

Abstract
Classical physiological studies using electrophysiological, biophysical, biochemical, and molecular techniques have created a detailed picture of molecular transport, bioenergetics, contractility and movement, and growth, as well as the regulation of these processes by external stimuli in cells and organisms. Newer systems biology approaches are beginning to provide deeper and broader understanding of these complex biological processes and their dynamic responses to a variety of environmental cues. In the past decade, advances in mass spectrometry–based proteomic technologies have provided invaluable tools to further elucidate these complex cellular processes, thereby confirming, complementing, and advancing common views of physiology. As one notable example, the application of proteomics to study the regulation of kidney function has yielded novel insights into the chemical and physical processes that tightly control body fluids, electrolytes, and metabolites to provide optimal microenvironments for various cellular and organ functions. Here, we systematically review, summarize, and discuss the most significant key findings from functional proteomic studies in renal epithelial physiology. We also identify further improvements in technological and bioinformatics methods that will be essential to advance precision medicine in nephrology.
I. FROM PHYSIOLOGY TO PROTEOMICS TO MECHANISMS
Life is dynamic. To survive, organisms must respond appropriately to fluctuating environments, both internal and external. These responses result from intricate coordinated changes in the quantities and activities of biomolecules within a living system, including proteins, DNA, RNA, lipids, and metabolites. These coordinated changes are orchestrated almost exclusively by the proteome, defined as the total repertoire of proteins (and their individual abundances) present within a cell/tissue/organism. Likewise, the observable phenotypes of any cell/tissue/organism are a direct manifestation of the respective proteomes. In humans, for example, the total number of protein-encoding genes is just over 20,000 (20,441 genes; http://www.ensembl.org/Homo_sapiens/Info/Annotation). However, almost 200,000 different gene transcripts (198,002) from the human genome have been identified, arising from processes like alternative mRNA splicing and encoding multiple isoforms of individual proteins. From such numbers, it would seem that the array of different combinations of proteins that could possibly be expressed in cells, and hence the array of different resulting cellular phenotypes, is essentially infinite. Surprisingly perhaps, this is not the case.
Adult humans (and other vertebrates) are comprised of a finite and rather limited number of distinct cell types, which can be distinguished on the basis of combined morphological, (ultra)structural, molecular biological and biochemical properties (6). In the most well-conceived and comprehensive analysis published to date, at least 411 distinct cell (pheno)types (and thus distinct proteomes) were identified for adult Homo sapiens (169). This number represented a minimum, due in part to the likelihood of incomplete identification of diversity among certain classes of cell types (primarily neurons, smooth muscle cells, and fibroblasts). A similar picture emerges from analysis at the organ system level: the adult human kidney, for example, contains at least 30 different cell types (5). The emerging science of single-cell “-omics” (genomics/epigenomics/transcriptomics/proteomics/metabolomics) holds the promise of an unbiased, high-resolution expanded classification of not only distinct cell (pheno)types, but also of stable cell states and even states of transition during dynamic responses to environmental changes or disease (68, 165). Nevertheless, even if the total number of unique cell types (i.e., unique proteomes) ultimately increases by an order of magnitude (or more), it will still represent a very small fraction of that which is theoretically possible. Thus, given the “relatively modest” number of distinct cell types and the rapid technological advances in high-throughput sequencing, proteomics, and bioinformatics, it seems possible that every distinct proteome defining its respective cellular phenotype could be fully characterized in the not-too-distant future.
In fact, two recent studies described attempts to identify every single protein corresponding to a protein-coding gene in the human body (85, 174). These draft “human tissue proteomes” resulted in a relatively comprehensive, albeit still incomplete (48), map of proteins obtained from nearly every human tissue. Despite the impressive successes of such modern systems biology studies, it should be pointed out that a significant fraction of the proteins (one-third or more) from the proteomes of every species still have no known functions (44a, 76, 116, 125). This situation represents a serious obstacle to the comprehensive classification of distinct cell types/states and full characterization of the respective proteomes that determine those phenotypes. But even more importantly, it is impossible to imagine how a complete understanding of physiological processes and regulation for any given species can be achieved in the absence of functional knowledge for more than one-third of that species’ proteome. Thus a global collaborative effort on functional studies of proteins at the proteome level is necessary to close the gap between breadth and depth of proteomic studies. One example of such a global project is The International Mouse Phenotyping Consortium, which began 7 yr ago with the goal of identifying the function of every mouse gene (using the C57BL/6 strain) through knockout technologies (https://www.mousephenotype.org) (39).
In the following sections, we summarize major aspects of 1) how protein functions are regulated at the molecular level, resulting in the orchestrated control of the internal milieu, and 2) the historical development of modern proteomic techniques for deciphering protein regulation and control of cellular physiology. Subsequently, we present a literature review in the field of kidney physiology to illustrate the use of proteomic technology for advancing knowledge in this field. Finally, we discuss important statistical and bioinformatics considerations related to appropriate interpretation of proteomics data.
A. Physiological Stimuli and Complex Dynamic Biological Responses
To understand how complex cellular processes are regulated at the molecular level, comprehensive systems biology approaches can be used for exploration of responses to specific stimuli. One such approach is proteomics, in which fundamental aspects affecting global protein function, abundance, posttranslational modifications (PTMs), protein intermolecular interactions, and localization, can be studied in an unbiased, large-scale manner (FIGURE 1).
FIGURE 1.

An overview of mechanisms regulating protein functions. Four major aspects controlling protein functions are illustrated. First, the abundance of any particular protein is tightly regulated and determined by the integration of transcriptional regulation, protein translation, and protein degradation processes. Second, numerous posttranslational modifications (PTMs) of proteins play major roles in controlling protein functions and subsequently help determine cellular responses to the changing environment. Acetylation, phosphorylation, and ubiquitinylation are depicted as examples of well-known PTMs. Third, protein intermolecular interactions, including protein-protein, protein-DNA/RNA, and protein-small molecule interactions, which define targets of protein functions, serve as another major regulatory point. Finally, the (re)localization of proteins to specific subcellular regions, such as nucleus, mitochondria, or plasma membrane, governs their appropriate site of action. See text for details.
1. Protein abundance
For any given protein, the total amount of that protein present under any specific conditions can affect its functional activities and the phenotype of its respective biological system. Thus, precise regulation of the copy number of proteins is essential. Proteins are continuously produced (via gene transcription and translation) and degraded in a regulated fashion (see FIGURE 1, top). The net result of these processes determines the abundance of any specific protein at any given time. It is worth noting that all of these processes (transcription, translation, and degradation) are themselves controlled by the activities of numerous proteins, the abundances of which are likewise regulated in a recursive fashion. The roles of multiple processes, in addition to transcriptional control (i.e., mRNA levels), in regulating protein abundance explain the discordant results between proteomics and transcriptomics studies in many instances (100). Therefore, the determination of protein abundance requires consideration of all of these mechanisms and is crucial for the accurate interpretation of quantitative proteomic analyses and ultimately for understanding the complex cellular physiology in response to specific stimuli.
2. Posttranslational modifications
Protein functions can be altered by either reversible or irreversible modifications to one or more of its constituent amino acids. These modifications lead to changes in the physicochemical, and ultimately biological, properties of proteins and greatly expand the combinatorial possibilities of protein structures beyond those afforded by the basic 20 amino acid building blocks. Currently, there are 480 different known protein modifications reported in the Uniprot database (9) (https://www.uniprot.org/docs/ptmlist). Some of these modifications have been extensively studied [e.g., phosphorylation (60), ubiquitinylation (166), and acetylation (32)], and their roles in physiological regulation in general are well understood (see FIGURE 1, bottom). Many other modifications, which have been difficult to study previously, including PTMs such as arginine methylation (94), succinylation (173), malonylation (178), and redox-mediated cysteine modifications such as glutathionylation and sulfenylation (58), are now gaining increased interest due to advances in the capabilities of proteomic strategies and technologies. Despite these advances, identification and study of the myriad possible modifications, which also often occur transiently and/or at extremely low levels, remain a major challenge to completely understanding the mechanism regulating cellular responses to any specific stimulus. Mass spectrometry-based proteomic analysis has provided a powerful and sensitive means for examining global changes in posttranslational protein modifications associated with physiological responses to alterations in the cellular environment.
3. Protein intermolecular interactions
Many different biomolecules are packed into the complex cellular and extracellular compartments of living systems. Such environments, unlike the purified protein system, promote physical interactions among proteins and other biomolecules such as DNAs, RNAs, small-molecule metabolites, as well as other proteins (see FIGURE 1, right). In fact, it is through these very interactions that the biological functions of proteins and their contributions to overall phenotype are manifested [with the possible exception of heretofore unproven quantum phenomena, which even Albert Einstein could not predict, calling them “spooky” (129)]. Changes in the conformational state of a protein (or even one of its subunits) induced by some stimulus may promote or disrupt these intermolecular interactions and, consequently, serve as a way to control functions of entire protein complexes. Stimuli that lead to derangements in some of these interactions could result in phenotypic changes associated with many diseases, including malignancy, autoimmune disorders, neurological disorders, and cardiovascular disease (cf. Refs. 97, 105). In any biological system, there is a vast array of protein intermolecular interactions occurring as spatially and temporally distinct, yet globally integrated, clusters, collectively referred to as the interactome. As will be illustrated in subsequent sections, mass spectrometry (MS)-based proteomics, in conjunction with immunoaffinity and/or other high-throughput techniques, have been invaluable for interactome analysis.
4. Protein localization
Yet another layer of complexity involving the contribution of protein functions to cellular phenotypes and physiology is regulation of the precise localization of proteins within appropriate cellular compartments (FIGURE 1, left). For many proteins, initial control of protein localization is directly encoded in their amino acid sequences (e.g., signal sequences and other targeting motifs) for directed transport into the correct compartment, such as nucleus, mitochondria, or endoplasmic reticulum (ER). To execute the appropriate physiological programs in response to stimuli, subsequent relocalization of proteins is regulated through many types of PTMs, altered interactions with other biomolecules, and physical movement via molecular motor-driven processes (15). Protein MS has been utilized to great effect in elucidating subcellular proteomes and mechanisms for regulating spatial (re)distribution of proteins as a function of physiological status.
B. Development of Modern High-Throughput Proteomics Techniques for Elucidating Physiological Mechanisms
Attempts to utilize MS for peptide/protein sequencing began as early as 1958; however, significant hurdles prevented routine sequence analysis, especially of complex samples, until the early 1990s (18). The most obvious question that arises regarding this topic is: “How is it even possible to sequence a protein using mass spectrometry?” First, the most fundamental concept related to this question is that every amino acid residue (except leucine and isoleucine) has a unique mass, and the mass spectrometer is the most accurate method for mass determination (technically, m/z, mass-to-charge ratio). Thus, if it were possible to fragment a peptide/protein randomly at each peptide bond, a statistical population representing every possible fragment would be produced. For this series of fragments, ordered by increasing or decreasing mass, the mass difference between any two consecutive fragments would specify the identity of the extra amino acid residue in the larger fragment (FIGURE 2). Sequence determination from such a series of ion fragments is called de novo sequencing. To measure mass (m/z), mass spectrometers require charged molecules (either positively or negatively charged ions) and a very high vacuum; in other words, the peptide/protein fragments must be ionized and in the gas phase. Ultimately, through the persistent systematic efforts of Klaus Biemann, Don Hunt, and their many talented coworkers and colleagues in this field, ionization and fragmentation methods were devised and subsequently refined for delivery of peptide/protein ions into the mass spectrometer and sequence determination. Another crucial development that dramatically accelerated progress in peptide/protein sequencing by MS was the coupling of multiple mass spectrometers, which greatly facilitated generation and detection of the requisite series of ions randomly fragmented at every peptide bond.
FIGURE 2.

Simplified fragmentation scheme and sequence determination for an example peptide using tandem mass spectrometry. Following ionization and introduction into the vacuum environment of the mass spectrometer system, protonation of the amide nitrogens in the peptide bonds occurs randomly across the population of gas-phase peptide molecules. The schematic structure for a singly-charged example peptide, protonated at the peptide bond between residues 3 and 4, is depicted at the top of the figure. Regardless of which peptide bond is protonated, the entire population of peptide molecules would be detected in the MS1 as the (M+H)+ precursor ion, with mass (m/z) 459. Subsequent transfer of the peptide precursor ions into the collision chamber results in random fragmentation at the peptide bonds (dashed lines), yielding two predominant series of fragment product ions (“b” and “y” ions, as described in the text). For the specific (M+H)+ precursor ion shown as an example in the figure, fragmentation at the indicated peptide bond would produce y1 ions from molecules in which the positive charge remains with the amide nitrogen, or b3 ions from molecules in which the positive charge segregates with the carbonyl carbon (forming an acylium ion) (top). Due to the random peptide bond protonation and fragmentation, a statistical population of all possible y and b ion pairs (top) is generated in the collision chamber. Subsequent analysis of these fragment product ions in the MS2 results in the MS2 spectrum depicted in the lower panel. Subtraction of m/z values for any two consecutive ions in the same series (either “y” or “b”) provides the residue mass (and hence, the identity) of the extra amino acid present in the larger fragment ion. In the example shown, (y3 – y2) = (402 – 288) = 114, identifying asparagine as the NH2-terminal residue in y3; similarly, (b3 – b2) = (285 – 172) = 113, identifying either leucine or isoleucine as the COOH-terminal residue in b3, and so forth. Ultimately, this process (called de novo sequencing) allows deduction of the complete sequence of the original peptide precursor ion. Notes: the diagrams and explanation presented here have been simplified somewhat: 1) for instance, using modern ionization methods and instrumentation, typically the peptide precursor ions (and fragment product ions) are multiply protonated, at the amide nitrogens in the peptide bonds plus the NH2-terminal amine group and/or the R groups of lysine/arginine; 2) as another example, the acylium ion at the COOH-terminus of b ions may undergo a structural rearrangement(s) [e.g., to form a cyclic oxazolone (25, 61, 134)]. Despite this additional “complexity” of multiple charge states and multiple possible structures for b ions, the actual masses of the peptide precursor and product ions are perfectly conserved, and thus the simplified conceptual description presented here is still completely valid.
In this tandem mass spectrometry (MS/MS) configuration, a peptide mass (m/z) can be measured in the first mass spectrometer (MS1), and this precursor ion can then be passed into a collision chamber (containing an optimal level of inert gas) to induce fragmentation at the weakest bonds (i.e., the peptide bonds). The series of fragment product ions generated by collision with the gas molecules [collision-induced dissociation (CID)] is subsequently passed into a second mass spectrometer (MS2) for m/z measurement of each product ion. During the evolution of MS protein sequencing technology, it was discovered that, not one, but two predominant sets of fragments are actually generated by CID, namely “b ions” (containing the NH2-terminal amino acid plus 1, 2, 3, etc., aa residues) and “y ions” (containing the COOH-terminal amino acid plus 1, 2, 3, etc., aa residues) (FIGURE 2). For a more complete overview of the principles of MS-based proteomics in general, the reader is directed to some recent reviews (12, 126, 152, 154).
More recently, numerous refinements and innovations have led to the current state of the art in high-throughput proteomics analysis. First, several new developments in sample preparation have aimed to streamline multiple sample preparation steps within an easily scalable platform, thus allowing rapid, parallel sample preparation (46, 74, 175). Second, novel liquid chromatographic techniques also have helped increase the number and reliability of peptide/protein identifications and quantification by implementing orthogonal strategies for prefractionation and/or enrichment while simultaneously minimizing sample loss (90, 150, 181). Third, advancements in ionization methods, fragmentation methods, and instrumentation have all dramatically enhanced modern proteomic analyses. The two most widely used advanced ionization methods are MALDI (matrix-assisted laser dissociation ionization) (81, 158) and ESI (electrospray ionization) (49). Although MALDI still has important applications in protein imaging (56, 88), the innovative coupling of microcolumn liquid chromatography (nano-LC) to ESI-MS/MS (75) provided an optimal platform that is still the basis for most high-throughput proteomic analyses. Additionally, new methods of fragmentation (such as higher-energy C-trap dissociation and electron transfer dissociation) have extended the sensitivity and range of proteomic analyses (122, 156). Finally, MS instrumentation itself has rapidly improved. New types of mass spectrometers have been developed, including the ion trap, as well as hybrid tandem mass spectrometers comprised of multiple types, most notably the “orbitrap” (a hybrid between ion trap and Fourier-transform ion cyclotron resonance mass spectrometers) (72). Development and coupling of additional robust automation technologies with the advances described above is expected to further increase throughput and reduce human errors, thereby paving the way toward even more routine comprehensive proteome characterization in the future.
The workflow for modern MS-based proteomic analyses is supported by three “pillars”: biochemical sample preparation, MS technique, and bioinformatics processing of the acquired data (including peptide matching algorithms for sequence determination, software for identification and quantification of proteins and PTMs, as well as tools for identifying functional molecular networks). These three areas are highly interrelated, and advances in one area have often inspired progress in the other. More details describing proteomic workflow, data acquisition, and peptide/protein quantification techniques are provided in Sect. II, additional concepts for understanding ms-based proteomics, below. Further reviews on MS principles and technical topics can be found elsewhere (3, 8, 12, 154).
To reach the ultimate goal of complete understanding of physiological mechanisms, however, further advances in integrating proteomics with other “omics” types of studies, such as genomics (“proteogenomics”), transcriptomics, and metabolomics, are occurring (7). Relatively few contemporary proteomic studies currently reach this level of integration, and even fewer such studies involve relevant experimental physiological settings or relate the findings to actual physiological functions within the same sample or system. In this regard, renal proteomics may serve as an example of how proteome studies, integrated with comprehensive physiological and systems biology approaches, can be utilized to significantly advance a research field for a better understanding of complex dynamic biological mechanisms (reviewed in Sect. III, application of functional proteomics to renal tubule physiology).
II. ADDITIONAL CONCEPTS FOR UNDERSTANDING MS-BASED PROTEOMICS
A. Essential Workflow for Typical High-Throughput MS-Based Proteomics
In a typical proteomic workflow (see FIGURE 3), starting with biochemical extraction from a cellular protein source of interest, the complex protein mixture is first subjected to proteolytic digestion (FIGURE 3A, top), a so-called bottom-up or shotgun proteomic strategy (in contrast to the less frequently utilized top-down approach, which analyzes the intact proteins). Prefractionation and/or enrichment could be performed either before or after proteolytic digestion. Prefractionation procedures include electrophoresis or chromatography [e.g., ion exchange, high-pH reversed-phase liquid chromatography (RPLC), or hydrophilic interaction liquid chromatography (LC)]. Enrichment procedures are mainly affinity based, including mono-specific interactions (e.g., antibodies) or group-specific interactions [e.g., immobilized metal affinity chromatography (IMAC) and metal oxide affinity chromatography for phosphoprotein/peptide enrichment]. The obvious advantage of prefractionation and/or enrichment is that it reduces sample complexity, thereby permitting an overall increase in the number of identified peptides and proteins and an increased detection of low-abundance peptide species (especially those containing PTMs). To eliminate interfering substances from samples before MS analysis, clean-up methods like StageTips, a C18-resin-based method, are usually executed (136). Subsequently, peptide mixtures are subjected to nano-flow RPLC, and the eluted peptides are directly ionized by ESI and introduced into the first mass analyzer for survey mass analysis (MS1) (FIGURE 3A, middle). The most abundant precursor peptides ion(s) eluting at a particular retention time (e.g., the precursor peptide ion with m/z = 859.9, illustrated as a green peak in FIGURE 3A, middle) is (are) automatically selected for fragmentation by CID and subsequently transferred to the second mass analyzer for m/z analysis of fragment product ions (MS2) (FIGURE 3A, bottom). This method for selecting precursor peptides to be fragmented is the most common and is called data-dependent acquisition (DDA) (see next section for additional details and descriptions of other methods).
FIGURE 3.

A typical workflow for high-throughput mass spectrometry (MS)-based proteomics. A, top: sample preparation for MS-based proteomics begins with a complex protein mixture of interest that is then subjected to proteolytic digestion. Optional steps, including prefractionation and/or enrichment of a specific subgroup of proteins/peptides, can be performed to increase sensitivity for specialized proteomics experiments. Bottom: the expected result from MS analysis is a large number of complex mass spectra that need to be further processed to identify peptide sequences. B, top: the most commonly used algorithm for peptide sequencing requires a proteome database specific for each experiment (e.g., ideally, the specific proteome for the cell/tissue types and species being investigated). Bottom: theoretical mass spectra generated from a list of possible peptide sequences are matched with the observed spectra, providing an automated interpretation of complex spectra obtained.
