

Abstract
Transcription is a fundamental cellular process and the first step in gene regulation. Although RNA polymerase is highly processive, in growing cells the progression of transcription can be hindered by obstacles on the DNA template, such as damaged DNA. Our recent findings highlight a trade-off between transcription fidelity and DNA break repair. While a lot of work has focused on the interaction between transcription and nucleotide excision repair, less is known about how transcription influences the repair of DNA breaks. We suggest that when the cell experiences stress from DNA breaks, the control of RNA polymerase processivity affects the balance between preserving transcription integrity and DNA repair. Here we discuss how the conflict between transcription and DNA double-strand break repair threatens the integrity of both RNA and DNA. In reviewing this field, we speculate on cellular paradigms where this equilibrium is well sustained, and instances where the maintenance of transcription fidelity is favored over genome stability.
Keywords: Transcription fidelity, genome stability, DNA break repair, DNA resection, RecBC
1. Introduction
In growing cells, transcription has to progress along DNA that is simultaneously undergoing other important processes, such as replication and DNA repair. The directed repair of DNA has long been recognized: the transcription-coupled nucleotide excision repair pathway (TC-NER) resolves UV-induced DNA damage and other bulky lesions in actively transcribed regions [1, 2]. Genetic defects that perturb the TC-NER pathway result in Cockayne Syndrome, a debilitating neurodevelopmental disorder[3]. However, bulky and helix-distorting lesions are not the only obstacles that RNA polymerase (RNAP) encounters on the DNA template. Much recent work has highlighted the interaction between RNAP and DNA double-strand breaks (DSB) ([4, 5] and references within). While it appears that RNAP should be released at a DSB without much consequence, as is the case in an in vitro transcription reaction, the effect of a DSB on transcription is not nearly as well defined within a living cell. DSB repair proteins have been found to be recruited more efficiently to transcribed regions[6]. On the other hand, whole genome sequencing methods have demonstrated a link between transcriptional activity itself and the generation of DNA breaks[7]. Though further studying the mechanistic of transcription-coupled DSB repair is important and should be a major focus of future research, one type of transcription-DSB repair conflict is receiving much less attention: what happens when the machineries performing the two processes independently of each other collide on the DNA? In the event that DSB repair proteins encounter an RNAP that is transcribing a nearby gene, how does the cell decide which process continues and which process is aborted? Moreover, in making this decision, how does the cell prioritize the fidelity of transcription over the integrity of DNA?
Extrinsic damaging agents such as ionizing radiation or chemical mutagens, as well as spontaneous incidents such as replication fork collapse, give rise to cellular DSBs[8, 9]. Repair of a DSB is uniquely challenging because both strands of helix being compromised and hence it requires homologous chromosomes or sister chromatids to accurately restore the sequence of the damaged DNA. Improper repair of DSBs can give rise to large-scale chromosomal rearrangements triggering global genomic instability[5, 9]. The devastating nature of DSBs make the decision between repair and transcription more complicated. Hence, we propose that there is a trade-off to preserving RNA in lieu of DNA in all organisms, and there exists a cellular decision that balances the repair of DNA with the fidelity of transcription (Figure 1).
Figure 1.
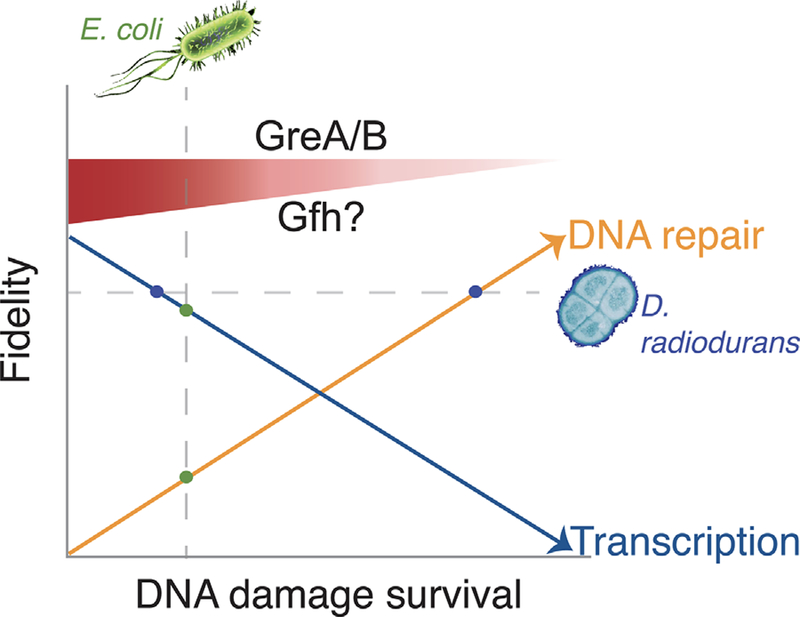
The cellular balance between transcription fidelity and DNA damage survival. GreA/B and possibly the D. radiodurans protein Gfh,[48,49] increase transcription fidelity at the cost of survival during DNA damage stress. In the absence of GreA/B, transcription fidelity is reduced, but survival after DNA damage is greatly improved. Under DNA damage stress, D. radiodurans is predicted to have a balance between maintaining transcription fidelity and performing DNA repair such that both RNA and DNA fidelity is high (blue dots), while in E. coli, the equilibrium is shifted toward transcription fidelity (green dots).
