

Abstract
Starvation significantly alters cellular physiology, and signs of aging have been reported to occur during starvation. Mitochondria are essential to the regulation of cellular energetics and aging. We sought to determine whether mitochondria exhibit signs of aging during starvation and whether quality control mechanisms regulate mitochondrial physiology during starvation. We describe effects of starvation on mitochondria in the first and third larval stages of the nematode Caenorhabditis elegans. When starved, C. elegans larvae enter developmental arrest. We observed fragmentation of the mitochondrial network, a reduction in mitochondrial DNA (mtDNA) copy number, and accumulation of DNA damage during starvation-induced developmental arrest. Mitochondrial function was also compromised by starvation. Starved worms had lower basal, maximal, and ATP-linked respiration. These observations are consistent with reduced mitochondrial quality, similar to mitochondrial phenotypes during aging. Using pharmacological and genetic approaches, we found that worms deficient for autophagy were short-lived during starvation and recovered poorly from extended starvation, indicating sensitivity to nutrient stress. Autophagy mutants unc-51/Atg1 and atg-18/Atg18 maintained greater mtDNA content than wild-type worms during starvation, suggesting that autophagy promotes mitochondrial degradation during starvation. unc-51 mutants also had a proportionally smaller reduction in oxygen consumption rate during starvation, suggesting that autophagy also contributes to reduced mitochondrial function. Surprisingly, mutations in genes involved in mitochondrial fission and fusion as well as selective mitophagy of damaged mitochondria did not affect mitochondrial content during starvation. Our results demonstrate the profound influence of starvation on mitochondrial physiology with organismal consequences, and they show that these physiological effects are influenced by autophagy.
Keywords: aging, autophagy, C. elegans, mitochondria, starvation
INTRODUCTION
Nutrient deprivation is a common environmental stress. Cells and entire animals must respond rapidly to changing nutrient conditions to maintain homeostasis. Metabolism, gene expression, and organelle structure and function all change in response to nutrient stress. Being central to cellular energy production, mitochondria are particularly important to adaptation to starvation. Healthy mitochondria exist in most cells as expansive networks with high membrane potential. Defects in mitochondrial maintenance have been implicated in metabolic and neurodegenerative diseases, as well as aging (8, 10, 23, 46, 49, 60). Damaged or aged mitochondria typically have reduced membrane potential, more rigid membranes, increased reactive oxygen species (ROS) production, mutations or lesions in mitochondria DNA (mtDNA), and reduced respiratory capacity (8, 23, 34, 50, 58–60). Yet how mitochondrial structure and function are remodeled during starvation, and by what regulatory mechanisms, is not well understood.
The nematode Caenorhabditis elegans is a simple animal model that is ideal to study effects of nutrient deprivation on mitochondria (62). C. elegans larvae adapt to and survive starvation by arresting development (4, 7, 19, 56). Developmental arrest is established via organismal signaling, gene regulatory, and metabolic changes. Arrest can occur at different stages during development, including but not limited to the first (L1) and third (L3) larval stages (2, 3, 6, 15, 47, 56). Worms arrest as L1 larvae when they hatch in the absence of food (2), and developing L2 larvae arrest as L3 larvae when food is withdrawn (56).
Historically, developmental arrest during starvation was thought to be an “ageless state” because time spent in larval arrest does not count against adult lifespan after recovery (19, 27). Recently, the paradigm of an ageless state has been challenged (53). Worms exhibit several age-associated phenotypes during L1 arrest including protein aggregation, ROS production, and mitochondrial fragmentation (53). With the exception of protein aggregation, these aging phenotypes are reversed during recovery from starvation in an endoplasmic reticulum-unfolded protein response-dependent fashion, possibly explaining the normal lifespan of worms arrested as larvae (53). These surprising findings merit additional characterization to substantiate aging during larval arrest. Thus we measured mitochondrial phenotypes associated with aging to determine whether mitochondria exhibit signs of aging during starvation. If such aging-related processes do occur, it is important to understand the mechanisms that regulate them.
Autophagy provides a mechanism to degrade cellular components to derive energy from macromolecules and organelles during starvation (29, 43, 61). Autophagy also contributes to cellular quality control during aging and directly impacts lifespan (12, 14, 17, 24, 31, 64). Here, we focus on mitophagy, the autophagic consumption of mitochondria. Three types of mitochondrial autophagy have been proposed (33). Type 1 and type 2 mitophagy involve sequestration of mitochondria in autophagosomes, whereas in type 3 mitophagy mitochondrially derived vesicles bud off of mitochondria and are trafficked to lysosomes independent of an autophagosome (33). Type 1 mitophagy is nonselective, as it does not specifically engulf damaged mitochondria but is more likely a clearance mechanism for excess mitochondria. Type 2 and type 3 mitophagy are initiated because of damaged mitochondria, for example mitochondria with reduced membrane potential, and rely on pink-1 (PTEN-induced putative kinase) and Parkin (33). Damaged mitochondria are selectively removed when pink-1 is stabilized on their outer membrane and recruits the E3 ubiquitin ligase, Parkin, which tags mitochondria for degradation (63).
We sought to characterize the influence of larval starvation on mitochondrial physiology in C. elegans. Our objectives were to determine whether mitochondria display signs of aging during starvation and to identify regulatory mechanisms required for these changes. We found that starvation in L1 or L3 stage larvae has extensive effects on mitochondria, including fragmentation, decreased mtDNA copy number, increased mtDNA damage, and reduced respiration. These effects are similar to those of aging in fed adults, suggesting common mechanisms. Further, we provide evidence that general autophagy promotes elimination of mitochondria and likely decreases whole animal respiratory capacity during starvation (33).
MATERIALS AND METHODS
Larval arrest.
To arrest worms as L1 larvae, synchronous populations of embryos were isolated from mixed-stage cultures of C. elegans by hypochlorite treatment. Embryos were cultured at a density of 1/μl in virgin S-basal (no ethanol or cholesterol) in 16-mm glass test tubes on a tissue culture roller drum at room temperature (~21°C). Embryos were hatched in the absence of food so that they enter L1 arrest.
To arrest worms as L3 larvae, embryos were isolated as with L1 arrest. Embryos were given 16–20 h to hatch and were then plated onto 10-cm nematode growth media (NGM) plates seeded with Escherichia coli OP50. Worms were allowed to develop for 24 h at 20°C, at which point they were washed from plates with virgin S-basal. Worms were washed at least three times by centrifugation in 15-ml conical tubes to remove residual bacteria. Worms were then suspended at a concentration of 1/μl in virgin S-basal, as with arrested L1 larvae.
Mitochondrial morphology.
The SJ4103 strain was used to visualize mitochondria in the body wall muscle of L3 larvae. Worms were subjected to L3 arrest as described above. Starved worms were sampled over time, and representative images were taken with a Hamamatsu ORCA R2 camera and Yokogawa spinning disk head mounted on a Zeiss AxioImager A1 microscope. The images were analyzed using IMARIS (Bitplane), and three-dimensional reconstructions were built from Z-stacks. Objects identified by the program were assessed for surface area, volume, and sphericity according to the default algorithms of the program.
Mitochondrial copy number and DNA damage.
