casein (Santa Cruz Biotech.; sc30042; 1:500) and rabbit anti-GFP (Abcam; ab6556-25; 1:500).
Quantitative PCR
RNA from mammospheres was extracted with Maxwell® 16 LEV simplyRNA cells kit (Promega) and reverse transcribed using random primers and ImProm-II reverse transcriptase (Promega), following manufacturer’s instructions. RT-PCR analyses were done on the Applied Biosystems 7500 Fast Real-Time PCR System with the fast-SYBR Green PCR kit (Applied Biosystems). The amount of each mRNA was normalized to the amount of GusB mRNA. Primers used for each gene are listed in the Key Resources Table.
Western Blot Analysis
Protein extracts from fresh mammary tissues or dissociated mammospheres (100,000 to 500,000 cells) were prepared in RIPA buffer (Tris-HCl 50 mM; NaCl 150 mM; 1% NP-40; EDTA 1 mM; 0.5% Sodium Deoxycholate; 0.1% SDS) supplemented with protease inhibitors (Roche). Proteins were quantified with the use of the DC Protein Assay (Biorad) in a 96-well format and the absorbance was measured at 750 nm with the GloMax® 96 Microplate Luminometer (Promega). SDS-PAGE was performed using the NuPage® Novex® Gel System apparatus (Invitrogen) at a constant current of 120 V for approximately 2 h. Samples were loaded on precast Nupage Novex 4%–12% Bis-Tris gels (Invitrogen) and the 1X NuPAGE® MOPS SDS was used as running buffer (Invitrogen). Following SDS-PAGE electrophoresis, proteins were transferred to nitrocellulose membranes (Protran; Schleicher & Schuell) by electroblotting for 1.5 h at 100 V and were then stained with Ponceau S to verify the efficiency of the transfer. Membranes were blocked for 1 h in blocking solution: 10% low fat milk in TBS-T (Tris Buffered Saline, 0.1% Tween 20) for all the antibodies used, except for anti-phospho-S15 p53 which was blocked with 5% BSA (Bovine serum albumin). All primary antibodies were used at a dilution of 1:500, except for the loading control (anti-Vinculin, 1:10,000). The membranes were washed 3 times in TBS-T (10 minutes each) and incubated with a secondary antibody linked to horseradish peroxidase for 1 h at room temperature. After 3 washes in TBS-T, the proteins were visualized using enhanced Clarity Western ECL Blotting Substrate (Biorad) and the ChemiDoc MP System (Biorad). Band densities were quantified by ImageJ software.
ChIP and ChIP-seq
ChIP and ChIP-seq experiments were performed as previously described (Tonelli et al., 2015) using anti-p53 (Rabbit Polyclonal, Novocastra NCL-L-p53-CM5p) and anti-c-Myc (Rabbit Polyclonal N-262, Santa Cruz Biotechnology sc-764) antibodies.
Multiplexed ChIP-seq libraries were sequenced on Illumina HiSeq2000. Fastq files of 50 bps single-end ChIP sequences were aligned to the murine reference genome (mm10) using bwa (version 0.6.2-r126) (Li and Durbin, 2010) with default setting. Bam files were processed by removing reads that: 1) did not align to the reference genome, 2) aligned to mitochondrial genome and Y chromosome, 3) did not pass quality controls, or 4) were duplicates (assumed to represent PCR artifactual events). The last step was done using samtools rmdup tools (version 0.1.18) (Li et al., 2009). Peak calling MACS (version 1.4) (Zhang et al., 2008) with default parameters (p ≤ 1e-5) was used to identify enriched regions of ChIP-seq binding using the input bam files as background. The peaks called for the 2 replicates for each experimental condition were then merged in a single dataset. Each set of bound regions is then annotated by the association with the most proximal gene, considered as the most probable target, with the GIN tool (Cesaroni et al., 2008) using the UCSC RefSeq table (http://hgdownload.cse.ucsc.edu/goldenPath/mm10/database/refGene.txt.gz).
RNA-seq and transcriptional profiling
Libraries of template RNA molecules were prepared from 0.5 to 2 μg high quality input RNA using the Illumina® TruSeq® RNA Sample Preparation Kit v2, following manufacturer’s instructions.
