

Abstract
Muscular dystrophies represent a large group of genetic disorders that significantly impair quality of life and often progress to premature death. There is no effective treatment for these debilitating diseases. Most therapies, developed to date, focus on alleviating the symptoms or targeting the secondary effects, while the underlying gene mutation is still present in the human genome. The discovery and application of programmable nucleases for site-specific DNA double-stranded breaks provides a powerful tool for precise genome engineering. In particular, the CRISPR/Cas system has revolutionized the genome editing field and is providing a new path for disease treatment by targeting the disease-causing genetic mutations. In this review, we provide a historical overview of genome-editing technologies, summarize the most recent advances, and discuss potential strategies and challenges for permanently correcting genetic mutations that cause muscular dystrophies.
I. INTRODUCTION
A. Skeletal Muscle Structure
From intense body movement in Greco-Roman wrestling to delicate vocal control in coloratura soprano, skeletal muscle supports a remarkably wide range of human activities. As one of the largest tissues, skeletal muscle accounts for ~40% of human body weight and is essential for physical support, locomotion, energy expenditure, and metabolism.
Skeletal muscle is a highly organized tissue and is composed of thousands of multinucleated myofibers, which are formed by fusion of mononucleated myoblasts during development and regeneration. Bundles of myofibers form a muscle fascicle, and groups of fascicles contribute to the structure of a skeletal muscle (FIGURE 1A). The functional unit of a myofiber is the sarcomere, which comprises actin thin filaments and myosin thick filaments. The sliding of the thin and thick filaments past each other generates a muscle contraction.
FIGURE 1.

Skeletal muscle structure. A: skeletal muscle is composed of thousands of multinucleated myofibers. Bundles of myofibers form muscle fascicles, and groups of fascicles contribute to skeletal muscle structure. B: satellite cells are adult skeletal muscle stem cells, which reside between the sarcolemma and basal lamina of myofibers.
B. Skeletal Muscle Regeneration and Satellite Cells
The adult musculature has a remarkable regenerative capacity, primarily due to the contribution of the skeletal muscle resident stem cells, known as satellite cells (48, 63, 454). Satellite cells reside between the sarcolemma and basal lamina of myofibers and are marked by expression of a paired-box transcription factor, Pax7 (350) (FIGURE 1B). Upon muscle injury, quiescent satellite cells become activated and undergo proliferation and differentiation, and finally form multinucleated myofibers by fusion. Activated satellite cells can also undergo asymmetric division, in which one daughter cell maintains a satellite stem cell fate and the other one acquires a myogenic commitment, becoming a satellite cell committed myogenic progenitor (76, 202, 358). Pax7 is the canonical biomarker for quiescent and activated satellite cells and is downregulated during myogenic differentiation. Genetic ablation experiments demonstrated that Pax7+ satellite cells are indispensable for adult skeletal muscle regeneration (215, 258, 283, 340).
Skeletal muscle regeneration is evolutionarily conserved among many bilaterians, requiring involvement of satellite cells or satellite-like cells (20). However, in certain bilateral species such as zebrafish and adult newt, myofiber dedifferentiation is also a unique mechanism for skeletal muscle regeneration. For example, extraocular muscle regeneration in adult zebrafish involves dedifferentiation of residual myocytes, which do not express Pax7 but express Mef2c (334). Similarly, limb muscle regeneration in the adult newt requires dedifferentiation of myocytes to Pax7-negative mononuclear cells (344).
C. Muscular Dystrophies
Despite the remarkable regenerative capacity of skeletal muscle, muscles are vulnerable to numerous disorders, including congenital myopathies, muscular dystrophies, and inflammatory myopathies. Muscular dystrophies are a large group of genetic disorders characterized by progressive weakness of multiple muscle groups. Owing to the advancement of genome research, the genetic causes of many muscular dystrophies have been identified, with many affecting sarcolemma-associated proteins, extracellular matrix proteins, glycosyltransferase enzymes, as well as nuclear proteins (174, 264). Depending on the mutation type and disease onset, muscular dystrophies can significantly impair the quality of life and cause premature death. There is no cure for these debilitating diseases. Initial efforts in gene therapy relied on gene replacement, but the source of the mutation remains present in the genome. Advancements in genome engineering technologies enable precise manipulation of the genetic mutations that cause muscular dystrophies and offer the prospect of a genetic therapy for the permanent correction of diverse genetic defects.
In this review, we provide a historical overview and consider the most recent advances in genome editing technologies. We also detail strategies of genome editing for the correction of genetic mutations that cause muscular dystrophies. Finally, we highlight current cell- and animal-based studies of muscular dystrophy and discuss current challenges and future perspectives of translating genome editing technologies to clinical applications.
II. GENOME AND EPIGENOME EDITING
A. History of Genome Editing: Meganuclease, ZFN, TALEN, and the CRISPR/Cas System
Three decades ago the laboratories of Mario Capecchi and Oliver Smithies independently developed methods for homologous recombination (HR)-mediated mammalian gene targeting technology by providing mammalian cells with exogenous plasmid DNA containing sequence homology to the endogenous genome (93, 250, 367, 393, 394). This HR-mediated technology allows precise gene knockout or correction of genetic mutations. HR-mediated embryonic stem (ES) cell gene targeting, together with mouse chimeras and germline transmission technologies developed by the laboratory of Martin Evans (49), paved the way for the generation of “knockin” and “knockout” animal models, which significantly expanded our knowledge of gene function and advanced many fields of biological research. However, because DNA double-strand breaks (DSBs) occur randomly in the genome, the frequency of HR-mediated gene targeting is low (between 10−6 and 10−4, depending on the length of sequence homology of the targeting vector) (86). Moreover, screening of correctly targeted clones requires positive-negative selection and/or Southern blot analysis, which is time consuming and labor intensive (57). Therefore, routine application of the conventional HR-mediated gene targeting technology for studying gene function was not feasible at that time.
In the early 1990s, it was discovered that HR-mediated gene targeting efficiency could be enhanced by more than 100-fold, when the DNA DSBs were initiated at the target region by providing mammalian cells with a rare-cutting meganuclease discovered in yeast (331). This discovery stimulated the development of programmable nucleases for creating site-specific DNA DSBs. Within the past two decades, four major classes of nucleases have been engineered, which are 1) meganucleases (366), 2) zinc-finger nucleases (ZFNs) (265, 406), 3) transcription activator-like effector nucleases (TALENs) (38, 74, 266, 275), and 4) CRISPR/Cas endonucleases (clustered regularly interspaced short palindromic repeats and CRISPR-associated proteins) (77, 168, 248, 466).
Permanent correction of genetic mutations that contribute to monogenic neuromuscular disorders offers the ultimate treatment for these diseases. Early attempts at genome editing for treatment of muscular dystrophies were challenged by low efficiency, cytotoxicity, and delivery issues (122, 217, 240, 303, 304, 316, 402, 443). The newly discovered CRISPR/Cas system has been effectively used in genome engineering and represents a new approach to therapeutic genome editing.
1. Meganucleases
Meganucleases are engineered homing endonucleases, which were initially discovered in archaea, bacteria, and unicellular eukaryotic genomes (375, 376). Unlike conventional type II restriction endonucleases that recognize short 4–8 base pairs (bp) of palindromic DNA sequences (315), meganucleases require extended DNA recognition sequences (typically 16–18 bp) to generate site-specific DNA DSBs (62). Meganucleases have been used to enhance HR-mediated gene targeting efficiency by introducing site-specific DNA DSBs in cultured mammalian cells and plants (72, 73, 94, 321, 331), but they have not been widely adopted for genome engineering because the DNA-recognition domain and the nuclease domain overlap (361, 366). This overlap may adversely affect the catalytic activity of the nuclease domain (14), making it very challenging to engineer the DNA recognition domain for specificity in new-sequence binding.
To address this issue, researchers began to focus on the type IIS restriction enzyme FokI, which has two separate domains for DNA recognition and cleavage (188, 218–220). They engineered novel chimeric FokI endonucleases with new DNA sequence specificities by swapping DNA-binding domains from other transcription factors, such as the Drosophila Ubx homeodomain (187), yeast Gal4 domain (189), zinc-finger protein (186), and TAL effector (74). The latter two chimeric FokI endonucleases paved the way for the development of ZFNs and TALENs, respectively.
2. ZFNs
ZFNs are chimeric endonucleases containing multiple Cys2-His2 zinc-finger domains at the amino terminus (NH2 terminus) for DNA-binding and a FokI nuclease domain at the carboxyl terminus (COOH terminus) for DNA cleavage (FIGURE 2A) (186). Each individual zinc-finger domain contains ~30 amino acids folded in a ββα arrangement and contacts 3 bp of DNA sequence (310). Each ZFN monomer consists of 3–6 individual zinc-finger domains, and thus can bind to 9–18 bp of DNA sequence. Two approaches have been applied to improve genome targeting specificity and expand the targeting range of ZFNs. The first one is to engineer the wild-type (WT) FokI nuclease to reduce the formation of cleavage-competent homodimers (265, 382). The engineered ZFNs require heterodimerization to form a functional nuclease, in which two monomers are separated by 5–7 bp of spacer (35). The second approach is to engineer zinc-finger domains for unique triplet DNA binding specificity by combinatorial library selection and/or oligomerized pool engineering (OPEN) (32, 95, 96, 128, 183, 237, 351).
FIGURE 2.

Programmable nucleases used for genome editing. A: a schematic illustration of a pair of zinc-finger nuclease (ZFN) monomers bound to DNA. ZFN is a chimeric endonuclease composed of multiple zinc finger domains (colored boxes) at the NH2 terminus for DNA binding and a Fok1 nuclease domain (green oval) at the COOH terminus for DNA cleavage. Dimerization of two Fok1 nucleases induces a DNA double-strand break (DSB) with 4 bp of 5′ overhang. B: a schematic illustration of a pair of transcription activator-like effector nucleases (TALENs) bound to DNA. TALEN is a chimeric endonuclease composed of multiple TALE repeats (colored rectangles) at the NH2 terminus and a Fok1 nuclease domain (green oval) at the COOH terminus. Each TALE repeat recognizes 1 bp of DNA, and the sequence specificity is determined by repeat-variable diresidues (RVD; shown in red). TALEN-mediated DNA DSBs are induced by dimerization of two Fok1 nucleases. C: a schematic illustration of the engineered CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats-CRISPR associated protein 9) system from Streptococcus pyogenes. In the CRISPR/Cas9 system, target recognition is mediated by DNA hybridization with a single guide RNA (sgRNA), which is an engineered RNA chimera composed of CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA). CRISPR/Cas9-mediated DNA DSB requires a protospacer adjacent motif (PAM; shown in red), and cleavage is induced at the nucleotide 3 bp proximal to the PAM. Red arrowheads indicate cleavage site.
These two approaches paved the way for modular assembly of customized ZFNs, which have been used for genome targeting in cultured cells, animals, and plants (116, 314, 352, 407). However, because of the context-dependent effects between adjacent zinc-finger domains, large-scale assembly of functional ZFNs remains challenging, and cytotoxicity caused by off-target effects is also a critical issue (115, 309, 343). Moreover, the genome targeting density of ZFNs is also limited because the engineered zinc-finger domains cannot target all 64 possible triplet DNA sequences, especially 5′-TNN-3′ sequences (N represents any nucleotide) (33). These obstacles prevent wide application of ZFNs for genome engineering.
3. TALENs
TALENs are chimeric endonucleases that contain multiple DNA-binding domains, known as transcription activator-like effectors (TALEs), at the NH2 terminus, and a FokI nuclease domain at the COOH terminus for DNA cleavage (FIGURE 2B) (74). Unlike the zinc-finger domain in ZFNs, which binds to a triplet DNA sequence, the TALE domain, consisting of 33–35 amino acids in tandem arrays, recognizes a single base pair (87, 242). The sequence specificity of each TALE repeat is determined by the 12th and 13th amino acids at the TALE domain, known as repeat variable diresidues (RVDs) (38, 275). Similar to ZFNs, functional TALENs require dimerization of the FokI nuclease domain with each TALE arm targeting 15–20 bp of DNA sequence separated by 12–21 bp of spacer. TALENs have been widely used to target genomes of various species including cultured cells, animals, and plants (116, 170, 352, 380). Although many cloning methods have been developed for the construction of functional TALENs, such as type II restriction enzyme-based Golden Gate assembly (58), solid-phase assembly (50, 327), and ligation-independent cloning (347), modular assembly of customized TALENs is still challenging and time consuming because each TALEN arm consists of up to 20 highly repetitive TALE arrays.
Despite the difficulty of assembling TALE arrays, TALENs still offer many advantages over other programmable nucleases. First, TALENs have the highest genome-targeting density compared with ZFNs and CRISPR/Cas because each TALE array recognizes DNA sequence at single nucleotide resolution (38, 275). Second, TALENs have minimal off-target effects because a functional TALEN requires dimerization of two TALEN pairs, which can bind 30–40 bp of DNA sequence (87, 182, 242). Therefore, TALENs offer benefits for genome engineering.
4. CRISPR/Cas system
The discovery of CRISPR can be dated back to 1987, when a Japanese research group identified a series of directed repeats interspaced with short spacer sequences in the genome of Escherichia coli, although the function of these repeats was unknown at that time (161). It was not until the mid 2000s that researchers discovered that these directed repeats are widely present in over 40% of sequenced bacteria and 90% of archaea genomes (270) and found that the short spacer sequences between the directed repeats are of plasmid and viral origin (41, 269, 319). After realizing that the CRISPR locus is actively transcribed and the protein product has potential nuclease and helicase activities, scientists proposed that the CRISPR/Cas system functions as an adaptive immune system in bacteria and archaea to defend against viral infection (23, 41, 52, 139, 163, 244, 253, 319). The CRISPR/Cas system can be grouped into two classes and six subtypes: the class 1 system encodes multiple effector proteins forming a Cascade complex (CRISPR-associated complex for antiviral defense) with their corresponding signature proteins, such as Cas3, Cas10, and Csf1 from type I, III, and IV CRISPR systems, respectively (243, 245, 246, 359). The class 2 system encodes a single Cas protein with multiple functions, including Cas9, Cpf1, and Cas13a/C2c2 from type II, V, and VI CRISPR systems (5, 102, 359, 466).
The mechanism of CRISPR immunity in bacteria and archaea varies between different CRISPR types, but generally can be divided into three stages, which are protospacer acquisition, precursor CRISPR RNA (pre-crRNA) processing, and crRNA-guided cleavage of exogenous nucleic acids (252, 440). Most CRISPR immunity requires a protospacer adjacent motif (PAM) located next to the crRNA target region in the exogenous invading genome (151, 440).
Owing to the simplicity of the class 2 CRISPR system in which only one RNA-guided endonuclease is required for nucleic acid cleavage, scientists engineered Cas9 endonuclease in conjunction with a hybrid crRNA-tracrRNA duplex, known as single guide RNA (sgRNA), for efficient site-specific genome cleavage in eukaryotic cells (FIGURE 2C) (77, 168, 248). Currently, the most widely used Cas9 endonuclease is from Streptococcus pyogenes with 5′-NGG-3′ or 5′-NAG-3′ PAM preference. Other Cas9 orthologs are also available for genome targeting, including Cas9 endonucleases from Staphylococcus aureus (322), Neisseria meningitides (150), and Streptococcus thermophilus (238, 279), although these Cas9 orthologs recognize longer and more complicated PAM sequences. Besides the type II CRISPR/Cas9 system, the most recently discovered type V and VI CRISPR effectors including Cpf1 (466) and Cas13a/C2c2 (5, 102) further expand the range of genome editing and nucleic acid detection. We will further discuss these two CRISPR effectors in section IID.
B. CRISPR/Cas-Mediated Genome Editing: C-NHEJ, HDR, and MMEJ
On average, each human cell undergoes ~50 spontaneous DNA DSBs during each cell cycle (416). DNA DSBs occur randomly, so the efficiency of HR-mediated gene targeting in the absence of programmable nucleases is extremely low (86). The RNA-guided CRISPR/Cas system significantly enhances and simplifies genome editing, in which the Cas9-sgRNA ribonucleoprotein complex binds to DNA by base-pairing with sgRNA, generating a site-specific DNA DSB adjacent to the PAM sequence. Depending on the cell cycle stage and repair machinery, the DNA DSBs can be repaired by error-prone nonhomologous end joining (NHEJ) or by accurate homology-directed repair (HDR). Additionally, there is a third DNA DSB repair pathway known as microhomology-mediated end joining (MMEJ), which is a subtype of alternative NHEJ (alt-NHEJ).
1. Classical NHEJ
Classical NHEJ (C-NHEJ) DNA repair machinery is triggered when a CRISPR/Cas-induced DNA DSB occurs in the absence of a repair template (FIGURE 3A). Although C-NHEJ is active in all stages of the cell cycle, it occurs preferentially during the G1 phase when the DNA-end resection activity is low (160). The end of a DNA DSB is recognized by Ku70/Ku80 heterodimers, which recruit and activate the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs). Depending on the nature of the break, the ends of DNA DSBs can be directly ligated by the DNA ligase IV-XRCC4 complex, or requires additional processing steps, such as end resection by Artmis, WRN, or APLF nucleases and nucleotide synthesis by DNA polymerases μ and λ (75, 83).
FIGURE 3.

DNA repair pathways involved in CRISPR/Cas-induced DNA double-strand break repair. A: classical nonhomologous end joining (c-NHEJ) is a Ku-dependent DNA repair pathway that is active throughout the cell cycle. In the absence of a donor template, c-NHEJ generates insertions or deletions (INDELs; shown in red) in the genome. B: when a DNA double-strand break (DSB) is induced in the S or G2 phase of the cell cycle, homology-directed repair (HDR) can be triggered if a donor template is present (magenta), leading to precise repair of the genome. C: a Ku-independent microhomology-mediated end joining (MMEJ) pathway can be used for DNA DSB repair if the DNA breakage site shares sequence homology. MMEJ-mediated repair generates INDELs in the genome (red).
DNA DSBs repaired by C-NHEJ usually generate insertions or deletions (INDELs). Depending on the location of the site-specific cleavage, C-NHEJ has been used for different purposes of genome editing. The most widely used application of C-NHEJ is gene disruption, because INDELs often cause a frameshift of an exon and subsequently disrupt gene function, resulting in a gene knockout. However, C-NHEJ can also cause exon skipping if the INDELs disrupt the splice acceptor site (217), or an exonic splice enhancer/silencer sequence (277), although the outcome of the latter scenario is less predictable. Depending on the reading frame of the skipped exon and adjacent exons, exon skipping can cause gene knockout when the newly spliced adjacent exons are out of frame. Conversely, exon skipping can also produce a truncated protein if the newly spliced adjacent exons are perfectly in frame with each other.
C-NHEJ was generally considered as an error-prone DSB repair pathway. However, some recent studies also demonstrated the precision of C-NHEJ (18, 251) and used this repair pathway for homology-independent targeted integration (HITI) of DNA fragments into postmitotic cells and animals, further expanding the application of CRISPR/Cas-mediated C-NHEJ in genome editing (381).
2. HDR
DNA DSBs can also be repaired by HDR during S and G2 phases of the cell cycle, when sister chromatids can be used as a template for HR (FIGURE 3B) (456). During HDR, the end of the DNA DSB is recognized by the MRE11-RAD50-NBS1 (MRN) complex, which undergoes initial DNA end resection induced by MRE11 (434), followed by extensive end resection induced by the EXO1-BLM complex (40), producing single-stranded DNA (ssDNA). The exposed ssDNA is coated by RPA until RAD51 detects the homology sequence, leading to strand invasion and Holliday junction formation (431). HDR is completed when the Holliday junction is either dissolved by the BLM/TOPOIII complex or resolved by GEN1 or SLX1/SLX4 nucleases (442).
Currently, the most widely used repair templates for HDR are double-stranded DNA (circular or linearized plasmid) and single-stranded oligodeoxynucleotides (ssODNs). Before programmable nucleases were employed in HR-mediated gene targeting, the length of sequence homology on the targeting vector for HDR could be up to 14 kb for efficient gene targeting (86). The development of programmable nucleases, especially the CRISPR/Cas system, significantly enhanced site-specific DNA DSBs and further reduced the length of sequence homology on the targeting vector to several hundred base pairs (255, 468).
A ssODN can also serve as a repair template for HDR, especially for introducing small DNA modifications (231, 329, 441, 469). Interestingly, asymmetrical ssODN complementary to the nontarget strand (the DNA strand that does not base pair with the CRISPR sgRNA) can drive the efficiency of HDR up to 60%, because the Cas9 endonuclease first releases the PAM-distal nontarget DNA strand, which is more available for ssODN binding (329). Due to the accuracy of HDR, it is possible that the Cas9 endonuclease will continuously generate DSBs at the target site as long as the PAM and the sgRNA target sequence remain intact, even when HDR is completed. Because of codon degeneracy, introducing a silent mutation at the third nucleotide of the triplet codon for the disruption of the PAM and/or sgRNA target sequence can effectively overcome the recleavage event (231, 469).
3. MMEJ
MMEJ is a Ku-independent alt-NHEJ repair pathway for DNA DSBs, which displays maximal activity in S phase (FIGURE 3C) (398). Similar to HDR, MMEJ undergoes initial DNA end resection induced by MRE11 but does not require extensive end resection induced by the EXO1-BLM complex (324, 398, 444). If microhomology is present, the exposed ssDNA ends generated by initial DNA end resection will anneal with each other and the gap between the newly annealed ssDNA will be filled by DNA polymerases θ (61, 460) and finally ligated by the LIG3-XRCC1 complex (17, 362, 421).
In the absence of template DNA, MMEJ is an error-prone DNA repair pathway because of INDEL formation (356, 398). However, several studies have adopted MMEJ for precise integration of exogenous reporter genes into the genome after TALEN or CRISPR/Cas9-mediated DNA DSBs (146, 285, 335). This method, known as Precise Integration into Target Chromosome (PITCh), requires three DNA DSBs, with one DSB located at the target locus in the genome, and the other two DSBs located at the 5′- and 3′-ends of a reporter cassette (e.g., GFP). The reporter cassette is cloned into a plasmid with 5–25 bp of sequence homology to the target locus, serving as the MMEJ repair template. Therefore, MMEJ provides an alternative method for precise genome editing similar to HDR.
C. Engineered Cas9 With Mutant Nuclease Domains
CRISPR/Cas9-mediated DNA DSBs are induced by two separate nuclease domains, in which the HNH nuclease domain cuts the target strand that hybridizes with the sgRNA and the RuvC nuclease domain cuts the nontarget strand (295). A single amino acid mutation at the RuvC-I domain (D10A) generates a Cas9 nickase that is only active for target strand cleavage (FIGURE 4A) (323). Double mutations at both RuvC-I and HNH nuclease domains (D10A, H840A) abolish the Cas9 nuclease activity, generating a deactivated Cas9 (dCas9) (FIGURE 4B) (168). A pair of Cas9 nickases can be used to induce DNA DSBs with high specificity, since only two adjacent DNA single-strand breaks can generate a DSB (323). Although dCas9 lacks its nuclease activity, it still has RNA-guided DNA binding activity and can be fused with different effectors, including cytidine deaminase, transcriptional activators/repressors, and epigenetic modifiers for different purposes, such as nucleotide conversion as well as genome and epigenome regulation (66, 126, 142, 179, 194, 196, 228, 274, 294). Unlike CRISPR/Cas9-mediated permanent genome alternation, CRISPR/dCas9-mediated genome and epigenome regulation does not modify the genome. Therefore, the CRISPR/dCas9 system provides a powerful tool for inducible and reversible control of gene expression and changing the epigenetic landscape without modifying endogenous genomic sequence.
FIGURE 4.

CRISPR/Cas9 with mutant nuclease domain. CRISPR/Cas9-mediated DNA double-strand break (DSB) is induced by two separate nuclease domains, in which the HNH domain cleaves the target strand and the RuvC domain cleaves the nontarget strand. A: D10A Cas9 nickase having a mutation in the RuvC domain is only active for target strand cleavage by the HNH nuclease domain. B: mutations at both RuvC and HNH nuclease domains (D10A, H840A) abolish the Cas9 nuclease activity, generating a deactivated Cas9 (dCas9). C: CRISPR/dCas9-mediated gene regulation is achieved by fusing dCas9 to transcriptional activation domains, transcriptional repression domains, or epigenetic modifiers.
1. Base editing: Cas9 nickase fused to cytidine deaminase
Base editing is a CRISPR/Cas-mediated genome editing technology in which the dCas9 or Cas9 nickase is fused to a cytidine deaminase for site-specific C-G to T-A conversion (194, 294) or fused to an engineered adenine deaminase for site-specific A-T to G-C conversion (123). Classically, CRISPR/Cas-mediated gene disruption introduces INDELs into the genome, which is imprecise and unpredictable, potentially leading to cytotoxicity by unintended alterations in the genome due to off-target effects. In contrast, gene knockout by Cas9 nickase-mediated base editing does not generate DNA DSBs. Several studies have applied this technology for precise gene knockout through site-specific introduction of premature stop codons (34, 203).
Based on the NCBI ClinVar database, more than 900 human genetic diseases are caused by T-to-C or A-to-G mutations, and these can be corrected by base editing (194, 206). Therefore, CRISPR/Cas9 nickase-mediated base editing represents a promising technology for therapeutic genome editing because its efficiency is higher than HDR, while INDEL formation is minimized since a DNA DSB is not required.