2. Maintaining transcription fidelity restricts DSB repair in Escherichia coli
Our work in E. coli has led to the surprising discovery that the maintenance of transcription fidelity can, at times, obstruct DNA break repair, and hence the preservation of genome stability[10]. In all organisms, misincorporation of incorrect ribonucleotides by RNAP can lead to transcription errors[11–13], which can affect cell function. Additionally, stalling or arrest of RNAP can prevent transcription elongation and hence the production of full-length transcripts[14, 15]. The transcription factors GreA and GreB in E. coli, and their eukaryotic homolog TFIIS maintain transcription fidelity both by reducing misincorporations and promoting transcription elongation[14, 16, 17]. Ribonucleotide misincorporations and physical obstacles on the DNA can cause RNAP to move backwards along the DNA, a process known as backtracking[18, 19]. When RNAP backtracks, the 3’ end of the nascent transcript slides out of the active site into a structure within the RNAP known as the secondary channel[20]. GreA and TFIIS can rescue RNAP backtracking by stimulating the cleavage of the extruded RNA, hence generating a new error-free 3’ end[16, 20]. This resets the transcription complex to allow for productive elongation to resume and RNAP to continue moving forward. Recently, it was demonstrated that UvrD, a protein known to function in the TC-NER pathway, stimulates backtracking in vitro and in vivo[19]. Based on in vitro transcription assays where addition of GreB reversed the effects of UvrD, and in vivo primer extension as well as drug-sensitivity assays in different deletion mutations, it was inferred that UvrD opposes the anti-backtracking function of GreA/B by pulling RNAP backwards at bulky lesions on the DNA[19]. UvrD acts in conjunction with the small molecule guanosine tetraphosphate (ppGpp) to promote RNAP backtracking and facilitate TC-NER[21].
We were interested in understanding how the modulation of transcription elongation would affect the repair of DNA breaks. We discovered that removal of GreA (by complete gene deletion), leading to the increase in backtracked RNAP complexes, improves the repair of DSBs (Figure 2A). Removal of the UvrD and ppGpp gene products, which reduces backtracking in vivo, had the opposite effect on DSB repair as expected since the two factors likely counteract the effects of GreA (Figure 2C). The enhanced kinetics of DSB repair due to loss of GreA and increased backtracking depends on the homology-directed repair pathway (HR). DNA breaks can be repaired by HR or by an alternate pathway called non-homologous end joining (NHEJ)[9, 22]. HR is a high fidelity pathway of DSB repair and unlike NHEJ is conserved across bacteria, archaea and eukaryotes. HR can be broadly divided into three steps: (1) DSB exonuclease degradation or resection of double-stranded ends to be repaired, (2) homologous pairing and strand exchange followed by gap filling by DNA polymerase and (3) resolution of recombination intermediates[9, 22, 23]. Using a sequencing-based method (XO-seq) to determine which of these steps was specifically impacted by the removal of GreA, we found that resection, the primary step of DSB repair, was reduced and propose a model whereby backtracked RNAP acts as a physical obstruction to resection[10]. In E. coli, the switch from DSB resection to the next step of recombination is facilitated by 8 nucleotide DNA sequences known as Chi sites (Figure 3A)[8, 24]. Chi sites modify RecBCD function in two important ways: first, RecBCD pauses and stops exonucleolytic degradation of DNA, and then the RecBCD complex loads the recombinase RecA onto the ssDNA that results from resection (Figure 3A)[23, 24]. Our results suggest that backtracked RNAP formed in the absence of GreA can act like a Chi site, enabling a switch from resection to RecA loading and hence improving DSB repair (Figure 3A). This raises an important and unexpected question: since the loss of GreA equips the cell to contend with DSBs more efficiently, why is the gene evolutionarily retained in the genome of most bacteria? We suggest that there is a trade-off between transcription fidelity and DNA repair. The maintenance of transcription fidelity is key to ensure immediate survival[25], as many essential gene products are present in low copy[26]. On the other hand, although damaged DNA is heritable, a stable proteome can allow for DNA repair using sister copies or chromosomes. Further, Chi sites are the only known factors that mediate the switch from resection to recombination in E. coli. We propose that backtracked RNAP, a conserved transcription conformation across species can also act universally to promote homologous recombination (Figure 3A). Below we provide some context to support our hypotheses.