All libraries were sequenced at 50bp pair-end reads on an Illumina HiSeq200. Sequences were aligned with Tophat2 (Kim et al., 2013) against the Mus musculus genome (UCSC, release mm10) and the RefSeq transcriptome references. Raw read counts were summarized using HTSeq (v0.5.3p9) (Anders et al., 2015) and then analyzed for differential expression (DE) with the R package DESeq2 (Anders and Huber, 2010) at gene level. For each contrast examined, we considered significantly regulated those genes that have a baseMean value > 10 and that show a differential expression (reported as log2 of fold change) associated to an adjusted p-value (q-value) ≤ 0.05.
Gene Set Enrichment Analysis (GSEA) was performed on GSEA v2.2.0 platform (https://www.broadinstitute.org/gsea/). To determine whether the MycER gene list (Myc-DEGs; n = 7,052) showed statistically significant concordant and/or discordant differences to the gene sets of p53-DEGs (n = 6,745) and the tumorigenic ErbB2-DEGs (n = 3,596) or Nut3-DEGs (n = 1,922) we used the pre-ranked tool. P-values were calculated by performing 1,000 random permutations of gene labels to create the ES (enriched score) null distribution. The normalized enrichment score (NES) was calculated considering separately up- and downregulated gene lists.
Pathway analyses were performed by challenging our gene set of 1,494 ErbB2 p53-Myc DEGS with the Molecular signature database (MSigDB v5.0) curated gene set C2 on the same GSEA platform, again considering separately up- and downregulated gene lists.
Hierarchical clustering and heatmaps were generated with different R packages using ClustVis (Metsalu and Vilo, 2015). Genes were clustered by Euclidean distance and average linkage, using relative expression values of the PKHhigh, PKH−-LTR-MycER and bulk LTR-MycER samples to the PKH− cells, which served as reference.
Quantification and Statistical Analysis
Summarized data are presented as mean ± standard deviation (SD). Statistical comparisons were carried out using two-tailed unpaired Student’s t tests, unless otherwise specified. The number of biological (n) or technical replicates, the type of statistical analyses performed and statistical significance are reported in the corresponding Figures and Figure legends.
Limiting dilution transplantation
Limiting dilution analysis was performed using the Extreme Limiting Dilution Analysis (ELDA) web tool (http://bioinf.wehi.edu.au/software/elda/) (Hu and Smyth, 2009). ELDA computes a 95% confidence interval for the active cell frequency in each population group and it implements a likelihood ratio test for the acceptance of the single-hit hypothesis (p-value). Slope: log-active cell fraction (equal to 1 when the single-hit hypothesis is confirmed). Fit: the single hit hypothesis is rejected when p ≤ 0.05.
Mammosphere and organoid self-renewal assays
Spheres and organoids were counted at the end of each passage using ImageJ (Schneider et al., 2012) and object threshold 100 μm. Cumulative sphere and cell curves were calculated as described in Method Details. Cumulative curves were plotted in a semi-logarithmic scale and approximated an exponential curve. Growth rates (GRs) were evaluated as the slope of the trend-line of the exponential curves. Data exponential regression resulted in the value of the coefficients of determination (R2). Statistical comparisons between growth curves shown in Figure 5B and 5C were performed by two-tailed paired t tests on the GR values calculated for each of the 4 independent experiments.
Signature validation on publicly available datasets
TCGA data pre-process
Breast cancer RNA-seq data were downloaded from cBioPortal database (Cerami et al., 2012, Gao et al., 2013) (http://www.cbioportal.org/), which includes data from the 2015 TCGA freeze on 1092 patients. Expression values are reported as normalized RSEM Z-score. Original gene annotation for the MitSig was ported from our mouse data to human homologous genes for 179 out of 189 genes using the Mouse Gene Database (MGI) available at http://www.informatics.jax.org/homology.shtml (Blake et al., 2017). Clinical data, including molecular subtype and estrogen receptor status, were downloaded from the Broad GDAC Firehose (https://gdac.broadinstitute.org/) and matched by barcode to the RNA-seq data [https://doi.org/10.7908/C11G0KM9].