Mitochondrial and nuclear genome copy number, as well as mtDNA damage, was measured as previously described (13). Worm lysate was obtained as explained previously (13). Samples were collected on days 1, 3, 6, 9, and 12 of L1 arrest from independent biological replicates. Five worms were picked into lysis buffer comprised of 60% molecular biology grade water, 35% 3.3× buffer (82.5 mM tricine pH 8, 264 mM potassium acetate, 36.2% wt/vol glycerol, 7.4% vol/vol DMSO, prepared in nuclease-free water), and 5% proteinase K. Worms were lysed at a concentration of 1 worm per 10 μl of lysis buffer. Mitochondrial and nuclear genome copy number were measured as described in Basic Protocol 2 of Ref. 13 using a mitochondrial gene plasmid and glp-1 lysate for standard curves. See Ref. 13 for primer and plasmid information. We measured mtDNA damage on the same sample lysates as described in Basic Protocol 1 after optimizing the number of cycles for wild-type (WT) L1 larvae (13). DNA lesions were normalized to day 1 of starvation.
Measuring oxygen consumption.
Oxygen consumption rates (OCR) were measured in the presence or absence of mitochondrial inhibitors in fed and starved worms using a Seahorse XFe24 Extracellular Flux Analyzer, as described previously (38). We adapted this protocol for L1 and L3 larvae, to investigate changes in respiration throughout starvation. Preparation of L3 samples was staggered such that all starvation time points were run on the same plate. Each time point was run in technical replicate wells, and multiple biological replicates were performed.
For L3 larvae, each well contained 250 worms for fed and starved 1-day conditions, 500 worms for starved 3- and 6-day conditions, and 1,000 worms for the starved 9- and 12-day conditions. Because of interference of the pharmacological inhibitors used, we ran one plate for the first set of inhibitors, carbonyl cyanide p-(tri-fluromethoxy)phenyl-hydrazone (FCCP) and sodium azide, and another plate immediately following for the second inhibitor, dicyclohexylcarbodiimide (DCCD). The inhibitors were prepared and used as previously described (38). Because response to the drugs differed from what is normally observed in well-fed adult nematodes, we slightly modified our data analysis from what was previously reported (38). We calculated total basal oxygen consumption as the average of OCR readings four through eight (of eight measurements). Maximal OCR was calculated by averaging measurements two through six after injection of the mitochondrial uncoupler FCCP. ATP-linked respiration was calculated by subtracting the average of OCR measurements two through four after injection of the complex V (ATP synthase) inhibitor DCCD from total basal OCR. Proton leak was determined by subtracting the average of four measurements after injection of sodium azide (complex IV inhibitor) from OCR after DCCD injection.
To measure total basal OCR in L1 larvae of WT and unc-51 mutants, similar methods were employed. Briefly, embryos were isolated from adult worms by standard hypochlorite treatment. Embryos were either hatched in the presence of E. coli OP50 on plates (fed) or suspended in virgin S-basal at a density of 1/μl (starved). Basal respiration was measured 24 h following the bleach. This allowed ~12 h for embryos to hatch and ~12 h for hatched L1 larvae to feed or acclimate to starvation conditions. Total basal OCR values were obtained as described for L3 worms, except that each well contained between 1,000 and 2,000 worms for both fed and starved (1 day) conditions. Either two or three biological replicates were performed, and multiple wells were included as technical replicates within each biological replicate.
ATP measurement.
Steady-state ATP levels were measured in arrested L3 larvae using the PE255 luciferase-expressing C. elegans strain as previously described (37). Time of larval starvation was staggered such that time points within a biological replicate were run on the same plate for ATP measurements. Sample and reagent preparation, measurement, and analysis were the same as detailed previously (37), except that, instead of 50 worms per well, we included 250 worms per well to adjust for lower ATP levels in smaller larvae.
Starvation survival.
To measure starvation survival, populations of arrested L1 or L3 larvae were obtained as described above. Worms were kept in 16-mm glass test tubes on a tissue culture roller drum at room temperature (~21°C). Survival was scored by plating 100 μl of suspended worms around the perimeter of a lawn of E. coli OP50 on a 6-cm NGM plate. The total number of worms plated (Tp) was counted. Two days later the total number of worms that were alive (TA) was counted. Survival was calculated as TA/TP. Curves were fit to starvation survival data by regression, and statistics were calculated using the worm survival package in R, as previously described (1, 22). Graphical representations of starvation survival in Fig. 3A depict individual data points and the curve fit to the data. For clarity, starvation survival plots in Fig. 6, A and C, only depict the curves fit to the data.
Fig. 3.

L3 larvae are more starvation resistant than L1 larvae. A: starvation survival is plotted for L1 and L3 larvae. L3 larvae survive starvation longer than L1 larvae. (P = 0.04, n = 3 biological replicates). B: worm length is plotted after growth upon feeding from the indicated time of L1 or L3 larval starvation. Larvae were recovered for 48 h after L1 starvation and for 24 h after L3 starvation because larvae in L3 arrest were fed for 24 h before starvation so that both were fed for 48 h total. Worms starved at the L1 stage are significantly shorter after 48 h of feeding (P = 0.0002, 1-way ANOVA, n = 3 biological replicates). Worms starved at the L3 stage are also significantly smaller after 24 h of recovery from extended starvation (P < 0.0001, 1-way ANOVA, n = 3 biological replicates). There is a significant interaction between the L1 and L3 reaction norms of size after recovery from starvation (P = 0.005, 2-way ANOVA, n = 3 biological replicates). Boxes are bounded by the upper and lower quartiles and also depict the median. Whiskers represent the minimum and maximum values within 1.5× interquartile range. Individual points depict outliers. C: percentage of worms reaching at least the L4 larval stage after 48 h of development from the indicated duration of L1 arrest is plotted. There is a marginally significant reduction in the number of worms that recover to at least the L4 stage (P = 0.07, Kruskal-Wallis test, n = 2). D: percentage of worms reaching at least the L4 stage after 24 h of feeding from the indicated duration of L3 larval arrest is plotted. Following starvation, there is a reduction in the number of worms that recover to at least the L4 stage (P < 0.0001, 1-way ANOVA, n = 2). The percentage of worms recovering to at least the L4 stage was significantly reduced (relative to recovery from 1 day of starvation) at days 9 and 12 (P < 0.001 in both cases, Tukey’s honestly significant difference test). Mean and SE values are depicted in C and D.
Fig. 6.

Blocking autophagy reduces starvation survival and delays recovery from extended starvation. A: L1 starvation survival is plotted for worms exposed to a range of concentrations of the autophagy inhibitor 3-methyladenine (3-MA). B: size after 48 h of feeding and growth following the indicated period of starvation is shown for worms exposed to a range of concentrations of 3-MA. C: L1 starvation survival is plotted for various genotypes. atg-18 and unc-51 mutants were short-lived during starvation (P = 7.4E-6, 0.02, respectively). pink-1 mutants were significantly long-lived during starvation (P = 0.001). D: size upon recovery from starvation is shown for the same genotypes as in C. atg-18 mutants were shorter upon recovery from starvation relative to wild-type (WT) (2-way ANOVA genotype comparison P = 2.3E-5, interaction term P = 0.06). Similarly, unc-51 mutants were also shorter than WT worms (2-way ANOVA, genotype comparison P = 1.9E-14). pink-1 mutants were significantly longer than WT (2-way ANOVA genotype comparison P = 0.002, interaction term P = 0.07). In B and D mean values and SE are shown.
Worm size measurements.
To measure size of worms following recovery from starvation, starved worms were plated on 10-cm NGM plates seeded with E. coli OP50 on the day indicated (Figs. 3A and 6, B and D). After 24 h for L3 or 48 h for L1 (L3 larvae were grown for 24 h before arrest, 48-h total growth), the worms were washed off the plates, washed with virgin S-basal, plated on unseeded 10-cm NGM plates, and imaged. Images of worms were taken on a ZeissDiscovery V20 stereomicroscope with motorized magnification. Magnification was adjusted depending on the size of the worms. All images were analyzed with the WormSizer plugin for FIJI (44).