A typical high-throughput proteomic analysis results in the generation of a vast number of MS2 fragmentation spectra (typically 25,000 spectra per 90-min LC-MS/MS run), prohibiting manual interpretation of sequence data. As an alternative to de novo sequencing (as described in the sect. IB), many automated search engines have been developed to permit sequence determination based on fragment product ion spectral matching. SEQUEST was the first such search algorithm (45), followed later by release of several other widely distributed search algorithms (34–36, 55, 128). These automated approaches all consist of three major components: 1) a proteome database; 2) a list of all predicted peptides and their m/z values following in silico proteolytic digestion; and 3) a peptide-spectral matching algorithm (FIGURE 3B). An ideal proteome database should contain all protein sequences from the species of interest. Consequently, comprehensive proteomics is possible nowadays only after the completion of genome sequencing for the experimental species being investigated (33, 155). Currently, well-accepted, open-access proteome databases include the National Center for Biotechnology Information Reference Sequence Database (RefSeq) (121), the Universal Protein Resource (UniProt) (161), and the Ensembl Project (4). All species-specific protein sequences in a given database are subsequently processed in silico to simulate the proteolytic digestion performed in the actual complex protein mixture being analyzed. This process results in a complete computer-generated list containing all predicted peptides and their respective masses (m/z) that would be produced following cleavage of all proteins in the database with a specific protease. In silico digested peptides with m/z values approximately equal to each detected precursor ion m/z value are selected as candidate sequences, and then theoretical MS2 spectra of these sequences are generated. As illustrated in FIGURE 3B, using the “green” precursor peptide ion with m/z 859.9 (from FIGURE 3A, middle) as an example, three candidate peptide sequences with essentially identical m/z values are identified from the complete list of in silico digested peptides. Matching between the observed MS2 spectrum and theoretical MS2 spectra of the candidate sequences is then performed (a process called “peptide-spectrum matching”) based on various scoring schemes to assess matching quality (106, 168). In the example shown, the second candidate peptide is identified with the highest probability, based on the degree of matching between the actual and predicted MS2 spectra (e.g., 12 of 13 predicted y ions and 6 of 13 predicted b ions detected in the actual MS2 spectrum of the example precursor peptide ion) (FIGURE 3, A AND B, bottom).
B. Data Acquisition Techniques for MS-Based Proteomics
Among the various factors influencing results of protein/peptide sequencing by MS, data acquisition techniques have been a major focus in the field; differences among these techniques can lead to dramatically different results in magnitude, depth, and overall reproducibility of peptide identifications, as well as precision of peptide quantification. These data acquisition techniques include DDA, data-independent acquisition (DIA), and targeted strategies. The choice among these options must be tailored to fit the experimental question being addressed (71).
The DDA strategy is typically used in discovery proteomics studies. In this technique, from the group of precursor ions present in an MS1 spectrum, the selection of a particular ion for further fragmentation is “dependent” on predetermined rules that are applied at the MS1 level. FIGURE 4A shows the commonly used rule for precursor ion selection, which is based on the relative signal intensity (among all precursor ions in a particular MS1 spectrum). In the example shown, the red ion is selected for fragmentation followed by an MS2 scan. Generally, a cycle of MS/MS analysis consists of an MS1 scan of the sample peptide mixture, followed by a rapid sequence of MS2 scans of the 10 most abundant precursor ions. Although the MS approach using DDA has an undemanding data analysis pipeline, and its instrumentation is easy to set up, the data-dependent nature of this approach obviously limits both completeness and reproducibility of peptide identification (i.e., not every precursor ion present in the sample is selected for fragmentation in each run, nor are the same precursor ions consistently selected for fragmentation between different runs).
FIGURE 4.

Data acquisition techniques for mass spectrometry (MS)-based proteomics. Several modes of data acquisition are illustrated. A: data-dependent acquisition (DDA) is the most common mode for a high-throughput proteomic experiment. This mode provides a high proteome coverage, generating sequences for several thousands of peptides and identification of their cognate proteins, but has limited sensitivity for very-low-abundance peptides. B: data-independent acquisition (DIA) is an emerging technique that strives to measure every peptide in the complex samples by coisolating and fragmenting several peptides together. The resulting spectra are more complicated than DDA spectra and require specialized software for interpretation. C: selected reaction monitoring (SRM) and parallel reaction monitoring (PRM) are collectively considered as techniques for targeted proteomic study. These high-sensitivity techniques can detect lower abundance peptides but require a list of predefined peptide sequences that will be selectively isolated by the first mass spectrometer (MS1) for subsequent fragmentation and analysis by MS2 [using either pre-selected “diagnostic” fragment product ions (SRM) or a full MS2 spectrum (PRM)].
To overcome the limitations of DDA, the DIA strategy has been developed to acquire the entire range of precursor ions (FIGURE 4B). The most well-known DIA technique called SWATH (sequential window acquisition of all theoretical fragment-ion spectra) divides an entire MS1 mass window into smaller consecutive mass windows (e.g., windows of 25 m/z units wide, as shown in FIGURE 4B, left) (57). All precursor ions in each mass window are isolated, fragmented, and analyzed simultaneously, creating a complex MS2 spectrum. This process is sequentially performed across the entire mass range, systematically collecting MS2 data from all detected precursors. The resulting fragment spectra are more complex than those obtained by DDA technique and require specialized algorithms to identify and quantify peptide sequences.
Another strategy developed to circumvent the reproducibility issue of the DDA technique is the so-called targeted strategy (FIGURE 4C) (54, 131). In contrast to DDA, the acquisition process for the targeted strategy is controlled by a predefined list of peptide masses of interest, thereby increasing the consistency of identifying the targeted peptides between experiments. A prior knowledge and selection of specific peptide masses (and hence, proteins) of interest is required for the utilization of this strategy, a task often completed using spectral libraries. Two common targeted methods are illustrated in FIGURE 4C. In the selected reaction monitoring method, a set of transition pairs (each pair consisting of m/z values for the selected precursor and one of its fragment ions) for each target is required for the setup; ideally, the expected chromatographic retention time of each transition pair can also be specified. During MS, the acquisition for each transition pair begins with an isolation of the pre-specified precursor ion using a very narrowly selective mass filter. After fragmentation, a second mass filter is applied on the resulting fragment ions to select for the pre-specified fragment ion. Alternatively, the parallel reaction monitoring method requires only a list of preselected precursor ion m/z values (plus optional expected retention times). The acquisition for each pre-specified precursor ion is similar to selected reaction monitoring method; however, after fragmentation, a full MS2 scan is performed for each targeted precursor ion. Targeted proteomics is reproducible and is robust even in cross-laboratory comparisons (1).
C. An Overview of Protein/Peptide Quantification Methods for MS-Based Proteomics
To elucidate complex dynamic biological responses to physiological stimuli, it is crucial to quantify various parameters of proteins, including abundance, PTMs, intermolecular interactions, and localization in a large-scale fashion, as explained in the first section of this review. The ultimate goal of quantitative proteomics analysis is to determine the copy number of every protein in samples, also known as “absolute quantification.” However, this goal is currently difficult to achieve in a high-throughput manner. More commonly, “relative quantification” techniques, which measure the amount of a particular protein in one condition relative to another condition, have been successfully utilized in numerous proteomic studies. Thus we will discuss the principle of relative quantification in proteomics in the following paragraphs.
Two strategies could be used for the relative quantification in MS-based proteomics studies: 1) stable isotopic labeling methods, and 2) label-free methods. For stable isotopic labeling methods, various combinations of heavy isotopes (such as deuterium, carbon-13, nitrogen-15, and oxygen-18) are employed to create predictable mass shifts of peptides, while preserving other physicochemical properties, either before or after proteolytic digestion. The labeling of each sample with a unique isotope combination allows mixing of samples from different conditions because the same peptide from each sample has a distinguishable mass observed in LC-MS/MS analysis. This technique eliminates the variation between each LC-MS/MS run and allows more accurate relative quantification. Several labeling reagents have been developed for quantitative proteomics including SILAC (stable isotope labeling by amino acid in cell culture) (123), dimethyl labeling (70), and isobaric labeling [such as tandem mass tag (TMT), (162) and isobaric tags for relative and absolute quantitation (iTRAQ) (143)]. FIGURE 5A illustrates examples of different stable isotopic labeling methods. In SILAC, special amino acids containing heavy isotopes are metabolically incorporated into proteins of each sample. Labeled samples are then mixed, processed, and subjected to LC-MS/MS analysis, thus minimizing variation from sample preparation procedures. Quantitative data could then be obtained from MS1 spectra based on relative ion intensity of light and heavy forms of individual peptides (FIGURE 5A, top right). On the other hand, dimethyl labeling and isobaric labeling employ chemical reactions at the peptide level to “mark” individual peptides from each sample at the peptide level. The different mass shifts between light, medium, and heavy dimethyl-labeled peptides in MS1 spectra allows comparison of precursor ion intensities for an individual peptide sequence between each sample (FIGURE 5A, middle right), similar to the SILAC method. Alternatively, isobaric labeling uses a set of special chemical tags that have exactly the same mass (i.e., isobaric), therefore producing sample-inseparable MS1 peaks. On fragmentation in the mass spectrometer, these isobaric tags are designed to generate mass-differing reporter product ions for which MS2 intensities are quantified (FIGURE 5A, bottom right). This strategy currently permits highly multiplexed proteomics analyses for up to 11 samples simultaneously (and potentially even more as new higher-resolution individual mass tags become available).
FIGURE 5.

Quantification methods for mass spectrometry (MS)-based proteomics. A: label-based quantification methods incorporate various labeling molecules with different isotopic mass properties into each sample. These techniques can be performed at either the protein or peptide level. The mass difference of labeled peptides is distinguishable by mass spectrometry, enabling the mixing of multiple samples for the same liquid chromatography-tandem mass spectrometry (LC-MS/MS) run. B: diagrams illustrate the principle of an extracted ion chromatogram (XIC). This reconstructed curve integrates ion intensity information from multiple spectra across the dimension of time [i.e., “elution time” or “retention time” (RT)] for a specific peptide. The area under an XIC curve can be used as a measure of abundance for that specific ion (i.e., peptide). C: diagrams illustrate a comparison of label-free vs. stable isotope-based quantification for two different samples (e.g., replicates, or control and experimentally treated samples). While sample preparation is much simpler for label-free methods, stable isotope-based methods reduce the total number of LC-MS/MS analyses required, as well as eliminating (or reducing) chromatographic variation between runs.
An additional factor complicating quantification in MS-based proteomics is the fact that individual peptides obviously do not elute instantaneously from the chromatography column; instead, peptides elute as “peaks” over a finite time interval. Thus measuring the amount of an individual peptide actually requires integration of its ion intensities over the duration of its chromatographic elution time [also called “retention time” (RT)]. The resulting chromatogram is called an extracted ion chromatogram (XIC) (115). FIGURE 5B illustrates the XIC reconstruction process for one precursor ion with m/z value m1 from six sequential MS1 spectra. This concept could be applied to both MS1 and MS2 level quantification techniques.
The label-free quantification approach compares the abundance of a particular precursor ion from one LC-MS/MS run to the corresponding ion from another run. The ease of sample preparation is clearly an advantage of the label-free approach. In addition, without any labeling reagent requirement, a limitless number of conditions can be compared. A diagram illustrating the concept of label-free quantification, in comparison to stable isotope-based quantification, is shown in FIGURE 5C. Two XICs of a precursor ion with m/z value m1 from LC-MS/MS run numbers 1 and 2 are compared for the label-free method (FIGURE 5C, left). Some degree of retention time shift can be observed in FIGURE 5C, demonstrating the expected run-to-run chromatographic variation. In contrast, label-based quantification allows simultaneous comparison of each XIC from multiple samples within a single LC-MS/MS run, thus eliminating the variation between runs (FIGURE 5C, right).
III. APPLICATION OF FUNCTIONAL PROTEOMICS TO RENAL TUBULE PHYSIOLOGY
The human kidney consists of ~1 million functional units, called nephrons (and the associated collecting ducts) (FIGURE 6). The nephron consists of a glomerulus, where the primary urine is filtered, and the renal tubules, with specialized “segments” organized in series. The nephron is followed by the collecting duct system, which carries out fine regulation of urine volume and composition. The renal tubule is responsible for multiple transport processes and consists of four major specialized segments: proximal tubule, loop of Henle, distal convoluted tubule (DCT), and collecting duct. When the primary urine flows through this system, solutes, metabolites, and electrolytes are reabsorbed or secreted, and these processes require tight regulation to maintain body homeostasis. To this end, different tubule epithelia express specific transporters, channels, and pumps that are distributed with segment-specific localization patterns, often in a polarized manner, along the tubule. Collectively, the regulation of these transport proteins is essential for maintaining body homeostasis in response to changing environmental conditions. Activities of these transport proteins are tightly controlled by gene expression, PTMs, and/or protein turnover. In the past decade, significant insights into the regulation of these proteins have been generated chiefly by applying MS-based proteomic analyses to renal tubule epithelia. The aim of this review section is to provide a systematic overview of insights into these processes obtained from proteomics-driven studies. FIGURE 6 shows the major physiological processes examined in the reviewed proteomic studies of the four major renal tubule segments. As can be seen in FIGURE 7A, the number of publications in this field has significantly increased since 2001, the year the human genome project was completed (92). Proteomic studies focused on the various kidney segments and urine have been performed (FIGURE 7B). To systematically summarize the studies involving renal tubule physiology, we grouped the studies according to the main renal segments examined (proximal tubule, the loop of Henle, distal tubule, or collecting duct). Some of these studies were selected for in-depth analysis in this review to highlight technological or biological advancements of our knowledge of kidney physiology. In each section, we first review the sample preparation protocols. Then we review profiling studies (which are not quantitative studies per se), followed by quantitative studies in response to physiological stimuli.
FIGURE 6.

Schematic diagram of the nephron and associated collecting ducts. For each renal tubule segment, the major physiological responses that have been studied using proteomic techniques are listed. BBMVs, brush-border membrane vesicles; PTH, parathyroid hormone.
FIGURE 7.
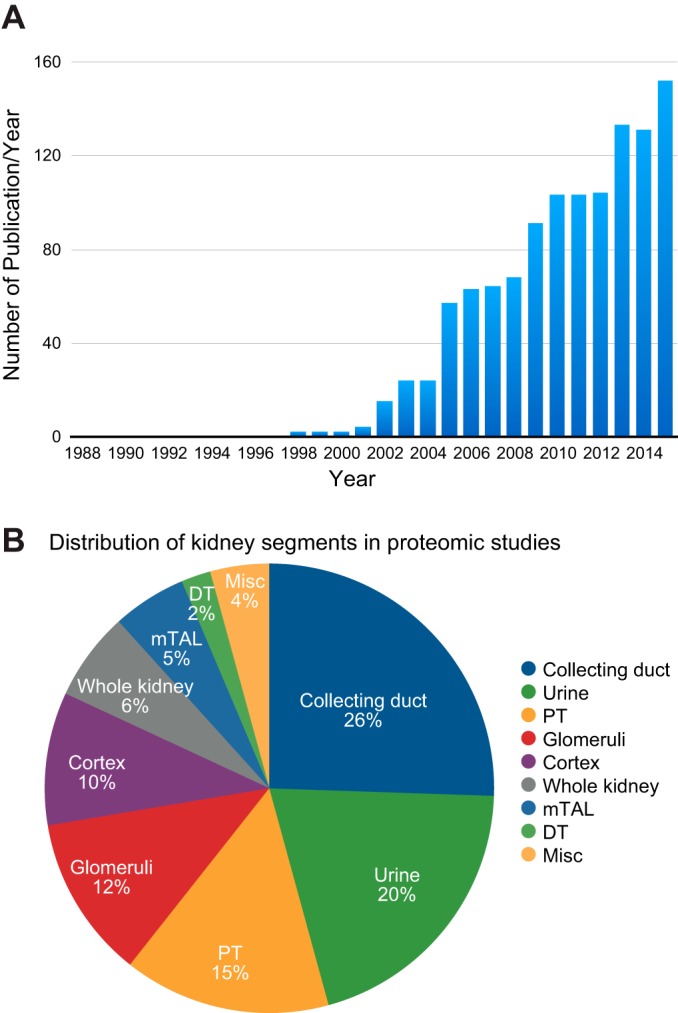
Analysis of kidney-related proteomic studies by category. A: the number of kidney-related proteomic studies appearing in each publication year. This analysis illustrates the rapid increase in proteomics research activity that coincided with completion of the human genome project. B: the distribution among kidney-related proteomic studies for the different anatomical components of the kidney (and urine). DT, distal tubules; mTAL, medullary thick ascending limb; PT, proximal tubules.
A. Response to Metabolic Acidosis and Angiotensin in the Proximal Tubule
1. Isolation
The proximal tubule is the renal tubule segment that reabsorbs the majority of solutes from the primary urine. The isolation of proximal tubules from native kidneys has been performed in many renal physiological studies and modelling. One of the commonly used isolation methods is Percoll density gradient centrifugation, in which the renal cortical tissue is removed by dissection and digested with collagenase to release the cortical tubules, which are then resuspended in a 42.5–45% Percoll solution and centrifuged. Approximately 95% purity of proximal tubules can be obtained with this method (28, 38, 52, 53, 99, 148, 170, 171). Recently, a rapid immunofluorescence staining technique coupled with laser microdissection was successfully used for isolating proximal tubule segments from mouse kidney tissues (111). Since proximal tubule cells are highly polarized epithelial cells expressing many solute transporters along their apical brush-border plasma membrane, the isolation of this membrane compartment has played a significant role in many earlier physiological studies of proximal tubules. To isolate brush-border membrane vesicles (BBMVs) from proximal tubules, the MgCl2 precipitation method has been used as a standard technique (16). Feric et al. (51) and Walmsley et al. (170, 171) applied this technique to obtain a highly enriched BBMV preparation for studying the BBMV proteome from rat proximal tubules. The mitochondrion is another subcellular fraction in proximal tubule cells that has been studied to a great extent because of its crucial role in the many active transport processes occurring in these cells. To enable more in-depth proteomic studies of this organelle, mitochondrial isolation from proximal tubule cells has been accomplished using differential and sucrose density centrifugation (52, 53).
2. Profiling
Several studies have been performed to reveal the proteome and subproteome of proximal tubule cells [including apical membrane (BBMV), mitochondrial, and cytosolic fractions] (53, 148, 171). These results were combined and deposited in a publicly accessible online database (https://hpcwebapps.cit.nih.gov/ESBL/Database/PCT/), which currently contains information for 1,660 proteins. To demonstrate the utility of this data set, Table 1 lists apical membrane transporters (identified via BBMV isolation) extracted from this database and sorted based on their abundance levels (calculated by normalized spectral counting). As expected based on previous knowledge in the renal physiology field, transport proteins for solutes, and water were the most abundant proteins detected in the brush border membrane. In addition, the observed order of relative abundance for specific transport proteins (neutral amino acids > basic amino acids > water > sodium-glucose > sodium-bicarbonate > sodium-phosphate > other solutes) provides additional insights that can be useful for further physiological studies and modeling. Feric et al. (51) carried out shotgun phospho-proteomic analysis of proximal tubule membrane proteins from normal rats, reporting 743 phospho-peptides, many of them novel and many of them in key transporter proteins of the proximal tubule. For example, two novel sites were identified in the COOH-terminal tail of Na-Pi2 (Slc34a1) and in various domains in the Na/H exchanger (NHE3) (Slc9a3).
Table 1.