GEO Affymetrix Array data pre-process
To perform the survival analysis, we took advantage of four different studies based on gene expression Affymetrix Array U133A data from 2005-2007 (GEO: GSE1456, GSE2034, GSE4922, GSE7390) for a total of 932 patients (reduced to 892 with matched clinical information). Molecular data were downloaded in the original CEL format, normalized with RMA and quantile normalization (Irizarry et al., 2003) and reported as log2 probe intensity. The annotation was performed using custom cdf file from brain array version 21 (https://mbni.org/www/) (Dai et al., 2005) to obtain a single match between the probe and the gene name (∼12,000 genes). Given the large number of samples, the aroma R package was used for the normalization step (Bengtsson et al., 2008). Out of the original 189 MitSig genes translated in 179 human genes, 161 were found in this array type.
Clinical information pre-process
The four studies reported various types of information regarding their participants (892 in total). With the exception of disease-free survival, none of the clinical data was available for all the four studies (see Table S4). Disease-free survival is reported as Status_Relapse: 0, censored, 1 any event including relapse, metastasis or death and Time_Relapse: number or fraction of months from entering the study until an event or censorship occurs. Estrogen receptor status was available for 3 out 4 of the studies and it was evinced from the molecular subtype for GEO: GSE1456 (Luminal tumors: receptor positive, Other: negative). Since each of the four clinical datasets reported information on different clinical variables, we could not perform a multivariate analysis. Thus, we carried out a Cox multiple regression analysis, including ER status, tumor grade, lymph node status, p53 status, age, and tumor size as possible covariates. The final number of samples retained was reduced to 885 by removing subjects with time-to-event equal to 0 and all the subjects with censorship or event over mean + 2 SD were censored at that time (considered outliers after ∼178 months to respect proportional hazards assumptions).
Statistical Analysis of TCGA and GEO data
Both TCGA and GEO matrices were divided into 2 groups (UP and DOWN) using hierarchical clustering with Euclidean distance and Ward’s linkage criterion. Non-parametric Dunnet’s Test was used to assess difference in the distribution of genetic characteristics and subtype between UP and DOWN MitSig groups in TCGA data. Survival data from the GEO datasets were analyzed with two standard techniques. In case of 2 categories (e.g., UP versus DOWN) Kaplan-Meier curves were drawn and Logrank test was used. For multivariate models, Cox Proportional Hazard Models were used and Shoenfeld residual tests were performed to check for proportional hazards assumptions.
Data and Software Availability
The accession number for the ChIP-seq and RNA-seq datasets reported in this paper is GEO: GSE87004.
Acknowledgments
We thank P. Dalton and S. Averaimo for editing the manuscript, M. Varasi and C. Mercurio for providing Nut3, B. Gallo for technical assistance, S. Ronzoni and J. Quarna for FACS, M. Garrè for confocal imaging, L. Giacò for bioinformatics support, and C. Recordati for histopathological analysis. We thank P.J. Park for supporting G.M. in the bioinformatics analysis. This work was funded by grants from WWCR, AIRC, ERC, and the Italian Ministry of Health to P.G.P. A. Santoro was supported by an FUV grant and T.V. by an FIRC grant. B.A. was funded by AIRC and the Italian Ministry of Health. F.N., A. Sabò, and L.C. were funded by AIRC.
Author contributions
Conceptualization: A. Santoro, T.V., P.G.P., C.E.P., and L.R.; Investigation: A. Santoro, T.V., X.A., C.E.P., L.R., and P.B.; Visualization: A. Santoro and T.V.; Writing: A. Santoro, T.V., and P.G.P.; Software, Formal Analysis, Data Curation: L. Luzi and G.M.; Resources: P.B., S.P., L. Lanfrancone, L.C., A. Sabò, F.N., M.C.M, G.I.D., and B.A.; Technical Assistance: E.D.; Revision: A. Santoro, T.V., L.M., P.G.P., F.N., M.C.M., L. Luzi, and B.A.; Supervision, Funding Acquisition: P.G.P.
Declaration of Interests
The authors declare no competing interests.