2. Transcriptional regulation: dCas9 fused to transcriptional activator/repressor
CRISPR/Cas9-mediated genome editing was developed to alter DNA sequences. In contrast, CRISPR/dCas9-based technology was developed to regulate gene expression at the transcriptional level without altering genome integrity. The general mechanism of CRISPR/dCas9-mediated transcriptional regulation is achieved by direct fusion or recruitment of transcriptional activators or repressors to the dCas9-sgRNA complex, forming a CRISPR activation or interference complex (CRISPRa or CRISPRi). In the presence of sequence-specific sgRNA, CRISPRa or CRISPRi is targeted to the transcription start site of a gene, thereby inducing or repressing gene expression, respectively (193, 391, 420).
Gene repression in eukaryotes requires that dCas9 be fused with a transcriptional repression domain, such as KRAB (Krüppel-associated box), MXI1 (MAX-interacting protein 1), or SID4X (four copies of mSin3 interaction domain) (FIGURE 4C) (126, 195, 392). Conversely, CRISPR/dCas9-mediated gene activation is achieved by fusion of transcriptional activation domain(s), such as VP64 (four copies of the Herpes Simplex Virus VP16 transcriptional activation domain), p65 (NF-Kb activation domain), and Rta (Epstein-Barr Virus-derived R transactivator) to the NH2 terminus and/or COOH terminus of dCas9, leading to gene activation in a variety of cell types (59, 66, 70, 236, 247, 313).
Several strategies have been exploited to further improve the potency of the CRISPRa system. One strategy is to use antibody/epitope-based recruitment of transcriptional activators, known as the SunTag system (387). Another strategy is to engineer the sgRNA scaffold for recruitment of the RNA binding protein fused with multiple transcriptional activators (196, 465). CRISPR/dCas9-based transcriptional activation can also be used for cell lineage reprogramming. As an example, dCas9 fused with two copies of the VP64 transcriptional activation domain can be directed to the Myod1 promoter, leading to reprogramming of fibroblasts to the myogenic lineage (59).
One of the applications of dCas9-based transcriptional regulation for the treatment of muscular dystrophies is to upregulate compensatory or paralogous proteins. Utrophin is an autosomal paralogue of dystrophin, and upregulation of utrophin can partially alleviate the dystrophic phenotype seen in the mouse model of Duchenne muscular dystrophy (DMD) (145, 396). Therefore, it would be interesting to evaluate the efficacy and efficiency of dCas9-based transcriptional upregulation of the endogenous utrophin gene for the treatment of DMD.
3. Epigenetic regulation: dCas9 fused to epigenetic modifiers
The CRISPR/dCas9 system can also be used for site-specific epigenome editing when fused with epigenetic modifiers, leading to histone code modification or changes in DNA methylation (FIGURE 4C). In an example of histone modification, dCas9 engineered from Neisseria meningitides was fused to histone demethylase LSD1 and used to target the enhancer regions of the Oct4, Sox2, and Tbx3 genes (179). This decreased the levels of H3K4me2 and H3K27ac and suppressed gene expression. Conversely, fusing dCas9 with the catalytic core of the histone acetyltransferase p300 induced robust transcriptional activation of the targeted genes (142). In addition to histone code modification, the DNA methylation pattern can also be altered. Fusing dCas9 with either Tet1 (Ten-eleven translocation methylcytosine dioxygenase 1) or Dnmt3a (DNA methyltransferase 3A) produced dCas9-Tet1 or dCas9-Dnmt3a, which can be used to target methylated or unmethylated promoter regions, leading to promoter activation or silencing (228, 274).
D. Novel CRISPR/Cas Systems: CRISPR/Cpf1 and CRISPR/Cas13a/C2c2
Currently, the most widely used forms of Cas9 and its orthologs are from the class 2 type II CRISPR/Cas system. Recent studies in class 2 type V and VI CRISPR/Cas systems revealed several new RNA-guided CRISPR effectors capable of nucleic acid cleavage and detection, such as Cpf1 (CRISPR from Prevotella and Francisella 1) and Cas13a (formerly C2c2). These new CRISPR/Cas systems further expand the genome editing range of the CRISPR system (5, 103, 111, 466).
1. CRISPR/Cpf1: more than an alternative to Cas9
CRISPR/Cpf1, from the class 2 type V CRISPR system, is a RNA-guided endonuclease capable of DNA cleavage (FIGURE 5A) (111, 466). Two Cpf1 orthologs, LbCpf1 (from Lachnospiraceae bacterium ND2006) and AsCpf1 (from Acidaminococcus sp. BV3L6), have been engineered for genome editing in a variety of systems, including mammalian cells, animals, and plants (106, 156, 166, 184, 185, 235, 281, 317, 397, 405, 464, 469). The CRISPR/Cpf1 system has many unique features compared with CRISPR/Cas9: 1) Cas9-mediated genome cleavage requires two RNA components consisting of a crRNA and a tracrRNA (which can be engineered as a single sgRNA hybrid), whereas Cpf1-mediated genome cleavage is tracrRNA-independent so it only requires a short crRNA. 2) The PAM sequence of Cpf1 is 5′-TTTN-3′, located at the 5′ end of a protospacer; in contrast, the 5′-NGG-3′ or 5′-NAG-3′ PAM for SpCas9 is located at the 3′ end of a protospacer. 3) Cas9-mediated DNA DSB is blunt-ended and proximal to the PAM site, whereas Cpf1-mediated DNA DSBs are cleaved as a staggered cut distal to the PAM site. 4) The pre-crRNA processing in the CRISPR/Cas9 system is catalyzed by an additional RNase III, whereas Cpf1 has intrinsic RNase activity and can directly process pre-crRNA by itself (111, 466).
FIGURE 5.

Novel CRISPR/Cas systems. Two novel class 2 CRISPR/Cas systems have been engineered for nucleic acid recognition and cleavage, such as Cpf1 (CRISPR from Prevotella and Francisella 1) and Cas13a (formerly C2c2). A: domain organization of the LbCpf1 protein discovered in Lachnospiraceae bacterium ND2006. All Cpf1 orthologs have two nuclease domains: 1) the RuvC domain which cleaves the nontarget DNA strand and 2) the Nuc domain which cleaves the target DNA strand. The LbCpf1 crRNA is shown hybridizing with its DNA target. The PAM is highlighted in red. Red arrowheads indicate cleavage site. B: domain organization of the LshCas13a protein discovered in Leptotrichia shahii. Cas13a has dual RNase activities, one specific for pre-crRNA processing and maturation, which is catalyzed by the helical-I domain, and the other one for RNA-guided single-stranded RNA (ssRNA) degradation, which is catalyzed by the HEPN1 and HEPN2 domains. The LshCas13a crRNA is shown hybridizing with its RNA target. CRISPR/Cas13a-mediated ssRNA cleavage is independent of a PAM; instead, it requires a 3′-protospacer flanking site (PFS; shown in red). C: domain organization of the SpCas9 protein discovered in Streptococcus pyogenes. The RuvC nuclease domain cuts the nontarget strand. The HNH nuclease domain cuts the target strand that hybridizes with the sgRNA. CTD, COOH-terminal domain.
Because of its T-rich PAM preference, Cpf1 represents an alternative to Cas9 for genome editing at AT-rich loci. In addition to the canonical 5′-TTTN-3′ PAM sequence, Cpf1 also recognizes 5′-CTTV-3′, 5′-TCTV-3′, 5′-TTCV-3′ (V represents A, G, or C) as noncanonical PAMs, because the PAM-binding channel of Cpf1 has conformational flexibility (449) that further expands the targeting range of the CRISPR/Cpf1 system. Another advantage of Cpf1 compared with Cas9 is the convenience of multiplex genome editing. CRISPR/Cas9-mediated multiplex genome editing requires multiple sgRNAs transcribed from separate promoters or additional RNA sequences for recognition and cleavage by other nucleases if multiple sgRNAs are transcribed from a single promoter (171, 336, 400, 445). However, CRISPR/Cpf1-mediated multiplex genome editing only requires a single promoter for the transcription of multiple crRNAs, because Cpf1 can process polycistronic crRNAs into individual ones using its own RNase activity, which significantly simplifies multiplex genome editing (467). Therefore, Cpf1 is more than an alternative to Cas9 in terms of genome and epigenome editing because it offers a broader range of editing options.
2. CRISPR/Cas13a/C2c2: programmable RNA-guided RNA-targeting CRISPR effector
Most CRISPR effectors discovered so far are RNA-guided deoxyribonucleases. To date, RNA-guided RNA-targeting activity has been shown in only three types of CRISPR systems: the type III-A, III-B systems and type VI CRISPR/Cas13a system (formerly C2c2) (5, 103, 140, 165, 178, 339, 372, 373, 386). Type III-A, III-B systems belong to the class 1 CRISPR family and require a multicomponent effector complex for RNA degradation, and so have limited application in RNA biology. In contrast, the type VI CRISPR/Cas13a system belongs to the class II CRISPR family and is a single-effector system for crRNA processing and RNA targeting (5, 102) (FIGURE 5B). These engineered Cas proteins further extend the CRISPR/Cas system from DNA editing to RNA editing (FIGURE 5).
Several Cas13 orthologs have been engineered for RNA recognition and cleavage both in vitro and in vivo (4, 80, 102, 129). For example, the Cas13a ortholog from Leptotrichia wadei (LwaCas13a) has been engineered for pathogenic virus and bacteria detection, mutation genotyping, and identification, which further expands the clinical application of the CRISPR/Cas13a system (129). In addition, LwaCas13a can be used in mammalian cells and plants for targeted RNA knockdown with high specificity, and its catalytically inactive form can also be applied for tracking RNA transcripts in vivo (4). Most recently, the Cas13b ortholog from Prevotella sp. P5–125 (PspCas13b) has been engineered for mammalian RNA targeting, and its catalytically inactive form fused with ADAR2 deaminase domain can also be applied for RNA base editing (80). Therefore, the recently characterized CRISPR/Cas13 system is becoming a powerful tool for studying RNA biology.
III. MYOEDITNG: PREVENTION OF MUSCULAR DYSTROPHIES
A. DMD: Dystrophin Gene Structure and Mutations
DMD is an X-linked recessive monogenic disease caused by mutations in the DMD gene, which encodes dystrophin (149). DMD is the most common type of monogenic muscular dystrophy, affecting ~1 in every 5,000 boys (135). DMD patients seem normal at birth, but within a few years they begin having trouble walking and lose ambulation between 7 and 12 yr of age. Cardiac and respiratory failure causes premature death, often by the early 30s.
Dystrophin is a key component of the dystrophin glycoprotein complex, which is a large multicomponent protein complex essential for sarcolemma integrity and stability (FIGURE 6) (120, 135). The structure of the full-length dystrophin protein can be organized into four major domains: 1) the NH2-terminal region containing an actin-binding domain; 2) the central region containing a stretch of 24 spectrin-like repeats, forming the rod domain, which is interrupted by 4 hinge regions; 3) the cysteine-rich domain which contains several subdomains, including a WW domain, two EF-hand-like domains, and a ZZ domain, which are important for interacting with β-dystroglycan, calmodulin, and ankyrin-B; and 4) the COOH-terminal domain which interacts with dystrobrevin and syntrophins (6, 120, 299). The DMD gene, comprised of 79 exons (FIGURE 7) (184, 264), gives rise to different isoforms of the dystrophin protein which are expressed in various tissues by tissue-specific promoters and/or alternative splicing (280). The large 427-kDa cytoskeletal protein that is primarily expressed in skeletal muscle and heart is transcribed from the Dp427m promoter.
FIGURE 6.

Structure of the dystrophin-glycoprotein complex (DGC). The main components of the DGC are the dystroglycan complex, sarcoglycan complex, and dystrophin. The DGC provides sarcolemma stability and integrity through interaction with laminin in the basement membrane on the extracellular matrix and actin in the cytoplasm. Other dystrophin-associated proteins include neuronal nitric oxide synthase (nNOS), dystrobrevins, syntrophins, and sarcospan. Mutations of the main components of the DGC cause muscular dystrophies, such as Duchenne or Becker muscular dystrophy (dystrophin mutation), and limb-girdle muscular dystrophy types 2C, 2D, 2E, and 2F (sarcoglycan mutations).
FIGURE 7.
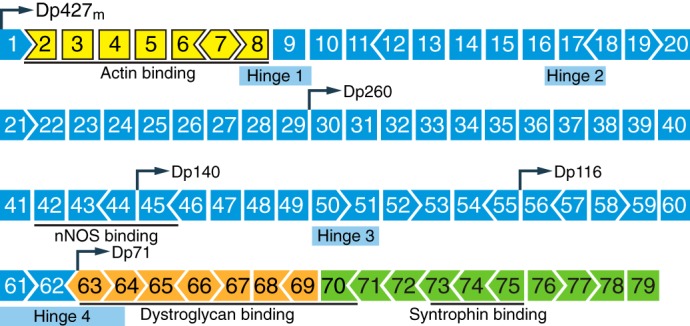
Structure of the dystrophin gene. The dystrophin gene has 79 exons. Different dystrophin isoforms can be transcribed from various promoters (demarcated as Dp, followed by a numeric number indicating isoform molecular weight in kilodaltons). The dystrophin protein expressed in skeletal muscle and heart is transcribed from the Dp427m promoter. Domains essential for binding with other DGC components or cytoskeletal proteins are underlined. Exons are color-coded according to the domain they encode: NH2 terminus (yellow), central rod domain (blue), cysteine-rich domain (orange), and COOH terminus (green).
More than 7,000 mutations have been identified in the DMD gene (36). These mutations can be categorized as deletion (68%), duplication (11%) of single or multiple exons, or small point mutations (20%), such as missense and nonsense substitutions (3, 36, 271). Mutations in the DMD gene are not uniformly distributed but cluster into hot spots, which are clustered within exons 2–20 and exons 45–55 (36). Approximately 15% of all exon deletion events and 50% of all exon duplication events are observed within exons 2–20, whereas 70% of all exon deletion events and 15% of all exon duplication events are observed within exons 45–55 (36, 451). In-frame deletion or duplication of exon(s) within the central region of the DMD gene retains the protein reading frame and generates either a truncated or extended dystrophin protein. These mutant dystrophin proteins retain their NH2 and COOH termini, which are essential for actin cytoskeleton and dystrophin glycoprotein complex interaction, leading to a milder form of muscular dystrophy, known as Becker muscular dystrophy (BMD) (2, 135). In contrast, out-of-frame deletion or duplication of exon(s) either disrupts the protein reading frame or generates a premature termination codon (PTC) and leads to DMD.
B. Animal Models of DMD
The most commonly used animal model for DMD is the mdx mouse, in which a C-to-T transition in exon 23 creates a nonsense mutation, leading to loss of full-length dystrophin expression (54, 360). The mdx mice do not develop severe DMD phenotypes, such as muscle wasting, scoliosis, and cardiomyopathy until reaching 15 mo of age. In contrast to DMD patients whose lifespan is significantly reduced, the lifespan of mdx mice is reduced by only 25% (60). Four chemically induced mdx strains have also been developed, known as mdx2cv, mdx3cv, mdx4cv, and mdx5cv, with a point mutation in intron 42, intron 65, exon 53, or exon 10, respectively (65). In addition to the mdx strains with point mutations, four additional DMD mouse models have been established with either exon 2 duplication, exon 45 deletion, exon 50 deletion, or exon 52 deletion (12, 417, 458).
Dystrophin-deficient mouse models generally do not develop severe pathological phenotypes as seen in DMD patients. Several double knockout (dKO) mouse models were generated, in which the Dmd gene was knocked out, along with additional genes required for sarcolemma integrity, stem cell maintenance, and muscle homeostasis (85, 130, 259, 278, 332). Genome editing technology also played a role in expanding the rodent models of DMD. For example, two DMD rat models were created by TALEN- or CRISPR/Cas-mediated targeting of the Dmd exon 23 or exons 3–6, leading to an exon 23 frame shifting or exon 3–6 deletion (208, 286). Most recently, CRISPR/Cas9 was used to create a mouse model lacks exon 50, representing the most common mutational “hot spot” in humans (7).
In addition to small rodent models, large animal models of DMD have been developed, including dogs (16, 197, 346, 365, 409, 418, 435), pigs (192, 353, 461), and non-human primates (69). Monkey models of DMD are still at F0 with mosaicism, which requires additional breeding to generate a pure background (69). Disease progression in some porcine models of DMD is so severe that the majority of the affected pigs die within the first week of life, which limits its application in therapeutic translation (353). In contrast, canine DMD models share more similar clinical phenotypes as seen in human patients, including limb muscle fibrosis, joint contracture, hypersalivation, and an early cardiac defect (259). Moreover, canine DMD models have fewer regenerated myofibers than mdx mice as indicated by central nucleation, which is histologically similar to human patients (81, 365). In addition, canine DMD models develop limb muscle weakness at 2–3 mo of age and have ~75% reduction of lifespan, showing similar disease progression as human patients (410). Therefore, the canine model of DMD seems superior to the other large animal models in regard to current availability, genetic background, and speed of disease progression.
C. Introduction of Myoediting
To date, more than 800 monogenic neuromuscular disorders with mutations in over 400 different genes have been recorded (177). The discovery and application of programmable nucleases for genome editing paves the way for permanent correction of these genetic diseases (79, 318, 320). Meganucleases, ZFNs, and TALENs have been reported to correct mutations responsible for certain muscular dystrophies including DMD, limb-girdle muscular dystrophy (LGMD), and myotonic dystrophy (DM) (122, 217, 240, 303, 304, 316, 402, 443). However, these early versions of programmable nucleases were not widely adopted for correcting mutations in various muscular dystrophies because of the low genome targeting density, difficulty of assembly of the functional nuclease domains, and cytotoxicity caused by off-target effects. The CRISPR/Cas system revolutionized the genome editing field and significantly simplified the process of permanent correction of monogenic neuromuscular disorders.
CRISPR/Cas-mediated genome editing in skeletal muscle and heart, which we termed myoediting (230, 231), can permanently correct various DMD mutations and restore dystrophin function. Initially, myoediting was performed in the germline of mdx mice, a mouse model of DMD with a nonsense mutation in exon 23. By injecting Cas9 mRNA, a sgRNA targeting the mutated exon 23, and a ssODN repair template into the zygotes of mdx mice, it was demonstrated that CRISPR/Cas9-mediated myoediting can successfully correct the Dmd mutation by HDR or NHEJ and restore dystrophin expression (231). However, germline editing in humans is currently not feasible, necessitating alternative strategies for therapeutic genome editing. Therefore, we and other groups used recombinant adeno-associated virus (rAAV) to deliver the CRISPR/Cas9 genome editing components to postnatal mdx mice for skipping or deleting the mutated exon in vivo (29, 104, 230, 289, 383). The rAAV-delivered CRISPR/Cas9-mediated postnatal genome editing successfully restored dystrophin expression and improved muscle function in mdx mice. These studies underscore the therapeutic potential of the CRISPR/Cas9 system for treating devastating muscle diseases.
The CRISPR/Cpf1 system was also used to correct DMD mutations in human induced pluripotent stem cells (iPSCs) and in mdx mice either by exon skipping or HDR (469), which further expands the range of CRISPR/Cas-mediated genome editing in AT-rich loci. Due to postmitotic and multinucleation features, skeletal muscle is ideal for therapeutic CRISPR/Cas9 genome editing because genomic correction of a subpopulation of nuclei leads to steady improvement of muscle function (29, 104, 230, 231, 289, 383, 469). Therefore, CRISPR/Cas-mediated myoediting represents a novel method for DMD treatment. In the following sections, different strategies of applying the CRISPR/Cas system for correcting DMD mutations will be discussed in detail. In addition, the potential of applying CRISPR/Cas-mediated genome editing for the correction of other muscular dystrophies will also be explored.
D. Strategies of CRISPR/Cas-Mediated DMD Correction
Initial efforts to apply programmable nucleases such as meganuclease, ZFN, and TALEN for precise genome editing provided many insights into the permanent correction of DMD mutations (217, 240, 303, 304, 316). The CRISPR/Cas system significantly simplified the genome editing process. To date, four strategies have been developed for CRISPR/Cas-mediated correction of DMD mutations, which are exon deletion, exon skipping, exon reframing, and exon knock-in.
1. Exon deletion
Approximately 80% of mutations found in the DMD gene are out-of-frame exon deletions or duplications, leading to reading frame incompatibility between adjacent exons (FIGURE 8A) (3, 36). The most traditional strategy to permanently restore the DMD open reading frame (ORF) is in-frame exon deletion, in which a pair of sgRNAs is used to generate two simultaneous DNA DSBs within the intron regions flanking the out-of-frame exon, leading to complete removal of a single exon or multiple exons to generate a compatible reading frame outcome with the adjacent exon (FIGURE 8B). CRISPR/Cas-mediated exon deletion is best suited for correcting DMD mutations caused by exon duplication and has been reported with high efficiency in human DMD myoblasts with exon 2 or exon 18–30 duplications (209, 437). Removal of the duplicated exons restores the DMD ORF and produces full-length dystrophin protein that is indistinguishable from wild-type or normal dystrophin, although small INDELs can be observed at the genomic level.
FIGURE 8.
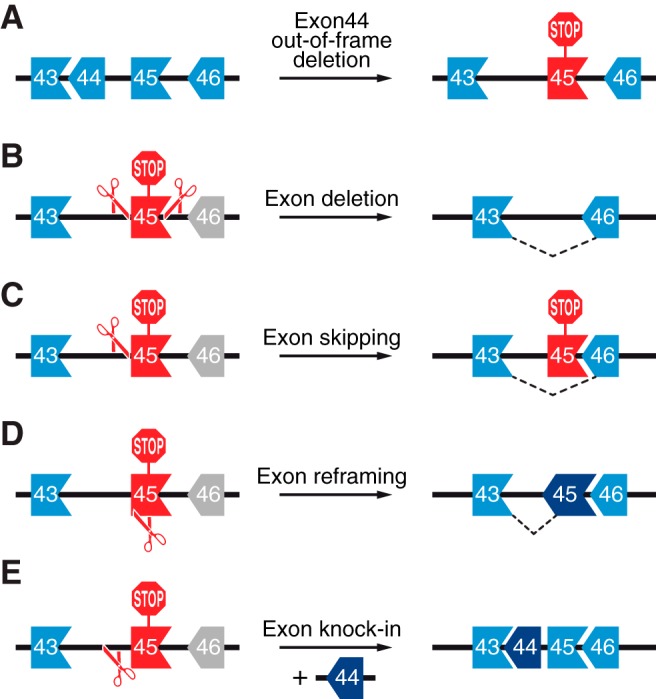
Strategies for CRISPR/Cas-mediated correction of DMD mutations. A: a schematic illustration showing arrangement of exons 43–46 of the DMD gene in terms of their reading frame compatibility. This genomic region is used here as an example to highlight the strategies for CRISPR/Cas9 correction of DMD mutations. An out-of-frame deletion of DMD exon 44 results in splicing of exon 43 to exon 45. This creates a premature stop codon in exon 45 (red STOP sign). B: exon deletion is used to restore the DMD reading frame. Two sgRNAs targeting introns 44 and 45 will generate two DNA DSBs flanking exon 45. This leads to excision of exon 45 and subsequent splicing of exon 43 to exon 46. C: exon skipping is mediated by a single sgRNA which targets the splice acceptor site of exon 45. The INDELs generated by NHEJ-mediated repair disrupt the splice acceptor site of exon 45, leading to splicing of exon 43 to exon 46. D: exon reframing is mediated by a single sgRNA targeting exon 45. The INDELs in exon 45 generated by NHEJ-mediated repair may restore the reading frame compatibility of exon 45 with exons 43 and 46. E: exon knock-in relies on HDR DNA repair pathway in the presence of a donor template. A single sgRNA targeting intron 44 will generate a DNA DSB and be repaired by HDR when exon 44 is used as a donor template, leading to exon 44 knock-in between exons 43 and 45.
DMD mutations caused by an out-of-frame exon deletion can be corrected by an in-frame exon deletion, producing a truncated dystrophin protein with internal deletions. For example, cultured myoblasts from DMD patients with an out-of-frame deletion of exons 48–50 have an incompatible reading frame when exon 47 is spliced with exon 51. A pair of sgRNAs targeting intron 50 and 51 was used to delete exon 51, restoring the reading frame between exon 47 and 52 (302). Similarly, the dystrophin reading frame incompatibility caused by an out-of-frame deletion of exons 45–52 has been corrected by exon 53 deletion, leading to splicing of exon 44 to exon 54 and subsequently restoring the dystrophin reading frame (239, 240). Multiple exons can also be deleted to restore the dystrophin reading frame. By using a pair of sgRNAs targeting intron 44 and 55, a large deletion extending from exon 45 to 55 was generated, leading to reading frame restoration of exon 44 to exon 56 (302, 457). Similarly, a large deletion extending from exons 44 to 54 was generated, by using a pair of sgRNAs targeting intron 43 and 54, leading to reading frame restoration between exon 43 and 55 (239, 240). Several in vivo studies in postnatal mdx or mdx4cv mice used exon deletion strategies to remove a single or multiple exons with a point mutation and thereby restored dystrophin expression and muscle function (29, 104, 230, 289, 383, 446).
Exon deletion is a promising strategy to correct mutations clustered in the second hot spot region (exons 45–55) because the spectrin-like repeats within the central rod domain are tolerant of large in-frame deletions (120, 135). However, special consideration should be given to mutations at the NH2 and COOH termini of dystrophin because these regions encode many essential domains known to interact with the actin cytoskeleton and dystrophin glycoprotein complex. For example, three different exon deletion strategies were applied to correct the DMD mutation caused by an out-of-frame deletion of exons 8–9, and different outcomes were observed in regard to dystrophin protein stability and function (205). Specifically, an in-frame deletion of exons 7–11 retained all three actin binding sites, but this truncated dystrophin was structurally unstable and showed minimal recovery of cardiomyocyte function in vitro. In contrast, in-frame deletion of exons 3–9 only retained actin binding site 1 but was the most effective strategy to restore functionality of human iPSC-derived cardiomyocytes. Reading frame restoration does not guarantee functional recovery, and hence, additional empirical analysis should be performed to further evaluate different correction strategies.
