

Abstract
The accurate diagnosis and clinical management of traumatic brain injury (TBI) is currently limited by the lack of accessible molecular biomarkers that reflect the pathophysiology of this heterogeneous disease. To address this challenge, we developed a microchip diagnostic that can characterize TBI more comprehensively using the RNA found in brain-derived extracellular vesicles (EVs). Our approach measures a panel of EV miRNAs, processed with machine learning algorithms to capture the state of the injured and recovering brain. Our diagnostic combines surface marker-specific nanomagnetic isolation of brain-derived EVs, biomarker discovery using RNA sequencing, and machine learning processing of the EV miRNA cargo to minimally invasively measure the state of TBI. We achieved an accuracy of 99% identifying the signature of injured vs. sham control mice using an independent blinded test set (N = 77), where the injured group consists of heterogeneous populations (injury intensity, elapsed time since injury) to model the variability present in clinical samples. Moreover, we successfully predicted the intensity of the injury, the elapsed time since injury, and the presence of a prior injury using independent blinded test sets (N = 82). We demonstrated the translatability in a blinded test set by identifying TBI patients from healthy controls (AUC = 0.9, N = 60). This approach, which can detect signatures of injury that persist across a variety of injury types and individual responses to injury, more accurately reflects the heterogeneity of human TBI injury and recovery than conventional diagnostics, opening new opportunities to improve treatment of traumatic brain injuries.
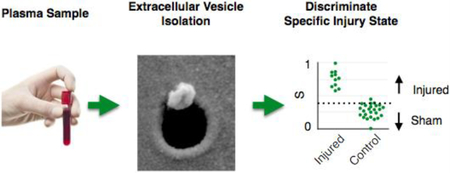
Graphical Abstract
We have developed a platform to isolate brain derived circulating extracellular vesicles (EVs) and measure multiple miRNA biomarkers packaged within these EVs, to classify specific states of traumatic brain injury.
Introduction
In the United States, 1.74 million people every year seek medical attention for traumatic brain injury (TBI), of which 80% are considered to have a mild TBI.(1) Even in those with mild injuries, approximately 10–20% of individuals will suffer long-term disability including seizures and emotional and behavioral issues.(2) One of the primary challenges in TBI care is appropriately classifying this heterogeneous injury and identifying patients at risk for these chronic impairments.(3) The neuropathology of TBI includes axonal shearing, inflammation, brain edema, and vascular injury, but the extent of these findings varies among patients.(4, 5) Conventional imaging studies, including magnetic resonance imaging and computed tomography, are commonly used to classify TBI, but do not reliably capture the full extent of the injury, particularly in those patients with mild injuries.(6,7) Currently, there are few molecular markers to assist in the assessment of an individual’s injury and subsequent recovery. Biomarkers are desperately needed in the field that correlate with these varied pathologies, track the progress of the disease, and predict outcome to facilitate accurate phenotyping of this heterogeneous disease.
Although there is great interest in developing biomarkers for both the diagnosis and management of TBI,(8,9) breakthroughs in this area have been limited. Most past biomarker work focuses on protein biomarkers in cerebrospinal fluid (CSF) or blood, including Tau(10, 11), calcium-binding protein S100B,(12) glial fibrillary acidic protein (GFAP)(13), neuron-specific enolase,(12, 13), ubiquitin carboxy-terminal hydrolase L1 (UCHL1),(13) myelin basic protein (MBP),(14) metabolites such as glucose(15), and miRNA(16). However, these efforts face detection sensitivity challenges because of the low concentration (fM-pM) of circulating molecular markers, as well as proteolytic degradation, clearance by the liver or kidney, and binding of potential biomarkers to carrier proteins.(9) Moreover, due to the diversity of head injury types and the complexity of cellular and molecular mechanisms underlying resilience to injury and recovery, it is difficult for any single biomarker to sufficiently characterize the complex states of the injured and recovering brain.(17)
Recently, the recognition that extracellular nanoscale vesicles (EVs), including exosomes and microvesicles, derived from brain cells that carry proteins and nucleic acid from their mother cells and cross the blood brain barrier (BBB), offer a new opportunity to evaluate molecular changes in neurons and glia after trauma.(18–20) In previous work, increases in circulating EVs have been observed in the blood of TBI patients following injury.(3, 21, 22) Despite their enormous potential, the use of EV biomarkers to improve patient care faces several challenges. Due to the nanoscale size of EVs, conventional size-based isolation is time consuming (> 6 hr), results in co-purification of cellular debris, and cannot selectively isolate specific sub-populations of EVs.(18, 23) While microfluidics can precisely sort and detect cells from complex media, applying these approaches to nanoscale EVs is limited by the low throughput and susceptibility to clogging of nanofluidics.
This work builds on the success of using microfluidic immunomagnetic sorting to isolate rare cells, achieving capture efficiency and selectivity not possible using conventional macroscale technologies.(27,28,38,39,65) In prior work, there has been great success using immunomagnetic sorting to isolate EVs, which due to the lack of magnetism of biological samples, can achieve surface-marker specific sorting without prior sample purification.(37,61–64) In this study, we use a new approach to nanofluidic sorting of brain-derived EVs, wherein millions of nanofluidic devices were incorporated onto a microchip platform and operated in parallel, increasing throughput by a million-fold and eliminating susceptibility to clogging from unprocessed clinical samples. By using nanoscale immunomagnetic traps, EVs can be sorted individually based on its labeling by a sufficient number of 50 nm Iron Oxide MNPs (Miltenyi) (70) (Fig. S12), analogous to the microfluidic devices used to selectively sort rare cells.(38,39,65) This capability distinguishes the ExoTENPO from previous microfluidic technologies and micro-scale bead systems, where the beads are larger then the EVs, that capture EVs onto functionalized surfaces(36) or beads(61,63), where the capture selectivity is set by the affinity ligand specificity. We applied our method to TBI, with three goals: 1. to develop a platform that could efficiently and practically profile biomarkers, 2. to use these novel miRNA biomarkers to accurately differentiate plasma or serum collected from uninjured and injured subjects, and 3. to use the EV biomarkers to predict injury intensity and injury history, key clinical variables that will guide therapy.