Published: January 15, 2019
Supplemental Information
References
- Abraham S.A., Hopcroft L.E., Carrick E., Drotar M.E., Dunn K., Williamson A.J., Korfi K., Baquero P., Park L.E., Scott M.T. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature. 2016;534:341–346. doi: 10.1038/nature18288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ades F., Zardavas D., Bozovic-Spasojevic I., Pugliano L., Fumagalli D., de Azambuja E., Viale G., Sotiriou C., Piccart M. Luminal B breast cancer: molecular characterization, clinical management, and future perspectives. J. Clin. Oncol. 2014;32:2794–2803. doi: 10.1200/JCO.2013.54.1870. [DOI] [PubMed] [Google Scholar]
- Akbani R., Ng P.K.S., Werner H.M.J., Shahmoradgoli M., Zhang F., Ju Z., Liu W., Yang J.Y., Yoshihara K., Li J. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat. Commun. 2014;5:3887. doi: 10.1038/ncomms4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annibali D., Whitfield J.R., Favuzzi E., Jauset T., Serrano E., Cuartas I., Redondo-Campos S., Folch G., Gonzàlez-Juncà A., Sodir N.M. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat. Commun. 2014;5:4632. doi: 10.1038/ncomms5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai T., Liu Y., Bae N., Nimer S.D. The p53 tumor suppressor protein regulates hematopoietic stem cell fate. J. Cell. Physiol. 2011;226:2215–2221. doi: 10.1002/jcp.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Porath I., Thomson M.W., Carey V.J., Ge R., Bell G.W., Regev A., Weinberg R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson, H., Simpson, K., Bullard, J., and Hansen, K. 2008. aroma.affymetrix: A Generic Framework in R for Analyzing Small to Very Large Affymetrix Data Sets in Bounded Memory. Tech Report 745, Department of Statistics, University of California (Berkeley)., https://statistics.berkeley.edu/sites/default/files/tech-reports/745.pdf.
- Bieging K.T., Mello S.S., Attardi L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer. 2014;14:359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake J.A., Eppig J.T., Kadin J.A., Richardson J.E., Smith C.L., Bult C.J., the Mouse Genome Database Group Mouse Genome Database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic Acids Res. 2017;45:D723–D729. doi: 10.1093/nar/gkw1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady C.A., Jiang D., Mello S.S., Johnson T.M., Jarvis L.A., Kozak M.M., Kenzelmann Broz D., Basak S., Park E.J., McLaughlin M.E. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasó-Maristany F., Filosto S., Catchpole S., Marlow R., Quist J., Francesch-Domenech E., Plumb D.A., Zakka L., Gazinska P., Liccardi G. PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat. Med. 2016;22:1303–1313. doi: 10.1038/nm.4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesaroni M., Cittaro D., Brozzi A., Pelicci P.G., Luzi L. CARPET: a web-based package for the analysis of ChIP-chip and expression tiling data. Bioinformatics. 2008;24:2918–2920. doi: 10.1093/bioinformatics/btn542. [DOI] [PubMed] [Google Scholar]
- Cheang M.C., Chia S.K., Voduc D., Gao D., Leung S., Snider J., Watson M., Davies S., Bernard P.S., Parker J.S. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst. 2009;101:736–750. doi: 10.1093/jnci/djp082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Ellison F.M., Keyvanfar K., Omokaro S.O., Desierto M.J., Eckhaus M.A., Young N.S. Enrichment of hematopoietic stem cells with SLAM and LSK markers for the detection of hematopoietic stem cell function in normal and Trp53 null mice. Exp. Hematol. 2008;36:1236–1243. doi: 10.1016/j.exphem.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianfrocca M., Goldstein L.J. Prognostic and predictive factors in early-stage breast cancer. Oncologist. 2004;9:606–616. doi: 10.1634/theoncologist.9-6-606. [DOI] [PubMed] [Google Scholar]
- Cicalese A., Bonizzi G., Pasi C.E., Faretta M., Ronzoni S., Giulini B., Brisken C., Minucci S., Di Fiore P.P., Pelicci P.G. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–1095. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- Colaluca I.N., Tosoni D., Nuciforo P., Senic-Matuglia F., Galimberti V., Viale G., Pece S., Di Fiore P.P. NUMB controls p53 tumour suppressor activity. Nature. 2008;451:76–80. doi: 10.1038/nature06412. [DOI] [PubMed] [Google Scholar]