2. Exon skipping
Exon skipping has been achieved using anti-sense oligonucleotide (AON)-based therapy (1). However, AON-based exon skipping corrects at the mRNA level, while retaining the mutant DMD in the genome. Thus this approach requires life-long treatment. In contrast, CRISPR/Cas-based exon skipping is achieved by NHEJ-mediated disruption of the splice acceptor or donor sequence at the genomic level, leading to permanent exon skipping and completely eliminating the source of the mutation. For example, human iPSCs derived from DMD patients with exon 44 deletion have an incompatible reading frame between exon 43 and 45 (FIGURE 8A). A single sgRNA was designed to specifically target the intron 44 and exon 45 boundary, thereby inducing a DNA DSB at the splice acceptor site of exon 45 (FIGURE 8C). The INDELs generated by NHEJ-based DSB repair disrupted the splice acceptor sequence of exon 45, leading to exon 45 skipping during mRNA splicing (217). Similarly, DMD mutations caused by an out-of-frame deletion of exons 48–50 or exons 45–52 have been corrected by skipping exon 51 or exon 53, respectively (239, 240, 469). Recently, exon skipping has also been used to correct Dmd in a mouse model representing the most commonly deleted hot spot mutation in humans (7). This mouse model has an out-of-frame deletion of exon 50, which generates a premature stop codon in exon 51. A single sgRNA was designed to target the exon 51 splice acceptor site, leading to exon 51 skipping. Therefore, using a single sgRNA-mediated exon skipping strategy, which abolishes either the splice acceptor site or splice donor site or allows for reframing, overcomes the necessity of double sgRNA-based exon deletion.
Usually, the single sgRNA-mediated exon skipping strategy generates a relatively small INDEL at the intron/exon boundary, destroying the exon splice acceptor or donor site but retaining the residual part of the exon sequence in the genome. If “AG” nucleotides are present in the residual part of the exon, they can serve as a pseudo-splice acceptor sequence, rendering the single sgRNA-mediated exon skipping ineffective. Therefore, additional experimental studies, such as reverse transcription polymerase chain reaction (RT-PCR) or Western blot analysis, should be performed to confirm exon skipping at the RNA and protein level.
3. Exon reframing
A NHEJ-based reframing strategy can also be applied to restore the dystrophin ORF, in which a single or a pair of sgRNAs are used to generate DNA DSBs within the exon region, leading to a targeted frameshift, since in theory, one-third of INDELs created by NHEJ should be in-frame (FIGURE 8D). Several studies have applied this strategy to restore the dystrophin reading frame by inducing targeted frameshifts in exons with an incompatible reading frame in regard to the adjacent exon, including exons 23, 45, 50, 51, 53, and 54 (29, 162, 217, 231, 239, 240, 302, 469). Unlike the exon deletion strategy, which excises a single or multiple exons, exon reframing only creates small INDELs, and hence, minimizes the length of the genomic deletion.
Both exon skipping and exon reframing strategies require using one sgRNA-mediated single cut in the genome. These two strategies are considered more efficient than using two sgRNA-mediated double cuts in the genome. This is because exon deletion by excision using two sgRNAs requires two cooperative DNA DSBs. However, two DNA DSBs do not always occur simultaneously since there is a possibility that a single DNA DSB can be rapidly rejoined by NHEJ-mediated DNA repair, leaving the second intronic DSB ineffective. In this situation, exon deletion cannot be achieved because of the latency between the two DNA DSBs. In contrast, one sgRNA-mediated single cut near the splice acceptor site can be sufficient to restore the DMD ORF. For example, if the INDEL disrupts the splice acceptor sequence, this could lead to exon skipping. Alternatively, if the INDEL does not disrupt the splice acceptor sequence, there is still a possibility that one-third of the INDELs within the exon could be in-frame, leading to exon reframing.
4. Exon knock-in
In general, DMD mutations corrected by exon deletion, skipping, or reframing strategies will generate truncated dystrophin proteins with internal deletions. In principle, DMD mutations can also be corrected by exon knock-in, leading to expression of full-length dystrophin protein (FIGURE 8E). Exon knock-in requires a DNA donor template and active cell cycle to induce HDR-mediated precise editing in the S and G2 phases. This repair strategy has been used in DMD patient-derived iPSCs to correct a mutation caused by an out-of-frame deletion of exon 45 (217). In addition, point mutations in mouse Dmd exon 23 and 53 have also been corrected by HDR-mediated precise editing (29, 231, 469, 471).
Mutations at specific regions in the NH2 and COOH termini of dystrophin generally are not feasible for exon deletion or skipping-based correction because essential domains known to interact with cytoskeletal actin or the sarcoglycan complex are encoded within these regions. Therefore, exon knock-in is required to correct these types of mutations. However, due to the postmitotic nature of mature skeletal muscle and cardiomyocytes, HDR efficiency remains low in CRISPR/Cas-mediated postnatal genome editing (29). Recently, precise genome editing in postmitotic cells and animals with high efficiency was reported (381). This technology, which was termed homology-independent targeted integration (HITI), only relies on the NHEJ pathway and can be used to precisely integrate DNA fragments into the mammalian genome, regardless of the cell cycle state, which may provide opportunities to correct certain DMD mutations by exon knock-in.
E. Facioscapulohumeral Muscular Dystrophy
1. Facioscapulohumeral muscular dystrophy type 1 and type 2
Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal dominant neuromuscular disease with an estimated prevalence ranging from 1:14,000 to 1:20,000 (109, 276, 298, 305, 390). Clinically, FSHD shows asymmetric regional muscle weakness in the face, shoulders, and upper arms. Symptoms progress to the trunk and leg muscles while the extraocular, pharyngeal, and cardiac muscles remain unaffected (389). In contrast to other muscular dystrophies that show a phenotype of severe myofiber degeneration, FSHD shows a minimal myopathic phenotype at the histopathological level, with evidence of endomysial inflammation specific to the perivascular region (10, 390). The mechanism of FSHD can be either genetic or epigenetic, leading to the classification of type 1 FSHD (FSHD1) and type 2 FSHD (FSHD2) (211, 333, 390).
FSHD1 represents 95% of the FSHD cases and is caused by contraction of D4Z4 macrosatellite repeats located in the subtelomeric region of chromosome 4, leading to DUX4-induced cytotoxicity in skeletal muscles (213, 390, 413, 433). Normal individuals carry 11–100 D4Z4 repeats on chromosome 4q35, while FSHD patients are limited to 1–10 copies of D4Z4 repeats (413). The reduced number of D4Z4 repeats alters the heterochromatic DNA structure at chromosome 4q35 and induces the expression of the DUX4 gene within the last D4Z4 repeat when a polyadenylation signal is present on the FSHD permissive 4qA haplotype. The FSHD nonpermissive 4qB haplotype lacks the exon distal to the D4Z4 repeat and its DUX4 transcript is destabilized (92, 213, 368). DUX4 encodes a double-homeobox transcription factor, which is normally only expressed in the testis (369, 459). When DUX4 is ectopically expressed in skeletal muscle, it activates various genes that are normally expressed only in the germline, stem cells, and the immune system, leading to oxidative stress, apoptosis, and inhibition of muscle regeneration (44, 124, 390). Unlike FSHD1 with D4Z4 repeats contraction, FSHD2 patients generally retain normal-sized D4Z4 repeat arrays but show strong reduction of D4Z4 methylation because of a mutation of the SMCHD1 gene (212). SMCHD1 is essential for hypermethylation of CpG islands, and its mutated form fails to methylate D4Z4 repeats, thereby causing DUX4 expression (212). Similar to FSHD1, DUX4 expression in FSHD2 requires the acquisition of a polyadenylation signal present on the FSHD permissive 4qA haplotype.
2. Animal models of FSHD
FSHD1 is caused by contraction of subtelomeric D4Z4 macrosatellite repeats, leading to DUX4-induced cytotoxicity in skeletal muscles (213, 390, 413, 433). Currently four DUX4 transgenic mouse models have been developed, which are D4Z4–2.5, D4Z4–12.5, iDUX4–2.7, and iDUX4-pA mice (43, 82, 201). Both D4Z4–2.5 and D4Z4–12.5 mouse models display abundant expression of DUX4 transcripts in the testis, consistent with germline DUX4 expression in humans. In addition to germline expression, DUX4 transcripts can also be detected in multiple tissues in D4Z4–2.5 mouse model, indicating that contraction of D4Z4 repeats leads to inefficient DUX4 repression in somatic tissues.
In contrast to D4Z4–2.5 and D4Z4–12.5 mouse models which uses an endogenous DUX4 promoter, two inducible DUX4 transgenic mouse models (iDUX4–2.7 and iDUX4-pA) were also generated in which the DUX4 expression is induced by doxycycline (43, 82). Low-level DUX4 expression in the iDUX4-pA mouse model without doxycycline induction results in progressive degenerative myopathy and other muscle phenotypes, such as inflammatory infiltration and fibrosis. In contrast, muscle-specific DUX4 induction in the iDUX4-pA mouse model leads to dystrophic phenotypes and impaired muscle regeneration after injury.
The FSHD pathogenic gene DUX4 is specific to primates, and retrotransposon-mediated expansion of its binding sites in the primate genome is not conserved in the murine system (124, 210, 459). Therefore, studies using both FSHD mouse models and human cell models, such as FSHD primary myoblasts and iPSCs, may provide more thorough information about disease progression and translational application.
3. CRISPR/Cas-mediated correction of FSHD1 and FSHD2
A CRISPR/dCas9-based gene editing strategy has been reported to reduce DUX4 expression in primary FSHD myocytes, in which dCas9 was fused to a KRAB transcriptional repressor and targeted to the DUX4 promoter or the first exon region, leading to transcriptional repression of DUX4 (144). However, CRISPR/dCas9-based transcriptional repression is transient while the 4q35 chromatin landscape and D4Z4 repeat number remain pathogenic, so any therapeutic benefit would be only temporary.
Two potential genome editing strategies might provide longer term benefit. The first strategy is CRISPR/dCas9-based DNA methylation. It has been reported that fusing dCas9 with a DNA methyltransferase (Dnmt3a) or with a hybrid form of two DNA methyltransferases (Dnmt3a-Dnmt3L) can create site-specific DNA methylation (228, 274, 374). It might be effective to use the dCas9-DNA methyltransferase system to revert the hypomethylated 4q35 chromatin landscape, leading to DUX4 silencing, since both FSHD1 and FSHD2 have reduced DNA methylation levels at the D4Z4 repeat arrays. The second strategy entails CRISPR/Cas9-based genome editing of the FSHD permissive 4qA haplotype (FIGURE 9A). The Cas9 nuclease can be directed to the 4qA haplotype by specific sgRNA(s) that target the polyadenylation signal region and induce DNA DSBs to either disrupt the polyadenylation sequence or excise the polyadenylation signal, thereby converting the permissive 4qA haplotype to the nonpermissive 4qB haplotype.
FIGURE 9.

Strategies for CRISPR/Cas-mediated correction of other muscular dystrophies. A: FSHD type I and type II are caused by acquisition of a poly-adenylation signal on the permissive 4qA haplotype, leading to DUX4 transcript stabilization. The Cas9 nuclease is directed to the 4qA haplotype by specific gRNA(s) targeting the polyadenylation signal and converts the permissive 4qA haplotype to the nonpermissive 4qB haplotype. The gray shaded area depicts D4Z4 macrosatellite repeats. PAS indicates poly-adenylation signal on the permissive 4qA haplotype. B: LGMD2C is caused by loss-of-function mutation of the SGCG gene. An out-of-frame deletion of exon 6 (shown by a dotted line around exon 6) results in splicing of exon 5 to exon 7, which creates a premature stop codon in exon 7 (red STOP sign). Two sgRNAs that target introns 3 and 7 will generate two DNA DSBs flanking exons 4–7. This leads to excision of exons 4–7 and subsequent splicing of exon 3 to exon 8 and permanent restoration of the SGCG reading frame, since deletion of exons 4–7 of the SGCG gene was shown to partially restore function of γ-sarcoglycan (121). C: myotonic dystrophy type I (DM1) is caused by trinucleotide CTG repeat expansion in the 3′ untranslated region (3′-UTR) of the DMPK gene. Two sgRNAs can be designed to generate two DNA DSBs that flank the CTG repeats, leading to deletion of the CTG repeats. D: myotonic dystrophy type 2 (DM2) is caused by a tetranucleotide CCTG repeat expansion in intron 1 of the CNBP gene. Two sgRNAs can be designed to generate two DNA DSBs that flank the CCTG repeats, leading to deletion of the CCTG repeats.
F. LGMD
LGMD is a general term for a highly heterogeneous group of autosomal neuromuscular diseases with variable disease phenotypes, ranging from progressive muscle weakness in proximal limbs with a normal life span to rapid disease progression in early childhood. LGMD can also affect distal limbs and the heart and cause life-threatening symptoms such as respiratory compromise and cardiac abnormalities (282, 292). To date, mutations in more than 30 loci have been reported to cause LGMD, including eight autosomal dominant forms (LGMD1A-1H) and 25 autosomal recessive forms (LGMD2A-2Y) (177). Owing to the scope of this review, only two subtypes of LGMD2 will be covered, which are dysferlinopathy and sarcoglycanopathies. The potential of applying CRISPR/Cas-mediated genome editing for the correction of LGMD2 subtypes will also be discussed.
1. Dysferlinopathy: LGMD2B and Miyoshi myopathy
Dysferlinopathy is caused by autosomal recessive mutation of the DYSE gene on chromosome 2p13 (25, 26, 226). Clinically, dysferlinopathy can be classified into LGMD2B and Miyoshi myopathy, depending on the muscle groups being affected. LGMD2B causes proximal muscle weakness in the pelvic and shoulder girdle muscle regions but spares other muscle groups (282, 293). In contrast, Miyoshi myopathy shows distal muscle weakness specifically affects the gastrocnemius and soleus muscles, without affecting other muscle groups (9).
The DYSE gene encodes a 230-kDa single-pass transmembrane protein, known as dysferlin, which is ubiquitously expressed in many tissues but enriched in striated muscles (25). Dysferlin binds to phospholipids in the presence of Ca2+ and is important for repairing membranes of skeletal muscle (22, 214). In addition, dysferlin also interacts with the dihydropyridine receptor (DHPR), caveolin-3, annexin A1, and desmoyokin (AHNAK) in t-tubule membranes, indicating its role in t-tubule maintenance (8, 153, 214, 256). Mutations in the DYSE gene are diverse, including missense and nonsense mutations, splice site and 3′-UTR mutations, and small insertions or deletions, leading to nonsense-mediated mRNA decay, protein misfolding, or mislocalization (158, 200, 430).
Dysferlin-deficient mice maintain an intact dystrophin glycoprotein complex but are defective in Ca2+-dependent membrane repair and display progressive dystrophic phenotypes, including sarcolemma lesions, muscle necrosis, inflammatory infiltration, and fatty tissue deposition (22, 147). AAV-based gene replacement therapy has been applied to treat dysferlinopathy. However, the size of the dysferlin cDNA (6.5 kb) exceeds the packaging limit of AAV. To address this issue, dysferlin cDNA was split into two cDNA fragments cloned into a dual-AAV system. After administration of the dual recombinant AAV vectors, full-length dysferlin transcripts were shown to be produced by either trans-splicing or homologous recombination (133, 232, 370). Although the AAV-based dysferlin replacement can restore muscle function, the endogenous DYSE mutations are still present in the genome. A recent study using TALENs and CRISPR/Cas9-genome editing successfully corrected the endogenous DYSE mutations in iPSCs derived from LGMD2B patients (402). After differentiation of corrected iPSCs into skeletal muscles, dysferlin expression was restored and correct protein localization was observed. This proof-of-concept study demonstrates the efficiency and accuracy of the CRISPR/Cas-genome editing system in permanent correction of monogenic neuromuscular disorders and represents an alternative method for the treatment of dysferlinopathy.
2. Sarcoglycanopathies: LGMD2C, 2D, 2E, and 2F
Sarcoglycanopathies can be classified as LGMD2C, 2D, 2E, or 2F, which are caused by loss-of-function mutations of the SGCG, SGCA, SGCB, or SGCD genes, respectively (293, 311). These genes encode γ-, α-, β-, and δ-sarcoglycans, which are single pass transmembrane proteins and together with ε- and ζ-sarcoglycans can form the sarcoglycan complex that resides in the sarcolemma (107, 260, 432). The sarcoglycan complex is a key component of the dystrophin glycoprotein complex, and its stability requires lateral association with the dystroglycan complex linked with dystrophin. Therefore, mutations in dystrophin can cause loss of the sarcolemma distribution of the sarcoglycan subunits, and patients affected with sarcoglycanopathies have similar phenotypes seen in DMD and BMD, displaying both muscular dystrophy and cardiomyopathy (24, 207, 261). Interestingly, mutations in any single sarcoglycan gene can lead to a significant reduction in or complete absence of the entire sarcoglycan complex (408). Many mouse models of sarcoglycanopathies have been developed (11, 78, 100, 101, 138, 428). These mouse models recapitulate many of the pathophysiological phenotypes seen in human patients, and hence serve as a reliable animal model of LGMD.
CRISPR/Cas9-mediated genome editing has successfully corrected SGCG and SGCA mutations in iPSCs derived from LGMD2C and LGMD2D patients (181, 402). However, these CRISPR/Cas9-mediated LGMD corrections rely on HDR, which is less efficient in postmitotic mature skeletal muscles. Interestingly, a truncated γ-sarcoglycan was engineered with in-frame deletion of exons 4–7 and shown to reduce pathophysiological phenotypes associated with LGMD2C with improvement in both skeletal muscle and heart function (121). Moreover, AON-mediated exon skipping was used to correct SGCG mutations in human cells. This proof-of-concept study challenges the conventional knowledge that exon skipping is restricted to correct DMD mutations and paves the way for the application of the CRISPR/Cas system to permanently correct SGCG mutations in LGMD2C by NHEJ-mediated exon skipping (FIGURE 9B).
G. Myotonic Dystrophy
1. Myotonic dystrophy type 1 and type 2
Myotonic dystrophy (DM) is an autosomal dominant neuromuscular disorder affecting ~1 in every 8,000 individuals (263, 404). Clinically, patients with DM suffer muscle degeneration leading to weakness and myotonia with other symptoms including cardiac conduction defects, cataracts, brain abnormalities, as well as gastrointestinal and endocrine disorders (132, 229, 268, 330, 436).
DM is classified into myotonic dystrophy type 1 (DM1) and type 2 (DM2). DM1, also known as Steinert’s disease, is caused by trinucleotide CTG repeat expansion in the 3′ untranslated region (3′-UTR) of the DMPK gene located on chromosome 19 (51, 114, 241). Healthy individuals carry 5–37 CTG repeats while repeat numbers greater than 37 are unstable and may further expand in length during cell division (15, 403, 404). DMPK mRNA transcripts with increased CUG expansion can form stable secondary structures, leading to increased steady-state expression of CELF1 (CUGBP Elav-like family member 1) by hyperphosphorylation, downregulation of DMPK itself, and aberrant splicing of other genes by sequestering splicing factors such as MBNL1 in the ribonuclear foci (97, 154, 167, 172, 204, 337). Dysregulation of CELF1 and MBNL1 splicing factors causes not only RNA toxicity and aberrant splicing, but also transcriptional dysregulation, mRNA instability, and microRNA dysregulation (45, 312, 325, 328, 415).
In contrast to DM1 with trinucleotide CTG repeat expansion in the 3′-UTR of the DMPK gene, DM2 is caused by a tetranucleotide CCTG repeat expansion in intron 1 of the CNBP gene in chromosome 3 (84, 225). Similar to DM1, the expansion of the CCUG repeats in CNBP mRNA transcripts also causes MBNL1 sequestration, leading to CNBP downregulation, RNA toxicity, and abnormal splicing of other genes (404). Because CNPB is required for cap-independent translation, other pathogenic effects such as abnormal protein translation or turnover may also contribute to DM2 (155, 341).
2. Animal models of DM
Approximately 20 mouse models of DM1 and DM2 have been established and are categorized into two groups: 1) mouse models that recapitulate the toxic RNA gain-of-function and 2) mouse models with abnormal splicing regulators (127). One of the most informative DM1 models is the HSALR transgenic line, in which 250 copies of the CTG repeats are placed in the 3′-UTR of the HSA gene that encodes the human α-skeletal actin. This mouse model develops severe myotonia, nuclear sequestration MBNL1, and aberrant splicing of multiple target transcripts (249).
Another transgenic mouse model, known as DM300–328, carries a large fragment of the human DMPK locus with expanded CTG repeats. This mouse model displays ribonuclear foci accumulation in multiple tissues, progressive muscle weakness, myotonia, tau protein distribution abnormalities, as well as defects in splicing and glucose metabolism, recapitulating many pathophysiological phenotypes seen in DM1 patients (136, 354, 355, 414). An inducible mouse model was also developed in which a floxed concatemer of three polyadenylation signals is inserted upstream of 960 copies of CTG repeats in exon 15 of the DMPK gene. This allows tissue-specific expression of the transgene by the Cre-lox system (300, 419).
In addition to mouse models that recapitulate toxic RNA gain-of-function, mouse models with abnormal splicing regulators were also developed, including Mbnl1 and Mbnl2 knockout mice and CELF1 overexpression mice (141, 148, 173, 198, 223, 233, 257, 395, 429). As with HSALR, the mouse model of DM1, a CCTG transgenic mouse model was developed to model DM2, in which the expanded (CCTG)121 repeats are introduced in intron 1 of the ZNF9 gene, displaying CELF1 upregulation, altered protein translation and degradation, and recapitulating muscle pathology, as seen in DM2 (338). DM mouse models with repeat expansion are more suitable for therapeutic application especially for CRISPR/Cas-mediated genome editing because the microsatellite repeats present in both DM1 and DM2 can conceivably be excised by NHEJ-mediated DNA repair.
3. TALENs and CRISPR/Cas-mediated correction of DM1 and DM2
In several studies, researchers have employed programmable nucleases for genome editing of DM1 and DM2. A TALEN-based system was used to generate site-specific DNA DSBs in the 3′-UTR of the DMPK gene and introduced multiple polyadenylation signals upstream of the trinucleotide CTG repeats by HDR, leading to early transcriptional termination and subsequently preventing the production of the toxic DMPK transcripts (122, 443). However, HDR-mediated knockin of polyadenylation signals requires a repair template and an active cell cycle, which may not be efficient for genome editing in postmitotic tissues such as skeletal muscle.
The CRISPR/Cas system and its derivatives have also been used to treat both DM1 and DM2 as well. For example, an engineered dCas9 fused to a PIN RNA endonuclease domain was shown to be active for RNA cleavage and could specifically eliminate microsatellite repeat expansions in DMPK and CNBP mRNAs, reducing pathophysiological phenotypes seen in DM1 and DM2 (27). Although bypassing the potential off-target genetic lesions caused by conventional CRISPR/Cas9-mediated DNA cleavage, this CRISPR/dCas9-based RNA-targeting system cannot permanently reduce the microsatellite repeats in DMPK and CNBP loci. Another study used CRISPR/Cas9-mediated DNA DSBs at the DMPK trinucleotide repeat region and successfully excised the entire expanded CTG/CAG repeats in human and mouse myoblasts. This approach successfully reverted the pathogenic hallmarks of DM1, including the cis epigenetic effects and the trans effects on the transcriptome and proteome (FIGURE 9C) (411). Therefore, CRISPR/Cas-mediated excision of expanded microsatellite repeats represents a promising strategy for permanent correction of DM1 and DM2 mutations (FIGURE 9, C and D) because it requires only NHEJ-mediated DNA repair, which is active throughout the cell cycle and hence can be used in many cell types.
IV. MYOEDITING IN “DISEASE-IN-A-DISH” AND ANIMAL MODELS
A. Human Induced Pluripotent Stem Cell Models of Muscular Dystrophies
The development of iPSCs has opened up new opportunities for disease modeling (307, 384, 385, 462). In principle, human iPSCs can self-renew indefinitely and can be differentiated into a variety of cell types, which become an inexhaustible and scalable source for stem cell research. Moreover, human iPSCs offer a similar genetic background to model human genetic diseases. For example, spinal muscular atrophy (SMA), caused by a mutation of the survival motor neuron 1 (SMN1) gene (53), cannot easily be modeled in mice. This is because humans have multiple copies of the paralogous SMN2 gene while mice do not carry the SMN2 gene (28). Therefore, additional knockout and transgenic strategies are required in animal models to model human SMA, which is time-consuming and expensive. In contrast, iPSCs derived from human SMA patients can be directly used for disease modeling and large-scale screening for drug discovery. Therefore, human iPSCs represent a complementary cellular source to the widely used animal models. Many human iPS cell lines have been established to model muscular dystrophies, including DMD, FSHD, LGMD, and DM. CRISPR/Cas has been utilized to correct a variety of DMD mutations found in human myoblasts and patient-derived iPS cell lines by different correction strategies (TABLE 1).
Table 1.