A list of apical membrane transporters reported in a proximal tubular cell proteome database
| Accession No. | Gene Name | Protein Name | Transporter Activity | Normalized Spectral Counts |
|---|---|---|---|---|
| Q794F9 | Slc3a2 | Solute carrier family 3 (amino acid transporter heavy chain), member 2 | Neutral amino acid | 85.39 |
| Q64319 | Slc3a1 | Solute carrier family 3 (amino acid transporter heavy chain), member 1 | l-cystine; amino acid; basic amino acid | 43.44 |
| P29975 | Aqp1 | Aquaporin 1 | Ammonium; carbon dioxide; glycerol; nitric oxide; potassium ion; water | 35.95 |
| P53792 | Slc5a2 | Solute carrier family 5 (sodium/glucose cotransporter), member 2 | Glucose-sodium symporter | 32.34 |
| Q9JI66 | Slc4a4 | Solute carrier family 4, sodium bicarbonate cotransporter, member 4 | Sodium-bicarbonate symporter | 22.24 |
| Q2A865 | Slc6a19 | Solute carrier family 6 (neutral amino acid transporter), member 19 | Neurotransmitter-sodium symporter; neutral amino acid | 20.50 |
| Q06496 | Slc34a1 | Solute carrier family 34 (type II sodium/phosphate cotransporter), member 1 | Sodium-phosphate symporter | 17.27 |
| P51907 | Slc1a1 | Solute carrier family 1 (neuronal/epithelial high affinity glutamate transporter, system Xag), member 1 | l-glutamate; cysteine; glutamate-sodium symporter; sodium-dicarboxylate symporter | 12.10 |
| D3Z9E5 | Slc5a8 | Solute carrier family 5 (sodium/monocarboxylate cotransporter), member 8 | Sodium-monocarboxylate symporter | 9.28 |
| Q9R1U7 | Slc22a8 | Solute carrier family 22 (organic anion transporter), member 8 | Organic anion; quaternary ammonium group | 6.85 |
| B1WBS5 | Slc5a10 | Solute carrier family 5 (sodium/sugar cotransporter), member 10 | Sodium/glucose cotransporter | 6.16 |
| P28570 | Slc6a8 | Solute carrier family 6 (neurotransmitter transporter), member 8 | Choline; creatine; creatine-sodium symporter; neurotransmitter-sodium symporter | 5.78 |
| Q8K4R8 | Slc34a3 | Solute carrier family 34 (type II sodium/phosphate cotransporter), member 3 | Sodium-phosphate symporter | 5.54 |
| Q62687 | Slc6a18 | Solute carrier family 6 (neutral amino acid transporter), member 18 | Amino acid; neurotransmitter-sodium symporter | 4.88 |
| Q80WK7 | Slc29a3 | Solute carrier family 29 (equilibrative nucleoside transporter), member 3 | Nucleoside | 4.21 |
| P53790 | Slc5a1 | Solute carrier family 5 (sodium/glucose cotransporter), member 1 | Glucose; glucose-sodium symporter | 4.02 |
| Q63089 | Slc22a1 | Solute carrier family 22 (organic cation transporter), member 1 | Acetylcholine; dopamine; monoamine; norepinephrine; organic cation; quaternary ammonium group; secondary active organic cation | 3.60 |
| P53987 | Slc16a1 | Solute carrier family 16 (monocarboxylate transporter), member 1 | Lactate | 2.69 |
| A2VD10 | Slc13a3 | Solute carrier family 13 (sodium-dependent dicarboxylate transporter), member 3 | l-Aspartate; citrate; dicarboxylic acid; high-affinity sodium-dicarboxylate symporter; organic acid-sodium symporter; sodium-dicarboxylate symporter; succinate | 2.22 |
| Q80W57 | Abcg2 | ATP-binding cassette, subfamily G (WHITE), member 2 | Drug | 2.02 |
| Q3ZAV1 | Slc22a12 | Solute carrier family 22 (organic anion/urate transporter), member 12 | Organic anion; urate | 1.81 |
See https://hpcwebapps.cit.nih.gov/ESBL/Database/PCT/ (53, 148, 171). The list is sorted based on the abundance levels of transporters (calculated from spectral counts normalized by amino acid length) for normal/control rats.
3. Proximal tubule response to metabolic acidosis
One of the major roles of proximal tubules is in the regulation of acid-base balance. Most of the filtered bicarbonate is effectively handled by a process called “bicarbonate reclamation” in proximal tubules, helping maintain serum bicarbonate levels within the normal range. A defect in this process can cause renal bicarbonate loss and the resultant metabolic acidosis. In situations that increase acid production in the body, proximal tubules will respond by enhancing plasma glutamine extraction and glutamine catabolism, promoting the generation of intracellular ammonium and bicarbonate ions that assist the excretion of excess acid. In the response to metabolic acidosis, the ammonium is selectively secreted into the urine, while the bicarbonate exits across the basolateral plasma membrane into the blood.
Previously, using traditional approaches, metabolic acidosis has been demonstrated to increase the expression levels of several transporters and enzymes in proximal tubules that facilitate glutamine uptake and catabolism, such as the basolateral glutamine transporter SLC38A3 (sodium-coupled neutral amino acid transporter 3), the apical NHE3, the mitochondrial aquaporin-8 (AQP8), phosphoenolpyruvate carboxykinase (PEPCK), glutaminase (GA), and glutamate dehydrogenase (37). Recently, a series of proteomic studies from the Curthoys laboratory has further expanded the molecular understanding of this complex adaptive response in proximal tubules as described below.
First, Curthoys et al. (38) used two-dimensional difference gel electrophoresis (2D-DIGE) to analyze changes in the total cellular proteome of renal proximal tubules from acidotic rats. Acidosis was induced in rats by stomach loading of NH4Cl, followed by its supplement in drinking water. After 2 h and 1, 3, and 7 days of the experiment, proximal tubules from acidotic rats were isolated by Percoll density gradient centrifugation (∼95% pure) and then studied using the 2D-DIGE technique. The results confirmed the adaptive increases in the protein abundance of GA and PEPCK in response to metabolic acidosis, as previously described (79, 164). In addition, 17 and 16 new proteins were found to be increased and decreased, respectively, after 7 days of metabolic acidosis. The time course analysis of these protein abundance changes allowed the investigators to classify the kinetic profiles of these altered proteins into two groups (i.e., those exhibiting rapid vs. gradual changes), thus assisting them in the formulation of a theoretical mechanism responsible for specific changes. The finding that all of the mRNAs encoding the gradually-changed proteins contain a putative pH response element within their 3′-untranslated region led to the conclusion that selective mRNA stabilization may be the primary mechanism for regulating levels of the proteins that were gradually increased in response to metabolic acidosis.
Second, Walmsley et al. (171) initiated a study at the subproteome level of proximal tubule cells by investigating the effect of metabolic acidosis on BBMV proteome using quantitative MS. Using the MgCl2 precipitation technique, BBMVs were isolated from proximal tubules (which were initially purified by Percoll density gradient centrifugation) from 1-, 3-, and 7-day acidotic rats and then subjected to a label-free LC-MS/MS analysis. In total, 298 proteins were identified, for which about one-fourth were predicted to have at least one transmembrane domain. The quantitative analysis of protein expression for control and each acidosis time point revealed several patterns of temporal changes in the protein abundance levels of apical membrane and associated proteins, reflecting the intricate dynamic molecular adaptation to acute and chronic phases of metabolic acidosis in proximal tubules. These changes were found in various groups of proteins that are essential to the adaptation, such as transporters (e.g., SLC5A2 and SLC5A8 were significantly increased), peptidases (e.g., γ-glutamyltranspeptidase and glutamate carboxypeptidase were significantly increased), glycolytic and gluconeogenic enzymes (most of them were significantly decreased), and trafficking proteins (e.g., myosin-9 and DAB2 were significantly increased and decreased, respectively).
In the third study of the series, mitochondrial proteins from proximal tubules were extracted and compared between control and 7-day chronic acidotic rats by two-dimensional LC-MS/MS (53). Based on spectral counting, the abundance levels of 33 proteins (from the total identification of 901 proteins) were reported to be significantly altered in the acidotic condition. This study, again, confirmed the upregulation of the two crucial enzymes in glutamine catabolism, GA, and glutamate dehydrogenase, in response to metabolic acidosis. In addition, the other regulated proteins discovered included not only several mitochondrial proteins, but also proteins from other organelles that were co-isolated with mitochondria (by differential and sucrose density centrifugation), such as peroxisomes and the ER. Among these regulated proteins, an increase in the expression level of mitochondrial carbonic anhydrase (CA5B) was of particular interest. This adaptation might help increase the translocation of bicarbonate ions generated in the mitochondria to the cytosol, thus facilitating the restoration of acid-base balance. Since acetylation of lysine in known to alter the abundance of mitochondrial proteins (188), the authors investigated lysine acetylation of mitochondrial proteins from the chronic acidotic rats using immunoblotting (probed with an anti-acetyl lysine antibody) and observed a marked increase in a number of these proteins. Subsequently, LC-MS/MS analysis identified a total of 39 lysine acetylation sites, 22 of which were novel sites. Future studies are required to investigate the role of lysine acetylation in the regulation of glutamine catabolism in mitochondria during metabolic acidosis.
In an attempt to complete the analysis on all of the relevant subcellular fractions from proximal tubule cells, the last study was conducted to perform a quantitative proteomic analysis of the cytosolic fraction isolated from proximal tubules of acidotic rats (148). This study identified a total of 461 proteins, 24 of which showed statistically significant changes in abundance in response to metabolic acidosis, as evaluated by spectral counting and average MS/MS total ion current. To better understand the complex molecular response to metabolic acidosis, the data from the apical membrane (BBMV), mitochondrial, and cytosolic proteome studies were combined and analyzed with the STRING database and network analysis tool for identifying/predicting protein-protein interactions (https://www.string-db.org/cgi/input.pl). For the upregulated proteins, clusters of interconnected proteins involved in ammoniagenesis, retinol metabolism, hydrogen ion transport, and fatty acid metabolism were identified. On the other hand, only a cluster of proteins involved in phenylalanine metabolism was detected for the downregulated proteins. The comprehensive list of proteins identified and quantified in this series of proteomic studies can be found at https://hpcwebapps.cit.nih.gov/ESBL/Database/PCT/. Although a significant amount of information has been revealed by these proteomic studies, future studies will still be needed to determine the precise role of each altered protein and the dynamic interplay between them in the adaptive response of proximal tubules to metabolic acidosis. Systems-level studies of the other modes of protein regulation in proximal tubules beyond abundance changes, e.g., PTMs (phosphorylation, acetylation, and ubiquitination), subcellular translocation, and protein-protein interactions, would help further elucidate this adaptive process.
4. Angiotensin and the proximal tubule
The renin-angiotensin-aldosterone system is a key regulator of blood pressure and sodium reabsorption in the kidney. The proximal tubule is one of the target sites of the activated peptide hormone angiotensin II (135). Many proteomic studies have been conducted to comprehend the molecular physiology of proximal tubule response to angiotensin stimulation in terms of total proteome and signaling phosphoprotein abundance changes, as well as response to the inhibition of angiotensin II production.
To comprehensively uncover proteome changes in response to angiotensin, Konvalinka et al. (89) incubated SILAC-labeled primary human proximal tubule cells with and without 10−7 M angiotensin II for 8 h. Two-dimensional LC-MS/MS analysis identified a total of 5,011 proteins, 53 and 30 of which were upregulated and downregulated, respectively. Gene ontology analysis revealed that biological processes, such as regulation of immune response, cell proliferation, and response to stress, were enriched in the upregulated proteins, while the regulatory process of lipoprotein particle clearance was enriched in the downregulated proteins. Importantly, heme oxygenase 1 was identified as the most significantly upregulated protein in response to angiotensin II stimulation, and this finding was validated via an in vivo experiment in angiotensin II type 1 receptor (AT1R) knockout mice. Heme oxygenase 1 in urine was also positively correlated with its kidney expression and could be a promising tool for noninvasively measuring angiotensin II activity in proximal tubules.
In another study, phosphorylation-mediated signaling in response to angiotensin II was examined by pathway-specific multi-immunoblotting to quantify 38 phosphorylation sites of signaling proteins in proximal tubules (99). Rats were treated with angiotensin II at a pressor or nonpressor dose for 2 wk in the presence or absence of an AT1R blocker, losartan, and then proximal tubules were freshly isolated and analyzed. The abundance of 14 phosphoproteins was demonstrated to be significantly altered by the pressor dose of angiotensin II, and these changes could be generally restored by losartan. With the nonpressor dose of angiotensin II, only seven phosphoproteins were reported to show changes in their abundance. Overall, this study revealed major responses to angiotensin II in signaling for protein kinase C, glycogen synthase kinase 3, and cAMP-dependent pathways in proximal tubules.
Leong et al. studied the effect of blocking angiotensin II formation (using captopril, an angiotensin-converting enzyme inhibitor) on the redistribution between different membrane domains of apical membrane-associated proteins in rat proximal tubules (98). After treatment with intravenous captopril for 20 min, a significant diuretic response, as demonstrated by an increase in urine output and proximal tubule flow, was induced in rats. Subsequently, rat kidney cortical tissues were homogenized and then fractionated by density gradient centrifugation to segregate the two apical membrane domains of proximal tubule cells, i.e., those located either at apical microvilli (low density) or intermicrovillar cleft (higher density). Based on immunoblot analysis, proximal tubule Na+ transporters, NHE3 (SLC9A3), and NaPi2 (SLC34A1), and their candidate regulators, NHERF-1 (SLC9A3R1) and unconventional myosin-VI (MYO6), were found to redistribute from low-density apical microvillus-enriched fractions into higher-density intermicrovillar cleft fractions after captopril administration. To discover more proteins that exhibited the same redistribution pattern as NHE3, an apical microvillus-enriched fraction that demonstrated a large redistribution of NHE3 was investigated by SDS-PAGE coupled with MALDI-time-of-flight MS (MALDI-TOF MS). Eight Coomassie blue-stained bands that showed a discernible difference in density between control and captopril-treated samples were detected, and the following proteins were identified in those bands: megalin, myosin II-A, clathrin, aminopeptidase N, dipeptidyl peptidase IV, ezrin, moesin, and vacuolar H+-ATPase subunit-β2. This redistribution of apical transporters and associated proteins in proximal tubules is potentially involved in the diuretic response and may contribute to the antihypertensive effect of angiotensin-converting enzyme inhibitors.
B. Response to Osmotic Stress and Hormonal Activation of cAMP in the Thick Ascending Limb of the Loop of Henle
1. Isolation
The loop of Henle is a major tubule segment regulating urinary concentrating ability of the kidney. Medullary thick ascending limbs (mTAL) of Henle’s loop cells can be isolated using the density gradient centrifugation method (47). Gunaratne et al. (62) slightly modified this method to isolate mTAL tubules from rat kidneys, which helped shorten the isolation duration, thus preserving tubule viability. Due to a high aerobic energy consumption for this cell type, furosemide was injected to the animals before mTAL isolation, which inhibited the activity of the sodium-potassium-chloride cotransporter (NKCC2), the main sodium transporter in mTAL, ultimately reducing ATP consumption by the Na-K-ATPase. In addition, oxygenated buffer was used during digestion with collagenase. The mTAL tubules were isolated by low-speed centrifugation, which separated the heavier mTAL tubules from other structures in the renal medulla. Alternatively, Zheleznova et al. (190) used a sieving approach with 100-µm nylon mesh to retain long mTAL tubules. This method allowed quick mTAL isolation with a purity of 93.6%. Another approach to isolate TAL segments is immunofluorescence laser microdissection (111). With a staining of cell-type specific markers, this technique could permit an isolation of target areas from tissue sections with very high purity.
2. Profiling
Proteomic profiling of native mTAL cells has been performed only on mitochondrial-and phosphoprotein-enriched fractions of mTAL cells; therefore, comprehensive proteomic studies of this segment have not yet been performed. In a study by Zheleznova et al. (190), mitochondria of mTAL cells were isolated from Dahl salt-sensitive rats and salt-resistant control rats and analyzed with LC-MS/MS. Ninety-six mTAL mitochondrial proteins were identified. Another study examined phosphoproteome of rat mTAL cells (62). A total of 654 phosphopeptides were identified (https://esbl.nhlbi.nih.gov/mtal-phospho/).
3. Response to osmotic stress
The tight regulation of cell volume is important for a cell to maintain its proper function. Extracellular osmolality is the major factor that affects cell volume. Cells in the renal medulla, including mTAL cells, are usually exposed to a high osmolality environment. Thus these cells must have special adaptations to conquer challenging osmotic conditions. Several studies by Dihazi et al. (17, 21, 41) used proteomic tools to explore these adaptations in an mTAL cell line derived from the outer medulla of a rabbit kidney. The cells were stressed with a 600 mosmol/kgH2O NaCl medium, compared with a control group cultivated in a standard medium (300 mosmol/kgH2O). Two-dimensional gel electrophoresis coupled with MALDI-TOF MS analysis was employed for protein quantification and identification. Consistent with preexisting knowledge (22), aldose reductase was identified among the upregulated proteins. This protein is known to mediate sorbitol synthesis; thus the increase in its abundance could explain the adaptive increase in intracellular osmolality of the mTAL cells. The proteomic study further identified 24 additional upregulated proteins, including proteins involved in energy metabolism, cytoskeleton-associated proteins, and molecular chaperone proteins. Moreover, four differentially expressed vimentin isoforms were also identified. In addition to osmotic stress due to NaCl, high glucose (30 vs. 5.5 mM for control cells) was also used to induce osmotic stress in the mTAL cells (21). This condition appeared to regulate different isoforms of vimentin in comparison to the high NaCl condition. Several ER-calcium binding chaperones, namely 78-kDa glucose regulated protein, Erp72, and calreticulin, were downregulated in response to NaCl-induced osmotic stress, and these changes were reversible after the high osmolality medium was exchanged back to a normal medium. The follow-up experiments suggested that the calcium binding and storage capacity of calreticulin is one of the key mechanisms that promotes the adaptive response of mTAL cells to osmotic stress (17).
4. Regulation of solute transport by phosphorylation-driven networks
Solute transport in the thick ascending limb of Henle’s loop is regulated by many hormones and intracellular chloride levels. Parathyroid hormone, calcitonin, glucagon, and vasopressin are known to trigger intracellular signaling networks in TAL cells via heterotrimeric G protein receptors that mediate an increase in the second-messenger cAMP (114). However, the downstream signaling pathways of these hormones are still elusive. To further elucidate the pathways involved, Gunaratne et al. (62) isolated mTAL tubules and incubated with a cocktail of these four hormones for 15 min. It was shown that the mixture of all four hormones induced the highest cAMP level, compared with levels with each hormone alone. The phosphoproteome of mTAL treated with the hormone mixture was compared with the control. Enrichment of phosphopeptides was done using Ga3+-IMAC. Label-free quantitative LC-MS/MS analysis found a total of 654 unique phosphopeptides; among these, 48 and 28 peptides were significantly increased and decreased, respectively. Bioinformatics analysis revealed many important findings. First, for upregulated phosphopeptides, most phosphorylations occurred at basophilic sites (a phosphorylated residue surrounded by basic amino acids, i.e., arginine, lysine, or histidine). On the other hand, a majority of downregulated phosphopeptides exhibited proline-directed sites (a phosphorylated residue surrounded by a proline, usually at positions +1 and −2). This study was one of the first to identify specific classes of kinases differentially regulated in the response to cAMP. Overall, the largest groups of phosphoproteins regulated by cAMP included transmembrane transporters, protein phosphatase regulators, and cytoskeletal binding proteins. For example, an interesting finding from the proteomic analysis was an increase in phosphorylation of a basophilic site, S552, of NHE3. This phosphorylated site is known to inhibit NHE3 transport activity in the proximal tubule (187). In an in vitro experiment to test the proteomic results, a basophilic kinase, protein kinase A, was shown to phosphorylate NKCC2 at S126 and S874, both of which were upregulated in the proteomic analysis. Finally, a minority of cAMP-regulated phosphorylated sites detected were neither basophilic nor proline-directed sites, such as T96 of NKCC2 (which was increased by cAMP activation). Another group of researchers used proteomic techniques to study the regulation of NKCC2 in response to hypotonic low-chloride conditions (138). They found that five phosphorylated sites on the NH2-terminal tail of NKCC2, i.e., S87, T91, T96, T101, and S126, were upregulated in NKCC2-overexpressing human embryonic kidney-293 cells when they were exposed to a hypotonic low-chloride medium. Mutation of T101 and S126 sites was demonstrated to significantly reduce the NKCC2 activity, supporting the physiological role of these phosphorylated residues. While the WNK/SPAK/OSR1 signaling pathway was shown to be responsible for T91, T96, and T101 phosphorylation, AMP-activated protein kinase potentially accounted for S126 phosphorylation in this hypotonic low-chloride condition, but a kinase for S87 was not identified. FIGURE 8 illustrates the proposed signaling network that regulates NKCC2 and NHE3, the key sodium transporters in TAL cells, from these proteomic studies. With a better insight in NKCC2 regulation, novel therapeutic agents could be developed to treat sodium-retention states that are resistant to the conventional diuretics.
FIGURE 8.