Strains.
The Bristol N2 strain was used and is annotated as WT. SJ4103 zcIs14[myo-3::GFP(mit)] was used for observations of mitochondria in body wall muscle tissue. PE255 feIs5[sur-5p::luciferase::GFP + rol-6(su1006)] was used for assaying ATP. The following strains and mutant alleles were used: VC893 atg-18(gk378), pink-1(tm1779), VC1024 pdr-1(gk448), CB369 unc-51(e369), DA631 eat-3(ad426), and CU6372 drp-1(tm1108).
Statistical analysis.
Statistical analysis was performed in Microsoft Excel and R studio. All references to n refer to the number of biological replicates included in statistical analysis. To assess significance of data over time during starvation, Bartlett’s test was initially run to test for equal variances. In cases where the null hypothesis of no difference in variance across samples was not rejected, a one-way ANOVA was run. In cases with significant one-way ANOVA results, a post hoc Tukey’s honestly significant difference test was performed to determine pairwise differences. When the null hypothesis of Bartlett’s test was rejected, we performed a Kruskal-Wallis test (Figs. 3D and 5, A–C). In cases of a significant P value, a post hoc Dunn’s test was performed to determine significant differences in pairwise comparisons.
Fig. 5.

Respiration and ATP content decrease during L3 larval starvation. A: basal oxygen consumption rate (OCR) of fed worms and over time during starvation is plotted (fed vs. starved P = 0.008, unpaired t-test, n = 4; during starvation P = 0.14, Kruskal-Wallis test, n = 5). B: maximal OCR of fed worms and over time during starvation is plotted (fed vs. starved P = 0.04, unpaired t-test, n = 4; during starvation P = 0.03, Kruskal-Wallis test, n = 5). A post hoc Dunn’s test indicates that maximal OCR at day 1 of starvation is significantly different than that at days 9 and 12 (P < 0.05 in each case). C: spare respiratory capacity of fed worms and over time during starvation is plotted. There was not a significant difference in spare capacity between fed worms and those starved for 1 day (P = 0.67, unpaired t-test, n = 4). There was a marginally significant decrease in spare respiratory capacity over time during starvation (P = 0.09, Kruskal-Wallis test, n = 5). D: ATP-linked respiration is plotted for fed worms and over time during starvation (fed vs. starved P = 0.006, unpaired t-test, n = 4; during starvation P = 0.0004, 1-way ANOVA, n = 5). Differences in ATP-linked respiration between day 1 and all other days drove this difference [P < 0.05 for each comparison, Tukey’s honestly significant difference (HSD) test]. E: ATP levels throughout L3 starvation are plotted (P = 0.001, 1-way ANOVA, n = 2 or 3). Post hoc Tukey’s HSD tests revealed that ATP levels at day 1 were significantly different than all other time points during starvation (in each case P < 0.01). No other individual comparisons were significantly different. Mean values and SE of biological replicates are shown in A–E. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
RESULTS
Mitochondria fragment during larval starvation.
We observed mitochondrial morphology during larval starvation. Body wall muscle cells of C. elegans have high mitochondrial density and are located superficially along the length of the body, facilitating visualization of mitochondria (Fig. 1A). The normally elongated mitochondrial networks in body wall muscle cells fragment over time during L3 larval starvation (Fig. 1, B–F). Observation of mitochondrial morphology is easier in L3 larvae than L1 larvae given larger cell size. We quantified several morphological characteristics of the mitochondrial network after 22–24 h of starvation. Both surface area and volume of individual mitochondria were significantly reduced (Fig. 1, G and H). Additionally, mitochondrial sphericity (a measure of how close an object approximates a sphere) appears to increase over time (Fig. 1I, P = 0.07). Overall, these observations indicate fragmentation of the mitochondrial network during larval starvation.
Fig. 1.

Mitochondria fragment during L3 larval starvation. A: a cartoon indicates the location of body wall muscle tissue in which mitochondria are visualized. B–F: representative images of mitochondria of body wall muscle in L3 larval worms at various times during starvation (0 h, 2 h, 16 h, 24 h, and 46 h). Scale bars in B–F are 15 μm. B’–F’: 4× zoomed section of image in B–F. G: surface area of individual mitochondria is plotted in fed worms (0 h of starvation) and after 22–24 h of starvation (P = 0.02, unpaired t-test, n = 3 biological replicates). H: volume of individual mitochondria is plotted after 0 h and 22–24 h of starvation (P = 0.02, unpaired t-test, n = 3 biological replicates). I: sphericity of individual mitochondria is plotted after 0 h and 22–24 h of starvation (P = 0.07, unpaired t-test, n = 3 biological replicates). In G–I mean values ± SE are shown. *P ≤ 0.05.
Mitochondrial DNA copy number decreases and lesions increase during starvation.
We measured mtDNA copy number during starvation in both L1 and L3 larvae. A PCR-based assay allows for quantitative assessment of the number of mitochondrial genomes (13). mtDNA copy number is normalized to nuclear DNA copy number. Nuclear DNA copy number provides an internal control for lysis efficiency and DNA integrity. Nuclear DNA copy number does not change significantly over time (data not shown), as expected, because developmentally arrested worms presumably maintain a fixed number of cells during starvation (2). Thus we conclude that changes in mtDNA copy number normalized to nuclear copy number are driven primarily by changes in the number of mitochondria or overall mitochondrial volume (mitochondrial content).
mtDNA copy number steadily decreases during starvation (Fig. 2). After 12 days of L1 starvation, the ratio of mitochondrial to nuclear DNA is reduced to 40% of that at day 1 (Fig. 2A). Mitochondrial DNA is also reduced in L3 larvae during starvation (Fig. 2B). Notably, L3 larvae have a higher mtDNA copy number than L1 larvae at day 1 of starvation, consistent with cellular growth and proliferation (62). The reduction of mtDNA over time during L3 starvation is qualitatively similar to the loss of mtDNA during L1 starvation; however, the loss of mtDNA after 12 days of starvation is not as severe in L3 larvae and is reduced to 62% of the day 1 baseline. Overall, these results suggest that mitochondrial content decreases during larval starvation.
Fig. 2.
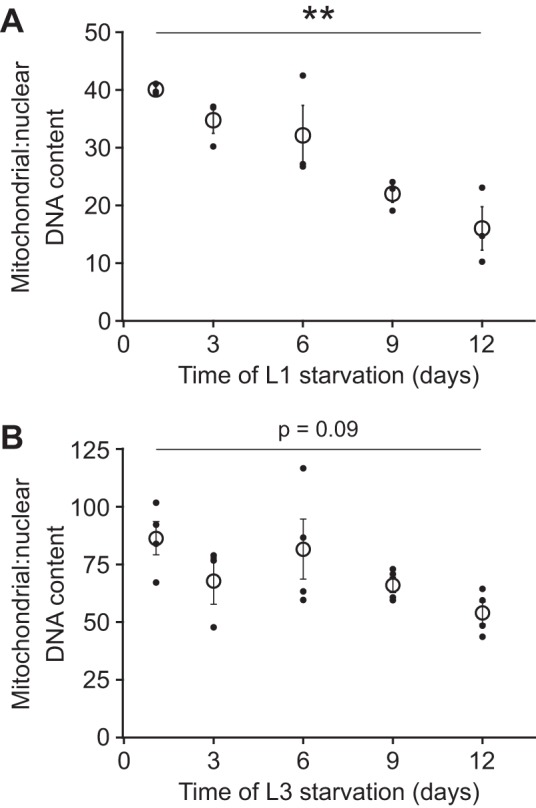
Mitochondrial DNA copy number decreases during starvation. A: mtDNA copy number relative to nuclear copy number is plotted over time during L1 starvation. There is a significant reduction in the relative concentration of mtDNA (P = 0.002, 1-way ANOVA, n = 3 biological replicates). A post hoc Tukey’s honestly significant difference (HSD) test indicated that mitochondrial:nuclear DNA content at day 1 was significantly different from day 9 (P < 0.05) and day 12 (P < 0.01). B: mtDNA copy number relative to nuclear DNA copy number is plotted over time during L3 starvation. There is a slight reduction of mtDNA content relative to nuclear DNA over time (P = 0.09, 1-way ANOVA, n = 4 biological replicates). Mitochondrial:nuclear DNA content at day 1 was significantly different from day 9 (P < 0.05, Tukey’s HSD test) and day 12 (P < 0.01, Tukey’s HSD test). Solid points are values from individual biological replicates, and open circles are means of these values. Error bars represent SE. **P ≤ 0.01.