CRISPR/Cas-mediated correction of DMD mutations in human cells
| Strategy | Cell Type | CRISPR/Cas Type | DMD Mutations | sgRNA Targeting Site | Genome Editing Outcome | Reference Nos. |
|---|---|---|---|---|---|---|
| Exon deletion | Myoblast | SpCas9 | Δ Ex45–52 | i52, i53 | Δ Ex53 | 239, 240 |
| Myoblast | SpCas9 | Δ Ex45–52 | i43, i54 | Δ Ex44–54 | 239, 240 | |
| Myoblast | SpCas9 | Δ Ex48–50 | i43, i54 | Δ Ex44–54 | 239, 240 | |
| Myoblast | SpCas9 | Δ Ex48–50 | i50, i51 | Δ Ex51 | 302 | |
| Myoblast | SpCas9 | Δ Ex48–50 | i44, i55 | Δ Ex45–55 | 302 | |
| Myoblast | SpCas9 | Dup. Ex2 | i2 | Δ Dup. Ex2 | 209 | |
| Myoblast | SpCas9 | Dup. Ex18–30 | i27 | Δ Ex28–30, Δ Dup. Ex18–27 | 437 | |
| iPSC | SpCas9 | Δ Ex46–47 | i44, i55 | Δ Ex45–55 | 457 | |
| iPSC | SpCas9 | Δ Ex46–51 | i44, i55 | Δ Ex45–55 | 457 | |
| iPSC | SpCas9 | Δ Ex8–9 | i2, i7 | Δ Ex3–7 | 205 | |
| iPSC | SpCas9 | Δ Ex8–9 | i5, i7 | Δ Ex6–7 | 205 | |
| iPSC | SpCas9 | Δ Ex8–9 | i6, i11 | Δ Ex7–11 | 205 | |
| Exon skipping | Myoblast | SpCas9 | Δ Ex48–50 | e51 | Skp. Ex51 | 239, 240 |
| Myoblast | SpCas9 | Δ Ex45–52 | e53 | Skp. Ex53 | 239, 240 | |
| iPSC | SpCas9 | Δ Ex44 | e45 SA | Skp. Ex45 | 217 | |
| iPSC | LbCpf1 | Δ Ex48–50 | i50, e51 | Skp. Ex51 | 469 | |
| Exon reframing | Myoblast | SpCas9 | Δ Ex45–52 | e53 | Ex53 reframing | 239, 240 |
| Myoblast | SpCas9 | Δ Ex48–50 | e51 | Ex51 reframing | 239, 240, 302 | |
| Myoblast | SpCas9 | Δ Ex51–53 | e50, e54 | Ex50, Ex54 reframing | 162 | |
| iPSC | SpCas9 | Δ Ex44 | e45 SA | Ex45 reframing | 217 | |
| iPSC | LbCpf1 | Δ Ex48–50 | e51 | Ex51 reframing | 469 | |
| Exon knock-in | iPSC | SpCas9 | Δ Ex44 | e45 SA | Ex44 knock-in | 217 |
i, intron; e, exon; Δ, deletion; Dup., duplication; SA, splice acceptor; Skp., skipping.
Because of different genetic backgrounds and reprogramming methods, different human iPS cell lines can be variable in terms of their differentiation properties and phenotypic output (39, 46). To address this issue, isogenic control cell lines can be generated by introducing mutations in WT iPSCs, mimicking mutational genotypes found in patients. Conversely, mutations in patient-derived iPSCs can be corrected by programmable nucleases. The direct comparison between genome-edited iPSCs with unedited isogenic control cell lines, in principle, can reduce genetic background variation and improve genotype-phenotype correlation. Moreover, long-term in vitro iPSC culturing may cause genomic abnormalities, and hence routine karyotyping is recommended to monitor genome stability (254).
B. In Vivo Genome Editing of Animal Models
The ultimate goal of therapeutic application of genome editing is to permanently correct mutations that contribute to human monogenic diseases. Programmable nucleases such as the CRISPR/Cas system have been demonstrated to be effective in precise correction of pathogenic mutations found in the human embryo (176, 221, 234, 388). Several proof-of-concept studies have shown the efficacy and efficiency of the CRISPR/Cas system in correcting Dmd mutations in different mouse models by germline editing and postnatal editing (TABLE 2). However, ethical issues, as well as public policies, restrict therapeutic application of human germline editing, leaving postnatal genome editing as the means to achieve the same goal.
Table 2.
CRISPR/Cas-mediated correction of Dmd mutations in mice
| Strategy | Mouse Model | CRISPR/Cas Type | DMD Mutations | sgRNA Targeting Site | Genome Editing Outcome | Reference Nos. |
|---|---|---|---|---|---|---|
| Exon deletion | mdx | SpCas9 and SaCas9 | Ex23 point mutation | i22, i23 | Δ Ex23 | 230, 289, 383 |
| mdx/Utr+/– | SpCas9 and SaCas9 | Ex23 point mutation | i20, i23 | Δ Ex21–23 | 104 | |
| mdx | SpCas9 | Ex23 point mutation | i20, i23 | Δ Ex21–23 | 446 | |
| mdx4cv | SpCas9 and SaCas9 | Ex53 point mutation | i51, i53 | Δ Ex52–53 | 29 | |
| Exon skipping | Δ Ex50 | SpCas9 | Δ Ex50 | e51 SA | Skp. Ex51 | 7 |
| Exon reframing | mdx | SpCas9 | Ex23 point mutation | e23 | Ex23 reframing | 231 |
| Δ Ex50 | SpCas9 | Δ Ex50 | e51 SA | Ex51 reframing | 7 | |
| mdx4cv | SpCas9 | Ex53 point mutation | e53 | Ex53 reframing | 29 | |
| Exon HDR | mdx | SpCas9 | Ex23 point mutation | e23 | Ex23 HDR | 231 |
| mdx | LbCpf1 | Ex23 point mutation | e23 | Ex23 HDR | 469 | |
| mdx4cv | SpCas9 | Ex53 point mutation | e53 | Ex53 HDR | 29 |
i, intron; e, exon; Δ, deletion; SA, splice acceptor; Skp., skipping.
Therapeutic genome editing requires delivering programmable nucleases and other genome editing components to target cells, which can be achieved ex vivo or in vivo. Ex vivo editing requires in vitro editing of the cellular genome, followed by transplantation of the targeted cell population to the original host. CRISPR/Cas-mediated ex vivo editing has been shown to be effective in the hematopoietic system (88, 125, 137, 345, 423). In theory, ex vivo genome editing can be applied to the skeletal muscle system because satellite cells serve as adult stem cells of skeletal muscle and are capable of surviving manipulation in vitro. However, skeletal muscle is the largest tissue in the human body, comprising ~40% of body weight. Transplantation of edited satellite cells or progenitor cells may improve local muscle function, but systemic functional recovery remains questionable (471). In addition to skeletal muscle, the heart is also affected in a variety of muscular dystrophies, but it is still under debate whether the heart contains stem cells or stem cell-like cell populations capable of transplantation and regeneration (21, 105, 284, 301, 378, 412). Taken together, ex vivo transplantation-based approaches are generally not feasible to treat muscular dystrophies, especially for those affecting both skeletal muscle and the heart. Therefore, in vivo postnatal genome editing turns out to be a more practical approach to permanently correct genetic mutations causing muscular dystrophies.
1. Delivery of genome editing components by nonviral vectors
Achieving in vivo postnatal genome editing requires an efficient and effective delivery system. Genome editing components can be physically or chemically delivered to target cells by nonviral vectors (FIGURE 10). For example, microinjection and electroporation have been demonstrated as effective methods to deliver CRISPR/Cas genome editing components to target cells (68, 190, 262, 296, 450). However, these physical approaches are widely used for germline editing or embryo manipulation but are not feasible for systemic delivery in the postnatal host. In contrast, hydrodynamic intravenous injection can achieve systemic delivery in multiple tissues, including liver, kidney, lung, skeletal muscle, and heart (42, 377). Some studies have applied this technology to deliver CRISPR/Cas genome editing components to postnatal mice for mutating cancer genes in the mouse liver, correcting a tyrosinemia mutation and disrupting hepatitis B virus (222, 447, 455, 470). However, hydrodynamic injection requires a large injection volume and high pressure, which may damage tissues or organs.
FIGURE 10.
Systemic delivery of genome editing components. Nonviral (lipid-based carriers or polymeric carriers) and viral (rAAV) vector-based delivery of genome editing components into target tissue. Following administration, the delivery vectors pass through the blood vessel by extravasation to reach their target tissues. In the tissue, they undergo cytoplasmic trafficking, endosomal escape, and nuclear entry to perform genome editing in the nucleus.
In addition to physical vectors, chemical vectors, such as polymeric carriers and lipid-based carriers, are also widely used for in vivo delivery. Polymeric carriers are cationic polymers that can condense negatively charged DNA or RNA, whereas lipid-based carriers can spontaneously assemble into liposomes consisting of nucleic acids and cationic or neutral lipids (453). The CRISPR/Cas system and other genome editing components can be chemically delivered into animals in the form of plasmid DNA, mRNA, or ribonucleoprotein complexes. Chemical vectors can protect nucleic acids or ribonucleoproteins from degradation by endonucleases or proteases in physiological fluids and extracellular space, improving stability and half-life. For example, chemically modified CRISPR/Cpf1 mRNA and crRNA have been reported to enhance genome editing efficiency (216). However, many challenges still remain, including efficient delivery to the tissue of interest, cellular internalization, and protection from the lysosomal degradation pathway (422, 453).
It has been reported that plasmid DNA can be retained in skeletal muscle for over a year after intramuscular injection, suggesting the likelihood of long-term gene expression and retention in postmitotic tissue (439). In contrast, localized nonviral delivery such as intramuscular injection cannot generate a systemic effect, which is not ideal for treating muscular dystrophies since skeletal muscle is one of the largest tissues in the human body and multiple muscle groups can be affected. Systemic nonviral delivery, such as intravenous or intraperitoneal injection, have extended the targeting range but may cause gene expression in nontargeted tissues. Therefore, delivery of genome editing components to animals by nonviral vectors still requires improvement and optimization before clinical translation.
2. Delivery of genome editing components by viral vectors
Many viral vectors have been used for gene therapy, including lentivirus, retrovirus, herpes simplex virus, poxvirus, adenovirus, adeno-associated virus, baculovirus, and Epstein-Barr virus (288, 426). Among these, adeno-associated virus (AAV) is the most promising viral vector for the delivery of genome editing components to specific tissues, such as muscle and heart. AAV is a nonenveloped DNA virus with an ~5 kb linear ssDNA genome (371). The genome of wild-type AAV has two major ORFs flanked by two inverted terminal repeats (ITRs), while in the recombinant AAV (rAAV), the viral ORFs encoding the replication and capsid proteins are replaced by the customized gene expression cassette (110). Both wild-type and rAAVs are nonpathogenic in humans or animals, and their propagation requires a helper virus, making them a safe delivery system for therapeutic genome editing (199, 342, 379).
The rAAVs have many appealing features, including broad spectrum tissue tropism with minimal integration risk and long-term transgene expression from the episomal genome after viral transduction (98, 297, 348). Currently, 13 AAV serotypes are widely available for gene delivery, and each of them shows different tissue tropism. AAV serotypes 1, 6, 8, and 9 have high tropism in skeletal muscle and heart (37, 64, 119, 131, 159, 306, 427, 463, 472). In addition, tissue tropism and transduction efficiency can also be improved by pseudo-typing. For example, AAV2 genomes pseudo-packaged into AAV5 capsids can enhance gene delivery to skeletal muscle, whereas improved cardiomyocyte transduction has been observed by pseudo-packaging AAV2 genomes into AAV6 capsids (99, 363). rAAV has been successfully used to as a delivery system to administer CRISPR/Cas9 and other genome editing components to postnatal mdx or mdx4cv mice and correct Dmd mutations by exon deletion or reframing strategies (29, 104, 230, 289, 383) (FIGURE 10). In addition, several other studies reported rAAV-delivered CRISPR/Cas9-mediated in vivo genome editing in mouse models of human Huntington disease and congenital muscular dystrophy (180, 273, 452). These studies demonstrated that the combination of a rAAV-based delivery system with CRISPR/Cas9-mediated postnatal genome editing is a compelling strategy to permanently correct mutations responsible for monogenic neuromuscular disorders. However, long-term benefits and effects in animal models still need to be examined to prepare for future clinical trials.
C. Challenges of Therapeutic Genome Editing
Despite many appealing features, the rAAV-based delivery system entails major challenges. rAAV has a limited packaging capacity which limits the packaging size of gene therapy components. Currently, in the CRISPR/Cas-based system, the size limitation has been addressed by various groups, whereby SpCas9 and SaCas9 have been efficiently packaged into different rAAV serotypes for correcting Dmd mutations in mice (29, 104, 230, 289, 383). To effectively deliver the rAAV-based genome editing components, tissue and cell type specificity is another parameter to be considered. Selection of the appropriate rAAV serotype is necessary for tissue tropism. Additionally, implementation of tissue-specific promoters can be used to regulate expression of the editing components. Recent administration of rAAV serotypes with muscle tropism and gene expression regulated under a muscle-specific promoter successfully delivered genome editing components to skeletal muscle and heart (7, 29, 143).
1. Immunogenicity
One of the greatest challenges of using rAAV as a delivery system is the immune response to the vector. Potential immunogenicity elicited by rAAV-based delivery of the CRISPR/Cas system can be evoked by 1) the restored protein product, 2) the CRISPR/Cas system, and 3) capsid proteins on the surface of rAAV virus.
Mutated genes in monogenic disorders encode abnormal proteins or cause a complete loss of protein. After CRISPR/Cas-mediated correction, epitopes derived from the newly restored protein may elicit immunogenicity. However, in the case of DMD, due to somatic mutation or alternative splicing, more than 50% of DMD patients display low level of dystrophin-positive revertant fibers (0.2–4%), which may mitigate a potential immune response (55, 290, 291). Indeed, in a gene transfer study, expression of murine full-length or mini-dystrophin in mdx mice did not evoke humoral or cytotoxic immune responses (108). Therefore, immunogenicity elicited by the rescued protein may not be a significant concern, at least in the case of DMD.
In regard to immunogenicity elicited by the CRISPR/Cas system, it was demonstrated that Cas9 endonucleases delivered by rAAV did evoke a humoral immune response in mice (71). However, they did not observe significant muscle cell damage or a repair response at 2 wk after rAAV administration. Another concern is if the rAAV needs to be readministered since once the SpCas9-mediated humoral immunity is established in the host, further application of SpCas9 for therapeutic genome editing may no longer be effective. Several potential strategies could be applied to address this issue, including 1) large-scale functional variant profiling of SpCas9 for epitope mutation; 2) replacing SpCas9 with other Cas endonucleases such as SaCas9, LbCpf1, and AsCpf1 after initial SpCas9 administration; and 3) performing plasmapheresis or transient immunosuppression to reduce the circulating antibody titer.
In addition to the immunogenicity response elicited by the CRISPR/Cas system, the humoral immune response evoked by rAAV is another challenge for in vivo therapeutic genome editing. Several studies in non-human primates have shown that high incidence of neutralizing antibodies (NAbs) after initial exposure to AAV can block AAV transduction, rendering gene delivery ineffective (164, 425). Moreover, the prevalence of AAV NAbs in human populations is also relatively high, ranging from 30 to 60% in AAV2, to 15–30% in AAV7, AAV8, and AAV9 serotypes (47, 56). Several strategies have been developed to overcome AAV-induced humoral immune responses, including 1) AAV capsid mutagenesis to alter NAb binding epitopes (287), 2) plasmapheresis to reduce AAV NAb titer (157, 272, 424), and 3) transient immunosuppression (267).
2. Off-target effects
Another concern around using programmable nucleases for therapeutic genome editing is the potential for mutagenesis caused by off-target effects. Initial programmable nuclease-like TALENs generally show minimal off-target effects because TALEN-mediated DNA DSBs require dimerization of two TALEN monomers and the dimerized TALEN pairs can recognize 30–40 bp of DNA sequence (182). In contrast, DNA DSBs induced by SpCas9, currently the most prevalently used Cas endonuclease, only requires a 20-nt sgRNA forming DNA-RNA duplex with the target DNA strand, which may increase mismatching frequency. Indeed, several early studies about SpCas9 specificity have shown that high-frequency mutagenesis caused by off-target effects is possible at mismatched sites (112, 152, 224). For example, INDELs caused by SpCas9 off-target cleavage can be detected at certain sites with up to five mismatches relative to the on-target site (112, 152, 308). Several approaches can be used to evaluate potential CRISPR/Cas-induced off-target effects. Computational prediction of off-target sites followed by DNA mismatch cleavage assay serves as a rapid and convenient method to evaluate sgRNA specificity. More recently, computational prediction of off-target sites followed by deep sequencing or unbiased whole-genome sequencing represents a more reliable method for systematic evaluation of CRISPR/Cas specificity (399).
Many strategies have been developed to reduce off-target effects caused by CRISPR/Cas9 nonspecific cleavage. Using truncated sgRNA with 2–3 nt deletion at the 5′-end can reduce the off-target-induced mutagenesis by 5,000-fold or more, and reduce the genome-wide off-target sites by 2- to 5-fold (113, 401). Another strategy is to use a pair of Cas9 nickases to generate two DNA nicks in close proximity, and this strategy has been shown to reduce off-target mutations by 50- to 1,500-fold (247, 323, 357). Other strategies such as titration of dosage for Cas9/sgRNA (152), fusion dCas9 to FokI nuclease (134, 400) have also been developed to reduce off-target effects.
Resolving the crystal structure of SpCas9 in complex with sgRNA and target DNA provided many insights into the potential improvement of Cas9 specificity through logistic engineering (295). The high-fidelity SpCas9 (SpCas9-HF1) was generated by alanine substitution to disrupt nonspecific interaction with the target DNA strand (191). Recent structural analysis demonstrated that binding SpCas9-HF1 to a substrate with even a single base pair mismatch at the PAM distal end completely abolishes stable docking of the HNH nuclease domain (67). In addition, enhanced SpCas9 (eSpCas9 1.1) was developed by neutralizing positively charged residues to weaken interactions with the non-target DNA strand (364). Additionally, a hyper-accurate Cas9 variant (HypaCas9) was developed in which the HNH nuclease activation is allosterically regulated by REC3, leading to high specificity of on-target cleavage (67). All of these engineered SpCas9 variants have been shown to significantly reduce off-target mutagenesis and have the potential to push therapeutic genome editing toward high specificity.
3. Long-term effects and benefits of postnatal genome editing
Postnatal genome editing can correct the genetic mutation in muscular dystrophies and, in the short term, leads to improved muscle function. At this time, little is known about the longevity of restored dystrophin protein in vivo. It is uncertain, for example, whether genome-edited, dystrophin-positive myofibers will be diluted out by dystrophin-negative myofibers generated from satellite cells carrying the DMD mutation. In contrast, cardiomyocytes in the adult human heart have a very low turnover rate (1% at the age of 25 to 0.45% at the age of 75) (30, 31). Therefore, therapeutic genome editing in the human heart should provide long-term clinical benefit. Indeed, several studies have demonstrated that CRISPR/Cas-mediated genome editing can restore cardiac function in dystrophic mice (104, 230).
V. CONCLUSIONS AND FUTURE PERSPECTIVES
In addition to mutations in the nuclear genome, mutations in the mitochondrial genome also cause primary mitochondrial DNA (mtDNA)-related diseases. Mutations in mitochondrial tRNAs or protein-coding genes can lead to MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes) or MERRF (myoclonus epilepsy and ragged red fibers) (89–91, 349). These genetic disorders not only affect muscle function but also result in pathogenesis in other systems, including brain, blood vessels, and the endocrine system. Currently, mitochondrial replacement therapy (MRT) is used to treat genetic disorders caused by mtDNA mutations, in which the meiotic spindle apparatus with chromosomes from an unfertilized maternal oocyte is transferred into a donor oocyte cytoplasm containing healthy mtDNA (438). However, healthy mtDNA replacement is not absolute, and <1% carryover of mutant mtDNA can be present after MRT. This carryover may lead to a gradual loss of healthy donor mtDNA and reversal to the maternal haplotype by genetic drift (175, 448). This brings up an interesting question of whether programmable nucleases can be used to eliminate mutant mtDNA. Indeed, ZFNs and TALENs have been applied to selectively degrade pathogenic mitochondrial genomes (19, 118, 326). Currently, efficient mitochondrial genome editing by the CRISPR/Cas system remains controversial (117, 169). Perhaps, efficient delivery of sgRNA into the mitochondrial matrix is an impediment for this application. We anticipate that in the near future, CRISPR/Cas-mediated genome editing can be further expanded to the mitochondrial genome.
To date, there are 840 neuromuscular diseases known to be caused by mutations in 465 different genes, with 72 mapped loci awaiting gene identification (177). These debilitating diseases cause early death or significantly impair the quality of life. Currently, there is no effective treatment for these diseases since most therapies developed to date focus on alleviating the symptoms or targeting the secondary effects, while the source of mutations is still present in the human genome. The discovery and application of programmable nucleases for site-specific DNA DSBs provide a powerful tool for precise genome engineering. In particular, the CRISPR/Cas system has revolutionized the genome editing field and provides a new path for disease treatment by removing the genetic mutations that cause disease rather than simply treating the symptoms.
GRANTS
This work was supported by Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center Grant U54 HD 087351 as well as Parent Project Muscular Dystrophy and Robert A. Welch Foundation Grant 1–0025 (to E. N. Olson).
DISCLOSURES
E. N. Olson and R. Bassel-Duby are consultants for Exonics Therapeutics. No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
We thank Jose Cabrera for assistance with graphics.
Present address of C. Long: Leon H. Charney Division of Cardiology, New York University School of Medicine, New York, NY 10016.
Address for reprint requests and other correspondence: E. N. Olson, Dept. of Molecular Biology, Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center and Hamon Center for Regenerative Science and Medicine, University of Texas Southwestern Medical Center, Dallas, TX 75390 (e-mail: Eric.Olson@UTSouthwestern.edu).