Signaling network regulating sodium-potassium-chloride cotransporter (NKCC2) and Na/H exchanger (NHE3) in thick ascending limb (TAL) cells. A proposed signaling network for various stimuli that upregulate NKCC2 and NHE3 activities in TAL cells is shown. Parathyroid hormone (PTH), glucagon, calcitonin, vasopressin, and β-adrenergic agonists mainly activate the adenylyl cyclase/cAMP pathway, which subsequently activates protein kinase A (PKA). Other downstream mediators of cAMP, which phosphorylate T96 of NKCC2 and S552 of NHE3, are still unidentified. The low-chloride hypoosmotic condition also activates NKCC2 through WNK/SPAK/OSR and AMPK pathways. S87 of NKCC2 is also found to be phosphorylated in the low-chloride condition; however, the kinase responsible for this phosphorylation is still unidentified.
C. Response to Aldosterone and Vasopressin in the Distal Convoluted Tubule and Connecting Tubule
1. Isolation
The distal tubule controls fine adjustment of renal salt excretion. Late segments of the DCT2, along with connecting tubules (CNT) and initial cortical collecting ducts (iCCD), were successfully isolated for proteomic study using a fluorescence-activated cell sorting (FACS) method (80). Kidneys from transgenic mice expressing enhanced green fluorescent protein (eGFP) driven by the TRPv5 promoter were harvested, and tubules were digested to single cells with trypsin. These cells were sieved through 40-μm mesh filters and isolated using a FACSAria III instrument (BD Biosciences). Approximately 1.4 million cells per mouse could be isolated. Even though this technique can help effectively isolate specific cells, it requires expression of transgenic fluorescence proteins.
2. Profiling
The MS-based proteomic analysis of the distal nephron isolated by FACS (>70% purity, based on TRPv5-eGFP positive cells) identified a total of 506 unique proteins (80). However, this study included only the late distal tubule segments (i.e., DCT2, CNTs, and early CCDs). Another proteomic study related to DCTs was recently reported (26). Using cultured mpkDCT cells (originally isolated from microdissected mouse DCT), this study provided a more comprehensive list of proteins associated with distal tubule-derived cells. In total, 6,330 proteins were identified, including 3,270 phosphoproteins. This database can be accessed via http://interpretdb.au.dk/database/mpkDCT/Total_Proteome.html for the total proteome and via http://interpretdb.au.dk/database/mpkDCT/Phosphoproteome.html for the phosphoproteome. Analysis of this proteome, together with the total mouse kinome from Kinbase, also provided a comprehensive DCT kinome, including 186 kinases.
3. Response to aldosterone
Aldosterone exerts its effects on the aldosterone-sensitive portions of the distal nephron, including DCT2, CNT, and CCD (108). Jensen et al. (80) studied the proteome changes at 24 h in the transgenic TRPv5-eGFP-positive distal tubule cells after a single subcutaneous injection with aldosterone (2 mg/kg). Using an isobaric tag for relative and absolute quantification (iTRAQ), only 20 proteins were identified as differentially expressed (mostly downregulated) after aldosterone treatment. DCT cells are among the most mitochondria rich in the kidney (42), consistent with the consensus that the DCT requires high levels of ATP consumption for active transport of electrolytes driven by the basolateral Na+-K+-ATPase (107). Nevertheless, only five of the identified proteins were mitochondrial enzymes; the remainder representing a diverse group of biological functions. In addition, even though the changes did not reach the threshold level for significance, a trend toward aldosterone regulation was observed for several proteins involved in glycolysis (upregulated) and glyconeogenesis (downregulated). Since, the iTRAQ method has been shown to suffer from a “ratio compression” problem in complex samples that may result in many changes not reaching statistical significance (124), pyruvate kinase (tentatively identified as upregulated) was selected for a confirmation study by immunohistochemistry, and an increase in abundance of this protein in the late distal tubule segments was confirmed. The protein database from this study can be accessed online via https://hpcwebapps.cit.nih.gov/ESBL/Database/STADT/.
4. Response to vasopressin
In addition to the well-known water permeability effect on collecting ducts, vasopressin also affects sodium transport in DCT (44). Cheng et al. (26) conducted a large-scale proteomic analysis of vasopressin-mediated signaling networks in mpkDCT cells. With SILAC-labeling followed by LC-MS/MS, mpkDCT cells treated with 1 nM dDAVP (a selective vasopressin analog) for 15 min were compared with the control. Eighty-six phosphorylation sites were upregulated, and 99 sites were downregulated. A set of responsible kinases could be predicted by analysis of amino acid sequence around these sites. Combining these data with the known regulatory pathways for the sodium-chloride cotransporter (NCC or SLC12A3) also produced a potential vasopressin signaling network in these cells. FIGURE 9 shows a simplified version of the modeled signaling pathway with cAMP/PKA, Ca-calmodulin/CAMKK, and phosphatidylinositol 3-kinase (PI3K) as key mediators downstream of the vasopressin receptor. The roles of these kinases were confirmed in ex vivo experiments with specific kinase inhibitors. Thus vasopressin acts at two levels to regulate NCC: 1) potentiation of NCC transport activity through PI3K-mediated phosphorylation; and 2) attenuation of ubiquitin-mediated NCC degradation through cAMP/PKA and CAMKK activation. Although not all parts of the network were validated, this study exemplifies the elucidation of complex signaling cascades using phosphoproteomics.
FIGURE 9.

Simplified signaling pathway for vasopressin-mediated sodium-chloride cotransporter (NCC) regulation in distal convoluted tubule (DCT) cells. Vasopressin stimulation of DCT cells activates the cAMP, Ca-calmodulin/CAMKK, and phosphatidylinositol 3-kinase (PI3K) second-messenger pathways, which subsequently activate PKA, SGK1, and Akt kinases. Final major effectors of NCC activity are WNK/SPAK/OSR (which phosphorylate NCC) and NEDD4L (which ubiquitinates NCC). Solid black lines represent pathways supported by experimental results for NCC phosphorylation in the presence of specific kinase inhibitors. Dashed lines represent effectors deduced from experiments with other stimuli such as aldosterone. Solid black and dashed gray lines designate stimulatory phosphorylation of NCC, whereas red dashed lines denote inhibitory regulation of NEDD4L, resulting in increased abundance and overall activity of NCC. V2R, vasopressin receptor 2.
D. Response to Vasopressin in the Collecting Duct
1. Isolation
The main physiological roles of the renal collecting duct are to concentrate urine and to control fine adjustment of renal salt and water homeostasis. This is mainly mediated via highly abundant transport proteins, such as urea transporters (e.g., Slc14a2), as well as the vasopressin-regulated water channel aquaporin-2 (AQP2), which translocate to the apical membrane of cells undergoing vasopressin exposure (127). Collecting ducts, especially inner medullary collecting ducts (IMCD), have been isolated for use in many proteomic studies. The method is based on the physical property that IMCD cells are heavier than other non-IMCD cells present in the renal inner medulla. In brief, renal inner medullas are removed by dissection, minced, and then digested with collagenase B and hyaluronidase. After that, the suspensions are subjected to low-speed centrifugation (50–80 g for 10–30 s). This method yields high purity IMCD tubules, characterized by enrichment of AQP2 and depletion of noncollecting duct markers, such as AQP1 (66, 67, 69, 167).
In addition to isolation of the IMCD tubules, further separation of subcellular membrane fractions from IMCD cells would be invaluable in deciphering the molecular physiology of IMCD. Such fractionation steps would allow subcellular localization of proteins and lead to a considerable increase in the depth of proteomic analyses. Some of the fractions that have been studied include AQP2-containing intracellular vesicles, both apical and basolateral plasma membrane fractions, and the nuclei.
In an early study of AQP2 trafficking, standard differential centrifugation was employed for separation of the plasma membrane fraction (17,000 g spin) from the intracellular vesicle fraction (200,000 g spin) (103). However, a subsequent study by Sachs et al. (144) suggested that this approach may not be valid, at least for IMCD. Using MS to compare the proteomes of multiple fractions obtained by sequential differential centrifugation of IMCD homogenates revealed that both the 17,000 g and 200,000 g fractions are highly heterogeneous and cannot be equated with “plasma membrane” and “intracellular vesicle” fractions, respectively. Thus alternative or augmented approaches to differential centrifugation are required for studying AQP2 trafficking.
To selectively isolate AQP2-containing intracellular vesicles, Barile et al. (13) generated biotinylated anti-AQP2 antibodies and mixed these antibodies with streptavidin-coated magnetic beads. The 200,000 g fraction from IMCD cells (including intracellular vesicles) was mixed with the antibody-coated beads to immunoisolate the AQP2-containing vesicles for proteomic analysis. Identification of multiple Rab GTPases confirmed the localization of AQP2 in various endosomal compartments.
For polarized epithelial cells, such as collecting duct cells, each region of the plasma membrane (apical and basolateral) contains a highly distinct set of proteins. As this difference is crucial for the collecting duct to function properly, effective separation techniques for proteins from these specific surfaces of the plasma membrane are of great importance. Yu et al. (183) first successfully biotinylated IMCD apical cell surfaces via perfusion of intact kidney medulla and used avidin affinity chromatography to enrich apical membrane proteins. To biotinylate the apical membrane, a special double-barreled pipette is required to deliver the fixative and biotinylation reagent. Second, for biotinylation of the basolateral membrane, IMCD suspensions were isolated, fixed, and incubated with biotinylation reagent. High-density membrane fractions were then prepared from biotinylated samples by differential centrifugation and enriched for biotinylated basolateral membrane proteins using CaptAvidin-agarose or streptavidin-agarose beads. Biotinylated apical and basolateral membrane proteins were then analyzed in proteomic experiments. A similar approach was applied to a collecting duct cell line (mpkCCD cells) to isolate apical plasma membrane proteins (101).
Isolations of nuclei were performed to study transcriptional regulation by vasopressin in native IMCD and mpkCCD cells, including effects on nucleus-cytoplasmic protein translocation and PTMs (19, 130, 149). In these studies, cellular fractionation into cytoplasm, soluble nuclear extract, and nuclear membrane pellet was accomplished using stepwise extraction by a commercial reagent kit.
2. Profiling
Proteome databases of IMCD and a collecting duct cell line (mpkCCD) are extensive. Published databases obtained from native kidney tissue include the IMCD proteome, apical membrane IMCD proteome, basolateral membrane IMCD proteome, nuclei IMCD proteome, IMCD phosphoproteome, and IMCD phosphotyrosinome (11, 13, 66, 67, 69, 130, 132, 144, 151, 160, 167, 183, 186). Currently, more than 8,000 proteins have been identified from the rat IMCD. The database can be accessed online via https://hpcwebapps.cit.nih.gov/ESBL/Database/IMCD_Proteome_Database. In addition, comprehensive proteomic analyses of subcellular fractions from mouse mpkCCD cells have also identified more than 8,000 proteins which, along with virtual Western blots, are publicly available at https://hpcwebapps.cit.nih.gov/ESBL/Database/mpkFractions/ (179, 180).
3. Response to vasopressin
Vasopressin is the key hormone in water homeostasis, which binds to cells of CNTs and collecting ducts expressing vasopressin type 2 receptor (gene symbol: Avpr2) (86). AQP2 (gene symbol: Aqp2) is the vasopressin-regulated water channel of the renal collecting duct and, as such, is central to the regulation of water excretion. As the circulating level of the peptide hormone, vasopressin, increases in the blood, water excretion decreases largely, but not solely, through the regulation of AQP2. We know from studies done in the 20th century that vasopressin regulates AQP2 in two main ways (87): 1) vasopressin-mediated regulation of trafficking of AQP2-containing membrane vesicles to and from the apical plasma membrane of collecting duct principal cells; and 2) vasopressin-mediated regulation of the total abundance of the AQP2 protein in collecting duct principal cells. Recently, advances in understanding vasopressin-mediated processes has been driven by a series of proteomic studies. FIGURE 10 outlines various aspects of the mechanisms involved in regulatory actions of vasopressin and the corresponding proteomic techniques that have been employed.
FIGURE 10.

Outline depicting proteomic studies designed to elucidate the mechanisms of vasopressin action on the collecting duct. Vasopressin effects on the collecting duct are traditionally divided into short-term effects and long-term effects. The numerous proteomic strategies that have been employed to decipher the detailed mechanism(s) of vasopressin are indicated in the green boxes. AQP2, aquaporin-2; MS, mass spectrometry; SILAC, stable isotope labeling by amino acid in cell culture.
a) short-term effects.
Activation of the vasopressin receptor in collecting ducts acutely triggers complex intracellular signaling cascades that control translocation of AQP2 molecules from intracellular vesicles to the apical plasma membrane. The vasopressin signal is rapidly transduced by PTMs, especially phosphorylation, of an extensive network of proteins. The overall response to vasopressin has been characterized in isolated perfused collecting ducts (147). The response is reproduced by addition of cyclic AMP (153) and occurs relatively rapidly, with an initial increase in water permeability ~40 s after vasopressin addition and a half-time for the full response in 8 min. Because of the dependence of this response on cyclic AMP, it has been widely assumed that the trafficking effects of vasopressin are mediated by one or both of the known cyclic AMP-dependent protein kinases (PKAcα or PKAcβ). Analysis of the phosphoproteome after dDAVP administration has provided valuable insight into vasopressin-signaling pathways.
Rinschen et al. (142) studied the phosphoproteome of mpkCCD cells after short-term treatment by dDAVP. Using SILAC labeling, followed by strong cation exchange fractionation and IMAC phosphopeptide enrichment, phosphoproteomes were compared between dDAVP-treated and control groups. A total of 3,869 phosphopeptides corresponding to 1,695 phosphoproteins were identified. Among vasopressin-regulated peptides, 273 were increased, whereas 254 were decreased. Gene ontology analysis of this data showed “transcriptional regulation,” “cell adhesion,” and “protein amino acid phosphorylation” to be prominent processes. Of interest, most upregulated phosphorylation sites were basophilic, which was consistent with vasopressin-mediated activation of kinases, such as PKA and CAMK2. Activation of CAMK2 is consistent with previous studies suggesting a link between calcium/calmodulin-mediated signaling and the regulation of water permeability (29, 31, 43, 63). Among downregulated phosphorylation sites, the predominant P-X-S-P sequences suggested that the activities of proline-directed kinases (e.g., MAPKs and cyclin-dependent kinases) were inhibited by vasopressin. To further expand the phosphoproteome database, Hoffert et al. (65) used the iTRAQ labeling technique to study dynamic changes in the phosphoproteome of native IMCD cells in response to vasopressin. IMCD tubule suspensions (see sect. IID1, above) were incubated with 1 nM dDAVP for 0.5, 2, 5, and 15 min. A total of 12,167 phosphopeptides were identified, and 273 phosphopeptides exhibited a significant change in abundance at a minimum of one time point. PANTHER pathway analysis of these changes revealed both known and unknown vasopressin signaling pathways, such as PI3K, PKB/Akt, MAPKs, Rho kinases, angiogenesis, Wnt, and heterotrimeric G protein Gαq- and Gαo-mediated pathways. For longitudinal analysis, the temporal pattern mining algorithm (https://hpcwebapps.cit.nih.gov/TPM/) (145) was used to cluster peptides with similar temporal profiles. Thirty clusters were identified, and gene ontology biological process analysis showed several processes regulated by vasopressin. One of the most prominent processes downregulated by short-term vasopressin signaling was apoptosis, involving altered phosphorylation of many proteins, such as Bok, Bad, and Krt8. This suggests a role for vasopressin in promoting cell survival of IMCDs.
Similarly, several phosphoproteomic studies of rat native IMCD cells have also been conducted. Hoffert et al. (66) isolated native IMCDs from rat kidneys and incubated with dDAVP for 10 min, then digested peptides were fractionated with strong cation exchange, and phosphopeptides were enriched with IMAC. These phosphopeptides were analyzed with LC-MS multisequential (LC-MSn) neutral loss scanning. A total of 223 unique phosphoproteins were identified. The most influential finding of this study was a cluster of four phosphorylation sites in the COOH-terminal tail of AQP2, i.e., at S256, S261, S264, and S269. Using more powerful MS techniques, Bansal et al. (11) later confirmed the essential findings of Hoffert et al. and demonstrated that vasopressin increases phosphorylation of the urea channel UT-A1 at three sites, S84, S486, and S499, while markedly expanding the number of known phosphorylation sites in the native IMCD. Additionally, comparison between dDAVP-treated and control groups found many new vasopressin-regulated phosphorylation sites. For example, S552 phosphorylation of β-catenin was significantly increased by dDAVP, as detected by both LC-MS/MS and Western blotting analysis. This phosphorylation site was previously shown to have a role in cell proliferation via promoting an interaction with the T-cell factor transcription factor (159). The identification of the four phosphorylation sites in AQP2 opened the door for future work to understand short-term regulation of trafficking by vasopressin, which we review in the following sections.
I) S256. S256 of AQP2 was originally predicted to be a vasopressin-regulated site by Kuwahara et al. (91) based on their initial identification of the full amino acid sequence of AQP2. Phosphorylation of this site was first demonstrated by Nishimoto et al. (118) using phospho-specific antibodies. Quantification of S256 phosphorylation in AQP2 shows that, even in the absence of vasopressin, phosphorylation of this site is high (177). Some studies have shown increases in phospho-occupancy at this site in response to vasopressin (66), others have shown decreases (177), and others no change (185). Time course studies using phospho-specific antibodies showed a vasopressin-induced increase in phosphorylation that occurs within 1 min of exposure to vasopressin (64). Mutational analysis shows that S256 must be phosphorylated for downstream sites S264 or S269 to become phosphorylated in response to vasopressin, indicating that S256 plays a permissive role in short-term regulation by vasopressin. Accordingly, mutation of S256 to other amino acids blocks AQP2 exocytosis (10, 102).
The amino acid sequence surrounding S256 of AQP2 (-R-R-R-Q-S*-V-E-) suggested to Kuwahara et al. (91) that it is phosphorylated by protein kinase A. However, in cultured mouse mpkCCD cells in which both genes encoding protein kinase A catalytic subunits were deleted, S256 still became phosphorylated in the presence of vasopressin (78), implying that S256 is phosphorylated by one or more basophilic protein kinases with specificities similar to protein kinase A. Kinase profiling with LC-MS/MS coupled with kinase inhibitor data (20) suggested that this kinase may be the delta isoform Ca2+/calmodulin-dependent protein kinase II (Camk2d). The involvement of this kinase fits with the observation that vasopressin increases intracellular calcium spike frequency in collecting duct cells (133), which is thought to be the result of protein kinase A-mediated phosphorylation of the Itpr3 (S934 and S1832) calcium release channel in the ER of collecting duct principal cells (78).
II) S261. In contrast to the other AQP2 phosphorylation sites, S261 phosphorylation decreases in response to vasopressin (66). Time course studies show that the decrease occurs over several minutes, i.e., much more slowly than the increase in S256 phosphorylation (64). The sequence surrounding this site (-V-E-L-H-S*-P-Q-) is consistent with phosphorylation by one of the proline-directed kinase family members, which would include both MAPKs and cyclin-dependent kinases. Bayesian analysis based on subcellular fraction in mpkCCD cells identified ERK1, ERK2, and p38α as the kinases most likely to phosphorylate this site. A previous study by Nedvetsky et al. (117) also implicated p38 in AQP2 phosphorylation at S261. A recent study by Cheung et al. (27) pinpointed the protein phosphatase PP2C in dephosphorylation of S261.
The role of S261 phosphorylation in AQP2 trafficking is controversial. Deen’s group has reported evidence for a role for S261 phosphorylation in AQP2 endocytosis (157). In contrast, Lu et al. (102) and Moeller et al. (113) reported no effect of S261 mutation to a nonphosphorylatable amino acid on AQP2 localization. Yui et al. (184) recently reported that S261 phosphorylation is important in AQP2 apical translocation.
III) S264. In IMCD, phosphorylation at S264 of AQP2 increases in response to vasopressin in a time course that lags behind that of S256 (64). However, S264 phospho-occupancy never increases above 4% of all AQP2 molecules in response to vasopressin (177), suggesting that phosphorylation at this site may be either unimportant or confined to some small subcellular compartment. Indeed, work by Fenton et al. (50) indicates that S264 phosphorylation is confined to clathrin-coated vesicles, early endosomal compartments, and recycling compartments. Its potential role in AQP2 trafficking needs more study.