Figure 2.
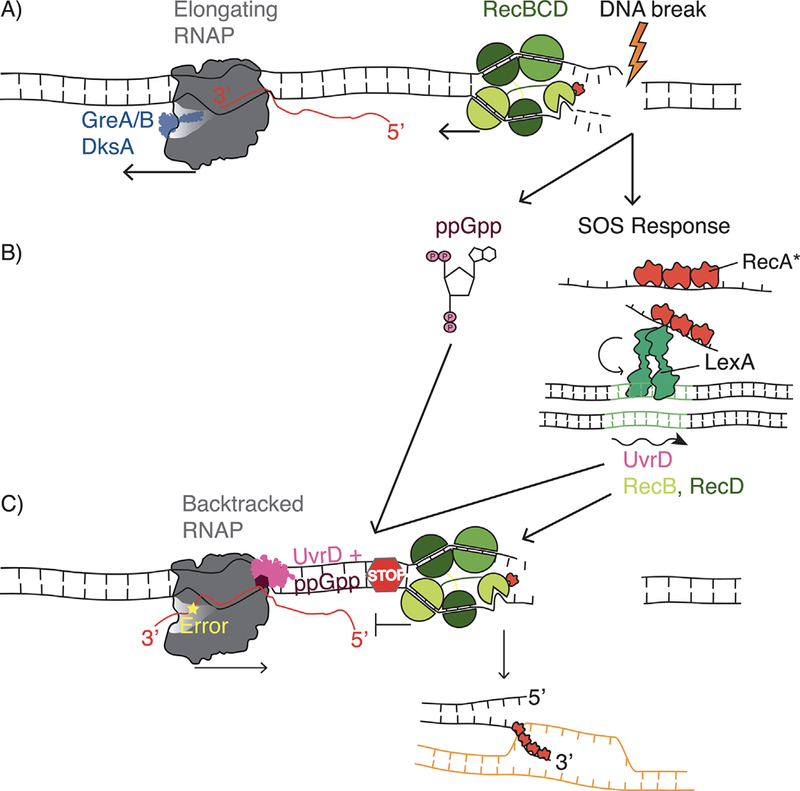
Collisions between backtracked RNA polymerase and RecBCD promote homologous recombination. Transcription by RNA polymerase (RNAP) and resection by RecBCD co-occur on the DNA template. RecBCD can initiate resection at a DNA break independent of ongoing transcription. A) GreA/B and DksA promote transcription elongation and prevent RNAP backtracking, allowing DNA degradation by RecBCD to continue. B) The SOS response in E. coli is activated by DNA breaks. ssDNA coated RecA filaments (RecA%) stimulates the autocatalytic cleavage of the transcriptional repressor LexA. Cleavage of LexA leads to the de-repression of genes in the SOS regulon, including RecBC and UvrD. C) A transcription error (yellow star) or UvrD along with ppGpp promote RNAP backtracking, increasing the chance for RecBCD to encounter backtracked RNAP. Stably bound backtracked RNAP stops RecBCD resection, which facilitates the switch to RecA loading and hence recombination.
Figure 3.
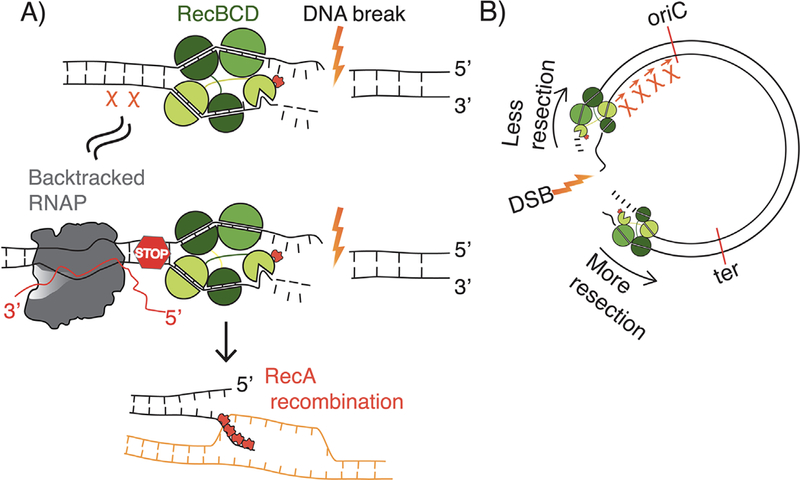
Backtracked RNA polymerase can act like a Chi site to reduce resection and promote recombination. A) Chi distribution across the E. coli genome is biased, with more correctly oriented Chi sites toward the origin (OriC), and fewer close to the terminus of replication (Ter). Because of this bias, resection by RecBCD is lower toward OriC compared to downstream of the DNA break site. B) RNAP can act like a Chi site. When RecBCD encounters a Chi site, the probability of RecBCD pausing increases. This pause results in a switch from exonucleolytic activity of RecBCD to the RecA loading activity. Thus, RecBCD loads RecA to allow for recombination at a Chi site. We hypothesize that backtracked RNAP acts analogously, promoting a RecBCD pause and stimulating recombination.