REFERENCES
- 1.Aartsma-Rus A. Overview on DMD exon skipping. Methods Mol Biol 867: 97–116, 2012. doi: 10.1007/978-1-61779-767-5_7. [DOI] [PubMed] [Google Scholar]
- 2.Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet 53: 145–151, 2016. doi: 10.1136/jmedgenet-2015-103387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 34: 135–144, 2006. doi: 10.1002/mus.20586. [DOI] [PubMed] [Google Scholar]
- 4.Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A, Lander ES, Voytas DF, Ting AY, Zhang F. RNA targeting with CRISPR-Cas13. Nature 550: 280–284, 2017. doi: 10.1038/nature24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DB, Shmakov S, Makarova KS, Semenova E, Minakhin L, Severinov K, Regev A, Lander ES, Koonin EV, Zhang F. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353: aaf5573, 2016. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nat Genet 3: 283–291, 1993. doi: 10.1038/ng0493-283. [DOI] [PubMed] [Google Scholar]
- 7.Amoasii L, Long C, Li H, Mireault AA, Shelton JM, Sanchez-Ortiz E, McAnally JR, Bhattacharyya S, Schmidt F, Grimm D, Hauschka SD, Bassel-Duby R, Olson EN. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci Transl Med 9: eaan8081, 2017. doi: 10.1126/scitranslmed.aan8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ampong BN, Imamura M, Matsumiya T, Yoshida M, Takeda S. Intracellular localization of dysferlin and its association with the dihydropyridine receptor. Acta Myol 24: 134–144, 2005. [PubMed] [Google Scholar]
- 9.Aoki M. Dysferlinopathy. In: GeneReviews, edited by Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N. Seattle, WA: Univ. of Washington, Seattle, 1993. [PubMed] [Google Scholar]
- 10.Arahata K, Ishihara T, Fukunaga H, Orimo S, Lee JH, Goto K, Nonaka I. Inflammatory response in facioscapulohumeral muscular dystrophy (FSHD): immunocytochemical and genetic analyses. Muscle Nerve Suppl 18, S13: S56–S66, 1995. doi: 10.1002/mus.880181312. [DOI] [PubMed] [Google Scholar]
- 11.Araishi K, Sasaoka T, Imamura M, Noguchi S, Hama H, Wakabayashi E, Yoshida M, Hori T, Ozawa E. Loss of the sarcoglycan complex and sarcospan leads to muscular dystrophy in beta-sarcoglycan-deficient mice. Hum Mol Genet 8: 1589–1598, 1999. doi: 10.1093/hmg/8.9.1589. [DOI] [PubMed] [Google Scholar]
- 12.Araki E, Nakamura K, Nakao K, Kameya S, Kobayashi O, Nonaka I, Kobayashi T, Katsuki M. Targeted disruption of exon 52 in the mouse dystrophin gene induced muscle degeneration similar to that observed in Duchenne muscular dystrophy. Biochem Biophys Res Commun 238: 492–497, 1997. doi: 10.1006/bbrc.1997.7328. [DOI] [PubMed] [Google Scholar]
- 13.Arnett AL, Konieczny P, Ramos JN, Hall J, Odom G, Yablonka-Reuveni Z, Chamberlain JR, Chamberlain JS. Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Mol Ther Methods Clin Dev 1: 14038, 2014. doi: 10.1038/mtm.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnould S, Chames P, Perez C, Lacroix E, Duclert A, Epinat JC, Stricher F, Petit AS, Patin A, Guillier S, Rolland S, Prieto J, Blanco FJ, Bravo J, Montoya G, Serrano L, Duchateau P, Pâques F. Engineering of large numbers of highly specific homing endonucleases that induce recombination on novel DNA targets. J Mol Biol 355: 443–458, 2006. doi: 10.1016/j.jmb.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 15.Ashizawa T, Sarkar PS. Myotonic dystrophy types 1 and 2. Handb Clin Neurol 101: 193–237, 2011. doi: 10.1016/B978-0-08-045031-5.00015-3. [DOI] [PubMed] [Google Scholar]
- 16.Atencia-Fernandez S, Shiel RE, Mooney CT, Nolan CM. Muscular dystrophy in the Japanese Spitz: an inversion disrupts the DMD and RPGR genes. Anim Genet 46: 175–184, 2015. doi: 10.1111/age.12266. [DOI] [PubMed] [Google Scholar]
- 17.Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem 279: 55117–55126, 2004. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 18.Auer TO, Duroure K, De Cian A, Concordet JP, Del Bene F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res 24: 142–153, 2014. doi: 10.1101/gr.161638.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bacman SR, Williams SL, Pinto M, Moraes CT. The use of mitochondria-targeted endonucleases to manipulate mtDNA. Methods Enzymol 547: 373–397, 2014. doi: 10.1016/B978-0-12-801415-8.00018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baghdadi MB, Tajbakhsh S. Regulation and phylogeny of skeletal muscle regeneration. Dev Biol 433: 200–209, 2018. doi: 10.1016/j.ydbio.2017.07.026. [DOI] [PubMed] [Google Scholar]
- 21.Balsam LB, Wagers AJ, Christensen JL, Kofidis T, Weissman IL, Robbins RC. Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature 428: 668–673, 2004. doi: 10.1038/nature02460. [DOI] [PubMed] [Google Scholar]
- 22.Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 423: 168–172, 2003. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- 23.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315: 1709–1712, 2007. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 24.Barresi R, Di Blasi C, Negri T, Brugnoni R, Vitali A, Felisari G, Salandi A, Daniel S, Cornelio F, Morandi L, Mora M. Disruption of heart sarcoglycan complex and severe cardiomyopathy caused by beta sarcoglycan mutations. J Med Genet 37: 102–107, 2000. doi: 10.1136/jmg.37.2.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, Richard I, Marchand S, Bourg N, Argov Z, Sadeh M, Mahjneh I, Marconi G, Passos-Bueno MR, Moreira ES, Zatz M, Beckmann JS, Bushby K. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet 20: 37–42, 1998. doi: 10.1038/1689. [DOI] [PubMed] [Google Scholar]
- 26.Bashir R, Strachan T, Keers S, Stephenson A, Mahjneh I, Marconi G, Nashef L, Bushby KM. A gene for autosomal recessive limb-girdle muscular dystrophy maps to chromosome 2p. Hum Mol Genet 3: 455–457, 1994. doi: 10.1093/hmg/3.3.455. [DOI] [PubMed] [Google Scholar]
- 27.Batra R, Nelles DA, Pirie E, Blue SM, Marina RJ, Wang H, Chaim IA, Thomas JD, Zhang N, Nguyen V, Aigner S, Markmiller S, Xia G, Corbett KD, Swanson MS, Yeo GW. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell 170: 899–912.e10, 2017. doi: 10.1016/j.cell.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bebee TW, Dominguez CE, Chandler DS. Mouse models of SMA: tools for disease characterization and therapeutic development. Hum Genet 131: 1277–1293, 2012. doi: 10.1007/s00439-012-1171-5. [DOI] [PubMed] [Google Scholar]
- 29.Bengtsson NE, Hall JK, Odom GL, Phelps MP, Andrus CR, Hawkins RD, Hauschka SD, Chamberlain JR, Chamberlain JS. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun 8: 14454, 2017. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisén J. Evidence for cardiomyocyte renewal in humans. Science 324: 98–102, 2009. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, Sjostrom SL, Szewczykowska M, Jackowska T, Dos Remedios C, Malm T, Andrä M, Jashari R, Nyengaard JR, Possnert G, Jovinge S, Druid H, Frisén J. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 161: 1566–1575, 2015. doi: 10.1016/j.cell.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 32.Bhakta MS, Henry IM, Ousterout DG, Das KT, Lockwood SH, Meckler JF, Wallen MC, Zykovich A, Yu Y, Leo H, Xu L, Gersbach CA, Segal DJ. Highly active zinc-finger nucleases by extended modular assembly. Genome Res 23: 530–538, 2013. doi: 10.1101/gr.143693.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhakta MS, Segal DJ. The generation of zinc finger proteins by modular assembly. Methods Mol Biol 649: 3–30, 2010. doi: 10.1007/978-1-60761-753-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Billon P, Bryant EE, Joseph SA, Nambiar TS, Hayward SB, Rothstein R, Ciccia A. CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol Cell 67: 1068–1079.e4, 2017. doi: 10.1016/j.molcel.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. FokI dimerization is required for DNA cleavage. Proc Natl Acad Sci USA 95: 10570–10575, 1998. doi: 10.1073/pnas.95.18.10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, Dawkins H, Lamont L, Roy AJ, Chamova T, Guergueltcheva V, Chan S, Korngut L, Campbell C, Dai Y, Wang J, Barišić N, Brabec P, Lahdetie J, Walter MC, Schreiber-Katz O, Karcagi V, Garami M, Viswanathan V, Bayat F, Buccella F, Kimura E, Koeks Z, van den Bergen JC, Rodrigues M, Roxburgh R, Lusakowska A, Kostera-Pruszczyk A, Zimowski J, Santos R, Neagu E, Artemieva S, Rasic VM, Vojinovic D, Posada M, Bloetzer C, Jeannet PY, Joncourt F, Díaz-Manera J, Gallardo E, Karaduman AA, Topaloğlu H, El Sherif R, Stringer A, Shatillo AV, Martin AS, Peay HL, Bellgard MI, Kirschner J, Flanigan KM, Straub V, Bushby K, Verschuuren J, Aartsma-Rus A, Béroud C, Lochmüller H. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat 36: 395–402, 2015. doi: 10.1002/humu.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blankinship MJ, Gregorevic P, Allen JM, Harper SQ, Harper H, Halbert CL, Miller AD, Chamberlain JS. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol Ther 10: 671–678, 2004. doi: 10.1016/j.ymthe.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 38.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326: 1509–1512, 2009. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 39.Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, Gnirke A, Eggan K, Meissner A. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 144: 439–452, 2011. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolderson E, Tomimatsu N, Richard DJ, Boucher D, Kumar R, Pandita TK, Burma S, Khanna KK. Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Res 38: 1821–1831, 2010. doi: 10.1093/nar/gkp1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151: 2551–2561, 2005. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- 42.Bonamassa B, Hai L, Liu D. Hydrodynamic gene delivery and its applications in pharmaceutical research. Pharm Res 28: 694–701, 2011. doi: 10.1007/s11095-010-0338-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bosnakovski D, Chan SSK, Recht OO, Hartweck LM, Gustafson CJ, Athman LL, Lowe DA, Kyba M. Muscle pathology from stochastic low level DUX4 expression in an FSHD mouse model. Nat Commun 8: 550, 2017. doi: 10.1038/s41467-017-00730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bosnakovski D, Xu Z, Gang EJ, Galindo CL, Liu M, Simsek T, Garner HR, Agha-Mohammadi S, Tassin A, Coppée F, Belayew A, Perlingeiro RR, Kyba M. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J 27: 2766–2779, 2008. doi: 10.1038/emboj.2008.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Botta A, Vallo L, Rinaldi F, Bonifazi E, Amati F, Biancolella M, Gambardella S, Mancinelli E, Angelini C, Meola G, Novelli G. Gene expression analysis in myotonic dystrophy: indications for a common molecular pathogenic pathway in DM1 and DM2. Gene Expr 13: 339–351, 2007. doi: 10.3727/000000006781510705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boulting GL, Kiskinis E, Croft GF, Amoroso MW, Oakley DH, Wainger BJ, Williams DJ, Kahler DJ, Yamaki M, Davidow L, Rodolfa CT, Dimos JT, Mikkilineni S, MacDermott AB, Woolf CJ, Henderson CE, Wichterle H, Eggan K. A functionally characterized test set of human induced pluripotent stem cells. Nat Biotechnol 29: 279–286, 2011. doi: 10.1038/nbt.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, Masurier C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther 21: 704–712, 2010. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 48.Brack AS, Rando TA. Tissue-specific stem cells: lessons from the skeletal muscle satellite cell. Cell Stem Cell 10: 504–514, 2012. doi: 10.1016/j.stem.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bradley A, Evans M, Kaufman MH, Robertson E. Formation of germ-line chimaeras from embryo-derived teratocarcinoma cell lines. Nature 309: 255–256, 1984. doi: 10.1038/309255a0. [DOI] [PubMed] [Google Scholar]
- 50.Briggs AW, Rios X, Chari R, Yang L, Zhang F, Mali P, Church GM. Iterative capped assembly: rapid and scalable synthesis of repeat-module DNA such as TAL effectors from individual monomers. Nucleic Acids Res 40: e117, 2012. doi: 10.1093/nar/gks624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 69: 385, 1992. [DOI] [PubMed] [Google Scholar]
- 52.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321: 960–964, 2008. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brzustowicz LM, Lehner T, Castilla LH, Penchaszadeh GK, Wilhelmsen KC, Daniels R, Davies KE, Leppert M, Ziter F, Wood D, Dubowitz V, Zerres K, Hausmanowa-Petrusewicz I, Ott J, Munsat TL, Gilliam TC. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-13.3. Nature 344: 540–541, 1990. doi: 10.1038/344540a0. [DOI] [PubMed] [Google Scholar]
- 54.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA 81: 1189–1192, 1984. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burrow KL, Coovert DD, Klein CJ, Bulman DE, Kissel JT, Rammohan KW, Burghes AH, Mendell JR; CIDD Study Group . Dystrophin expression and somatic reversion in prednisone-treated and untreated Duchenne dystrophy. Neurology 41: 661–666, 1991. doi: 10.1212/WNL.41.5.661. [DOI] [PubMed] [Google Scholar]
- 56.Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis 199: 381–390, 2009. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet 6: 507–512, 2005. doi: 10.1038/nrg1619. [DOI] [PubMed] [Google Scholar]
- 58.Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39: e82, 2011. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chakraborty S, Ji H, Kabadi AM, Gersbach CA, Christoforou N, Leong KW. A CRISPR/Cas9-based system for reprogramming cell lineage specification. Stem Cell Rep 3: 940–947, 2014. doi: 10.1016/j.stemcr.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chamberlain JS, Metzger J, Reyes M, Townsend D, Faulkner JA. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J 21: 2195–2204, 2007. doi: 10.1096/fj.06-7353com. [DOI] [PubMed] [Google Scholar]
- 61.Chan SH, Yu AM, McVey M. Dual roles for DNA polymerase theta in alternative end-joining repair of double-strand breaks in Drosophila. PLoS Genet 6: e1001005, 2010. doi: 10.1371/journal.pgen.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chandrasegaran S, Carroll D. Origins of Programmable Nucleases for Genome Engineering. J Mol Biol 428, 5 Pt B: 963–989, 2016. doi: 10.1016/j.jmb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chang NC, Rudnicki MA. Satellite cells: the architects of skeletal muscle. Curr Top Dev Biol 107: 161–181, 2014. doi: 10.1016/B978-0-12-416022-4.00006-8. [DOI] [PubMed] [Google Scholar]
- 64.Chao H, Liu Y, Rabinowitz J, Li C, Samulski RJ, Walsh CE. Several log increase in therapeutic transgene delivery by distinct adeno-associated viral serotype vectors. Mol Ther 2: 619–623, 2000. doi: 10.1006/mthe.2000.0219. [DOI] [PubMed] [Google Scholar]
- 65.Chapman VM, Miller DR, Armstrong D, Caskey CT. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc Natl Acad Sci USA 86: 1292–1296, 1989. doi: 10.1073/pnas.86.4.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, P R Iyer E, Lin S, Kiani S, Guzman CD, Wiegand DJ, Ter-Ovanesyan D, Braff JL, Davidsohn N, Housden BE, Perrimon N, Weiss R, Aach J, Collins JJ, Church GM. Highly efficient Cas9-mediated transcriptional programming. Nat Methods 12: 326–328, 2015. doi: 10.1038/nmeth.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, Sternberg SH, Joung JK, Yildiz A, Doudna JA. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550: 407–410, 2017. doi: 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen S, Lee B, Lee AY, Modzelewski AJ, He L. Highly Efficient Mouse Genome Editing by CRISPR Ribonucleoprotein Electroporation of Zygotes. J Biol Chem 291: 14457–14467, 2016. doi: 10.1074/jbc.M116.733154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen Y, Zheng Y, Kang Y, Yang W, Niu Y, Guo X, Tu Z, Si C, Wang H, Xing R, Pu X, Yang SH, Li S, Ji W, Li XJ. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum Mol Genet 24: 3764–3774, 2015. doi: 10.1093/hmg/ddv120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, Rangarajan S, Shivalila CS, Dadon DB, Jaenisch R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res 23: 1163–1171, 2013. doi: 10.1038/cr.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chew WL, Tabebordbar M, Cheng JK, Mali P, Wu EY, Ng AH, Zhu K, Wagers AJ, Church GM. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat Methods 13: 868–874, 2016. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chiurazzi M, Ray A, Viret JF, Perera R, Wang XH, Lloyd AM, Signer ER. Enhancement of somatic intrachromosomal homologous recombination in Arabidopsis by the HO endonuclease. Plant Cell 8: 2057–2066, 1996. doi: 10.1105/tpc.8.11.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Choulika A, Perrin A, Dujon B, Nicolas JF. Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol Cell Biol 15: 1968–1973, 1995. doi: 10.1128/MCB.15.4.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186: 757–761, 2010. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204, 2010. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Conboy MJ, Karasov AO, Rando TA. High incidence of non-random template strand segregation and asymmetric fate determination in dividing stem cells and their progeny. PLoS Biol 5: e102, 2007. doi: 10.1371/journal.pbio.0050102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823, 2013. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Coral-Vazquez R, Cohn RD, Moore SA, Hill JA, Weiss RM, Davisson RL, Straub V, Barresi R, Bansal D, Hrstka RF, Williamson R, Campbell KP. Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell 98: 465–474, 1999. doi: 10.1016/S0092-8674(00)81975-3. [DOI] [PubMed] [Google Scholar]
- 79.Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med 21: 121–131, 2015. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, Zhang F. RNA editing with CRISPR-Cas13. Science 358: 1019–1027, 2017. doi: 10.1126/science.aaq0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cozzi F, Cerletti M, Luvoni GC, Lombardo R, Brambilla PG, Faverzani S, Blasevich F, Cornelio F, Pozza O, Mora M. Development of muscle pathology in canine X-linked muscular dystrophy. II. Quantitative characterization of histopathological progression during postnatal skeletal muscle development. Acta Neuropathol 101: 469–478, 2001. [DOI] [PubMed] [Google Scholar]
- 82.Dandapat A, Bosnakovski D, Hartweck LM, Arpke RW, Baltgalvis KA, Vang D, Baik J, Darabi R, Perlingeiro RC, Hamra FK, Gupta K, Lowe DA, Kyba M. Dominant lethal pathologies in male mice engineered to contain an X-linked DUX4 transgene. Cell Reports 8: 1484–1496, 2014. doi: 10.1016/j.celrep.2014.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining. Transl Cancer Res 2: 130–143, 2013. doi: 10.3978/j.issn.2218-676X.2013.04.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W, Schneider C, Koch MC, Beilman GJ, Harrison AR, Dalton JC, Ranum LP. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology 60: 657–664, 2003. doi: 10.1212/01.WNL.0000054481.84978.F9. [DOI] [PubMed] [Google Scholar]
- 85.Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 90: 717–727, 1997. doi: 10.1016/S0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- 86.Deng C, Capecchi MR. Reexamination of gene targeting frequency as a function of the extent of homology between the targeting vector and the target locus. Mol Cell Biol 12: 3365–3371, 1992. doi: 10.1128/MCB.12.8.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Deng D, Yan C, Pan X, Mahfouz M, Wang J, Zhu JK, Shi Y, Yan N. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science 335: 720–723, 2012. doi: 10.1126/science.1215670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.DeWitt MA, Magis W, Bray NL, Wang T, Berman JR, Urbinati F, Heo SJ, Mitros T, Muñoz DP, Boffelli D, Kohn DB, Walters MC, Carroll D, Martin DI, Corn JE. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Transl Med 8: 360ra134, 2016. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.DiMauro S. Mitochondrial diseases. Biochim Biophys Acta 1658: 80–88, 2004. doi: 10.1016/j.bbabio.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 90.DiMauro S, Hirano M. Melas. In: GeneReviews, edited by Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N. Seattle, WA: Univ. of Washington, Seattle, 1993. [Google Scholar]
- 91.DiMauro S, Hirano M. Merrf. In: GeneReviews, edited by Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N. Seattle, WA: Univ. of Washington, Seattle, 1993. [Google Scholar]
- 92.Dixit M, Ansseau E, Tassin A, Winokur S, Shi R, Qian H, Sauvage S, Mattéotti C, van Acker AM, Leo O, Figlewicz D, Barro M, Laoudj-Chenivesse D, Belayew A, Coppée F, Chen YW. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci USA 104: 18157–18162, 2007. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Doetschman T, Gregg RG, Maeda N, Hooper ML, Melton DW, Thompson S, Smithies O. Targetted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature 330: 576–578, 1987. doi: 10.1038/330576a0. [DOI] [PubMed] [Google Scholar]
- 94.Donoho G, Jasin M, Berg P. Analysis of gene targeting and intrachromosomal homologous recombination stimulated by genomic double-strand breaks in mouse embryonic stem cells. Mol Cell Biol 18: 4070–4078, 1998. doi: 10.1128/MCB.18.7.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dreier B, Beerli RR, Segal DJ, Flippin JD, Barbas CF III. Development of zinc finger domains for recognition of the 5′-ANN-3′ family of DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem 276: 29466–29478, 2001. doi: 10.1074/jbc.M102604200. [DOI] [PubMed] [Google Scholar]
- 96.Dreier B, Fuller RP, Segal DJ, Lund CV, Blancafort P, Huber A, Koksch B, Barbas CF III. Development of zinc finger domains for recognition of the 5′-CNN-3′ family DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem 280: 35588–35597, 2005. doi: 10.1074/jbc.M506654200. [DOI] [PubMed] [Google Scholar]
- 97.Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, Hall MP, Shiue L, Swanson MS, Thornton CA, Ares M Jr. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol 17: 187–193, 2010. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Duan D, Sharma P, Yang J, Yue Y, Dudus L, Zhang Y, Fisher KJ, Engelhardt JF. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J Virol 72: 8568–8577, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Duan D, Yan Z, Yue Y, Ding W, Engelhardt JF. Enhancement of muscle gene delivery with pseudotyped adeno-associated virus type 5 correlates with myoblast differentiation. J Virol 75: 7662–7671, 2001. doi: 10.1128/JVI.75.16.7662-7671.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Duclos F, Straub V, Moore SA, Venzke DP, Hrstka RF, Crosbie RH, Durbeej M, Lebakken CS, Ettinger AJ, van der Meulen J, Holt KH, Lim LE, Sanes JR, Davidson BL, Faulkner JA, Williamson R, Campbell KP. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J Cell Biol 142: 1461–1471, 1998. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Durbeej M, Cohn RD, Hrstka RF, Moore SA, Allamand V, Davidson BL, Williamson RA, Campbell KP. Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol Cell 5: 141–151, 2000. doi: 10.1016/S1097-2765(00)80410-4. [DOI] [PubMed] [Google Scholar]
- 102.East-Seletsky A, O’Connell MR, Burstein D, Knott GJ, Doudna JA. RNA Targeting by Functionally Orthogonal Type VI-A CRISPR-Cas Enzymes. Mol Cell 66: 373–383.e3, 2017. doi: 10.1016/j.molcel.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JH, Tjian R, Doudna JA. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538: 270–273, 2016. doi: 10.1038/nature19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.El Refaey M, Xu L, Gao Y, Canan BD, Adesanya TMA, Warner SC, Akagi K, Symer DE, Mohler PJ, Ma J, Janssen PML, Han R. In Vivo Genome Editing Restores Dystrophin Expression and Cardiac Function in Dystrophic Mice. Circ Res 121: 923–929, 2017. doi: 10.1161/CIRCRESAHA.117.310996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ellison GM, Vicinanza C, Smith AJ, Aquila I, Leone A, Waring CD, Henning BJ, Stirparo GG, Papait R, Scarfò M, Agosti V, Viglietto G, Condorelli G, Indolfi C, Ottolenghi S, Torella D, Nadal-Ginard B. Adult c-kit(pos) cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell 154: 827–842, 2013. doi: 10.1016/j.cell.2013.07.039. [DOI] [PubMed] [Google Scholar]
- 106.Endo A, Masafumi M, Kaya H, Toki S. Efficient targeted mutagenesis of rice and tobacco genomes using Cpf1 from Francisella novicida. Sci Rep 6: 38169, 2016. doi: 10.1038/srep38169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ettinger AJ, Feng G, Sanes JR. epsilon-Sarcoglycan, a broadly expressed homologue of the gene mutated in limb-girdle muscular dystrophy 2D. J Biol Chem 272: 32534–32538, 1997. doi: 10.1074/jbc.272.51.32534. [DOI] [PubMed] [Google Scholar]
- 108.Ferrer A, Wells KE, Wells DJ. Immune responses to dystropin: implications for gene therapy of Duchenne muscular dystrophy. Gene Ther 7: 1439–1446, 2000. doi: 10.1038/sj.gt.3301259. [DOI] [PubMed] [Google Scholar]
- 109.Flanigan KM, Coffeen CM, Sexton L, Stauffer D, Brunner S, Leppert MF. Genetic characterization of a large, historically significant Utah kindred with facioscapulohumeral dystrophy. Neuromuscul Disord 11: 525–529, 2001. doi: 10.1016/S0960-8966(01)00201-2. [DOI] [PubMed] [Google Scholar]
- 110.Flotte TR, Berns KI. Adeno-associated virus: a ubiquitous commensal of mammals. Hum Gene Ther 16: 401–407, 2005. doi: 10.1089/hum.2005.16.401. [DOI] [PubMed] [Google Scholar]
- 111.Fonfara I, Richter H, Bratovič M, Le Rhun A, Charpentier E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532: 517–521, 2016. doi: 10.1038/nature17945. [DOI] [PubMed] [Google Scholar]