- D’Alesio C., Punzi S., Cicalese A., Fornasari L., Furia L., Riva L., Carugo A., Curigliano G., Criscitiello C., Pruneri G. RNAi screens identify CHD4 as an essential gene in breast cancer growth. Oncotarget. 2016;7:80901–80915. doi: 10.18632/oncotarget.12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M., Wang P., Boyd A.D., Kostov G., Athey B., Jones E.G., Bunney W.E., Myers R.M., Speed T.P., Akil H. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deome K.B., Faulkin L.J., Jr., Bern H.A., Blair P.B. Development of mammary tumors from hyperplastic alveolar nodules transplanted into gland-free mammary fat pads of female C3H mice. Cancer Res. 1959;19:515–520. [PubMed] [Google Scholar]
- Desmedt C., Piette F., Loi S., Wang Y., Lallemand F., Haibe-Kains B., Viale G., Delorenzi M., Zhang Y., d’Assignies M.S., TRANSBIG Consortium Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin. Cancer Res. 2007;13:3207–3214. doi: 10.1158/1078-0432.CCR-06-2765. [DOI] [PubMed] [Google Scholar]
- Fan C., Oh D.S., Wessels L., Weigelt B., Nuyten D.S., Nobel A.B., van’t Veer L.J., Perou C.M. Concordance among gene-expression-based predictors for breast cancer. N. Engl. J. Med. 2006;355:560–569. doi: 10.1056/NEJMoa052933. [DOI] [PubMed] [Google Scholar]
- Friedmann-Morvinski D., Bushong E.A., Ke E., Soda Y., Marumoto T., Singer O., Ellisman M.H., Verma I.M. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338:1080–1084. doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendoo D.M., Ratanasirigulchai N., Schröder M.S., Paré L., Parker J.S., Prat A., Haibe-Kains B. Genefu: an R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics. 2016;32:1097–1099. doi: 10.1093/bioinformatics/btv693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godar S., Ince T.A., Bell G.W., Feldser D., Donaher J.L., Bergh J., Liu A., Miu K., Watnick R.S., Reinhardt F. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjantonakis A.K., Gertsenstein M., Ikawa M., Okabe M., Nagy A. Generating green fluorescent mice by germline transmission of green fluorescent ES cells. Mech. Dev. 1998;76:79–90. doi: 10.1016/s0925-4773(98)00093-8. [DOI] [PubMed] [Google Scholar]
- Ho J.S., Ma W., Mao D.Y., Benchimol S. p53-Dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol. Cell. Biol. 2005;25:7423–7431. doi: 10.1128/MCB.25.17.7423-7431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Smyth G.K. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U., Speed T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Ivshina A.V., George J., Senko O., Mow B., Putti T.C., Smeds J., Lindahl T., Pawitan Y., Hall P., Nordgren H. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006;66:10292–10301. doi: 10.1158/0008-5472.CAN-05-4414. [DOI] [PubMed] [Google Scholar]
- Kendrick H., Regan J.L., Magnay F.A., Grigoriadis A., Mitsopoulos C., Zvelebil M., Smalley M.J. Transcriptome analysis of mammary epithelial subpopulations identifies novel determinants of lineage commitment and cell fate. BMC Genomics. 2008;9:591. doi: 10.1186/1471-2164-9-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerosuo L., Piltti K., Fox H., Angers-Loustau A., Häyry V., Eilers M., Sariola H., Wartiovaara K. Myc increases self-renewal in neural progenitor cells through Miz-1. J. Cell Sci. 2008;121:3941–3950. doi: 10.1242/jcs.024802. [DOI] [PubMed] [Google Scholar]
- Kim J., Woo A.J., Chu J., Snow J.W., Fujiwara Y., Kim C.G., Cantor A.B., Orkin S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143:313–324. doi: 10.1016/j.cell.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse J.P., Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurenti E., Wilson A., Trumpp A. Myc’s other life: stem cells and beyond. Curr. Opin. Cell Biol. 2009;21:844–854. doi: 10.1016/j.ceb.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., He Y., Dubois W., Wu X., Shi J., Huang J. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol. Cell. 2012;46:30–42. doi: 10.1016/j.molcel.