IV) S269. Phosphorylation of AQP2 at S269 is strongly regulated by vasopressin (64). In IMCD, AQP2 S269 phospho-occupancy was seen to increase from 3 to 26% of all AQP2 molecules in response to vasopressin (177), an order of magnitude increase. Immunogold localization in collecting duct cells (64, 112) demonstrated that AQP2 phosphorylated at S269 is limited to the apical plasma membrane with virtually no intracellular labeling. This observation lead to the hypothesis that S269 phosphorylation regulates AQP2 abundance in the apical plasma membrane by blocking or inhibiting AQP2 endocytosis. S269 is the third from last amino acid in AQP2 and is part of a putative PDZ ligand (172). Wang et al. (172) proposed that S269 phosphorylation inhibits endocytosis of AQP2 by inhibiting its ability to bind to the PDZ-protein, Sipa1l1 (rodents; SIPA1L1, most other species). Sipa1l1 is a Rap-GAP, i.e., it stimulates the GTPase activity of Rap, a small GTP-binding protein that regulates the Raf-MEK-ERK signaling pathway by activating Raf. S1611 of Sipa1l1 was identified by phosphoproteomics in mpkCCD cells as a protein kinase A target that may regulate Sipa1l1 GAP activity (78). Previously, Noda et al. (120) used MALDI-TOF MS to identify a Sipa1l1 paralog, SPA1 (gene symbol: Sipa1) as an AQP2 interacting protein. AQP2 trafficking to the apical membrane was inhibited when SPA-1 was mutated so that it lacked Rap1GAP activity (119). Collectively, these results demonstrate a substantial role for vasopressin-induced phosphorylation of S269 in the vesicular trafficking and localization of AQP2, likely via regulation of specific AQP2 protein-protein interactions.
Although the cAMP/PKA pathway is a major mediator of vasopressin signaling, pathways controlled by other kinases may also be involved in regulating vasopressin signaling. To address this possibility, Bradford et al. collected extensive previously published data and estimated probabilities (using Bayes’ theorem) for involvement of each kinase in the entire rat proteome in phosphorylation of AQP2 S256 (as a surrogate for vasopressin signaling) (20). Next, various specific kinase inhibitors were applied to isolated IMCDs before incubation with dDAVP, followed by Western blotting to quantify phosphorylated AQP2 S256. The results from this inhibitor experiment were combined with the initial probability estimates. Based on these collective analyses, the top-ranked kinases for vasopressin-mediated AQP2 S256 phosphorylation were CAMK2, followed by PKA, and PKB/Akt. In a follow-up experiment, dephosphorylated IMCD lysates were treated with three basophilic recombinant kinases, i.e., PKA, CAMK2, or serum/glucocorticoid-regulated kinase (SGK, which exhibits similar kinase substrate specificity to PKB/Akt), then a phosphoproteomic analysis was performed using LC-MS/MS. The results confirmed that, while all three of these kinases could phosphorylate AQP2 at S256, both CAMK2 and PKA were more potent than serum/glucocorticoid-regulated kinase, in agreement with the predicted probability ranking.
The second component of the short-term vasopressin response is the regulation of AQP2 trafficking. Tracking the intracellular trafficking of AQP2 in response to vasopressin is crucial to fully understand molecular mechanisms of vasopressin actions. Quantitative MS, combined with biochemical procedures that isolate particular subcellular fractions, has the potential to identify the movement of particular proteins from one location to another in cells. In an experiment to establish AQP2 subcellular localization in the unstimulated IMCD cells, Barile et al. (13) isolated AQP2-containing vesicles by immunoaffinity purification as described above and fractionated these vesicles by SDS-PAGE into 35 gel fractions. In-gel digestion followed by LC-MS/MS analysis identified 181 proteins, many of which are markers of subcellular membrane domains, such as members of the Rab family and SNARE proteins. This finding demonstrated that, in the basal state, AQP2 resides mainly in endosomes, the trans-Golgi network, and rough ER. In a targeted analysis of Rab proteins following treatment with vasopressin, some evidence was obtained, suggesting redistribution of AQP2 from late endosomes into recycling endosomes. To fully elucidate the dynamic intracellular trafficking of AQP2 in response to vasopressin, further studies are needed using this immune-isolation approach, coupled with unbiased quantitative LC-MS/MS analysis.
The ultimate result of vasopressin-stimulated trafficking is the increase in insertion of AQP2 into the apical plasma membrane. Using biotinylation of apical surface proteins, Loo et al. (101) studied changes in the apical membrane proteome following dDAVP stimulation in SILAC-labeled mpkCCD cells. Membrane abundance of 100 proteins was found to be altered by vasopressin. Among these proteins, AQP2 exhibited the highest increase in abundance, followed by Mal2. Importantly, Mal2 protein is a component of lipid rafts and functions in transcytosis (an intracellular transport pathway used to deliver membrane-bound proteins from the basolateral to the apical surface). Gene ontology analysis of these 100 proteins revealed “negative regulation of cellular component organization,” “actin filament capping,” “regulation of actin filament polymerization,” and “regulation of cytoskeleton organization” to be the predominantly enriched processes. Using live-cell imaging, apical F-actin relocalization to basolateral regions (peripheralization) was demonstrated in response to dDAVP, highlighting the major role of F-actin dynamics in AQP2 translocation.
The effect of vasopressin on protein-protein interactions in the collecting duct was also recently studied. By focusing on the UT-A1, a known urea transporter regulated by vasopressin, Chou et al. (30) used chemical cross-linking, followed by immunoprecipitation of UT-A1 in native rat IMCD cells, incubated with or without dDAVP for 15 min to study its interacting partner proteins. LC-MS/MS analysis revealed 25 partner proteins significantly changed in its binding with UT-A1 in response to dDAVP. These included two upregulated proteins, Nae1 and Uba3, which are involved in the activation of Nedd8, a ubiquitin-like protein. Among 23 significantly decreased proteins were several actin cytoskeleton-related proteins, Na-K-ATPase subunits, and phosphorylase b kinase regulatory subunit-β. The decreased interaction between UT-A1 and Na-K-ATPase subunits, which reside mainly on basolateral membrane, also support a notion that UT-A1 basolateral localization is physiologically regulated, at least in part, by vasopressin.
b) long-term effects.
A major well-established, long-term effect of vasopressin stimulation on collecting ducts is an increase in AQP2 protein abundance (40). This could be due to increased AQP2 synthesis, decreased AQP2 degradation, or a combination of these processes. Application of various proteomic techniques to study long-term vasopressin stimulation has contributed to a more comprehensive understanding of the mechanisms involved in regulation of AQP2 abundance.
In the first such study, van Balkom et al. (167) examined long-term vasopressin effects on the rat IMCD proteome using 2D-DIGE coupled with MALDI-TOF MS. Another early study employed the complementary technique of one-dimensional electrophoresis prefractionation, followed by LC-MS/MS with isotope-coded affinity tagging to study the effect of long-term vasopressin stimulation in rat IMCD (132). In these two studies, partially overlapping subsets of 43 and 33 proteins, respectively, were identified that increased, decreased, or shifted position. Among these proteins, cytoskeletal proteins and proteins involved in energy metabolism, response to stress, intracellular transport, vesicular trafficking, and translational elongation were predominant groups of proteins identified. From these studies, it is clear that AQP2 is not the only protein that exhibits changes in abundance resulting from long-term vasopressin treatment. Further proteomic analyses in mpkCCD cells have allowed even deeper profiling of global changes in protein abundance (83).
To study the potential role of long-term vasopressin stimulation on regulation of protein abundance by transcriptional mechanisms, Schenk et al. (149) quantified protein abundance changes in fractions of cytoplasmic extract, nuclear extract, and nuclear pellet from mpkCCD cells following 30 and 60 min of dDAVP treatment. The aim of this study was to identify proteins that translocated between these compartments (e.g., transcription factors and their regulators) in response to vasopressin treatment. Using SILAC labeling, a total of 3,987 proteins were identified in the nuclear fractions, 65 of which were significantly changed in abundance, indicating altered translocation of these proteins. Among the transcription factors identified, JunB, Elf3, and Gatad2b were significantly increased, while Hmbox1 was decreased. Nedd4 was the only posttranslational modifier found to be increased. The Mediator complex (a multiprotein complex that acts as a transcriptional coactivator of RNA polymerase II) was also increased, suggesting that the complex was assembled in cytoplasm and translocated together to the nucleus in response to vasopressin stimulation. Both total and phosphorylated forms of β-catenin were also increased in the nuclear fractions. Similarly, Bolger et al. (19) studied the nuclear phosphoproteome of mpkCCD cells in response to 30 min of dDAVP treatment. A total of 1,251 phosphorylation sites were identified in the nuclear fractions, 39 of which were significantly changed. Bioinformatics analysis of these regulated phosphorylation sites showed cell-cell junction and transcriptional regulation proteins to be the most common functional clusters. Phosphorylation at S552 of β-catenin was increased (known to increase β-catenin transcriptional activity), whereas phosphorylation at S73 of c-Jun was decreased after dDAVP treatment (suggesting decreased transcriptional activity of c-Jun). Follow-up studies used chromatin immunoprecipitation employing a β-catenin antibody coupled to LC-MS/MS to identify β-catenin interacting proteins in the nuclei of native IMCD cells from rats (77). Among the interacting proteins was Taf1, which plays a central role in transcriptional initiation and is part of the transcriptional preinitiation complex that is regulated by various transcription factors through the action of the mediator complex. Finally, in a very recent study using a higher-sensitivity mass spectrometer, Pickering et al. (130) reported a total of 5,048 proteins from nuclear fractions of rat native IMCD following vasopressin treatment, of which 369 were transcription factors. dDAVP treatment resulted in altered abundance of 99 proteins in nuclear pellets and 88 proteins in nuclear extract fractions, including 7 downregulated histone proteins in the nuclear pellets. These changes in histone proteins possibly reflect vasopressin-mediated chromatin remodeling.
While it is clear from the preceding review that long-term effects of vasopressin are mediated by changes in transcription, other post-transcriptional mechanisms such as selective translational regulation and/or selective protein degradation could also play a role. To address these possibilities, Khositseth et al. performed a proteome-transcriptome comparison for mpkCCD cells, using SILAC labeling and microarray analysis to evaluate the long-term effects in response to dDAVP (83). After the quantification of 2,990 proteins, 188 were found to be altered in abundance. Importantly, less than one-third of these proteins (notably including AQP2) were found to exhibit corresponding changes in both protein abundances and mRNA levels. These results demonstrated a significant role for posttranscriptional regulation of protein abundance as part of the long-term effects of vasopressin regulation.
To further study the potential role of long-term vasopressin stimulation on posttranscriptional regulation of protein abundance, specifically focusing on protein turnover mechanisms, another proteomics technique was used to determine vasopressin effects on protein half-lives. Sandoval et al. (146) applied dynamic SILAC labeling to mpkCCD cells to determine the half-lives for all cellular proteins. dDAVP treatment significantly affected half-lives of only 5 out of 2,172 proteins identified, including AQP2, which increased nearly 50% (from 9.8 to 14.2 h). Therefore, vasopressin not only increases AQP2 transcription (83, 182), but it also decreases AQP2 degradation as well. Mathematical modeling of these data also provided information about translation rates for 1,620 of the identified cellular proteins. Among 151 proteins found to have altered translation rates after vasopressin treatment, AQP2 exhibited the largest increase (~10-fold). Collectively, these results, combined with those from studies described above, demonstrated that the long-term effects of vasopressin on AQP2 abundance are mediated by multiple regulatory mechanisms at the levels of transcription, translation, and degradation. A further interesting finding from the Sandoval et al. study (146) was that the translation rates of seven glutathione S-transferase proteins (all from the cytoplasmic GST superfamily) were also increased by vasopressin treatment, resulting in an overall increase in glutathionylated proteins. The role of vasopressin in the increased activity of this antioxidation pathway remains to be elucidated.
One interesting aspect in vasopressin physiology is the “vasopressin escape” phenomenon: despite the presence of continuous high vasopressin level, the tubule cells exhibit a diminishing response to the hormone (i.e., water diuresis occurs). Hoorn et al. (69) employed a proteomic approach to study this phenomenon by first infusing rats with dDAVP for 72 h, followed by dividing the rats into two groups (water-loading and water-restricting groups). IMCDs were isolated and analyzed with 2D-DIGE/MALDI-TOF and Western blotting. A total of 22 proteins with significant changes were identified. After subjecting these identified regulated proteins to bioinformatics pathway analysis (Ingenuity Pathway Analysis), additional proteins representing key nodes in signaling networks were predicted to be involved in vasopressin escape. For example, c-Myc, c-Fos, and c-Jun were transcription factors predicted to exhibit changes in abundance regulated by vasopressin escape, along with additional regulatory proteins, including c-src, heat shock protein 70, and RACK1. Altered abundance for all of these proteins were validated by Western blotting in this study, suggesting that a complex signaling network mediated by these proteins is involved in the vasopressin escape phenomenon.
IV. BIOINFORMATICS TECHNIQUES IN SYSTEMS BIOLOGY STUDIES
A. Drawing Physiological Conclusions From Proteomics Data
Successful collection of proteomic data is only the first half of a proteomics study. The second half is the extraction of physiological insights from the data. Here we provide some guidelines on how to do this. The starting point is an expression matrix (often in the form of a spreadsheet) with a list of proteins and/or peptides mapped to quantitative data. Such matrices are typically derived from data analysis by different search algorithms. An important step is data filtering. A hypothetical data set may contain 9,000 proteins, each with a ratio representing protein abundance under the experimental condition, divided by protein abundance under the control condition. Such a data set will include many low signal-to-noise proteins whose quantifications are imprecise. For a valid downstream analysis of these data, the low signal-to-noise proteins must be filtered out. For this, a measure of relative protein abundance or signal-to-noise ratio is needed in addition to the experimental-to-control ratio. A cutoff threshold for the abundance variable can be chosen to optimize the balance between false positives and false negatives. This can be done in two ways. First, if enough control observations are made or specific control vs. control experiments have been performed, the variability among controls can be calculated for different abundance threshold cutoffs. This gives the intrinsic variability of the method, which can be used to calculate a 95% or 99% confidence interval for control vs. control observations at different thresholds. These confidence intervals then can be used to estimate whether experimental vs. control ratios are likely to be due to the intrinsic variability of the method or to the experimental variable. In this approach, a global false discovery rate will be 1% if the 99% confidence interval is chosen and 5% if the 95% confidence interval is chosen. Second, the empirical Bayes method can be employed if it is expected that relatively few proteins will be changed as a result of the experimental perturbation. With this method, the distributions of experimental-to-control ratios are used in lieu of the control-to-control ratios described in the first method. Regardless of the method chosen, our experience is that data set filtering is crucial to being able to extract mechanistic conclusions from proteomics data.
Another way to assess whether measured ratios are likely to be reflective of real responses is to use P values derived from statistical testing for each protein over multiple replicates. If the P value is evaluated relative to a threshold (e.g., P < 0.05), a problem arises, i.e., that multiple applications of the test will result in multiple false positives. For example, if abundance ratios for 5,000 proteins are measured in 4 experimental/control pairs and a t-test is applied to each protein with a P < 0.05 threshold, there will be on the order of 0.05 × 5,000 = 250 additional false positives simply as a consequence of multiple applications of the statistical test itself. Many investigators create a correction to the P value to account for multiple tested, using for example, the Benjamini-Hochberg correction. However, this correction amounts to little more than “window dressing.” A more realistic way to deal with the problem is to recognize the P value is simply a continuously distributed descriptive statistic reflecting signal-to-noise (59) and not as a measure of truth. In practice, the use of P values or false discovery rate corrected P values plotted against the logarithm of experimental-to-control ratios (a so-called volcano plot) gives the user a global view of which measured parameters exhibited reproducible changes and which parameters remained unchanged in proteomic experiments (FIGURE 11). However, we can never establish absolute certainty of such a change.
FIGURE 11.

A volcano plot as a visual tool for globally evaluating results from quantitative proteomic experiments. A special kind of scatterplot called “volcano plot” can help visualize the result of a high-throughput study. The x-axis represents the effect size in terms of logarithm (typically base 2) of experimental-to-control expression ratio. The y-axis represents the negative logarithm of P values, which indicate statistical significance. In this example plot, appropriate cutoff values for expression ratios (vertical blue dotted lines; e.g., >50% change) and false discovery rate (FDR) corrected P values (horizontal red dotted line) are shown and used to highlight possible significant and biologically relevant changes. For this example, three proteins (pink data points) are identified as being downregulated in response to the experimental treatment, suggesting a possible molecular mechanism and/or hypothesis for additional studies.
If we cannot prove that an observed change in the abundance of any particular protein is not a false positive, then why do proteomics at all? This limitation may be alarming to biochemists who are interested only in a particular protein, but should not be problematic to physiologists who are chiefly interested in gaining a mechanistic understanding of biological processes. Biological processes depend on coordinated changes in multiple proteins. In general, for such a process, a change in a single protein associated with the process does not demonstrate much about the process, but if all or most of the protein changes involved in a process are found, a high level of confidence for the process can be inferred. An example is from a study by Yang et al. (179) quantifying protein abundance changes in response to the hormone vasopressin in several protein fractions derived from differential centrifugation of cultured collecting duct cells. It was observed that 64 separate ribosomal subunits underwent a small nominal shift from the 200,000 g pellet (containing free ribosomes) to the 17,000 g pellet (containing the rough ER) in response to vasopressin. The increase in the 17,000 g pellet amounted to only ~15–20% for each ribosomal protein subunit. If analyzed individually, the small change in each subunit may have been dismissed. Analyzed together, the same small change in all but 2 of 66 ribosomal proteins leads to a calculated P value of 10−34, supporting the hypothesis that the entire ribosome translocated from the cytoplasm to the rough ER in response to vasopressin. This modest translocation of the cellular ribosomal pool is responsible for the massive vasopressin-stimulated increase in translation of the integral membrane protein AQP2 in the rough ER (146). Thus analysis of proteomic data sets to study physiological processes depends on detection of multiple coordinated changes in an independently curated group of proteins that together would be unlikely to be due to random variation.
Consequently, successful application of proteomics to physiological investigations depends on the availability of prior data to identify sets of proteins that are coordinately involved in particular physiological processes, (e.g., processes defined by gene ontology terms). These data sets can be mapped to the new proteomics data and the question can be asked: Are (increases/decreases/changes) in proteins in the functional dataset more frequent than (increases/decreases/changes) among all proteins expressed in the cell?” This is a question that can be answered through use of standard statistical tests such as χ2 testing or Fishers’ exact testing.
1. The folly of validation
In the old days, when DNA microarrays were first introduced in biology, the arrays consisted of cDNAs immobilized on glass slides. The relative abundances of various transcripts were estimated based on their ability to hybridize with individual cDNA features. False positives were frequent, and it was well recognized that individual observations needed to be verified in individual experiments, a process that came to be known as validation. As effective ways of eliminating false positives became available for oligonucleotide arrays (e.g., commercial Affymetrix expression arrays), the reliability of microarrays dramatically increased. However, the tradition of validation remained attached to microarray procedures and eventually to RNAseq and to proteomics. In our opinion, the concept of validation should be abandoned in favor of the concept of reproducibility. With regard to proteomics, there appears to be a propensity to validate LC-MS/MS results with Western blotting, a technique that is decidedly inferior, both for specificity and for quantification. It has even been convincingly argued that, given the limitations of Western blotting, all results from Western blotting experiments should be “validated” with protein MS. Indeed, many investigators now recognize that a better way to assure reproducibility in proteomics studies is to use targeted proteomics to increase the number of times a particular quantification is observed (2).
2. The folly of fold change
A large number of software programs that carry out differential expression analysis used the concept of fold change as a means of quantification. Unfortunately, fold change is an ambiguous term and should be avoided. Originally, fold change was calculated as
However, the writers of many differential expression analysis programs have used a different definition, namely,
For very large changes, these two measures are very nearly equal. However, a problem arises when changes are small. Logically, if there is no change, the fold change should be 0, not 1. A reduction to one-half is given as a fold change of 0.5, which the uninitiated might expect to represent an increase, not a decrease. Because of the ambiguities that have arisen with the term fold change, we recommend that, instead, users should report the logarithm of the ratio of the experimental value to the control value. Most investigators choose the base 2 logarithm for this purpose.