- 112.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31: 822–826, 2013. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32: 279–284, 2014. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fu YH, Pizzuti A, Fenwick RG Jr, King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, de Jong P, , et al. . An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 255: 1256–1258, 1992. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- 115.Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, Nowrouzi A, Bartholomae CC, Wang J, Friedman G, Holmes MC, Gregory PD, Glimm H, Schmidt M, Naldini L, von Kalle C. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 29: 816–823, 2011. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- 116.Gaj T, Gersbach CA, Barbas CF III. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31: 397–405, 2013. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gammage PA, Moraes CT, Minczuk M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet 34: 101–110, 2018. doi: 10.1016/j.tig.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gammage PA, Rorbach J, Vincent AI, Rebar EJ, Minczuk M. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol Med 6: 458–466, 2014. doi: 10.1002/emmm.201303672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci USA 99: 11854–11859, 2002. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gao QQ, McNally EM. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr Physiol 5: 1223–1239, 2015. doi: 10.1002/cphy.c140048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gao QQ, Wyatt E, Goldstein JA, LoPresti P, Castillo LM, Gazda A, Petrossian N, Earley JU, Hadhazy M, Barefield DY, Demonbreun AR, Bönnemann C, Wolf M, McNally EM. Reengineering a transmembrane protein to treat muscular dystrophy using exon skipping. J Clin Invest 125: 4186–4195, 2015. doi: 10.1172/JCI82768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gao Y, Guo X, Santostefano K, Wang Y, Reid T, Zeng D, Terada N, Ashizawa T, Xia G. Genome Therapy of Myotonic Dystrophy Type 1 iPS Cells for Development of Autologous Stem Cell Therapy. Mol Ther 24: 1378–1387, 2016. doi: 10.1038/mt.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR. Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage. Nature 551: 464–471, 2017. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Geng LN, Yao Z, Snider L, Fong AP, Cech JN, Young JM, van der Maarel SM, Ruzzo WL, Gentleman RC, Tawil R, Tapscott SJ. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell 22: 38–51, 2012. doi: 10.1016/j.devcel.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Genovese P, Schiroli G, Escobar G, Tomaso TD, Firrito C, Calabria A, Moi D, Mazzieri R, Bonini C, Holmes MC, Gregory PD, van der Burg M, Gentner B, Montini E, Lombardo A, Naldini L. Targeted genome editing in human repopulating haematopoietic stem cells. Nature 510: 235–240, 2014. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154: 442–451, 2013. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gomes-Pereira M, Cooper TA, Gourdon G. Myotonic dystrophy mouse models: towards rational therapy development. Trends Mol Med 17: 506–517, 2011. doi: 10.1016/j.molmed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gonzalez B, Schwimmer LJ, Fuller RP, Ye Y, Asawapornmongkol L, Barbas CF III. Modular system for the construction of zinc-finger libraries and proteins. Nat Protoc 5: 791–810, 2010. doi: 10.1038/nprot.2010.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, Myhrvold C, Bhattacharyya RP, Livny J, Regev A, Koonin EV, Hung DT, Sabeti PC, Collins JJ, Zhang F. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356: 438–442, 2017. doi: 10.1126/science.aam9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell 90: 729–738, 1997. doi: 10.1016/S0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 131.Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG, Russell DW, Chamberlain JS. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med 10: 828–834, 2004. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Groh WJ, Groh MR, Saha C, Kincaid JC, Simmons Z, Ciafaloni E, Pourmand R, Otten RF, Bhakta D, Nair GV, Marashdeh MM, Zipes DP, Pascuzzi RM. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med 358: 2688–2697, 2008. doi: 10.1056/NEJMoa062800. [DOI] [PubMed] [Google Scholar]
- 133.Grose WE, Clark KR, Griffin D, Malik V, Shontz KM, Montgomery CL, Lewis S, Brown RH Jr, Janssen PM, Mendell JR, Rodino-Klapac LR. Homologous recombination mediates functional recovery of dysferlin deficiency following AAV5 gene transfer. PLoS One 7: e39233, 2012. doi: 10.1371/journal.pone.0039233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol 32: 577–582, 2014. doi: 10.1038/nbt.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Guiraud S, Aartsma-Rus A, Vieira NM, Davies KE, van Ommen GJ, Kunkel LM. The Pathogenesis and Therapy of Muscular Dystrophies. Annu Rev Genomics Hum Genet 16: 281–308, 2015. doi: 10.1146/annurev-genom-090314-025003. [DOI] [PubMed] [Google Scholar]
- 136.Guiraud-Dogan C, Huguet A, Gomes-Pereira M, Brisson E, Bassez G, Junien C, Gourdon G. DM1 CTG expansions affect insulin receptor isoforms expression in various tissues of transgenic mice. Biochim Biophys Acta 1772: 1183–1191, 2007. doi: 10.1016/j.bbadis.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 137.Gundry MC, Brunetti L, Lin A, Mayle AE, Kitano A, Wagner D, Hsu JI, Hoegenauer KA, Rooney CM, Goodell MA, Nakada D. Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell Reports 17: 1453–1461, 2016. doi: 10.1016/j.celrep.2016.09.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hack AA, Ly CT, Jiang F, Clendenin CJ, Sigrist KS, Wollmann RL, McNally EM. Gamma-sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin. J Cell Biol 142: 1279–1287, 1998. doi: 10.1083/jcb.142.5.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Haft DH, Selengut J, Mongodin EF, Nelson KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLOS Comput Biol 1: e60, 2005. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139: 945–956, 2009. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hao M, Akrami K, Wei K, De Diego C, Che N, Ku JH, Tidball J, Graves MC, Shieh PB, Chen F. Muscleblind-like 2 (Mbnl2) -deficient mice as a model for myotonic dystrophy. Dev Dyn 237: 403–410, 2008. doi: 10.1002/dvdy.21428. [DOI] [PubMed] [Google Scholar]
- 142.Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33: 510–517, 2015. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Himeda CL, Chen X, Hauschka SD. Design and testing of regulatory cassettes for optimal activity in skeletal and cardiac muscles. Methods Mol Biol 709: 3–19, 2011. doi: 10.1007/978-1-61737-982-6_1. [DOI] [PubMed] [Google Scholar]
- 144.Himeda CL, Jones TI, Jones PL. CRISPR/dCas9-mediated Transcriptional Inhibition Ameliorates the Epigenetic Dysregulation at D4Z4 and Represses DUX4-fl in FSH Muscular Dystrophy. Mol Ther 24: 527–535, 2016. doi: 10.1038/mt.2015.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hirst RC, McCullagh KJ, Davies KE. Utrophin upregulation in Duchenne muscular dystrophy. Acta Myol 24: 209–216, 2005. [PubMed] [Google Scholar]
- 146.Hisano Y, Sakuma T, Nakade S, Ohga R, Ota S, Okamoto H, Yamamoto T, Kawahara A. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Sci Rep 5: 8841, 2015. doi: 10.1038/srep08841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ho M, Post CM, Donahue LR, Lidov HG, Bronson RT, Goolsby H, Watkins SC, Cox GA, Brown RH Jr. Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum Mol Genet 13: 1999–2010, 2004. doi: 10.1093/hmg/ddh212. [DOI] [PubMed] [Google Scholar]
- 148.Ho TH, Bundman D, Armstrong DL, Cooper TA. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum Mol Genet 14: 1539–1547, 2005. doi: 10.1093/hmg/ddi162. [DOI] [PubMed] [Google Scholar]
- 149.Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51: 919–928, 1987. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 150.Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, Thomson JA. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA 110: 15644–15649, 2013. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157: 1262–1278, 2014. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31: 827–832, 2013. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Huang Y, Laval SH, van Remoortere A, Baudier J, Benaud C, Anderson LV, Straub V, Deelder A, Frants RR, den Dunnen JT, Bushby K, van der Maarel SM. AHNAK, a novel component of the dysferlin protein complex, redistributes to the cytoplasm with dysferlin during skeletal muscle regeneration. FASEB J 21: 732–742, 2007. doi: 10.1096/fj.06-6628com. [DOI] [PubMed] [Google Scholar]
- 154.Huichalaf C, Sakai K, Jin B, Jones K, Wang GL, Schoser B, Schneider-Gold C, Sarkar P, Pereira-Smith OM, Timchenko N, Timchenko L. Expansion of CUG RNA repeats causes stress and inhibition of translation in myotonic dystrophy 1 (DM1) cells. FASEB J 24: 3706–3719, 2010. doi: 10.1096/fj.09-151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Huichalaf C, Schoser B, Schneider-Gold C, Jin B, Sarkar P, Timchenko L. Reduction of the rate of protein translation in patients with myotonic dystrophy 2. J Neurosci 29: 9042–9049, 2009. doi: 10.1523/JNEUROSCI.1983-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hur JK, Kim K, Been KW, Baek G, Ye S, Hur JW, Ryu SM, Lee YS, Kim JS. Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat Biotechnol 34: 807–808, 2016. doi: 10.1038/nbt.3596. [DOI] [PubMed] [Google Scholar]
- 157.Hurlbut GD, Ziegler RJ, Nietupski JB, Foley JW, Woodworth LA, Meyers E, Bercury SD, Pande NN, Souza DW, Bree MP, Lukason MJ, Marshall J, Cheng SH, Scheule RK. Preexisting immunity and low expression in primates highlight translational challenges for liver-directed AAV8-mediated gene therapy. Mol Ther 18: 1983–1994, 2010. doi: 10.1038/mt.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ikezoe K, Furuya H, Ohyagi Y, Osoegawa M, Nishino I, Nonaka I, Kira J. Dysferlin expression in tubular aggregates: their possible relationship to endoplasmic reticulum stress. Acta Neuropathol 105: 603–609, 2003. doi: 10.1007/s00401-003-0686-1. [DOI] [PubMed] [Google Scholar]
- 159.Inagaki K, Fuess S, Storm TA, Gibson GA, Mctiernan CF, Kay MA, Nakai H. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther 14: 45–53, 2006. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, Haber JE, Foiani M. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431: 1011–1017, 2004. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169: 5429–5433, 1987. doi: 10.1128/jb.169.12.5429-5433.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Iyombe-Engembe JP, Ouellet DL, Barbeau X, Rousseau J, Chapdelaine P, Lagüe P, Tremblay JP. Efficient Restoration of the Dystrophin Gene Reading Frame and Protein Structure in DMD Myoblasts Using the CinDel Method. Mol Ther Nucleic Acids 5: e283, 2016. doi: 10.1038/mtna.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jansen R, Embden JD, Gaastra W, Schouls LM. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43: 1565–1575, 2002. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 164.Jiang H, Couto LB, Patarroyo-White S, Liu T, Nagy D, Vargas JA, Zhou S, Scallan CD, Sommer J, Vijay S, Mingozzi F, High KA, Pierce GF. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood 108: 3321–3328, 2006. doi: 10.1182/blood-2006-04-017913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jiang W, Samai P, Marraffini LA. Degradation of Phage Transcripts by CRISPR-Associated RNases Enables Type III CRISPR-Cas Immunity. Cell 164: 710–721, 2016. doi: 10.1016/j.cell.2015.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jiang Y, Qian F, Yang J, Liu Y, Dong F, Xu C, Sun B, Chen B, Xu X, Li Y, Wang R, Yang S. CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat Commun 8: 15179, 2017. doi: 10.1038/ncomms15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jin J, Wang GL, Salisbury E, Timchenko L, Timchenko NA. GSK3beta-cyclin D3-CUGBP1-eIF2 pathway in aging and in myotonic dystrophy. Cell Cycle 8: 2356–2359, 2009. doi: 10.4161/cc.8.15.9248. [DOI] [PubMed] [Google Scholar]
- 168.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821, 2012. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jo A, Ham S, Lee GH, Lee YI, Kim S, Lee YS, Shin JH, Lee Y. Efficient Mitochondrial Genome Editing by CRISPR/Cas9. BioMed Res Int 2015: 305716, 2015. doi: 10.1155/2015/305716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14: 49–55, 2013. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kabadi AM, Ousterout DG, Hilton IB, Gersbach CA. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res 42: e147, 2014. doi: 10.1093/nar/gku749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci USA 105: 20333–20338, 2008. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. A muscleblind knockout model for myotonic dystrophy. Science 302: 1978–1980, 2003. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 174.Kanagawa M, Toda T. The genetic and molecular basis of muscular dystrophy: roles of cell-matrix linkage in the pathogenesis. J Hum Genet 51: 915–926, 2006. doi: 10.1007/s10038-006-0056-7. [DOI] [PubMed] [Google Scholar]
- 175.Kang E, Wu J, Gutierrez NM, Koski A, Tippner-Hedges R, Agaronyan K, Platero-Luengo A, Martinez-Redondo P, Ma H, Lee Y, Hayama T, Van Dyken C, Wang X, Luo S, Ahmed R, Li Y, Ji D, Kayali R, Cinnioglu C, Olson S, Jensen J, Battaglia D, Lee D, Wu D, Huang T, Wolf DP, Temiakov D, Belmonte JC, Amato P, Mitalipov S. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature 540: 270–275, 2016. doi: 10.1038/nature20592. [DOI] [PubMed] [Google Scholar]
- 176.Kang X, He W, Huang Y, Yu Q, Chen Y, Gao X, Sun X, Fan Y. Introducing precise genetic modifications into human 3PN embryos by CRISPR/Cas-mediated genome editing. J Assist Reprod Genet 33: 581–588, 2016. doi: 10.1007/s10815-016-0710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kaplan JC, Hamroun D, Rivier F, Bonne G. The 2017 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul Disord 26: 895–929, 2017. doi: 10.1016/j.nmd.2016.11.010. [DOI] [PubMed] [Google Scholar]
- 178.Kazlauskiene M, Tamulaitis G, Kostiuk G, Venclovas Č, Siksnys V. Spatiotemporal Control of Type III-A CRISPR-Cas Immunity: Coupling DNA Degradation with the Target RNA Recognition. Mol Cell 62: 295–306, 2016. doi: 10.1016/j.molcel.2016.03.024. [DOI] [PubMed] [Google Scholar]
- 179.Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, Maehr R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods 12: 401–403, 2015. doi: 10.1038/nmeth.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kemaladewi DU, Maino E, Hyatt E, Hou H, Ding M, Place KM, Zhu X, Bassi P, Baghestani Z, Deshwar AG, Merico D, Xiong HY, Frey BJ, Wilson MD, Ivakine EA, Cohn RD. Correction of a splicing defect in a mouse model of congenital muscular dystrophy type 1A using a homology-directed-repair-independent mechanism. Nat Med 23: 984–989, 2017. doi: 10.1038/nm.4367. [DOI] [PubMed] [Google Scholar]
- 181.Kim EY, Page P, Dellefave-Castillo LM, McNally EM, Wyatt EJ. Direct reprogramming of urine-derived cells with inducible MyoD for modeling human muscle disease. Skelet Muscle 6: 32, 2016. doi: 10.1186/s13395-016-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet 15: 321–334, 2014. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- 183.Kim S, Lee MJ, Kim H, Kang M, Kim JS. Preassembled zinc-finger arrays for rapid construction of ZFNs. Nat Methods 8: 7, 2011. doi: 10.1038/nmeth0111-7a. [DOI] [PubMed] [Google Scholar]
- 184.Kim SK, Kim H, Ahn WC, Park KH, Woo EJ, Lee DH, Lee SG. Efficient Transcriptional Gene Repression by Type V-A CRISPR-Cpf1 from Eubacterium eligens. ACS Synth Biol 6: 1273–1282, 2017. doi: 10.1021/acssynbio.6b00368. [DOI] [PubMed] [Google Scholar]
- 185.Kim Y, Cheong SA, Lee JG, Lee SW, Lee MS, Baek IJ, Sung YH. Generation of knockout mice by Cpf1-mediated gene targeting. Nat Biotechnol 34: 808–810, 2016. doi: 10.1038/nbt.3614. [DOI] [PubMed] [Google Scholar]
- 186.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA 93: 1156–1160, 1996. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kim YG, Chandrasegaran S. Chimeric restriction endonuclease. Proc Natl Acad Sci USA 91: 883–887, 1994. doi: 10.1073/pnas.91.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kim YG, Li L, Chandrasegaran S. Insertion and deletion mutants of FokI restriction endonuclease. J Biol Chem 269: 31978–31982, 1994. [PubMed] [Google Scholar]
- 189.Kim YG, Smith J, Durgesha M, Chandrasegaran S. Chimeric restriction enzyme: Gal4 fusion to FokI cleavage domain. Biol Chem 379: 489–495, 1998. doi: 10.1515/bchm.1998.379.4-5.489. [DOI] [PubMed] [Google Scholar]
- 190.Kimura Y, Hisano Y, Kawahara A, Higashijima S. Efficient generation of knock-in transgenic zebrafish carrying reporter/driver genes by CRISPR/Cas9-mediated genome engineering. Sci Rep 4: 6545, 2014. doi: 10.1038/srep06545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529: 490–495, 2016. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Klymiuk N, Blutke A, Graf A, Krause S, Burkhardt K, Wuensch A, Krebs S, Kessler B, Zakhartchenko V, Kurome M, Kemter E, Nagashima H, Schoser B, Herbach N, Blum H, Wanke R, Aartsma-Rus A, Thirion C, Lochmüller H, Walter MC, Wolf E. Dystrophin-deficient pigs provide new insights into the hierarchy of physiological derangements of dystrophic muscle. Hum Mol Genet 22: 4368–4382, 2013. doi: 10.1093/hmg/ddt287. [DOI] [PubMed] [Google Scholar]
- 193.Komor AC, Badran AH, Liu DR. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 168: 20–36, 2017. doi: 10.1016/j.cell.2016.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533: 420–424, 2016. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Konermann S, Brigham MD, Trevino A, Hsu PD, Heidenreich M, Cong L, Platt RJ, Scott DA, Church GM, Zhang F. Optical control of mammalian endogenous transcription and epigenetic states. Nature 500: 472–476, 2013. doi: 10.1038/nature12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517: 583–588, 2015. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kornegay JN, Bogan JR, Bogan DJ, Childers MK, Li J, Nghiem P, Detwiler DA, Larsen CA, Grange RW, Bhavaraju-Sanka RK, Tou S, Keene BP, Howard JF Jr, Wang J, Fan Z, Schatzberg SJ, Styner MA, Flanigan KM, Xiao X, Hoffman EP. Canine models of Duchenne muscular dystrophy and their use in therapeutic strategies. Mamm Genome 23: 85–108, 2012. doi: 10.1007/s00335-011-9382-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Koshelev M, Sarma S, Price RE, Wehrens XH, Cooper TA. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum Mol Genet 19: 1066–1075, 2010. doi: 10.1093/hmg/ddp570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet 15: 445–451, 2014. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Krahn M, Béroud C, Labelle V, Nguyen K, Bernard R, Bassez G, Figarella-Branger D, Fernandez C, Bouvenot J, Richard I, Ollagnon-Roman E, Bevilacqua JA, Salvo E, Attarian S, Chapon F, Pellissier JF, Pouget J, Hammouda H, Laforêt P, Urtizberea JA, Eymard B, Leturcq F, Lévy N. Analysis of the DYSF mutational spectrum in a large cohort of patients. Hum Mutat 30: E345–E375, 2009. doi: 10.1002/humu.20910. [DOI] [PubMed] [Google Scholar]
- 201.Krom YD, Thijssen PE, Young JM, den Hamer B, Balog J, Yao Z, Maves L, Snider L, Knopp P, Zammit PS, Rijkers T, van Engelen BG, Padberg GW, Frants RR, Tawil R, Tapscott SJ, van der Maarel SM. Intrinsic epigenetic regulation of the D4Z4 macrosatellite repeat in a transgenic mouse model for FSHD. PLoS Genet 9: e1003415, 2013. doi: 10.1371/journal.pgen.1003415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 129: 999–1010, 2007. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kuscu C, Parlak M, Tufan T, Yang J, Szlachta K, Wei X, Mammadov R, Adli M. CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations. Nat Methods 14: 710–712, 2017. doi: 10.1038/nmeth.4327. [DOI] [PubMed] [Google Scholar]
- 204.Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell 28: 68–78, 2007. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kyrychenko V, Kyrychenko S, Tiburcy M, Shelton JM, Long C, Schneider JW, Zimmermann WH, Bassel-Duby R, Olson EN. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2: e95918, 2017. doi: 10.1172/jci.insight.95918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44, D1: D862–D868, 2016. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res 94: 1023–1031, 2004. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- 208.Larcher T, Lafoux A, Tesson L, Remy S, Thepenier V, François V, Le Guiner C, Goubin H, Dutilleul M, Guigand L, Toumaniantz G, De Cian A, Boix C, Renaud JB, Cherel Y, Giovannangeli C, Concordet JP, Anegon I, Huchet C. Characterization of dystrophin deficient rats: a new model for Duchenne muscular dystrophy. PLoS One 9: e110371, 2014. doi: 10.1371/journal.pone.0110371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lattanzi A, Duguez S, Moiani A, Izmiryan A, Barbon E, Martin S, Mamchaoui K, Mouly V, Bernardi F, Mavilio F, Bovolenta M. Correction of the Exon 2 Duplication in DMD Myoblasts by a Single CRISPR/Cas9 System. Mol Ther Nucleic Acids 7: 11–19, 2017. doi: 10.1016/j.omtn.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Leidenroth A, Hewitt JE. A family history of DUX4: phylogenetic analysis of DUXA, B, C and Duxbl reveals the ancestral DUX gene. BMC Evol Biol 10: 364, 2010. doi: 10.1186/1471-2148-10-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lek A, Rahimov F, Jones PL, Kunkel LM. Emerging preclinical animal models for FSHD. Trends Mol Med 21: 295–306, 2015. doi: 10.1016/j.molmed.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lemmers RJ, Tawil R, Petek LM, Balog J, Block GJ, Santen GW, Amell AM, van der Vliet PJ, Almomani R, Straasheijm KR, Krom YD, Klooster R, Sun Y, den Dunnen JT, Helmer Q, Donlin-Smith CM, Padberg GW, van Engelen BG, de Greef JC, Aartsma-Rus AM, Frants RR, de Visser M, Desnuelle C, Sacconi S, Filippova GN, Bakker B, Bamshad MJ, Tapscott SJ, Miller DG, van der Maarel SM. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet 44: 1370–1374, 2012. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, Miller DG, Tapscott SJ, Tawil R, Frants RR, van der Maarel SM. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 329: 1650–1653, 2010. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lennon NJ, Kho A, Bacskai BJ, Perlmutter SL, Hyman BT, Brown RH Jr. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J Biol Chem 278: 50466–50473, 2003. doi: 10.1074/jbc.M307247200. [DOI] [PubMed] [Google Scholar]
- 215.Lepper C, Partridge TA, Fan CM. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 138: 3639–3646, 2011. doi: 10.1242/dev.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Li B, Zhao W, Luo X, Zhang X, Li C, Zeng C, Dong Y. Engineering CRISPR-Cpf1 crRNAs and mRNAs to maximize genome editing efficiency. Nat Biomed Eng 1: 0066, 2017. doi: 10.1038/s41551-017-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T, Tanaka M, Amano N, Watanabe A, Sakurai H, Yamamoto T, Yamanaka S, Hotta A. Precise correction of the dystrophin gene in Duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep 4: 143–154, 2015. doi: 10.1016/j.stemcr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Li L, Chandrasegaran S. Alteration of the cleavage distance of Fok I restriction endonuclease by insertion mutagenesis. Proc Natl Acad Sci USA 90: 2764–2768, 1993. doi: 10.1073/pnas.90.7.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Li L, Wu LP, Chandrasegaran S. Functional domains in Fok I restriction endonuclease. Proc Natl Acad Sci USA 89: 4275–4279, 1992. doi: 10.1073/pnas.89.10.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Li L, Wu LP, Clarke R, Chandrasegaran S. C-terminal deletion mutants of the FokI restriction endonuclease. Gene 133: 79–84, 1993. doi: 10.1016/0378-1119(93)90227-T. [DOI] [PubMed] [Google Scholar]
- 221.Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z, Lv J, Xie X, Chen Y, Li Y, Sun Y, Bai Y, Songyang Z, Ma W, Zhou C, Huang J. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 6: 363–372, 2015. doi: 10.1007/s13238-015-0153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lin SR, Yang HC, Kuo YT, Liu CJ, Yang TY, Sung KC, Lin YY, Wang HY, Wang CC, Shen YC, Wu FY, Kao JH, Chen DS, Chen PJ. The CRISPR/Cas9 System Facilitates Clearance of the Intrahepatic HBV Templates In Vivo. Mol Ther Nucleic Acids 3: e186, 2014. doi: 10.1038/mtna.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet 15: 2087–2097, 2006. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 224.Lin Y, Cradick TJ, Brown MT, Deshmukh H, Ranjan P, Sarode N, Wile BM, Vertino PM, Stewart FJ, Bao G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res 42: 7473–7485, 2014. doi: 10.1093/nar/gku402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293: 864–867, 2001. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 226.Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, Serrano C, Urtizberea JA, Hentati F, Hamida MB, Bohlega S, Culper EJ, Amato AA, Bossie K, Oeltjen J, Bejaoui K, McKenna-Yasek D, Hosler BA, Schurr E, Arahata K, de Jong PJ, Brown RH Jr. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 20: 31–36, 1998. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- 227.Liu N, Garry GA, Li S, Bezprozvannaya S, Sanchez-Ortiz E, Chen B, Shelton JM, Jaichander P, Bassel-Duby R, Olson EN. A Twist2-dependent progenitor cell contributes to adult skeletal muscle. Nat Cell Biol 19: 202–213, 2017. doi: 10.1038/ncb3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, Jaenisch R. Editing DNA Methylation in the Mammalian Genome. Cell 167: 233–247.e17, 2016. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Logigian EL, Blood CL, Dilek N, Martens WB, Moxley RT IV, Wiegner AW, Thornton CA, Moxley RT III. Quantitative analysis of the “warm-up” phenomenon in myotonic dystrophy type 1. Muscle Nerve 32: 35–42, 2005. doi: 10.1002/mus.20339. [DOI] [PubMed] [Google Scholar]
- 230.Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351: 400–403, 2016. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 345: 1184–1188, 2014. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Lostal W, Bartoli M, Bourg N, Roudaut C, Bentaïb A, Miyake K, Guerchet N, Fougerousse F, McNeil P, Richard I. Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transfer. Hum Mol Genet 19: 1897–1907, 2010. doi: 10.1093/hmg/ddq065. [DOI] [PubMed] [Google Scholar]
- 233.Lueck JD, Mankodi A, Swanson MS, Thornton CA, Dirksen RT. Muscle chloride channel dysfunction in two mouse models of myotonic dystrophy. J Gen Physiol 129: 79–94, 2007. doi: 10.1085/jgp.200609635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ma H, Marti-Gutierrez N, Park SW, Wu J, Lee Y, Suzuki K, Koski A, Ji D, Hayama T, Ahmed R, Darby H, Van Dyken C, Li Y, Kang E, Park AR, Kim D, Kim ST, Gong J, Gu Y, Xu X, Battaglia D, Krieg SA, Lee DM, Wu DH, Wolf DP, Heitner SB, Belmonte JCI, Amato P, Kim JS, Kaul S, Mitalipov S. Correction of a pathogenic gene mutation in human embryos. Nature 548: 413–419, 2017. doi: 10.1038/nature23305. [DOI] [PubMed] [Google Scholar]