Our approach measures multiple EV miRNA biomarkers to capture a more comprehensive view of the injured and recovering brain than is possible with a single biomarker. (Fig. 1A) Conventional methods relying on only a single molecular biomarker are often not sufficiently specific and only give a limited view of the recovering brain.(8,59) Because EVs package multiple biomarkers from the injured and recovering brain, there is an opportunity to measure a panel of molecular biomarkers to more fully describe the complex states of the recovering brain. Several groups have measured multiple miRNAs (71,72) or protein markers (13,66) and combined these multiple measures algorithmically to diagnose TBI. In fact, this year the FDA has approved a diagnostic that can identify patients with intracranial lesions following a head injury without the need for a computed tomography (CT) x-ray scan, using UCH-L1 and GFAP concentration in the blood.(U.S. Food & Drug Administration, 2018). However, despite these recent successes, because of the wide variety of injury types and severities endemic to TBI, and the unique and dynamically changing combination of multiple endophenotypes of an injured and recovering brain(4,5), it remains challenging to adequately map biomarkers to an individual’s unique state of injury and recovery to improve clinical outcomes. To analyze these multiple molecular biomarkers, we used machine learning algorithms to reduce a panel of EV miRNA biomarkers into a set of optimized linear discriminators. By choosing relevant states of injury and training the machine learning algorithm to find patterns that optimally classify this set, we can outperform single markers. In this study, we isolated brain-derived EVs based on their expression of the AMPA receptor subunit (GluR2), performed small RNA sequencing, measured multiple miRNA biomarkers packaged in these EVs, and then combined these biomarkers using Linear Discriminant Analysis (LDA) to specifically classify the injured and recovering brain.(Fig. 1A) We used an established murine model for blast injury, which produced a mild TBI with neurobehavioral impairments 5 days after injury(24), to develop and characterize this method. Previous work characterized biomechanics of the blast overpressure injury (52), the design of the device used to create the blast overpressure loading (53), the deficits that appeared following mild blast exposure (54), the time course of blood-brain barrier opening after exposure (55), and the difference between mild and moderate blast overpressure exposure (56). Using blast injury, we demonstrated successful classification of injured mice versus sham controls, identified key pathways activated in the injured brain, and successfully classified between different pressures of blast injury, time points post injury, and histories of previous injuries. We additionally validated the translatability of this approach to humans by using the biomarkers identified in our mouse study and successfully classified (AUC = 0.9) a cohort of human clinical samples (N = 60), identifying uninjured controls versus those that experienced a diverse set of TBIs with an Abbreviated Injury Score (AIS) 2–5, using < 1 mL of serum.
Fig. 1. Immunomagnetic isolation of brain-derived EVs using TENPO.

A. The workflow of our machine learning-based TBI diagnostics that uses brain-derived EVs isolated using TENPO. B. EVs are immunomagnetically labeled using biotin anti-GluR2 antibody and anti-biotin magnetic nanoparticles (MNP). C. Finite element simulation of the magnetic field gradient generated by the TENPO’s permalloy-coated nanopores. The highest magnetic field gradient is at the edges of the pores where the labelled EVs are captured. D. The TENPO consists of millions of magnetic nanopores (600 nm diameter), which are incorporated into an acrylic reservoir. Fluid is pulled through the chip from the output using a syringe pump. The scale bar is 1 μm. E. Size distribution of EVs measured with DLS. Input is mixed cortical neuron/astrocyte cultured medium and isolate are GluR2+ EVs eluted from TENPO. The input has peaks at 8.7 nm, 78.8 nm, and 615 nm whereas the isolate has a major peak at 141.8 nm. F. Scanning electron microscopy (SEM) image of an EV captured using TENPO. The captured particle has a morphology consistent with EVs labeled with magnetic nanoparticles. G. Total EV miRNA yield (ng) comparison between centrifugation method (122.7 ng) and TENPO (216.2 ng). EVs are isolated from 20ml of mixed cortical neuron/astrocyte cultured medium.
Results
Surface marker-specific nanomagnetic isolation of brain-derived EVs
We used a nanofluidic architecture to precisely isolate and enrich for specific subpopulations of circulating EVs directly from unprocessed serum or plasma, based on the targeted EVs’ positive expression of a surface marker. Immunomagnetic sorting has the advantage that biological material is inherently not magnetic, enabling high contrast between targeted EVs and background material.(25) We used an antibody for the AMPA receptor subunit, glutamate receptor 2 (GluR2) because GluR2 is known to be found preferentially in brain-derived EVs.(22, 26) We labeled EVs with magnetic nanoparticles (r = 25 nm) functionalized with affinity ligands to label GluR2+ surface markers.(Fig. 1B) Our EV sorting Track-Etched magnetic NanoPOre (TENPO) chip consists of an ion track-etched polycarbonate membrane with 600 nm diameter pores coated with a soft magnetic film (Ni80Fe20) (Fig. 1C), rotating conventional microfluidic sorting by 90° to form magnetic traps at the edges of pores instead of in channels. Using TENPO, we can isolate EV sub-populations directly from V ≅ 10 mL of serum or plasma in less than 30 minutes, using either pan-exosome surface markers or brain-specific surface markers. Importantly, this architecture is insensitive to clogging from protein aggregates, cell debris, and lipoproteins present in serum or plasma samples (73, 74) because blockage of any individual pore causes the flow to be diverted to neighboring pores and does not affect overall device behavior (Fig. S13), enabling the highly specific EV capture in serum or plasma demonstrated throughout this paper. Finite element simulations were used to model the magnetophoretic force, and the strong trapping forces at the edge of the pore motivated us to make the pores as small as possible, without inadvertently trapping objects based on size, to bring targeted EVs close to the regions of strong forces.(Fig. 1C) Moreover, because the flow velocity is minimized by increasing the number of pores, we were motivated to maximize the pore density as much as possible without leading to a significant number of overlapping pores. The capture rate, i.e. the fraction of captured targeted EVs, decreases as a function of flow rate but can be fully recovered by stacking multiple TENPO membranes in series, enabling high capture rate at flow rates as high as ɸ = 10 mL/hr. (Fig. S1)
The TENPO uses a track etching process for manufacturing, a process that is widely available and therefore makes TENPO suitable for translation from the laboratory environment to practical clinical settings. In this work, we build off of our previous work in which we used magnetic micropores with a diameter d = 5 μm to isolate bacteria(27) and d = 30 μm to isolate mammalian cells(25). Track etching allows this approach to be scaled to the nanoscale (d= 600 nm) to tailor it to the isolation of EVs(59), without requiring electron beam lithography.(Fig. 1D) The orders of magnitude decrease in cost relative to conventional nanolithography comes at the expense of not controlling the pores’ location, which is not a critical feature for this application. These TENPO membranes are incorporated into a microfluidic device using laser micromachined layers of adhesive-coated polymer sheets.(27) In this paper, we used TENPO with a pore size d = 600 nm, a membrane area A = 15.2 cm2, a pore density ρ > 107 pores / cm2, and N = 6 membranes in series.
To characterize the performance of the TENPO for isolating brain-derived EVs, we first used an in vitro cell culture model of TBI.(28) To validate that the vesicles isolated by our device have a size consistent with EVs, we measured the size distribution of particles in the medium from mixed cortical neuron/astrocyte cultured cells and compared it to TENPO isolated vesicles from that medium. The GluR2+ EVs in 20 mL of medium were magnetically labeled and run through our device at ɸ = 10 mL/hr. After the isolated vesicles were eluted (System Biosciences), their size distribution was profiled and compared to that of the input using Dynamic Light Scattering (DLS). We found that our device isolated particles with a size distribution <d> = 140 nm.(Fig. 1E) Additionally, we fixed the vesicles directly on our device after capture and imaged them using scanning electron microscopy (SEM) (Electron Microscopy Resource Laboratory, University of Pennsylvania).(Fig. 1F) We observed 150–200 nm vesicles, captured at the edge of the pores consistent with the trapping mechanism of TENPO. Additionally, we performed Western blot analysis on the captured material to validate for the presence of exosomal markers (TSG101, Alix, CD9) and GluR2, our capture marker. (Fig. S2)
For downstream analysis, we first lysed the vesicles directly on the chip to minimize loss, collected the lysate, and then extracted the RNA off chip. By using TENPO isolation, we achieved 1.8x improvement in RNA yield from 20 ml of cortical neuron/astrocyte cultured medium compared to a centrifugal technique (Total exosome isolation kit, Life technologies).(29)(Fig. 1G) The details of this extraction is described in the Methods. The captured vesicles had properties consistent with that of the presence of exosomes, but because we used a brain-specific surface marker and not an exosome specific marker, co-purification of microvesicles is likely. For the diagnostic application that is the focus of this paper, co-purification of brain-derived microvesicles does not affect the results. We quantified the specificity of ExoTENPO capture by comparing the quantity of EV RNA capture from mixed cortical neuron/astrocyte cultured medium using the ExoTENPO, comparing GluR2 versus a control wherein the antibodies were replaced with an isotype control (biotin mouse IgG1 k isotype, Biolegend). There was >500% RNA yield (P < 0.005) using the GluR2+ markers as affinity ligands compared to the isotype control, confirming the specificity of the ExoTENPO (Fig. S14).