3. The role and consequence of transcription fidelity factors across species
While E. coli has two Gre factors – GreA and GreB, bacterial species including Bacillus subtilis, Deinococcus radiodurans, Streptococcus sp. and others with small genomes only encode one Gre factor[27]. In E. coli, deletion of both gre genes results in reduced survival at higher temperatures, whereas in bacteria with minimal genomes such as Mycoplasma genetalium and Mycoplasma pneumoniae, the only gre gene, greA, is essential[28, 29]. The synthetic 531 kb minimal genome, encoding the basic set of genes required for cellular existence, contains greA, underscoring the essentiality of this factor and its importance in prokaryotes[30]. The Gre factors belong to a family of proteins that interact with the secondary channel of RNAP. This family also includes DksA, which functions both during transcription initiation and elongation[31, 32]. Additionally, DksA has also shown to reduce misincorporations that lead to transcription errors in vitro and in vivo[33, 34]. DksA is incapable of transcript cleavage and the mechanism through which DksA acts to lower transcription errors appears to be distinct from the Gre factors[33]. While the mechanistic details of DksA’s effects on transcription errors are still being explored, the existence of two separate mechanisms to mediate transcription fidelity underscores its cellular importance. Further, in wild-type E. coli, the cumulative (base substitutions and indels) in vivo transcription error rate/ transcribed nucleotide was found to be 9.94 × 10−5 [35, 36], far higher than the cumulative in vivo mutation rate of 2.2 × 10−10 mutations/ nucleotide/ generation determined in the same strain[37].
The eukaryotic transcript cleavage factor, TFIIS, is functionally similar to Gre in restarting backtracked RNAPII complexes, and in the preservation of transcription fidelity[16, 38]. Hence, transcript cleavage and the maintenance of transcription fidelity are conserved across all kingdoms of life. In Gre and TFIIS mutants, misincorporation events increase by ~2- and ~7-fold over that observed in wild-type strains respectively[12]. Although mRNA is considered to be transient, the consequences of transcription errors produce heritable phenotypes and can contribute to stochastic variability between bacterial cells[39, 40]. Increased transcription error rates have also been associated with human disease and aging. Human genes with poly-A or poly-T tracts that cause transcription slippage are prone to transcription errors that increase with age[41]. Similar age related accumulation of transcription errors has also been demonstrated in yeast cells encoding a low fidelity RNAP[42]. A few cases where disease causation stemmed from transcription errors have also been reported[43]. There are still a lot of unknowns regarding the cellular programs that maintain transcription fidelity that require further exploration. Such investigations could reveal the mechanisms and extent to which RNA errors contribute to disease.
4. Is RNAP processivity modulated during DNA damage stress?
The finding that the absence of GreA improves DSB repair raises questions about the regulation of Gre factor expression during DNA damage. Further, the improvement of repair by increased RNAP backtracking warrants an investigation of the specific control of RNAP processivity after DNA damage stress. One possibility is that transcription factor-induced RNAP backtracking is upregulated to promote DNA break repair. DSBs trigger the DNA damage (SOS) response in E. coli (Figure 2B). The SOS response is induced when the cell senses the presence of genomic lesions that need to be repaired[44]. SOS activation leads to the expression of several genes required to inhibit cell division and repair damaged DNA[45]. The Gre factors are not a part of the SOS regulon, and their specific upregulation under different types of stress has not yet been studied. However, uvrD is induced by the SOS response, and it is likely that the increased UvrD levels favors dimerization that stimulates UvrD’s helicase activity, which maybe necessary to promote backtracking[19, 46]. ppGpp synthesis is also increased under conditions of increased DNA stress, although independently of SOS[21], suggesting that factors that induce RNAP backtracking are specifically functional during DNA damage stress (Figure 2B).