3. Making your data available to other investigators
Most journals that publish studies based on protein MS require that the data be deposited in a publicly accessible repository such as the PRIDE (PRoteomics IDEntifications) Database at https://www.ebi.ac.uk/pride/archive/ maintained by the European Molecular Biology Laboratory. In general, such databases include important metadata describing experiments as well as the raw spectral files, but not curated information. Since a large part of the added value of a data set is in the advanced bioinformatics, authors should consider including their analytical methodologies, scripts, and algorithms in the deposited data. However, similar government-supported repositories have lost support and disappeared in the past, and it would be wise to deposit the curated data elsewhere to ensure its continued availability (104). Many journals allow processed, curated data to be included with the supporting information included with published papers. Beyond this, increased visibility and accessibility can be obtained if investigators place curated data on the internet in the form of user-friendly web pages (see for example, https://hpcwebapps.cit.nih.gov/ESBL/Database/). Such pages can be constructed fairly simply without web programming skills by saving the relevant tab of a well annotated spreadsheet as an .html document and posting on an institutional website [see for example, https://hpcwebapps.cit.nih.gov/ESBL/Database/PodocytePhosphoproteome/index.htm, a database referring to a phosphoproteomic study in podocytes (140a)]. More sophisticated sites can be built with fairly fundamental web programming skills learnable in a few days, or through use of special open-source tools (see for example, https://hpcwebapps.cit.nih.gov/ESBL/Database/IMCD_Proteome_Database/).
V. CONCLUSIONS
Proteomic profiling of the main renal tubule segments of the kidney has provided unique insights into the tight regulation of solute and electrolyte transport processes and metabolism. With the rapidly expanding amount of data from these high-throughput studies, finding comprehensive information for individual proteins or genes of interest can be quite tedious. As a result, Zhao et al. (189) developed a tool called BIG (Biological Information Gatherer; https://big.nhlbi.nih.gov/index.jsp), which helps summarize all data about each individual protein or gene in the comprehensive “Renal Epithelial Transcriptome and Proteome Databases” (Epithelial Systems Biology Laboratory, NHLBI) and reports the output in readable sentences.
Despite the significant progress presented in the preceding review, these studies represent an oversimplification of renal tubule physiology, since only the four main renal tubule segments of the kidney (FIGURE 6) can be practically isolated for proteomic studies. In actuality, 14 discrete renal tubule segments have been manually dissected under the microscope for transcriptomic analysis (96), undoubtedly including many differentiated epithelial cell types not previously characterized by proteomic analysis. Therefore, further technical developments are needed to facilitate higher resolution isolation of distinct renal tubule cell types for proteomic analysis. One promising approach would be the use of a tubule segment-specific GFP expression system, based on transcriptomics-derived knowledge of gene-specific expression in the 14 discrete renal tubule segments, to isolate specific fluorescently tagged tubule segments by FACS. In addition, deeper profiling of these distinct renal tubule cell types will provide even more detailed understanding of renal physiology. Such profiling could be further enhanced through the use of orthogonal fractionation techniques, such as capillary electrophoresis/isoelectric focusing (73) or high-pH RPLC (14) upstream of standard (i.e., low-pH) RPLC-MS/MS.
The “classical” strategies for studying kidney physiology have been augmented by high-throughput proteomic techniques, which have vastly increased the amount of new information available. However, many such studies simply conclude with a list of protein changes in response to experimental manipulations, along with bioinformatics analyses intended to reveal important new physiological insights into kidney function. Unfortunately, the results from these studies have often not led to follow-up investigations involving new critical testable hypotheses. In addition to increased knowledge of normal kidney physiology, more studies are also needed to advance understanding of renal pathophysiology. For example, two recent proteomic-driven studies revealed a prominent role for selective autophagy in the downregulation of specific proteins during development of nephrogenic diabetes insipidus due to electrolyte imbalance disorders [hypokalemia (84) and hypercalcemia (82)]. Another proof-of-concept study recently demonstrated an application of proteomic analysis in exploring the molecular pathophysiology in an animal model of chronic tubulointerstitial fibrosis (137). In a final illustrative example, Merkulova et al. (109) used proteomic analysis plus genetic knock-down experiments to elucidate the interactome of kidney V-ATPase, a protein that plays a key role in renal acid excretion and blood pH homeostasis, identifying several novel interacting proteins regulating V-ATPase functions. The findings help to explain the pathophysiological roles of V-ATPase in various diseases.
Finally, complete understanding of renal physiology will require the study of the entire nephron, wherein studies of both glomerular and renal tubule physiology are complementary. Another layer of complexity in renal physiology and pathophysiology is in the heterogeneity of kidney tissues at the single-cell/tubule/nephron level. The field of renal single-cell biology has been pioneered by single-cell and single-tubule transcriptomic analyses (24, 95). Although more technically challenging, a recent study has successfully investigated kidney proteomes from mice with proteinuric kidney disease at the single-nephron level (68). In contrast to the situation with renal tubule physiology, proteomic studies involving glomerular biology are often disease focused (139), and phosphoproteomic studies have only been performed very recently (140, 141), 9 yr after the first phosphoproteomic study in renal tubules by Hoffert et al. (66). In addition, the complete understanding of renal physiology will also require more comprehensive, multidimensional investigations of the cellular proteome for each of the more than 26 different kidney cell types (5). Such multidimensional analyses would include the dynamic changes in phosphorylation and other PTMs [ubiquitination (166) and acetylation (176)], together with explorations of protein-protein interactions and protein translocations by advanced proteomic and biochemical techniques (93). The advanced experimental approaches suggested here would generate a detailed multidimensional protein atlas of the human kidney, serving as a key resource for precision medicine and novel targeted strategies for treating kidney diseases. It is highly expected that, when combined with multi-omics and molecular microscope approaches, the proteomic study from renal biopsy(s) of an individual with kidney disease will help deliver accurate diagnosis and personalized treatment to the patient, the concept that is initiated by the Kidney Precision Medicine Project (https://www.niddk.nih.gov/research-funding/research-programs/kidney-precision-medicine-project-kpmp). Last but not least, FIGURE 12 summarizes steps toward new knowledge, knowledge gaps, and potential advances and solutions for proteomic studies, ranging from the creation of a proteomic list to bioinformatic interrogation, inferential interpretation, and formulation of hypotheses and new experiments. The authors anticipate that further partnership between renal physiology and proteome science will continue to be fruitful and productive.
FIGURE 12.

Steps toward new knowledge, knowledge gaps, and potential advances and solutions. The creation of a proteomic list can be improved regarding both qualitative (multidimensional analyses and single-cell/tubule/nephron studies) and quantitative (fractionation techniques and instrumentation) aspects. Bioinformatic interrogation of biological databases cannot be taken as a static process, since these databases are continuously evolving and not always accurate. Thus standardization and quality control of data curation in a dynamic manner are required (191). Functional enrichment analysis is a popular method to identify over- or underrepresented protein groups. This kind of analysis is prone to several sources of bias that can confound the data interpretation, especially in the selection of background comparison list and in the statistical interpretation of high-throughput data (163). Inferential interpretation leading to the formulation of hypotheses and new experiments is a highly complex process. Machine-learning techniques, such as deep learning, have gained much attention over the last few years. Even though these techniques cannot yield an explicit hypothesis or understanding of the underlying mechanisms, they help us draw biologically relevant conclusions from the omics data (23). Lastly, new experiments to validate the previous findings and test new hypotheses are essential, the task that always requires high effort. Automated systems and cloud-based biology are undertaken as a future direction (167a).
GRANTS
M. M. Rinschen was supported by the Deutsche Forschungsgemeinschaft (RI 2811/1 and 2811/2). This work was supported by the Chulalongkorn Academic Advancement into Its 2nd Century Project (CUAASC) and a Kidney Foundation of Thailand research grant.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
Address for reprint requests and other correspondence: T. Pisitkun, National Heart, Lung and Blood Institute, NHLBI 10/6N312, 31 Center Dr., Bethesda, MD 20892 (e-mail: pisitkut@nhlbi.nih.gov) and M. M. Rinschen, University Hospital Cologne, Joseph-Stelzmann-Str. 26, 50931 Cologne, Germany (e-mail: markus.rinschen@uk-koeln.de).
M. M. Rinschen and K. Limbutara contributed equally to this work.
REFERENCES
- 1.Abbatiello SE, Schilling B, Mani DR, Zimmerman LJ, Hall SC, MacLean B, Albertolle M, Allen S, Burgess M, Cusack MP, Gosh M, Hedrick V, Held JM, Inerowicz HD, Jackson A, Keshishian H, Kinsinger CR, Lyssand J, Makowski L, Mesri M, Rodriguez H, Rudnick P, Sadowski P, Sedransk N, Shaddox K, Skates SJ, Kuhn E, Smith D, Whiteaker JR, Whitwell C, Zhang S, Borchers CH, Fisher SJ, Gibson BW, Liebler DC, MacCoss MJ, Neubert TA, Paulovich AG, Regnier FE, Tempst P, Carr SA. Large-scale interlaboratory study to develop, analytically validate and apply highly multiplexed, quantitative peptide assays to measure cancer-relevant proteins in plasma. Mol Cell Proteomics 14: 2357–2374, 2015. doi: 10.1074/mcp.M114.047050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aebersold R, Burlingame AL, Bradshaw RA. Western blots versus selected reaction monitoring assays: time to turn the tables? Mol Cell Proteomics 12: 2381–2382, 2013. doi: 10.1074/mcp.E113.031658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature 537: 347–355, 2016. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- 4.Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Gil L, Girón CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Juettemann T, Keenan S, Laird MR, Lavidas I, Maurel T, McLaren W, Moore B, Murphy DN, Nag R, Newman V, Nuhn M, Ong CK, Parker A, Patricio M, Riat HS, Sheppard D, Sparrow H, Taylor K, Thormann A, Vullo A, Walts B, Wilder SP, Zadissa A, Kostadima M, Martin FJ, Muffato M, Perry E, Ruffier M, Staines DM, Trevanion SJ, Cunningham F, Yates A, Zerbino DR, Flicek P; Ensembl 2017 . Nucleic Acids Res 45: D635–D642, 2017. doi: 10.1093/nar/gkw1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Awqati Q, Oliver JA. Stem cells in the kidney. Kidney Int 61: 387–395, 2002. doi: 10.1046/j.1523-1755.2002.00164.x. [DOI] [PubMed] [Google Scholar]
- 6.Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson JD. Molecular Biology of the Cell. New York: Garland, 1989. [Google Scholar]
- 7.Alfaro JA, Sinha A, Kislinger T, Boutros PC. Onco-proteogenomics: cancer proteomics joins forces with genomics. Nat Methods 11: 1107–1113, 2014. [Erratum in Nat Methods 12: 160, 2015.] doi: 10.1038/nmeth.3138. [DOI] [PubMed] [Google Scholar]
- 8.Altelaar AF, Munoz J, Heck AJ. Next-generation proteomics: towards an integrative view of proteome dynamics. Nat Rev Genet 14: 35–48, 2013. doi: 10.1038/nrg3356. [DOI] [PubMed] [Google Scholar]
- 9.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LS. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res 32: D115–D119, 2004. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arthur J, Huang J, Nomura N, Jin WW, Li W, Cheng X, Brown D, Lu HJ. Characterization of the putative phosphorylation sites of the AQP2 C terminus and their role in AQP2 trafficking in LLC-PK1 cells. Am J Physiol Renal Physiol 309: F673–F679, 2015. doi: 10.1152/ajprenal.00152.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bansal AD, Hoffert JD, Pisitkun T, Hwang S, Chou CL, Boja ES, Wang G, Knepper MA. Phosphoproteomic profiling reveals vasopressin-regulated phosphorylation sites in collecting duct. J Am Soc Nephrol 21: 303–315, 2010. doi: 10.1681/ASN.2009070728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bantscheff M, Lemeer S, Savitski MM, Kuster B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem 404: 939–965, 2012. doi: 10.1007/s00216-012-6203-4. [DOI] [PubMed] [Google Scholar]
- 13.Barile M, Pisitkun T, Yu MJ, Chou CL, Verbalis MJ, Shen RF, Knepper MA. Large scale protein identification in intracellular aquaporin-2 vesicles from renal inner medullary collecting duct. Mol Cell Proteomics 4: 1095–1106, 2005. doi: 10.1074/mcp.M500049-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batth TS, Francavilla C, Olsen JV. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J Proteome Res 13: 6176–6186, 2014. doi: 10.1021/pr500893m. [DOI] [PubMed] [Google Scholar]
- 15.Bauer NC, Doetsch PW, Corbett AH. Mechanisms regulating protein localization. Traffic 16: 1039–1061, 2015. doi: 10.1111/tra.12310. [DOI] [PubMed] [Google Scholar]
- 16.Biber J, Stieger B, Stange G, Murer H. Isolation of renal proximal tubular brush-border membranes. Nat Protoc 2: 1356–1359, 2007. doi: 10.1038/nprot.2007.156. [DOI] [PubMed] [Google Scholar]
- 17.Bibi A, Agarwal NK, Dihazi GH, Eltoweissy M, Van Nguyen P, Mueller GA, Dihazi H. Calreticulin is crucial for calcium homeostasis mediated adaptation and survival of thick ascending limb of Henle’s loop cells under osmotic stress. Int J Biochem Cell Biol 43: 1187–1197, 2011. doi: 10.1016/j.biocel.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 18.Biemann K. Laying the groundwork for proteomics: mass spectrometry from 1958 to 1988. J Proteomics 107: 62–70, 2014. doi: 10.1016/j.jprot.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 19.Bolger SJ, Hurtado PA, Hoffert JD, Saeed F, Pisitkun T, Knepper MA. Quantitative phosphoproteomics in nuclei of vasopressin-sensitive renal collecting duct cells. Am J Physiol Cell Physiol 303: C1006–C1020, 2012. doi: 10.1152/ajpcell.00260.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradford D, Raghuram V, Wilson JLL, Chou C-L, Hoffert JD, Knepper MA, Pisitkun T. Use of LC-MS/MS and Bayes’ theorem to identify protein kinases that phosphorylate aquaporin-2 at Ser256. Am J Physiol Cell Physiol 307: C123–C139, 2014. doi: 10.1152/ajpcell.00377.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buchmaier BS, Bibi A, Müller GA, Dihazi GH, Eltoweissy M, Kruegel J, Dihazi H. Renal cells express different forms of vimentin: the independent expression alteration of these forms is important in cell resistance to osmotic stress and apoptosis. PLoS One 8: e68301, 2013. [Erratum in PLoS One 13: e0196935, 2018.] doi: 10.1371/journal.pone.0068301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burg MB. Role of aldose reductase and sorbitol in maintaining the medullary intracellular milieu. Kidney Int 33: 635–641, 1988. doi: 10.1038/ki.1988.46. [DOI] [PubMed] [Google Scholar]
- 23.Bzdok D, Altman N, Krzywinski M. Points of significance: statistics versus machine learning. Nat Methods 15: 233–234, 2018. doi: 10.1038/nmeth.4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Păunescu TG, Merkulova M, Breton S, Verlander JW, Wall SM, Brown D, Burg MB, Knepper MA. Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc Natl Acad Sci USA 114: E9989–E9998, 2017. doi: 10.1073/pnas.1710964114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X, Turecček F. Simple b ions have cyclic oxazolone structures. A neutralization-reionization mass spectrometric and computational study of oxazolone radicals. J Am Soc Mass Spectrom 16: 1941–1956, 2005. doi: 10.1016/j.jasms.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 26.Cheng L, Wu Q, Kortenoeven ML, Pisitkun T, Fenton RA. A Systems level analysis of vasopressin-mediated signaling networks in kidney distal convoluted tubule cells. Sci Rep 5: 12829, 2015. doi: 10.1038/srep12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheung PW, Ueberdiek L, Day J, Bouley R, Brown D. Protein phosphatase 2C is responsible for VP-induced dephosphorylation of AQP2 serine 261. Am J Physiol Renal Physiol 313: F404–F413, 2017. doi: 10.1152/ajprenal.00004.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheung PY, Lai WP, Lau HY, Lo SC, Wong MS. Acute and chronic effect of dietary phosphorus restriction on protein expression in young rat renal proximal tubules. Proteomics 2: 1211–1219, 2002. doi:. [DOI] [PubMed] [Google Scholar]
- 29.Chou CL, Christensen BM, Frische S, Vorum H, Desai RA, Hoffert JD, de Lanerolle P, Nielsen S, Knepper MA. Non-muscle myosin II and myosin light chain kinase are downstream targets for vasopressin signaling in the renal collecting duct. J Biol Chem 279: 49026–49035, 2004. doi: 10.1074/jbc.M408565200. [DOI] [PubMed] [Google Scholar]
- 30.Chou CL, Hwang G, Hageman DJ, Han L, Agrawal P, Pisitkun T, Knepper MA. Identification of UT-A1 and AQP2 interacting proteins in rat inner medullary collecting duct. Am J Physiol Cell Physiol, 314: C99–C117, 2018. doi: 10.1152/ajpcell.00082.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chou CL, Yip KP, Michea L, Kador K, Ferraris JD, Wade JB, Knepper MA. Regulation of aquaporin-2 trafficking by vasopressin in the renal collecting duct. Roles of ryanodine-sensitive Ca2+ stores and calmodulin. J Biol Chem 275: 36839–36846, 2000. doi: 10.1074/jbc.M005552200. [DOI] [PubMed] [Google Scholar]
- 32.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325: 834–840, 2009. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 33.Collins FS, Morgan M, Patrinos A. The Human Genome Project: lessons from large-scale biology. Science 300: 286–290, 2003. doi: 10.1126/science.1084564. [DOI] [PubMed] [Google Scholar]
- 34.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26: 1367–1372, 2008. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 35.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805, 2011. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 36.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20: 1466–1467, 2004. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 37.Curthoys NP, Moe OW. Proximal tubule function and response to acidosis. Clin J Am Soc Nephrol 9: 1627–1638, 2014. doi: 10.2215/CJN.10391012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Curthoys NP, Taylor L, Hoffert JD, Knepper MA. Proteomic analysis of the adaptive response of rat renal proximal tubules to metabolic acidosis. Am J Physiol Renal Physiol 292: F140–F147, 2007. doi: 10.1152/ajprenal.00217.2006. [DOI] [PubMed] [Google Scholar]
- 39.Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, Meehan TF, Weninger WJ, Westerberg H, Adissu H, Baker CN, Bower L, Brown JM, Caddle LB, Chiani F, Clary D, Cleak J, Daly MJ, Denegre JM, Doe B, Dolan ME, Edie SM, Fuchs H, Gailus-Durner V, Galli A, Gambadoro A, Gallegos J, Guo S, Horner NR, Hsu CW, Johnson SJ, Kalaga S, Keith LC, Lanoue L, Lawson TN, Lek M, Mark M, Marschall S, Mason J, McElwee ML, Newbigging S, Nutter LM, Peterson KA, Ramirez-Solis R, Rowland DJ, Ryder E, Samocha KE, Seavitt JR, Selloum M, Szoke-Kovacs Z, Tamura M, Trainor AG, Tudose I, Wakana S, Warren J, Wendling O, West DB, Wong L, Yoshiki A, MacArthur DG, Tocchini-Valentini GP, Gao X, Flicek P, Bradley A, Skarnes WC, Justice MJ, Parkinson HE, Moore M, Wells S, Braun RE, Svenson KL, de Angelis MH, Herault Y, Mohun T, Mallon AM, Henkelman RM, Brown SD, Adams DJ, Lloyd KC, McKerlie C, Beaudet AL, Bućan M, Murray SA; International Mouse Phenotyping Consortium; Jackson Laboratory; Infrastructure Nationale PHENOMIN, Institut Clinique de la Souris (ICS); Charles River Laboratories; MRC Harwell; Toronto Centre for Phenogenomics; Wellcome Trust Sanger Institute; RIKEN BioResource Center . High-throughput discovery of novel developmental phenotypes. Nature 537: 508–514, 2016. doi: 10.1038/nature19356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DiGiovanni SR, Nielsen S, Christensen EI, Knepper MA. Regulation of collecting duct water channel expression by vasopressin in Brattleboro rat. Proc Natl Acad Sci USA 91: 8984–8988, 1994. doi: 10.1073/pnas.91.19.8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dihazi H, Asif AR, Agarwal NK, Doncheva Y, Müller GA. Proteomic analysis of cellular response to osmotic stress in thick ascending limb of Henle’s loop (TALH) cells. Mol Cell Proteomics 4: 1445–1458, 2005. doi: 10.1074/mcp.M400184-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Dørup J. Ultrastructure of distal nephron cells in rat renal cortex. J Ultrastruct Res 92: 101–118, 1985. doi: 10.1016/0889-1605(85)90132-6. [DOI] [PubMed] [Google Scholar]