The difference between the rates of mtDNA copy number decline in starved L1 and L3 larvae is likely due to differences in starvation sensitivity at different larval stages. L3 larvae survive starvation longer than L1 larvae (Fig. 3A) and recover to be larger and more advanced in developmental stage after 48 h of cumulative developmental time while feeding (Fig. 3, B–D). Thus the greater loss of mtDNA copy number in starved L1 larvae compared with L3 larvae is consistent with L1 larvae generally being more sensitive to starvation than L3 larvae. It is possible that L3 larvae survive starvation longer owing to a hormetic response resulting from brief L1 starvation for synchronization (see materials and methods). Alternatively, nutrient stores accumulated during 24-h feeding as L1 and L2 larvae could render L3 larvae more resistant to starvation.
We quantified the number of mtDNA lesions during L1 starvation with a PCR-based assay. A hallmark of aged mitochondria is accumulation of mtDNA damage (58). We observed a significant increase in mtDNA lesions over time (Fig. 4A). We did not detect an increase in nuclear DNA damage until 12 days of starvation (Fig. 4B). We suspect that necrosis or other cellular damage occurring during advanced starvation contributes to this late increase in nuclear DNA lesions (54). In contrast, increased mtDNA damage was observed earlier during starvation when necrosis is not evident and viability remains high. Thus mitochondria appear to be specifically susceptible to DNA damage during starvation.
Fig. 4.
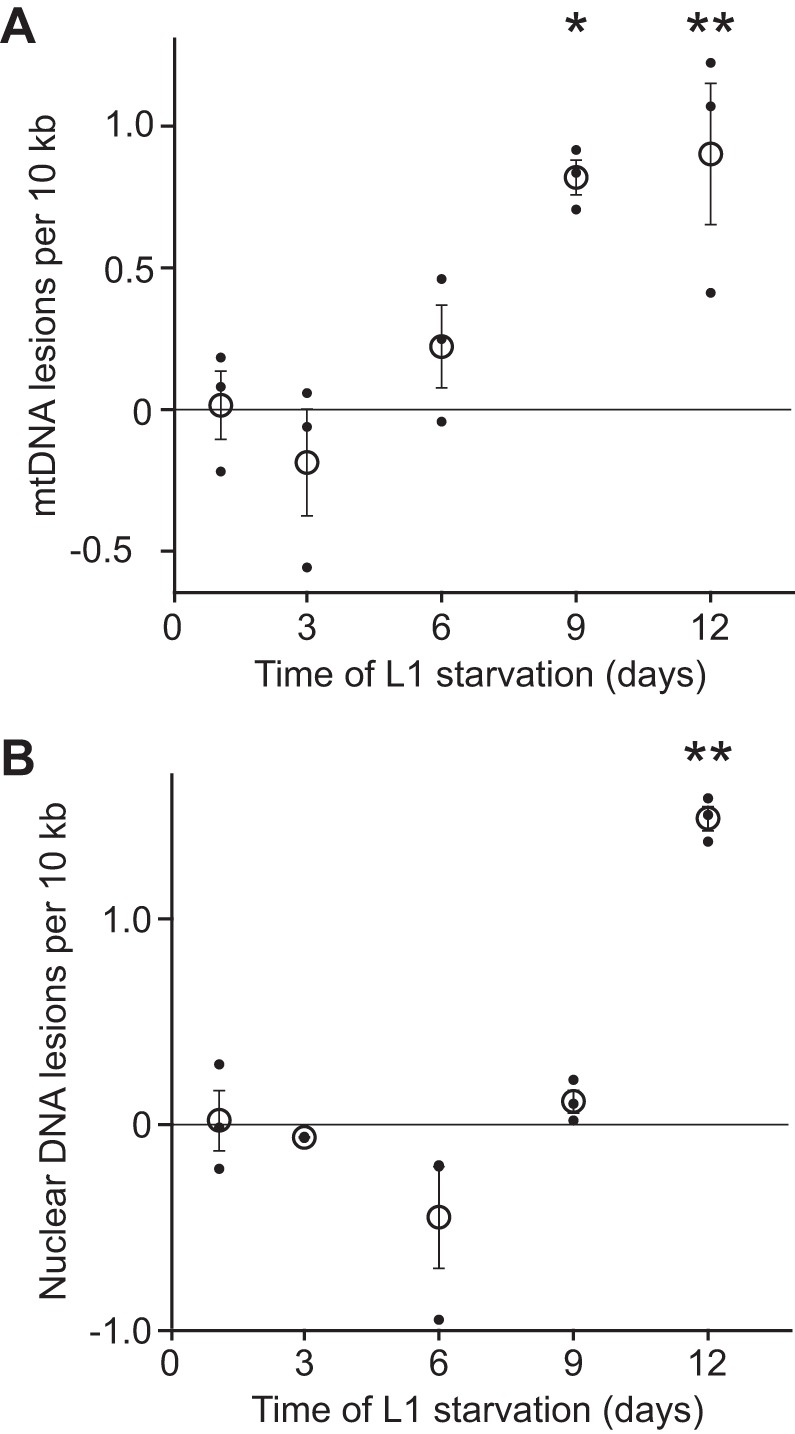
Mitochondria accumulate DNA damage during starvation. A: number of mtDNA lesions per 10 kb is plotted throughout L1 starvation. There is a significant increase in the number of lesions over time (P = 0.003, 1-way ANOVA, n = 3). At days 9 and 12 of starvation worms had a significant increase in mtDNA lesions. P < 0.05, P < 0.01 respectively [Tukey’s honestly significant difference (HSD) test, n = 3]. B: number of nuclear DNA lesions per 10 kb is plotted throughout L1 starvation. There is a significant difference in the number of lesions over time (P < 0.001, 1-way ANOVA, n = 3). However, this difference is driven only by a significant increase in nuclear DNA lesions at day 12 compared with day 1 (P < 0.01, Tukey’s HSD test, n = 3). Solid points are values from individual experiments, and open circles are means of these values. Error bars represent SE. *P ≤ 0.05; **P ≤ 0.01.
Mitochondria have reduced function during starvation.