Another mechanism that supports the idea that RNAP processivity is modulated when cells experience DNA damage has also emerged from the study of extremophile bacteria. The capacity of D. radiodurans to endure high doses of radiation is correlated with the intracellular level of manganese ions (Mn2+)[47]. The reason why the bacterium accumulates Mn2+ is not entirely clear. Gfh proteins are a family of Gre homologs that also interact with the RNAP secondary channel and are uniquely found in extremophile bacterial genomes (e.g., D. radiodurans and Thermus aquaticus)[48]. These proteins stimulate RNAP pausing in the presence of Mn2+ ions, which is required at high levels to survive radiation exposure[49]. In addition, one of these Gfh members is induced during stress[49]. Thus, there exists a complicated interplay between Mn2+ ion concentration after DNA damage, transcription processivity, and the repair outcome that needs to be investigated further. Understanding how factors that control transcription processivity are modulated under genotoxic stress could provide insight into the mechanisms that maintain the balance between transcription fidelity and DNA repair (Figure 1).
5. Error free transcripts allow DNA damage survival through proteome protection
Our studies suggest that in a wild-type E. coli cell experiencing DNA damage stress due to the generation of DNA breaks, the equilibrium between transcription fidelity and genome stability is inclined in favor of the former. The physiology of the extremophile bacterium D. radiodurans provides an example of a case where the balance is more level, and both RNA and DNA integrity may be preserved. D. radiodurans can withstand extreme doses of ionizing radiation, a potent DSB inducing agent. It has been suggested that mechanisms that protect the proteome from oxidation contribute significantly to the survival of D. radiodurans upon exposure to radiation[50]. Since the repair of DNA involves pathways often consisting of multiple constituents, repair may be not possible without all of the functional repair components. Interestingly, the RNAP from D. radiodurans is more efficient at intrinsic transcript cleavage, the reaction that rescues backtracking[51]. It is possible that transcript cleavage activity reduces transcription errors and results in the translation of error-free proteins from high fidelity transcripts. Errors in transcription can overwhelm protein quality control systems and ultimately reduce the lifespan of yeast cells[42]. Although introducing amino acid substitutions in the E. coli RNAP, based on residues found specifically in D. radiodurans RNAP, enhances the RNA cleavage activity of the E. coli enzyme, it remains to be seen how this affects radiation resistance[52]. We predict that the accuracy of transcription could provide advantages for DNA damage endurance by ensuring the production of fully functional proteins that are needed for survival.
6. Is the eukaryotic THO resolve conflicts similarly to GreA?
As described above, research in bacteria has contributed to our insights on how the two nucleic acids can be protected to different levels under DNA damage stress. The conservation of the basic processes of transcription, DNA repair and DNA damage responses across organisms suggests that similar decisions between preservation of transcription fidelity and genome stability are needed to be made in eukaryotes. We have noticed several similarities between greA mutants and the phenotypes of some eukaryotic mutants that promote transcription-associated recombination (TAR). TAR or the transcription dependent increase in recombination is a phenomenon best described in eukaryotes[53, 54]. Mutations in the THO complex of yeast increase TAR[53]. THO is a complex of four proteins involved in transcription elongation and mRNA export[55]. Deletions in components of the THO complex produce transcription elongation defects but also a hyper-recombination phenotype[56]. This phenotype is similar to that observed in greA null mutant[10, 32, 57]. The obvious question of the role of the THO complex in transcription fidelity is yet to be addressed. This line of inquiry may also determine the nature of the balance between transcription fidelity and genome stability in eukaryotes.
7. backtracked RNAP controls DNA resection
The first step of DSB repair is DNA resection. Resection involves unwinding of dsDNA and degradation of the 5’ terminating strand to generate 3’ ssDNA substrates to load recombination proteins. In E. coli, the tri-subunit complex RecBCD performs resection: RecBCD binds to the DSB end and uses helicase and exonucleases activities to unwind and degrade DNA (Figure 2A)[23]. RecBCD is a highly processive motor machine that can move along the DNA at high speeds for long distances before dissociating[58]. Without any interference, this degradative capacity could be extremely destructive to the E. coli genome as observed in a recombinase defective mutant [59]. In E. coli, Chi sites restrict degradation by causing RecBCD to pause, which allows for the recombination protein RecA to be loaded onto the generated 3’ ssDNA[60] (Figure 3A). Chi recognition by RecBCD occurs only 20–40% of the time in vitro[61]. While the levels of Chi recognition are harder to measure in living cells, the predicted probability of recognition of a single Chi site is approximately 40% in vivo[62]. It is thus possible that a back-up mechanism exists to keep RecBCD degradation in check.
Single molecule approaches have been used to investigate conflicts between E. coli RNAP and RecBCD on labeled DNA assembled into curtains[58]. Most elongating RNAP complexes were pushed by RecBCD for a long distance (~10 kb) and were eventually ejected from the DNA. Only 15% of collisions resulted in RecBCD stalling, suggesting that elongating transcription complexes by themselves cannot limit RecBCD resection. Additionally, these experiments show that encounters between elongation complexes and RecBCD don’t cause RNAP backtracking. However, when stalled RNAP was used for the encounter instead of an actively transcribing holoenzyme, the number of RNAP pushed by RecBCD was reduced[58]. Stalled RNAP represents a precursor to backtracked RNAP, which form much more stable complexes on the DNA[20]. The effect of backtracked RNAP on RecBCD degradation can be tested with the DNA curtain approach by engineering backtracking prone regions into the DNA template, or adding purified factors such as UvrD to enhance backtracking and generate arrays of backtracked complexes if necessary.