RNA sequencing of brain-derived EV miRNAs from a murine blast injury model for biomarker identification
To accomplish our first objective and identify miRNA biomarkers to classify injury state in mice, we first sequenced EV miRNAs from both mice that were injured using our blast model (415kPa, N = 5 pooled) and mice that were uninjured (N = 5 pooled).(Fig. 2A) For each animal, 400–600 μL of plasma was processed. We obtained 15–25 ng of total RNA from each animal and pooled the samples to maximize the sensitivity of sequencing, at the expense of being able to resolve animal-to-animal variability in the sequencing data. NEBNext Small RNA Library Prep Set for Illumina (BioLabs) was used to make a library and then sequenced on a NextSeq500.
Fig. 2. miRNA sequencing of brain-derived EVs from injured and control mice.

A. Brain-derived EV miRNAs were isolated using GluR2+ labeling and TENPO isolation and sequenced from pooled blast injured (415kPa) and control mice. DESeq normalized expression levels of individual miRNAs were plotted using a heat map. B. The five most abundantly expressed miRNAs from control and injured groups are plotted. C. Top 10 upregulated and downregulated miRNAs after the injury. D. 7 miRNAs biomarkers were selected and validated using qPCR. We used RNU6 for normalization. Expression levels from qPCR were compared to RNA sequencing. (R2 = 0.83 without and R2 = 0.62 with an outlier) Error bar indicates standard deviation of three technical replicates.
The top three most abundant miRNAs were the same in the injured and uninjured samples, miR-486b-5p, miR-486a-5p, and let-7i-5p, but their relative abundance was significantly different between injured and sham states (p < 0.05; Fig. 2B). We selected eight biomarkers to use in our multiplexed assay, (Fig. 2C) based on a combination of their abundance and either a maximally positive or negative differential expression in the injured versus the sham control samples. We required all EV miRNA biomarkers to have a total raw read count > 50. From the EV miRNA that met this criteria, we selected three biomarkers that were up-regulated in injured versus sham (miR-129–5p, miR-212–5p, miR-9–5p) and we selected four biomarkers that were down-regulated in injured versus sham (miR-152–5p, miR-21, miR-374b-5p, miR-664–3p). Following the sequencing experiment, we isolated EVs from a separate group of N = 10 injured mice and N = 10 sham mice and measured the eight candidate markers with qPCR. We found a positive correlation between qPCR and the sequencing data (R2 = 0.83 with a single outlier (miR-152–5p) removed and R2 = 0.62 with the outlier) (Fig. 2D).
We performed a bioinformatics analysis on the full set of sequencing data to explore the signaling pathways that were activated after TBI. We performed a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis using 53 microRNAs with fold change (DESeq normalized data) > 3. Among significantly enriched pathways (N=62; FDR corrected p-value < 0.05), we highlight the eight relevant brain injury-related pathways (Table 1) as well as the uncurated list (Fig. S3). Some of these pathways were supported in previous studies in the literature(30, 31). We summarized target genes corresponding the microRNAs and the pathways.(Fig. S4)
Table 1.
KEGG pathway analysis for blast injured (415kPa) mice versus sham mice. 8 different pathways related to traumatic brain injury.
| KEGG pathway | p-value | #genes | #miRNAs |
|---|---|---|---|
| Axon guidance | 0.0000000633 | 69 | 39 |
| Long-term potentiation | 0.0000589 | 39 | 29 |
| Glutamatergic synapse | 0.0000589 | 54 | 34 |
| Oxytocin signaling pathway | 0.0000671 | 77 | 39 |
| GABAergic synapse | 0.001291407 | 34 | 27 |
| Dopaminergic synapse | 0.001778928 | 60 | 30 |
| Neurotrophin signaling pathway | 0.001809901 | 57 | 33 |
| Cholinergic synapse | 0.03748049 | 51 | 31 |
Workflow of using machine learning algorithms to diagnose injury based on EV miRNA signatures
The work flow of our machine learning-based EV diagnostic is shown in Fig. 3A. To develop our LDA-based classification, we used qPCR to measure Ct values of EV miRNA biomarkers in each individual subject within a training cohort wT, for which each subject’s true state (e.g. injured or uninjured is known) is known a priori. As an example case to demonstrate the value of this approach, we measured a training set consisting of a cohort of mice that experienced a blast injury (415kPa) (N = 5) and a cohort of mice that were uninjured (N = 5). Expression levels were calculated by normalizing to the RNA gene RNU6. Leave-one-out cross validation (N-1) method was used for analysis of the training set data. The performance of the training sets are included in Fig. S5. This training set is fed into an LDA algorithm (Matlab) to generate an LDA vector x. This vector x is used to calculate an LDA score (w · x) for each mouse that can be used to optimally classify the mice, where every mouse with an LDA score above a threshold value Ψ, defined by the training data, is classified as belonging to one group and every mouse below it is classified as belonging to the other group. (Fig S5A) This threshold can be chosen to tradeoff sensitivity and specificity. We next evaluate the LDA algorithm by performing a set of measurements on a user-blinded test set of subjects wB, where we are a priori blinded to each subject’s true injury state. Here, we applied a model averaging approach (bootstrapping) that considers multiple subsets of features to help overcome the limitation of the small size of datasets inherent to animal studies. The use of blinded test sets allowed us to validate our classification subjects while avoiding the effects of overfitting. In our example case, we used N = 36 mice in the blinded test set.
Fig. 3. Machine learning based diagnostics for blast injured mice.

A. A training set of data wT, where the true state of the mice is a priori known, is fed into an LDA algorithm to generate an LDA vector x. This LDA vector is then evaluated using a blinded test set (wB · x), where the true state of the mice was not a priori known, to classify samples into different groups. B. The blinded test set is plotted by their LDA score (wB · x), where the mice can be classified into the correct group by choosing a threshold value ψ. C. A confusion matrix was generated to summarize the results of classifying the blinded test set. (N = 36).
For each of these mice, an LDA score S was calculated (Fig. 3B) and based on this score a classification was predicted. (Fig. 3C) Our LDA method classified N = 36 mice in the blinded test set with accuracy of 94%, outperforming any of the individual EV miRNA that we measured. Amongst the panel of miRNAs that we measured, several miRNAs were expressed with a statistically significant difference between these two groups (Fig. S6), e.g. miR-9–5p (p < 0.05). Here, we normalized the RNU6. However, due to mouse-to-mouse variability within the groups, there was no single EV biomarker that could accurately classify all of the mice. For example, the best performing biomarker miR-9–5p achieved only an AUC = 0.74.