Fragmented mitochondria and reduced mtDNA copy number during starvation suggest that mitochondrial function decreases. We assessed several parameters of mitochondrial function in L3 larvae. We chose to use L3 larvae for this analysis because they are larger than L1 larvae and have higher mitochondrial content and increased respiratory capacity. We measured whole animal oxygen consumption as a proxy for respiration and found that basal oxygen consumption rate (OCR) is significantly reduced in starved worms relative to fed counterparts (Fig. 5A). Basal oxygen consumption is a gross measure of mitochondrial activity. However, we were curious whether mitochondrial respiration is simply downregulated during starvation or whether the respiratory capacity of mitochondria is compromised. We used FCCP to uncouple mitochondria and measure maximal OCR, which provides a measure of total potential mitochondrial respiration. Starved worms have a lower maximal OCR than fed worms (Fig. 5B). Respiratory capacity continues to decrease throughout starvation, consistent with fewer and increasingly fragmented mitochondria. Alternatively, it is possible that the decrease in respiratory capacity results from substrate limitation rather than mitochondrial dysfunction; clearly, substrate availability is expected to decrease during starvation. Similar to maximal oxygen consumption, spare capacity tends to decrease over time during starvation (Fig. 5C). ATP-linked respiration, a specific measure of the contribution of respiration to ATP production, is also reduced during starvation (Fig. 5D). Consistent with these observations, ATP levels also decrease during extended starvation (Fig. 5E). We did not detect measurable levels of proton leak during L3 starvation, suggesting a negligible effect on basal metabolic rate. Together these results show that whole animal mitochondrial function is reduced during starvation. Decreased respiration is likely due to a combination of reduced mitochondrial content, increased fragmentation, and decreased substrate availability.
Autophagy is required for starvation resistance.
Autophagy is essential for cells to recycle material for energy during nutrient deprivation (40). It has been proposed that either too much or too little autophagy will limit starvation survival, suggesting that autophagy mediates a tradeoff between starvation survival and recovery (21). That is, by liberating energy, autophagy is thought to promote survival but at the cost of recovery rate due to loss of cellular material. We used pharmacological and genetic methods to test the involvement of autophagy in starvation resistance. The drug 3-methyladenine (3-MA) is a potent inhibitor of autophagy with demonstrated efficacy in C. elegans (5, 57). Worms exposed to increasing concentrations of 3-MA were increasingly short-lived during L1 larval starvation (Fig. 6A). Concentrations of 3-MA as low as 0.5 mM significantly reduced starvation survival (Fig. 6A, P = 0.05). 3-MA (50 mM) provided the most significant reduction in survival by reducing median survival by 73%. However, such high concentrations of the drug may also have off-target effects.
Worms are developmentally delayed when recovering from extended L1 starvation, with the degree of delay depending on the duration of starvation (16, 18, 32, 53). Thus size after 48 h of recovery is a second phenotype by which we assess starvation resistance. In addition to reducing starvation survival, worms exposed to 3-MA were smaller after 48 h of recovery, with a strong dose-dependent effect (Fig. 6B). Notably, no concentration of 3-MA that we tested provided a significant increase of starvation survival or recovery rate. This aspect of our results is inconsistent with an autophagy-mediated tradeoff between starvation survival and recovery rate. Nonetheless, our results clearly demonstrate that autophagy contributes to starvation resistance.
We complemented pharmacological analysis of autophagy with genetic analysis. The genes atg-18 and unc-51 are involved in autophagy in C. elegans (5, 28, 42). atg-18 encodes a WD40 repeat-containing protein that is homologous to the autophagy gene Atg18 in budding yeast (35, 41, 42). unc-51 encodes a serine/threonine kinase homologous to the yeast autophagy protein Atg1 (39, 41, 48). unc-51 is required for initiation of autophagy, and atg-18 is required for vesicle retrieval and recycling (40). The genes pink-1 (PTEN-induced putative kinase) and pdr-1 (Parkin) are specifically involved in mitophagy to degrade damaged mitochondria. We also analyzed drp-1 (dynamin-related protein) involved in mitochondrial fission, and eat-3/Opa1, involved in mitochondrial fusion (20, 30). Thus phenotypic analysis of mutations affecting these genes should allow us to differentiate contributions to starvation resistance of general autophagy (atg-18 and unc-51), selective mitophagy of damaged mitochondria (pink-1 and pdr-1), and mitochondrial fission (drp-1) and fusion (eat-3) processes.
We measured L1 starvation survival and recovery of mutants for each of these genes. atg-18 mutants were dramatically short-lived during starvation (Fig. 6C, P = 7.4E-6), and unc-51 mutants had a modest but significant reduction in starvation survival (P = 0.02). These results are consistent with the limited survival of worms exposed to 3-MA during starvation (Fig. 6A) and the reported role of autophagy in starvation survival (21). drp-1, eat-3, and pdr-1 each did not significantly impact starvation survival (Fig. 6C, P = 0.18, 0.26, 0.28, respectively). Surprisingly, pink-1 mutants were long-lived during L1 larval starvation (Fig. 6C, P = 0.001). Overall, these results suggest that autophagy, but not mitochondrial fission and fusion or damage-induced mitophagy, promotes starvation survival.
We also measured size after 48 h of recovery from starvation in the same panel of mutants. The short-lived atg-18 mutant recovers poorly from short periods of starvation (Fig. 6D, 2-way ANOVA genotype comparison P = 2.3E-5, interaction term P = 0.06), further corroborating pharmacological analysis with 3-MA. unc-51 mutants have generally stunted growth (Fig. 6D, 2-way ANOVA, genotype comparison P = 1.9E-14), thereby complicating interpretation of the effect of starvation on growth rate. Notably, pink-1 mutant worms, which we found to be long-lived during starvation (Fig. 6C), also displayed improved recovery from starvation (Fig. 6D), further suggesting that they are starvation resistant. pdr-1 mutants were not significantly different from wild-type (WT) worms in size after recovery from starvation (2-way ANOVA, genotype comparison P = 0.15, interaction term P = 0.85). drp-1 and eat-3 mutants had reduced growth rates with and without starvation (2-way ANOVA genotype comparison P = 0.0002, 9.5E-5, respectively) with no detectable interaction between genotype and condition (2-way ANOVA, P = 0.31, 0.67, respectively), consistent with no detectable effect of these mutations on starvation resistance (Fig. 6C). These results suggest that disrupting autophagy compromises recovery from starvation and that perturbation of mitochondrial fission and fusion dynamics impedes growth generally but not specifically in response to starvation.
Autophagy is required for mtDNA copy number reduction during starvation.
We hypothesized that the reduction of mitochondria during starvation is due at least in part to autophagic degradation. To test this hypothesis, we measured the ratio of mitochondrial:nuclear DNA copy number during starvation in mutants affecting autophagy, selective mitophagy, and mitochondrial fission and fusion. After 12 days of starvation, drp-1, eat-3, pink-1, and pdr-1 mutations each did not significantly impact mtDNA content (Fig. 7A). However, unc-51 mutants retained significantly more mtDNA relative to nuclear DNA (P = 0.03). Because atg-18 mutants did not survive until day 12 of L1 starvation, we measured mtDNA copy number at day 6. Similar to unc-51 mutants, atg-18 mutants had significantly higher mtDNA copy number after 6 days of starvation (P = 0.03, Fig. 7B). We confirmed these results by measuring ratios of mitochondrial:nuclear DNA throughout starvation (Fig. 7C). Both atg-18 and unc-51 mutants retained higher levels of mitochondrial DNA compared with WT controls. These results suggest that nonselective autophagy contributes to reduction of mtDNA content during starvation.
Fig. 7.