8. Are Backtracked RNAPs served as an alternate to Chi sites?
We propose that backtracked RNAP could assist in preventing RecBCD from destroying the genome during resection. Chi organization in the E. coli genome is striking: 75.5% of Chi sites are oriented toward the single origin of replication (oriC) in E. coli[63, 64]. The proposed reason for the directionally of Chi sites is to help rescue replication forks that have collapsed along their journey from oriC to ter [8, 65] (Figure 3B). Transcriptional units also show specific bias in relation to oriC. In E.coli, 55% of all genes and 70% of essential genes are encoded on the leading strand and hence co-oriented with the direction of replication[66]. This directionality plays an important role in reducing head-on replication transcription conflicts that lead to genomic instability[67]. We can speculate that the levels of transcription near and directed away from the origin lead to more backtracking, and could serve to reduce RecBCD resection and promote RecA loading. Such a role for backtracked RNAP would preserve the coding regions of essential genes co-oriented with transcription. Our results with XO-Seq suggest that backtracking is a significant barrier for resection in Chi-free regions[10]. However, it is still unclear if conflicts between RecBCD and backtracked RNAP will have different consequences in the head-on vs. co-directional orientations.
Our work shows that backtracked RNAP can limit resection and promote recombination[10]. This physiological mechanism to protect genome degradation can be useful in Chi free regions that are more prone to degradation by resection and genomic instability. The transcription of foreign DNA and hence the production of backtracked RNAP on these templates could also protect such invasive DNA and thereby promote horizontal gene transfer and evolution of the bacterial genome.
Resection in eukaryotes is a two-step process. An initial resection step is performed by MRN/X and CtIP/Sae2, after which long-range resection is catalyzed by additional helicase/nuclease complexes[68, 69]. Identification of factors that terminate resection in eukaryotes, where there are no Chi signals, has been the focus of a lot of current research. One proposed model of resection cessation is that chromatin and nucleosomes act as a roadblock to obstruct the resecting enzyme[70]. The idea that backtracked RNAP can limit resection is consistent with this hypothesis. Since the basic fundamental operations of break repair and transcription are conserved across species, it is possible that backtracked RNAP may act as a resection regulator in all kingdoms of life.
9. Conclusions
DNA is an arena where several intertwined fundamental biological activities such as replication, DNA repair, transcription and other aspects of gene regulation co-occur. Hence, collisions between these processes are unavoidable. Therefore, rules have to be established to ensure harmonious cohabitation. In this review, we discussed the collusion between DNA repair and transcription fidelity: the secret agreement between these processes to avoid collision that probably occurred early on in evolution. While the details of this agreement appear to differ across species, how these ancient conflicts have been resolved is still largely a mystery. Yet, the high fidelity of these processes suggests that the integrity of one may depend on the other. Clearly, further investigation will be required to shed light on the intricacies of avoidance of DNA transaction accidents.
Acknowledgement
We would like to thank Catherine Bradley and Herman Dierick for their comments and insights.