- 235.Ma S, Liu Y, Liu Y, Chang J, Zhang T, Wang X, Shi R, Lu W, Xia X, Zhao P, Xia Q. An integrated CRISPR Bombyx mori genome editing system with improved efficiency and expanded target sites. Insect Biochem Mol Biol 83: 13–20, 2017. doi: 10.1016/j.ibmb.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 236.Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. CRISPR RNA-guided activation of endogenous human genes. Nat Methods 10: 977–979, 2013. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, Unger-Wallace E, Sander JD, Müller-Lerch F, Fu F, Pearlberg J, Göbel C, Dassie JP, Pruett-Miller SM, Porteus MH, Sgroi DC, Iafrate AJ, Dobbs D, McCray PB Jr, Cathomen T, Voytas DF, Joung JK. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell 31: 294–301, 2008. doi: 10.1016/j.molcel.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Magadán AH, Dupuis ME, Villion M, Moineau S. Cleavage of phage DNA by the Streptococcus thermophilus CRISPR3-Cas system. PLoS One 7: e40913, 2012. doi: 10.1371/journal.pone.0040913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Maggio I, Liu J, Janssen JM, Chen X, Gonçalves MA. Adenoviral vectors encoding CRISPR/Cas9 multiplexes rescue dystrophin synthesis in unselected populations of DMD muscle cells. Sci Rep 6: 37051, 2016. doi: 10.1038/srep37051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Maggio I, Stefanucci L, Janssen JM, Liu J, Chen X, Mouly V, Gonçalves MA. Selection-free gene repair after adenoviral vector transduction of designer nucleases: rescue of dystrophin synthesis in DMD muscle cell populations. Nucleic Acids Res 44: 1449–1470, 2016. doi: 10.1093/nar/gkv1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barceló J, O’Hoy K, et. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science 255: 1253–1255, 1992. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 242.Mak AN, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 335: 716–719, 2012. doi: 10.1126/science.1216211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Makarova KS, Aravind L, Wolf YI, Koonin EV. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol Direct 6: 38, 2011. doi: 10.1186/1745-6150-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1: 7, 2006. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9: 467–477, 2011. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJ, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, van der Oost J, Backofen R, Koonin EV. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13: 722–736, 2015. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31: 833–838, 2013. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science 339: 823–826, 2013. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289: 1769–1772, 2000. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 250.Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature 336: 348–352, 1988. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 251.Maresca M, Lin VG, Guo N, Yang Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res 23: 539–546, 2013. doi: 10.1101/gr.145441.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Marraffini LA. CRISPR-Cas immunity in prokaryotes. Nature 526: 55–61, 2015. doi: 10.1038/nature15386. [DOI] [PubMed] [Google Scholar]
- 253.Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322: 1843–1845, 2008. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Martins-Taylor K, Xu RH. Concise review: genomic stability of human induced pluripotent stem cells. Stem Cells 30: 22–27, 2012. doi: 10.1002/stem.705. [DOI] [PubMed] [Google Scholar]
- 255.Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol 33: 538–542, 2015. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Matsuda C, Hayashi YK, Ogawa M, Aoki M, Murayama K, Nishino I, Nonaka I, Arahata K, Brown RH Jr. The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle. Hum Mol Genet 10: 1761–1766, 2001. doi: 10.1093/hmg/10.17.1761. [DOI] [PubMed] [Google Scholar]
- 257.Matynia A, Ng CH, Dansithong W, Chiang A, Silva AJ, Reddy S. Muscleblind1, but not Dmpk or Six5, contributes to a complex phenotype of muscular and motivational deficits in mouse models of myotonic dystrophy. PLoS One 5: e9857, 2010. doi: 10.1371/journal.pone.0009857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.McCarthy JJ, Mula J, Miyazaki M, Erfani R, Garrison K, Farooqui AB, Srikuea R, Lawson BA, Grimes B, Keller C, Van Zant G, Campbell KS, Esser KA, Dupont-Versteegden EE, Peterson CA. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development 138: 3657–3666, 2011. doi: 10.1242/dev.068858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.McGreevy JW, Hakim CH, McIntosh MA, Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech 8: 195–213, 2015. doi: 10.1242/dmm.018424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.McNally EM, Ly CT, Kunkel LM. Human epsilon-sarcoglycan is highly related to alpha-sarcoglycan (adhalin), the limb girdle muscular dystrophy 2D gene. FEBS Lett 422: 27–32, 1998. doi: 10.1016/S0014-5793(97)01593-7. [DOI] [PubMed] [Google Scholar]
- 261.McNally EM, Pytel P. Muscle diseases: the muscular dystrophies. Annu Rev Pathol 2: 87–109, 2007. doi: 10.1146/annurev.pathol.2.010506.091936. [DOI] [PubMed] [Google Scholar]
- 262.Meca-Cortés O, Guerra-Rebollo M, Garrido C, Borrós S, Rubio N, Blanco J. CRISPR/Cas9-Mediated Knockin Application in Cell Therapy: A Non-viral Procedure for Bystander Treatment of Glioma in Mice. Mol Ther Nucleic Acids 8: 395–403, 2017. doi: 10.1016/j.omtn.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Meola G, Cardani R. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta 1852: 594–606, 2015. doi: 10.1016/j.bbadis.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 264.Mercuri E, Muntoni F. Muscular dystrophies. Lancet 381: 845–860, 2013. doi: 10.1016/S0140-6736(12)61897-2. [DOI] [PubMed] [Google Scholar]
- 265.Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, Beausejour CM, Waite AJ, Wang NS, Kim KA, Gregory PD, Pabo CO, Rebar EJ. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25: 778–785, 2007. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- 266.Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29: 143–148, 2011. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 267.Mingozzi F, Chen Y, Edmonson SC, Zhou S, Thurlings RM, Tak PP, High KA, Vervoordeldonk MJ. Prevalence and pharmacological modulation of humoral immunity to AAV vectors in gene transfer to synovial tissue. Gene Ther 20: 417–424, 2013. doi: 10.1038/gt.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Minnerop M, Weber B, Schoene-Bake JC, Roeske S, Mirbach S, Anspach C, Schneider-Gold C, Betz RC, Helmstaedter C, Tittgemeyer M, Klockgether T, Kornblum C. The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain 134: 3530–3546, 2011. doi: 10.1093/brain/awr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Mojica FJ, Díez-Villaseñor C, García-Martínez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60: 174–182, 2005. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- 270.Mojica FJ, Díez-Villaseñor C, Soria E, Juez G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol Microbiol 36: 244–246, 2000. doi: 10.1046/j.1365-2958.2000.01838.x. [DOI] [PubMed] [Google Scholar]
- 271.Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 2: 90–95, 1988. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- 272.Monteilhet V, Saheb S, Boutin S, Leborgne C, Veron P, Montus MF, Moullier P, Benveniste O, Masurier C. A 10 patient case report on the impact of plasmapheresis upon neutralizing factors against adeno-associated virus (AAV) types 1, 2, 6, and 8. Mol Ther 19: 2084–2091, 2011. doi: 10.1038/mt.2011.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Monteys AM, Ebanks SA, Keiser MS, Davidson BL. CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo. Mol Ther 25: 12–23, 2017. doi: 10.1016/j.ymthe.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Morita S, Noguchi H, Horii T, Nakabayashi K, Kimura M, Okamura K, Sakai A, Nakashima H, Hata K, Nakashima K, Hatada I. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat Biotechnol 34: 1060–1065, 2016. doi: 10.1038/nbt.3658. [DOI] [PubMed] [Google Scholar]
- 275.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science 326: 1501, 2009. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 276.Mostacciuolo ML, Pastorello E, Vazza G, Miorin M, Angelini C, Tomelleri G, Galluzzi G, Trevisan CP. Facioscapulohumeral muscular dystrophy: epidemiological and molecular study in a north-east Italian population sample. Clin Genet 75: 550–555, 2009. doi: 10.1111/j.1399-0004.2009.01158.x. [DOI] [PubMed] [Google Scholar]
- 277.Mou H, Smith JL, Peng L, Yin H, Moore J, Zhang XO, Song CQ, Sheel A, Wu Q, Ozata DM, Li Y, Anderson DG, Emerson CP, Sontheimer EJ, Moore MJ, Weng Z, Xue W. CRISPR/Cas9-mediated genome editing induces exon skipping by alternative splicing or exon deletion. Genome Biol 18: 108, 2017. doi: 10.1186/s13059-017-1237-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Mourkioti F, Kustan J, Kraft P, Day JW, Zhao MM, Kost-Alimova M, Protopopov A, DePinho RA, Bernstein D, Meeker AK, Blau HM. Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nat Cell Biol 15: 895–904, 2013. doi: 10.1038/ncb2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Müller M, Lee CM, Gasiunas G, Davis TH, Cradick TJ, Siksnys V, Bao G, Cathomen T, Mussolino C. Streptococcus thermophilus CRISPR-Cas9 Systems Enable Specific Editing of the Human Genome. Mol Ther 24: 636–644, 2016. doi: 10.1038/mt.2015.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2: 731–740, 2003. doi: 10.1016/S1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- 281.Murovec J, Pirc Ž, Yang B. New variants of CRISPR RNA-guided genome editing enzymes. Plant Biotechnol J 15: 917–926, 2017. doi: 10.1111/pbi.12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Murphy AP, Straub V. The Classification, Natural History and Treatment of the Limb Girdle Muscular Dystrophies. J Neuromuscul Dis 2, s2: S7–S19, 2015. doi: 10.3233/JND-150105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 138: 3625–3637, 2011. doi: 10.1242/dev.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, Rubart M, Pasumarthi KB, Virag JI, Bartelmez SH, Poppa V, Bradford G, Dowell JD, Williams DA, Field LJ. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature 428: 664–668, 2004. doi: 10.1038/nature02446. [DOI] [PubMed] [Google Scholar]
- 285.Nakade S, Tsubota T, Sakane Y, Kume S, Sakamoto N, Obara M, Daimon T, Sezutsu H, Yamamoto T, Sakuma T, Suzuki KT. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat Commun 5: 5560, 2014. doi: 10.1038/ncomms6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Nakamura K, Fujii W, Tsuboi M, Tanihata J, Teramoto N, Takeuchi S, Naito K, Yamanouchi K, Nishihara M. Generation of muscular dystrophy model rats with a CRISPR/Cas system. Sci Rep 4: 5635, 2014. doi: 10.1038/srep05635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Nance ME, Duan D. Perspective on Adeno-Associated Virus Capsid Modification for Duchenne Muscular Dystrophy Gene Therapy. Hum Gene Ther 26: 786–800, 2015. doi: 10.1089/hum.2015.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Nayerossadat N, Maedeh T, Ali PA. Viral and nonviral delivery systems for gene delivery. Adv Biomed Res 1: 27, 2012. doi: 10.4103/2277-9175.98152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, Madhavan S, Pan X, Ran FA, Yan WX, Asokan A, Zhang F, Duan D, Gersbach CA. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351: 403–407, 2016. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Nicholson LV, Davison K, Johnson MA, Slater CR, Young C, Bhattacharya S, Gardner-Medwin D, Harris JB. Dystrophin in skeletal muscle. II. Immunoreactivity in patients with Xp21 muscular dystrophy. J Neurol Sci 94: 137–146, 1989. doi: 10.1016/0022-510X(89)90224-4. [DOI] [PubMed] [Google Scholar]
- 291.Nicholson LV, Johnson MA, Bushby KM, Gardner-Medwin D, Curtis A, Ginjaar IB, den Dunnen JT, Welch JL, Butler TJ, Bakker E. Integrated study of 100 patients with Xp21 linked muscular dystrophy using clinical, genetic, immunochemical, and histopathological data. Part 1. Trends across the clinical groups. J Med Genet 30: 728–736, 1993. doi: 10.1136/jmg.30.9.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Nigro V, Aurino S, Piluso G. Limb girdle muscular dystrophies: update on genetic diagnosis and therapeutic approaches. Curr Opin Neurol 24: 429–436, 2011. doi: 10.1097/WCO.0b013e32834aa38d. [DOI] [PubMed] [Google Scholar]
- 293.Nigro V, Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol 33: 1–12, 2014. [PMC free article] [PubMed] [Google Scholar]
- 294.Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, Mochizuki M, Miyabe A, Araki M, Hara KY, Shimatani Z, Kondo A. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353: aaf8729, 2016. doi: 10.1126/science.aaf8729. [DOI] [PubMed] [Google Scholar]
- 295.Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156: 935–949, 2014. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, Kang Y, Zhao X, Si W, Li W, Xiang AP, Zhou J, Guo X, Bi Y, Si C, Hu B, Dong G, Wang H, Zhou Z, Li T, Tan T, Pu X, Wang F, Ji S, Zhou Q, Huang X, Ji W, Sha J. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156: 836–843, 2014. doi: 10.1016/j.cell.2014.01.027. [DOI] [PubMed] [Google Scholar]
- 297.Nonnenmacher M, Weber T. Intracellular transport of recombinant adeno-associated virus vectors. Gene Ther 19: 649–658, 2012. doi: 10.1038/gt.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain 132: 3175–3186, 2009. doi: 10.1093/brain/awp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.O’Brien KF, Kunkel LM. Dystrophin and muscular dystrophy: past, present, and future. Mol Genet Metab 74: 75–88, 2001. doi: 10.1006/mgme.2001.3220. [DOI] [PubMed] [Google Scholar]
- 300.Orengo JP, Chambon P, Metzger D, Mosier DR, Snipes GJ, Cooper TA. Expanded CTG repeats within the DMPK 3′ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc Natl Acad Sci USA 105: 2646–2651, 2008. doi: 10.1073/pnas.0708519105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal-Ginard B, Bodine DM, Leri A, Anversa P. Bone marrow cells regenerate infarcted myocardium. Nature 410: 701–705, 2001. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 302.Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, Gersbach CA. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun 6: 6244, 2015. doi: 10.1038/ncomms7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Ousterout DG, Kabadi AM, Thakore PI, Perez-Pinera P, Brown MT, Majoros WH, Reddy TE, Gersbach CA. Correction of dystrophin expression in cells from Duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol Ther 23: 523–532, 2015. doi: 10.1038/mt.2014.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Ousterout DG, Perez-Pinera P, Thakore PI, Kabadi AM, Brown MT, Qin X, Fedrigo O, Mouly V, Tremblay JP, Gersbach CA. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol Ther 21: 1718–1726, 2013. doi: 10.1038/mt.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Padberg GW, Frants RR, Brouwer OF, Wijmenga C, Bakker E, Sandkuijl LA. Facioscapulohumeral muscular dystrophy in the Dutch population. Muscle Nerve Suppl 18, S13: S81–S84, 1995. doi: 10.1002/mus.880181315. [DOI] [PubMed] [Google Scholar]
- 306.Pan X, Yue Y, Zhang K, Hakim CH, Kodippili K, McDonald T, Duan D. AAV-8 is more efficient than AAV-9 in transducing neonatal dog heart. Hum Gene Ther Methods 26: 54–61, 2015. doi: 10.1089/hgtb.2014.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451: 141–146, 2008. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 308.Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31: 839–843, 2013. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods 8: 765–770, 2011. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Pavletich NP, Pabo CO. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science 252: 809–817, 1991. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- 311.Pegoraro E, Hoffman EP. Limb-Girdle Muscular Dystrophy Overview. In: GeneReviews, edited by Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K. Seattle, WA: Univ. of Washington, Seattle, 1993. [PubMed] [Google Scholar]
- 312.Perbellini R, Greco S, Sarra-Ferraris G, Cardani R, Capogrossi MC, Meola G, Martelli F. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul Disord 21: 81–88, 2011. doi: 10.1016/j.nmd.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 313.Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Thakore PI, Glass KA, Ousterout DG, Leong KW, Guilak F, Crawford GE, Reddy TE, Gersbach CA. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods 10: 973–976, 2013. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Perez-Pinera P, Ousterout DG, Gersbach CA. Advances in targeted genome editing. Curr Opin Chem Biol 16: 268–277, 2012. doi: 10.1016/j.cbpa.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Pingoud A, Wilson GG, Wende W. Type II restriction endonucleases–a historical perspective and more. Nucleic Acids Res 42: 7489–7527, 2014. doi: 10.1093/nar/gku447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Popplewell L, Koo T, Leclerc X, Duclert A, Mamchaoui K, Gouble A, Mouly V, Voit T, Pâques F, Cédrone F, Isman O, Yáñez-Muñoz RJ, Dickson G. Gene correction of a duchenne muscular dystrophy mutation by meganuclease-enhanced exon knock-in. Hum Gene Ther 24: 692–701, 2013. doi: 10.1089/hum.2013.081. [DOI] [PubMed] [Google Scholar]
- 317.Port F, Bullock SL. Augmenting CRISPR applications in Drosophila with tRNA-flanked sgRNAs. Nat Methods 13: 852–854, 2016. doi: 10.1038/nmeth.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Porteus MH. Towards a new era in medicine: therapeutic genome editing. Genome Biol 16: 286, 2015. doi: 10.1186/s13059-015-0859-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151: 653–663, 2005. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- 320.Prakash V, Moore M, Yáñez-Muñoz RJ. Current Progress in Therapeutic Gene Editing for Monogenic Diseases. Mol Ther 24: 465–474, 2016. doi: 10.1038/mt.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Puchta H, Dujon B, Hohn B. Two different but related mechanisms are used in plants for the repair of genomic double-strand breaks by homologous recombination. Proc Natl Acad Sci USA 93: 5055–5060, 1996. doi: 10.1073/pnas.93.10.5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520: 186–191, 2015. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380–1389, 2013. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol 16: 819–824, 2009. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- 325.Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, Wahbi K, Day JW, Fujimura H, Takahashi MP, Auboeuf D, Dreumont N, Furling D, Charlet-Berguerand N. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol 18: 840–845, 2011. doi: 10.1038/nsmb.2067. [DOI] [PubMed] [Google Scholar]
- 326.Reddy P, Ocampo A, Suzuki K, Luo J, Bacman SR, Williams SL, Sugawara A, Okamura D, Tsunekawa Y, Wu J, Lam D, Xiong X, Montserrat N, Esteban CR, Liu GH, Sancho-Martinez I, Manau D, Civico S, Cardellach F, Del Mar O’Callaghan M, Campistol J, Zhao H, Campistol JM, Moraes CT, Izpisua Belmonte JC. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 161: 459–469, 2015. doi: 10.1016/j.cell.2015.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30: 460–465, 2012. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Rhodes JD, Lott MC, Russell SL, Moulton V, Sanderson J, Wormstone IM, Broadway DC. Activation of the innate immune response and interferon signalling in myotonic dystrophy type 1 and type 2 cataracts. Hum Mol Genet 21: 852–862, 2012. doi: 10.1093/hmg/ddr515. [DOI] [PubMed] [Google Scholar]
- 329.Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol 34: 339–344, 2016. doi: 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- 330.Rönnblom A, Forsberg H, Danielsson A. Gastrointestinal symptoms in myotonic dystrophy. Scand J Gastroenterol 31: 654–657, 1996. doi: 10.3109/00365529609009145. [DOI] [PubMed] [Google Scholar]
- 331.Rouet P, Smih F, Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol 14: 8096–8106, 1994. doi: 10.1128/MCB.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Sacco A, Mourkioti F, Tran R, Choi J, Llewellyn M, Kraft P, Shkreli M, Delp S, Pomerantz JH, Artandi SE, Blau HM. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 143: 1059–1071, 2010. doi: 10.1016/j.cell.2010.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Sacconi S, Salviati L, Desnuelle C. Facioscapulohumeral muscular dystrophy. Biochim Biophys Acta 1852: 607–614, 2015. doi: 10.1016/j.bbadis.2014.05.021. [DOI] [PubMed] [Google Scholar]
- 334.Saera-Vila A, Kasprick DS, Junttila TL, Grzegorski SJ, Louie KW, Chiari EF, Kish PE, Kahana A. Myocyte Dedifferentiation Drives Extraocular Muscle Regeneration in Adult Zebrafish. Invest Ophthalmol Vis Sci 56: 4977–4993, 2015. doi: 10.1167/iovs.14-16103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Sakuma T, Nakade S, Sakane Y, Suzuki KT, Yamamoto T. MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat Protoc 11: 118–133, 2016. doi: 10.1038/nprot.2015.140. [DOI] [PubMed] [Google Scholar]
- 336.Sakuma T, Nishikawa A, Kume S, Chayama K, Yamamoto T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci Rep 4: 5400, 2014. doi: 10.1038/srep05400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Salisbury E, Sakai K, Schoser B, Huichalaf C, Schneider-Gold C, Nguyen H, Wang GL, Albrecht JH, Timchenko LT. Ectopic expression of cyclin D3 corrects differentiation of DM1 myoblasts through activation of RNA CUG-binding protein, CUGBP1. Exp Cell Res 314: 2266–2278, 2008. doi: 10.1016/j.yexcr.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Salisbury E, Schoser B, Schneider-Gold C, Wang GL, Huichalaf C, Jin B, Sirito M, Sarkar P, Krahe R, Timchenko NA, Timchenko LT. Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. Am J Pathol 175: 748–762, 2009. doi: 10.2353/ajpath.2009.090047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Samai P, Pyenson N, Jiang W, Goldberg GW, Hatoum-Aslan A, Marraffini LA. Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell 161: 1164–1174, 2015. doi: 10.1016/j.cell.2015.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Sambasivan R, Yao R, Kissenpfennig A, Van Wittenberghe L, Paldi A, Gayraud-Morel B, Guenou H, Malissen B, Tajbakhsh S, Galy A. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 138: 3647–3656, 2011. doi: 10.1242/dev.067587. [DOI] [PubMed] [Google Scholar]
- 341.Sammons MA, Antons AK, Bendjennat M, Udd B, Krahe R, Link AJ. ZNF9 activation of IRES-mediated translation of the human ODC mRNA is decreased in myotonic dystrophy type 2. PLoS One 5: e9301, 2010. doi: 10.1371/journal.pone.0009301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Samulski RJ, Muzyczka N. AAV-Mediated Gene Therapy for Research and Therapeutic Purposes. Annu Rev Virol 1: 427–451, 2014. doi: 10.1146/annurev-virology-031413-085355. [DOI] [PubMed] [Google Scholar]
- 343.Sander JD, Ramirez CL, Linder SJ, Pattanayak V, Shoresh N, Ku M, Foden JA, Reyon D, Bernstein BE, Liu DR, Joung JK. In silico abstraction of zinc finger nuclease cleavage profiles reveals an expanded landscape of off-target sites. Nucleic Acids Res 41: e181, 2013. doi: 10.1093/nar/gkt716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Sandoval-Guzmán T, Wang H, Khattak S, Schuez M, Roensch K, Nacu E, Tazaki A, Joven A, Tanaka EM, Simon A. Fundamental differences in dedifferentiation and stem cell recruitment during skeletal muscle regeneration in two salamander species. Cell Stem Cell 14: 174–187, 2014. doi: 10.1016/j.stem.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 345.Sather BD, Romano Ibarra GS, Sommer K, Curinga G, Hale M, Khan IF, Singh S, Song Y, Gwiazda K, Sahni J, Jarjour J, Astrakhan A, Wagner TA, Scharenberg AM, Rawlings DJ. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci Transl Med 7: 307ra156, 2015. doi: 10.1126/scitranslmed.aac5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Schatzberg SJ, Olby NJ, Breen M, Anderson LV, Langford CF, Dickens HF, Wilton SD, Zeiss CJ, Binns MM, Kornegay JN, Morris GE, Sharp NJ. Molecular analysis of a spontaneous dystrophin ‘knockout’ dog. Neuromuscul Disord 9: 289–295, 1999. doi: 10.1016/S0960-8966(99)00011-5. [DOI] [PubMed] [Google Scholar]
- 347.Schmid-Burgk JL, Schmidt T, Kaiser V, Höning K, Hornung V. A ligation-independent cloning technique for high-throughput assembly of transcription activator-like effector genes. Nat Biotechnol 31: 76–81, 2013. doi: 10.1038/nbt.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Schnepp BC, Jensen RL, Chen CL, Johnson PR, Clark KR. Characterization of adeno-associated virus genomes isolated from human tissues. J Virol 79: 14793–14803, 2005. doi: 10.1128/JVI.79.23.14793-14803.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet 13: 878–890, 2012. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell 102: 777–786, 2000. doi: 10.1016/S0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 351.Segal DJ, Dreier B, Beerli RR, Barbas CF III. Toward controlling gene expression at will: selection and design of zinc finger domains recognizing each of the 5′-GNN-3′ DNA target sequences. Proc Natl Acad Sci USA 96: 2758–2763, 1999. doi: 10.1073/pnas.96.6.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Segal DJ, Meckler JF. Genome engineering at the dawn of the golden age. Annu Rev Genomics Hum Genet 14: 135–158, 2013. doi: 10.1146/annurev-genom-091212-153435. [DOI] [PubMed] [Google Scholar]
- 353.Selsby JT, Ross JW, Nonneman D, Hollinger K. Porcine models of muscular dystrophy. ILAR J 56: 116–126, 2015. doi: 10.1093/ilar/ilv015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Sergeant N, Sablonnière B, Schraen-Maschke S, Ghestem A, Maurage CA, Wattez A, Vermersch P, Delacourte A. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum Mol Genet 10: 2143–2155, 2001. doi: 10.1093/hmg/10.19.2143. [DOI] [PubMed] [Google Scholar]
- 355.Seznec H, Agbulut O, Sergeant N, Savouret C, Ghestem A, Tabti N, Willer JC, Ourth L, Duros C, Brisson E, Fouquet C, Butler-Browne G, Delacourte A, Junien C, Gourdon G. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum Mol Genet 10: 2717–2726, 2001. doi: 10.1093/hmg/10.23.2717. [DOI] [PubMed] [Google Scholar]
- 356.Sfeir A, Symington LS. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem Sci 40: 701–714, 2015. doi: 10.1016/j.tibs.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, Skarnes WC. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 11: 399–402, 2014. doi: 10.1038/nmeth.2857. [DOI] [PubMed] [Google Scholar]
- 358.Shinin V, Gayraud-Morel B, Gomès D, Tajbakhsh S. Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat Cell Biol 8: 677–682, 2006. doi: 10.1038/ncb1425. [DOI] [PubMed] [Google Scholar]