Designing machine learning for reproducibility
To prevent the effects of overfitting and correctly apply machine learning algorithms in this study, we applied several well established strategies.(60) Most importantly, to evaluate each classification, we created an independent user-blinded test set, which consisted of samples from different subjects than used in the training set, to test our diagnostic performance. For the independent test set, we received blinded samples with unknown labels, processed them using our platform, and predicted their labels using the models generated by training sets. The predicted labels were then sent and compared to true labels to prevent any issue with overfitting. Additionally, we took the following steps to mitigate overfitting. We first restricted the number of features to eight miRNAs to ensure that the number of features was fewer than that of our smallest training set. In the application of machine learning to each training set, we first evaluated the predictive performance using a leave-one-out cross validation (N-1) to attempt to predict each model’s performance when used on an independent, user-blinded test data. We also validated the specificity of the predictive power of our analysis by comparing the performance of each classification to a data set where a training set of scrambled group labels was created. We applied bootstrapping, a model averaging approach that considers multiple subsets of features to better represent the outcome and help overcome the limitation of the small size of datasets. Additionally, we restricted ourselves in this study to LDA, and did not use the increasingly popular deep learning or neural networks that rely on layers of interconnected, nonlinear data transformations. While these models can be highly effective, they generally require very large datasets during training to avoid overfitting.(60) Additionally, in this study we include all raw data to both allow others to confirm our findings (Supplementary Data 1) and to encourage the community to use our data to further improve TBI diagnostics.
Classification of heterogeneous injuries and sham controls
In an actual clinical setting, there is a much greater diversity in the injuries that a subject can sustain than in the simple example demonstrated above. To evaluate whether our approach could diagnose TBI in animals with a variable set of injuries, we created a blinded test cohort of N = 77 mice with variable injury intensity and a variable window of time (Δt) between the injury and the sample collected, as well as sham controls. (Fig. 4A) In this user-blinded test set, we included two different blast pressures (high blast injury-415 kPa and low blast injury-215 kPa) and multiple times elapsed since injury Δt = 1hr, Δt = 1day, Δt = 4 days, and Δt = 14 days. When we measured blood samples from each of these mice using our chip and compared our prediction for each mouse’s injury state (injured or uninjured) to the true state of the mice, a high accuracy of 99% was achieved. For this classification, we used a training set (N = 30) that consisted of two groups: N = 5 sham controls and a diverse set of injured mice (N = 25) including N = 5 low blast Δt = 1hr, N = 5 low blast Δt = 1day, N = 5 low blast Δt = 4 days, N = 5 low blast Δt = 14 days, N = 5 high blast Δt = 1hr. (Fig. S5B)
Fig. 4. Diagnosis of heterogeneous injury with variable intensity and time elapsed since injury from sham controls.

A. Classification of injured vs. sham control mice in a user blinded test set (N = 77), which includes variable intensity and time elapsed since injury Δt, achieving an Area Under the Curve AUC = 1 and accuracy = 0.99. B. Discrimination of injured vs. sham mice as a function of time elapsed since injury Δt. C. Comparison of AUC of our approach and simply counting GluR2+ EVs as a function of time elapsed since injury Δt.
We next evaluated how long after an injury the EV miRNA signature measured by our chip persists. To address this question, we evaluated the specificity and sensitivity of our classification of mice as a function of the time elapsed since their injury Δt. We considered a user blinded test set of mice (N = 66) that received a low level of blast injury (215kPa), where blood was collected from cohorts of mice at either Δt of 1 hour, 1 day, 4 days, and 14 days after the injury. When we considered all of the time points together, an AUC = 1 for discriminating injured versus sham was achieved (p < 0.001, Fisher’s Exact Test). (Fig. 4B) We compared our chip’s performance to simply counting GluR2+ EVs (Fig. 4C) (22), and we found that our approach of using EV miRNA signatures outperformed EV counting at every time point after injury Δt. For this experiment, a single training set (N = 25) that consisted of two groups was created, including injured mice that were collected at multiple time points after injury (N = 5 Δt = 1hr, N = 5 Δt = 1day, N = 5 Δt = 4 days, N = 5 Δt = 14 days) and sham controls (N = 5). (Fig. S5C)
Discrimination of injury intensity, time elapsed since injury, and history of prior injuries using EV miRNA biomarkers
Beyond identifying the presence or absence of an injury, an important goal for a TBI clinical biomarker is to characterize features of the injury that include its intensity, the time since the injury occurred, and whether the injury was isolated or in combination with an additional previous injury. To this end, we next evaluated whether we could retrain our LDA approach, using the same EV miRNA biomarkers identified in our sequencing experiment, to predict the injury intensity, the time elapsed since the injury Δt, and injury history. Each of these classifications was performed by creating a separately trained model and was evaluated using an independent user-blinded test set.(60) Here, we chose the sample size for multiple injury states that could both demonstrate statistically significant results (p < 2 × 10−3 for the data shown in Fig. S5D and Fig. S5E) and be consistent with the ethics rules of our IACUC protocol to minimize the number of mice used.
We first demonstrated the ability to classify the intensity of injury between mice with a low level of blast injury (215 kPa) and with a higher level of blast injury (415 kPa) from sham controls. We used a classification scheme based on the blast pressure level itself rather than any specific histological or behavioral differences associated with two levels of blast injury. In an independent blinded test set (N = 43 mice), we successfully classified two different levels of injuries from sham controls. (Accuracy = 88%, Chi Square test, p-value = 1.1 × 10−11) (Fig. 5A) The training set included a total of 19 animals (N = 7 sham mice, N = 6 with high blast injury, and N = 6 with low blast injury) collected Δt = 1 hr after injury. (Fig. S5D) To validate the specificity of our signatures, we scrambled the training set as a control experiment and found that the accuracy dropped to 26%, similar to random guessing (33%). Sample size, limited by number of sacrificed mice, can affect power of classification. We examined the effect of sample size on cross-validation accuracy by including our left-out test data in the training set and examining cross-validation accuracy as a function of sample size. Cross-validation accuracy increased as a function of sample size, again indicating that data contains meaningfully separable signals. (Fig. S8)
Fig. 5. Discrimination of injury intensity, time elapsed since injury, and history of injury using EV miRNA signatures in a murine blast injury model.

A. Classification of intensity of injury for sham mice and mice with a low blast (LB) injury (215kPa) and a high blast (HB) injury (415k Pa) at Δt = 1hr after injury. B. Classification of time elapsed since injury (Δt), for mice with LB injury. C. Schematic of history of injury study. Black dots correspond to the time of injury, and the colored coded dots correspond to the time the sample was taken. D. Classification of sham mice and mice with a history of injury c and without a history of injury b (single injury).
We next evaluated the ability to detect the time elapsed Δt since the injury. In an independent user blinded dataset (N = 41), we were able to discriminate between mice that had an injury with an elapsed time between injury and the sample being taken of: Δt = 1 hr, 1 day, 4 days, and 14 days.(Fig. 5B) An accuracy of 71% was achieved for this multi label classification. (Chi Square test, p-value = 2.6 × 10−7) To validate the specificity of our signatures, we scrambled the training set as a control experiment and found the accuracy dropped to 24%, similar to random guessing (25%). The training set was comprised of a total of 23 animals across four groups; Δt = 1 hr (N = 6), Δt = 1 day (N = 6), Δt = 4 days (N = 5), and Δt = 14 days (N = 6). (Fig. S5G) All injured mice in these experiments had a low blast (215kPa) injury.