Autophagy is required for reduction of mtDNA copy number during starvation. A: ratio of mitochondrial to nuclear DNA copy number at day 12 of L1 starvation is plotted for a panel of mutants and normalized as a percentage of wild-type (WT). unc-51 mutants had increased mtDNA copy number (P = 0.03, unpaired t-test, n = 3). B: ratio of mitochondrial:nuclear DNA copy number at day 6 of L1 starvation of atg-18 mutants is shown as percentage of WT. atg-18 mutants had elevated mtDNA copy number (P = 0.03, unpaired t-test, n = 3). C: mtDNA copy number relative to nuclear copy number is plotted over time during L1 starvation for WT, atg-18 mutants, and unc-51 mutants. There is a significant impact of genotype in both cases (atg-18 P = 0.01, 2-way ANOVA, and unc-51 P = 0.002, 2-way ANOVA, n = 3). D: basal oxygen consumption rate (OCR) is plotted for L1 stage WT and unc-51 mutant worms in fed and 1-day-starved conditions. There is a significant interaction between genotype and nutrient condition (P = 0.01, 2-way ANOVA interaction term, n = 2 or 3). Graphs in A–D depict mean values from biological replicates, and error bars represent SE. *P ≤ 0.05; **P ≤ 0.01.
We measured whole animal basal oxygen consumption to determine how mitochondrial function is affected in autophagy mutants. Because atg-18 mutants are generally sick, we only measured respiration in unc-51 mutants. Despite being small and difficult to measure, we used L1 larvae to avoid complications from delayed development of unc-51 mutant larvae. Fed WT L1 larvae had higher basal OCR compared with unc-51 mutants (Fig. 7D, P = 0.09, unpaired t-test, n = 2). This result is consistent with lower substrate availability and slow growth and limited movement of unc-51 mutants (48). However, OCR was not affected by genotype during starvation (P = 0.79, unpaired t-test, n = 3). We reasoned that, because unc-51 mutant worms retained higher mitochondrial content during starvation (Fig. 7, A and C), these results may be due to them having less of a reduction in respiration in response to starvation. Indeed, basal OCR in starved WT worms is reduced to 12% of that of fed worms, whereas basal OCR in starved unc-51 mutant worms was 26% that of fed worms. This difference in the relative reduction of OCR is statistically significant (Fig. 7D, P = 0.01, 2-way ANOVA), consistent with retention of mitochondria during starvation in the unc-51 mutant. Alternatively, the mutant worms may have reached a physiological minimum for respiration, making the relative decline less relevant. However, it is unclear what would define such a minima other than viability itself or what would prevent worms from crossing such a threshold into inviability. We conclude that autophagy contributes to reduction of both mitochondrial content and function during larval starvation.
DISCUSSION
Our primary objective was to characterize effects of larval starvation on mitochondrial physiology. We document a variety of effects that are qualitatively similar to the effects of aging in fed adults. Furthermore, we show that autophagy promotes starvation resistance without an apparent autophagy-mediated tradeoff between starvation survival and growth rate upon recovery from starvation. In addition, we provide evidence that nonselective autophagy in particular promotes reduction of mitochondrial content and function during starvation.
Mitochondria exhibit aging phenotypes during starvation-induced larval arrest.
We were motivated to determine whether mitochondria exhibit signs of aging during larval arrest in C. elegans. We confirm the report of mitochondrial fragmentation during larval starvation with quantitative analysis of mitochondria in the body wall muscle of arrested L3 larvae (Fig. 1). Furthermore, we found that mitochondria show additional aging phenotypes during larval starvation. In addition to fragmentation of the mitochondrial network, there is an overall reduction in copy number of mtDNA, an increase in mtDNA damage, and reduction in respiratory capacity. Reduction of mitochondrial content, detected as decreased mtDNA copy number, likely contributes to reduction of whole animal respiratory capacity, but OCR was reduced earlier and to a greater degree than reduction of mitochondrial copy number alone can account for (Figs. 2 and 5). We therefore conclude that there are likely many parameters, beyond just the copy number of mitochondrial genomes, that influence mitochondrial function. Together these results confirm and extend on the reported age-associated phenotypes that occur during larval arrest (53), with an emphasis on mitochondrial physiology. Reversal of several age-associated phenotypes has been reported to occur in L1 larvae during recovery from L1 arrest (53), but future work is required to determine the extent to which the mitochondrial phenotypes reported here are reversed by feeding. Together these studies demonstrate the value of using starvation-induced larval arrest as a developmental model for aging (2).
Beyond induction of autophagy, the mechanistic causes of the mitochondrial phenotypes we describe remain unclear. Nutrient deprivation is itself a significant stress, with an apparent impact on the integrity of macromolecules (15). Thus development of aging-associated phenotypes simply from the passing of time is difficult to discern from consequences of starvation. Extensive morphological remodeling of mitochondria during starvation is consistent with their central role in cellular energetics. However, it is surprising to see accumulation of mtDNA damage relatively early in starvation, given that nuclear DNA does not accumulate detectable damage until later stages of starvation as lethality sets in (Fig. 4). It is possible that increased reactive oxygen species (ROS) levels could lead to such a decline in mitochondrial quality, or vice versa. ROS have been shown to increase during starvation (53). ROS also activate autophagy during starvation (52, 55). Thus it is possible that ROS production provides a common mechanism to explain increased mtDNA damage, reduced mitochondrial function, and the induction of autophagy in starved worms. However, the source of elevated ROS during starvation is unknown.
Autophagy reduces mitochondrial content during starvation.
Autophagy promotes starvation resistance. We used pharmacological and genetic approaches to show that worms deficient for autophagy are short-lived during starvation and recover poorly upon feeding (Fig. 6). Autophagy is particularly increased in intestine, muscle, and neurons during starvation in C. elegans (9). The intestine is a primary energy storage site, muscles have high mitochondrial content, and neurons have high energetic demands during starvation (45). We therefore speculate that autophagic recycling of organelles, membranes, and proteins for energy most likely explains its contribution to starvation resistance. Mitochondria represent a significant amount of biomass for possible conversion to energy during nutrient stress, and their fragmentation may reflect breakdown for this purpose. It remains to be determined whether other organelles are similarly affected during starvation.
Disruption of autophagy does not uncouple starvation survival and recovery rate. Reduced starvation survival typically correlates with limited growth during recovery although uncoupling of the two phenotypes has been reported (53). It was possible that disrupting autophagy could uncouple these phenotypes. Hypothetically, worms with blocked autophagy could recover more rapidly than controls if internal nutrient stores and organelles had not been consumed during starvation. However, this does not appear to be the case (Fig. 6). Rather, the ability to survive and recover from starvation appears to be closely coupled even with disruption of autophagy, suggesting that blocking autophagy causes an overall reduction in starvation resistance. It has been proposed that autophagy is tightly regulated such that either too much or too little autophagy during starvation reduces survival (21). We did not attempt to increase autophagy during starvation, but we did not observe any significant increase in survival from blocking autophagy to varying degrees (Fig. 6). However, the increased starvation resistance of pink-1 mutants is surprising. Intriguingly, the pdr-1/Parkin mutant displayed a trend toward increased survival and recovery, similar to pink-1, but this was not statistically significant. Interestingly, pink-1 and pdr-1 mutants are reported to be sensitive to other stresses and toxin exposures (36, 49). It is possible that the relative rates of selective and nonselective autophagy vary by context, depending on energetic demands and the amount or type of damage to different organelles. Nonetheless, the correlation between starvation survival and recovery rate held even with the starvation-resistant phenotype of pink-1 mutants.