Commonly used abbreviations
- (RNAP)
RNA polymerase
- (DSB)
double-strand DNA breaks
- (TC-NER)
transcription-coupled nucleotide excision repair
- (HR)
homology-directed repair
- (Chi)
Cross-over hotspot instigator
- (ppGpp)
guanosine tetraphosphate
References
- [1].Mellon I, Hanawalt PC, Nature. 1989, 342, 95. [DOI] [PubMed] [Google Scholar]
- [2].Hanawalt PC, Spivak G, Nat Rev Mol Cell Biol. 2008, 9, 958. [DOI] [PubMed] [Google Scholar]
- [3].Karikkineth AC, Scheibye-Knudsen M, Fivenson E, Croteau DL, Bohr VA, Ageing Res Rev. 2017, 33, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].D’Alessandro G, d’Adda di Fagagna F, J Mol Biol. 2017, 429, 3215. [DOI] [PubMed] [Google Scholar]
- [5].Gaillard H, Aguilera A, Annu Rev Biochem. 2016, 85, 291. [DOI] [PubMed] [Google Scholar]
- [6].Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP, Legube G, Nat Struct Mol Biol. 2014, 21, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hoffman EA, McCulley A, Haarer B, Arnak R, Feng W, Genome Res. 2015, 25, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kowalczykowski SC, Trends Biochem Sci. 2000, 25, 156. [DOI] [PubMed] [Google Scholar]
- [9].Helleday T, Lo J, van Gent DC, Engelward BP, DNA Repair (Amst). 2007, 6, 923. [DOI] [PubMed] [Google Scholar]
- [10].Sivaramakrishnan P, Sepulveda LA, Halliday JA, Liu J, Nunez MAB, Golding I, Rosenberg SM, Herman C, Nature. 2017, 550, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Erie DA, Hajiseyedjavadi O, Young MC, von Hippel PH, Science. 1993, 262, 867. [DOI] [PubMed] [Google Scholar]
- [12].James K, Gamba P, Cockell SJ, Zenkin N, Nucleic Acids Res. 2017, 45, 1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nudler E, Mustaev A, Lukhtanov E, Goldfarb A, Cell. 1997, 89, 33. [DOI] [PubMed] [Google Scholar]
- [14].Borukhov S, Sagitov V, Goldfarb A, Cell. 1993, 72, 459. [DOI] [PubMed] [Google Scholar]
- [15].Toulme F, Mosrin-Huaman C, Sparkowski J, Das A, Leng M, Rahmouni AR, EMBO J. 2000, 19, 6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Koyama H, Ito T, Nakanishi T, Sekimizu K, Genes Cells. 2007, 12, 547. [DOI] [PubMed] [Google Scholar]
- [17].Kireeva ML, Nedialkov YA, Cremona GH, Purtov YA, Lubkowska L, Malagon F, Burton ZF, Strathern JN, Kashlev M, Mol Cell. 2008, 30, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nudler E, Cell. 2012, 149, 1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Epshtein V, Kamarthapu V, McGary K, Svetlov V, Ueberheide B, Proshkin S, Mironov A, Nudler E, Nature. 2014, 505, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Komissarova N, Kashlev M, Proc Natl Acad Sci U S A. 1997, 94, 1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kamarthapu V, Epshtein V, Benjamin B, Proshkin S, Mironov A, Cashel M, Nudler E, Science. 2016, 352, 993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Persky NS, Lovett ST, Crit Rev Biochem Mol Biol. 2008, 43, 347. [DOI] [PubMed] [Google Scholar]
- [23].Dillingham MS, Kowalczykowski SC, Microbiol Mol Biol Rev. 2008, 72, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Smith GR, Microbiol Mol Biol Rev. 2012, 76, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Radman M, DNA Repair (Amst). 2016, 44, 186. [DOI] [PubMed] [Google Scholar]
- [26].Taniguchi Y, Choi PJ, Li GW, Chen H, Babu M, Hearn J, Emili A, Xie XS, Science. 2010, 329, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kulish D, Lee J, Lomakin I, Nowicka B, Das A, Darst S, Normet K, Borukhov S, J Biol Chem. 2000, 275, 12789. [DOI] [PubMed] [Google Scholar]
- [28].Glass JI, Assad-Garcia N, Alperovich N, Yooseph S, Lewis MR, Maruf M, Hutchison CA 3rd, Smith HO, Venter JC, Proc Natl Acad Sci U S A. 2006, 103, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zenkin N, Yuzenkova Y, Biomolecules. 2015, 5, 1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hutchison CA 3rd, Chuang RY, Noskov VN, Assad-Garcia N, Deerinck TJ, Ellisman MH, Gill J, Kannan K, Karas BJ, Ma L, Pelletier JF, Qi ZQ, Richter RA, Strychalski EA, Sun L, Suzuki Y, Tsvetanova B, Wise KS, Smith HO, Glass JI, Merryman C, Gibson DG, Venter JC, Science. 2016, 351, aad6253. [DOI] [PubMed] [Google Scholar]
- [31].Paul BJ, Barker MM, Ross W, Schneider DA, Webb C, Foster JW, Gourse RL, Cell. 2004, 118, 311. [DOI] [PubMed] [Google Scholar]