We next demonstrated the ability of our approach to detect a history of prior injuries in our murine injury model. We evaluated the following groups of mice, a: sham controls, b: mice that were measured 1 hour after a single blast injury (215kPa), and c: mice that were measured 1 hour after a second blast injury (215kPa) that had received the same injury 24 hours prior. (Fig. 5C) Using independent blinded test sets, we demonstrated that we could classify mice with a history of injury (c), mice that were injured only once (b), and sham controls (a) (Accuracy = 88%, N = 42) (Fig. 5D). Here, we show both a 3D plot and a 2D projection of the same plot to provide two different perspectives. The training set was comprised of a total of 18 animals across three groups (N = 7 sham, N = 6 single injury, N = 7 double injury). (Fig. S5E)
Detection of EV miRNA signatures of injury in human samples
To evaluate our approach in human clinical samples, we used the same miRNAs determined from our preclinical model experiments to predict the injured/uninjured state for individual samples from a blinded test cohort of 60 human subjects. Amongst the panel of miRNA that we measured, several were differentially expressed between the injured and control groups, e.g. miR-129–5p, miR-9–5p (p <0.05), but no single miRNA could classify the individual subjects into the correct groups due to variable in expression between individual subjects. (Fig. S7) Therefore, as in our mouse studies, we used an LDA analysis to detect EV miRNA signatures in TBI patients. The training set included 15 total patients (N = 5 TBI patients and N = 10 healthy controls). (Fig. S5F) The user-blinded test set included those with TBIs with an Abbreviated Injury Score (AIS) of 2–5 (N = 32) and healthy controls (N = 28) (Fig. 6A). We performed a receiver operator characteristic (ROC) analysis on our predictions and achieved an AUC = 0.9 (Fig. 6B). In addition, we separately analyzed whether we could classify patients with TBIs that also had systemic injuries from healthy controls (AUC = 0.94, N = 44) and patients with TBIs that either had no or minor systemic injury from healthy controls (AUC = 0.996, N = 37). (Fig. 6C, Fig. S9) These results demonstrate that our diagnostic is, in fact, more successful at detecting TBI where there is no or minor systemic injury. These results support our hypothesis that the EVs that our chip analyzes are brain-derived. We note that clinical samples used here were collected 0.4–120 hours after injury (median = 41 hours), a heterogeneity which can be observed in standard clinical settings. We compared the predictive power of our diagnostic with that of digital ELISA, an emerging gold standard in TBI blood-based diagnostics. (57, 58) Using the SIngle MOlecule Array (SIMOA; Quanterix) platform we measured the protein biomarkers Tau and NF-L. SIMOA allows ultra-sensitive detection of individual proteins (LOD < 1.75 pg/ml for four protein markers) with 1000-fold more sensitivity than conventional ELISA. (57, 58) We measured the concentrations (pg/ml) of four protein markers from N = 15 healthy controls and N = 36 TBI patients, a representative subset of the same clinical cohort we used for our study. Though average protein levels were higher in TBI patients than in healthy controls for all four protein biomarkers, none of the markers was significantly different between TBI patients and healthy controls (p > 0.05), likely due to the variability of the injury severity (AIS 2–5) and time elapsed since the injury (0.4–120 hours) within our patient cohort.(Fig. S10) The AUCs from the protein markers ranged from 0.66–0.88, which were lower than what we have achieved (AUC = 0.9) using our EV diagnostic. Moreover, our machine learning based analysis was unable to successfully combine the four protein markers for an improved result (AUC ~ 0.5) due to the high variability of the protein concentrations (Fig. S10A) within each group (healthy control, TBI patients) measured using the SIMOA platform, failing to create a representative training set for algorithm development.
Fig. 6. Diagnosis of TBI using clinical samples.

A. The results of Linear Discriminant Analysis on a user blinded test set (N = 60), including variable injury levels (AIS 2–5), multiple time points after injury 0.4–120 hours after injury (median = 41 hours), and healthy controls. B. A Receiver Operator Characteristic (ROC) curve showing the tradeoff between sensitivity and specificity for this diagnosis. An AUC = 0.9 was achieved. Inset: A confusion matrix summarizing the results of the study. C. AUC comparison of two different subgroups (#1: patients with no/minor systemic injury, #2: patients with systemic injury) to total population (#3).
Discussion
In this work we demonstrate that brain-derived EVs contain miRNA signatures that can be used to classify specific states of traumatic brain injury and to identify possible signaling pathways activated in the brain after injury. We showed that the precision of nanofluidic immunomagnetic EV isolation can be applied to the enrichment of brain-derived EVs directly from serum or plasma and that this method can be successfully applied in both a murine model and human samples. Importantly, this work serves as a demonstration of the potential of identifying and using a panel of biomarkers to define an injury signature rather than searching for a single marker that can distinguish complex states of injury and recovery with reasonable specificity. We believe that by building upon the approach described in this paper, there is enormous potential to use this technique to better understand subtypes of TBI-related injury, recovery, and therapeutic response. Moreover, because our TENPO approach miniaturizes EV isolation into a handheld device and speeds up the isolation from half a day to less than an hour, there is great potential to translate this technology into a point-of-care device by pairing it with a miniaturized miRNA detection. (32, 33) In its most successful form, this technology can be used in the home for monitoring recovery and treatment effectiveness. (1) Additionally, a point-of-care device can be deployed to military and sports settings where rapid evaluation can help better evaluate and guide those with injuries to the help that they need.
In comparison to prior work isolating EVs, our work has several key advantages. Relative to size-based techniques, TENPO can isolate subsets of EVs that originate from a specific tissue.(18) In this paper, this ability allows us to enrich for brain-derived nanoscale vesicles from the vast background of other materials including other exosomes, microvesicles, and cellular debris, and resolve the signatures of TBI injury and recovery. The TENPO differentiates itself from prior published(34, 35, 36, 37) and commercial work (e.g. Dynabeads, ThermoFisher) where surface marker specific isolation of EVs is used. Chiefly, prior works isolated EVs onto bulk functionalized surfaces, where specificity is defined by the capture antibody. In the TENPO, because the feature size matches that of the EVs, we can emulate established microfluidics approaches to sort cells (38, 39) and precisely balance the drag force and the magnetic capture force. Thus, analogous to flow cytometry, we can define a threshold number of magnetic nanoparticle labels above which an EV will be captured on our device at a given flow rate. In comparison to previously published work that measured protein or miRNA biomarkers from serum or plasma to predict TBI injury states (13, 45–48), we can achieve comparable or improved diagnostic performance (AUC of 0.9) on a more heterogeneous population of TBI patients (0.4–120 hours after injury), which better reflect clinical use. (Fig. S11) Moreover, our device differentiates itself in its capability to specifically classify the injury intensity, time elapsed since injury, and the history of injury.