Nonselective autophagy, as opposed to selective mitophagy, regulates mitochondrial content during starvation. Selective mitophagy (type 2 and 3) removes mtDNA with lesions after UV-induced mitochondrial damage in a manner that requires pink-1, drp-1, and eat-3 (5, 33, 41). Here, we show that general autophagy is required for the reduction of mtDNA during starvation, as unc-51 and atg-18 mutants each retain more mtDNA during starvation than WT (Fig. 7, A–C). unc-51 mutant larvae also had less of a reduction in basal respiration in response to starvation than WT, consistent with greater retention of mitochondria. In general, fusion of the mitochondrial network provides a mechanism to exclude mitochondria from autophagosomes and prevent degradation (11, 51). We report that mutants for mitochondrial fission (drp-1) and fusion (eat-3) did not significantly impact mtDNA levels during starvation. This result suggests that regulated mitochondrial dynamics are dispensable for this autophagic process. Similarly, mutants disrupting damage-induced mitophagy (pink-1 and pdr-1/Parkin) did not significantly affect mtDNA levels during starvation. These observations are consistent with nonselective (type 1) mitophagy, the autophagosomal degradation of mitochondria independent of pink-1 and Parkin (33), reducing mitochondrial content and whole animal respiration. This conclusion is surprising given clear evidence of mitochondrial damage and compromised function. However, the pink-1 and Parkin selective mitophagy pathway responds primarily to mitochondrial membrane potential, which we did not measure (33, 63). Perhaps starved larvae somehow maintain membrane potential. Alternatively, selective mitophagy may be inhibited during starvation to avoid degrading too many mitochondria, which would impact the ability to grow upon recovery.
Our results demonstrate pervasive effects of larval starvation on mitochondrial physiology with organismal consequences, and they suggest that autophagy contributes to these effects. Nonselective autophagy also regulates sequestration of mitochondria to vacuoles during starvation in yeast (26). Similar degradation of mitochondria in starved hepatocytes co-occurs with fragmentation of the mitochondrial network (25). These results along with ours suggest a conserved role of nonselective autophagy during starvation among eukaryotes.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants R01GM117408 (L. R. Baugh), P42ES010356, R01ES028218 (J. N. Meyer), and R35 MIRA GM118049 (D. R. Sherwood). Some strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by the NIH Office of Research Infrastructure Programs (P40OD010440).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.D.H., T.C.L., D.R.S., J.N.M., and L.R.B. conceived and designed research; J.D.H., T.C.L., C.S., and D.F.M. performed experiments; J.D.H., T.C.L., D.F.M., J.N.M., and L.R.B. analyzed data; J.D.H., T.C.L., D.R.S., J.N.M., and L.R.B. interpreted results of experiments; J.D.H. and T.C.L. prepared figures; J.D.H. drafted manuscript; J.D.H., T.C.L., D.F.M., D.R.S., J.N.M., and L.R.B. edited and revised manuscript; J.D.H., T.C.L., C.S., D.F.M., D.R.S., J.N.M., and L.R.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Adam Schindler for advice on L3 arrest methods. We thank Ian Ryde and Tony Luz for assistance with mtDNA analysis and Seahorse methods.
REFERENCES
- 1.Artyukhin AB, Schroeder FC, Avery L. Density dependence in Caenorhabditis larval starvation. Sci Rep 3: 2777, 2013. doi: 10.1038/srep02777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baugh LR. To grow or not to grow: nutritional control of development during Caenorhabditis elegans L1 arrest. Genetics 194: 539–555, 2013. doi: 10.1534/genetics.113.150847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baugh LR, Demodena J, Sternberg PW. RNA Pol II accumulates at promoters of growth genes during developmental arrest. Science 324: 92–94, 2009. doi: 10.1126/science.1169628. [DOI] [PubMed] [Google Scholar]
- 4.Baugh LR, Sternberg PW. DAF-16/FOXO regulates transcription of cki-1/Cip/Kip and repression of lin-4 during C. elegans L1 arrest. Curr Biol 16: 780–785, 2006. doi: 10.1016/j.cub.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 5.Bess AS, Crocker TL, Ryde IT, Meyer JN. Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans. Nucleic Acids Res 40: 7916–7931, 2012. doi: 10.1093/nar/gks532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burnell AM, Houthoofd K, O’Hanlon K, Vanfleteren JR. Alternate metabolism during the dauer stage of the nematode Caenorhabditis elegans. Exp Gerontol 40: 850–856, 2005. doi: 10.1016/j.exger.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Cassada RC, Russell RL. The dauerlarva, a post-embryonic developmental variant of the nematode Caenorhabditis elegans. Dev Biol 46: 326–342, 1975. doi: 10.1016/0012-1606(75)90109-8. [DOI] [PubMed] [Google Scholar]
- 8.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell 125: 1241–1252, 2006. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 9.Chapin HC, Okada M, Merz AJ, Miller DL. Tissue-specific autophagy responses to aging and stress in C. elegans. Aging (Albany NY) 7: 419–434, 2015. doi: 10.18632/aging.100765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chistiakov DA, Sobenin IA, Revin VV, Orekhov AN, Bobryshev YV. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed Res Int 2014: 238463, 2014. doi: 10.1155/2014/238463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: 589–598, 2011. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gomes LC, Odedra D, Dikic I, Pohl C. Autophagy and modular restructuring of metabolism control germline tumor differentiation and proliferation in C. elegans. Autophagy 12: 529–546, 2016. doi: 10.1080/15548627.2015.1136771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez-Hunt CP, Rooney JP, Ryde IT, Anbalagan C, Joglekar R, Meyer JN. PCR-based analysis of mitochondrial DNA copy number, mitochondrial DNA damage, and nuclear DNA damage. Curr Protoc Toxicol 67: 20.11.1–20.11.25, 2016. doi: 10.1002/0471140856.tx2011s67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet 4: e24, 2008. doi: 10.1371/journal.pgen.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hibshman JD, Doan AE, Moore BT, Kaplan RE, Hung A, Webster AK, Bhatt DP, Chitrakar R, Hirschey MD, Baugh LR. daf-16/FoxO promotes gluconeogenesis and trehalose synthesis during starvation to support survival. eLife 6: e30057, 2017. doi: 10.7554/eLife.30057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hibshman JD, Hung A, Baugh LR. Maternal diet and insulin-like signaling control intergenerational plasticity of progeny size and starvation resistance. PLoS Genet 12: e1006396, 2016. [Erratum in PLoS Genet 14: e1007639, 2018.] doi: 10.1371/journal.pgen.1006396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jia K, Hart AC, Levine B. Autophagy genes protect against disease caused by polyglutamine expansion proteins in Caenorhabditis elegans. Autophagy 3: 21–25, 2007. doi: 10.4161/auto.3528. [DOI] [PubMed] [Google Scholar]
- 18.Jobson MA, Jordan JM, Sandrof MA, Hibshman JD, Lennox AL, Baugh LR. Transgenerational effects of early life starvation on growth, reproduction, and stress resistance in Caenorhabditis elegans. Genetics 201: 201–212, 2015. doi: 10.1534/genetics.115.178699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson TE, Mitchell DH, Kline S, Kemal R, Foy J. Arresting development arrests aging in the nematode Caenorhabditis elegans. Mech Ageing Dev 28: 23–40, 1984. doi: 10.1016/0047-6374(84)90150-7. [DOI] [PubMed] [Google Scholar]