- [32].Tehranchi AK, Blankschien MD, Zhang Y, Halliday JA, Srivatsan A, Peng J, Herman C, Wang JD, Cell. 2010, 141, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Satory D, Gordon AJ, Wang M, Halliday JA, Golding I, Herman C, Nucleic Acids Res. 2015, 43, 10190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Roghanian M, Zenkin N, Yuzenkova Y, Nucleic Acids Res. 2015, 43, 1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Traverse CC, Ochman H, Proc Natl Acad Sci U S A. 2016, 113, 3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Traverse CC, Ochman H, MBio. 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lee H, Popodi E, Tang H, Foster PL, Proc Natl Acad Sci U S A. 2012, 109, E2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jeon C, Agarwal K, Proc Natl Acad Sci U S A. 1996, 93, 13677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gordon AJ, Halliday JA, Blankschien MD, Burns PA, Yatagai F, Herman C, PLoS Biol. 2009, 7, e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gordon AJ, Satory D, Halliday JA, Herman C, PLoS Genet. 2013, 9, e1003595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Paoloni-Giacobino A, Rossier C, Papasavvas MP, Antonarakis SE, Hum Genet. 2001, 109, 40. [DOI] [PubMed] [Google Scholar]
- [42].Vermulst M, Denney AS, Lang MJ, Hung CW, Moore S, Moseley MA, Thompson JW, Madden V, Gauer J, Wolfe KJ, Summers DW, Schleit J, Sutphin GL, Haroon S, Holczbauer A, Caine J, Jorgenson J, Cyr D, Kaeberlein M, Strathern JN, Duncan MC, Erie DA, Nat Commun. 2015, 6, 8065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].van Leeuwen FW, de Kleijn DP, van den Hurk HH, Neubauer A, Sonnemans MA, Sluijs JA, Koycu S, Ramdjielal RD, Salehi A, Martens GJ, Grosveld FG, Peter J, Burbach H, Hol EM, Science. 1998, 279, 242. [DOI] [PubMed] [Google Scholar]
- [44].Michel B, PLoS Biol. 2005, 3, e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fernandez De Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, Ohmori H, Woodgate R, Mol Microbiol. 2000, 35, 1560. [DOI] [PubMed] [Google Scholar]
- [46].Nguyen B, Ordabayev Y, Sokoloski JE, Weiland E, Lohman TM, Proc Natl Acad Sci U S A. 2017, 114, 12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Daly MJ, Gaidamakova EK, Matrosova VY, Vasilenko A, Zhai M, Venkateswaran A, Hess M, Omelchenko MV, Kostandarithes HM, Makarova KS, Wackett LP, Fredrickson JK, Ghosal D, Science. 2004, 306, 1025. [DOI] [PubMed] [Google Scholar]
- [48].Agapov A, Esyunina D, Pupov D, Kulbachinskiy A, Biochem J 2016, 473, 4493. [DOI] [PubMed] [Google Scholar]
- [49].Esyunina D, Agapov A, Kulbachinskiy A, Proc Natl Acad Sci U S A. 2016, 113, 8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Krisko A, Radman M, Cold Spring Harb Perspect Biol. 2013, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Esyunina DM, Kulbachinskiy AV, Biochemistry (Mosc). 2015, 80, 1271. [DOI] [PubMed] [Google Scholar]
- [52].Esyunina D, Turtola M, Pupov D, Bass I, Klimasauskas S, Belogurov G, Kulbachinskiy A, Nucleic Acids Res. 2016, 44, 1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gottipati P, Helleday T, Mutagenesis. 2009, 24, 203. [DOI] [PubMed] [Google Scholar]
- [54].Aguilera A, EMBO J. 2002, 21, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rondon AG, Jimeno S, Garcia-Rubio M, Aguilera A, J Biol Chem. 2003, 278, 39037. [DOI] [PubMed] [Google Scholar]
- [56].Garcia-Rubio M, Chavez S, Huertas P, Tous C, Jimeno S, Luna R, Aguilera A, Mol Genet Genomics. 2008, 279, 123. [DOI] [PubMed] [Google Scholar]
- [57].Borukhov S, Polyakov A, Nikiforov V, Goldfarb A, Proc Natl Acad Sci U S A. 1992, 89, 8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Finkelstein IJ, Visnapuu ML, Greene EC, Nature. 2010, 468, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kuzminov A, Stahl FW, Genes Dev. 1999, 13, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Anderson DG, Kowalczykowski SC, Cell. 1997, 90, 77. [DOI] [PubMed] [Google Scholar]
- [61].Taylor AF, Smith GR, Proc Natl Acad Sci U S A. 1992, 89, 5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Cockram CA, Filatenkova M, Danos V, El Karoui M, Leach DR, Proc Natl Acad Sci U S A. 2015, 112, E4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y, Science. 1997, 277, 1453. [DOI] [PubMed] [Google Scholar]
- [64].Arnold D. A. a. K., C. S, in In eLS, (Ed.), 2001. [Google Scholar]
- [65].Kuzminov A, Mol Microbiol. 1995, 16, 373. [DOI] [PubMed] [Google Scholar]
- [66].Rocha EP, Microbiology. 2004, 150, 1609. [DOI] [PubMed] [Google Scholar]
- [67].Merrikh H, Zhang Y, Grossman AD, Wang JD, Nat Rev Microbiol. 2012, 10, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Symington LS, Cold Spring Harb Perspect Biol. 2014, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhu Z, Chung WH, Shim EY, Lee SE, Ira G, Cell. 2008, 134, 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Myler LR, Finkelstein IJ, Prog Biophys Mol Biol. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]