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Kon N., Jiang L., Tan M., Ludwig T., Zhao Y., Baer R., Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim E., Wu D., Pal B., Bouras T., Asselin-Labat M.L., Vaillant F., Yagita H., Lindeman G.J., Smyth G.K., Visvader J.E. Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways. Breast Cancer Res. 2010;12:R21. doi: 10.1186/bcr2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Elf S.E., Miyata Y., Sashida G., Liu Y., Huang G., Di Giandomenico S., Lee J.M., Deblasio A., Menendez S. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins C.P., Brown-Swigart L., Evan G.I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–1334. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Meletis K., Wirta V., Hede S.M., Nistér M., Lundeberg J., Frisén J. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133:363–369. doi: 10.1242/dev.02208. [DOI] [PubMed] [Google Scholar]
- Metsalu T., Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015;43:W566–W570. doi: 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller F.R., Santner S.J., Tait L., Dawson P.J. MCF10DCIS.com xenograft model of human comedo ductal carcinoma in situ. J. Natl. Cancer Inst. 2000;92:1185–1186. doi: 10.1093/jnci/92.14.1185a. [DOI] [PubMed] [Google Scholar]
- Miller L.D., Smeds J., George J., Vega V.B., Vergara L., Ploner A., Pawitan Y., Hall P., Klaar S., Liu E.T., Bergh J. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc. Natl. Acad. Sci. USA. 2005;102:13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H., Spike B.T., Wahl G.M., Levine A.J. Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc. Natl. Acad. Sci. USA. 2010;107:22745–22750. doi: 10.1073/pnas.1017001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W.J., Sinn E., Pattengale P.K., Wallace R., Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54:105–115. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- Murphy D.J., Junttila M.R., Pouyet L., Karnezis A., Shchors K., Bui D.A., Brown-Swigart L., Johnson L., Evan G.I. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell. 2008;14:447–457. doi: 10.1016/j.ccr.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panciera T., Azzolin L., Fujimura A., Di Biagio D., Frasson C., Bresolin S., Soligo S., Basso G., Bicciato S., Rosato A. Induction of Expandable Tissue-Specific Stem/Progenitor Cells through Transient Expression of YAP/TAZ. Cell Stem Cell. 2016;19:725–737. doi: 10.1016/j.stem.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E.Y., Chang E., Lee E.J., Lee H.W., Kang H.G., Chun K.H., Woo Y.M., Kong H.K., Ko J.Y., Suzuki H. Targeting of miR34a-NOTCH1 axis reduced breast cancer stemness and chemoresistance. Cancer Res. 2014;74:7573–7582. doi: 10.1158/0008-5472.CAN-14-1140. [DOI] [PubMed] [Google Scholar]
- Parker J.S., Mullins M., Cheang M.C., Leung S., Voduc D., Vickery T., Davies S., Fauron C., He X., Hu Z. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasi C.E., Dereli-Öz A., Negrini S., Friedli M., Fragola G., Lombardo A., Van Houwe G., Naldini L., Casola S., Testa G. Genomic instability in induced stem cells. Cell Death Differ. 2011;18:745–753. doi: 10.1038/cdd.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawitan Y., Bjöhle J., Amler L., Borg A.L., Egyhazi S., Hall P., Han X., Holmberg L., Huang F., Klaar S. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005;7:R953–R964. doi: 10.1186/bcr1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pece S., Tosoni D., Confalonieri S., Mazzarol G., Vecchi M., Ronzoni S., Bernard L., Viale G., Pelicci P.G., Di Fiore P.P. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Poli V., Fagnocchi L., Fasciani A., Cherubini A., Mazzoleni S., Ferrillo S., Miluzio A., Gaudioso G., Vaira V., Turdo A. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat. Commun. 2018;9:1024. doi: 10.1038/s41467-018-03264-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinson D.A., Dillon C.P., Kwiatkowski A.V., Sievers C., Yang L., Kopinja J., Rooney D.L., Zhang M., Ihrig M.M., McManus M.T. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat. Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- Sachdeva M., Zhu S., Wu F., Wu H., Walia V., Kumar S., Elble R., Watabe K., Mo Y.Y. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc. Natl. Acad. Sci. USA. 2009;106:3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackleton M., Vaillant F., Simpson K.J., Stingl J., Smyth G.K., Asselin-Labat M.-L., Wu L., Lindeman G.J., Visvader J.E. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- Soady K.J., Kendrick H., Gao Q., Tutt A., Zvelebil M., Ordonez L.D., Quist J., Tan D.W.-M., Isacke C.M., Grigoriadis A., Smalley M.J. Mouse mammary stem cells express prognostic markers for triple-negative breast cancer. Breast Cancer Res. 2015;17:31. doi: 10.1186/s13058-015-0539-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiriou C., Pusztai L. Gene-expression signatures in breast cancer. N. Engl. J. Med. 2009;360:790–800. doi: 10.1056/NEJMra0801289. [DOI] [PubMed] [Google Scholar]
- Soucek L., Whitfield J.R., Sodir N.M., Massó-Vallés D., Serrano E., Karnezis A.N., Swigart L.B., Evan G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27:504–513. doi: 10.1101/gad.205542.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soussi T., Wiman K.G. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell. 2007;12:303–312. doi: 10.1016/j.ccr.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Stingl J., Eirew P., Ricketson I., Shackleton M., Vaillant F., Choi D., Li H.I., Eaves C.J. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- Straight A.F., Cheung A., Limouze J., Chen I., Westwood N.J., Sellers J.R., Mitchison T.J. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Termén S., Tan E.J., Heldin C.H., Moustakas A. p53 regulates epithelial-mesenchymal transition induced by transforming growth factor β. J. Cell. Physiol. 2013;228:801–813. doi: 10.1002/jcp.24229. [DOI] [PubMed] [Google Scholar]
- Tonelli C., Morelli M.J., Bianchi S., Rotta L., Capra T., Sabò A., Campaner S., Amati B. Genome-wide analysis of p53 transcriptional programs in B cells upon exposure to genotoxic stress in vivo. Oncotarget. 2015;6:24611–24626. doi: 10.18632/oncotarget.5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosoni D., Zecchini S., Coazzoli M., Colaluca I., Mazzarol G., Rubio A., Caccia M., Villa E., Zilian O., Di Fiore P.P., Pece S. The Numb/p53 circuitry couples replicative self-renewal and tumor suppression in mammary epithelial cells. J. Cell Biol. 2015;211:845–862. doi: 10.1083/jcb.201505037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschaharganeh D.F., Xue W., Calvisi D.F., Evert M., Michurina T.V., Dow L.E., Banito A., Katz S.F., Kastenhuber E.R., Weissmueller S. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell. 2014;158:579–592. doi: 10.1016/j.cell.2014.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev L.T., Vu B.T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Vater C.A., Bartle L.M., Dionne C.A., Littlewood T.D., Goldmacher V.S. Induction of apoptosis by tamoxifen-activation of a p53-estrogen receptor fusion protein expressed in E1A and T24 H-ras transformed p53-/- mouse embryo fibroblasts. Oncogene. 1996;13:739–748. [PubMed] [Google Scholar]
- Ventura A., Kirsch D.G., McLaughlin M.E., Tuveson D.A., Grimm J., Lintault L., Newman J., Reczek E.E., Weissleder R., Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Vita M., Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 2006;16:318–330. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Vousden K.H., Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Wang Y., Klijn J.G., Zhang Y., Sieuwerts A.M., Look M.P., Yang F., Talantov D., Timmermans M., Meijer-van Gelder M.E., Yu J. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- Watt F.M., Frye M., Benitah S.A. MYC in mammalian epidermis: how can an oncogene stimulate differentiation? Nat. Rev. Cancer. 2008;8:234–242. doi: 10.1038/nrc2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W., Zender L., Miething C., Dickins R.A., Hernando E., Krizhanovsky V., Cordon-Cardo C., Lowe S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.P., Liao Y., Xia W., Zou Y., Spohn B., Hung M.C. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol. 2001;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the ChIP-seq and RNA-seq datasets reported in this paper is GEO: GSE87004.