This work builds upon the work of others that have identified biomarkers to measure TBI(9–17). We believe that a robust diagnostic could be developed by combining currently identified TBI biomarkers to generate a signature using machine learning algorithms, rather than evaluating the performance of each biomarker individually. Importantly, our method can distinguish specific states of brain injury, both in terms of intensity/mechanism and in temporal course. In our open-ended search for biomarkers by performing RNA sequencing, the best miRNA marker was unable to accurately classify individual subjects due to a. the heterogeneity of GluR2+ EVs within a patient and b. the heterogeneity of GluR2+ EVs across patients. To accurately classify patients despite this heterogeneity, we measured and combined multiple miRNAs to find a signature that could correctly classify each subject (AUC > 0.9). We believe that this work demonstrates the potential of machine learning based diagnostics for accurately predicting and tracking complex states of TBI. We note here that we utilized one of the simplest learning methods due to the limited sample sizes, but with more extensive data other more complex machine learning models might be fitted with greater prediction accuracy, especially for quantitative variables like time since injury.
This work demonstrated that GluR2+ EVs could be used to classify specific states of injury. In future work, we pan to further develop TENPO to isolate multiple subsets of EVs, based on multiple surface markers, that can more comprehensively sample the multiple, heterogeneous EV subpopulations emitted by different cell types in the brain following an injury (77). For example, ACSA-2+ expression can be used to enrich for astrocyte derived EVs (75) and CD184+/CD44+ for glial cells (76).
In this study, we used miRNA biomarkers identified from a single murine injury to identify miRNA implicated in injury and recovery. The TENPO framework we describe can be adapted to examine multiple sub-populations of EVs, allowing an increasingly comprehensive picture of the injured and recovering brain to be captured. Building upon this work, we plan to sequence a variety of injury types, severities, and states of recovery in both cohorts of mice and humans. Our method combines three different components—novel isolation technology for brain-derived EVs, biomarker discovery via small RNA sequencing, and machine learning based diagnostics—which enabled us to develop potential diagnostics for TBI. Additionally, performance of TENPO isolation can be further enhanced by increasing the magnetization, and the specificity, of the magnetic labels, by doing a combination of: a. use MNPs with 10x larger saturation magnetization moments ms (e.g. MnFe) rather than iron oxide,(67) b. use smaller MNPs d < 30 nm rather than the 50 nm diameter particles used in this study,(68) to overcome steric hinderance and allow a greater number of particles to bind to each EV, c. use a magnetic amplification to increase the number of MNPs per EV at the cost of increased assay complexity.(69) Since EVs are shed from almost all types of cells, our platform could be further utilized in different brain-related diseases or even in other diseases.
Materials and methods
Experimental design.
The main objective is to isolate specific EV sub-populations from clinical samples and to analyze their miRNA cargo to diagnose traumatic brain injury. We evaluated the performance of the TENPO using magnetic beads and finite element simulation and validated the performance of RNA sequencing using qPCR. The capability of the TENPO to isolate specific EVs from complex samples and extract RNA for downstream analysis was first tested using cell culture models and sham serum and plasma. Subsequently, the capability to use our platform for the diagnosis of traumatic brain injury was first tested using the blast injury mouse model and subsequently a cohort of clinical samples. All the test sets for the machine learning algorithm were blinded and independently created to prevent overfitting and truly evaluate the performance of our platform.
TENPO fabrication.
The TENPO membranes were fabricated using thermal evaporation. On the surface of a track-etched polycarbonate membrane with 600nm pores (Whatman), 200 nm layer of permalloy (Ni80Fe20) was thermally evaporated (Kurt Lesker PVD-75, Singh Nanofabrication Facility, University of Pennsylvania) and then 30 nm layer of gold was subsequently deposited to prevent oxidation. Moisture-resistant polyester film (McMaster-Carr, 0.004” thick) and solvent-resistant tape (McMaster-Carr) were cut using laser micromachining (Universal Laser VLS 3.50) to incorporate TENPO membranes into a microfluidic device. An optically clear cast acrylic sheet (McMaster-Carr) was used as a reservoir for the input, and a polydimethylsiloxane (PDMS) piece was used for the output, pressure-fit to tygon tubing to connect to a negative pressure supply (Programmable Syringe Pump, Braintree Scientific).
Dynamic light scattering (DLS).
To obtain size distribution of nanoparticles (EVs), we used DLS (Zetasizer, Malvern) where we loaded 300–400 μl of samples with the correct settings (e.g. refractive index, viscosity). All the measurements were performed as triplicates.
RNA sequencing.
NEBNext Small RNA Library Prep Set for Illumina (BioLabs) was used to make a library. We first isolated RNA on chip using Total Exosomal RNA Isolation Kit (Life Technologies). Then, we measure the RNA amount using Qubit (Life Technologies) and as recommended by the protocol, the samples with more than 100 ng of RNA were selected for usage. Then, quality control check was performed on a BioAnalyzer using a DNA 1000 chip. For size selection, AMPure XP beads were used (Beckman Coulter). 140–150 bp sizes were selected using the beads and the sizes were confirmed by the BioAnalyzer using High Sensitivity Chip. The RNA-seq libraries were pooled together and the final concentration was quantified using a KAPA Library Quantification Kit (KAPA Biosystems). The libraries were sequenced using a NextSeq 500/550 kit (FC-404–2005, Illumina) on a NextSeq500 (75 base pair length).
Small RNA-seq analysis.
miRNA expression was found with mirDeep2 (40) and Bowtie (41) (mm10), using miRBase version 21 (42). Read counts from miRNA families (miRNAs with the same seed sequence) were combined. Quantified miRNA expression values were normalized by DESeq2 (43). Using DIANA mirPath v. 2.0 (44), we identified the pathways of target genes regulated by miRNAs.
qPCR.
miScript SYBR Green PCR kit (Qiagen) and miScript primers (Qiagen) were used. Master mix that consists of miScript SYBR Green, miScript primer, universal primer, and water were made at 5:1:1:2 ratio and 9 μl of the master mix was added to each well, followed by 1 μl of cDNA. 40 cycles were run with a default setting using CFX384 Touch Real-Time PCR machine (Bio Rad). Triplicates were done for each sample and negative template control (NTC) was used to check any contamination. The melting curves were first checked before the analysis.
Linear Discriminant analysis (LDA).
Using Matlab (R2015b), multiple features (genes) from multiple groups (intensity of injury, time points after injury, history of injury) were simplified for classification using LDA. The confusion matrix and the LDA plot were made using results from Matlab. Leave-one-out cross validation (N-1) method was used for analysis of the training set data. An independent de novo user blinded test set was used for the final evaluation. For stepwise regression, we used combntns function in MATLAB to generate every single combination from the panel of biomarkers.
EV isolation (TENPO).
Anti-biotin ultrapure microbeads (Miltenyi Biotec) and biotinylated antibodies were used for magnetic labeling. For the antibody, biotin anti-human, mouse GluR1/GluR2 antibody (Bioss) was used. First, biotinylated antibodies were added to the sample and incubated for 20 mins at room temperature with shaking. Then, anti-biotin ultrapure microbeads were added to the samples and incubated for 20 mins at room temperature with shaking. Then the samples were added to the reservoir of the TENPO chip and negative pressure was applied by a programmable syringe pump (Braintree). As the samples were pulled through the chip, magnetically labeled EVs were captured at the edge of the pores of the chip.
EV miRNA isolation.