- 359.Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, Zhang F, Koonin EV. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol Cell 60: 385–397, 2015. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 244: 1578–1580, 1989. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 361.Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, Pâques F. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther 11: 11–27, 2011. doi: 10.2174/156652311794520111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Simsek D, Brunet E, Wong SY, Katyal S, Gao Y, McKinnon PJ, Lou J, Zhang L, Li J, Rebar EJ, Gregory PD, Holmes MC, Jasin M. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet 7: e1002080, 2011. doi: 10.1371/journal.pgen.1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Sipo I, Fechner H, Pinkert S, Suckau L, Wang X, Weger S, Poller W. Differential internalization and nuclear uncoating of self-complementary adeno-associated virus pseudotype vectors as determinants of cardiac cell transduction. Gene Ther 14: 1319–1329, 2007. doi: 10.1038/sj.gt.3302987. [DOI] [PubMed] [Google Scholar]
- 364.Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science 351: 84–88, 2016. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Smith BF, Yue Y, Woods PR, Kornegay JN, Shin JH, Williams RR, Duan D. An intronic LINE-1 element insertion in the dystrophin gene aborts dystrophin expression and results in Duchenne-like muscular dystrophy in the corgi breed. Lab Invest 91: 216–231, 2011. doi: 10.1038/labinvest.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Smith J, Grizot S, Arnould S, Duclert A, Epinat JC, Chames P, Prieto J, Redondo P, Blanco FJ, Bravo J, Montoya G, Pâques F, Duchateau P. A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucleic Acids Res 34: e149, 2006. doi: 10.1093/nar/gkl720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Smithies O, Gregg RG, Boggs SS, Koralewski MA, Kucherlapati RS. Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination. Nature 317: 230–234, 1985. doi: 10.1038/317230a0. [DOI] [PubMed] [Google Scholar]
- 368.Snider L, Asawachaicharn A, Tyler AE, Geng LN, Petek LM, Maves L, Miller DG, Lemmers RJ, Winokur ST, Tawil R, van der Maarel SM, Filippova GN, Tapscott SJ. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum Mol Genet 18: 2414–2430, 2009. doi: 10.1093/hmg/ddp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Snider L, Geng LN, Lemmers RJ, Kyba M, Ware CB, Nelson AM, Tawil R, Filippova GN, van der Maarel SM, Tapscott SJ, Miller DG. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet 6: e1001181, 2010. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Sondergaard PC, Griffin DA, Pozsgai ER, Johnson RW, Grose WE, Heller KN, Shontz KM, Montgomery CL, Liu J, Clark KR, Sahenk Z, Mendell JR, Rodino-Klapac LR. AAV.Dysferlin Overlap Vectors Restore Function in Dysferlinopathy Animal Models. Ann Clin Transl Neurol 2: 256–270, 2015. doi: 10.1002/acn3.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Srivastava A, Lusby EW, Berns KI. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J Virol 45: 555–564, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Staals RH, Zhu Y, Taylor DW, Kornfeld JE, Sharma K, Barendregt A, Koehorst JJ, Vlot M, Neupane N, Varossieau K, Sakamoto K, Suzuki T, Dohmae N, Yokoyama S, Schaap PJ, Urlaub H, Heck AJ, Nogales E, Doudna JA, Shinkai A, van der Oost J. RNA targeting by the type III-A CRISPR-Cas Csm complex of Thermus thermophilus. Mol Cell 56: 518–530, 2014. doi: 10.1016/j.molcel.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Staals RHJ, Agari Y, Maki-Yonekura S, Zhu Y, Taylor DW, van Duijn E, Barendregt A, Vlot M, Koehorst JJ, Sakamoto K, Masuda A, Dohmae N, Schaap PJ, Doudna JA, Heck AJR, Yonekura K, van der Oost J, Shinkai A. Structure and activity of the RNA-targeting Type III-B CRISPR-Cas complex of Thermus thermophilus. Mol Cell 52: 135–145, 2013. doi: 10.1016/j.molcel.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Stepper P, Kungulovski G, Jurkowska RZ, Chandra T, Krueger F, Reinhardt R, Reik W, Jeltsch A, Jurkowski TP. Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase. Nucleic Acids Res 45: 1703–1713, 2017. doi: 10.1093/nar/gkw1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Stoddard BL. Homing endonuclease structure and function. Q Rev Biophys 38: 49–95, 2005. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 376.Stoddard BL. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure 19: 7–15, 2011. doi: 10.1016/j.str.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Suda T, Liu D. Hydrodynamic gene delivery: its principles and applications. Mol Ther 15: 2063–2069, 2007. doi: 10.1038/sj.mt.6300314. [DOI] [PubMed] [Google Scholar]
- 378.Sultana N, Zhang L, Yan J, Chen J, Cai W, Razzaque S, Jeong D, Sheng W, Bu L, Xu M, Huang GY, Hajjar RJ, Zhou B, Moon A, Cai CL. Resident c-kit(+) cells in the heart are not cardiac stem cells. Nat Commun 6: 8701, 2015. doi: 10.1038/ncomms9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Sun JY, Anand-Jawa V, Chatterjee S, Wong KK. Immune responses to adeno-associated virus and its recombinant vectors. Gene Ther 10: 964–976, 2003. doi: 10.1038/sj.gt.3302039. [DOI] [PubMed] [Google Scholar]
- 380.Sun N, Zhao H. Transcription activator-like effector nucleases (TALENs): a highly efficient and versatile tool for genome editing. Biotechnol Bioeng 110: 1811–1821, 2013. doi: 10.1002/bit.24890. [DOI] [PubMed] [Google Scholar]
- 381.Suzuki K, Tsunekawa Y, Hernandez-Benitez R, Wu J, Zhu J, Kim EJ, Hatanaka F, Yamamoto M, Araoka T, Li Z, Kurita M, Hishida T, Li M, Aizawa E, Guo S, Chen S, Goebl A, Soligalla RD, Qu J, Jiang T, Fu X, Jafari M, Esteban CR, Berggren WT, Lajara J, Nuñez-Delicado E, Guillen P, Campistol JM, Matsuzaki F, Liu GH, Magistretti P, Zhang K, Callaway EM, Zhang K, Belmonte JC. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540: 144–149, 2016. doi: 10.1038/nature20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Szczepek M, Brondani V, Büchel J, Serrano L, Segal DJ, Cathomen T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol 25: 786–793, 2007. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- 383.Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, Cong L, Zhang F, Vandenberghe LH, Church GM, Wagers AJ. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351: 407–411, 2016. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861–872, 2007. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 385.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663–676, 2006. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 386.Tamulaitis G, Kazlauskiene M, Manakova E, Venclovas Č, Nwokeoji AO, Dickman MJ, Horvath P, Siksnys V. Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol Cell 56: 506–517, 2014. doi: 10.1016/j.molcel.2014.09.027. [DOI] [PubMed] [Google Scholar]
- 387.Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159: 635–646, 2014. doi: 10.1016/j.cell.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Tang L, Zeng Y, Du H, Gong M, Peng J, Zhang B, Lei M, Zhao F, Wang W, Li X, Liu J. CRISPR/Cas9-mediated gene editing in human zygotes using Cas9 protein. Mol Genet Genomics 292: 525–533, 2017. doi: 10.1007/s00438-017-1299-z. [DOI] [PubMed] [Google Scholar]
- 389.Tawil R, Van Der Maarel SM. Facioscapulohumeral muscular dystrophy. Muscle Nerve 34: 1–15, 2006. doi: 10.1002/mus.20522. [DOI] [PubMed] [Google Scholar]
- 390.Tawil R, van der Maarel SM, Tapscott SJ. Facioscapulohumeral dystrophy: the path to consensus on pathophysiology. Skelet Muscle 4: 12, 2014. doi: 10.1186/2044-5040-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Thakore PI, Black JB, Hilton IB, Gersbach CA. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat Methods 13: 127–137, 2016. doi: 10.1038/nmeth.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Thakore PI, D’Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, Reddy TE, Crawford GE, Gersbach CA. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods 12: 1143–1149, 2015. doi: 10.1038/nmeth.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51: 503–512, 1987. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- 394.Thomas KR, Folger KR, Capecchi MR. High frequency targeting of genes to specific sites in the mammalian genome. Cell 44: 419–428, 1986. doi: 10.1016/0092-8674(86)90463-0. [DOI] [PubMed] [Google Scholar]
- 395.Timchenko NA, Patel R, Iakova P, Cai ZJ, Quan L, Timchenko LT. Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. J Biol Chem 279: 13129–13139, 2004. doi: 10.1074/jbc.M312923200. [DOI] [PubMed] [Google Scholar]
- 396.Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature 384: 349–353, 1996. doi: 10.1038/384349a0. [DOI] [PubMed] [Google Scholar]
- 397.Tóth E, Weinhardt N, Bencsura P, Huszár K, Kulcsár PI, Tálas A, Fodor E, Welker E. Cpf1 nucleases demonstrate robust activity to induce DNA modification by exploiting homology directed repair pathways in mammalian cells. Biol Direct 11: 46, 2016. doi: 10.1186/s13062-016-0147-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, Razavian N, Berns MW, Wu X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci USA 110: 7720–7725, 2013. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Tsai SQ, Joung JK. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat Rev Genet 17: 300–312, 2016. doi: 10.1038/nrg.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, Goodwin MJ, Aryee MJ, Joung JK. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32: 569–576, 2014. doi: 10.1038/nbt.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, Aryee MJ, Joung JK. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33: 187–197, 2015. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Turan S, Farruggio AP, Srifa W, Day JW, Calos MP. Precise Correction of Disease Mutations in Induced Pluripotent Stem Cells Derived From Patients With Limb Girdle Muscular Dystrophy. Mol Ther 24: 685–696, 2016. doi: 10.1038/mt.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Turner C, Hilton-Jones D. The myotonic dystrophies: diagnosis and management. J Neurol Neurosurg Psychiatry 81: 358–367, 2010. doi: 10.1136/jnnp.2008.158261. [DOI] [PubMed] [Google Scholar]
- 404.Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol 11: 891–905, 2012. doi: 10.1016/S1474-4422(12)70204-1. [DOI] [PubMed] [Google Scholar]
- 405.Ungerer J, Pakrasi HB. Cpf1 Is A Versatile Tool for CRISPR Genome Editing Across Diverse Species of Cyanobacteria. Sci Rep 6: 39681, 2016. doi: 10.1038/srep39681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435: 646–651, 2005. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 407.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11: 636–646, 2010. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 408.Vainzof M, Passos-Bueno MR, Canovas M, Moreira ES, Pavanello RC, Marie SK, Anderson LV, Bonnemann CG, McNally EM, Nigro V, Kunkel LM, Zatz M. The sarcoglycan complex in the six autosomal recessive limb-girdle muscular dystrophies. Hum Mol Genet 5: 1963–1969, 1996. doi: 10.1093/hmg/5.12.1963. [DOI] [PubMed] [Google Scholar]
- 409.Valentine BA, Cooper BJ, Cummings JF, deLahunta A. Progressive muscular dystrophy in a golden retriever dog: light microscope and ultrastructural features at 4 and 8 months. Acta Neuropathol 71: 301–310, 1986. doi: 10.1007/BF00688053. [DOI] [PubMed] [Google Scholar]
- 410.Valentine BA, Cooper BJ, de Lahunta A, O’Quinn R, Blue JT. Canine X-linked muscular dystrophy. An animal model of Duchenne muscular dystrophy: clinical studies. J Neurol Sci 88: 69–81, 1988. doi: 10.1016/0022-510X(88)90206-7. [DOI] [PubMed] [Google Scholar]
- 411.Van Agtmaal EL, André LM, Willemse M, Cumming SA, van Kessel IDG, van den Broek WJAA, Gourdon G, Furling D, Mouly V, Monckton DG, Wansink DG, Wieringa B. CRISPR/Cas9-Induced (CTG⋅CAG)nRepeat Instability in the Myotonic Dystrophy Type 1 Locus: Implications for Therapeutic Genome Editing. Mol Ther 25: 24–43, 2017. doi: 10.1016/j.ymthe.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SC, Middleton RC, Marbán E, Molkentin JD. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 509: 337–341, 2014. doi: 10.1038/nature13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Van Deutekom JC, Bakker E, Lemmers RJ, van der Wielen MJ, Bik E, Hofker MH, Padberg GW, Frants RR. Evidence for subtelomeric exchange of 3.3 kb tandemly repeated units between chromosomes 4q35 and 10q26: implications for genetic counselling and etiology of FSHD1. Hum Mol Genet 5: 1997–2003, 1996. doi: 10.1093/hmg/5.12.1997. [DOI] [PubMed] [Google Scholar]
- 414.Vignaud A, Ferry A, Huguet A, Baraibar M, Trollet C, Hyzewicz J, Butler-Browne G, Puymirat J, Gourdon G, Furling D. Progressive skeletal muscle weakness in transgenic mice expressing CTG expansions is associated with the activation of the ubiquitin-proteasome pathway. Neuromuscul Disord 20: 319–325, 2010. doi: 10.1016/j.nmd.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 415.Vihola A, Bachinski LL, Sirito M, Olufemi SE, Hajibashi S, Baggerly KA, Raheem O, Haapasalo H, Suominen T, Holmlund-Hampf J, Paetau A, Cardani R, Meola G, Kalimo H, Edström L, Krahe R, Udd B. Differences in aberrant expression and splicing of sarcomeric proteins in the myotonic dystrophies DM1 and DM2. Acta Neuropathol 119: 465–479, 2010. doi: 10.1007/s00401-010-0637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci USA 100: 12871–12876, 2003. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Vulin A, Wein N, Simmons TR, Rutherford AM, Findlay AR, Yurkoski JA, Kaminoh Y, Flanigan KM. The first exon duplication mouse model of Duchenne muscular dystrophy: a tool for therapeutic development. Neuromuscul Disord 25: 827–834, 2015. doi: 10.1016/j.nmd.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 418.Walmsley GL, Arechavala-Gomeza V, Fernandez-Fuente M, Burke MM, Nagel N, Holder A, Stanley R, Chandler K, Marks SL, Muntoni F, Shelton GD, Piercy RJ. A duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient cavalier king charles spaniels is amenable to exon 51 skipping. PLoS One 5: e8647, 2010. doi: 10.1371/journal.pone.0008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Wang GS, Kearney DL, De Biasi M, Taffet G, Cooper TA. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J Clin Invest 117: 2802–2811, 2007. doi: 10.1172/JCI32308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Wang H, La Russa M, Qi LS. CRISPR/Cas9 in Genome Editing and Beyond. Annu Rev Biochem 85: 227–264, 2016. doi: 10.1146/annurev-biochem-060815-014607. [DOI] [PubMed] [Google Scholar]
- 421.Wang H, Rosidi B, Perrault R, Wang M, Zhang L, Windhofer F, Iliakis G. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res 65: 4020–4030, 2005. doi: 10.1158/0008-5472.CAN-04-3055. [DOI] [PubMed] [Google Scholar]
- 422.Wang HX, Li M, Lee CM, Chakraborty S, Kim HW, Bao G, Leong KW. CRISPR/Cas9-Based Genome Editing for Disease Modeling and Therapy: Challenges and Opportunities for Nonviral Delivery. Chem Rev 117: 9874–9906, 2017. doi: 10.1021/acs.chemrev.6b00799. [DOI] [PubMed] [Google Scholar]
- 423.Wang J, Exline CM, DeClercq JJ, Llewellyn GN, Hayward SB, Li PW, Shivak DA, Surosky RT, Gregory PD, Holmes MC, Cannon PM. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol 33: 1256–1263, 2015. doi: 10.1038/nbt.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Wang L, Calcedo R, Bell P, Lin J, Grant RL, Siegel DL, Wilson JM. Impact of pre-existing immunity on gene transfer to nonhuman primate liver with adeno-associated virus 8 vectors. Hum Gene Ther 22: 1389–1401, 2011. doi: 10.1089/hum.2011.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Wang L, Calcedo R, Wang H, Bell P, Grant R, Vandenberghe LH, Sanmiguel J, Morizono H, Batshaw ML, Wilson JM. The pleiotropic effects of natural AAV infections on liver-directed gene transfer in macaques. Mol Ther 18: 126–134, 2010. doi: 10.1038/mt.2009.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Wang L, Li F, Dang L, Liang C, Wang C, He B, Liu J, Li D, Wu X, Xu X, Lu A, Zhang G. In Vivo Delivery Systems for Therapeutic Genome Editing. Int J Mol Sci 17: E626, 2016. doi: 10.3390/ijms17050626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, Chen C, Li J, Xiao X. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol 23: 321–328, 2005. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- 428.Wansapura JP, Millay DP, Dunn RS, Molkentin JD, Benson DW. Magnetic resonance imaging assessment of cardiac dysfunction in δ-sarcoglycan null mice. Neuromuscul Disord 21: 68–73, 2011. doi: 10.1016/j.nmd.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Ward AJ, Rimer M, Killian JM, Dowling JJ, Cooper TA. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum Mol Genet 19: 3614–3622, 2010. doi: 10.1093/hmg/ddq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Wenzel K, Carl M, Perrot A, Zabojszcza J, Assadi M, Ebeling M, Geier C, Robinson PN, Kress W, Osterziel KJ, Spuler S. Novel sequence variants in dysferlin-deficient muscular dystrophy leading to mRNA decay and possible C2-domain misfolding. Hum Mutat 27: 599–600, 2006. doi: 10.1002/humu.9424. [DOI] [PubMed] [Google Scholar]
- 431.West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol 4: 435–445, 2003. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 432.Wheeler MT, Zarnegar S, McNally EM. Zeta-sarcoglycan, a novel component of the sarcoglycan complex, is reduced in muscular dystrophy. Hum Mol Genet 11: 2147–2154, 2002. doi: 10.1093/hmg/11.18.2147. [DOI] [PubMed] [Google Scholar]
- 433.Wijmenga C, Hewitt JE, Sandkuijl LA, Clark LN, Wright TJ, Dauwerse HG, Gruter AM, Hofker MH, Moerer P, Williamson R, van Ommen G-JB, Padberg GW, Frants RR. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet 2: 26–30, 1992. doi: 10.1038/ng0992-26. [DOI] [PubMed] [Google Scholar]
- 434.Williams RS, Williams JS, Tainer JA. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol 85: 509–520, 2007. doi: 10.1139/O07-069. [DOI] [PubMed] [Google Scholar]
- 435.Winand N, Pradham D, Cooper B. Molecular characterization of severe Duchenne-type muscular dystrophy in a family of Rottweiler dogs. In: Molecular Mechanisms of Neuromuscular Disease. Tucson, AZ: Muscular Dystrophy Association, 1994. [Google Scholar]
- 436.Winblad S, Lindberg C, Hansen S. Temperament and character in patients with classical myotonic dystrophy type 1 (DM-1). Neuromuscul Disord 15: 287–292, 2005. doi: 10.1016/j.nmd.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 437.Wojtal D, Kemaladewi DU, Malam Z, Abdullah S, Wong TW, Hyatt E, Baghestani Z, Pereira S, Stavropoulos J, Mouly V, Mamchaoui K, Muntoni F, Voit T, Gonorazky HD, Dowling JJ, Wilson MD, Mendoza-Londono R, Ivakine EA, Cohn RD. Spell Checking Nature: Versatility of CRISPR/Cas9 for Developing Treatments for Inherited Disorders. Am J Hum Genet 98: 90–101, 2016. doi: 10.1016/j.ajhg.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Wolf DP, Hayama T, Mitalipov S. Mitochondrial genome inheritance and replacement in the human germline. EMBO J 36: 2177–2181, 2017. doi: 10.15252/embj.201797606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Wolff JA, Ludtke JJ, Acsadi G, Williams P, Jani A. Long-term persistence of plasmid DNA and foreign gene expression in mouse muscle. Hum Mol Genet 1: 363–369, 1992. doi: 10.1093/hmg/1.6.363. [DOI] [PubMed] [Google Scholar]
- 440.Wright AV, Nuñez JK, Doudna JA. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 164: 29–44, 2016. doi: 10.1016/j.cell.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 441.Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S, Yan Z, Li D, Li J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 13: 659–662, 2013. doi: 10.1016/j.stem.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 442.Wyatt HD, West SC. Holliday junction resolvases. Cold Spring Harb Perspect Biol 6: a023192, 2014. doi: 10.1101/cshperspect.a023192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Xia G, Gao Y, Jin S, Subramony SH, Terada N, Ranum LP, Swanson MS, Ashizawa T. Genome modification leads to phenotype reversal in human myotonic dystrophy type 1 induced pluripotent stem cell-derived neural stem cells. Stem Cells 33: 1829–1838, 2015. doi: 10.1002/stem.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol 16: 814–818, 2009. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Xie K, Minkenberg B, Yang Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci USA 112: 3570–3575, 2015. doi: 10.1073/pnas.1420294112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Xu L, Park KH, Zhao L, Xu J, El Refaey M, Gao Y, Zhu H, Ma J, Han R. CRISPR-mediated Genome Editing Restores Dystrophin Expression and Function in mdx Mice. Mol Ther 24: 564–569, 2016. doi: 10.1038/mt.2015.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, Cai W, Yang G, Bronson R, Crowley DG, Zhang F, Anderson DG, Sharp PA, Jacks T. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514: 380–384, 2014. doi: 10.1038/nature13589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Yamada M, Emmanuele V, Sanchez-Quintero MJ, Sun B, Lallos G, Paull D, Zimmer M, Pagett S, Prosser RW, Sauer MV, Hirano M, Egli D. Genetic Drift Can Compromise Mitochondrial Replacement by Nuclear Transfer in Human Oocytes. Cell Stem Cell 18: 749–754, 2016. doi: 10.1016/j.stem.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Yamano T, Zetsche B, Ishitani R, Zhang F, Nishimasu H, Nureki O. Structural Basis for the Canonical and Non-canonical PAM Recognition by CRISPR-Cpf1. Mol Cell 67: 633–645.e3, 2017. doi: 10.1016/j.molcel.2017.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154: 1370–1379, 2013. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Yang J, Li SY, Li YQ, Cao JQ, Feng SW, Wang YY, Zhan YX, Yu CS, Chen F, Li J, Sun XF, Zhang C. MLPA-based genotype-phenotype analysis in 1053 Chinese patients with DMD/BMD. BMC Med Genet 14: 29, 2013. doi: 10.1186/1471-2350-14-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Yang S, Chang R, Yang H, Zhao T, Hong Y, Kong HE, Sun X, Qin Z, Jin P, Li S, Li XJ. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J Clin Invest 127: 2719–2724, 2017. doi: 10.1172/JCI92087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet 15: 541–555, 2014. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 454.Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev 93: 23–67, 2013. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, Koteliansky V, Sharp PA, Jacks T, Anderson DG. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol 32: 551–553, 2014. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.You Z, Bailis JM. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol 20: 402–409, 2010. doi: 10.1016/j.tcb.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Young CS, Hicks MR, Ermolova NV, Nakano H, Jan M, Younesi S, Karumbayaram S, Kumagai-Cresse C, Wang D, Zack JA, Kohn DB, Nakano A, Nelson SF, Miceli MC, Spencer MJ, Pyle AD. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 18: 533–540, 2016. doi: 10.1016/j.stem.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Young CS, Mokhonova E, Quinonez M, Pyle AD, Spencer MJ. Creation of a Novel Humanized Dystrophic Mouse Model of Duchenne Muscular Dystrophy and Application of a CRISPR/Cas9 Gene Editing Therapy. J Neuromuscul Dis 4: 139–145, 2017. doi: 10.3233/JND-170218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Young JM, Whiddon JL, Yao Z, Kasinathan B, Snider L, Geng LN, Balog J, Tawil R, van der Maarel SM, Tapscott SJ. DUX4 binding to retroelements creates promoters that are active in FSHD muscle and testis. PLoS Genet 9: e1003947, 2013. doi: 10.1371/journal.pgen.1003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Yu AM, McVey M. Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res 38: 5706–5717, 2010. doi: 10.1093/nar/gkq379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Yu HH, Zhao H, Qing YB, Pan WR, Jia BY, Zhao HY, Huang XX, Wei HJ. Porcine Zygote Injection with Cas9/sgRNA Results in DMD-Modified Pig with Muscle Dystrophy. Int J Mol Sci 17: E1668, 2016. doi: 10.3390/ijms17101668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science 318: 1917–1920, 2007. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 463.Yue Y, Shin JH, Duan D. Whole body skeletal muscle transduction in neonatal dogs with AAV-9. Methods Mol Biol 709: 313–329, 2011. doi: 10.1007/978-1-61737-982-6_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Zaidi SS, Mahfouz MM, Mansoor S. CRISPR-Cpf1: A New Tool for Plant Genome Editing. Trends Plant Sci 22: 550–553, 2017. doi: 10.1016/j.tplants.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 465.Zalatan JG, Lee ME, Almeida R, Gilbert LA, Whitehead EH, La Russa M, Tsai JC, Weissman JS, Dueber JE, Qi LS, Lim WA. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 160: 339–350, 2015. doi: 10.1016/j.cell.2014.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163: 759–771, 2015. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, Wu WY, Scott DA, Severinov K, van der Oost J, Zhang F. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol 35: 31–34, 2017. doi: 10.1038/nbt.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Zhang JP, Li XL, Li GH, Chen W, Arakaki C, Botimer GD, Baylink D, Zhang L, Wen W, Fu YW, Xu J, Chun N, Yuan W, Cheng T, Zhang XB. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol 18: 35, 2017. doi: 10.1186/s13059-017-1164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Zhang Y, Long C, Li H, McAnally JR, Baskin KK, Shelton JM, Bassel-Duby R, Olson EN. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci Adv 3: e1602814, 2017. doi: 10.1126/sciadv.1602814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Zhen S, Hua L, Liu YH, Gao LC, Fu J, Wan DY, Dong LH, Song HF, Gao X. Harnessing the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated Cas9 system to disrupt the hepatitis B virus. Gene Ther 22: 404–412, 2015. doi: 10.1038/gt.2015.2. [DOI] [PubMed] [Google Scholar]
- 471.Zhu P, Wu F, Mosenson J, Zhang H, He TC, Wu WS. CRISPR/Cas9-Mediated Genome Editing Corrects Dystrophin Mutation in Skeletal Muscle Stem Cells in a Mouse Model of Muscle Dystrophy. Mol Ther Nucleic Acids 7: 31–41, 2017. doi: 10.1016/j.omtn.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Zincarelli C, Soltys S, Rengo G, Koch WJ, Rabinowitz JE. Comparative cardiac gene delivery of adeno-associated virus serotypes 1-9 reveals that AAV6 mediates the most efficient transduction in mouse heart. Clin Transl Sci 3: 81–89, 2010. doi: 10.1111/j.1752-8062.2010.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]