- 43.Ecelbarger CA, Chou CL, Lolait SJ, Knepper MA, DiGiovanni SR. Evidence for dual signaling pathways for V2 vasopressin receptor in rat inner medullary collecting duct. Am J Physiol Renal Physiol 270: F623–F633, 1996. doi: 10.1152/ajprenal.1996.270.4.F623. [DOI] [PubMed] [Google Scholar]
- 44.Elalouf JM, Roinel N, de Rouffignac C. Effects of antidiuretic hormone on electrolyte reabsorption and secretion in distal tubules of rat kidney. Pflugers Arch 401: 167–173, 1984. doi: 10.1007/BF00583877. [DOI] [PubMed] [Google Scholar]
- 44a.Ellens KW, Christian N, Singh C, Satagopam VP, May P, Linster CL. Confronting the catalytic dark matter encoded by sequenced genomes. Nucleic Acids Res 45: 11495–11514, 2017. doi: 10.1093/nar/gkx937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5: 976–989, 1994. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 46.Erde J, Loo RR, Loo JA. Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. J Proteome Res 13: 1885–1895, 2014. doi: 10.1021/pr4010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eveloff J, Haase W, Kinne R. Separation of renal medullary cells: isolation of cells from the thick ascending limb of Henle’s loop. J Cell Biol 87: 672–681, 1980. doi: 10.1083/jcb.87.3.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ezkurdia I, Calvo E, Del Pozo A, Vázquez J, Valencia A, Tress ML. The potential clinical impact of the release of two drafts of the human proteome. Expert Rev Proteomics 12: 579–593, 2015. doi: 10.1586/14789450.2015.1103186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science 246: 64–71, 1989. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 50.Fenton RA, Moeller HB, Hoffert JD, Yu MJ, Nielsen S, Knepper MA. Acute regulation of aquaporin-2 phosphorylation at Ser-264 by vasopressin. Proc Natl Acad Sci USA 105: 3134–3139, 2008. doi: 10.1073/pnas.0712338105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feric M, Zhao B, Hoffert JD, Pisitkun T, Knepper MA. Large-scale phosphoproteomic analysis of membrane proteins in renal proximal and distal tubule. Am J Physiol Cell Physiol 300: C755–C770, 2011. doi: 10.1152/ajpcell.00360.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Freund DM, Prenni JE, Curthoys NP. Proteomic profiling of the mitochondrial inner membrane of rat renal proximal convoluted tubules. Proteomics 13: 2495–2499, 2013. doi: 10.1002/pmic.201200558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Freund DM, Prenni JE, Curthoys NP. Response of the mitochondrial proteome of rat renal proximal convoluted tubules to chronic metabolic acidosis. Am J Physiol Renal Physiol 304: F145–F155, 2013. doi: 10.1152/ajprenal.00526.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, Domon B. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol Cell Proteomics 11: 1709–1723, 2012. doi: 10.1074/mcp.O112.019802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH. Open mass spectrometry search algorithm. J Proteome Res 3: 958–964, 2004. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 56.Gessel MM, Norris JL, Caprioli RM. MALDI imaging mass spectrometry: spatial molecular analysis to enable a new age of discovery. J Proteomics 107: 71–82, 2014. doi: 10.1016/j.jprot.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 11: O111.016717, 2012. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Go YM, Jones DP. The redox proteome. J Biol Chem 288: 26512–26520, 2013. doi: 10.1074/jbc.R113.464131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goodman SN. Toward evidence-based medical statistics. 1: The P value fallacy. Ann Intern Med 130: 995–1004, 1999. doi: 10.7326/0003-4819-130-12-199906150-00008. [DOI] [PubMed] [Google Scholar]
- 60.Grimsrud PA, Swaney DL, Wenger CD, Beauchene NA, Coon JJ. Phosphoproteomics for the masses. ACS Chem Biol 5: 105–119, 2010. doi: 10.1021/cb900277e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grzetic J, Oomens J. Spectroscopic evidence for an oxazolone structure in anionic b-type peptide fragments. J Am Soc Mass Spectrom 23: 290–300, 2012. doi: 10.1007/s13361-011-0297-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gunaratne R, Braucht DW, Rinschen MM, Chou CL, Hoffert JD, Pisitkun T, Knepper MA. Quantitative phosphoproteomic analysis reveals cAMP/vasopressin-dependent signaling pathways in native renal thick ascending limb cells. Proc Natl Acad Sci USA 107: 15653–15658, 2010. doi: 10.1073/pnas.1007424107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hoffert JD, Chou CL, Fenton RA, Knepper MA. Calmodulin is required for vasopressin-stimulated increase in cyclic AMP production in inner medullary collecting duct. J Biol Chem 280: 13624–13630, 2005. doi: 10.1074/jbc.M500040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoffert JD, Fenton RA, Moeller HB, Simons B, Tchapyjnikov D, McDill BW, Yu MJ, Pisitkun T, Chen F, Knepper MA. Vasopressin-stimulated increase in phosphorylation at Ser269 potentiates plasma membrane retention of aquaporin-2. J Biol Chem 283: 24617–24627, 2008. doi: 10.1074/jbc.M803074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoffert JD, Pisitkun T, Saeed F, Song JH, Chou C-L, Knepper MA. Dynamics of the G protein-coupled vasopressin V2 receptor signaling network revealed by quantitative phosphoproteomics. Mol Cell Proteomics 11: M111.014613, 2012. doi: 10.1074/mcp.M111.014613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoffert JD, Pisitkun T, Wang G, Shen RF, Knepper MA. Quantitative phosphoproteomics of vasopressin-sensitive renal cells: regulation of aquaporin-2 phosphorylation at two sites. Proc Natl Acad Sci USA 103: 7159–7164, 2006. doi: 10.1073/pnas.0600895103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoffert JD, van Balkom BW, Chou CL, Knepper MA. Application of difference gel electrophoresis to the identification of inner medullary collecting duct proteins. Am J Physiol Renal Physiol 286: F170–F179, 2004. doi: 10.1152/ajprenal.00223.2003. [DOI] [PubMed] [Google Scholar]
- 68.Höhne M, Frese CK, Grahammer F, Dafinger C, Ciarimboli G, Butt L, Binz J, Hackl MJ, Rahmatollahi M, Kann M, Schneider S, Altintas MM, Schermer B, Reinheckel T, Göbel H, Reiser J, Huber TB, Kramann R, Seeger-Nukpezah T, Liebau MC, Beck BB, Benzing T, Beyer A, Rinschen MM. Single-nephron proteomes connect morphology and function in proteinuric kidney disease. Kidney Int 93: 1308–1319, 2018. doi: 10.1016/j.kint.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 69.Hoorn EJ, Hoffert JD, Knepper MA. Combined proteomics and pathways analysis of collecting duct reveals a protein regulatory network activated in vasopressin escape. J Am Soc Nephrol 16: 2852–2863, 2005. doi: 10.1681/ASN.2005030322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hsu JL, Huang SY, Chow NH, Chen SH. Stable-isotope dimethyl labeling for quantitative proteomics. Anal Chem 75: 6843–6852, 2003. doi: 10.1021/ac0348625. [DOI] [PubMed] [Google Scholar]
- 71.Hu A, Noble WS, Wolf-Yadlin A. Technical advances in proteomics: new developments in data-independent acquisition. F1000 Res 5: 419, 2016. doi: 10.12688/f1000research.7042.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu Q, Noll RJ, Li H, Makarov A, Hardman M, Graham Cooks R. The Orbitrap: a new mass spectrometer. J Mass Spectrom 40: 430–443, 2005. doi: 10.1002/jms.856. [DOI] [PubMed] [Google Scholar]
- 73.Hühner J, Lämmerhofer M, Neusüß C. Capillary isoelectric focusing-mass spectrometry: Coupling strategies and applications. Electrophoresis 36: 2670–2686, 2015. doi: 10.1002/elps.201500185. [DOI] [PubMed] [Google Scholar]
- 74.Humphrey SJ, Azimifar SB, Mann M. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat Biotechnol 33: 990–995, 2015. doi: 10.1038/nbt.3327. [DOI] [PubMed] [Google Scholar]
- 75.Hunt DF, Henderson RA, Shabanowitz J, Sakaguchi K, Michel H, Sevilir N, Cox AL, Appella E, Engelhard VH. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science 255: 1261–1263, 1992. doi: 10.1126/science.1546328. [DOI] [PubMed] [Google Scholar]
- 76.Hutchison CA III, Chuang RY, Noskov VN, Assad-Garcia N, Deerinck TJ, Ellisman MH, Gill J, Kannan K, Karas BJ, Ma L, Pelletier JF, Qi ZQ, Richter RA, Strychalski EA, Sun L, Suzuki Y, Tsvetanova B, Wise KS, Smith HO, Glass JI, Merryman C, Gibson DG, Venter JC. Design and synthesis of a minimal bacterial genome. Science 351: aad6253, 2016. doi: 10.1126/science.aad6253. [DOI] [PubMed] [Google Scholar]
- 77.Hwang JR, Chou CL, Medvar B, Knepper MA, Jung HJ. Identification of β-catenin-interacting proteins in nuclear fractions of native rat collecting duct cells. Am J Physiol Renal Physiol 313: F30–F46, 2017. doi: 10.1152/ajprenal.00054.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Isobe K, Jung HJ, Yang CR, Claxton J, Sandoval P, Burg MB, Raghuram V, Knepper MA. Systems-level identification of PKA-dependent signaling in epithelial cells. Proc Natl Acad Sci USA 114: E8875–E8884, 2017. doi: 10.1073/pnas.1709123114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Iynedjian PB, Ballard FJ, Hanson RW. The regulation of phosphoenolpyruvate carboxykinase (GTP) synthesis in rat kidney cortex. The role of acid-base balance and glucocorticoids. J Biol Chem 250: 5596–5603, 1975. [PubMed] [Google Scholar]
- 80.Jensen TB, Pisitkun T, Hoffert JD, Jensen UB, Fenton RA, Praetorius HA, Knepper MA, Praetorius J. Assessment of the effect of 24-hour aldosterone administration on protein abundance in fluorescence-sorted mouse distal renal tubules by mass spectrometry. Nephron Physiol 121: p9–p15, 2012. doi: 10.1159/000346832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal Chem 60: 2299–2301, 1988. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 82.Khositseth S, Charngkaew K, Boonkrai C, Somparn P, Uawithya P, Chomanee N, Payne DM, Fenton RA, Pisitkun T. Hypercalcemia induces targeted autophagic degradation of aquaporin-2 at the onset of nephrogenic diabetes insipidus. Kidney Int 91: 1070–1087, 2017. doi: 10.1016/j.kint.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 83.Khositseth S, Pisitkun T, Slentz DH, Wang G, Hoffert JD, Knepper MA, Yu M-J. Quantitative protein and mRNA profiling shows selective post-transcriptional control of protein expression by vasopressin in kidney cells. Mol Cell Proteomics 10: M110.004036, 2011. doi: 10.1074/mcp.M110.004036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Khositseth S, Uawithya P, Somparn P, Charngkaew K, Thippamom N, Hoffert JD, Saeed F, Michael Payne D, Chen SH, Fenton RA, Pisitkun T. Autophagic degradation of aquaporin-2 is an early event in hypokalemia-induced nephrogenic diabetes insipidus. Sci Rep 5: 18311, 2015. doi: 10.1038/srep18311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, Thomas JK, Muthusamy B, Leal-Rojas P, Kumar P, Sahasrabuddhe NA, Balakrishnan L, Advani J, George B, Renuse S, Selvan LD, Patil AH, Nanjappa V, Radhakrishnan A, Prasad S, Subbannayya T, Raju R, Kumar M, Sreenivasamurthy SK, Marimuthu A, Sathe GJ, Chavan S, Datta KK, Subbannayya Y, Sahu A, Yelamanchi SD, Jayaram S, Rajagopalan P, Sharma J, Murthy KR, Syed N, Goel R, Khan AA, Ahmad S, Dey G, Mudgal K, Chatterjee A, Huang TC, Zhong J, Wu X, Shaw PG, Freed D, Zahari MS, Mukherjee KK, Shankar S, Mahadevan A, Lam H, Mitchell CJ, Shankar SK, Satishchandra P, Schroeder JT, Sirdeshmukh R, Maitra A, Leach SD, Drake CG, Halushka MK, Prasad TS, Hruban RH, Kerr CL, Bader GD, Iacobuzio-Donahue CA, Gowda H, Pandey A. A draft map of the human proteome. Nature 509: 575–581, 2014. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kishore BK, Mandon B, Oza NB, DiGiovanni SR, Coleman RA, Ostrowski NL, Wade JB, Knepper MA. Rat renal arcade segment expresses vasopressin-regulated water channel and vasopressin V2 receptor. J Clin Invest 97: 2763–2771, 1996. doi: 10.1172/JCI118731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Knepper MA. Molecular physiology of urinary concentrating mechanism: regulation of aquaporin water channels by vasopressin. Am J Physiol Renal Physiol 272: F3–F12, 1997. doi: 10.1152/ajprenal.1997.272.1.F3. [DOI] [PubMed] [Google Scholar]
- 88.Kompauer M, Heiles S, Spengler B. Atmospheric pressure MALDI mass spectrometry imaging of tissues and cells at 1.4-μm lateral resolution. Nat Methods 14: 90–96, 2017. doi: 10.1038/nmeth.4071. [DOI] [PubMed] [Google Scholar]
- 89.Konvalinka A, Zhou J, Dimitromanolakis A, Drabovich AP, Fang F, Gurley S, Coffman T, John R, Zhang SL, Diamandis EP, Scholey JW. Determination of an angiotensin II-regulated proteome in primary human kidney cells by stable isotope labeling of amino acids in cell culture (SILAC). J Biol Chem 288: 24834–24847, 2013. doi: 10.1074/jbc.M113.485326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kulak NA, Geyer PE, Mann M. Loss-less nano-fractionator for high sensitivity, high coverage proteomics. Mol Cell Proteomics 16: 694–705, 2017. doi: 10.1074/mcp.O116.065136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kuwahara M, Fushimi K, Terada Y, Bai L, Marumo F, Sasaki S. cAMP-dependent phosphorylation stimulates water permeability of aquaporin-collecting duct water channel protein expressed in Xenopus oocytes. J Biol Chem 270: 10384–10387, 1995. doi: 10.1074/jbc.270.18.10384. [DOI] [PubMed] [Google Scholar]
- 92.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann Y, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, , et al.; International Human Genome Sequencing Consortium . Initial sequencing and analysis of the human genome. Nature 409: 860–921, 2001. [Erratum in Nature 411: 720, 2001; Nature 412: 565, 2001.] doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 93.Larance M, Lamond AI. Multidimensional proteomics for cell biology. Nat Rev Mol Cell Biol 16: 269–280, 2015. doi: 10.1038/nrm3970. [DOI] [PubMed] [Google Scholar]
- 94.Larsen SC, Sylvestersen KB, Mund A, Lyon D, Mullari M, Madsen MV, Daniel JA, Jensen LJ, Nielsen ML. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci Signal 9: rs9, 2016. doi: 10.1126/scisignal.aaf7329. [DOI] [PubMed] [Google Scholar]
- 95.Lee JW, Alsady M, Chou CL, de Groot T, Deen PMT, Knepper MA, Ecelbarger CM. Single-tubule RNA-Seq uncovers signaling mechanisms that defend against hyponatremia in SIADH. Kidney Int 93: 128–146, 2018. doi: 10.1016/j.kint.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015. doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell 152: 1237–1251, 2013. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Leong PK, Devillez A, Sandberg MB, Yang LE, Yip DK, Klein JB, McDonough AA. Effects of ACE inhibition on proximal tubule sodium transport. Am J Physiol Renal Physiol 290: F854–F863, 2006. doi: 10.1152/ajprenal.00353.2005. [DOI] [PubMed] [Google Scholar]
- 99.Li XC, Zhuo JL. Phosphoproteomic analysis of AT1 receptor-mediated signaling responses in proximal tubules of angiotensin II-induced hypertensive rats. Kidney Int 80: 620–632, 2011. doi: 10.1038/ki.2011.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu Y, Beyer A, Aebersold R. On the dependency of cellular protein levels on mRNA abundance. Cell 165: 535–550, 2016. doi: 10.1016/j.cell.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 101.Loo CS, Chen CW, Wang PJ, Chen PY, Lin SY, Khoo KH, Fenton RA, Knepper MA, Yu MJ. Quantitative apical membrane proteomics reveals vasopressin-induced actin dynamics in collecting duct cells. Proc Natl Acad Sci USA 110: 17119–17124, 2013. doi: 10.1073/pnas.1309219110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lu HJ, Matsuzaki T, Bouley R, Hasler U, Qin QH, Brown D. The phosphorylation state of serine 256 is dominant over that of serine 261 in the regulation of AQP2 trafficking in renal epithelial cells. Am J Physiol Renal Physiol 295: F290–F294, 2008. doi: 10.1152/ajprenal.00072.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marples D, Knepper MA, Christensen EI, Nielsen S. Redistribution of aquaporin-2 water channels induced by vasopressin in rat kidney inner medullary collecting duct. Am J Physiol Cell Physiol 269: C655–C664, 1995. doi: 10.1152/ajpcell.1995.269.3.C655. [DOI] [PubMed] [Google Scholar]
- 104.Martens L. Resilience in the proteomics data ecosystem: how the field cares for its data. Proteomics 13: 1548–1550, 2013. doi: 10.1002/pmic.201300118. [DOI] [PubMed] [Google Scholar]
- 105.Martin L, Chang HY. Uncovering the role of genomic “dark matter” in human disease. J Clin Invest 122: 1589–1595, 2012. doi: 10.1172/JCI60020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Matthiesen R. Algorithms for database-dependent search of MS/MS data. Methods Mol Biol 1007: 119–138, 2013. doi: 10.1007/978-1-62703-392-3_5. [DOI] [PubMed] [Google Scholar]
- 107.McCormick JA, Ellison DH. Distal convoluted tubule. Compr Physiol 5: 45–98, 2015. doi: 10.1002/cphy.c140002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Meneton P, Loffing J, Warnock DG. Sodium and potassium handling by the aldosterone-sensitive distal nephron: the pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol 287: F593–F601, 2004. doi: 10.1152/ajprenal.00454.2003. [DOI] [PubMed] [Google Scholar]
- 109.Merkulova M, Păunescu TG, Azroyan A, Marshansky V, Breton S, Brown D. Mapping the H(+) (V)-ATPase interactome: identification of proteins involved in trafficking, folding, assembly and phosphorylation. Sci Rep 5: 14827, 2015. doi: 10.1038/srep14827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Micanovic R, Khan S, El-Achkar TM. Immunofluorescence laser micro-dissection of specific nephron segments in the mouse kidney allows targeted downstream proteomic analysis. Physiol Rep 3: e12306, 2015. doi: 10.14814/phy2.12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moeller HB, Knepper MA, Fenton RA. Serine 269 phosphorylated aquaporin-2 is targeted to the apical membrane of collecting duct principal cells. Kidney Int 75: 295–303, 2009. doi: 10.1038/ki.2008.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Moeller HB, MacAulay N, Knepper MA, Fenton RA. Role of multiple phosphorylation sites in the COOH-terminal tail of aquaporin-2 for water transport: evidence against channel gating. Am J Physiol Renal Physiol 296: F649–F657, 2009. doi: 10.1152/ajprenal.90682.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Morel F, Chabardès D, Imbert-Teboul M, Le Bouffant F, Hus-Citharel A, Montégut M. Multiple hormonal control of adenylate cyclase in distal segments of the rat kidney. Kidney Int Suppl 11: S55–S62, 1982. [PubMed] [Google Scholar]
- 115.Murray KK, Boyd RK, Eberlin MN, Langley GJ, Li L, Naito Y. Definitions of terms relating to mass spectrometry (IUPAC Recommendations 2013). Pure Appl Chem 85: 1515–1609, 2013. doi: 10.1351/PAC-REC-06-04-06. [DOI] [Google Scholar]
- 116.Nadzirin N, Firdaus-Raih M. Proteins of unknown function in the Protein Data Bank (PDB): an inventory of true uncharacterized proteins and computational tools for their analysis. Int J Mol Sci 13: 12761–12772, 2012. doi: 10.3390/ijms131012761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nedvetsky PI, Tabor V, Tamma G, Beulshausen S, Skroblin P, Kirschner A, Mutig K, Boltzen M, Petrucci O, Vossenkämper A, Wiesner B, Bachmann S, Rosenthal W, Klussmann E. Reciprocal regulation of aquaporin-2 abundance and degradation by protein kinase A and p38-MAP kinase. J Am Soc Nephrol 21: 1645–1656, 2010. doi: 10.1681/ASN.2009111190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nishimoto G, Zelenina M, Li D, Yasui M, Aperia A, Nielsen S, Nairn AC. Arginine vasopressin stimulates phosphorylation of aquaporin-2 in rat renal tissue. Am J Physiol Renal Physiol 276: F254–F259, 1999. doi: 10.1152/ajprenal.1999.276.2.F254. [DOI] [PubMed] [Google Scholar]
- 119.Noda Y, Horikawa S, Furukawa T, Hirai K, Katayama Y, Asai T, Kuwahara M, Katagiri K, Kinashi T, Hattori M, Minato N, Sasaki S. Aquaporin-2 trafficking is regulated by PDZ-domain containing protein SPA-1. FEBS Lett 568: 139–145, 2004. doi: 10.1016/j.febslet.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 120.Noda Y, Horikawa S, Katayama Y, Sasaki S. Identification of a multiprotein “motor” complex binding to water channel aquaporin-2. Biochem Biophys Res Commun 330: 1041–1047, 2005. doi: 10.1016/j.bbrc.2005.03.079. [DOI] [PubMed] [Google Scholar]
- 121.O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, Astashyn A, Badretdin A, Bao Y, Blinkova O, Brover V, Chetvernin V, Choi J, Cox E, Ermolaeva O, Farrell CM, Goldfarb T, Gupta T, Haft D, Hatcher E, Hlavina W, Joardar VS, Kodali VK, Li W, Maglott D, Masterson P, McGarvey KM, Murphy MR, O’Neill K, Pujar S, Rangwala SH, Rausch D, Riddick LD, Schoch C, Shkeda A, Storz SS, Sun H, Thibaud-Nissen F, Tolstoy I, Tully RE, Vatsan AR, Wallin C, Webb D, Wu W, Landrum MJ, Kimchi A, Tatusova T, DiCuccio M, Kitts P, Murphy TD, Pruitt KD. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44: D733–D745, 2016. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods 4: 709–712, 2007. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 123.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1: 376–386, 2002. doi: 10.1074/mcp.M200025-MCP200. [DOI] [PubMed] [Google Scholar]
- 124.Ow SY, Salim M, Noirel J, Evans C, Rehman I, Wright PC. iTRAQ underestimation in simple and complex mixtures: “the good, the bad and the ugly”. J Proteome Res 8: 5347–5355, 2009. doi: 10.1021/pr900634c. [DOI] [PubMed] [Google Scholar]
- 125.Pandey AK, Lu L, Wang X, Homayouni R, Williams RW. Functionally enigmatic genes: a case study of the brain ignorome. PLoS One 9: e88889, 2014. doi: 10.1371/journal.pone.0088889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Patterson SD, Aebersold RH. Proteomics: the first decade and beyond. Nat Genet 33, Suppl: 311–323, 2003. doi: 10.1038/ng1106. [DOI] [PubMed] [Google Scholar]
- 127.Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE. Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol 10: 135–146, 2015. doi: 10.2215/CJN.05760513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20: 3551–3567, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 129.Perry P. 3 of Nature’s Greatest Mysteries May Be Solved Thanks to Quantum Biology. Big Think (Online). https://bigthink.com/philip-perry/3-of-natures-greatest-mysteries-may-be-solved-thanks-to-quantum-biology. [12 Sept 2017].