The second part of the ExoRNeasy serum/plasma kit (Qiagen) was used for RNA extraction from isolated EVs. We isolated EVs on chip using TENPO, then directly applied QIAzol lysis reagent (Qiagen) on chip. We collected the lysate then used the second part of the ExoRNeasy serum/plasma kit to extract RNA from the lysate off chip. The EV miRNA was eluted in a small volume (~30 μl) and it was stored at −80C or processed immediately for further analysis.
Blast injury.
Adult male (12–14 weeks old) C57BL/6J mice (Charles River, MA) were exposed to a blast overpressure insult that mimicked blast-induced traumatic brain injury. (24) Anesthetized mice (isoflurane 3.0% induction, 1.5–2.0% maintenance) were used as sham controls. Anesthetized mice were prepared for injury by placing sound insulating foam into each ear canal. Mice were loaded into a holder positioned 1 cm away from the end of the shock tube, positioned with their snouts facing the shock tube. Head motion was limited with a metal rod encircling the snout and placing a a cervical collar between the occiput and shoulders. A single overpressure of either 215 +/− 18kPa or 415 +/− 41 kPa was delivered and the animal was immediately removed from the holder assembly. Throughout these experiments, we adhered strictly to the guidelines from the University of Pennsylvania office of University Laboratory Animal Resources (ULAR) using approved protocols, and animal care and use was in accordance with the guidelines specified by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania.
Human sample collection.
Clinical samples were collected under Institutional Review Board approved protocols at the University of Texas Southwestern and the University of Pennsylvania. Blood samples were drawn from consented patients within 120 hours of their injury. Serum samples were immediately aliquoted and frozen at −80 °C until further analyses.
Scanning Electron Microscopy (SEM).
We fixed the EVs captured on TENPO using a fixing reagent (2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1 sodium cacodylate buffer, pH 7.4). SEM images were taken at the Electron Microscopy Resource Laboratory, University of Pennsylvania.
Western blot analysis.
Lysates from mouse EVs were analyzed using Western blot analysis as we described.49–51 Briefly, proteins were resolved on a 10% sodium dodecyl sulfate (SDS) polyacrylamide gel and transferred to a polyvinylidene difluoride membrane using transfer buffer (5.8% tris-base, 29% glycine, 20% methanol, 45.2% distilled water) for 1 hour. Blots were blocked for 1 hour at room temperature in 5% bovine serum albumin (BSA) dissolved in TBST (500mM tris-base, 60mM KCl and 2.8M NaCl, pH 7.4 with 0.1% Tween). Blots were then incubated with primary antibodies diluted in TBST containing 3% BSA overnight at 4°C. ALIX and GluR1/R2 primary antibodies were used at a dilution of 1:500 and TSG101 and CD9 primary antibodies were used at a dilution of 1:1000. Blots were incubated with secondary antibodies at 1:5000 dilution in TBST containing 5% milk for 1 hour at room temperature. Membranes were developed using the enhanced chemiluminescence system (GE Healthcare; Piscataway, NJ) and Syngene G:BOX iChemi XR system and GeneSnap software (Version 7.09.a, Syngene, Frederick, MD). Primary antibodies included Purified Anti-ALIX Antibody (634502, BioLegend, San Diego, CA), Anti-TSG101 Antibody (ab125011, Abcam, Cambridge, MA), Biotin anti-mouse CD9 Antibody (124804, BioLegend, San Diego, CA), GluR1 + GluR2 polyclonal antibody, biotin conjugated (bs-10042R-Biotin, Bioss, Woburn, MA). Secondary antibodies included Goat anti-Rat IgG (H+L) Secondary Antibody, HRP (31470, Thermo Fisher), Anti-rabbit HRP (Jackson labs, 711-036-152), and Anti-mouse HRP (Jackson labs, 715-035-150).
Statistical Analysis.
To generate a predictive model in our analysis of both human and blast injury mouse subjects, we used a cohort of training set data and linear discriminant analysis, carried out using Matlab. We tested our predictive models using an independent, user blinded test set to avoid the effects of data overfitting. We evaluated the predictive value using Fisher’s Exact Test. No outlier analysis was performed. Sample size was chosen using Fisher’s Exact Test to measure the p-value of our device’s classification.
Data Availability.
In this study, we include all raw data to both allow others to confirm our findings (Supplementary Data 1) and to encourage the community to use our data to further improve TBI diagnostics.
Supplementary Material
Acknowledgements
We thank Tawny Meredith-Duliba and Erika Silverman from the Diaz-Arrastia lab at Penn for collecting and processing clinical samples that were used for this study. Funding was provided by the New Jersey Commission on Brain Injury Research (CSCR14IRG005) to DI, DM, and BF, NIH NS 088276 to DM, Allen Foundation to DM and DI, U01 NS086090 to RDA, the Pennsylvania Department of Health Commonwealth Universal Research Enhancement Program, the National Institute of Health: 1R21CA182336–01A1 to DI and JK, and DI was supported by an American Cancer Society - CEOs Against Cancer - CA Division Research Scholar Grant, (RSG-15-227-01-CSM), the National Science Foundation’s CAREER Award (#1554200), and The Hartwell Individual Research Award.
References
- 1.Ma VY, Chan L, Carruthers KJ, Archives of physical medicine and rehabilitation, 95, 986–995, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stern RA, Riley DO, Daneshvar DH, Nowinski CJ, Cantu RC, McKee AC. Pm&r 3, S460–S467, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Maas AI, Menon DK, Lingsma HF, Pineda JA, Sandel ME, and Manley GT., Journal of neurotrauma, 29, 32–46, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, and Stewart W., Brain, 136, 28–42, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golding EM., Brain research reviews, 38, 377–388, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Bruce ED, Konda S, Dean DD, Wang EW, Huang JH, and Little DM., Neuroscience, 66, 103–113, 2015. [DOI] [PubMed] [Google Scholar]
- 7.Kochanek PM, Berger RP, Bayr H, Wagner AK, Jenkins LW, Clark RSB. Current opinion in critical care, 14, 135–141, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Jeter CB, Hergenroeder GW, Hylin MJ, Redell JB, Moore AN, et al. Journal of neurotrauma 30, 657–670, 2013. [DOI] [PubMed] [Google Scholar]
- 9.Zetterberg H, Smith DH, Blennow K. Nature Reviews Neurology, 9, 201–210, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, et al. , Journal of Neuropathology & Experimental Neurologym, 68, 709–735, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bulut M, Koksal O, Dogan S, Bolca N, Ozguc H, et al. , Advances in therapy, 23, 12–22, 2006. [DOI] [PubMed] [Google Scholar]
- 12.De K, Leffers P, Menheere PPCA, Meerhoff S, Twijnstra A, Acta neurologica scandinavica,103, 175–179, 2001. [DOI] [PubMed] [Google Scholar]
- 13.Diaz-Arrastia R, Wang KKW, Papa L, Sorani MD, Yue JK, et al. , Journal of neurotrauma,31, 19–25, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger RP, Adelson PD, Pierce MC, Dulani T, Cassidy LD, et al. , Journal of Neurosurgery: Pediatrics, 103, 61–68, 2005. [DOI] [PubMed] [Google Scholar]