- 20.Kanazawa T, Zappaterra MD, Hasegawa A, Wright AP, Newman-Smith ED, Buttle KF, McDonald K, Mannella CA, van der Bliek AM. The C. elegans Opa1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet 4: e1000022, 2008. doi: 10.1371/journal.pgen.1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang C, You YJ, Avery L. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev 21: 2161–2171, 2007. doi: 10.1101/gad.1573107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaplan REW, Chen Y, Moore BT, Jordan JM, Maxwell CS, Schindler AJ, Baugh LR. dbl-1/TGF-β and daf-12/NHR signaling mediate cell-nonautonomous effects of daf-16/FOXO on starvation-induced developmental arrest. PLoS Genet 11: e1005731, 2015. doi: 10.1371/journal.pgen.1005731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kauppila TES, Kauppila JHK, Larsson NG. Mammalian mitochondria and aging: an update. Cell Metab 25: 57–71, 2017. doi: 10.1016/j.cmet.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 24.Khan LA, Yamanaka T, Nukina N. Genetic impairment of autophagy intensifies expanded polyglutamine toxicity in Caenorhabditis elegans. Biochem Biophys Res Commun 368: 729–735, 2008. doi: 10.1016/j.bbrc.2008.01.150. [DOI] [PubMed] [Google Scholar]
- 25.Kim I, Lemasters JJ. Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. Am J Physiol Cell Physiol 300: C308–C317, 2011. doi: 10.1152/ajpcell.00056.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kissová I, Salin B, Schaeffer J, Bhatia S, Manon S, Camougrand N. Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy 3: 329–336, 2007. doi: 10.4161/auto.4034. [DOI] [PubMed] [Google Scholar]
- 27.Klass MR. Aging in the nematode Caenorhabditis elegans: major biological and environmental factors influencing life span. Mech Ageing Dev 6: 413–429, 1977. doi: 10.1016/0047-6374(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 28.Kovacs AL, Zhang H. Role of autophagy in Caenorhabditis elegans. FEBS Lett 584: 1335–1341, 2010. doi: 10.1016/j.febslet.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 40: 280–293, 2010. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell 4: 815–826, 1999. doi: 10.1016/S1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- 31.Lapierre LR, Meléndez A, Hansen M. Autophagy links lipid metabolism to longevity in C. elegans. Autophagy 8: 144–146, 2012. doi: 10.4161/auto.8.1.18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee I, Hendrix A, Kim J, Yoshimoto J, You YJ. Metabolic rate regulates L1 longevity in C. elegans. PLoS One 7: e44720, 2012. [Erratum in PLoS One 8: doi: 10.1371/annotation/c69de5f4-dd02-4f92-9fc7-9a6a660a075e, 2013.] doi:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lemasters JJ. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol 2: 749–754, 2014. doi: 10.1016/j.redox.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lemire B. Mitochondrial genetics. WormBook 1–10, 2005. doi: 10.1093/bmb/ldt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu Q, Yang P, Huang X, Hu W, Guo B, Wu F, Lin L, Kovács AL, Yu L, Zhang H. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell 21: 343–357, 2011. doi: 10.1016/j.devcel.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 36.Luz AL, Godebo TR, Smith LL, Leuthner TC, Maurer LL, Meyer JN. Deficiencies in mitochondrial dynamics sensitize Caenorhabditis elegans to arsenite and other mitochondrial toxicants by reducing mitochondrial adaptability. Toxicology 387: 81–94, 2017. doi: 10.1016/j.tox.2017.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luz AL, Lagido C, Hirschey MD, Meyer JN. In vivo determination of mitochondrial function using luciferase-expressing Caenorhabditis elegans: contribution of oxidative phosphorylation, glycolysis, and fatty acid oxidation to toxicant-induced dysfunction. Curr Protoc Toxicol 69: 1–22, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luz AL, Smith LL, Rooney JP, Meyer JN. Seahorse Xfe24 extracellular flux analyzer-based analysis of cellular respiration in Caenorhabditis elegans. Curr Protoc Toxicol 66: 1–15, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 192: 245–250, 1997. doi: 10.1016/S0378-1119(97)00084-X. [DOI] [PubMed] [Google Scholar]
- 40.Meléndez A, Levine B. Autophagy in C. elegans. WormBook 1–26, 2009. doi: 10.1895/wormbook.1.147.1. [DOI] [PubMed] [Google Scholar]
- 41.Meléndez A, Tallóczy Z, Seaman M, Eskelinen E-L, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301: 1387–1391, 2003. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- 42.Minnerly J, Zhang J, Parker T, Kaul T, Jia K. The cell non-autonomous function of ATG-18 is essential for neuroendocrine regulation of Caenorhabditis elegans lifespan. PLoS Genet 13: e1006764, 2017. [Erratum in PLoS Genet 13: e1006764, 2017.] doi: 10.1371/journal.pgen.1006764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15: 1101–1111, 2004. doi: 10.1091/mbc.e03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore BT, Jordan JM, Baugh LR. WormSizer: high-throughput analysis of nematode size and shape. PLoS One 8: e57142, 2013. doi: 10.1371/journal.pone.0057142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mullaney BC, Ashrafi K. C. elegans fat storage and metabolic regulation. Biochim Biophys Acta 1791: 474–478, 2009. doi: 10.1016/j.bbalip.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell 148: 1145–1159, 2012. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Riordan VB, Burnell AM. Intermediary metabolism in the dauer larva of the nematode Caenorhabditis elegans-1. Glycolysis, gluconeogenesis, oxidative phosphorylation and the tricarboxylic acid cycle. Comp Biochem Physiol B Comp Biochem 92: 233–238, 1989. doi: 10.1016/0305-0491(89)90271-X. [DOI] [Google Scholar]
- 48.Ogura K, Wicky C, Magnenat L, Tobler H, Mori I, Müller F, Ohshima Y. Caenorhabditis elegans unc-51 gene required for axonal elongation encodes a novel serine/threonine kinase. Genes Dev 8: 2389–2400, 1994. doi: 10.1101/gad.8.20.2389. [DOI] [PubMed] [Google Scholar]
- 49.Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521: 525–528, 2015. doi: 10.1038/nature14300. [DOI] [PubMed] [Google Scholar]
- 50.Porter C, Hurren NM, Cotter MV, Bhattarai N, Reidy PT, Dillon EL, Durham WJ, Tuvdendorj D, Sheffield-Moore M, Volpi E, Sidossis LS, Rasmussen BB, Børsheim E. Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am J Physiol Endocrinol Metab 309: E224–E232, 2015. doi: 10.1152/ajpendo.00125.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 108: 10190–10195, 2011. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodríguez-Vargas JM, Ruiz-Magaña MJ, Ruiz-Ruiz C, Majuelos-Melguizo J, Peralta-Leal A, Rodríguez MI, Muñoz-Gámez JA, de Almodóvar MR, Siles E, Rivas AL, Jäättela M, Oliver FJ. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res 22: 1181–1198, 2012. doi: 10.1038/cr.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roux AE, Langhans K, Huynh W, Kenyon C. Reversible age-related phenotypes induced during larval quiescence in C. elegans. Cell Metab 23: 1113–1126, 2016. doi: 10.1016/j.cmet.2016.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ 15: 105–112, 2008. doi: 10.1038/sj.cdd.4402231. [DOI] [PubMed] [Google Scholar]
- 55.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26: 1749–1760, 2007. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schindler AJ, Baugh LR, Sherwood DR. Identification of late larval stage developmental checkpoints in Caenorhabditis elegans regulated by insulin/IGF and steroid hormone signaling pathways. PLoS Genet 10: e1004426, 2014. doi: 10.1371/journal.pgen.1004426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 79: 1889–1892, 1982. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA 91: 10771–10778, 1994. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci USA 102: 5618–5623, 2005. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell 61: 654–666, 2016. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol 119: 301–311, 1992. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsang WY, Lemire BD. The role of mitochondria in the life of the nematode, Caenorhabditis elegans. Biochim Biophys Acta 1638: 91–105, 2003. [DOI] [PubMed] [Google Scholar]
- 63.Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RLA, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrané J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107: 378–383, 2010. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yen WL, Klionsky DJ. How to live long and prosper: autophagy, mitochondria, and aging. Physiology (Bethesda) 23: 248–262, 2008. doi: 10.1152/physiol.00013.2008. [DOI] [PubMed] [Google Scholar]