- 130.Pickering CM, Grady C, Medvar B, Emamian M, Sandoval PC, Zhao Y, Yang CR, Jung HJ, Chou CL, Knepper MA. Proteomic profiling of nuclear fractions from native renal inner medullary collecting duct cells. Physiol Genomics 48: 154–166, 2016. doi: 10.1152/physiolgenomics.00090.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods 9: 555–566, 2012. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- 132.Pisitkun T, Bieniek J, Tchapyjnikov D, Wang G, Wu WW, Shen RF, Knepper MA. High-throughput identification of IMCD proteins using LC-MS/MS. Physiol Genomics 25: 263–276, 2006. doi: 10.1152/physiolgenomics.00214.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pisitkun T, Jacob V, Schleicher SM, Chou CL, Yu MJ, Knepper MA. Akt and ERK1/2 pathways are components of the vasopressin signaling network in rat native IMCD. Am J Physiol Renal Physiol 295: F1030–F1043, 2008. doi: 10.1152/ajprenal.90339.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Polfer NC, Oomens J, Suhai S, Paizs B. Spectroscopic and theoretical evidence for oxazolone ring formation in collision-induced dissociation of peptides. J Am Chem Soc 127: 17154–17155, 2005. doi: 10.1021/ja056553x. [DOI] [PubMed] [Google Scholar]
- 135.Quan A, Baum M. Regulation of proximal tubule transport by angiotensin II. Semin Nephrol 17: 423–430, 1997. [PubMed] [Google Scholar]
- 136.Rappsilber J, Ishihama Y, Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75: 663–670, 2003. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 137.Rattanasinganchan P, Sopitthummakhun K, Doi K, Hu X, Payne DM, Pisitkun T, Leelahavanichkul A. A folic acid-induced rat model of renal injury to identify biomarkers of tubulointerstitial fibrosis from urinary exosomes. Asian Biomed 10: 491–502, 2016. [Google Scholar]
- 138.Richardson C, Sakamoto K, de los Heros P, Deak M, Campbell DG, Prescott AR, Alessi DR. Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J Cell Sci 124: 789–800, 2011. doi: 10.1242/jcs.077230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rinschen MM, Benzing T, Limbutara K, Pisitkun T. Proteomic analysis of the kidney filtration barrier–Problems and perspectives. Proteomics Clin Appl 9: 1053–1068, 2015. doi: 10.1002/prca.201400201. [DOI] [PubMed] [Google Scholar]
- 140.Rinschen MM, Pahmeyer C, Pisitkun T, Schnell N, Wu X, Maaß M, Bartram MP, Lamkemeyer T, Schermer B, Benzing T, Brinkkoetter PT. Comparative phosphoproteomic analysis of mammalian glomeruli reveals conserved podocin C-terminal phosphorylation as a determinant of slit diaphragm complex architecture. Proteomics 15: 1326–1331, 2015. doi: 10.1002/pmic.201400235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140a.Rinschen MM, Schroeter CB, Koehler S, Ising C, Schermer B, Kann M, Benzing T, Brinkkoetter PT. Quantitative deep mapping of the cultured podocyte proteome uncovers shifts in proteostatic mechanisms during differentiation. Am J Physiol Cell Physiol 311: C404–C417, 2016. doi: 10.1152/ajpcell.00121.2016. [DOI] [PubMed] [Google Scholar]
- 141.Rinschen MM, Wu X, König T, Pisitkun T, Hagmann H, Pahmeyer C, Lamkemeyer T, Kohli P, Schnell N, Schermer B, Dryer S, Brooks BR, Beltrao P, Krueger M, Brinkkoetter PT, Benzing T. Phosphoproteomic analysis reveals regulatory mechanisms at the kidney filtration barrier. J Am Soc Nephrol 25: 1509–1522, 2014. doi: 10.1681/ASN.2013070760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rinschen MM, Yu MJ, Wang G, Boja ES, Hoffert JD, Pisitkun T, Knepper MA. Quantitative phosphoproteomic analysis reveals vasopressin V2-receptor-dependent signaling pathways in renal collecting duct cells. Proc Natl Acad Sci USA 107: 3882–3887, 2010. doi: 10.1073/pnas.0910646107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 3: 1154–1169, 2004. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 144.Sachs AN, Pisitkun T, Hoffert JD, Yu MJ, Knepper MA. LC-MS/MS analysis of differential centrifugation fractions from native inner medullary collecting duct of rat. Am J Physiol Renal Physiol 295: F1799–F1806, 2008. doi: 10.1152/ajprenal.90510.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Saeed F, Pisitkun T, Knepper M, Hoffert JD. Mining Temporal Patterns from iTRAQ Mass Spectrometry (LC-MS/MS) Data. : The Proceedings of the ISCA 3rd International Conference on Bioinformatics and Computational Biology (BiCoB), March 23–25, 2011, New Orleans, LA Saarbrucken, Germany: dblp, 2011, p. 152–159. [Google Scholar]
- 146.Sandoval PC, Slentz DH, Pisitkun T, Saeed F, Hoffert JD, Knepper MA. Proteome-wide measurement of protein half-lives and translation rates in vasopressin-sensitive collecting duct cells. J Am Soc Nephrol 24: 1793–1805, 2013. doi: 10.1681/ASN.2013030279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sands JM, Nonoguchi H, Knepper MA. Vasopressin effects on urea and H2O transport in inner medullary collecting duct subsegments. Am J Physiol Renal Physiol 253: F823–F832, 1987. doi: 10.1152/ajprenal.1987.253.5.F823. [DOI] [PubMed] [Google Scholar]
- 148.Schauer KL, Freund DM, Prenni JE, Curthoys NP. Proteomic profiling and pathway analysis of the response of rat renal proximal convoluted tubules to metabolic acidosis. Am J Physiol Renal Physiol 305: F628–F640, 2013. doi: 10.1152/ajprenal.00210.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schenk LK, Bolger SJ, Luginbuhl K, Gonzales PA, Rinschen MM, Yu MJ, Hoffert JD, Pisitkun T, Knepper MA. Quantitative proteomics identifies vasopressin-responsive nuclear proteins in collecting duct cells. J Am Soc Nephrol 23: 1008–1018, 2012. doi: 10.1681/ASN.2011070738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shishkova E, Hebert AS, Coon JJ. Now, more than ever, proteomics needs better chromatography. Cell Syst 3: 321–324, 2016. doi: 10.1016/j.cels.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Simons BL, Wang G, Shen RF, Knepper MA. In vacuo isotope coded alkylation technique (IVICAT); an N-terminal stable isotopic label for quantitative liquid chromatography/mass spectrometry proteomics. Rapid Commun Mass Spectrom 20: 2463–2477, 2006. doi: 10.1002/rcm.2615. [DOI] [PubMed] [Google Scholar]
- 152.Siuzdak G. The emergence of mass spectrometry in biochemical research. Proc Natl Acad Sci USA 91: 11290–11297, 1994. doi: 10.1073/pnas.91.24.11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Star RA, Nonoguchi H, Balaban R, Knepper MA. Calcium and cyclic adenosine monophosphate as second messengers for vasopressin in the rat inner medullary collecting duct. J Clin Invest 81: 1879–1888, 1988. doi: 10.1172/JCI113534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Steen H, Mann M. The ABC’s (and XYZ’s) of peptide sequencing. Nat Rev Mol Cell Biol 5: 699–711, 2004. doi: 10.1038/nrm1468. [DOI] [PubMed] [Google Scholar]
- 155.Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A, Coulson A, D’Eustachio P, Fitch DH, Fulton LA, Fulton RE, Griffiths-Jones S, Harris TW, Hillier LW, Kamath R, Kuwabara PE, Mardis ER, Marra MA, Miner TL, Minx P, Mullikin JC, Plumb RW, Rogers J, Schein JE, Sohrmann M, Spieth J, Stajich JE, Wei C, Willey D, Wilson RK, Durbin R, Waterston RH. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol 1: e45, 2003. doi: 10.1371/journal.pbio.0000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci USA 101: 9528–9533, 2004. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tamma G, Robben JH, Trimpert C, Boone M, Deen PM. Regulation of AQP2 localization by S256 and S261 phosphorylation and ubiquitination. Am J Physiol Cell Physiol 300: C636–C646, 2011. doi: 10.1152/ajpcell.00433.2009. [DOI] [PubMed] [Google Scholar]
- 158.Tanaka K, Waki H, Ido Y, Akita S, Yoshida Y, Yoshida T, Matsuo T. Protein and polymer analyses up to m/z 100 000 by laser ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 2: 151–153, 1988. doi: 10.1002/rcm.1290020802. [DOI] [Google Scholar]
- 159.Taurin S, Sandbo N, Yau DM, Sethakorn N, Dulin NO. Phosphorylation of beta-catenin by PKA promotes ATP-induced proliferation of vascular smooth muscle cells. Am J Physiol Cell Physiol 294: C1169–C1174, 2008. doi: 10.1152/ajpcell.00096.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tchapyjnikov D, Li Y, Pisitkun T, Hoffert JD, Yu MJ, Knepper MA. Proteomic profiling of nuclei from native renal inner medullary collecting duct cells using LC-MS/MS. Physiol Genomics 40: 167–183, 2010. doi: 10.1152/physiolgenomics.00148.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.The UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res 45: D158–D169, 2017. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75: 1895–1904, 2003. [Erratum in Anal Chem 75: 4942, 2003; Anal Chem 78: 4235, 2006.] doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 163.Timmons JA, Szkop KJ, Gallagher IJ. Multiple sources of bias confound functional enrichment analysis of global -omics data. Genome Biol 16: 186, 2015. doi: 10.1186/s13059-015-0761-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tong J, Harrison G, Curthoys NP. The effect of metabolic acidosis on the synthesis and turnover of rat renal phosphate-dependent glutaminase. Biochem J 233: 139–144, 1986. doi: 10.1042/bj2330139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Trapnell C. Defining cell types and states with single-cell genomics. Genome Res 25: 1491–1498, 2015. doi: 10.1101/gr.190595.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Udeshi ND, Mertins P, Svinkina T, Carr SA. Large-scale identification of ubiquitination sites by mass spectrometry. Nat Protoc 8: 1950–1960, 2013. doi: 10.1038/nprot.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.van Balkom BW, Hoffert JD, Chou CL, Knepper MA. Proteomic analysis of long-term vasopressin action in the inner medullary collecting duct of the Brattleboro rat. Am J Physiol Renal Physiol 286: F216–F224, 2004. doi: 10.1152/ajprenal.00307.2003. [DOI] [PubMed] [Google Scholar]
- 167a. Van Der Mersch V. Exploring the Cloud Laboratory: Advances in Biotech & Science-as-a-Service (Online). Nordic APIS; https://nordicapis.com/exploring-the-cloud-laboratory-advances-in-biotech-science-as-a-service. [15 April 2018]. [Google Scholar]
- 168.Verheggen K, Raeder H, Berven FS, Martens L, Barsnes H, Vaudel M. Anatomy and evolution of database search engines-a central component of mass spectrometry based proteomic workflows. Mass Spectrom Rev 2017: 1–15, 2017. doi: 10.1002/mas.21543. [DOI] [PubMed] [Google Scholar]
- 169.Vickaryous MK, Hall BK. Human cell type diversity, evolution, development, and classification with special reference to cells derived from the neural crest. Biol Rev Camb Philos Soc 81: 425–455, 2006. doi: 10.1017/S1464793106007068. [DOI] [PubMed] [Google Scholar]
- 170.Walmsley SJ, Broeckling C, Hess A, Prenni J, Curthoys NP. Proteomic analysis of brush-border membrane vesicles isolated from purified proximal convoluted tubules. Am J Physiol Renal Physiol 298: F1323–F1331, 2010. doi: 10.1152/ajprenal.00711.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Walmsley SJ, Freund DM, Curthoys NP. Proteomic profiling of the effect of metabolic acidosis on the apical membrane of the proximal convoluted tubule. Am J Physiol Renal Physiol 302: F1465–F1477, 2012. doi: 10.1152/ajprenal.00390.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang PJ, Lin ST, Liu SH, Kuo KT, Hsu CH, Knepper MA, Yu MJ. Vasopressin-induced serine 269 phosphorylation reduces Sipa1l1 (signal-induced proliferation-associated 1 like 1)-mediated aquaporin-2 endocytosis. J Biol Chem 292: 7984–7993, 2017. doi: 10.1074/jbc.M117.779611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Weinert BT, Schölz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, Choudhary C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Reports 4: 842–851, 2013. doi: 10.1016/j.celrep.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 174.Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, Mathieson T, Lemeer S, Schnatbaum K, Reimer U, Wenschuh H, Mollenhauer M, Slotta-Huspenina J, Boese JH, Bantscheff M, Gerstmair A, Faerber F, Kuster B. Mass-spectrometry-based draft of the human proteome. Nature 509: 582–587, 2014. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 175.Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362, 2009. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 176.Wu Q, Xu W, Cao L, Li X, He T, Wu Z, Li W. SAHA treatment reveals the link between histone lysine acetylation and proteome in nonsmall cell lung cancer A549 Cells. J Proteome Res 12: 4064–4073, 2013. doi: 10.1021/pr4004079. [DOI] [PubMed] [Google Scholar]
- 177.Xie L, Hoffert JD, Chou CL, Yu MJ, Pisitkun T, Knepper MA, Fenton RA. Quantitative analysis of aquaporin-2 phosphorylation. Am J Physiol Renal Physiol 298: F1018–F1023, 2010. doi: 10.1152/ajprenal.00580.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, Boeke JD, Zhao Y. Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics 11: 100–107, 2012. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yang CR, Raghuram V, Emamian M, Sandoval PC, Knepper MA. Deep proteomic profiling of vasopressin-sensitive collecting duct cells. II. Bioinformatic analysis of vasopressin signaling. Am J Physiol Cell Physiol 309: C799–C812, 2015. doi: 10.1152/ajpcell.00214.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yang CR, Tongyoo P, Emamian M, Sandoval PC, Raghuram V, Knepper MA. Deep proteomic profiling of vasopressin-sensitive collecting duct cells. I. Virtual Western blots and molecular weight distributions. Am J Physiol Cell Physiol 309: C785–C798, 2015. doi: 10.1152/ajpcell.00213.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yang F, Shen Y, Camp DG II, Smith RD. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev Proteomics 9: 129–134, 2012. doi: 10.1586/epr.12.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yu MJ, Miller RL, Uawithya P, Rinschen MM, Khositseth S, Braucht DW, Chou CL, Pisitkun T, Nelson RD, Knepper MA. Systems-level analysis of cell-specific AQP2 gene expression in renal collecting duct. Proc Natl Acad Sci USA 106: 2441–2446, 2009. doi: 10.1073/pnas.0813002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yu MJ, Pisitkun T, Wang G, Shen RF, Knepper MA. LC-MS/MS analysis of apical and basolateral plasma membranes of rat renal collecting duct cells. Mol Cell Proteomics 5: 2131–2145, 2006. doi: 10.1074/mcp.M600177-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yui N, Ando F, Sasaki S, Uchida S. Ser-261 phospho-regulation is involved in pS256 and pS269-mediated aquaporin-2 apical translocation. Biochem Biophys Res Commun 490: 1039–1044, 2017. doi: 10.1016/j.bbrc.2017.06.162. [DOI] [PubMed] [Google Scholar]
- 185.Yui N, Sasaki S, Uchida S. Aquaporin-2 Ser-261 phosphorylation is regulated in combination with Ser-256 and Ser-269 phosphorylation. Biochem Biophys Res Commun 482: 524–529, 2017. doi: 10.1016/j.bbrc.2016.11.118. [DOI] [PubMed] [Google Scholar]
- 186.Zhao B, Knepper MA, Chou C-L, Pisitkun T. Large-scale phosphotyrosine proteomic profiling of rat renal collecting duct epithelium reveals predominance of proteins involved in cell polarity determination. Am J Physiol Cell Physiol 302: C27–C45, 2012. doi: 10.1152/ajpcell.00300.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhao H, Wiederkehr MR, Fan L, Collazo RL, Crowder LA, Moe OW. Acute inhibition of Na/H exchanger NHE-3 by cAMP. Role of protein kinase a and NHE-3 phosphoserines 552 and 605. J Biol Chem 274: 3978–3987, 1999. doi: 10.1074/jbc.274.7.3978. [DOI] [PubMed] [Google Scholar]
- 188.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science 327: 1000–1004, 2010. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhao Y, Yang CR, Raghuram V, Parulekar J, Knepper MA. BIG: a large-scale data integration tool for renal physiology. Am J Physiol Renal Physiol 311: F787–F792, 2016. doi: 10.1152/ajprenal.00249.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zheleznova NN, Yang C, Ryan RP, Halligan BD, Liang M, Greene AS, Cowley AW Jr. Mitochondrial proteomic analysis reveals deficiencies in oxygen utilization in medullary thick ascending limb of Henle in the Dahl salt-sensitive rat. Physiol Genomics 44: 829–842, 2012. doi: 10.1152/physiolgenomics.00060.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zou D, Ma L, Yu J, Zhang Z. Biological databases for human research. Genomics Proteomics Bioinformatics 13: 55–63, 2015. doi: 10.1016/j.gpb.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]