- 15.Wagner AK, Bayir H, Ren D, Puccio A, Zafonte RD, et al. , Journal of neurotrauma, 21, 125–136, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Redell JB, Moore AN, Ward III, Norman H, Hergenroeder GW, et al. , Journal of neurotrauma,27, 2147–2156, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pineda JA, Wang KKW, Hayes RL, Brain Pathology, 14, 202–209, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ko J, Carpenter E, Issadore D, Analyst, 1412, 450–460, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao H, Chung J, Balaj L, Charest A, Bigner DD, et al. , Nature medicine 18, 1835–1840, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nilsson J, Skog J, Nordstrand A, Baranov V, Mincheva-Nilsson L, et al. , British journal of cancer, 100, 1603–1607, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor DD, Gercel-Taylor C., Phil. Trans. R. Soc. B, 369, 20130503, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ko J, Hemphill MA, Gabrieli D, Wu L, Yelleswarapu V, et al. , Scientific Reports, 6, 31215, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hyun KA, Kim J, Gwak H, Jung HI., Analyst,141, 382–392, 2016. [DOI] [PubMed] [Google Scholar]
- 24.Beamer M, Tummala SR, Gullotti D, Kopil C, Gorka S, et al. , Experimental neurology; 283, 16–28, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muluneh M, Issadore D., Advanced drug delivery reviews, 66,101–109, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faure J, Lachenal G, Court M, Hirrlinger J, Chatellard-Causse C, et al. , Molecular and Cellular Neuroscience, 31, 642–648, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Muluneh M, Shang S, Issadore D., Advanced healthcare materials, 3, 1078–1085, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ko J, Yelleswarapu V, Singh A, Shah N, Issadore D., Lab on a Chip, 16, 3049–3057, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lusardi TA, Wolf JA, Putt ME, Smith DH, Meaney DF., Journal of neurotrauma, 21, 61–72, 2004. [DOI] [PubMed] [Google Scholar]
- 30.Balakathiresan N, Bhomia M, Chandran R, Chavko M, McCarron RM., Journal of neurotrauma, 29, 1379–1387, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma A, Chandran R, Barry ES, Bhomia M, Hutchison MA et al. , PloS one, 9, e112019, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niemz A, Ferguson TM, Boyle DS., Trends in biotechnology, 29, 240–250, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ping J, Vishnubhotla R, Vrudhula A, Johnson ATC., ACS nano, 10, 8700–8704, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Im H, Shao H, Park YI, Peterson VM, Castro CM., Nature biotechnology, 32, 490–495, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raposo G, Stoorvogel W., J Cell Biol, 200, 373–383, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanwar SS, Dunlay CJ, Simeone DM, Nagrath S., Lab on a Chip, 14, 1891–1900, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He M, Crow J, Roth M, Zeng Y, Godwin AK., Lab on a Chip, 14, 3773–3780, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adams JD, Kim U, Soh HT., Proceedings of the National Academy of Sciences, 105, 18165–18170, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xia N, Hunt TP, Mayers BT, Alsberg E, Whitesides GM, et al. , Biomedical Microdevices, 8, 299–308, 2006. [DOI] [PubMed] [Google Scholar]
- 40.Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N., Nucleic Acids Res, 40, 37–52, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langmead B, Salzberg SL., Nat Methods, 9, 357–359, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kozomara A, Griffiths-Jones S., Nucleic Acids Res, 42, D68–73, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Love MI, Huber W, Anders S., Genome biology, 15, 550, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vlachos I-S, et al. , Nucleic acids research, 40, W498–W504, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shahim P, Tegner Y, Wilson D-H, Randall J, Skillbäck T, Pazooki D, Kallberg B, Blennow K, Zetterberg H., JAMA Neurol, 71, 684–692, 2014. [DOI] [PubMed] [Google Scholar]
- 46.Mondello S, Akinyi L, Buki A, et al. , Neurosurgery, 70, 666–675, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Papa L et al. , Annals of Emergency Medicine, 59, 471–483, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Papa L et al. , J. Trauma Acute Care Surg, 72, 1335–44, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hadzimichalis NM, et al. , Schizophrenia Research, 24, 248–250, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carrel D, et al. , The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 29, 8248–8258, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hernandez K, et al. , Frontiers in Cellular Neuroscience, 10, 6, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gullotti DM, et al. , J. Biomech. Eng, 136, 91004, 2014. [DOI] [PubMed] [Google Scholar]
- 53.Panzer MB, Matthews KA, Yu AW, Morrison B, Meaney DF, Bass CR., Frontiers in Neurology, 3, PMID DOI: 10.3389/fneur.2012.00046, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patel TP, Gullotti DM, Hernandez P, O’Brien WT, Capehart BP, Morrison B, Bass C, Eberwine JE, Abel T, Meaney DF. Frontiers in Behavioral Neuroscience, 8, 349, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hue CD et al. , J. Neurotrauma, 33, 1202, 2016. [DOI] [PubMed] [Google Scholar]
- 56.Beamer M, Tummala SR, Gullotti D, Kopil K, Gorka S, Ted A, Bass CR, Morrison B, Cohen AS, Meaney DF., Experimental Neurology. PMID DOI: 10.1016/j.expneurol.2016; 05: 025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, Piech T, Patel PP, Chang L, Rivnak AJ, Ferrell EP, Randall JD, Provuncher GK, Walt DR, Duffy DC., Nat. Biotechnol, 28, 595–599, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rissin DM, Fournier DR, Piech T, Kan CW, Campbell TG, Song L, Chang L, Rivnak AJ, Patel PP, Provuncher GK, Ferrell EP, Howes SC, Pink BA, Minnehan KA, Wilson DH, Duffy DC., Anal. Chem, 83, 2279–2285, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ko J, et al. , ACS Nano, 10, 1021/acsnano.7b05503, 2017. [Google Scholar]
- 60.Ko J, Baldassano S-N, Loh P, Kording K, Litt B, Issadore D., Lab on a Chip, DOI: 10.1039/C7LC00955K, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clayton A, et al. Journal of immunological methods, 2471–2, 163–174, 2001. [DOI] [PubMed] [Google Scholar]
- 62.Shao H, et al. Nature Communications, 6, 6999, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao Z, et al. Lab on a Chip, 163, 489–496, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jeong S, et al. ACS Nano, 102, 1802–1809, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ozkumur E, Science Translational Medicine, 5179, 179ra47–179ra47, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Battista D Frontiers in Neurology 6, 110, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haun JB, et al. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology, 23: 291–304, 2010. [DOI] [PubMed] [Google Scholar]
- 68.Lee J, et al. Nature Medicine 131, 95, 2007. [DOI] [PubMed] [Google Scholar]
- 69.Liong M, et al. Nature communications, 4, 1752, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schreiber S, et al. Small 42, 270–278, 2008. [DOI] [PubMed] [Google Scholar]
- 71.Sharma A, et al. PLOS One, 2014. [Google Scholar]
- 72.Bhomia M, et al. Scientific Reports 6, 28148, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Muller L, et al. J. Immunol. Methods 411, 55–65, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yuana Y, et al. J. Extracell. Vesicles 2, 21494, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feldmann M, et al. Journal of Biological Methods, 1(2), 2014. [Google Scholar]
- 76.Yuan SH, et al. PLOS One, 6(3), 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Willms E, et al. Scientific reports, 6, 22519, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
In this study, we include all raw data to both allow others to confirm our findings (Supplementary Data 1) and to encourage the community to use our data to further improve TBI diagnostics.